

SUMMARY
SARS-CoV-2 lineages have diverged into highly prevalent variants termed Variants of Concern (VOCs). Here, we characterized emerging SARS-CoV-2 spike polymorphisms in vitro and in vivo to understand their impact on transmissibility, virus pathogenicity and fitness. We demonstrate that the substitution S:655Y, represented in the Gamma and Omicron VOCs, enhances viral replication and spike protein cleavage. The S:655Y substitution was transmitted more efficiently than its ancestor S:655H in the hamster infection model and was able to outcompete S:655H in the hamster model and in a human primary airway system. Finally, we analyzed a set of emerging SARS-CoV-2 variants to investigate how different sets of mutations may impact spike processing. All VOCs tested exhibited increased spike cleavage and fusogenic capacity. Taken together, our study demonstrates that the spike mutations present in VOCs that become epidemiologically prevalent in humans, are linked to an increase in spike processing and virus transmission.
Keywords: SARS-CoV-2, spike cleavage, H655Y mutation, variants of concern, syncytia formation
Graphical Abstract
eTOC:
Escalera et al., show that spike mutation H655Y which is present in SARS-CoV-2 variants Gamma and Omicron, enhances spike protein cleavage, cell-cell fusion, and transmission in the hamster model. Additionally, SARS-CoV-2 variants of concern are shown to have independently acquired mutations associated with a gain in spike cleavage and syncytia formation.
INTRODUCTION
SARS-CoV-2 has been spreading worldwide causing millions of infections and deaths since its emergence in Wuhan, China, in late 2019. Apart from humans, ferrets, cats, dogs, Syrian golden hamsters, and nonhuman primates are also susceptible to SARS-CoV-2 infection and transmission (Fenollar et al., 2021; Shi et al., 2020). In addition, cases of viral spread in mink farms and mink-to-human cross-species transmission have been reported (Hammer et al., 2021; Oude Munnink et al., 2021). The spike (S) glycoprotein of SARS-CoV-2 is the main determinant of host tropism and susceptibility, and the main target of antibody responses (Tong et al., 2021). Therefore, the emergence of adaptive mutations present in the spike protein can strongly affect host tropism and viral transmission (Johnson et al., 2020; Starr et al., 2020). The S protein is composed of two subunits: S1 which contains the receptor binding domain (RBD) that initiates infection by binding to the angiotensin converting enzyme 2 (ACE2) receptor present in the host cell surface; and the S2 subunit that mediates fusion between viral and cellular membranes (Duan et al., 2020; Huang et al., 2020). To fuse with the host cell, the S protein needs to be cleaved by cellular proteases at the S1/S2 and S2’ sites. Importantly, the S1/S2 site of SARS-CoV-2 viruses contains a multibasic furin motif (681PRRXR685) absent in other beta coronaviruses (Coutard et al., 2020; Hoffmann et al., 2020) that can be processed by furin proteases, but also by transmembrane serine proteases such as TMPRSS2, or by cathepsins present in the endosomes (Bestle et al., 2020; Hoffmann et al., 2020; Matsuyama et al., 2010; Ord et al., 2020; Tang et al., 2021). The S1/S2 cleavage exposes the S2’ site, and a second cleavage of the S2’ is needed to release an internal fusion peptide that mediates membrane fusion (Xia et al., 2020).
Since 2019, several SARS-CoV-2 lineages have emerged leading to the divergence of an extensive subset of SARS-CoV-2 variants termed Variants of Concern (VOCs). This has led to the natural selection of several mutations in the spike protein with different functional consequences, some of them unknown. As SARS-CoV-2 variants are arising, more research is needed to understand what the drivers of evolutionary changes are over time, the potential impact on epidemiology, antigenicity, escape from neutralizing antibodies induced by previous infection and vaccination and virus fitness. The first widely adaptative substitution described was the spike protein amino acid change S: D614G which became dominant in March 2020 and is present in most of the variants currently circulating worldwide. This substitution is known to enhance viral replication in the upper respiratory tract as well as in vivo transmission (Korber et al., 2020; Plante et al., 2021). Several other polymorphisms became dominant in late 2020. The N501Y substitution convergently evolved in early emerging VOCs Alpha (B.1.1.7), Beta (B.1.351), Gamma (P.1) and Omicron (B.1.1.529) variants and has been associated with an enhanced spike affinity for the cellular ACE2 receptor (Liu et al., 2021; Yi et al., 2020). This mutation is located in the receptor-binding motif (RBM) of the RBD, the primary target of many neutralizing antibodies. Importantly, accumulation of mutations in the RBD can decrease neutralizing antibody responses elicited by infection or vaccination against ancestral SARS-CoV-2 variants (Aydillo et al., 2021). Similarly, the later SARS-CoV-2 Kappa (B.1.617.1) and Delta (B.1.617.2) variants have also shown a significantly reduced sensitivity to convalescent and immune sera (Edara et al., 2021; Mlcochova et al., 2021a; Planas et al., 2021b). Other mutations outside the RBD have also become prevalent. A clear example is the polymorphism found at position S:681 in the furin cleavage site, which includes P681H and P681R in the Alpha/Omicron and Kappa/Delta variants, respectively. Some preliminary reports have pointed to an enhancement in virus transmissibility associated with this polymorphism, perhaps due to an increase of spike cleavage (Peacock et al., 2021). Additionally, several other mutations have been identified at the edge of the furin cleavage site. This is the case of the H655Y substitution found in the Gamma (P.1) and Omicron variants (B.1.1.529). This mutation was associated with changes in antigenicity by conferring escape from human monoclonal antibodies (Baum et al., 2020). Moreover, it has also been found to be selected in animal models after experimental infection in vivo (Braun et al., 2021; Rathnasinghe et al., 2021), indicating a potential role in host replication, transmissibility, and pathogenicity.
Here, we characterized emerging SARS-CoV-2 spike polymorphisms in vitro and in vivo to understand their impact on transmissibility, virus pathogenicity and fitness. Using the mink model of COVID-19, we found that the S:H655Y substitution was acquired in vivo after infection with the WA1 isolate (USA-WA1/2020). To investigate the advantage conferred by S:H655Y, we analyzed the kinetics, spike processing by cellular proteases and syncytium formation ability of a panel of SARS-CoV-2 variants harboring 655Y, including human isolates derived from patients seeking care at the Mount Sinai Health System in New York (NY) City which was one of the major early epicenters of COVID-19 pandemic. Our results demonstrated that the 655Y polymorphism enhances spike cleavage and viral growth. Furthermore, the S:655Y substitution was transmitted more efficiently than its ancestor S:655H in the hamster infection model and was able to overcome S:655H in the human airway epithelial system. Finally, and in the context of the current epidemiological situation, we analyzed a set of emerging SARS-CoV-2 variants to investigate how different sets of mutations may impact spike processing. We demonstrated that novel circulating VOCs that became more prevalent have independently acquired mutations associated with a gain in spike cleavage and syncytia formation. Taken together, our study shows a link between an increased spike processing and increased virus transmission due to the spike mutations present in SARS-CoV-2 variants that become epidemiologically more prevalent in humans.
RESULTS
SARS-CoV-2 VARIANTS HARBORING 655Y SHOW AN ENHANCED SPIKE PROTEIN CLEAVAGE AND FUSOGENIC ABILITY IN VITRO
Minks have been suggested to play a role in the initial local spread and evolution of SARS-CoV-2 variants in different countries in Europe (Hammer et al., 2021; Oude Munnink et al., 2021). While minks are susceptible to SARS-CoV-2, they are also capable for zoonotic transmission of SARS-CoV-2 to because of the similarity of the ACE2 receptor between minks and humans. We used the mink model to investigate the replication and pathogenicity of the WA1 (USA-WA1/2020) isolate of SARS-CoV-2, as a representative of the first original human viruses that initiated the SARS-CoV-2 pandemic. This variant corresponds to one of the first USA isolates and does not contain any changes on the S protein when compared to the initial isolates from Wuhan, such as the Wuhan-1 virus. For this purpose, six minks were intranasally infected with 106 pfu of WA1 isolate resulting in productive viral replication in the upper respiratory tract with infectious virus recovered from nasal washes at days 1, 3 and 5 post-infection (Supplementary Figure 1A–B). At day 4 post-inoculation, infectious virus was detected by plaque assays from left cranial lung and nasal turbinates but not from any of the other tissues analyzed (Supplementary Figure 1C). We then selected small and large viral plaques in the Vero-E6 cell-based plaque assays from infected mink lung specimens and performed next generation sequencing of the genome from the plaque-isolated viruses. As compared to the Wuhan-1 and WA1 reference sequences, all mink-derived viral isolates encoded the H655Y amino acid substitution within the spike (S) (Supplementary Figure 1D). Additionally, the three viral isolates with the small plaque phenotype encoded the T259K amino acid substitution while the three viral isolates with the large plaque phenotype encoded the R682W amino acid substitution. It is known that S:682W/Q substitution in the furin cleavage site region may emerge after subsequent passages in VeroE6 cells (Lamers et al., 2021; Liu et al., 2020). Next-generation sequencing analysis of the viral quasispecies population in mink lung specimens and WA1 stock demonstrated the presence of around 30% of the S:682W/Q substitution indicating that this mutation was selected during the course of the VeroE6 infections and stock generation and not during the infection in minks. On the other hand, S:655Y was present in less than 10% of the WA1 original stock but it was dominant in all the mink isolates in 99% of the viral RNA obtained from the lungs suggesting that this mutation was selected and may confer an advantage in the mink host.
To understand the magnitude and the spread of the 655Y polymorphism over time, we investigated the frequency of S:655Y in sequences sampled worldwide since the initial outbreak to the end of the first wave of SARS-CoV-2 (Figure 1A). For this, 7,059 sequences sampled from GISAID up to September 2020 were used. Human variants harboring the 655Y mutation were spread throughout the phylogenetic tree and distributed in all clades with no differences according to temporal distribution, suggesting that the 655Y mutation arose independently multiple times. Remarkably, the S:H655Y polymorphism was also found among the initial variants introduced in New York (NY) City in March 2020. To determine the replication phenotype, we decided to investigate this NY 655Y variant (NY7) together with some of its contemporaneous SARS-CoV-2 isolates circulating in New York during the early pandemic outbreak (Gonzalez-Reiche et al., 2020). To this end, we isolated 12 viruses based on their genotypes (Gonzalez-Reiche et al., 2020), including NY7 which carries the S:655Y mutation for culture directly from nasopharyngeal specimens obtained from COVID-19 infected patients. Of note, the dominant 614G spike polymorphism was present in seven (NY1, NY2, NY3, NY4, NY9, NY10 and NY12; 58%) of the selected human SARS-CoV-2 (hCoV) NY isolates consistent with its early emergence and rapid spread worldwide (Korber et al., 2020; Plante et al., 2021). Confirmation sequencing of the isolates showed that 682W/Q substitutions appeared in four (33%) viruses after initial isolation and culturing in VeroE6 cells. However, this mutation was absent in the original nasal swabs collected from the corresponding COVID-19 infected patients. This is consistent with in vitro adaptative mutations previously described (Lamers et al., 2021). Moreover, a five amino acid sequence (Δ675–679) flanking the furin cleavage site was deleted in five (42%) of the isolates as compared to the sequence from the original specimen. This deletion has been previously reported to be a common in vitro mutation selected in Vero cells (Liu et al., 2020). Amino acids substitutions of the S protein of these initial human isolates compared to the Wuhan-1 reference are shown Supplementary Figure 2A and Supplementary Table 2. We next studied the replication kinetics of the NY SARS-CoV-2 isolates by comparing their multicycle growth curves at an MOI of 0.01 in VeroE6 and human Caco-2 cells. As expected, NY2, NY4 and NY9 containing the 682Q/W showed an advantage in growth at 48 hours post-infection (p.i.) in VeroE6, while no differences could be found in Caco-2 cells (Supplementary Figure 2B–E). Remarkably, NY7 (S:655Y) showed higher growth at 48 hours p.i in Caco-2 cells (Supplementary Figure 2C and E) when compared to the rest of these early SARS-CoV-2 isolates. These results support our conclusion that the 655Y polymorphism conferred a viral advantage. To investigate the spike cleavage efficiency of the 655Y versus other human isolates, we performed infections in VeroE6, and supernatants were analyzed by Western blot for the S2 domain of the S protein. Importantly, two bands were clearly visible for the NY7 (S:655Y) (Figure 1B), corresponding to both the cleaved (95 kDa) and uncleaved (180 kDa) form of the S protein. In contrast, subtle bands for S2 were detected in NY6, NY10 and NY12 and only the uncleaved S form was detected in the other early human isolates. Consistently, quantification of full-length and cleaved spike protein showed that around 80% of the NY7 (S:655Y) spike was in its S2 cleaved form (Figure 1C), indicating that the 655Y polymorphism may facilitate S protein processing. To confirm these results in a primary human cell model, we assessed the spike protein processing of the twelve early NY human isolates in pneumocyte-like cells. This system has been previously described as a robust model to study SARS-CoV-2 infection (Riva et al., 2020; White et al., 2021). Human pneumocyte-like cells were differentiated from human induced pluripotent stem cell and infected with the NY SARS-CoV-2 isolates. Cell extracts were collected at 48 hours p.i and analyzed by Western blot (Figure 1D). As expected, NY7 (S:655Y) showed an increase cleavage efficiency with 70% of cleaved spike protein (Figure 1E). Additionally, we observed S2 cleaved protein in NY12 consistent with the S2 subtle bands detected in VeroE6 cells. This isolate only contains the S:D614G mutation. Although other NY variants used in this study also harbor the D614G, no enhancement in spike cleavage was observed. This can be explained by the acquisition of additional spike mutations during in vitro culture (Δ675–679, 682Q/W) in the isolates containing the D614G, which could be masking the effect of D614G in spike processing.
Figure 1. Characterization of spike protein processing of human SARS-CoV-2 isolates from New York (NY).
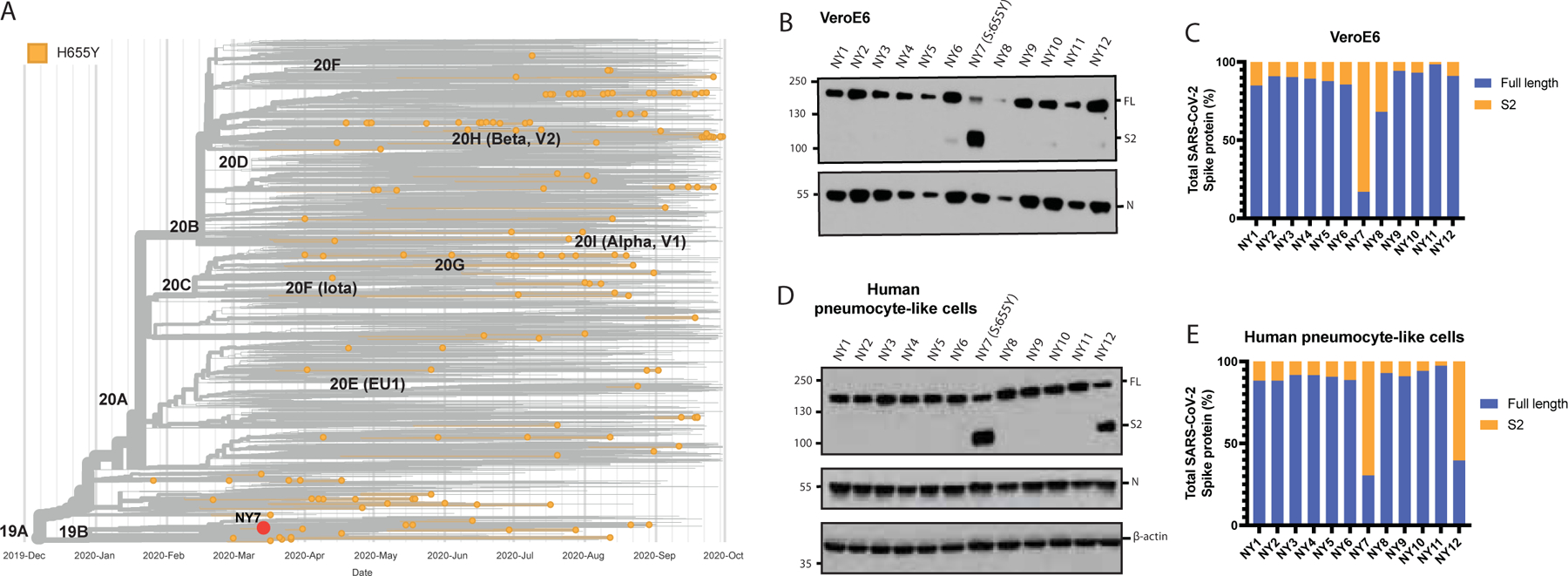
A) Time-calibrated phylogenetic analysis of the global distribution of H655Y substitution during the early SARS-CoV-2 outbreak. The phylogenetic tree was generated with Nextstrain with 7059 genomes sampled for representation of the H655Y substitution over time of worldwide data deposited in the GISAID database from December 2019 to September 2020. B) Western blotting of spike protein cleavage of the twelve human SARS-CoV-2 viruses isolated from nasal swabs of COVID-19 infected patients and collected during the first pandemic wave in NY. Infections were performed in VeroE6 cells at an MOI of 0.01 and supernatants were collected at 48 hours p.i. Full length (FL) spike protein (180 kDa), S2 cleaved spike (95 kDa) and Nucleocapsid (N, 50 kDa) were detected using specific antibodies. Levels of N protein were used as loading control. C) Quantification of full length and cleaved spike protein of the early human NY isolates in VeroE6 cells. Spike protein levels were normalized to nucleocapsid expression. D) Western blotting of spike protein cleavage of the twelve human SARS-CoV-2 NY viruses in human pneumocyte-like cells. Cells were infected with 3000 pfu per well of the corresponding viral isolate and cell extracts were collected at 48 hours p.i. Full length (FL) spike protein (180 kDa), S2 cleaved spike (95 kDa), Nucleocapsid (N, 50 kDa) and β-actin (45 kDa) were detected using specific antibodies. Levels of N and β-actin protein were used as loading control. E) Quantification of full length and cleaved spike protein of the early human NY isolates in human pneumocyte-like cells. Spike protein levels were normalized to nucleocapsid expression.
