

Abstract
Purpose:
Gene fusions involving R-spondin (RSPOfp) and RNF43 mutations have been shown to drive Wnt-dependent tumor initiation in colorectal cancer (CRC). Herein, we aimed to characterize the molecular features of RSPOfp/RNF43mutated (mut) compared to wildtype CRCs to gain insights into potential rationales for therapeutic strategies.
Experimental design:
A discovery cohort was classified for RSPOfp/RNF43 status using DNA/RNA sequencing and immunohistochemistry. An independent cohort was used to validate our findings.
Results:
The discovery cohort consisted of 7,245 CRC samples. RSPOfp and RNF43 mutations were detected in 1.3% (n=94) and 6.1% (n=443) of cases. We found 5 RSPO fusion events that had not previously been reported (e.g. IFNGR1-RSPO3). RNF43-mut tumors were associated with right-sided primary tumors. No RSPOfp tumors had RNF43 mutations. In comparison to wildtype CRCs, RSPOfp tumors were characterized by a higher frequency of BRAF, BMPR1A and SMAD4 mutations. APC mutations were observed in only a minority of RSPOfp-positive compared to wildtype cases (4.4 vs. 81.4%). Regarding RNF43 mutations, a higher rate of KMT2D and BRAF mutations were detectable compared to wildtype samples. While RNF43 mutations were associated with a microsatellite instability (MSI-H)/mismatch repair deficiency (dMMR) phenotype (64.3%), and a TMB ≥10 mt/Mb (65.8%), RSPOfp was not associated with MSI-H/dMMR. The validation cohort replicated our genetic findings.
Conclusions:
This is the largest series of RSPOfp/RNF43-mut CRCs reported to date. Comprehensive molecular analyses asserted the unique molecular landscape associated with RSPO/RNF43 and suggested potential alternative strategies to overcome the low clinical impact of Wnt-targeted agents and immunotherapy.
Keywords: Wnt, colorectal cancer, RNF43, RSPO, R-spondin, molecular profile
INTRODUCTION:
Colorectal cancer (CRC) remains one of the major causes of cancer specific morbidity and mortality worldwide (1, 2). Despite therapeutic improvements, the prognosis of patients with metastatic disease remains poor with a 5-year overall survival (OS) rate of approximately 14% (1). Thus, new therapeutic strategies are urgently needed to improve survival.
Activation of the Wnt/β-catenin pathway, mostly facilitated by genetic mutations encoding for the adenomatous polyposis coli (APC) protein, can initiate tumorigenesis in CRC (3, 4). In vitro experiments have determined that the restoration of functional APC leads to tumor regression even in CRC cells with additional oncogenic mutations (i.e. TP53 or KRAS) (5). Therefore, Wnt/β-catenin signaling represents a major oncogenic driver in CRC. Over the last years, different genetic alterations activating the Wnt signaling pathway have been discovered. Seshagiri and colleagues (6) described chromosomal rearrangements involving members of the R-spondin family (RSPO) in CRC for the first time, which can be observed in up to 8% of CRCs. Studies suggest that such RSPO translocations alone are sufficient to initiate carcinogenesis (7), as are mutations in RNF43, a negative feedback regulator of the Wnt/β-catenin pathway. RSPO molecules bind to the G-protein coupled receptor (LGR) family (LGR4/5/6) which contains a leucine-rich repeat segment resulting in an up-regulation of the Wnt/β-catenin pathway by sequestering the E3 ubiquitin ligase RNF43 (8). Mutations in RNF43 have been described in a variety of malignancies, including CRC and gastric cancer, with a frequency of up to 20% (9-15). Interestingly, the frequency of RNF43 mutations was noted to be even higher in microsatellite-unstable cancers (11). Most of the loss-of-function (LOF) mutations in RNF43 have been determined to lead to an increased cell surface abundance of the Wnt receptor Frizzled, rendering the cells dependent upon Wnt/β-catenin signaling (16). Therefore, these cells are suggested to be more sensitive to inhibition of the porcupine homolog (PORCN) protein (16), a posttranslational modificator of the Wnt protein (17).
