

Abstract
Cancer immunotherapy can result in lasting tumor regression, but predictive biomarkers of treatment response remain ill-defined. Here, we performed single-cell proteomics, transcriptomics, and genomics on matched untreated and interleukin-2 (IL-2) injected metastases from patients with melanoma. Lesions that completely regressed following intralesional IL-2 harbored increased fractions and densities of non-proliferating CD8+ T cells lacking expression of PD-1, LAG-3 and TIM-3 (PD-1−LAG-3−TIM-3−). Untreated lesions from patients who subsequently responded with complete eradication of all tumor cells in all injected lesions (individuals referred to herein as “extreme responders”) were characterized by proliferating CD8+ T cells with an exhausted phenotype (PD-1+LAG-3+TIM-3+), stromal B-cell aggregates, and expression of IFNγ and IL-2 response genes. Loss of membranous MHC class I expression in tumor cells of untreated lesions was associated with resistance to IL-2 therapy. We validated this finding in an independent cohort of metastatic melanoma patients treated with intralesional or systemic IL-2. Our study suggests that intact tumor cell antigen presentation is required for melanoma response to IL-2 and describes a multi-dimensional and spatial approach to develop immuno-oncology biomarker hypotheses using routinely collected clinical biospecimens.
Keywords: Immunotherapy, biomarker, single-cell proteomics, melanoma, antigen presentation
INTRODUCTION
Up to 20% of patients with metastatic cutaneous melanoma present with “in-transit” metastasis, a form of locoregional recurrence (1). Therapeutic options for in-transit melanoma include surgical excision, isolated limb perfusion, radiation, systemic therapies, and intralesional therapies, including interleukin-2 (IL-2), which was the first FDA-approved immunotherapeutic agent (2, 3). Today, IL-2 is regaining popularity as an immuno-oncology agent as a number of pharmaceutical companies have IL-2 candidates in clinical trials for a variety of solid cancers (4). Compared to systemic IL-2 administration, intralesional IL-2 injection reduces systemic toxicity while maximizing intratumoral IL-2 concentration. Prior studies have reported that 41–96% of injected lesions show a complete response to IL-2 (3, 5). Although responding lesions have been shown to harbor increased densities of CD8+ T cells (6, 7), more detailed molecular and cellular effects of intralesional IL-2 and biomarkers of response are unknown. In this study, we used single-cell proteomics and bulk transcriptomics and genomics to identify changes in the tumor microenvironment and biomarkers of response associated with intralesional IL-2 therapy for patients with melanoma while describing a multi-dimensional approach for biomarker hypothesis development.
Materials and Methods
Patients and tissue.
This study includes all patients with in-transit melanoma who presented for intralesional IL-2 therapy at Memorial Sloan Kettering Cancer Center (MSKCC) from 2015–2017 (n=7) and who met the criteria of having pretreatment (untreated) and IL2-injected tissue and subsequent IL2-response data at the level of each injected lesion (Supplementary Table S1). All patients signed statements of informed consent under protocols approved by the MSKCC Institutional Review Board, and the study was conducted in accordance with the Declaration of Helsinki. All lesions were surgically resected (Charlotte E. Ariyan) and immediately formalin-fixed and paraffin-embedded (FFPE). All biospecimens were obtained as part of routine clinical care with standard FFPE tissue processing in the MSKCC surgical pathology lab (CLIA accredited). FFPE tissue blocks were maintained in the MSKCC Department of Pathology temperature controlled storage units until use. For each specimen, adjacent tissue sections were freshly cut for hematoxylin and eosin (H&E) (1 section, 5-microns), multiplexed immunofluorescence (IF) (Cell Dive, Cytvia, Issaquah, WA) (1 section, 5-microns), NanoString (10 sections, 10-microns), MSK-IMPACT (20 sections, 5-microns), and immunohistochemistry (IHC) for MHC class I (1 section, 4-microns). Cell Dive validation IHCs were completed on 28 additional 4-micron thick tissue sections from lesion 3_2. Multiplexed IF (validation assay) was completed on one 4-micron thick tissue section. H&Es were reviewed by a board-certified Dermatopathologist (Travis J. Hollmann) for classification of treatment response as either complete response (CR) or non-CR based on the presense or absence of tumor cells, respectively.
Our validation cohort includes 19 patients with metastatic melanoma who received intralesional IL-2 or high-dose systemic IL-2 and who met the criteria of having pretreatment (untreated) tissue with no prior therapies and subsequent IL2-response data at the level of each injected lesion for intralesional IL-2 therapy and at the level of the patient for high-dose systemic IL-2 therapy. All patients signed statements of informed consent, and the study was conducted in accordance with the Declaration of Helsinki. All lesions were surgically resected and immediately FFPE using standard FFPE tissue processing. “CR” denotes full regression of the tumor following IL-2 therapy, whereas “non-CR” denotes remaining tumor cells following IL-2 therapy.
Targeted RNA sequencing using NanoString.
NanoString was performed for all lesions in the initial discovery cohort with the exception of lesions 3_2 and 6_2, which were excluded due to low RNA quantity post-extraction. RNA extraction was performed after macrodissection to exclude necrotic and normal skin regions. FFPE sections were deparaffinized using the mineral oil method. Briefly, 800μL mineral oil (Thermo Fisher catalog# AC415080010) was mixed with the sections and the sample was incubated at 65°C for 10 minutes. Phases were separated by centrifugation in 360μL Buffer PKD (DNeasy Blood & Tissue Kit, QIAGEN catalog# 69504), and Proteinase K (600 mAU/mL) (DNeasy Blood & Tissue Kit, QIAGEN catalog# 69504) was added for digestion. After a three-step incubation (65°C for 45’, 80°C for 15’, 65°C for 30’) and additional centrifugation, the aqueous phase containing RNA was removed and DNase treated (DNeasy Blood & Tissue Kit, QIAGEN catalog# 69504). The RNA was then extracted using the RNeasy FFPE Kit (QIAGEN catalog #73504) according to the manufacturer’s instructions. A minimum of 100 ng of total RNA per sample was used to measure the expression of 770 immune-related genes and 20 internal reference genes (PanCancer IO 360 gene expression panel) using NanoString Technologies’ nCounter platform. Normalization using the internal reference genes was performed using nSolver. Differential expression analysis was run using the DESeq2 Bioconductor package.
Targeted DNA sequencing using MSK-IMPACT.
MSK-IMPACT was performed for all tumor-containing untreated and non-CR lesions in the initial discovery cohort with the exception of patient 6 due to tissue availability. DNA extraction was performed after macrodissection to exclude necrotic and normal skin regions. FFPE tissue was deparaffinized using heat treatment (90°C for 10’ in 480μL PBS and 20μL 10% Tween 20), centrifugation (10,000xg for 15’), and ice chill. Paraffin and supernatant were removed, and the pellet was washed with 1mL of 100% ethanol followed by an incubation overnight in 400μl of 1M NaSCN for rehydration and impurity removal. Tissues were subsequently digested with 40μl of Proteinase K (600 mAU/ml) in 360μl Buffer ATL (DNeasy Blood & Tissue Kit, QIAGEN catalog# 69504) at 55°C. DNA isolation proceeded with the DNeasy Blood & Tissue Kit (QIAGEN catalog# 69504) according to the manufacturer’s protocol modified by replacing buffer AW2 with 80% ethanol. DNA was eluted in 0.5X Buffer AE heated to 55°C. Next-generation sequencing of patient-matched tumor/normal DNA was performed in the MSKCC Integrated Genomics Operation core facility using MSK-IMPACT 468, which has been previously described (8). A genetically matched normal was used for all cases. The current analysis framework can be found at https://github.com/mskcc/roslin-variant/wiki/Roslin-Output-v2.5.
H&E staining.
H&E staining was performed for all lesions (initial discovery and validation cohorts) using the Ventana Symphony automated H&E stainer with standard clinical protocol. Tissue sections were baked for one hour at 60°C, hydrated, stained with hematoxylin (Leica catalog# 3801560), stained with bluing reagent (Leica catalog# 3802918), stained with eosin counsterstain (Leica catalog# 3801600), rinsed, dehydrated, and coverslipped.
Multiplexed immunofluorescence (Cell Dive).