To confirm whether the 655Y mutation was solely responsible for the increased S cleavage, we analyzed the replication and cleavage efficiency of a panel of SARS-CoV-2 viruses, all bearing the 655Y substitution but containing additional substitutions across the genome. We included two of the isolated mink variants (MiA1 and MiA2); NY7 and another human isolate derived from a COVID-19 patient infected in February 2021 (NY13, S:614G, 655Y); and a WA1–655Y variant isolated after wild type WA1 infection in VeroE6 cells and subsequent plaque purification. Additionally, the WA1 reference and NY6 were used as controls since they lack the 655Y substitution. It should be noted that NY6 has a five amino acid deletion before the furin cleavage site (Figure 2A and Supplementary Figure 2A). We assessed differences in replication and S processing of this panel of viruses by comparing growth in both VeroE6 and Vero-TMPRSS2 cells. As shown in Figure 2B, WA1–655Y infection yielded higher titers in both VeroE6 and Vero-TMPRSS2 cells as compared to infection by WA1 (statistically significant in VeroE6 at 24h p.i., Supplementary Table 4). These isolates only differ in the position 655Y while the rest of the genome is isogenic, supporting that 655Y spike polymorphism enhances viral replication and growth. Next, viral supernatants were used to analyze the plaque phenotype in VeroE6 and Vero-TMPRSS2 and to compare S protein expression levels after infection. In general, all isolates showed higher plaque size in the presence of TMPRSS2, consistent with enhancement of cell entry (Supplementary Figure 3A). However, differences were found by Western blot and only the isolates bearing 655Y showed enhanced spike cleavage in both VeroE6 and Vero-TMPRSS2 (Figure 2C). More than 90% of the total spike protein from these 655Y variants corresponded to the S2 cleaved form of the spike. In contrast, NY6 and W A1 controls showed poor cleavage efficiency (Figure 2D and E). Finally, we performed infections with a representative panel of viruses containing the 655Y (NY7, NY13, WA1–655Y, NY6, WA1) in human pneumocytes-like cells to assess viral growth and spike protein processing. Consistent with our previous findings, WA1–655Y demonstrated higher replication efficiency in our human airway epithelial system compared to WA1 wild type (Figure 2F, Supplementary Table 4). Moreover, all isolates encoding the 655Y spike mutation exhibited enhanced spike cleavage as shown by Western Blot (Figure 2G and H). This demonstrates that the S:655Y polymorphism plays a crucial role in SARS-CoV-2 spike protein processing and cell entry in human pneumocyte-like cells.
Figure 2. The H655Y amino acid substitution enhances spike cleavage and viral growth.
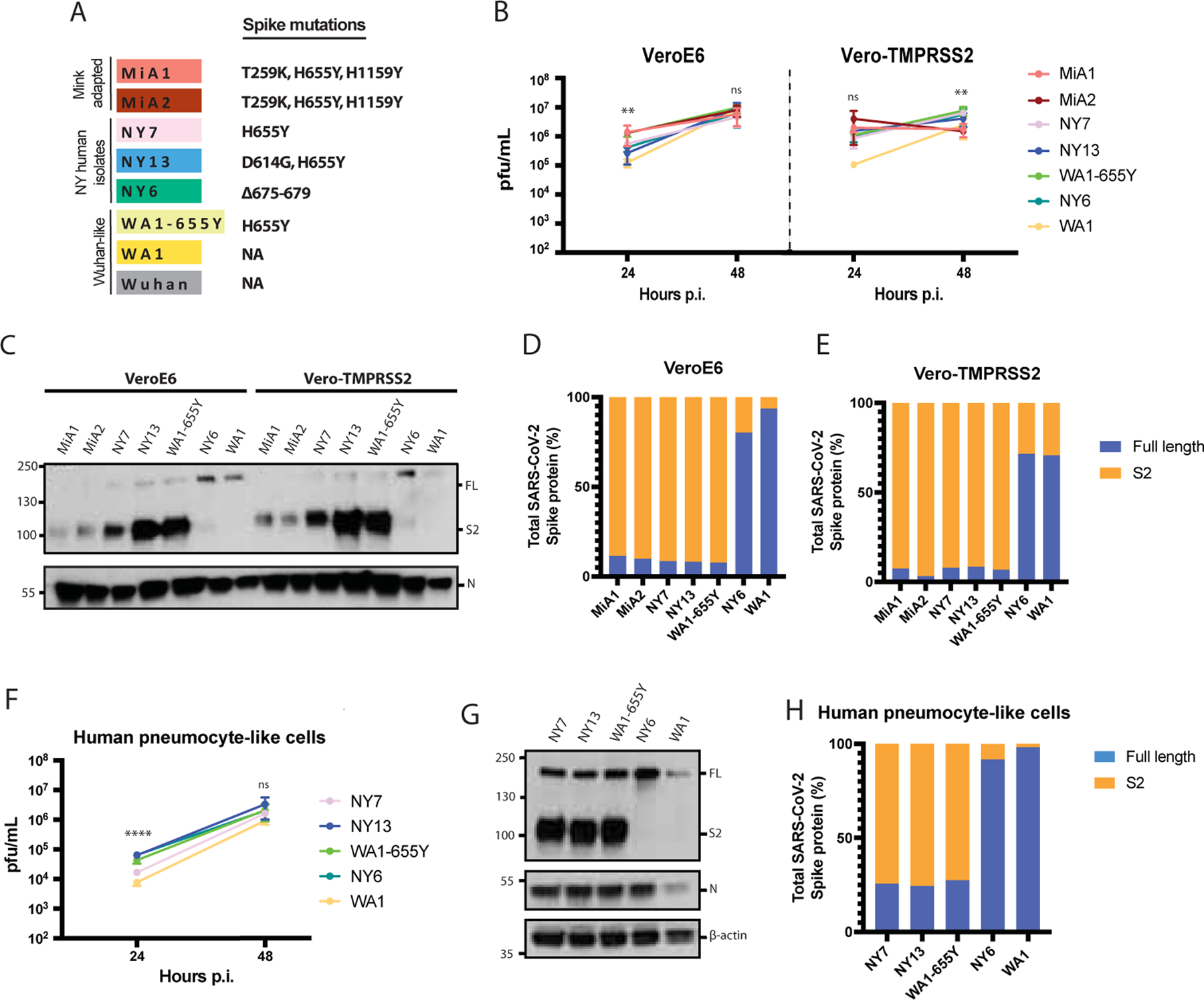
A) Spike polymorphisms present in the mink-adapted variants, early human SARS-CoV-2 New York (NY) isolates, WA1–655Y and WA1 wild type viruses. Wuhan1 is included as a reference. B) Replication kinetics of early SARS-CoV-2 viruses in VeroE6 and Vero-TMPRSS2 cells. Infections were performed at an MOI of 0.01. Viral titers were determined by plaque assay at the indicated hours post-infection and expressed as PFU per milliliter. Shown are the means and SDs from three replicates. ANOVA test for multiple comparison was used to compare mean differences within different isolates and time points. Viral isolates were compared two by two using the Tukey’s correction. Statistical significance was considered when p≤ 0.05 (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns, not significant). Color codes relate to the isolates shown in A. C) Western blotting of S protein from supernatants of VeroE6 and Vero-TMPRSS2 infected cells. Infections were performed at an MOI of 0.01 and viral supernatants were collected at 48 hours post-infection (p.i.). Full length (FL) spike protein (180 kDa), S2 cleaved spike (95 kDa) and Nucleocapsid (N, 50 kDa) were detected using specific antibodies. Levels of N protein were used as loading control. D-E) Quantification of full length and cleaved spike protein of the indicated viruses in VeroE6 and Vero-TMPRSS2 cells. Spike protein levels were normalized to nucleocapsid expression. F) Replication kinetics of early SARS-CoV-2 viruses in human pneumocyte-like cells. Cells were infected with 3000 pfu per well of the corresponding viral isolate. Viral titers were determined by plaque assay at the indicated hours post-infection and expressed as PFU per milliliter. Shown are the means and SDs from three replicates ANOVA test for multiple comparison was used to com p are mean differences within different isolates and time points. Viral isolates compared two by two using the Tukey’s correction. Statistical significance was considered when p ≤ 0.05 (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns, not significant). G) Western blotting of S protein in human pneumocyte-like cells infected with 3000 pfu per well of the corresponding early SARS-CoV-2 virus. Cell extracts were collected at 48 hours post-infection (p.i.). Full length (FL) spike protein (180 kDa), S2 cleaved spike (95 kDa), Nucleocapsid (N, 50 kDa) and β-actin (45 kDa) were detected using specific antibodies. Levels of N and β-actin protein were used as loading control. E) Quantification of full length and cleaved spike protein of the indicated viruses in human pneumocyte-like cells. Spike protein levels were normalized to nucleocapsid expression.
Syncytia formation has been described as one of the hallmarks of SARS-CoV-2 infection and pathogenesis in the lungs. It is mediated by the interaction between the spike protein expressed on the cell surface of infected cells and the ACE2 receptor (Lin et al., 2021). To investigate whether 655Y spike polymorphism enhances cell-cell fusion, we infected Vero-TMPRSS2 at an MOI of 0.01 with the mink (MiA1) and human isolates (NY7, NY13 and NY6) and used specific antibodies to detect the S and N protein, as well as nuclei staining with DAPI after 24 p.i. by immunofluorescence microscopy. As shown in Figure 3A, the isolates harboring the 655Y polymorphism showed a slight increase in syncytia formation as compared to NY6 (S:Δ675–679) and WA1 isolates. Next, we quantified cell–cell fusion using a split-GFP complementation plasmid-based system (Mlcochova et al., 2021b; Planas et al., 2021a). We co-transfected Vero-TMPRSS2 cells with each of the spike protein together GPF1–10 or GFP11 plasmids. For this, we cloned the spike protein of representative viruses from our 655Y panel in the pCAGGS expression vector: NY7, WA1–655Y, NY6 and WA1 wild type. Additionally, the WA1 spike protein with a deleted furin cleavage site (S: ΔTNSPRRARSVAS) was also included since this multibasic site has been suggested to be responsible for SARS-CoV-2 mediated syncytia. After 24h, the Vero-TMPRSS2 transfected cells were pooled and plated to allow fusion for 24h (Figure 3B). Additionally, spike protein expression was also quantified using an antibody specific against S2 domain (Supplementary Figure 4A). Figure 3C shows GFP signal from each spike variant normalized to the DAPI positive cells and spike expression levels. Differences in fusogenicity were then calculated relative to WA1 wild type spike. We observed that spike proteins harboring the 655Y were more fusogenic and induce higher levels of syncytia than WA1 and NY6 spikes. As expected, the WA1 spike with a deleted furin cleavage site resulted in significantly less fusion, indicating that an intact furin cleavage site is required for syncytia formation.
Figure 3. The 655Y spike polymorphism increases cell-cell fusion.
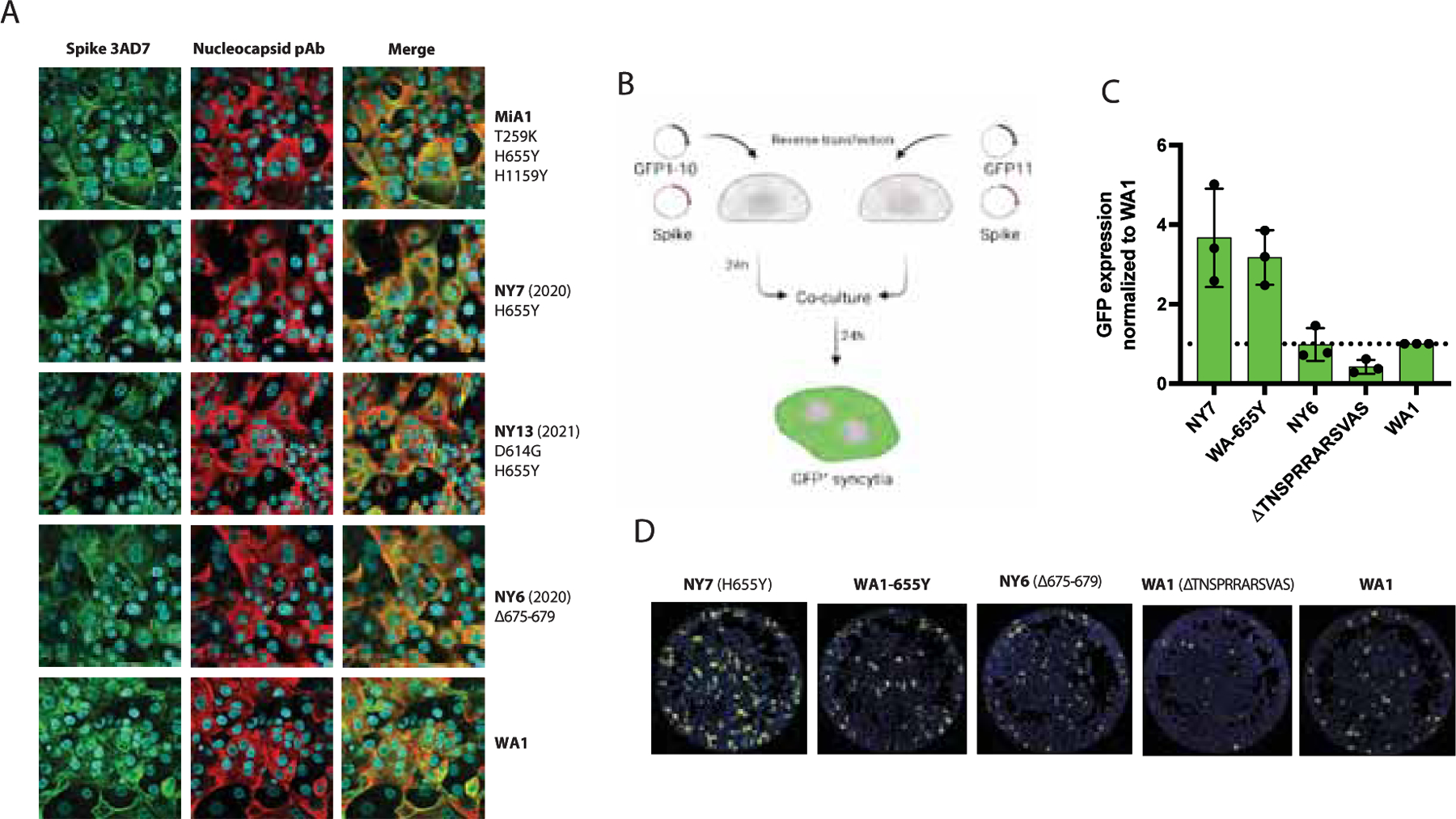
A) Immunofluorescence of SARS-CoV-2 S and N protein localization in Vero-TMPRSS2 infected cells at an MOI of 0.01 and 24 hours p.i. Spike protein was detected using a specific monoclonal antibody 3AD7 (green), N protein was detected using a polyclonal antiserum (red) and 4’,6-diamidino-2-phenylindole (DAPI) was used to stain the nucleus. B) Schematic representation of the split-GFP fusion assay. C) Quantification of cell-cell fusion represented as GFP expression produced by each spike variant over WA1 wild type spike. GFP signals were normalized to spike expression and DAPI counts. Shown are the means and SD of three independent experiments. D) Images showing GFP positive syncytia formation obtained by Celigo Image Cytometer. Cell nuclei was stained using DAPI.