RSPOfp-positive/RNF43-mutated (mut) tumors represent a distinct genetic subgroup of CRCs. However, gene alterations co-occurring in this subgroup are largely unknown. Thus, we set up this study to define the molecular profile of RSPO/RNF43-positive CRC that may provide important insights how Wnt/β-catenin pathway deregulation drives tumor growth in CRC. For this, we performed extensive genomic and transcriptomic sequencing, as well as immunohistochemistry (IHC), to compare molecular profiles of RSPO/RNF43-positive vs. wildtype (WT) cases, and detected clusters of gene mutation associations as well as several relations with microsatellite instability (MSI-H) and tumor mutation burden (TMB).
MATERIALS AND METHODS:
Sample characterization of the discovery cohort
Colorectal carcinoma specimens of 7,245 patients were submitted to Caris Life Sciences (Phoenix, AZ, USA) for genomic profiling. These cases were retrospectively reviewed, and gene sequencing, amplification and protein expression data were analyzed. The pathology report was included with the specimens and H&E slides were prepared for each tumor sample to be reviewed by board-certified pathologists to confirm the diagnosis of CRC. Tumors with a histologic diagnosis that was not concordant with the diagnosis of CRC were excluded from this analysis. During the recruitment period, tests have varied since there were different requests by the treating physicians and the testing technologies continuously evolved over time. The next generation sequencing (NGS) platform for tumors tested in 2015 or earlier used the MiSeq platform (45 genes included) while those tested after 2015 were sequenced with the NextSeq platform (592 genes included). In keeping with 45 CFR 46.101(b) this study was performed utilizing retrospective, de-identified clinical data. Therefore, this study is considered IRB exempt and no patient consent was necessary from the subjects. Thus, only basic demographic information was available. Patients were stratified into RSPOfp or RNF43 positive and negative cases. RNF43 mutations included only pathogenic or presumed pathogenic mutations. Tumors with benign RNF43 mutations, presumed benign RNF43 mutations, or RNF43 variants of unknown significance were categorized as RNF43-WT. Germline testing could not be performed due to the lack of access to germline DNA.
Samples of the validation cohort
A total of 816 cases of CRCs were recruited between January 2016 and December 2017 at the Singapore General Hospital, Singapore. A local Ethics Committee approval was obtained. Molecular profiling was analyzed for RNF43 mutations (excluding the specific G569fs variant) and co-mutations in Wnt and MEK signaling pathways as well as MSI-H or mismatch repair deficiency (dMMR) (MSI-H/dMMR). RSPO fusions were not characterized.
Analyses performed
Immunohistochemistry (IHC) was performed on 1,258 tumor samples on formalin-fixed paraffin-embedded (FFPE) sections on glass slides for the discovery cohort. Four micrometer sections were mounted on slides and stained using an automated system (Benchmark, Ventana Medical Systems, Tucson, AZ; Autostainer, DAKO, Carpinteria, CA) according to manufacturer's instructions, and were optimized and validated per CLIA/CAP and ISO requirements. All proteins of interest were evaluated on tumor cells. An intensity score (0 = no staining; 1+ = weak staining; 2+ = moderate staining; 3+ = strong staining) and a proportion score to determine the percentage of cells staining positive (0-100%) was used. The primary antibody used to detect PD-L1 expression was SP142 (Spring Biosciences, CA, USA). The staining was deemed positive if its intensity on the membrane of the tumor cells was ≥2+ and the percentage of positively stained cells was ≥5%. Results were classified as positive or negative by using previously defined thresholds specific to each marker, based on published clinical literature that associates biomarker status to specific treatment response. The primary antibody used for PD-L1 testing was MRQ-22 (Ventana) and staining was scored as positive if the number of PD-L1 positive cells was >1 cell per high power field. A single board-certified pathologist independently evaluated immunohistochemical results.
NGS was performed on FFPE tumor samples using the NextSeq platform (Illumina, Inc., San Diego, CA). A custom-designed SureSelect XT assay was used to enrich 592 whole-gene targets (Agilent Technologies, Santa Clara, CA). All variants were detected with >99% confidence based on allele frequency and amplicon coverage with an average sequencing depth of coverage of >500 and with an analytic sensitivity of 5%. Genetic variants identified were interpreted by board-certified molecular geneticists and categorized as ‘pathogenic,’ ‘presumed pathogenic,’ ‘variant of unknown significance,’ ‘presumed benign,’ or ‘benign,’ according to the American College of Medical Genetics and Genomics (ACMG) standards. When assessing mutation frequencies of individual genes, ‘pathogenic,’ and ‘presumed pathogenic’, were defined as mutations while ‘benign’ or ‘presumed benign’ variants and ‘variants of unknown significance’ were defined as wild type.