Multiplexed IF (Cell Dive) was performed for all lesions in the initial discovery cohort. Multiple primary antibodies clones were evaluated for each antigen by IHC on normal human multi-tissue controls (tonsil, placenta, skin, colon, kidney, pancreas, testicle, lung, and spleen) (Supplementary Fig. S1, Supplementary Table S2). The normal human controls were processed with standard FFPE tissue processing in the MSKCC surgical pathology lab (CLIA accredited), and FFPE tissue blocks were maintained in the MSKCC Department of Pathology temperature controlled storage units until use. All patients signed statements of informed consent under protocols approved by the MSKCC Institutional Review Board, and the study was conducted in accordance with the Declaration of Helsinki. The most optimal clone was conjugated to Cy2, Cy3, or Cy5 Bis NHS Ester dyes (GE catalog# PA22000, PA13000, PA25000, respectively) using a previously published protocol (9). Each conjugated primary antibody was evaluated at three different dilutions to the unconjugated antibody. The staining protocol consists of incubating slides for 60 minutes with the conjugated primary antibody and a 3% BSA in PBS diluent at room temperature followed by 3 rounds of 5 minutes washes in 1X PBS. Each epitope was tested for stability to alkaline H2O2-based signal inactivation by exposing adjacent sections of a multi-tissue control to 0, 1, 5, and 10 cycles of alkaline H2O2 followed by staining with the antibody (9).
The multi-tissue control was included on each slide for quality control. Tissue sections were baked for one hour at 60°C, deparaffinized, hydrated, processed through a 2-step antigen retrieval process (step 1: citrate-based pH 6.0 , Vector catalog# H-3300; step 2: EDTA-based pH 8.5, Sigma catalog# T6066-100G/Biorad catalog# 161-0729/Sigma catalog# P9416) using a previously published protocol (9), and blocked overnight using normal donkey serum (Jackson ImmunoResearch catalog# 017-000-121). The tissue was then stained with DAPI for 15 minutes (Thermo Scientific catalog# D3571) and washed 3 rounds for 5 minutes with 1X PBS. A whole slide image was acquired for field of view (FOV) selection. FOVs were placed on the tumor-stroma interface, tumor center, and regressed tumor. Tissue sections then underwent 16 cycles of background imaging, staining, imaging, and signal inactivation. Images were acquired using the Cytell Cell Imaging System (Cytvia, Issaquah, WA). Image App software was used for image acquisition and registration (using DAPI), and this functionality is fully incorporated into the commercial Cell Dive product (Leica Microsystems). An acquired background image following each cycle of dye inactivation was used to subtract autofluorescence from the subsequent stain round resulting in autofluorescence removed images.
Multiplexed immunofluorescence (validation assay).
Multiplexed IF (validation assay) was performed for all lesions in the initial discovery cohort. Tissue sections were processed using the protocol described for IHC through antigen retrieval followed by 3 sequential cycles of staining (CD4: 0.7 microgram/mL, CD8: 0.125 microgram/mL, TIM-3: 0.15 microgram/mL) with each round including a 30-minute combined block and primary antibody incubation (Akoya opal antibody diluent/block catalog#ARD1001). The CD4, CD8, and TIM-3 antibodies used can be found in Supplementary Table S2. CD4 and CD8 detection was performed using 10-minute incubation with a goat anti-mouse Poly HRP secondary antibody (Invitrogen catalog# B40961). The HRP-conjugated secondary antibody polymer was detected using fluorescent tyramide signal amplification using Opal dyes 520 (CD4), 570 (CD8), 650 (TIM-3) (Akoya catalog# FP1487001KT, catalog# FP1488001KT, and catalog# FP1496001KT, respectively). The covalent tyramide reaction was followed by heat-induced stripping of the primary/secondary antibody complex using Akoya AR9 buffer (catalog# AR900250ML) and Leica Bond ER2 (90% ER2 and 10% AR9) at 100°C for 20 minutes preceding the next cycle. After 3 sequential rounds of staining, sections were stained with Hoechst (Invitrogen catalog# 33342) and mounted with ProLong Gold antifade reagent mounting medium (Invitrogen catalog# P36930). Whole slide images were acquired using Zeiss AXIO scanner. Indica Labs’ HALO Image Analysis software was used for image analysis. Necrotic regions were excluded.
Immunohistochemistry.
IHC for MHC class I was performed for all lesions (initial discovery and validation cohorts) and for the 28 markers in the multiplexed IF panel (Supplementary Table S2) for lesion 3_2 using an automated staining system (Leica Bond RX) with 3,3’ Diaminobenzidine (DAB) detection (Leica Bond Polymer Refine Detection catalog# DS9800). Tissue sections were baked for 3 hours at 62°C in vertical slide orientation with subsequent deparaffinization performed on the Leica Bond RX. Antigen retrieval was conducted for 30 minutes using Leica Bond epitope retrieval solution 2 (ER2) (EDTA, pH 9.0) (catalog# AR9640) followed by incubation of the primary antibody at previously optimized concentrations for 30 minutes (a list of the primary antibodies used can be found in Supplementary Table S2) followed by incubation of the secondary antibody (Leica Bond Polymer Refine Detection catalog# DS9800). MHC class I staining was scored for the percentage of membrane-positive tumor cells within the entire tissue section in 5% increments (0% to 100%). Slides were scored blindly by Travis J. Hollmann and Maryam Pourmaleki. IHC staining for the 28 markers for lesion 3_2 was visually inspected alongside the multiplexed IF (Cell Dive) staining for the 28 markers for lesion 3_2 by Travis J. Hollmann and Maryam Pourmaleki to ensure accuracy of the multiplexed IF method.
Multiplexed immunofluorescence analysis (Cell Dive).
Image analysis:
HALO was used for image visualization and analysis. For each FOV, images for the 28 markers and DAPI were stacked. Markers with technical issues or non-specific staining in either a single FOV or the lesion were excluded. High intensity artifacts were annotated for exclusion. The tumor-stroma interface was manually annotated for each interface FOV using the marker SOX-10, which labels the nuclei of tumor cells. Annotation coordinates were exported for downstream analysis. Nuclear segmentation parameters and thresholds were set for each lesion and optimized using 2 FOVs.
Cell loss computation:
DAPI images (first and last cycles 1 and 32) were processed with intensity normalization and histogram matching. Sum of squared differences was used to generate a pixel level bit mask image highlighting areas of cell loss/drift between images. The bit mask and cell coordinates were used to calculate a loss/drift percentage for each cell
Data processing:
Each cell was assigned a unique ID. Cells in regions with artifacts, in the 20-micron border region of each FOV, and with greater than 10% loss/drift of pixels were removed from analysis. For each interface FOV, we created a quad tree consisting of pixel coordinates of the annotated tumor interface using R-package SearchTrees v0.5.2. For each cell, we identified the nearest point on the tumor interface using k-nearest neighbor lookup on the tree and measured the distance from the cell to that point (0.293 microns per pixel conversion). Each cell falling within +/− 360-microns of the tumor interface was assigned to a 10-micron interval. Distance in microns between all unique pairs of cells in each FOV was calculated for cell neighborhood analyses. Intensity values for each marker were normalized by dividing the intensities for each marker by the value of the threshold. Intensity values below the threshold (less than 1) were flattened to 1. The log of the intensity values for each marker was divided by the width of the log intensity (97.5-percentile value) distribution across the FOV. Cells were assigned to a cell type using positive and negative combinations of cell identity markers. A method was created to reset problematic thresholds.
t-SNE analysis:
We performed dimensionality reduction on the full set of marker intensities using the Rtsne package. The normalized/transformed intensities were projected onto 2 dimensions using the t-SNE method with a perplexity of 250 and 5,000 iterations.
Statistical analysis:
We transformed fractions to log odds and used R function wilcox.test() with the default two-sided option to compute significance of the differences and effect sizes as log odds ratios (OR). We used the same method log-transformed densities, reporting effect sizes as fold changes. P-values were adjusted for multiple testing with Bonferroni adjustment.
Statistical integration of overall and intra-patient analyses:
From filtered cell fractions for overall CR versus untreated and overall non-CR versus untreated comparisons, we filtered cell fractions with at least one significant overall OR (p-adjusted<0.05). Harmonic mean p-value (HMP) was calculated for each intra-patient comparison and adjusted using Bonferroni test. The union of untreated lesion FOVs was used for patients with 2 untreated lesions (#4, #6).
Tumor MHC class I neighborhoods:
Immune cells were grouped by being in MHC-I–low, MHC-I–mixed, MHC-I–high, tumor-free, and isolated neighborhoods (30-micron radius) based on neighborhoods having a maximum of 25% MHC-I+ tumor cells, between 25–75% MHC-I+ tumor cells, a minimum of 75% MHC-I+ tumor cells, no tumor cells, or no neighbors, respectively.
Identification of B-cell aggregates.