THE 655Y SPIKE POLYMORPHISM INCREASES TRANSMISSIBILITY IN VIVO AND SHOWS ENHANCED PHENOTYPE IN HUMAN PRIMARY AIRWAY MODEL
Syrian golden hamsters are a recognized rodent model to investigate infection and transmission of SARS-CoV-2 (Rosenke et al., 2020; Sia et al., 2020). To test whether the S:655Y polymorphism enhances viral transmission, five pairs of Syrian golden hamsters were placed in individual cages to perform viral competition experiments. For this, one hamster of each pair was infected intranasally with 105 pfu of a mix of WA1 and WA1–655Y viruses at a one-to-one ratio (Figure 4A). Viral input proportion was confirmed by next-generation sequencing (Supplementary Table 6). Direct infected (DI) and direct contact (DC) hamsters were euthanized after day 5 and 7 post-infection, respectively, and lungs and nasal turbinates were harvested for subsequent viral titer quantification. In addition, nasal washes were collected on day 2 and 4 in both DI and DC, and on day 6 p.i. of DC animals. One of the DI hamsters died after nasal wash collection at day 2, leaving 4 animals subjected to follow up in the DI group. Hamsters were also monitored daily for body weight loss. After 2 days p.i. DC hamsters exhibited a decrease in weight indicating early viral transmission from infected animals (Figure 4B). This observation was further supported by detection of infectious virus in nasal washes at 2 days p.i in both DI and DC hamsters. At day 4 p.i., viral titers were detected in 3 out of 4 animals and at day 6 p.i., viral replication was not detected in two of the DC nasal wash samples (Figure 4C). In general, we observed a decrease over time in the infectious virus present in nasal washes from DI and DC animals. We then determined the relative abundance of S:655Y on the viral RNA present in the nasal washes by next-generation sequencing (Figure 4D). The consensus RNA sequence from all DI hamsters contained the S:655Y polymorphism suggesting that WA1–655Y virus was able to overcome the wild type WA1 isolate during infection. Similarly, S:655Y was present in all the nasal washes collected from four DC hamsters indicating an advantage conferred by this mutation in viral transmission. However, one DC animal (hamster 4-C) showed a decrease of 655Y abundance over time. Interestingly, this hamster lost less weight when compared to the rest of the animals. Next, we analyzed the viral growth in lungs and nasal turbinates from DI (collected at day 5 p.i.) and DC (collected at day 7 p.i.) hamsters (Figure 4E). No differences were found in viral titers in the tissues from both animal groups. However, we observed that three DC hamsters had lower lung titers compared to the rest of the animals. These same hamsters also exhibited low viral loads in the nasal turbinates. We then sequenced the viral RNA present in these tissues (Figure 4F–G). The RNA from two lungs and two nasal turbinates from DI and DC hamsters could not be amplified by specific PCR for downstream sequencing. Figure 4F–G shows that all lungs and nasal turbinate tissues from DI animals analyzed had the S:655Y mutation. In addition, S:655Y was present in the nasal turbinates and lungs of 3 out of 4 DC animals (75%) (Supplementary Table 6). Collectively, our results suggest that the S:655Y polymorphism enhances viral transmission in vivo.
Figure 4. The 655Y polymorphism prevails over the 655H in the transmission in vivo model.
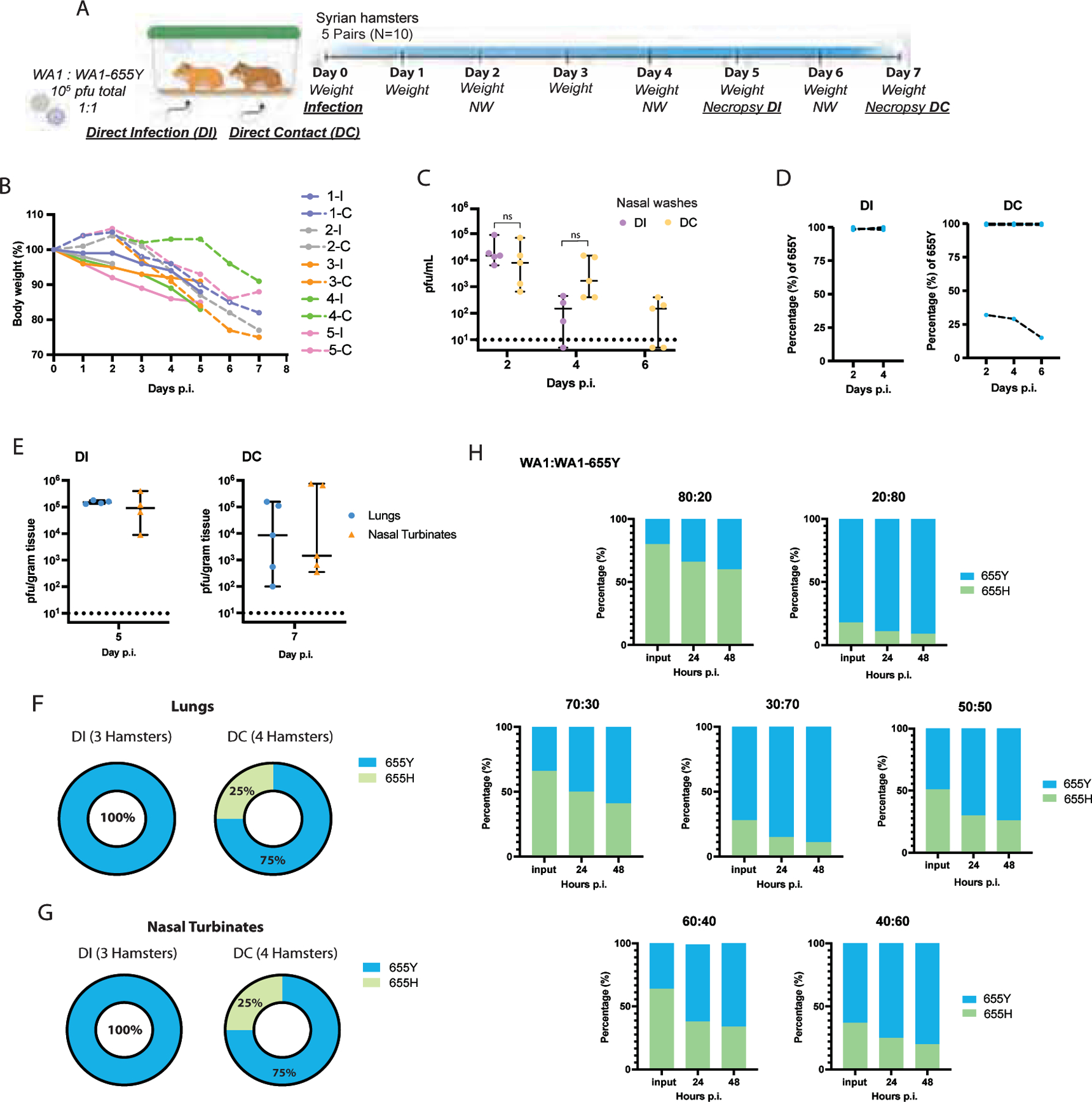
A) Ten 3-weeks-old female Syrian hamsters were placed in pairs. Only one hamster per cage was infected intranasally with a total of 105 pfu of SARS-CoV-2 WA1 and WA1–655Y isolates in a one-to-one ratio. Nasal washes were collected at day 2, 4 and 6 post-infection (p.i.). Lungs and nasal turbinates were harvested from direct-infected (DI) and direct-contact (DC) hamsters at day 5 and 7 p.i., respectively. B) Body weight change of individual hamsters over time. C) Viral titers of nasal washes expressed as PFU per milliliter. Shown are the medians with 95% CI. Mann-Whitney t test was performed to compare differences within each group. Statistical significance was considered when p ≤ 0.05 (ns, not significant). D) Relative abundance of 655Y mutation in the RNA from nasal washes in the DI and DC hamsters. The y axis shows the percentage of 655Y polymorphism in the total good quality sequencing reads from each biological RNA sample and the x axis indicates the day p.i. samples were collected. E) Viral titers of lungs and nasal turbinates expressed as PFU per gram of tissue. Shown are the medians with 95% CI. Mann-Whitney t test was performed to compare differences within each group. Statistical significance was considered when p ≤ 0.05 (ns, not significant). Titers of DI and DC hamsters are shown at day 5 and 7 p.i., respectively. F-G) Proportion of hamsters with 655Y (blue) and H (green) in the nasal turbinates and lungs from DI and DC as confirmed by next generation sequencing. H) Competition experiments between SARS-CoV-2 WA1 and WA1–655Y isolates in human pneumocyte-like cells at different ratios. Proportion of 655Y (blue) and H (green) expressed as percentage are shown for the input and after 24 and 48 hours p.i. Shown are the medians of three independent experiments.
Next, we performed viral competition experiments (Figure 4H) to validate the advantage conferred by the 655Y polymorphism in human pneumocyte-like cells. Cells were inoculated with a total of 2000 pfu per well of the WA1 and WA1–655Y mixture at different ratios (80:20; 20:80, 70:30; 30:70, 60:40; 40:60 and 50:50). Supernatants were collected after 24- and 48-hours p.i and the RNA from these samples was extracted to evaluate the abundance of S:655Y mutation by next-generation sequencing. Viral inputs were also sequence confirmed. After 24 hours p.i., the S:655Y polymorphism became dominant over the isogenic S:655H in all viral ratios we tested. Consistent with our in vivo transmission experiments, most of the viral RNA present in the supernatants of pneumocyte-like infected cells after 48 hours p.i. harbored the 655Y mutation when viral isolates were inoculated at a 50:50 ratio (one to one ratio). Taken together, our viral competition and transmission experiments showed that S:655Y became predominant in both DI and DC animals. Similarly, this mutation was able to overcome the ancestor S:655H in the human airway epithelial system in all viral ratios tested indicating that S:655Y plays a role in SARS-CoV-2 viral transmissibility and replication fitness.
SARS-CoV-2 VARIANTS EVOLVE TO ACQUIRE AN INCREASED SPIKE CLEAVAGE AND FUSOGENIC ABILITY
Current circulating SARS-CoV-2 VOCs bear novel spike polymorphisms that correlate with an enhanced human transmission (Liu et al., 2021) and reduced antibody neutralization (Edara et al., 2021; Harvey et al., 2021). Interestingly, both the Gamma variant (lineage P.1) which emerged in Brazil in November 2020, and the Omicron variant (B.1.1.529) which emerged in South Africa at the end of November 2021 harbor the amino acid spike substitution H655Y. In the context of the evolving epidemiological situation, we decided to investigate whether the co-emergence of different selective mutations in some representative VOC had a similar phenotype to SARS-CoV-2 viruses harboring the S:655Y. We first estimated the amino acid substitution frequencies around the cleavage site region (655 to 701) from globally available data (2,072,987 sequences deposited in GISAID Database up to 28 June 2021). As expected, the P681H/R, H655Y and A701V substitutions showed high prevalence since they are harbored by the main VOCs lineages. Additionally, the less prevalent Q675H and Q677H were also found to be a highly variable position and present in widely circulating variants (Rego et al., 2021). We then spatially and structurally mapped these amino acid changes within and surrounding the furin cleavage sequence of the S protein (Figure 5A). The 655 position was located in close proximity to the furin cleavage site. Next, we performed a phylogenetic analysis of sequences sampled worldwide from February 2020 to June 2021 to illustrate the temporal distribution and phylogenetic relationship of the high prevalent S mutations (Figure 5B). For this, a sample set of 13,847 sequences deposited in GISAID up to June 2021 were analyzed. While the H665Y frequency was higher in the Gamma lineage (P.1), it could be found also in 19B clade (Figure 5C), in line with our identified NY7 isolate. The P681H substitution located in the furin cleavage site of the spike protein was identified in the Alpha variant that emerged in September 2020. Interestingly, this mutation was also found in the Theta variant, first detected in February 2021 (Figure 5D). In contrast, Kappa and Delta variants harbor polymorphism P681R (Figure 5D). Finally, the frequency of A701V mutation was higher in Beta and Iota variants which emerged in October and November 2020, respectively (Figure 5E).
Figure 5. Global epidemiology of SARS-CoV-2 variants of concern (VOCs).
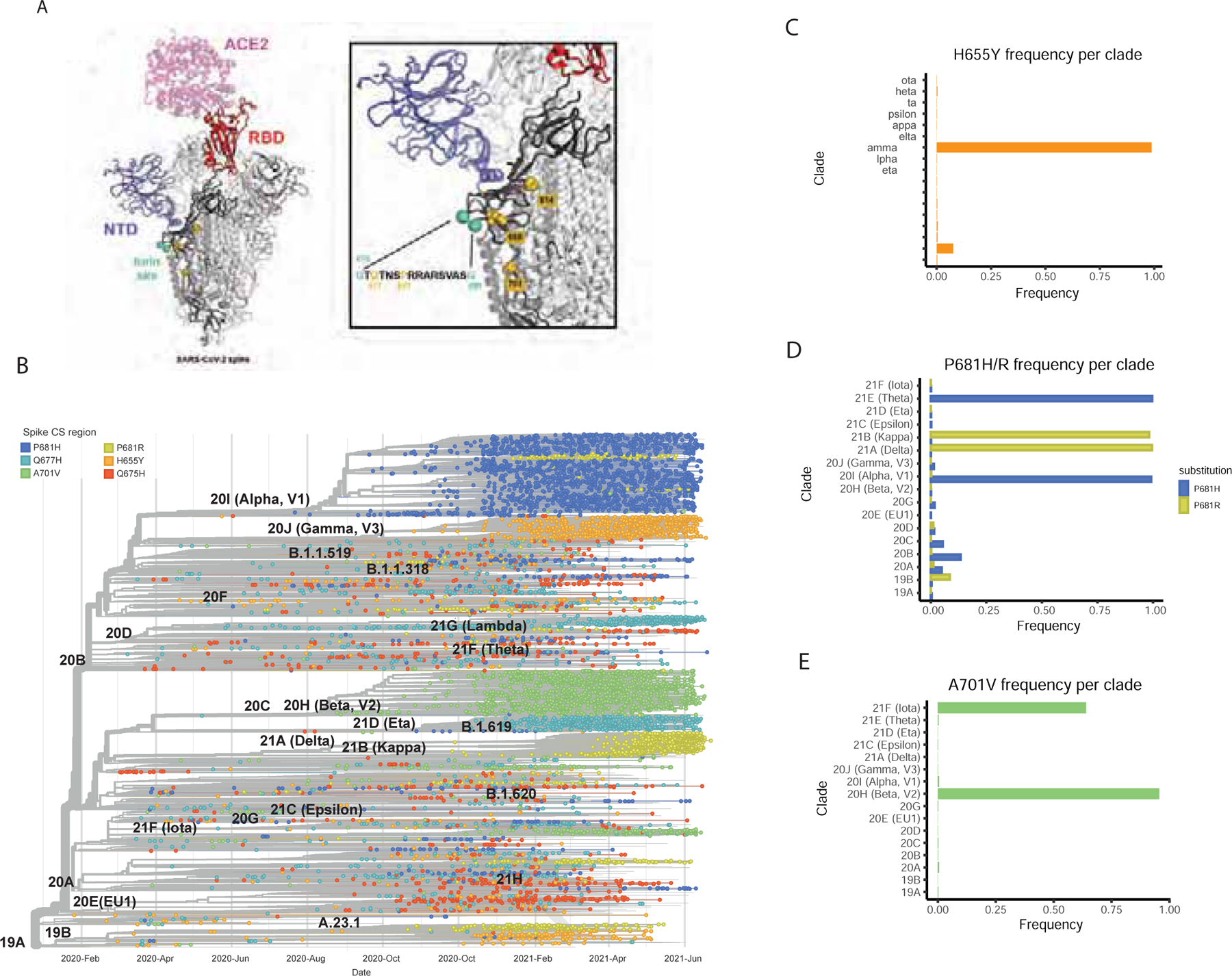
The amino acid substitution frequencies around the cleavage site region (655 to 701) from globally available data (2,072,987 sequences deposited in GISAID Database as of 28 June 2021) was estimated. A) Shows the high prevalent mutations identified mapped onto the structure of the S glycoprotein. The model was generated by superposition of PDB 6M0J and 7C2L (Chi et al., 2020; Lan et al., 2020). One RBD in the up conformation (red) is bound with ACE2 receptor (pink). The NTD is colored blue, the amino-acid substitutions are shown as gold spheres and the furin cleavage loop (disordered and therefore missing in most atomic models) is flanked with cyan spheres. One spike protomer is shown in bold colors while the other two are colored white. A zoomed-in image of the region of interest and the sequence of the furin site loop is also shown. Amino-acid residues of interest are highlighted in gold. B) Time-calibrated phylogenetic tree of SARS-CoV-2 circulating variants illustrating the temporal distribution and phylogenetic relationships of the most prevalent S mutations along the S1/S2 region (highlighted in color). The phylogenetic tree was generated using NextStrain and analysis was performed using a sample of 13,847 genomes focused on the most prevalent substitutions between S:655 and S:701 between February 2020 and June 2021 from GISAID database. C-E) Frequency per clade of H655Y, P681H/R and A701V spike polymorphisms.