A combination of multiple test platforms was used to determine the MSI or MMR status of the tumors profiled, including fragment analysis (FA, Promega, Madison, WI), IHC (MLH1, M1 antibody; MSH2, G2191129 antibody; MSH6, 44 antibody; and PMS2, EPR3947 antibody [Ventana Medical Systems, Inc., Tucson, AZ, USA]) and NGS (for tumors tested with NextSeq platform, 7,000 target microsatellite loci were examined and compared to the reference genome hg19 from the University of California).
Statistics
Statistical comparisons were performed with the Chi-square test and the Mann-Whitney U test when appropriate. A two-sided p-value < 0.05 was considered as statistically significant. P-values were further corrected for multiple comparison using the Benjamini-Hochberg method to avoid type I error, and an adjusted p-value (q-value) of <0.05 was considered as a significant difference.
Real-world overall survival information was obtained from insurance claims data in an updated larger cohort (incorporating the initially described discovery cohort) and calculated from first specimen collection to last contact. Kaplan-Meier estimates were calculated for the molecularly defined patient cohorts.
Availability of data and materials
The deidentified sequencing data are owned by Caris Life Sciences. The datasets generated during and analyzed during the current study are available from the authors upon reasonable request and with permission of Caris Life Sciences. Qualified researchers may contact the corresponding author with their request.
RESULTS
Patients` characteristics and prognosis
In total, 7,245 CRC patients were tested for alterations in RSPO and RNF43 (see Table 1). Of those, 443 (6.1%) and 94 patients (1.3%) showed a RNF43 mutation or a RSPOfp, respectively. RSPO3 fusions were more frequently detected than RSPO2 translocations (89 vs. 5 cases). Patients with a RSPOfp were younger than patients harboring a RNF43 mutation (61 vs. 69 years, p=0.0003). No difference in distribution by gender was noted between RSPOfp and wildtype (WT) cases (p=n.s.). For RNF43 mutations, a significant female predominance was observed compared to male patients (p<0.001). Furthermore, we found a higher percentage of cases with RNF43 mutations in right-sided than in left-sided CRC (14.3 vs. 3.1%, p<0.001). However, no site-specific difference was observed for RSPOfp.
Table 1:
Characteristics of the discovery cohort.
| Characteristic |
RSPO fusion positive |
RNF43-mut |
RNF43 and RSPO wildtype |
|
|---|---|---|---|---|
| Total – no. (%) | 94 (1.3) | 443 (6.1) | 6,708 (92.6) | |
| Age (years) | Median Age | 61 | 69 | 62 |
| Range | 36-90 | 18-93 | 15-98 | |
| Sex – no. (%) | Female | 46 (49) | 263 (59) | 2,922 (44) |
| Male | 48 (51) | 180 (41) | 3,786 (56) | |
| Tumor Location – no. (%) | Left | 23 (25) | 66 (15) | 2,156 (32) |
| Rectal | 29 (31) | 39 (9) | 1,640 (24) | |
| Right | 24 (26) | 232 (52) | 1,612 (24) | |
| Transverse | 8 (9) | 46 (10) | 293 (4) | |
| Unclear | 10 (11) | 60 (13.5) | 1,007 (15) |
The most frequently detected RSPOfp was the PTPRK-RSPO3 fusion protein (n=89). Of note, we detected 5 fusion partners (CPSF1, CDH17, MATN2 and ADAM9) which had not been described before (see Supplementary Table 1). The most frequently detected point mutations in RNF43 was G659fs, followed by R117fs and P660fs (see Supplementary Table 2).