Clusters of B cells were identified using the set of B-cell pairs with less than 30 microns between them. Starting with one pair as a cluster, all B-cell neighbors of both cells in the pair were added to the cluster, followed by neighbors of neighbors. Clusters of 20 B cells or more were labeled B-cell aggregates.
CD8+ T-cell neighborhoods.
Immune cells were grouped by being in CD8+ T-cell triple-negative (TN), single-positive (SP), double-positive (DP), or triple-positive (TP) neighborhoods using PD-1, LAG-3, and TIM-3 based on neighborhoods having exclusively one degree of CD8+ T-cell exhaustion (TN, SP, DP, or TP). All other immune cells were excluded from the analysis. Log OR of each cell fraction in groups SP, DP, TP versus TN was computed. Cell fractions were tested for monotonicity as exhaustion progresses using R package Kendall.
Data and materials availability.
All data supporting findings of this study are available online at https://zenodo.org/record/4300912#.YcUQgH3MLFR. All code and detailed computational methods for multiplexed IF and NanoString analysis are available at https://github.com/mskcc/Halo_Melanoma_IL2.
RESULTS
We identified seven patients with multiple contemporaneous in-transit melanoma metastases, of which at least one metastasis was surgically removed without prior IL-2 injection (termed untreated) and at least one metastasis had received IL-2 injections prior to resection (Fig. 1A). This cohort of matched untreated and IL2-injected lesions provided an opportunity to investigate pretreatment molecular and cellular makeup as well as IL2-associated changes within the tumor that are associated with tumor response to therapy. For each lesion, we assessed the co-expression of 28 proteins at single-cell resolution (using multiplexed immunofluorescence, multiplexed IF (9, 10), the abundance of 770 immune response–related transcripts (bulk RNA), and the presence or absence of genetic alterations in 468 cancer genes (bulk DNA) in immediately adjacent FFPE tissue sections (Fig. 1B).
Fig. 1. Multi-dimensional assessment of in-transit melanoma metastases treated with intralesional IL-2.
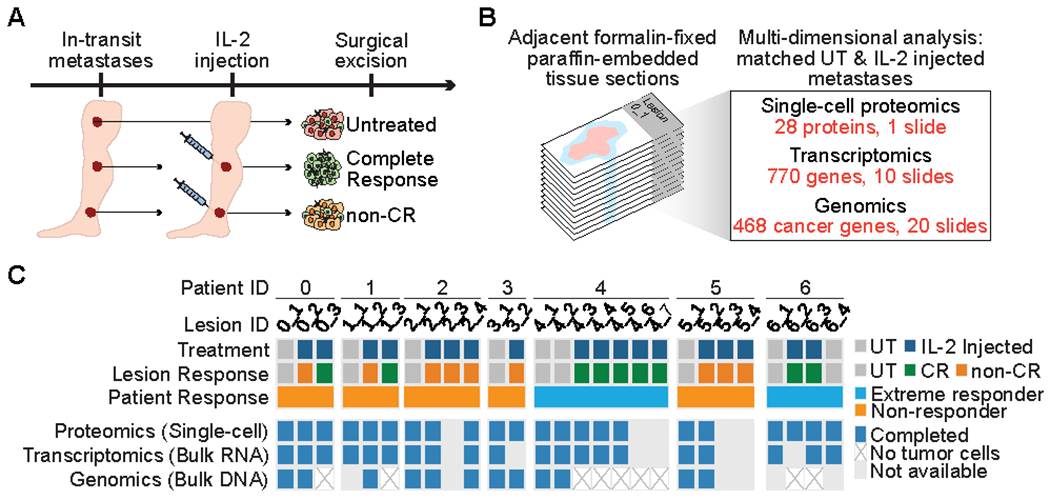
(A) Study design. Multiple cutaneous in-transit metastases were excised from each melanoma patient, including at least one untreated (UT) and one IL2-injected lesion. Treatment response for IL2-injected lesions was classified as complete response (CR) or non-CR. (B) Allocation of consecutive tumor tissue sections for molecular analyses. (C) Summary of molecular analyses completed for each lesion. “Non-responder” refers to all non-/mixed responder patients.
In total, 9/18 (50%) IL2–injected lesions responded completely to intralesional IL-2 (termed CR), defined by the absence of tumor cells on histopathological review. The other 9/18 IL IL2–injected lesions contained abundant residual tumor cells (termed non-CR; Fig. 1C, Supplementary Table S1). Two patients (termed extreme responders) experienced complete regression of all IL2–injected lesions, whereas all other patients (termed non-/mixed responders) had at least one IL2–resistant lesion. Metastases from a given patient harbored similar mutations and copy number profiles (Supplementary Fig. S2, A and B), supporting their origin from the same primary tumor clone. When compared to a larger cohort of cutaneous melanomas that were sequenced using MSK-IMPACT (8), our cohort showed a similar distribution of genetic alterations (Supplementary Fig. S2, B and C).
Our selection of markers for single-cell proteomic analysis included markers for both cell identity (e.g., tumor cell) and cell function (e.g., antigen presentation) (Supplementary Fig. S1A, Supplementary Table S2). For each marker, we evaluated specificity and sensitivity of the fluorescent dye-conjugated antibody through staining of normal human tissues (Supplementary Fig. S1, B and C), epitope stability to H2O2-based signal inactivation through repeated dye inactivation cycles (Supplementary Fig. S1D), and multiplexed IF staining specificity through staining of adjacent tumor sections with standard IHC (Supplementary Fig. S1E). Using combinations of positivity and negativity for cell identity markers, we defined 16 immune cell types, tumor cells, adipocyte/Langerhans cells, epithelial cells, and nerve cells (Supplementary Table S3).
To identify functionally distinct subpopulations of tumor and immune cells, we annotated each cell type with combinations of 15 cell-function markers, ultimately resulting in 664 distinct “cell states”. For comparisons between samples, the relative abundance of these subpopulations was expressed as cell fractions (e.g., fraction of Ki67+ CD8+ T cells over all CD8+ T cells) and cell densities (e.g., Ki67+ CD8+ T cells per millimeter squared), ultimately amounting to 685 distinct cell fractions and 664 distinct cell densities (Supplementary Fig. S3, Supplementary Table S4).
Following cell segmentation, marker thresholding, and removal of cells displaced during any staining cycle, we identified a total of 2,572,629 cells within 333 high-dimensional FOVs from 22 excised lesions in total (untreated, CR, and non-CR) of which 928,592 were immune cells (Supplementary Fig. S3B, Supplementary Table S5). Examination of multiple FOVs from each lesion allowed us to survey a much broader tumor area than typically examined using tissue microarrays (Supplementary Fig. S4A and S4B, Supplementary Table S5). We observed considerable heterogeneity in the composition of immune-cell infiltrates within FOVs from the same lesion (Supplementary Fig. S4C). At the single-cell level, the expression of some markers appeared mostly restricted to specific immune-cell types (e.g., expression of the IL-2 receptor alpha chain CD25 in CD4+ regulatory T cells), whereas other markers (e.g., Ki67) were expressed in all immune-cell types (Supplementary Fig. S3C). Within cell types, certain cell-function markers were more broadly expressed than others (e.g., expression of TIM-3 and CD27 in T cells and B cells) (Supplementary Fig. S3D–G).
To gain insight into the molecular processes that are associated with a complete lesion response to intralesional IL-2, we determined the frequency of each cell state in untreated and IL2–injected lesions (Fig. 2A, Supplementary Table S6). Compared to untreated lesions, CR lesions showed increased fractions of CD8+ T cells over all T cell subtypes and all immune cells, a well-documented effect of IL-2 (6, 7). CR lesions also showed increased fractions of PD-1−LAG-3−TIM-3− CD8+ T cells, CD4+ (helper) T cells, CD4+ regulatory T cells, B cells, and natural killer (NK) cells (Fig. 2A, Supplementary Fig. S5). CR lesions also showed the lowest fractions of TIM-3+ cells (across most immune-cell types), a finding that we validated using an independent multiplexed IF platform (Fig. 2A, Supplementary Fig. S6A and S6B). This is reminiscent of the reported association between TIM-3 and treatment resistance in other immuno-oncology contexts (11, 12). Non-CR lesions, on the other hand, showed an admixture of immune and tumor cells and no statistically significant change in CD8+ T-cell infiltration compared to untreated lesions (Fig. 2A). Interestingly, non-CR lesions did harbor increased numbers of CD25+ CD4+ regulatory T cells, suggesting that failure to upregulate the IL-2 receptor alpha (CD25), a well-documented effect of IL-2 (13), is not the cause of IL-2 resistance in these lesions. We observed no differences in immune-cell populations between multiple untreated lesions from the same patient (Supplementary Fig. S7A and 7B, Supplementary Table S7). Overall, there was considerable heterogeneity in the IL-2 response between patients (Supplementary Fig. S8, Supplementary Table S6). For example, non-CR lesions from two patients showed significantly increased fractions of PD-1−LAG-3−TIM-3− CD8+ T cells. However, at the level of the cohort, this trend did not reach statistical significance.