Next, we analyzed the in vitro phenotype of some of the most prevalent SARS-CoV-2 VOCs. Multiple protein sequence alignment of the VOCs used are shown in Figure 6A. In contrast to the NY isolates previously analyzed, VOCs showed a considerable number of unique changes across the whole spike protein. However, small differences in replication kinetics and plaque phenotypes were observed in Vero-TMPRSS2 cells (Figure 6B and Supplementary Figure 3B). Conversely, bigger differences were observed when replication efficiency was determined in VeroE6 cells at 24 hours post-infection (p.i.) (Figure 6B). Additionally, viral supernatants were titrated on both VeroE6 and Vero-TMRPSS2 cells (Supplementary Figure 3B). Substantial differences in plaque phenotypes were observed, especially for the Kappa (B.1.617.1) and Delta (B.1.617.2) variants, when TMRPSS2 was present. Thus, these late VOCs that emerged late in the COVID-19 pandemic might strongly depend on the presence of TMRPSS2 to establish optimal infection in vitro. Last, we investigated the extent of the spike cleavage of these VOCs. For this, infections were performed in VeroE6 and Vero-TMPRSS2 at an MOI of 0.01 and viral supernatants were analyzed after 48 hours post-infection (p.i.) by Western blot (Figure 6C). N protein was used as a control for viral replication and loading. Similarly, WA1 was included as a reference since no selective mutations are found in the S protein. Figure 6C shows similar cleaved and uncleaved S protein levels for all the SARS-CoV-2 VOCs in the presence of TMRPSS2 expressed in Vero-TMPRSS2 cells. In contrast, only Beta (B.1.351) and Gamma (P.1), exhibited an increased spike cleavage when the infections were performed in wild type VeroE6 cells. Interestingly, the spike and nucleocapsid expression of Kappa (B.1.617.1) and Delta (B.1.617.2) variants was not detectable by Western blot analysis of VeroE6 supernatants. However, when we performed same experiments in cells extracts, we were able to detect spike protein of both Delta and Kappa variants (data not shown). When we quantified the expression levels of full-length and cleaved spike protein, we observed that Gamma variant had the highest cleavage efficiency in VeroE6 cells. Similarly, Kappa and Delta variants showed the greater levels of cleaved spike in Vero-TMPRSS2 cells compared to the WA1 (Figure 6D). Next, we evaluated the replication kinetics and spike protein processing of the VOCs in the human pneumocyte-like primary cell system. For this, cells were inoculated with 3000 pfu per well of the circulating SARS-CoV-2 VOCs. Viral supernatants were collected after 24- and 48- hours p.i. to determine viral titers by plaque assay; and cell extracts were collected after 48 hours p.i for Western Blot analysis. As shown in Figure 6E, Alpha, Beta and Gamma variants exhibited similar replication efficiency. However, Kappa and Delta variant yielded lower titers in the human epithelial airway. Nonetheless, when we assessed the extent of the spike protein processing, all VOCs showed an enhanced spike cleavage compared to WA1 isolates (Figure 6F and G).
Figure 6. SARS-CoV-2 VOCs evolve to a convergent phenotype associated to an increase on S cleavage.
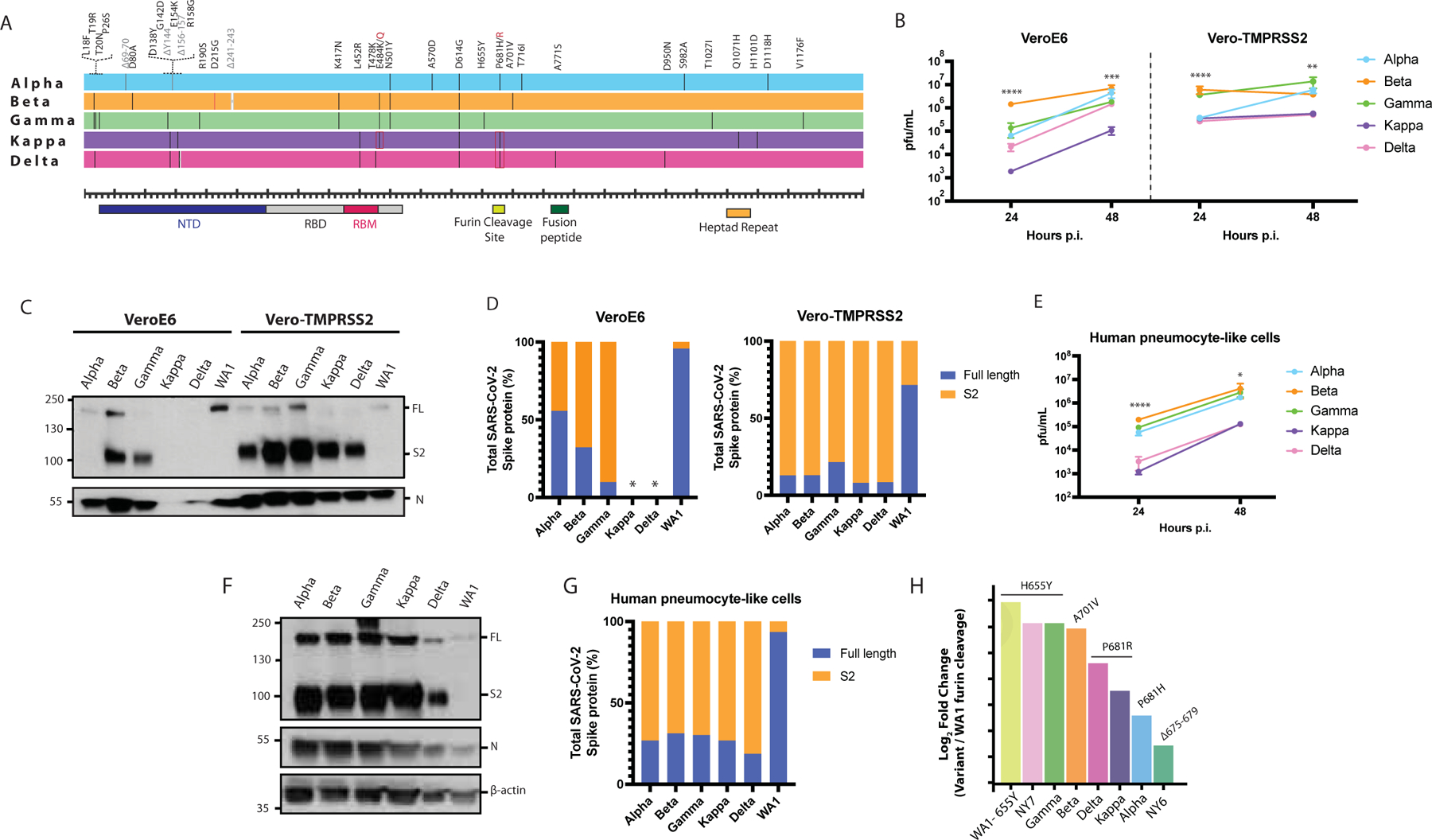
A) Multiple alignments of the S protein of the indicated SARS-CoV-2 VOCs. Diagram shows the corresponding S amino acid substitutions mapped to the S gene. B) Viral growth of SARS-CoV-2 variants in VeroE6 and Vero-TMPRSS2 cells. Infections were performed at an MOI of 0.01. Viral titers were determined by plaque assay at the indicated hours post-infection and expressed as PFU per milliliter Shown are the means and SDs from three replicates. ANOVA test for multiple comparison was used to compare mean differences within different isolates and time points. Viral isolates were compared two by two using the Tukey’s correction. Statistical significance was considered when p ≤ 0.05 (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns, not significant). Color codes relate to the isolates shown in A. C) Western blotting of spike cleavage in supernatants from VeroE6 and Vero-TMPRSS2 infected cells at an MOI of 0.01. Viral supernatants were collected at 48 hours post-infection. Full length (FL) S protein (180 kDa), S2 cleaved spike (95 kDa) and Nucleocapsid (N, 50 kDa) were detected using specific antibodies. Levels of N protein were used as loading control. D) Quantification of full length and cleaved spike protein of the VOCs in VeroE6 and Vero-TMPRSS2 cells. Spike protein levels were normalized to nucleocapsid expression. Asterisk (*) indicates under limit of detection. E) Replication kinetics of SARS-CoV-2 VOCs in human pneumocyte-like cells. Cells were infected with 3000 pfu per well of the corresponding viral isolate. Viral titers were determined by plaque assay at the indicated hours post-infection and expressed as PFU per milliliter. Shown are the means and SDs from three replicates. ANOVA test for multiple comparison was used to compare mean differences within different isolates and time points. Viral isolates were compared two by two using the Tukey’s correction. Statistical significance was considered when p ≤ 0.05 (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns, not significant). F) Western blotting of S protein in human pneumocyte-like cells infected with 3000 pfu per well of the corresponding VOC. Cell extracts were collected at 48 hours post-infection (p.i.). Full length (FL) spike protein (180 kDa), S2 cleaved spike (95 kDa), Nucleocapsid (N, 50 kDa) and β-actin (45 kDa) were detected using specific antibodies. Levels of N and β-actin protein were used as loading control. G) Quantification of full length and cleaved spike protein of the VOCs in human pneumocyte-like cells. Spike protein levels were normalized to nucleocapsid expression. H) Quantification of the cleavage efficiency by mass spectrometry. Vero-TMPRSS2 cells were infected at an MOI of 0.1 with the indicated VOCs and NY7 (S:H655Y) and W A 1–655Y isolates. WA1 and NY6 were used as controls. Cells extracts were collected after 24 hours post-infection. Cleavage efficiency was determined by measuring the abundance of the resulting peptide (SVASQSIIAYTMSLGAE) after cleavage at the terminal arginine of the furin cleavage site. Total spike, ORF3a and N protein were used as internal standard to normalize across variants. The y axis shows the log2 of fold change between cleaved peptide abundance for each variant normalized by WA1 control.
The canonical cleavage at the S1/S2 site occurs at the last arginine (R) of the multibasic PRRAR motif and is performed by furin proteases at this specific residue. Thus, we next quantified the abundance of furin-cleaved peptide of VOCs in Vero-TMPRSS2 cell supernatants by targeted mass spectrometry. Vero-TMPRSS2 cells were infected at an MOI of 0.1 with the indicated VOCs and NY7 (S:H655Y) and WA1–655Y isolates. WA1 and NY6 were used as controls. Cell extracts were collected after 24 hours post-infection and samples were prepared. The abundance of the C-terminal peptide resulting from endogenous furin cleavage at the terminal arginine (PRRAR \ SVASQSIIAYTMSLGAE) was quantified as a proxy of cleavage efficiency since this peptide is common for all SARS-CoV-2 VOCs, except for the Beta that contained a V instead of A at the end of the peptide (SVASQSIIAYTMSLGVE). Fold change peptide-level abundance for each variant compared to WA1 control was calculated and plotted in Figure 6H. Isolates WA1–655Y, NY7 and Gamma, all of them harboring the 655Y mutation, and Beta harboring 701V, showed the higher abundance of furin-cleaved peptide. Conversely, lower levels of C-terminal cleaved peptide were found for the VOCs harboring the 681H/R amino acid change suggesting that an introduction of an amino acid change at this position might modify the canonical cleavage residue at the last R of the furin cleavage site.
Finally, we assessed the fusogenic capacity of the S protein of VOCs by immunofluorescence microscopy of infected Vero-TMPRSS2 cells and quantified by using the split-GFP complementation system. We found that all the variants induced strong syncytium formation (Figure 7). Interestingly, extensive fusogenic capacity was also exhibited by Delta and Kappa variants consistent with abundant cleaved S form found by Western blot (Figure 6C–D, F–G). Because cleavage at the multibasic furin motif is believed to be required for optimal syncytium formation (Hoffmann et al., 2020), we finally compared the ability to induce cell fusion by the Kappa variant and a mutated form lacking amino acids at the furin cleavage site (S: Δ678–682). This Δ678–682 Kappa was obtained after consecutive passage and culturing in VeroE6 cells. As shown in Figure 7B, a loss of fusogenic activity was observed when compared to the intact Kappa VOC. These results were consistent with our previous findings showing that the fusogenic ability of the WA1 spike protein is compromised when the furin cleavage site is deleted (S: ΔTNSPRRARSVAS) (Figure 3C and D). When we quantified the cell fusion mediated by the S protein of the VOCs, we observed that all VOCs S proteins were between 2 and 6-fold more fusogenic than WA1 S (Figure 7C). Additionally, we evaluated the potential role of specific mutations present in the S of VOCs in mediating cell-cell fusion. For this, we generated VOCs spike plasmids containing the ancestor amino acids present in WA1 instead of the amino acid present in the original isolate: H681P in Alpha spike, V701A in Beta, Y655H in Gamma and R681P in Delta spike. Backmutation of S:681H to 681P resulted in a significant reduction in fusion activity compared to the original Alpha spike, yet it remained more fusogenic than WA1 wild type. For Beta, Gamma and Delta spikes, we also observed a slightly decreased in fusogenicity when the ancestral WA1 mutation was present, suggesting that these specific polymorphisms located within or in the close proximity to the furin cleavage site region may play a role in SARS-CoV-2 induced syncytia. To evaluate the individual gain-of-function of each of these polymorphisms, we introduced these individual mutations into the WA1 spike backbone bearing the D614G (WA1614G), since 614G became prevalent and is present in all the VOCs studied here. We observed that the D614G mutation alone (WA1614G) increased fusogenicity compared to the WA1 spike. This is consistent with the advantage in spike processing observed in human lung cells in vitro (Figure 1D). Since it is clear now that D614G also contributes to some extent to spike processing, the impact of single point mutations like H655Y could be masked when 614G was present. Therefore, this indicates all these mutations individually contribute to enhance fusion and their combination might even increase spike processing and cell fusion, especially in conditions with limiting host protease activity, which may occur in vivo.
Figure 7. SARS-CoV-2 VOCs exhibit enhanced cell-cell fusion.
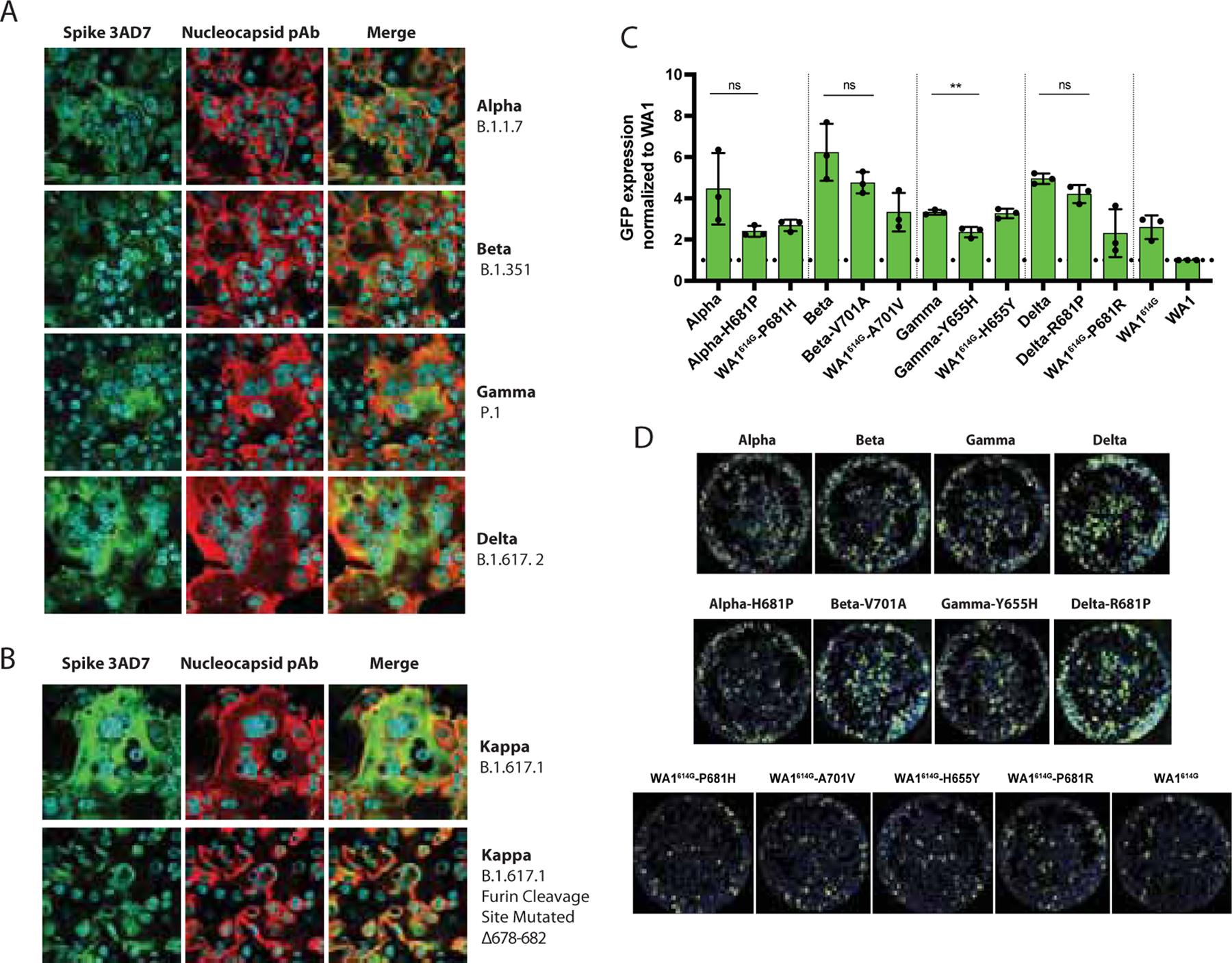
A-B) Immunofluorescence of SARS-CoV-2 S and N protein in Vero-TMPRSS2 infected cells at an MOI of 0.01 and 24 hours p.i. for the indicated VOCs. Spike protein was detected using a specific monoclonal antibody 3AD7 (green), N protein was detected using a polyclonal antiserum (red) and 4’,6-diamidin o -2-phenylindole (DAPI) was used to stain the nucleus. C) Quantification of cell-cell fusion represented as GFP expression showed by each spike VOC over WA1 wild type spike. GFP signals were normalized to spike expression and DAPI counts. Shown are the means and SD of three independent experiments. Two-tailed t test was performed to compare mean differences between each VOC S and corresponding reverse mutant. Statistical significance was considered when p ≤ 0.05 (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns, not significant). D) Images showing GFP positive syncytia formation obtained by Celigo Image Cytometer. Cell nuclei was stained using DAPI.