Until now, the prognostic relevance of RNF43 mutations and RSPOfp remains largely unclear. Therefore, we performed survival analyses using real-world data obtained from insurance claims. CRC patients harboring a RNF43 mutation or a RSPOfp are associated with a poor survival compared to WT cases (Figure 1AB). Moreover, in the MSS sub-cohort patients harboring RSPOfp or RNF43 mutations were characterized by poor survival (RSPOfp vs. WT: HR 0.61, 95%CI 0.47-0.79, p<0.001; RNF43-mut vs. WT: HR 0.65, 95%CI 0.57-0.75, p<0.001).
Figure 1: Real-world overall survival stratified by RSPO/RNF43 status.

A) Comparison of RSPOfp vs. RNF43/RSPOfp-WT patients (RSPOfp vs. WT: HR 0.62, 95%CI 0.48–0.81, p<0.001).
B) Comparison of RNF43-mut vs. RNF43/RSPOfp-WT patients (HR 0.86, 95%CI 0.78–0.94, p<0.001).
Molecular landscape of RSPO fusion proteins and RNF43 mutations
RSPOfp-positive CRCs were associated with a higher rate of co-incident mutations in BRAF (35.9 vs. 6.3%), SMAD4 (30.0 vs. 13.5%), BMPR1A (5.4 vs. 0.2%), AKT1 (3.3 vs. 0.4%) and ERBB3 (5.4 vs. 1.6%, all q<0.05) compared to WT cases (Figure 2A).
Figure 2: Molecular landscape of the discovery cohort.
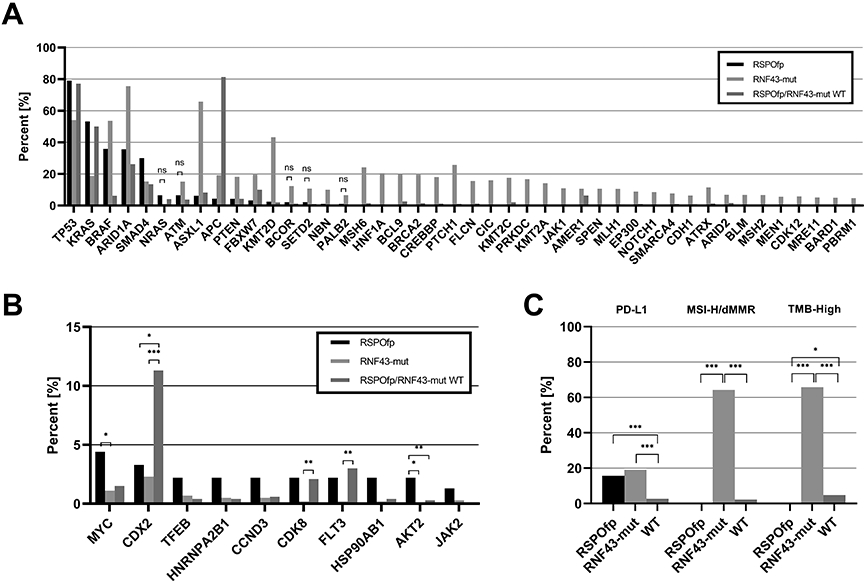
A) Comparison of the genetic landscape of RNF43-mut, RSPOfp and RNF43/RSPOfp-WT tumors. Shown are mutations that are significantly different between RSPOfp vs. RNF43-mut tumors (all q<0.05; ns: p<0.05, but q<0.05 was not reached).
B) Copy number alterations in RNF43-mut, RSPOfp and RNF43/RSPO-WT tumors. *q<0.05, **q<0.01, ***q<0.001.
C) MSI, TMB and PDL-1 status in RNF43-mut, RSPOfp and RNF43/RSPOfp-WT tumors. None of the RSPOfp patients showed a TMB >10 mt/Mb or a MSI-H/dMMR status.*q<0.05, **q<0.01, ***q<0.001.
Compared to RNF43-mut cancers, co-incident mutations in TP53 (79.1%), KRAS (53.3%), and SMAD4 (30.0%) occurred more frequently in RSPOfp-positive cancers (RNF43: 54.1%, 18.8% and 15.3%, respectively; all q<0.05). Importantly, in RSPOfp-positive CRCs we discovered no concomitant RNF43 mutations. In contrast, tumors containing RNF43 mutations exhibited a different molecular landscape as compared to RSPOfp-positive tumors: ARID1A (75.6 vs. 35.7%), ASXL1 (65.8 vs. 6.3%), BRAF (53.6 vs. 35.9%), KMT2D (43.3 vs. 2.5%), and PTEN (18.2 vs. 4.3%) gene alterations were more frequently detected (all q<0.05). Of note, APC mutations were observed in 19.3% of RNF43-mut cases, in 4.4% of RSPOfp-positive tumors and in 81.4% of WT cases (all q<0.05). Regarding BRAF mutations the most prevalent genetic variant was the V600E mutation (78.8%).