Fig. 2. Immune-cell states and gene-expression signatures following tumor-cell eradication by IL-2.
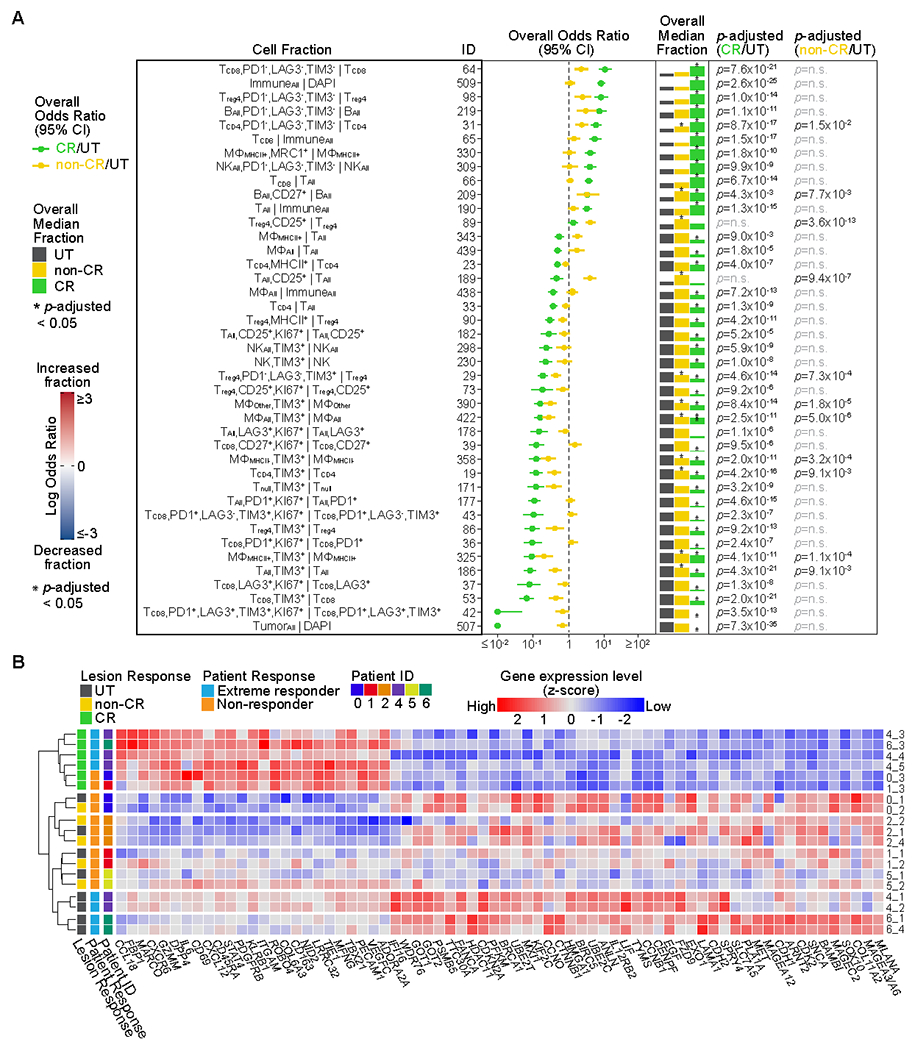
(A) Shown are cell fractions (rows) with significant changes following IL-2 injection. The forest plot shows the overall effect size (odds ratio) and 95% confidence interval (CI) of each cell fraction across all patients for complete response (CR) (n=101 fields of view, FOVs) versus untreated (n=112 FOVs) and non-CR (n=120 FOVs) versus untreated. The overall median fraction, scaled to 1 for the largest fraction, is shown for untreated, non-CR, and CR lesions. Significant results, determined using a two-sided Wilcoxon test adjusted by Bonferroni correction, are indicated with an asterisk above the median fraction with p-adjusted noted (n.s., not significant). See also Supplementary Table S6. See Supplementary Table S3 for full cell type names. (B) The heatmap indicates scaled RNA expression values for differentially expressed genes (p-adjusted<0.0001) in CR (n=6) versus untreated (n=9) and/or non-CR (n=5) versus untreated, sorted by CR versus untreated fold change. See also Supplementary Table S9.
We also analyzed the density of distinct cell states in CR, non-CR, and untreated lesions and again observed increased densities of CD8+ T cells and PD-1−LAG-3−TIM-3− CD8+ T cells, CD4+ (helper) T cells, CD4+ regulatory T cells, and B cells in CR lesions and increased densities of CD25+ CD4+ regulatory T cells in non-CR lesions, both compared to untreated lesions (Supplementary Fig. S9, Supplementary Table S8). We also observed an increase in the density of B cells and a decrease in the density of proliferating (Ki67+) macrophages in CR lesions compared to untreated lesions (Supplementary Fig. S9, Supplementary Table S8).
Our examination of immune-cell states at the single-cell level suggested that effective antitumor immunity consequent to IL-2 injection was characterized by the presence of non-proliferating T cells with a non-exhausted phenotype (PD-1−LAG-3−TIM-3−). At the level of the transcriptome, we identified 70 genes that were differentially expressed in CR lesions compared to untreated lesions (Fig. 2B, Supplementary Table S9). This “IL2–response signature” included the upregulation of 25 genes associated with T-cell activation (e.g., ADORA2A, CD69, DPP4, GZMM, STAT4, TBX21) and immune-cell localization to tumors (e.g., CCL18, MARCO, CXCR6, GZMM, DPP4, CD69). Hierarchical clustering of the differentially expressed genes grouped untreated and non-CR lesions from the same patient, suggesting IL-2 injection in the non-CR lesions failed to cause transcriptional reprogramming towards antitumor immunity. Interestingly, the clustering also grouped the 4 untreated lesions from the two patients who experienced complete regression of all injected lesions (extreme responders), indicating the presence of a transcriptional state that favors IL-2 response.
One of the key goals of our analysis was to identify molecular or cellular changes in untreated lesions that might predict a complete lesion response to subsequent IL-2 therapy. We therefore compared untreated lesions from “extreme responders” to the untreated lesions from patients for whom none or only some of the lesions had responded to IL-2 (non-/mixed responders) (Fig. 3A). Untreated lesions from extreme responders harbored a higher fraction of CD8+ T cells and a lower fraction of CD4+ T cells and MHC-II− macrophages (Fig. 3B, Supplementary Table S10). Untreated lesions from extreme responders also had higher fractions of proliferating T cell populations, CD27+ “activated” T- and NK-cell populations, B7-H3+ macrophage populations and tumor cells, and PD-L1−B7-H3−IDO-1− macrophage populations and tumor cells. Untreated lesions from extreme responders also harbored a higher fraction of PD-1+LAG-3+TIM-3+ (exhausted) CD8+ T cells and both exhausted and proliferating CD8+ T cells, suggesting they were tumor reactive. We also observed a higher density of B cells in untreated lesions from extreme responders (Supplementary Fig. S10, Supplementary Table S11).
Fig. 3. Pre-treatment tumor MHC-I expression is associated with complete tumor response to IL-2.