Altogether, our results are consistent with the notion that current highly transmissible circulating VOCs have evolved independently to acquire mutations associated with increased spike protein processing, transmission and fusogenic potential. Importantly, additional VOCs are rapidly emerging and acquiring an increased number of mutations in the spike. This is the case of the Omicron variant which harbors not only S:H655Y but also N679K and P681H mutations in the furin cleavage site. Here, we provided evidence that some of these specific polymorphisms enhance viral growth, spike cleavage and syncytia formation and therefore, confer an advantage in viral transmission.
DISCUSSION
Emerging SARS-CoV-2 VOCs con ta in novel spike polymorphisms with unclear functional consequences on epidemiology, viral fitness, and antigenicity. In this study, we evaluated the impact of different spike mutations on viral infection, pathogenicity, and in vivo transmission. We found that in the mink animal model the 655Y spike substitution is selected after infection with the WA1 isolate. Phylogenetic analysis of genome sequences collected worldwide showed an early sporadic appearance of S:655Y during the first pandemic wave in New York in March 2020, and the presence of this mutation in several posterior lineages, including SARS-CoV-2 Gamma variant and the Omicron variant (B.1.1.529, first reported in South Africa in November 2021), pointing to a potential role in adaptation and evolution. To better understand the impact of this polymorphism, we isolated and in vitro characterized a panel of SARS-CoV-2 viruses bearing the 655Y spike mutation. Our results demonstrated that S:655Y enhances the viral growth and the spike protein processing required for optimal cell entry and viral-host membrane fusion. In addition, we performed viral competition and transmission experiments in the hamster animal model and human epithelial airway system and showed that S:655Y became predominant in both direct infected and direct contact animals and in the human lung primary cell model. Finally, we showed that VOCs converge to gain spike cleavage efficiency and fusogenic potential.
Here, we demonstrate that viruses containing the H655Y polymorphism confer a growth advantage in both VeroE6, Vero-TMPRSS2 cells and human pneumocyte-like cells. Interestingly, the early human isolate NY7 harboring the 655Y mutation also showed higher replication in human Caco-2 cells. However, it is known that other mutations outside of the S gene could be impacting viral replication and infection (Banerjee et al., 2021; Begum et al., 2020). Therefore, we confirmed the S:655Y mutation alone was responsible for the enhanced growth and spike cleavage phenotype when comparing WA1 wild type and WA1–655Y isolates. These variants have the same viral amino acid sequence except for the amino acid present at position 655 of the spike. Since most of the isolates used in this study contain a constellation of mutations across the genome that could increase viral fitness, comparison of both viruses in parallel allowed to detect differences in growth and spike cleavage that can be attributed only to 655Y polymorphism. The S:655Y is present in the S1 spike domain outside of the RBD and has been associated with a decrease of the neutralizing activity when targeted by some monoclonal antibodies (Baum et al., 2020). However, H655Y has been also naturally selected in cats and mice suggesting a beneficial impact of this substitution in widen viral host range and susceptibility (Braun et al., 2021; Rathnasinghe et al., 2021). Our data further supports this argument because we also found that S:655Y is selected after replication in minks, a natural host for SARS-CoV-2. Besides, when we assessed the viral transmission efficiency of 655Y versus the ancestor 655H in competition experiments in the hamster model, we also found that 655Y becomes more prevalent, as the bulk of infectious viruses recovered from the infected animals harbored this mutation, except for one hamster. This indicates that S:655Y can overcome S:655H in vivo. To recreate SARS-CoV-2 natural infection in the human epithelial airway, we additionally performed viral competition experiments in human pneumocyte-like cells and demonstrated that S:655Y exhibits a higher replication fitness and prevails over its ancestor S:655H.
Intense worldwide surveillance has established that SARS-CoV-2 variants are constantly emerging. In particular, the spike protein has shown high plasticity (Starr et al., 2020). Most of the spike mutations associated with a decrease in neutralization by antibodies against earlier viruses are located in the RBD or N-terminal domain (NTD), which are critical for binding and interacting with the ACE2 cellular receptor. While mutations at these domains may impact SARS-CoV-2 vaccine efficacy, it is also vital to characterize other mutations that might explain the gain in transmissibility observed for the VOCs. Since the Gamma variant that emerged in November 2020 al so harbors the 655Y polymorphism (Figure 6A), we decided to investigate its phenotype in vitro. Similar to the earlier S:655Y isolates, this variant also exhibited an increase in spike processing efficiency. More importantly, this phenotype was also confirmed in all emerging VOCs analyzed when infections were performed in the Vero-TMPRSS2 and human pneumocyte-like cells in vitro indicating that additional mutations within S confer this advantage. Most likely, the spike mutations P681H in Alpha variant -first identified in United Kingdom- and P681R harbored by Kappa and Delta variants -first emerged in India- allowed this enhanced S cleavage. Interestingly, for these three variants, optimal cleavage appeared to be dependent on TMPRSS2 protease activity present in the human epithelial airway (Figure 6C and F). To note, the Omicron variant not only contains the 655Y but also 681H and 614G. Additionally, the N679K polymorphism is also present. Therefore, this combination of mutations points to an enhancement on transmissibility.
To confirm the cleavage at the putative furin cleavage site, we determined the relative abundance of the furin cleaved peptide produced after the 685-terminal arginine. We observed higher amount of cleavage at this position as compared to the previous circulating viruses, although lower amounts were detected in Alpha, Kappa and Delta variants as compared to the viruses harboring the 655Y mutation. This suggests that a change in residue 681 may introduce an additional cleavage site, perhaps recognized by TMPRSS2 protease that enhances spike cleavage of these variants and produces an additional cleavage peptide different in size and amino acid sequences. Further research is needed to confirm the existence of a recognition site for additional proteases different than furin in the amino acid motif SH/RRRAR when the P681R/H mutation is present. In any case, all the VOCs analyzed proved to be strong syncytia inducers which could potentially indicate a role in pathogenesis and lung damage mediated by TMPRSS2 activity after infection in humans (Buchrieser et al., 2020). On the other hand, the Beta variant, which was first identified in South Africa in October 2020, does not contain a change in the furin cleavage site or in the spike position 655, but instead a change in the residue found at position 701. Although this residue is found around 20 amino acids away from the furin cleavage motif, we found similar results when the extent of the spike processing and fusogenicity was investigated (Figure 6C and F, Figure 7). Additionally, we found that P681R/H, H655Y and A701V amino acid substitutions were important fusion inducers since reversion of these positions to the WA1 ancestral sequence decreased spike fusogenicity as shown in a plasmid-based cell-cell fusion assay (Figure 7C–D). It is important to note that the VOCs investigated in here independently acquired S mutations around the furin cleavage site that became epidemiologically more prevalent in humans. When we investigated the spatial distribution by superimposition of the crystal structure of the S protein, we found that these highly prevalent polymorphisms were all located in close proximity to the furin site loop (Figure 5A). Any substitution in this protein domain is likely to have an effect on the structural integrity and dynamics, potentially impacting the accessibility of the polybasic site to the relevant protease and likely facilitating the recognition by furin as well as cell-cell mediated fusion.
In summary, our study demonstrates that the 655Y spike polymorphism, present in the Gamma and now Omicron VOC, is a key determinant of SARS-CoV-2 infection and transmission. The selection and increasing frequency of S:655Y in the human population and following SARS-CoV-2 infection of different animal models such as mice and minks suggests this mutation is associated with an improvement of viral fitness and adaptation to diverse hosts through an increased cleavage of the spike protein. Additionally, we provide evidence of adaptative mutations that SARS-CoV-2 V OCs have been acquired and are associated with an increased spike protein processing and fusogenic potential. This has significant implications in the understanding of the viral determinants that can impact viral transmissibility, viral evolution, and possibly SARS-CoV-2 antigenicity and pathogenicity.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact:
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Adolfo Garcia-Sastre (adolfo.garcia-sastre@mssm.edu)
Materials availability:
All materials and plasmids generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement (MTA).
Data availability:
All data is available in the manuscript or the supplementary materials. The targeted mass spectrometry data including raw files, Skyline document and resulting transition lists have been deposited at ProteomeXchange (PXD027641). Next generation sequencing raw data for the competition experiments in hamsters and pneumocytes have been deposited in SRA. BioProjectID PRJNA793894 and BioSample accessions SAMN24592477 to SAMN24592565. Reagents used are almost exclusively commercially available and non-proprietary. Any additional information required to reanalyze the data reported in this work paper is available from th e Lead Contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines:
VeroE6 and Caco-2 cell lines were originally purchased from the American Type Culture Collection (ATCC). VeroE6-TMPRSS2 cell line was purchased from BPS Bioscience. A master cell bank was created for each cell line and early-passage cells were thawed in every experimental step. VeroE6 and Caco-2 cell lines were maintained in Dulbecco’s modified Eagle’s medium (DMEM) with glucose, L-glutamine, and sodium pyruvate (Corning, 10–017-CV) supplemented with 10% fetal bovine serum (FBS, Gibco, 16140071), non-essential amino acids (Corning, 25–025-CI), penicillin (100 UI/mL), streptomycin (100 UI/mL) (Corning, 30–002-CI) and normocin (100 ug/mL) (InvivoGen, ant-nr-1) to prevent mycoplasma infection. VeroE6-TMPRSS2 cell line was cultured in the same growth media described above and further supplemented with sodium pyruvate (Corning, 25–000-CI) and puromycin (3 ug/mL) (InvivoGen, ant-pr-1). All cell lines were grown at 37°C in 5% CO2.
Plasmids:
Spike expression plasmids were constructed by extracting viral RNA from the corresponding SARS-CoV-2 viral stocks using E.Z.N.A Viral RNA kit (Omega Bio-Tek, R6874) and further amplification of full-length spike using the SuperScript IV One-Step RT-PCR System (Thermo Fisher, 12594025). Spike cDNA fragments were subcloned into the pCAGGS vector using In-Fusion HD cloning kit (Takara, 638948). Mutations in the S coding sequence were introduced via PCR with overlapping primers and inserted into pCAGGS using the In-Fusion HD cloning kit (Takara, 638948). All plasmids were subjected to sequencing analysis (Psomagen) to verify the integrity of the construct and the presence of the desired mutations. Sequence data were analyzed using SnapGene 5.3.2 software.
Viruses:
All experiments in this study were performed in the BSL-3 facility following Icahn School of Medicine biosafety guidelines. Human SARS-CoV-2: nasopharyngeal swab specimens were collected as part of the routine SARS-CoV-2 surveillance conducted by the Mount Sinai Pathogen Surveillance program (IRB approved, HS#13–00981). Specimens were selected for viral culture on Vero-E6 cells based on the complete viral genome sequence information (Gonzalez-Reiche et al., 2020). Human isolates NY1 to NY12 were obtained from the nasopharyngeal swabs of patients infected with SARS-CoV-2 in March 2020 while NY13 (PV28021) was cultured from the nasopharyngeal swab of a patient infected in February 2021. All nasal swabs were kindly provided by Dr. Viviana Simon. GISAID accession numbers of these isolates are shown in Supplementary Table 1. Viral transport media was incubated with VeroE6 cells until cytopathic effect was observed and supernatants from infected cells were used for plaque purification of clonal population as previously described (Aydillo et al., 2020). SARS-CoV-2 isolates USA-WA1/2020 (NR-52281), hCoV-19/England/204820464/2020 (NR-54000, Alpha) and hCoV-19/Japan/TY7–503/2021 (NR-54982, Gamma) were obtained from BEI resources. hCoV-19/USA/MD-HP01542/2021 JHU (Beta variant) was a gift from Dr. Andrew Pekosz. Kappa and Delta variants were kindly provided by Dr. Mehul S. Suthar and Dr. Viviana Simon, respectively. Animal SARS-CoV-2: Mink SARS-CoV-2 variants (MiA-1 and MiA-2) were isolated during the mink experiment described below. After plaque assay analysis of the left cranial lung collected from a WA1 infected mink at 4 days p.i., two plaque phenotypes were observed. Only the small plaque phenotype viruses were grown and used in this study. USA-WA1/2020-H655Y (WA1–655Y) was kindly provided by Dr. Jürgen Richt. USA-WA1/2020 - H655Y (WA1-H655Y) was isolated via plaque purification in VeroE6 cells. Briefly, wild-type USA - WA1/2020 isolate was passaged once in VeroE6 cells, which contained a minor population of the H655Y mutation (0.7%) as confirmed by next generation sequencing. The passage 1 stock was then subjected to plaque purification on VeroE6 cells. Plaques were picked and expanded via two passages on VeroE6 cells to obtain a viral stock with 100% H655Y mutation and no other non-synonymous mutations >5% compared to the wild-type WA1/2020 reference. All the viral stocks were produced by infecting VeroE6 or Vero-TMPRSS2 cells at a MOI of 0.01. Infected cells were maintained in infection media ( DMEM with glucose, L-glutamine, and sodium pyruvate supplemented with 2% FBS, non-essential amino acids, HEPES, penicillin (100 UI/mL) and streptomycin (100 UI/mL)) at 37°C in 5% CO2. Infected cells were monitored by microscopy and cell-infected supernatants were collected at day 2 post-infection when cytopathic effect was observed. Viral supernatants were clarified of cell debris by spin down followed by centrifugation at 2000 × g for 20 min in Amicon Ultra-15 centrifugal filters (Sigma, 100 kDa cutoff) to concentrate the viral stocks. Aliquots were stored at −80°C until titration by plaque assay. All SARS-CoV-2 variants were sequence-confirmed before performing the experiments.
Animal models: Female golden Syrian hamster of approximately 4-weeks-old were purchased from Charles River Laboratories (Crl:LVG(SYR)). Hamsters were housed in pairs under specific pathogen-free conditions in ventilated cages. Female American Mink (Neovison vison) of approximately 6-months-old were sourced by Triple F Farms (Gillett, PA). All mink were individually housed, given ad libitum access to food and water, and maintained on a 12-hour light/dark cycle. Experiments with infected SARS-CoV-2 hamsters and mink were performed in an ABSL-3 facility of the Emerging Pathogens Institute at the Icahn School of Medicine at Mount Sinai (ISMMS). All animal studies were approved by the Institutional Animal Care and Use Committee (IACUC) of ISMMS.