Copy number alterations (CNAs) in MYC and AKT2 genes were differently distributed between RSPOfp-positive tumors compared to RNF43-mut tumors (4.4 vs. 1.1%, and 2.2 vs. 0.0%, respectively; all q<0.05). Amongst others, CNAs in CDX2 gene were found more often in WT cases (11.3%) than in RSPOfp-positive (3.3%) or RNF43-mut (2.3%) samples (all q<0.05) (Figure 2B).
Validation cohort
An independent validation cohort was used to confirm our findings in terms of RNF43 mutations. The retrospective use of a next generation sequencing panel without analyses on fusions prohibited further validation of the findings generated in the RSPOfp subset of the discovery cohort. The validation cohort consisted of 816 CRC patients (Table 2). This cohort was obtained from a time period between 2016 and 2017 and was retrospectively analyzed. The data was mined for molecular status of RNF43 and other mutations including APC, KRAS, BRAF, NRAS, and other genes. MSI-H/dMMR was also included in this analysis. In line with the findings from our discovery cohort, the incidence of RNF43 mutations was similar (7.97% vs. 6.1%, p=n.s.). Moreover, the co-activation of Wnt and MAPK signaling (including, APC, KRAS, BRAF and NRAS) was strongly associated with RNF43 mutations (in total: 88%); 12% had no detectable coincident mutations. Of the RNF43-mut cases, 11% showed a MSI-H/dMMR status, 64% showed a MSS/pMMR status, while 25% had no data for MSI-H/dMMR status available.
Table 2:
Characteristics of the validation cohort.
| Characteristics | No. (%) |
|---|---|
| CRC cases | 816 |
| RNF43 mutation status | |
| Mutated | 65 (7.97) |
| Wildtype | 751 (92.03) |
| Comutations in APC, KRAS, BRAF and | 57 (88) |
| NRAS | |
| No comutation in the respective genes | 8 (12) |
| Microsatellite status | |
| MSI-H/dMMR (%) | 7 (11) |
| MSS/pMMR (%) | 42 (64) |
| Not reported (%) | 16 (25) |
RNF43 mutations are associated with microsatellite instability
Next, we analyzed biomarkers associated with a predictive value for response to immune checkpoint inhibitors. In samples harboring a RSPO fusions, no individual with a MSI-H/dMMR genotype was detected (0.0%), compared to a MSI-H/dMMR rate of 64.3% in RNF43-mut samples (q<0.001), and 2.3% in WT tumors (q<0.001) (Figure 2C). Moreover, in RSPOfp-positive tumors no case presented with a tumor mutational burden (TMB) of ≥10 mt/Mb. However, in RNF43-mut samples, 65.8% had a TMB ≥10 mt/Mb (q<0.001), which was also higher than for WT cases (4.8%, q<0.001). Positive staining for PD-L1 was detected in 15.7% of RSPOfp-positive specimens, 19.1% of RNF43-mut samples, and 2.7% of WT cases (q<0.001 for RSPOfp vs. WT, and RNF43-mut vs. WT).
Since MSI-H/dMMR status may trigger secondary mutations, we performed a subgroup analysis in MSS cases. A higher prevalence of females and right-sided primary locations in the MSS subset of patients harboring a RNF43 mutation was observed. Comparing the molecular profile of the RSPOfp-positive and the MSS/RNF43-mut cases, no differences in the frequency of TP53 mutations (79.1 vs. 85.2%, p=n.s.) and BRAF mutations (35.9 vs. 43.7%, p=n.s.) were observed. However, there were more KRAS mutations in the RSPOfp-positive group than in the MSS/RNF43-mut group (53.3 vs. 24.2%, q<0.001). Moreover, the rate of APC mutations in the MSS/RNF43-mut subgroup was only 11.5%, compared to 81.6% in MSS/WT cases (q<0.01) (Figure 3A). Interestingly, in the MSS/RNF43-mut subgroup, ARID1A and ASXL1 mutations were identified in 22.7% and 2.2%, respectively, compared to 75.6% and 65.8% in the overall RNF43-mut cohort.