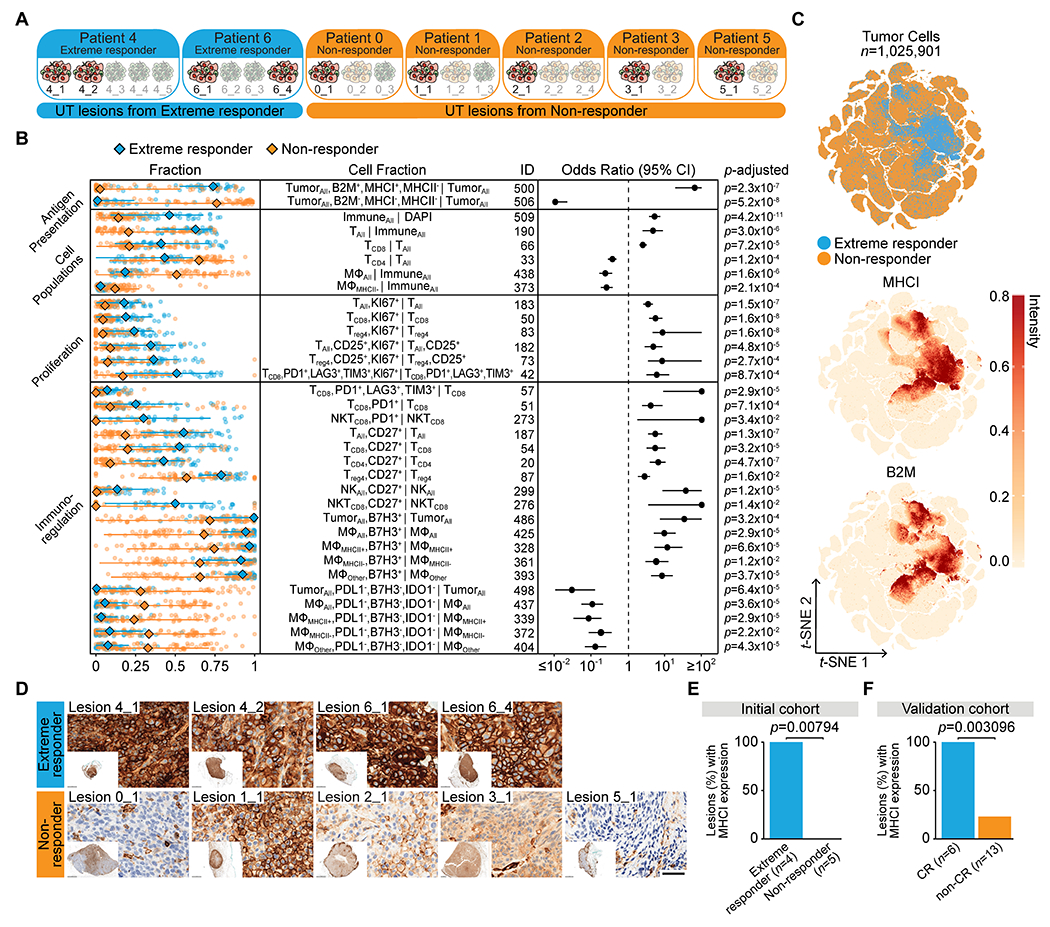
(A) Grouping of untreated (UT) lesions from extreme responder and non-/mixed responder patients (labeled “non-responder” throughout figures). (B) Shown are cell fractions (rows) with significant differences in untreated lesions from extreme responders (n=38 fields of view, FOVs) versus non-/mixed responders (n=74 FOVs). The left panel shows fractions in each FOV, with overall median, minimum, and maximum (each point represents an FOV). The forest plot shows effect size (odds ratio) and 95% confidence interval (CI) of each cell fraction with p-adjusted noted (two-sided Wilcoxon test adjusted by Bonferroni correction). See also Supplementary Table S10. See Supplementary Table S3 for full cell type names. (C) t-SNE of untreated lesion tumor cells colored by patient response and normalized intensity of MHC-I and B2M. (D) MHC-I IHC of untreated lesions (scale bar, 50-microns). (E) Bar graph showing the percent of untreated lesions with expression of membranous MHC-I in greater than 75% of tumor cells in the initial cohort (extreme responder (n=4) and non-responder (n=5)) and (F) in the validation cohort (CR, complete responder (n=6) and non-CR, non-complete responder (n=13)) based on IHC staining (Fisher’s exact test, two-sided, exact p-value noted).
One of the most consistent differences between untreated lesions from extreme responders compared to non-/mixed responders was the higher fraction of B2M+MHC-I+MHC-II− tumor cells (ID: 500) and lower fraction and lower density of B2M−MHC-I−MHC-II− tumor cells (ID:506) (Fig. 3B). In fact, there was near complete overlap between the lack of MHC-I and B2M expression on tumor cells and subsequent IL-2 response failure at the single-cell level (Fig. 3C). We confirmed this finding by IHC staining for MHC-I (Fig. 3D). All untreated lesions from extreme responders exhibited membranous MHC-I positivity in at least 75% of tumor cells, whereas all untreated lesions from non-/mixed responders exhibited membranous MHC-I positivity (if any) in less than 75% of tumor cells (Fig. 3E). We also found higher fractions of B2M+MHC-I+MHC-II+ macrophage/monocytes in untreated lesions from extreme responders, suggesting MHC-I expression in both tumor cells and macrophages is associated with IL-2 response (Supplementary Fig. S11, Supplementary Table S10).
To confirm the association between tumor MHC-I expression and complete response to IL-2, we performed IHC staining for MHC-I in tumor biopsies from 19 previously untreated patients with metastatic melanoma who subsequently received IL-2 therapy (Table 1, Supplementary Table S12). We again found that all patients who had a CR exhibited membranous MHC-I positivity in the vast majority (at least 75%) of tumor cells (Fig. 3F). Conversely, lack of membranous tumor MHC-I expression was strongly associated with no or incomplete tumor response (non-CR) (Supplementary Table S13).
Table 1. Patient characteristics.
Clinical information for the metastatic melanoma patients in the initial cohort (n=7) and in the validation cohort (n=19) treated with either intralesional interleukin-2 (IL-2) or high-dose systemic IL-2. Patient response was classified as complete responder (CR) (termed “extreme responder” in the initial cohort) or non-complete responder (non-CR) (termed “non-/mixed responder” in the initial cohort).
| Cohort | Institution | Patient ID | Age | Gender | Lesion Site | IL-2 Treatment | Patient Response |
|---|---|---|---|---|---|---|---|
| Initial | MSKCC | 4 | 42 | M | Skin | Intralesional | CR |
| Initial | MSKCC | 6 | 74 | F | Skin | Intralesional | CR |
| Initial | MSKCC | 0 | 56 | M | Soft tissue | Intralesional | non-CR |
| Initial | MSKCC | 1 | 81 | F | Skin | Intralesional | non-CR |
| Initial | MSKCC | 2 | 67 | F | Soft tissue | Intralesional | non-CR |
| Initial | MSKCC | 3 | 86 | F | Skin | Intralesional | non-CR |
| Initial | MSKCC | 5 | 69 | M | Skin | Intralesional | non-CR |
|
| |||||||
| Validation | Calgary | v_01 | 69 | F | Skin | Intralesional | CR |
| Validation | Calgary | v_02 | 86 | F | Skin | Intralesional | CR |
| Validation | Calgary | v_03 | 93 | M | Skin | Intralesional | CR |
| Validation | NIH | v_04 | 52 | M | Soft tissue | Systemic | CR |
| Validation | NIH | v_05 | 43 | M | Skin | Systemic | CR |
| Validation | NIH | v_06 | 45 | F | Lymph node | Systemic | CR |
| Validation | NIH | v_07 | 54 | M | Skin | Systemic | non-CR |
| Validation | NIH | v_08 | 63 | F | Skin | Systemic | non-CR |
| Validation | NIH | v_09 | 52 | M | Soft tissue | Systemic | non-CR |
| Validation | NIH | v_10 | 27 | M | Lymph node | Systemic | non-CR |
| Validation | NIH | v_11 | 48 | M | Soft tissue | Systemic | non-CR |
| Validation | NIH | v_12 | 48 | M | Lung | Systemic | non-CR |
| Validation | NIH | v_13 | 37 | M | Lymph node | Systemic | non-CR |
| Validation | MDACC | v_14 | 62 | M | Lung | Systemic | non-CR |
| Validation | MDACC | v_15 | 76 | F | Skin | Systemic | non-CR |
| Validation | MDACC | v_16 | 65 | M | Lymph node | Systemic | non-CR |
| Validation | MDACC | v_17 | 54 | M | Lymph node | Systemic | non-CR |
| Validation | JHMI | v_18 | 53 | F | Skin | Systemic | non-CR |
| Validation | JHMI | v_19 | 59 | M | Skin | Systemic | non-CR |
Abbreviations: MSKCC, Memorial Sloan Kettering Cancer Center; Calgary, University of Calgary; NIH, National Institutes of Health; MDACC, MD Anderson Cancer Center; JHMI, Johns Hopkins Medical Institute.
Lack of membranous tumor MHC-I expression was not associated with mutations in B2M or other antigen presentation–pathway related genes (Supplementary Fig. S12A). Untreated lesions from extreme responders showed significantly increased bulk RNA levels of B2M, HLAB, and HLAC (Supplementary Fig. S12, B–F), consistent with our findings at the protein level. Loss of B2M RNA expression in bulk tumor was positively correlated with loss of MHC-I protein expression on the tumor cell membrane (Supplementary Fig. S12G).