METHOD DETAILS
Human pneumocyte-like cell generation:
Human embryonic stem cell lines hPSC1 (H9, WiCell) were cultured with mTeSR (Stemcell Technologies, 85850) on Vitronectin XF (Stemcell Technologies, 07180)-coated tissue culture plates. Cells were split in a ratio of 1:6 to 1:12 every 4–6 days using Gentle dissociation reagent (Stemcell Technologies, 07174). Pneumocyte differentiation was induced as previously described (Riva et al., 2020; White et al., 2021). Briefly, cells were collected at 70–80% confluency and approximately, 2 million cells per 10 cm2 were plated on 12-well Vitronectin-coated tissue culture plates in mTeSR. The next day, definitive endoderm differentiation was induced as previously described (Jacob et al., 2019). Cells were split after 4 days and further induced to differentiate based on an adapted alveolar differentiation protocol (Ghaedi et al., 2013) in Iscove’s modified Dulbecco’s medium (IMDM, Life Technologies, 31980030) supplemented with 10% FBS (Sigma, F4135), 2 mM L-glutamine (Life Technologies 25030081), 0.5 μM all-trans-retinoic acid (Sigma, R2626), 10 ng/ml FGF-10 (R&D Systems, 345-FG-025), 10 ng/ml EGF (R&D Systems, 236-EG-01M), 100 ng/ml Wnt3a (R&D Systems, 5036-WN-010), 10 ng/ml KGF (R&D Systems, 251-KG-050) and 5 ng/ml BMP-4 (R&D Systems, 314-BP-010). Viral infections were performed on day 11 of differentiation.
Infection of cell cultures:
Approximately 3.2 × 105 VeroE6 or Vero-TMPRSS2 or Caco-2 were seeded in a 12 well-plate and cultured at 37°C in 5% CO2 for 16 hours. Cells were infected with the corresponding SARS-CoV-2 isolate at an MOI of 0.01. Alternatively, human pneumocyte - like cells were infected with 3000 plaque forming units (PFU) per well in 12 well-plate or 2000 PFU/well in a 24 well-plate. Cells were incubated with the virus for 1 hour and then, cells were washed with PBS to ensure removal of non-attached virus. After infection, cells were maintained in infection media or the pneumocyte supplemented IMDM media (described above). Supernatants or cell extracts were collected at the indicated time points and stored at −80°C for plaque assay analysis and virus quantification.
Western blotting:
VeroE6, Vero-TMPRSS2 cells and human pneumocyte-like cells were infected with the indicated SARS-CoV-2 isolates, similar to the description above. Viral supernatants and cell extracts were collected at 24- and 48-hours post-infection. Supernatants were clarified by low-speed spin. Viral supernatants and cell extracts were mixed with RIPA buffer (Sigma, R0278) containing EDTA-free protease inhibitor cocktail (Roche, 04693132001) and 10% SDS (Invitrogen, AM9822) to a final concentration of 1%. Then, samples were boiled for 10 minutes at 100°C and centrifuged for 10 minutes at 4°C and maximum speed. Viral supernatants were subjected to SDS-PAGE protein electrophoresis using precast 10% TGX gels (BioRad, 4561036). Gels were run at 120 V and subsequently transferred to polyvinylidene fluoride (PVDF) membranes (BioRad, 704156) using BIO-RAD semi-dry transfer system. Then, membranes were fixed with 100% methanol for 1 minute and blocked with 5 % non-fat dry milk-containing Tris-buffered saline with Tween-20 (TBST) with 0.1% Tween-20 for 1 hour in shaking and room temperature (RT). Next, membranes were incubated with primary antibodies overnight at 4°C followed by incubation with secondary antibodies in a 3% milk diluted in TBST for 1 hour at RT. Primary antibodies against SARS-CoV-2 Spike S2 protein (ThermoFisher, MA5–35946) and nucleocapsid (Novus Biologicals, NB100–56576) were purchased from the indicated suppliers and used at a dilution of 1:3000 and 1:2000, respectively. Anti-mouse secondary IgG-HRP antibody (Abcam, 6823) was used at a dilution 1:5000 to detect SARS-CoV-2 Spike protein and anti-rabbit secondary IgG-HRP antibody (Kindle Biosciences, R1006) at 1:2000 to detect SARS-CoV-2 nucleocapsid. β-actin was detected using IgG-HRP antibody (Cell Signaling; 5125) at dilution of 1:1000. Western blot quantification was performed using ImageJ and the level of full length and cleaved spike protein were normalized to nucleocapsid protein.
Plaque assay:
To determine viral titers, 3.2 × 105 VeroE6 or VeroE6-TMPRSS2 were seeded in a 12 well-plate the day before plaque assay was performed. Briefly, ten-fold serial dilutions were performed in infection media for SARS-CoV-2 and inoculated onto confluent VeroE6 or VeroE6-TMPRSS2 cell monolayer. After one-hour adsorption, supernatants were removed, and cells monolayers were overlaid with minimum essential media (MEM) containing 2% FBS and purified agar (OXOID) at a final concentration of 0.7%. Cells were then incubated 3 days at 37°C. Cells were fixed overnight with 10% formaldehyde for inactivation of potential SARS-CoV-2 virus. Overlay was removed and cells were washed once with PBS. Plaques were visualized by immunostaining. Briefly, cells were blocked in 5 % milk diluted in TBST. After 1-hour, anti-mouse SARS-CoV-2- NP antibody (1C7C7, kindly provided by Dr. Moran) was added at a dilution of 1:1000 in 1% milk-TBST and incubated for 1 hour at RT. Then, cells were washed two times in PBS and stained with goat anti-mouse secondary IgG-HRP antibody (abcam, 6823) at a dilution of 1:5000 in 1% milk-TBST and incubated for 1 hour at RT. Finally, cells were washed three times and the plaques were developed with TrueBlue substrate (KPL-Seracare, 5510–0030). Viral titers were calculated considering number of cells per well and expressed as plaque forming units (PFU)/ml.
RT-qPCR for viral infectivity analysis:
To quantify the levels of SARS-CoV-2 RNA after infection in VeroE6 and Caco-2 cells, we used the CDC 2019-nCoV real-time RT-qPCR protocol, with modifications. Primers and probes were purchased from the indicated supplier (Integrated DNA Technologies, 10006713, RUO Kit) and consisted of two 2019-nCoV-specific sets (N1, N2). Names and sequences of the primers used are shown in Supplementary Table 3. Assays were run in a 384-well format using the QuantiFast Pathogen RT-PCR + IC Kit (QIAGEN, 211454). USA/WA1/2020 SARS-CoV-2 RNA (20,000 genome copies per reaction) and nuclease-free water were included as controls. Reactions were performed in duplicate using the following cycling conditions on the Roche LightCycler 480 Instrument II (Roche Molecular Systems, 05015243001): 50 °C for 20 min, 95 °C for 1 s, 95 °C for 5 min, followed by 40 cycles of 95 °C for 15 s and 60 °C for 45 s. To determine the limit of detection for SARS-CoV-2, we used a commercially available plasmid control (Integrated DNA Technologies, 10006625). Infectivity was calculated as a ratio between genomic RNA calculated by RT-qPCR and PFU values determined by plaque assay analysis.
Immunofluorescence:
VeroE6-TMPRSS2 cells were seeded at a concentration of 3.2 × 105 cells per well in a 12 well glass-bottom plate and cultured at 37°C in 5% CO2 for 16 hours. Cells were then infected with the corresponding SARS-CoV-2 variant at an MOI of 0.01 and maintained in infection media. After 24 post-infection, cells were fixed with 10% methanol-free formaldehyde and incubated with primary antibodies against spike KL-S-3A7 (Amanat et al., 2021) and nucleoprotein polyclonal anti-serum (Spiegel et al., 2005) diluted in 1% bovine serum albumin (BSA) for 1 hour at RT. Then, cells were washed and stained with secondary antibodies anti-mouse Alexa Fluor-488 (ThermoFisher, A21202) and anti-Rabbit Alexa Fluor 568 (ThermoFisher, A11011) in 1% BSA for 1h at RT. DAPI (4’,6-diamidino-2-phenylindole) was used to visualize the nucleus.
Cell-cell fusion assay:
This assay is based on a split-GFP system (pQCXIP-GFP1–10 and pQCXIP-GFP11) where GFP signals are produced upon cell fusion (Kodaka et al., 2015). The split GFP plasmids: pQCXIP-BSR-GFP1–10 and pQCXIP-GFP11 were a gift from Yutaka Hata (Addgene plasmid #68715, https://www.addgene.org/68715/ and #68716 https://www.addgene.org/68716/). Briefly, 200,000 Vero-TMPRSS2 cells were co-transfected with 500 ng pCAGGS- spike and 500 ng pQCXIP-GFP110 or pQCXIP-GFP11 using TransIT-LT1 (Takara; MIR2300). The next day, cells were detached, pooled and reseeded in black 96-well or 24-well plates (Greiner, M0562–32EA) at 25,000 and 200,000 total cells/well, respectively. After 24 h, cells were fixed with 4% paraformaldehyde in PBS for 10 min. To measure spike expression, cells were permeabilized with 0.1% Triton X-100 for 15 min and stained with primary antibody anti-S2 (Sino Biological, 40590-D001) and secondary Alexa-Fluor 594 goat anti-human antibody (Thermo Fisher, A-11014). DAPI was used to visualize the nucleus. Images of the whole wells were obtained and analyzed using the Celigo Image Cytometer (Nexcelom). GFP signals were normalized to spike expression and DAPI counts to represent fusion activity.
Hamster infections:
Only one hamster per cage was inoculated intranasally with a total of 105 pfu of a mix of WA1 and WA1–655Y SARS-CoV-2 viruses in a one-to-one ratio administered in 100 ul of PBS. Animals were monitored daily for body weight loss. On days 2, 4, 6 post-infection, animals were anesthetized with 100 mg/kg Ketamine and 20 mg/kg Xylazine and nasal washes were collected in 200 ul PBS. Directly infected (DI) and direct contact (DC) hamsters were humanely euthanized for collection of lungs and nasal turbinates on day 5 and 7 post-infection, respectively. Anesthetized hamsters were euthanized by intracardiac injection of sodium pentobarbital (Sleepaway - Zoetis) euthanasia solution. Samples were collected for viral quantification by plaque assay and next-generation sequencing.
Mink infections:
Six minks were infected with an infectious dose of 106 pfu of WA1 isolate administered intranasally in a 1 mL volume. Three minks were mock-infected to serve as healthy controls. Minks were anesthetized by intramuscular administration of 30 mg/kg Ketamine and 2 mg/kg Xylazine prior to intranasal infection, collection of specimens, or euthanasia. Nasal washes, rectal swabs, and oropharyngeal swabs were collected on days 1, 3, and 5 post-infection. On days 4 and 7 post-infection, three mink per day were humanely euthanized for collection of tissue specimens for viral quantification by plaque assay and sequencing. Body weights of mink were collected days 0, 1, 3, 4, 5 and 7 post-infection. Anesthetized minks were euthanized by intracardiac injection of sodium pentobarbital (Sleepaway - Zoetis) euthanasia solution.
Sample preparation for targeted proteomics:
VeroE6-TMPRSS2 cells were seeded in a 6 well-plate and infected with the corresponding SARS-CoV-2 variants at an MOI of 0.1. Cells were lysated after 24 hours with RIPA buffer containing EDTA-free protease inhibitor cocktail and 10% SDS to a final concentration of 1%. Then, samples were boiled for 10 minutes at 100°C. Cells were centrifuged at 12k rpm on a tabletop centrifuge at RT for 20 minutes to remove insoluble debris and separated into three samples to assess technical reproducibility. 50 ul for each sample were loaded in a 1:4 ratio (v/v) with urea buffer (8M urea, 100 mM AmBic pH 8.1) on a Microcon 30 kDa MWCO (Millipore, Sigma) and centrifuged to dryness at 9500 rpm for 15 minutes at RT, until all sample was loaded. The filters were washed three times with 200 ul of urea buffer using similar centrifugation parameter as the sample loading. 100 ul of reduction buffer (8 M urea, 100 mM AmBic pH 8.1, 5 mM TCEP) was added and the samples were incubated at 37°C for 20 minutes to reduce the cysteines. Chloroacetamide (CAA) was added to 10 mM final concentration and the samples were incubated for 30 minutes in the dark at RT. The filters were washed 3 times with 200 ul of urea buffer and 3 times with 200 ul of digestion buffer (50 mM AmBiC). GluC was added to samples in a 1:100 ratio (w/w) and the filters were incubated on a shaker for 16 hours at 37°C and 450 rpm. Peptides were collected by centrifugation and the filters were washed twice with 100 ul of LC-MS grade water. Desalting was done using the Nest group microspin C18 columns. Activation of the resin was done with 1 column volume (CV) of MeCN and the columns were equilibrated with 2 CV of 0.1% FA in water. Samples were loaded and flowthrough was loaded again before washing the columns with 3 CV of 0.1% FA in water. Peptide elution was done with 2 CV of 50% MeCN in 0.1% FA and 1 CV of 80% MeCN in 0.1% FA. Following collection, the peptides were dried under the vacuum. Samples were resuspended at 1 ug/ul in 0.1% FA and approximately 1 ug was injected into the mass spectrometer.
Mass spectrometry:
All samples were acquired on a Thermo Q Exactive (Thermo Fisher) connected to a nanoLC easy 1200 (Thermo Fisher). Samples containing mutation in the region of interest (furin cleavage) as well as one representative sample having conservation in the furin cleavage site were analyzed by Data Dependent Acquisition (DDA) MS to obtain fragments library to design the targeted assays. For DDA the peptides were separated in 120 minutes with the following gradient: 4% B (0.1% FA in MeCN) to 18% B for 65 minutes, followed by another linear gradient from 18% to 34% of B and lastly the organic solvent was increased to 95% in 5 minutes and kept for 5 minutes to wash the column. The mass spectrometer was operated in positive mode, each MS1 scan was performed with a resolution of 70000 at 200 m/z. Peptide ions were accumulated for 100 ms or until the ion population reached an AGC of 3e6. The top 15 most abundant precursors were fragmented using high-collisional-induced dissociation (HCD) with a normalized energy of 27 using an isolation window of 2.2 Da and a resolution of 17500 (200 m/z). For targeted analysis, the samples were separated in 62 minutes to concentrate the analytes in narrower peaks and increase signal. The gradient employed was from 3% B to 34% in 40 minutes then B was increased to 42% in 10 minutes and then finally to 95% in 5 minutes. As for the DDA the column was washed for 5 minutes at 95% B before the next run. The mass spectrometer was operated in positive mode and targeted acquisition. Specifically, one MS1 scan (70000 resolution, 1e6 AGC, 100 ms IT) and seven unscheduled targeted MS2 scan were performed per cycle. Each MS2 was acquired at 35000 resolution, with a AGC of 2e5 a maximum IT of 110 ms and an isolation window of 2.0 m/z. Isolated ions were fragmented using HCD at 27 NCE.
Illumina sequencing:
Viral RNA present in the nasal washes of the hamsters and in the supernatants from the competition assay in pneumocytes were extracted using E.Z.N.A Viral RNA kit (Omega Bio-Tek, R6874) following manufacturer’s instructions. Viral RNA from hamster nasal turbinates and lungs was isolated Direct-zol RNA Miniprep kit (Zymo Research, R2050) using manufacturer’s instructions. Samples for sequencing were prepared using whole-genome amplification with custom designed tiling primers (Quick et al., 2017) and the Artic Consortium protocol (https://artic.network/ncov-2019), with modifications. Briefly, cDNA synthesis was performed with random hexamers and ProtoScript II (New England Biolabs, E6560) using 7 μL of RNA according to manufacturer’s recommendations. The RT reaction was incubated for 30 minutes at 48°C, followed by enzyme inactivation at 85°C for 5 minutes. Targeted amplification was performed as previously described (Gonzalez-Reiche et al., 2020). Next, amplicons were visualized on a 2% agarose gel and cleaned with Ampure XT beads. Amplicon libraries were prepared using the Nextera XT DNA Sample Preparation kit (Illumina, FC-131–1096), as recommended by the manufacturer. Finally, to assembly SARS-CoV-2 genomes a custom reference-based analysis pipeline was used (https://github.com/mjsull/COVID_pipe). For the hamster samples and the inoculum, in addition to whole genome sequencing, the same workflow was used to sequence a 2,223 bp amplicon targeting the S1 to S1/S2 region (nucleotide positions 21,386 to 23,609) to quantify the variant frequency at the position S:655 position.
Oxford Nanopore sequencing:
The frequency of variants at position S:655 in the hamster and pneumocyte samples was further confirmed with Oxford Nanopore (ONT) sequencing in a MinION Mk1C instrument. The same cDNA used for Illumina sequencing was used to amplify a fragment of 356bp along the spike region of interest (positions 23,468 to 23,821) using the Artic primer pair nCoV-2019_78 V3 (https://artic.network/ncov-2019). The purified amplicons were barcoded with the Native Barcode expansion kit (Oxford Nanopore, EXP-NBD196) and PCR-free libraries were prepared using the ligation sequencing (Oxford Nanopore, SQK-LSK109). A total of 20 fmol of the multiplexed library was sequenced on a Flongle flowcell (Oxford Nanopore, FLO-FLG001) in a single 8-hour run. Sequencing data acquisition was done with the ONT software MinKNOW v4.3.7. Basecalling and demultiplexing was done with Guppy 5.0.12 in high accuracy basecalling mode. The Genome assembly was done with the Artic pipeline (artic-network/artic-ncov2019) with default parameters, where reads were aligned to the reference genome Wuhan-Hu-1 (MN908947.3) using minimap2 (2.17-r941) (Li, 2018), consensus variants were called with Nanopolish (0.13.2). Final read coverage for the targeted region ranged from 80 to 3251x, (median coverage of 1578x).
QUANTIFICATION AND STATISTICAL ANALYSIS
Multiple alignments of spike proteins: multiple alignment of the spike protein of mink and human NY variants (Supplementary Figure 1D and 2A) was performed using COBALT multiple alignment tool (Papadopoulos and Agarwala, 2007). Spike amino acid sequence of SARS-CoV-2 ancestor (MN908947.3) was used to compare and identify the spike mutation of these variants. For the analysis of high prevalent amino acid changes within and surrounding the furin cleavage of the spike protein of current circulating VOCs, amino acid substitution frequencies around the cleavage site region (655 to 701) were estimated from globally available data (Available from GISAID [PMID: 31565258] as of 2021-06-28 with 2,072,987 records). The downloaded data was processed through the augur pipeline with Nextstrain v7 for SARS-CoV-2 (https://github.com/nextstrain/ncov) for sequence alignment and curation with default parameters (Hadfield et al., 2018). Multiple sequence alignment of the spike amino acid sequences was parsed and sliced for the region of interest in Biophyton v.75 with BioAlignIO package. In order to determine the more prevalent substitutions, sequences with ambiguous consensus calls were removed, and residues with substitutions along the 655–701 region present in frequencies of 0.05% or less across the entire dataset were masked. The 5% cutoff value was chosen based on the frequency distribution of substitutions per site on a histogram that revealed that variants in most positions occur in frequencies of less than 0.2%. The final list of most variable positions included residues 655, 675, 677, 681 and 701. The relative proportion of their occurrence time and Nextstrain clade (https://nextstrain.org/blog/2021-01-06-updated-SARS-CoV-2-clade-naming) was plotted in R with ggplot2 (Wickham) v3.3.5.
Phylogenetic analysis: phylogenetic analysis was performed using the same dataset from above to build a time-calibrated phylogenetic tree with Nextstrain, to visualize the distribution of H655Y (up to 2020-09-30) and other prevalent substitutions along the 655 - 701 region (up to 2021-06-23). A global subsampling scheme with a focus on the variable residues was done to ensure their representation by geographical region and over time. The final builds contained 7,059 (early) and 13,847 (late) sequences including the early New York isolates.
Mass spectrometry analysis : For the mass spectrometry analysis, DDA data was searched with MSFragger (Kong et al., 2017) using a FASTA file combining the human proteome, the SARS-Cov2 proteome, all the variants and the C-term and N-term cleaved Spike protein entry for each variant. The Speclib workflow was used to generate a library which was imported into Skyline (MacLean et al., 2010) for selection of peptides and internal controls. Overall, 3 versions of the C-term furin cleaved peptide (SVASQSIIAYTMSLGAE) with two charge states (2+ and 3+) and the oxidated methionine were used. 4 other peptides were included as controls: 2 from the C-term spike fragment to be used as proxy for total spike quantity and 2 from ORF3a and N protein to be used as internal standard to normalize across variants. Following acquisition, the PRM data was imported into the Skyline document with the following transition settings: MS1 filtering was enabled, and MS/MS filtering was changed to targeted using Orbitrap as mass analyzer (35000 resolution) and high selectivity extraction. A minimum of 6 transitions and a maximum of 18 having m/z > precursors were selected for data analysis. After manual peak boundaries selection and elimination of interferences the transition results were exported. Transitions where the signal/background ratio was less than 5 were removed to ensure robust quantitative accuracy. The transitions were summed within the same charge state and the 2+ unmodified SVASQSIIAYTMSLGAE was used for quantification. The data was normalized using median centering of the other Spike peptide (ILPVSMTKTSVD) as internal standard. Following normalization, log2 fold change was calculated by averaging the intensities for the furin-cleaved peptide per variant and divide them by the one from the WA1 variant used here as control sample. Resulting ratios were logged and used for visualization and statistical analysis.
Statistical analysis: statistical analysis was performed using GraphPad Prism. Two-way ANOVA with Holm-Šídák posttest and one-way ANOVA with the Tukey’s correction was used for multiple comparisons. Two-tailed t test was performed to compare mean differences in cell fusion between each VOC S and corresponding reverse mutant. Mann-Whitney t test was performed to compare differences in viral titers in nasal washes, nasal turbinates and lungs of direct infected and contact hamsters. Statistical significance was considered when p ≤ 0.05 (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns, not significant).
Supplementary Material
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| SARS-CoV-2 Spike Protein S2 mAb (1A9) | Thermo Fisher Scientific | Thermo Fisher Scientific Cat# MA5–35946, RRID: AB_2866558 |
| SARS-CoV-2 Nucleocapsid Protein antibody | Novus Biologicals | Novus Cat# NB100–56576, RRID: AB_838838 |
| Anti-mouse IgG-HRP antibody | Abcam | Abcam Cat# ab6823, RRID: AB_955395 |
| Anti-rabbit IgG-HRP antibody | Kindle Biosciences | Kindle Biosciences Cat# R1006, RRID: AB_2800464 |
| β-Actin IgG-HRP mAb (13E5) | Cell Signaling Technology | Cell Signaling Technology Cat# 5125, RRID: AB_1903890 |
| Anti-mouse SARS-CoV-2 NP antibody | Center for Therapeutic Antibody Development (CTAD), Icahn School of Medicine at Mount Sinai | Cat# NP1C7C7 |
| SARS-CoV-2 Spike KL-S-3A7 antibody | Icahn School of Medicine at Mount Sinai | Amanat et al., 2021 |
| SARS-CoV-2 nucleoprotein polyclonal anti-serum | Spiegel et al., 2005 | NA |
| Anti-mouse Alexa Fluor-488 antibody | Thermo Fisher Scientific | Thermo Fisher Scientific Cat# A-21202, RRID: AB_141607 |
| Anti-rabbit Alexa Fluor-568 antibody | Thermo Fisher Scientific | Thermo Fisher Scientific Cat# A-11011, RRID: AB_143157 |
| SARS-CoV-2 Spike S2 Chimeric mAb | Sino Biological | Sino Biological Cat# 40590-D001, RRID:AB_2857932 |
| Anti-human Alexa-Fluor 594 antibody | Thermo Fisher Scientific | Thermo Fisher Scientific Cat# A-11014, RRID: AB_2534081 |
| Bacterial and virus strains | ||
| SARS-CoV-2 isolate USA-WA1/2020 | BEI resources | Sequence ID: NR-52281 |
| hCoV-19/England/204820464/2020 | BEI resources | Sequence ID: NR-54000 |
| hCoV-19/Japan/TY7–503/2021 | BEI resources | Sequence ID: NR-54982 |
| hCoV-19/USA/MD-HP01542/2021 JHU | BEI resources Dr. (Andrew Pekosz) | Sequence ID: NR-55351 |
| hCoV-19/USA/CA-Stanford-15_S02/2021 | BEI resources (Dr.Mehul Suthar) | Sequence ID: NR-55486 |
| SARS-CoV-2 Delta variant | Isolated from the nasopharyngeal swab of a COVID-19 infected patient (Dr. Viviana Simon) | Isolate ID: PV29995 |
| Mink SARS-CoV-2 variants (MiA-1 and MiA-2) | This study | NA |
| SARS-CoV-2 isolate USA-WA1/2020-H655Y (WA1–655Y) | Dr. Jürgen Richt | NA |
| Biological samples | ||
| Nasopharyngeal swab specimens from COVID-19 infected patients | Gonzalez-Reiche et al., 2020 | Supplementary Table 1 |
| Chemicals, peptides, and recombinant proteins | ||
| mTeSR medium | Stemcell Technologies | Cat# 85850 |
| Vitronectin XF | Stemcell Technologies | Cat# 07180 |
| all-trans-retinoic acid | Sigma | Cat# R2626 |
| FGF-10 | R&D Systems | Cat# 345-FG-025 |
| EGF | R&D Systems | Cat# 236-EG-01M |
| Wnt3a | R&D Systems | Cat# 5036-WN-010 |
| KGF | R&D Systems | Cat# 251-KG-050 |
| BMP-4 | R&D Systems | Cat# 314-BP-010 |
| Critical commercial assays | ||
| E.Z.N.A Viral RNA kit | Omega Bio-Tek | Cat# R6874 |
| SuperScript IV One-Step RT-PCR System | Thermo Fisher Scientific | Cat# 12594025 |
| In-Fusion HD cloning kit | Takara | Cat# 638948 |
| QuantiFast Pathogen RT-PCR + IC Kit | Qiagen | Cat# 211454 |
| TransIT-LT1 | Takara | Cat# MIR2300 |
| Direct-zol RNA Miniprep kit | Zymo Research | Cat# R2050 |
| ProtoScript II First Strand cDNA Synthesis Kit | New England Biolabs | Cat# E6560 |
| Nextera XT DNA Sample Preparation kit | Illumina | Cat# FC-131–1096 |
| Native Barcode expansion kit | Oxford Nanopore | Cat# EXP-NBD196 |
| Ligation sequencing kit | Oxford Nanopore | Cat# SQK-LSK109 |
| Flongle flowcell | Oxford Nanopore | Cat# FLO-FLG001 |
| Deposited data | ||
| NGS sequencing data | This study | SRA BioProjectID: PRJNA793894 |
| Mass spectrometry | This study | ProteomeXchange: PXD027641 |
| Experimental models: Cell lines | ||
| VeroE6 | ATCC | ATCC Cat# CRL-1586, RRID: CVCL_0574 |
| VeroE6-TMPRSS2 | BPS Bioscience | Cat# 78081 |
| Caco-2 | ATCC | ATCC Cat# HTB-37, RRID: CVCL_0025 |
| Human pneumocyte-like cells | Icahn School of Medicine at Mount Sinai | Riva et al., 2020; White et al., 2021 |
| Experimental models: Organisms/strains | ||
| American Mink (Neovison vison) | Triple F Farms | NA |
| Golden Syrian Hamster | Charles River | Charles River Cat# Crl:LVG(SYR) |
| Oligonucleotides | ||
| RT-qPCR primers RUO Kit | Integrated DNA Technologies | Cat# 10006713 |
| Artic primer pair nCoV-2019_78 V3 | Artic Consortium | https://artic.network/ncov-2019 |
| Recombinant DNA | ||
| pCAGGS expression vector | Dr.Adolfo Garcia-Sastre’s laboratory stock | NA |
| pQCXIP-BSR-GFP1–10 | Kodaka et al., 2015 | Addgene plasmid # 68715; RRID: Addgene_68715 |
| pQCXIP-GFP11 | Kodaka et al., 2015 | Addgene plasmid # 68716; RRID: Addgene_68716 |
| Software and algorithms | ||
| Graphpad Prism | Graphpad | https://www.graphpad.com/scientific-software/prism/ |
| SnapGene 5.3.2 | SnapGene | https://www.snapgene.com/ |
| ImageJ | NIH | https://imagej.nih.gov/ij/download.html |
| Artic Consortium protocol | Artic Consortium | https://artic.network/ncov-2019 |
| SARS-CoV-2 genome assembly | GitHub | https://github.com/mjsull/COVID_pipe |
| ONT software MinKNOW v4.3.7 | Oxford Nanopore | https://nanoporetech.com/ |
| Guppy 5.0.12 | Oxford Nanopore | https://community.nanoporetech.com/downloads/guppy/release_notes |
| Minimap2 2.17-r941 | Li, 2018 | https://github.com/lh3/minimap2 |
| Nanopolish 0.13.2 | GitHub | https://github.com/jts/nanopolish |
| COBALT multiple alignment | Papadopoulos and Agarwala, 2007 | https://www.ncbi.nlm.nih.gov/tools/cobalt/re_cobalt.cgi |
| Nextstrain v7 for SARS-CoV-2 | Hadfield et al., 2018 | https://github.com/nextstrain/ncov |
| Biophyton v.75 | Python Software Foundation | https://biopython.org/wiki/Download |
| ggplot2 v3.3.5 | Wickham, 2016 | https://ggplot2.tidyverse.org. |
| MSFragger | Kong et al., 2017 | https://msfragger.nesvilab.org/ |
Highlights:
The S:655Y mutation in SARS-CoV-2 enhances spike protein cleavage and fusogenicity
The S:655Y mutation increases transmissibility in vivo
S:655Y was able to outcompete ancestral S:655H in vivo
SARS-CoV-2 VOCs evolve to acquire an increased spike cleavage and fusogenic ability
ACKNOWLEDGEMENTS
We thank Richard Cadagan for technical assistance. We also thank Florian Krammer for providing the monoclonal mouse anti-spike KL-S-3A7 antibody used in this study and Andrew Pekosz and Mehul S. Suthar for providing Beta and Kappa VOCs, respectively. We also want to thank Lisa Miorin and Michael Schotsaert for training and support. This research was partly funded by CRIPT (Center for Research on Influenza Pathogenesis and Transmission), a NIAID funded Center of Excellence for Influenza Research and Response (CEIRR, contract #75N93021C00014) (AGS), NCI SeroNet grant U54CA260560 (AGS), NIAID grants U19AI135972 and U19AI142733 (AGS), DARPA grant HR0011-19-2-0020 (AGS), JPB Foundation (AGS), Open Philanthropy Project (research grant 2020-215611 (5384) (AGS), anonymous donors to AGS, NBAF Transition Funds from the State of Kansas (JAR), NIAID Centers of Excellence for Influenza Research and Surveillance under contract number HHSN 272201400006C (JAR), AMP Core of the Center for Emerging and Zoonotic Infectious Diseases of the National Institute of General Medical Sciences (NIGMS) of the National Institutes of Health under award number P20GM130448 (JAR) and Department of Homeland Security Center of Excellence for Emerging and Zoonotic Animal Diseases under grant number HSHQDC 16-A-B0006 (JAR). AGR is funded by Marion Alban MSCIC Scholars Award and the 2020 Robin Chemers Neustein Postdoctoral fellowship. ML is funded by a fellowship of the Belgian American Education Foundation
DECLARATION OF INTERESTS
The A.G.-S. laboratory has received research support from Pfizer, Senhwa Biosciences, Kenall Manufacturing, Avimex, Johnson & Johnson, Dynavax, 7Hills Pharma, N-fold LLC, Pharmamar, ImmunityBio, Accurius, Nanocomposix, Hexamer and Merck, outside of the reported work. A.G.-S. has consulting agreements for the following companies involving cash and/or stock: Vivaldi Biosciences, Contrafect, 7Hills Pharma, Avimex, Vaxalto, Pagoda, Accurius, Esperovax, Farmak, Applied Biological Laboratories and Pfizer, outside of the reported work. A.G.-S. is inventor on patents and patent applications on the use of antivirals and vaccines for the treatment and prevention of virus infections, owned by the Icahn School of Medicine at Mount Sinai, New York. The Icahn School of Medicine at Mount Sinai has filed a patent application relating to SARS-CoV-2 serological assays, which lists Viviana Simon as co-inventor. Mount Sinai has spun out a company, Kantaro, to market serological tests for SARS-CoV-2.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Amanat F, Strohmeier S, Lee WH, Bangaru S, Ward AB, Coughlan L, and Krammer F (2021). Murine Monoclonal Antibodies against the Receptor Binding Domain of SARS-CoV-2 Neutralize Authentic Wild-Type SARS-CoV-2 as Well as B.1.1.7 and B.1.351 Viruses and Protect In Vivo in a Mouse Model in a Neutralization-Dependent Manner. mBio, e0100221. 10.1128/mBio.01002-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aydillo T, Gonzalez-Reiche AS, Aslam S, van de Guchte A, Khan Z, Obla A, Dutta J, van Bakel H, Aberg J, García-Sastre A, et al. (2020). Shedding of Viable SARS-CoV-2 after Immunosuppressive Therapy for Cancer. N Engl J Med 383, 2586–2588. 10.1056/NEJMc2031670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aydillo T, Rombauts A, Stadlbauer D, Aslam S, Abelenda-Alonso G, Escalera A, Amanat F, Jiang K, Krammer F, Carratala J, and García-Sastre A (2021). Immunological imprinting of the antibody response in COVID-19 patients. Nat Commun 12, 3781. 10.1038/s41467-021-23977-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee S, Seal S, Dey R, Mondal KK, and Bhattacharjee P (2021). Mutational spectra of SARS-CoV-2 orf1ab polyprotein and signature mutations in the United States of America. J Med Virol 93, 1428–1435. 10.1002/jmv.26417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baum A, Fulton BO, Wloga E, Copin R, Pascal KE, Russo V, Giordano S, Lanza K, Negron N, Ni M, et al. (2020). Antibody cocktail to SARS-CoV-2 spike protein prevents rapid mutational escape seen with individual antibodies. Science 369, 1014–1018. 10.1126/science.abd0831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begum F, Mukherjee D, Das S, Thagriki D, Tripathi PP, Banerjee AK, and Ray U (2020). Specific mutations in SARS-CoV2 RNA dependent RNA polymerase and helicase alter protein structure, dynamics and thus function: Effect on viral RNA replication. 10.1101/2020.04.26.063024. [DOI] [Google Scholar]