Our data indicated that expression of MHC-I protein on the tumor cell membrane, which is required for antigen presentation, was associated with a higher fraction of exhausted and proliferating CD8+ T cells. To explore this relationship in greater detail, we compared the functional state of immune cells in the immediate “neighborhood” (i.e, within 30 μm) of MHC-I–high and MHC-I–low tumor cells (Supplementary Fig. S13, Supplementary Table S14). MHC-I-–high neighborhoods harbored higher fractions of T cells and T-cell populations positive for CD27, PD-1, LAG-3, TIM-3, and MHC-II, while MHC-I–low neighborhoods harbored higher fractions of NK cells, consistent with the known inhibitory effect of MHC-I on NK cells (14) and a dominant innate immune response in MHC-I–low lesions.
We next examined the expression of immune-related genes in untreated lesions from extreme responders and non-/mixed responders where we found differential expression of 96 genes. Untreated lesions from extreme responders showed uregulation of several genes associated with IFNγ and IFNα signaling, antigen presentation, IL-2 response, tertiary lymphoid structures (TLS), and T-cell dysfunction (Fig. 4A, Supplementary Table S15). Some of these gene-expression signatures (e.g., IFNγ signature and TLS) have been associated with clinical response to immune checkpoint blockade (ICB) in melanoma (15, 16). We did not observe significant expression changes in genes that identify immune cell populations, suggesting that upregulation of these gene-expression signatures in extreme responders was not simply a reflection of higher immune cell counts (Supplementary Fig. S14). Given that tumor mutation burden (TMB) has been predictive of response to ICB (17), we determined TMB in our untreated lesions, but we found no instances of hypermutation (Supplementary Table S16).
Fig. 4. Activated tumor microenvironment prior to IL-2 treatment characterizes extreme responders.
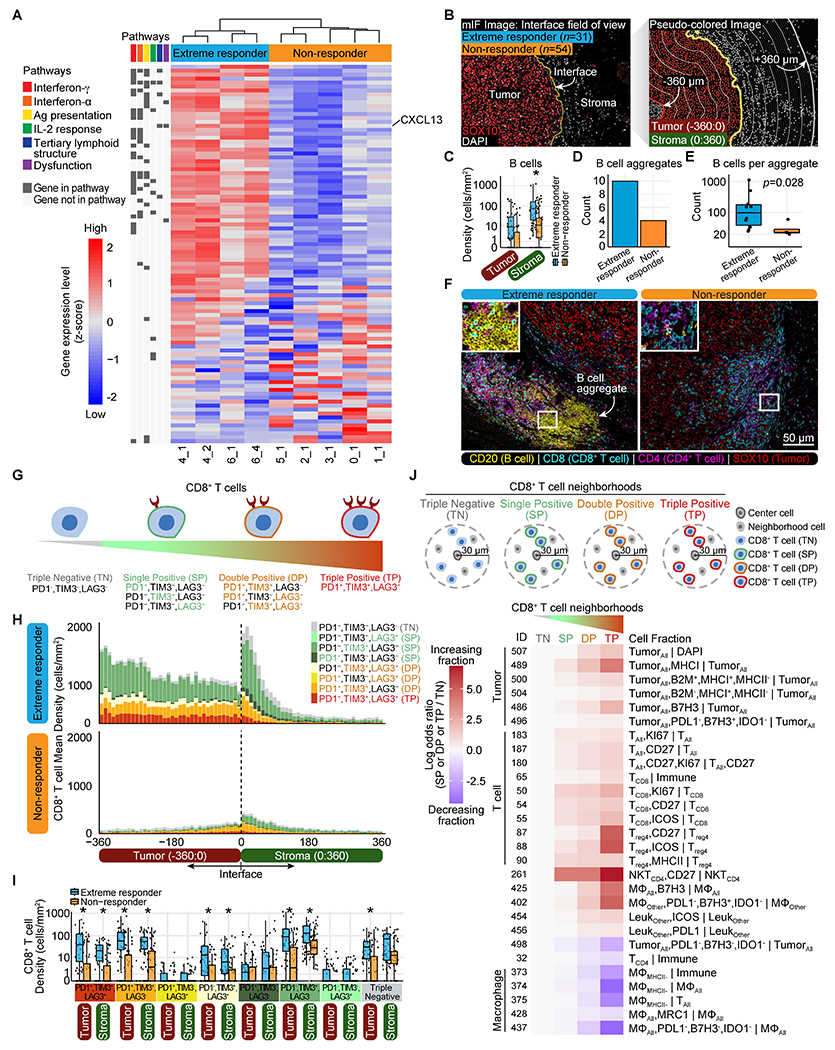
(A) The heatmap indicates scaled RNA expression values for differentially expressed genes (p-adjusted<0.05) in untreated lesions from extreme responders (n=4) versus non-/mixed responders (labeled “non-responder” throughout figures) (n=5), sorted by fold change. See also Supplementary Table S15. (B) Schematic of tumor interface analysis. mIF, multiplexed IF. (C) Box plots showing B-cell density in tumor and stroma of untreated lesions (minimum, median, and maximum with each point representing a field of view, FOV). Significant results, determined using a two-sided Wilcoxon test adjusted by Bonferroni correction, are indicated with an asterisk (p-adjusted<0.05). See also Supplementary Table S17. (D) Total B-cell aggregate counts in untreated lesions. (E) Total count of B cells per aggregate (Wilcoxon rank sum test, exact p-value noted) in untreated lesions (extreme responder (n=10) and non-responder (n=4)). (F) Representative multiplexed IF images from an untreated lesion of an extreme responder (6_4) and non-/mixed responder (1_1) showing B cell aggregates. (G) Cartoon of CD8+ T-cell states. (H) Mean density of CD8+ T cells expressing all combinations of PD-1/TIM-3/LAG-3 in untreated lesions in 10-micron intervals from −360:360-microns. See also Supplementary Table S18. (I) Box plots showing density of CD8+ T cells expressing all combinations of PD-1/TIM-3/LAG-3 in tumor and stroma of untreated lesions (minimum, median, and maximum with each point representing an FOV). See also Supplementary Table S18. (J) CD8+ T-cell neighborhood definitions. The heatmap indicates effect size (odds ratio) of each cell fraction (rows) in untreated lesions for SP/DP/TP neighborhoods normalized against TN neighborhoods. See also Supplementary Table S19.
One of the TLS-related and upregulated genes in untreated lesions from extreme responders was CXCL13, a B-cell attractant that is secreted by dysfunctional CD8+ T cells (18, 19) and has been linked to clinical response to ICB (20). We therefore examined the spatial distribution of B cells and exhausted CD8+ T cells relative to the tumor–stroma interface (+/− 360 μm) in untreated lesions (Fig. 4B). We had previously observed increased densities of B cells in untreated lesions from extreme responders (Supplementary Fig. S10). Upon examining the spatial distribution of the B cells, we found that these B cells were predominantely in the stroma of extreme responders following intralesional IL-2 (Fig. 4C, Supplementary Table S17). Although our review of H&E stains of the untreated lesions did not identify TLS, using multiplexed IF we observed a greater number of CD20+ B-cell aggregates, which were surrounded by both CD8+ and CD4+ T cells, in untreated lesions from extreme responders (Fig. 4, D–F, Supplementary Table S17).
Next we examined the degree of CD8+ T-cell exhaustion (Fig. 4G) as a function of their spatial distribution relative to the tumor–stroma interface (Fig. 4H, Supplementary Table S18). In untreated lesions, we observed increasing fractions of PD-1+LAG-3+TIM-3+ exhausted CD8+ T cells, as CD8+ T cells approach the tumor interface from both the stroma (–360 μm) and from within the tumor (+360 μm, Supplementary Fig. S15A and S15B, Supplementary Table S18). We observed increased densities of several CD8+ T-cell populations (i.e., expressing different combinations of PD-1, LAG-3, and TIM-3) in both the tumor (−360:0 μm) and stroma regions (0:360 μm) of extreme responders compared to non-/mixed responders (Fig. 4I, Supplementary Table S18).
To identify potential contributors of CD8+ T-cell exhaustion, we also characterized the cellular neighborhood of CD8+ T cells as they progress from a triple-negative (PD-1−LAG-3−TIM-3−) to triple-positive (PD-1+LAG-3+TIM-3+) state (Fig. 4J, Supplementary Table S19). As CD8+ T cells become exhausted (PD-1+LAG-3+TIM-3+), their neighborhood is characterized by increasing fractions of B2M+MHC-I+MHC-II− tumor cells, proliferating/activated (Ki67+CD27+ICOS+) T and NK cells, B7-H3+ macrophages and tumor cells, and decreasing fractions of CD4+ T cells, MHC-II− macrophages, and PD-L1−B7-H3−IDO-1− macrophage and tumor cells in their cellular neighborhoods.