- Bestle D, Heindl MR, Limburg H, Van Lam van T, Pilgram O, Moulton H, Stein DA, Hardes K, Eickmann M, Dolnik O, et al. (2020). TMPRSS2 and furin are both essential for proteolytic activation of SARS-CoV-2 in human airway cells. Life Sci Alliance 3. 10.26508/lsa.202000786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun KM, Moreno GK, Halfmann PJ, Hodcroft EB, Baker DA, Boehm EC, Weiler AM, Haj AK, Hatta M, Chiba S, et al. (2021). Transmission of SARS-CoV-2 in domestic cats imposes a narrow bottleneck. PLoS Pathog 17, e1009373. 10.1371/journal.ppat.1009373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchrieser J, Dufloo J, Hubert M, Monel B, Planas D, Rajah MM, Planchais C, Porrot F, Guivel-Benhassine F, Van der Werf S, et al. (2020). Syncytia formation by SARS-CoV-2-infected cells. EMBO J 39, e106267. 10.15252/embj.2020106267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi X, Yan R, Zhang J, Zhang G, Zhang Y, Hao M, Zhang Z, Fan P, Dong Y, Yang Y, et al. (2020). A neutralizing human antibody binds to the N-terminal domain of the Spike protein of SARS-CoV-2. Science 369, 650–655. 10.1126/science.abc6952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coutard B, Valle C, de Lamballerie X, Canard B, Seidah NG, and Decroly E (2020). The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antiviral Res 176, 104742. 10.1016/j.antiviral.2020.104742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan L, Zheng Q, Zhang H, Niu Y, Lou Y, and Wang H (2020). The SARS-CoV-2 Spike Glycoprotein Biosynthesis, Structure, Function, and Antigenicity: Implications for the Design of Spike-Based Vaccine Immunogens. Front Immunol 11, 576622. 10.3389/fimmu.2020.576622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edara VV, Lai L, Sahoo MK, Floyd K, Sibai M, Solis D, Flowers MW, Hussaini L, Ciric CR, Bechnack S, et al. (2021). Infection and vaccine-induced neutralizing antibody responses to the SARS-CoV-2 B.1.617.1 variant. bioRxiv. 10.1101/2021.05.09.443299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenollar F, Mediannikov O, Maurin M, Devaux C, Colson P, Levasseur A, Fournier PE, and Raoult D (2021). Mink, SARS-CoV-2, and the Human-Animal Interface. Front Microbiol 12, 663815. 10.3389/fmicb.2021.663815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghaedi M, Calle EA, Mendez JJ, Gard AL, Balestrini J, Booth A, Bove PF, Gui L, White ES, and Niklason LE (2013). Human iPS cell-derived alveolar epithelium repopulates lung extracellular matrix. J Clin Invest 123, 4950–4962. 10.1172/jci68793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Reiche AS, Hernandez MM, Sullivan MJ, Ciferri B, Alshammary H, Obla A, Fabre S, Kleiner G, Polanco J, Khan Z, et al. (2020). Introductions and early spread of SARS-CoV-2 in the New York City area. Science 369, 297–301. 10.1126/science.abc1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadfield J, Megill C, Bell SM, Huddleston J, Potter B, Callender C, Sagulenko P, Bedford T, and Neher RA (2018). Nextstrain: real-time tracking of pathogen evolution. Bioinformatics 34, 4121–4123. 10.1093/bioinformatics/bty407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammer AS, Quaade ML, Rasmussen TB, Fonager J, Rasmussen M, Mundbjerg K, Lohse L, Strandbygaard B, Jorgensen CS, Alfaro-Nunez A, et al. (2021). SARS-CoV-2 Transmission between Mink (Neovison vison) and Humans, Denmark. Emerg Infect Dis 27, 547–551. 10.3201/eid2702.203794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey WT, Carabelli AM, Jackson B, Gupta RK, Thomson EC, Harrison EM, Ludden C, Reeve R, Rambaut A, Consortium C-GU, et al. (2021). SARS-CoV-2 variants, spike mutations and immune escape. Nat Rev Microbiol 19, 409–424. 10.1038/s41579-021-00573-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann M, Kleine-Weber H, and Pohlmann S (2020). A Multibasic Cleavage Site in the Spike Protein of SARS-CoV-2 Is Essential for Infection of Human Lung Cells. Mol Cell 78, 779–784 e775. 10.1016/j.molcel.2020.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Yang C, Xu XF, Xu W, and Liu SW (2020). Structural and functional properties of SARS-CoV-2 spike protein: potential antivirus drug development for COVID-19. Acta Pharmacol Sin 41, 1141–1149. 10.1038/s41401-020-0485-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob A, Vedaie M, Roberts DA, Thomas DC, Villacorta-Martin C, Alysandratos KD, Hawkins F, and Kotton DN (2019). Derivation of self-renewing lung alveolar epithelial type II cells from human pluripotent stem cells. Nat Protoc 14, 3303–3332. 10.1038/s41596-019-0220-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson BA, Xie X, Kalveram B, Lokugamage KG, Muruato A, Zou J, Zhang X, Juelich T, Smith JK, Zhang L, et al. (2020). Furin Cleavage Site Is Key to SARS-CoV-2 Pathogenesis. bioRxiv. 10.1101/2020.08.26.268854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodaka M, Yang Z, Nakagawa K, Maruyama J, Xu X, Sarkar A, Ichimura A, Nasu Y, Ozawa T, Iwasa H, et al. (2015). A new cell-based assay to evaluate myogenesis in mouse myoblast C2C12 cells. Exp Cell Res 336, 171–181. 10.1016/j.yexcr.2015.06.015. [DOI] [PubMed] [Google Scholar]
- Kong AT, Leprevost FV, Avtonomov DM, Mellacheruvu D, and Nesvizhskii AI (2017). MSFragger: ultrafast and comprehensive peptide identification in mass spectrometry-based proteomics. Nat Methods 14, 513–520. 10.1038/nmeth.4256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korber B, Fischer WM, Gnanakaran S, Yoon H, Theiler J, Abfalterer W, Hengartner N, Giorgi EE, Bhattacharya T, Foley B, et al. (2020). Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell 182, 812–827 e819. 10.1016/j.cell.2020.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamers MM, Mykytyn AZ, Breugem TI, Wang Y, Wu DC, Riesebosch S, van den Doel PB, Schipper D, Bestebroer T, Wu NC, and Haagmans BL (2021). Human airway cells prevent SARS-CoV-2 multibasic cleavage site cell culture adaptation. Elife 10. 10.7554/eLife.66815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan J, Ge J, Yu J, Shan S, Zhou H, Fan S, Zhang Q, Shi X, Wang Q, Zhang L, and Wang X (2020). Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 581, 215–220. 10.1038/s41586-020-2180-5. [DOI] [PubMed] [Google Scholar]
- Li H (2018). Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100. 10.1093/bioinformatics/bty191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin L, Li Q, Wang Y, and Shi Y (2021). Syncytia formation during SARS-CoV-2 lung infection: a disastrous unity to eliminate lymphocytes. Cell Death Differ 28, 2019–2021. 10.1038/s41418-021-00795-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Liu J, Plante KS, Plante JA, Xie X, Zhang X, Ku Z, An Z, Scharton D, Schindewolf C, et al. (2021). The N501Y spike substitution enhances SARS-CoV-2 transmission. bioRxiv. 10.1101/2021.03.08.434499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Zheng H, Lin H, Li M, Yuan R, Peng J, Xiong Q, Sun J, Li B, Wu J, et al. (2020). Identification of Common Deletions in the Spike Protein of Severe Acute Respiratory Syndrome Coronavirus 2. J Virol 94. 10.1128/JVI.00790-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, Frewen B, Kern R, Tabb DL, Liebler DC, and MacCoss MJ (2010). Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968. 10.1093/bioinformatics/btq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuyama S, Nagata N, Shirato K, Kawase M, Takeda M, and Taguchi F (2010). Efficient activation of the severe acute respiratory syndrome coronavirus spike protein by the transmembrane protease TMPRSS2. J Virol 84, 12658–12664. 10.1128/jvi.01542-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mlcochova P, Kemp S, Dhar MS, Papa G, Meng B, Mishra S, Whittaker C, Mellan T, Ferreira I, Datir R, et al. (2021a). SARS-CoV-2 B.1.617.2 Delta variant emergence, replication and sensitivity to neutralising antibodies. bioRxiv, 2021.2005.2008.443253. 10.1101/2021.05.08.443253. [DOI] [Google Scholar]
- Mlcochova P, Kemp SA, Dhar MS, Papa G, Meng B, Ferreira I, Datir R, Collier DA, Albecka A, Singh S, et al. (2021b). SARS-CoV-2 B.1.617.2 Delta variant replication and immune evasion. Nature 599, 114–119. 10.1038/s41586-021-03944-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ord M, Faustova I, and Loog M (2020). The sequence at Spike S1/S2 site enables cleavage by furin and phospho-regulation in SARS-CoV2 but not in SARS-CoV1 or MERS-CoV. Sci Rep 10, 16944. 10.1038/s41598-020-74101-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oude Munnink BB, Sikkema RS, Nieuwenhuijse DF, Molenaar RJ, Munger E, Molenkamp R, van der Spek A, Tolsma P, Rietveld A, Brouwer M, et al. (2021). Transmission of SARS-CoV-2 on mink farms between humans and mink and back to humans. Science 371, 172–177. 10.1126/science.abe5901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos JS, and Agarwala R (2007). COBALT: constraint-based alignment tool for multiple protein sequences. Bioinformatics 23, 1073–1079. 10.1093/bioinformatics/btm076. [DOI] [PubMed] [Google Scholar]
- Peacock TP, Sheppard CM, Brown JC, Goonawardane N, Zhou J, Whiteley M, Consortium PV, de Silva TI, and Barclay WS (2021). The SARS-CoV-2 variants associated with infections in India, B.1.617, show enhanced spike cleavage by furin. bioRxiv, 2021.2005.2028.446163. 10.1101/2021.05.28.446163. [DOI] [Google Scholar]
- Planas D, Bruel T, Grzelak L, Guivel-Benhassine F, Staropoli I, Porrot F, Planchais C, Buchrieser J, Rajah MM, Bishop E, et al. (2021a). Sensitivity of infectious SARS-CoV-2B.1.1.7 and B.1.351 variants to neutralizing antibodies. Nat Med 27, 917–924. 10.1038/s41591-021-01318-5. [DOI] [PubMed] [Google Scholar]
- Planas D, Veyer D, Baidaliuk A, Staropoli I, Guivel-Benhassine F, Rajah MM, Planchais C, Porrot F, Robillard N, Puech J, et al. (2021b). Reduced sensitivity of SARS-CoV-2 variant Delta to antibody neutralization. Nature. 10.1038/s41586-021-03777-9. [DOI] [PubMed] [Google Scholar]
- Plante JA, Liu Y, Liu J, Xia H, Johnson BA, Lokugamage KG, Zhang X, Muruato AE, Zou J, Fontes-Garfias CR, et al. (2021). Spike mutation D614G alters SARS-CoV-2 fitness. Nature 592, 116–121. 10.1038/s41586-020-2895-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quick J, Grubaugh ND, Pullan ST, Claro IM, Smith AD, Gangavarapu K, Oliveira G, Robles-Sikisaka R, Rogers TF, Beutler NA, et al. (2017). Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nat Protoc 12, 1261–1276. 10.1038/nprot.2017.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathnasinghe R, Jangra S, Cupic A, Martinez-Romero C, Mulder LCF, Kehrer T, Yildiz S, Choi A, Mena I, De Vrieze J, et al. (2021). The N501Y mutation in SARS-CoV-2 spike leads to morbidity in obese and aged mice and is neutralized by convalescent and post-vaccination human sera. medRxiv. 10.1101/2021.01.19.21249592. [DOI] [Google Scholar]
- Rego N, Fernández-Calero T, Arantes I, Noya V, Mir D, Brandes M, Zanetti J, Arleo M, Pereira E, Possi T, et al. (2021). Spatiotemporal dissemination pattern of SARS-CoV-2 B1.1.28-derived lineages introduced into Uruguay across its southeastern border with Brazil. medRxiv, 2021.2007.2005.21259760. 10.1101/2021.07.05.21259760. [DOI] [Google Scholar]
- Riva L, Yuan S, Yin X, Martin-Sancho L, Matsunaga N, Pache L, Burgstaller-Muehlbacher S, De Jesus PD, Teriete P, Hull MV, et al. (2020). Discovery of SARS-CoV-2 antiviral drugs through large-scale compound repurposing. Nature 586, 113–119. 10.1038/s41586-020-2577-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenke K, Meade-White K, Letko M, Clancy C, Hansen F, Liu Y, Okumura A, Tang-Huau TL, Li R, Saturday G, et al. (2020). Defining the Syrian hamster as a highly susceptible preclinical model for SARS-CoV-2 infection. Emerg Microbes Infect 9, 2673–2684. 10.1080/22221751.2020.1858177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Wen Z, Zhong G, Yang H, Wang C, Huang B, Liu R, He X, Shuai L, Sun Z, et al. (2020). Susceptibility of ferrets, cats, dogs, and other domesticated animals to SARS-coronavirus 2. Science 368, 1016–1020. 10.1126/science.abb7015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sia SF, Yan LM, Chin AWH, Fung K, Choy KT, Wong AYL, Kaewpreedee P, Perera R, Poon LLM, Nicholls JM, et al. (2020). Pathogenesis and transmission of SARS-CoV-2 in golden hamsters. Nature 583, 834–838. 10.1038/s41586-020-2342-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiegel M, Pichlmair A, Martínez-Sobrido L, Cros J, García-Sastre A, Haller O, and Weber F (2005). Inhibition of Beta interferon induction by severe acute respiratory syndrome coronavirus suggests a two-step model for activation of interferon regulatory factor 3. J Virol 79, 2079–2086. 10.1128/jvi.79.4.2079-2086.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starr TN, Greaney AJ, Hilton SK, Ellis D, Crawford KHD, Dingens AS, Navarro MJ, Bowen JE, Tortorici MA, Walls AC, et al. (2020). Deep Mutational Scanning of SARS-CoV-2 Receptor Binding Domain Reveals Constraints on Folding and ACE2 Binding. Cell 182, 1295–1310 e1220. 10.1016/j.cell.2020.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang T, Jaimes JA, Bidon MK, Straus MR, Daniel S, and Whittaker GR (2021). Proteolytic Activation of SARS-CoV-2 Spike at the S1/S2 Boundary: Potential Role of Proteases beyond Furin. ACS Infect Dis 7, 264–272. 10.1021/acsinfecdis.0c00701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong P, Gautam A, Windsor I, Travers M, Chen Y, Garcia N, Whiteman NB, McKay LGA, Lelis FJN, Habibi S, et al. (2021). Memory B cell repertoire for recognition of evolving SARS-CoV-2 spike. bioRxiv. 10.1101/2021.03.10.434840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White KM, Rosales R, Yildiz S, Kehrer T, Miorin L, Moreno E, Jangra S, Uccellini MB, Rathnasinghe R, Coughlan L, et al. (2021). Plitidepsin has potent preclinical efficacy against SARS-CoV-2 by targeting the host protein eEF1A. Science 371, 926–931. 10.1126/science.abf4058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H ggplot2: Elegant Graphics for Data Analysis. https://ggplot2.tidyverse.org. Springer, New York; 2016. [Google Scholar]
- Xia S, Zhu Y, Liu M, Lan Q, Xu W, Wu Y, Ying T, Liu S, Shi Z, Jiang S, and Lu L (2020). Fusion mechanism of 2019-nCoV and fusion inhibitors targeting HR1 domain in spike protein. Cell Mol Immunol 17, 765–767. 10.1038/s41423-020-0374-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi C, Sun X, Ye J, Ding L, Liu M, Yang Z, Lu X, Zhang Y, Ma L, Gu W, et al. (2020). Key residues of the receptor binding motif in the spike protein of SARS-CoV-2 that interact with ACE2 and neutralizing antibodies. Cell Mol Immunol 17, 621–630. 10.1038/s41423-020-0458-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data is available in the manuscript or the supplementary materials. The targeted mass spectrometry data including raw files, Skyline document and resulting transition lists have been deposited at ProteomeXchange (PXD027641). Next generation sequencing raw data for the competition experiments in hamsters and pneumocytes have been deposited in SRA. BioProjectID PRJNA793894 and BioSample accessions SAMN24592477 to SAMN24592565. Reagents used are almost exclusively commercially available and non-proprietary. Any additional information required to reanalyze the data reported in this work paper is available from th e Lead Contact upon request.