DISCUSSION
Clinical response to ICB has been extensively studied and associated with both tumor cell–intrinsic factors (including TMB, HLA expression, and PD-L1 expression) (17, 21) and tumor microenvironment factors (including TLS and dysfunctional T cells) (16, 18, 22). There are presently no known biomarkers of response for IL-2. Our data shows that the absence of membranous MHC-I in tumor cells is associated with the failure to respond to IL-2. This finding emerged from our multi-dimensional analysis of in-transit metastases from 7 melanoma patients receiving intralesional IL-2 and was subsequently confirmed in an independent multi-institutional validation cohort of 19 patients with metastatic melanoma. Of note, melanoma patients in our validation cohort had received IL-2 therapy in either an intralesional or high-dose systemic formulation, broadening the impact of this biomarker to melanoma patients receiving systemic IL-2. Expression of MHC-I on the tumor-cell membrane in melanoma has also been reported to be associated with clinical response to anti–CTLA-4, but not anti–PD-1 ICB (23).
In addition to widespread tumor-cell MHC-I expression, untreated lesions from “extreme responders” showed hallmarks of a tumor-reactive microenvironment, with stromal B-cell aggregates, increased expression of IFNγ, IFNα, and IL-2 response–related genes, and CD8+ T cells with an “exhausted” phenotype. The immune-cell infiltrate after complete eradication of all tumor cells, on the other hand, was characterized by CD8+ T cells with a “non-exhausted” phenotype. Given the limitations of our study design, we were unable to determine the molecular basis of this “switch” in CD8+ T-cell phenotype. It might represent an IL2-induced reversal of a pre-existent exhausted state, novel trafficking of naïve CD8+ T cells into these lesions, local expansion of naïve CD8+ T cells in these lesions, or a combination thereof (24–26). Further studies with sequential tumor biopsies may be able to address this important mechanistic question.
Our study combined in situ single-cell profiling with bulk RNA and DNA profiling from adjacent unstained tissue sections of FFPE tumor, a biospecimen source that is widely available for the majority of cancer patients. This approach allowed us to identify therapy-associated cell states and gene-expression signatures and generate immunotherapy response biomarker hypotheses. Since the single-cell data architecture, spatial analytics, and biostatistical outputs developed in our work are platform agnostic and expandable to any number of cell phenotypes, our approach could be helpful to guide the clinical development of other immunotherapies for cancer.
Supplementary Material
Synopsis:
By integrating single-cell proteomic and bulk genomic technology to analyze routinely collected cancer biospecimens, the authors identify molecular and cellular changes associated with intralesional IL-2 therapy in melanoma and provide a template for immunotherapy biomarker hypothesis development.
Acknowledgments:
We thank Hedvig Hricak, Anup Sood, Jedd Wolchok, and members of the Mellinghoff, Hollmann, and Socci research groups for their helpful suggestions and support with this project. We thank Steven A. Rosenberg and James C. Yang (National Cancer Institute, National Institutes of Health), Russell Witt (University of Texas MD Anderson Cancer Center), and Elizabeth Rush and Cindy Sander (University of Pittsburgh) for their help with obtaining patients for the validation cohort. We acknowledge the use of the Integrated Genomics Operation Core, funded by the NCI Cancer Center Support Grant (CCSG, P30 CA08748), Cycle for Survival, and the Marie-Josée and Henry R. Kravis Center for Molecular Oncology.
Financial support:
National Institutes of Health grant 1 R35 NS105109 03 (IKM)
National Institutes of Health grant T32 GM132083 (M. Pourmaleki)
National Institutes of Health grant P30CA008748 (MSK Cancer Center Support Grant)
Geoffrey Beene Cancer Research Center (IKM)
Cycle for Survival (IKM)
Parker Institute for Cancer Immunotherapy (CEA, YL, TJH)
National Institutes of Health grant 1 R01 CA219896-01A1 (JAW)
Melanoma Research Alliance (JAW)
American Association for Cancer Research SU2C SU2C-AACR-IRG-19-17 (JAW)
MD Anderson Cancer Center’s Melanoma Moon Shots Program (JAW)
National Institutes of Health grant T32 CA 009599 (MGW)
MD Anderson Cancer Support grant P30 CA016672 (MGW)
P50 CA254865-01A1 (JMK)
Melanoma and Skin Cancer SPORE (JMK)
National Cancer Institute Center for Cancer Research (SLG)
Conflict of interests:
TJH receives funding from Bristol Myers Squibb and Calico labs. IKM has received research funding from General Electric, Agios, and Lilly and honoraria from Roche for a presentation. IKM has served in advisory roles for Agios, Amgen, Debiopharm, Novartis, Puma Biotechnology, and Voyager Therapeutics. JAW has received compensation for speaker’s bureau and honoraria from Imedex, Dava Oncology, Omniprex, Illumina, Gilead, PeerVies, MedImmune, and Bristol-Myers-Squibb. JAW serves as a consultant/advisory board member for Roche/Genentech, Novartis, AstraZeneca, GlaxoSmithKline (GSK), BMS, Merck, Biothera Pharmaceuticals, and Micronoma. JMK has received compensation for speaker’s bureau and honoraria from Bristol Myers Squibb. JMK serves as a consultant/advisory board member for Amgen, Ankyra Therapeutics, Axio Research/Instil Bio, Becker Pharmaceutical Consulting, Checkmate Pharmaceuticals, DermTech, Elsevier, Fenix Group International, Harbour BioMed, Immunocore LLC, Intellisphere, LLC/Cancer Network, Iovance Biotherapeutics, IQVIA, Istari Oncology, Merck, Millennium Pharmaceuticals/Takeda Pharmaceutical, Natera, Inc., Novartis Pharmaceuticals, OncoCyte Corporation, OncoSec Medical Inc., Pfizer, Replimune, Scopus BioPharma, and SR One Capital Management. SLG has a clinical trial agreement with Clinigen.
References
- 1.Meier F, Will S, Ellwanger U, Schlagenhauff B, Schittek B, Rassner G, Garbe C, Metastatic pathways and time courses in the orderly progression of cutaneous melanoma. Br. J. Dermatol 147, 62–70 (2002). [DOI] [PubMed] [Google Scholar]
- 2.Coit DG, Thompson JA, Algazi A, Andtbacka R, Bichakjian CK, Carson WE, Daniels GA, DiMaio D, Ernstoff M, Fields RC, Fleming MD, Gonzalez R, Guild V, Halpern AC, Hodi FS, Joseph RW, Lange JR, Martini MC, Materin MA, Olszanski AJ, Ross MI, Salama AK, Skitzki J, Sosman J, Swetter SM, Tanabe KK, Torres-Roca JF, Trisal V, Urist MM, McMillian N, Engh A, Melanoma, Version 2.2016, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. JNCCN. 14, 450–473 (2016). [DOI] [PubMed] [Google Scholar]
- 3.Byers BA, Temple-Oberle CF, Hurdle V, McKinnon JG, Treatment of in-transit melanoma with intra-lesional interleukin-2: a systematic review. J. Surg. Oncol 110, 770–775 (2014). [DOI] [PubMed] [Google Scholar]
- 4.Mullard A, Restoring IL-2 to its cancer immunotherapy glory. Nat. Rev. Drug Discov 20, 163–165 (2021). [DOI] [PubMed] [Google Scholar]
- 5.Read T, Lonne M, Sparks DS, David M, Wagels M, Schaider H, Soyer HP, Smithers BM, A systematic review and meta-analysis of locoregional treatments for in-transit melanoma. J. Surg. Oncol 119, 887–896 (2019). [DOI] [PubMed] [Google Scholar]
- 6.Radny P, Caroli UM, Bauer J, Paul T, Schlegel C, Eigentler TK, Weide B, Schwarz M, Garbe C, Phase II trial of intralesional therapy with interleukin-2 in soft-tissue melanoma metastases. Br. J. Cancer. 89, 1620–1626 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hassan S, Petrella TM, Zhang T, Kamel-Reid S, Nordio F, Baccarelli A, Sade S, Naert K, Habeeb AA, Ghazarian D, Wright FC, Pathologic complete response to intralesional interleukin-2 therapy associated with improved survival in melanoma patients with in-transit disease. Ann. Surg. Oncol 22, 1950–1958 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, Srinivasan P, Gao J, Chakravarty D, Devlin SM, Hellmann MD, Barron DA, Schram AM, Hameed M, Dogan S, Ross DS, Hechtman JF, DeLair DF, Yao J, Mandelker DL, Cheng DT, Chandramohan R, Mohanty AS, Ptashkin RN, Jayakumaran G, Prasad M, Syed MH, Rema AB, Liu ZY, Nafa K, Borsu L, Sadowska J, Casanova J, Bacares R, Kiecka IJ, Razumova A, Son JB, Stewart L, Baldi T, Mullaney KA, Al-Ahmadie H, Vakiani E, Abeshouse AA, Penson AV, Jonsson P, Camacho N, Chang MT, Won HH, Gross BE, Kundra R, Heins ZJ, Chen H-W, Phillips S, Zhang H, Wang J, Ochoa A, Wills J, Eubank M, Thomas SB, Gardos SM, Reales DN, Galle J, Durany R, Cambria R, Abida W, Cercek A, Feldman DR, Gounder MM, Hakimi AA, Harding JJ, Iyer G, Janjigian YY, Jordan EJ, Kelly CM, Lowery MA, Morris LGT, Omuro AM, Raj N, Razavi P, Shoushtari AN, Shukla N, Soumerai TE, Varghese AM, Yaeger R, Coleman J, Bochner B, Riely GJ, Saltz LB, Scher HI, Sabbatini PJ, Robson ME, Klimstra DS, Taylor BS, Baselga J, Schultz N, Hyman DM, Arcila ME, Solit DB, Ladanyi M, Berger MF, Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med 23, 703–713 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gerdes MJ, Sevinsky CJ, Sood A, Adak S, Bello MO, Bordwell A, Can A, Corwin A, Dinn S, Filkins RJ, Hollman D, Kamath V, Kaanumalle S, Kenny K, Larsen M, Lazare M, Li Q, Lowes C, McCulloch CC, McDonough E, Montalto MC, Pang Z, Rittscher J, Santamaria-Pang A, Sarachan BD, Seel ML, Seppo A, Shaikh K, Sui Y, Zhang J, Ginty F, Highly multiplexed single-cell analysis of formalin-fixed, paraffin-embedded cancer tissue. Proc. Natl. Acad. Sci 110, 11982–11987 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sood A, Miller AM, Brogi E, Sui Y, Armenia J, McDonough E, Santamaria-Pang A, Carlin S, Stamper A, Campos C, Pang Z, Li Q, Port E, Graeber TG, Schultz N, Ginty F, Larson SM, Mellinghoff IK, Multiplexed immunofluorescence delineates proteomic cancer cell states associated with metabolism. JCI Insight. 1, 87030 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Acharya N, Sabatos-Peyton C, Anderson AC, Tim-3 finds its place in the cancer immunotherapy landscape. J. Immunother. Cancer. 8, e000911 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wolf Y, Anderson AC, Kuchroo VK, TIM3 comes of age as an inhibitory receptor. Nat. Rev. Immunol 20, 173–185 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cesana G, Iodice G, Cohen S, Moroziewicz D, Mitcham J, Stoutenburg J, Cheung K, Hesdorffer C, Kim-Schulze S, Kaufman H, Characterization of CD4+CD25+ Regulatory T Cells in Patients Treated With High-Dose Interleukin-2 for Metastatic Melanoma or Renal Cell Carcinoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol 24, 1169–77 (2006). [DOI] [PubMed] [Google Scholar]
- 14.Raulet DH, Gasser S, Gowen BG, Deng W, Jung H, Regulation of Ligands for the NKG2D Activating Receptor. Annu. Rev. Immunol 31, 413–441 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grasso CS, Tsoi J, Onyshchenko M, Abril-Rodriguez G, Ross-Macdonald P, Wind-Rotolo M, Champhekar A, Medina E, Torrejon DY, Shin DS, Tran P, Kim YJ, Puig-Saus C, Campbell K, Vega-Crespo A, Quist M, Martignier C, Luke JJ, Wolchok JD, Johnson DB, Chmielowski B, Hodi FS, Bhatia S, Sharfman W, Urba WJ, Slingluff CL, Diab A, Haanen JBAG, Algarra SM, Pardoll DM, Anagnostou V, Topalian SL, Velculescu VE, Speiser DE, Kalbasi A, Ribas A, Conserved Interferon-γ Signaling Drives Clinical Response to Immune Checkpoint Blockade Therapy in Melanoma. Cancer Cell. 38, 500–515.e3 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cabrita R, Lauss M, Sanna A, Donia M, Skaarup Larsen M, Mitra S, Johansson I, Phung B, Harbst K, Vallon-Christersson J, van Schoiack A, Lövgren K, Warren S, Jirström K, Olsson H, Pietras K, Ingvar C, Isaksson K, Schadendorf D, Schmidt H, Bastholt L, Carneiro A, Wargo JA, Svane IM, Jönsson G, Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature. 577, 561–565 (2020). [DOI] [PubMed] [Google Scholar]
- 17.Yarchoan M, Albacker LA, Hopkins AC, Montesion M, Murugesan K, Vithayathil TT, Zaidi N, Azad NS, Laheru DA, Frampton GM, Jaffee EM, PD-L1 expression and tumor mutational burden are independent biomarkers in most cancers. JCI Insight. 4 (2019), doi: 10.1172/jci.insight.126908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van der Leun AM, Thommen DS, Schumacher TN, CD8+ T cell states in human cancer: insights from single-cell analysis. Nat. Rev. Cancer. 20, 218–232 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li H, van der Leun AM, Yofe I, Lubling Y, Gelbard-Solodkin D, van Akkooi ACJ, van den Braber M, Rozeman EA, Haanen JBAG, Blank CU, Horlings HM, David E, Baran Y, Bercovich A, Lifshitz A, Schumacher TN, Tanay A, Amit I, Dysfunctional CD8 T Cells Form a Proliferative, Dynamically Regulated Compartment within Human Melanoma. Cell. 176, 775–789.e18 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Litchfield K, Reading JL, Puttick C, Thakkar K, Abbosh C, Bentham R, Watkins TBK, Rosenthal R, Biswas D, Rowan A, Lim E, Bakir MA, Turati V, Guerra-Assunção JA, Conde L, Furness AJS, Saini SK, Hadrup SR, Herrero J, Lee S-H, Loo PV, Enver T, Larkin J, Hellmann MD, Turajlic S, Quezada SA, McGranahan N, Swanton C, Meta-analysis of tumor- and T cell-intrinsic mechanisms of sensitization to checkpoint inhibition. Cell. 184, 596–614.e14 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chowell D, Morris LGT, Grigg CM, Weber JK, Samstein RM, Makarov V, Kuo F, Kendall SM, Requena D, Riaz N, Greenbaum B, Carroll J, Garon E, Hyman DM, Zehir A, Solit D, Berger M, Zhou R, Rizvi NA, Chan TA, Patient HLA class I genotype influences cancer response to checkpoint blockade immunotherapy. Science. 359, 582–587 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miller BC, Sen DR, Al Abosy R, Bi K, Virkud YV, LaFleur MW, Yates KB, Lako A, Felt K, Naik GS, Manos M, Gjini E, Kuchroo JR, Ishizuka JJ, Collier JL, Griffin GK, Maleri S, Comstock DE, Weiss SA, Brown FD, Panda A, Zimmer MD, Manguso RT, Hodi FS, Rodig SJ, Sharpe AH, Haining WN, Subsets of exhausted CD8 + T cells differentially mediate tumor control and respond to checkpoint blockade. Nat. Immunol 20, 326–336 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rodig SJ, Gusenleitner D, Jackson DG, Gjini E, Giobbie-Hurder A, Jin C, Chang H, Lovitch SB, Horak C, Weber JS, Weirather JL, Wolchok JD, Postow MA, Pavlick AC, Chesney J, Hodi FS, MHC proteins confer differential sensitivity to CTLA-4 and PD-1 blockade in untreated metastatic melanoma. Sci. Transl. Med 10 (2018), doi: 10.1126/scitranslmed.aar3342. [DOI] [PubMed] [Google Scholar]
- 24.Spolski R, Li P, Leonard WJ, Biology and regulation of IL-2: from molecular mechanisms to human therapy. Nat. Rev. Immunol 18, 648–659 (2018). [DOI] [PubMed] [Google Scholar]
- 25.Boyman O, Sprent J, The role of interleukin-2 during homeostasis and activation of the immune system. Nat. Rev. Immunol 12, 180–190 (2012). [DOI] [PubMed] [Google Scholar]
- 26.Kalia V, Sarkar S, Regulation of Effector and Memory CD8 T Cell Differentiation by IL-2—A Balancing Act. Front. Immunol 9, 2987 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.