Figure 3: Molecular landscape of the MSS subcohort.
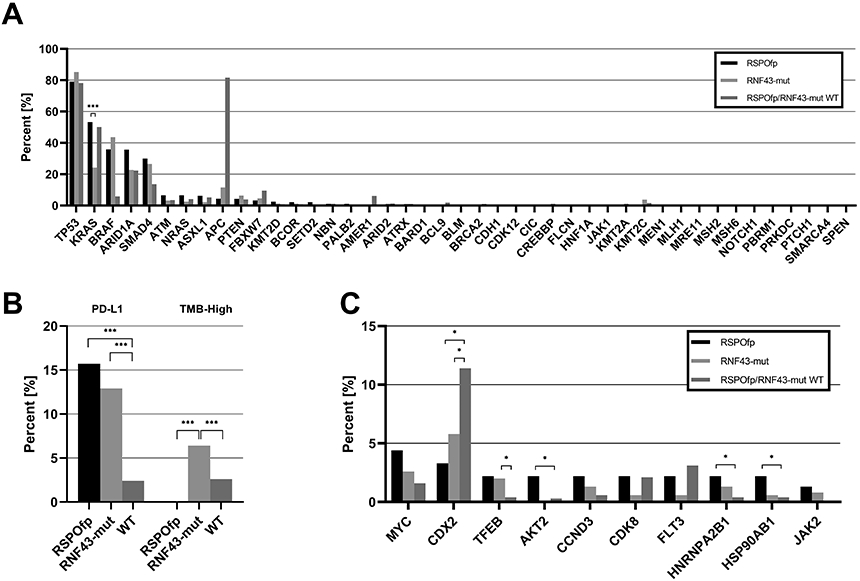
A) Genetic landscape comparison between RSPOfp vs. RNF43-mut tumors in the MSS subgroup. KRAS mutation was the only statistically different genetic alteration. ***q<0.001.
B) TMB and PDL-1 status in RNF43-mut, RSPOfp and RNF43/RSPOfp-WT CRCs. None of the RSPOfp patients showed a TMB >10 mt/Mb or a MSI-H/dMMR status.*q<0.05, **q<0.01, ***q<0.001.
C) Copy number alterations in RNF43 mutations, RSPO rearrangements and RNF43/RSPO-WT samples. *q<0.05.
In the MSS/RNF43-mut subgroup (n=158) and MSS/RNF43/RSPOfp WT cases (n=6,533), only 6.4% of the RNF43-mut samples and 2.6% of the WT samples had a TMB ≥10 mt/Mb (q<0.001). Furthermore, PD-L1 positive staining was observed in 12.9% of the MSS/RNF43-mut subgroup and in 2.4% of the MSS WT samples (q<0.001) (Figure 3B). In terms of CNA within the MSS subgroup, we observed more frequent CDX2 CNAs within the RNF43/RSPO WT (11%) compared to RSPOfp-positive (3%) and RNF43-mut (6%) cancers (all, q<0.05). In contrast, CNAs in TFEB, AKT2, HNRNPA2B1 as well as in HSP90AB1 were frequently less detected in RNF43/RSPO WT compared to RNF43/RSPO-positive tumors (all, q<0.05) (Figure 3C).
Regarding the MSI-H/dMMR subcohort it revealed that patients harbouring RNF43 mutations are characterized by increased frequencies of BRAF, KMT2D, HNF1A and BRCA2 mutations (all, q<0.001). In contrast, a lower prevalence of APC, KRAS, CTNNB1 and PIK3CA mutations compared to RNF43 WT patients was observed (all, q<0.05) (Supplementary Figure 1).
Since literature is conflicting regarding the functional loss of the specific RNF43 G659fs variant we evaluated the subset of RNF43 G659fs patients. Of note, virtually all of these cases showed a MSI-H/dMMR (99.2%) and a high TMB (99.6%) status. A comparison of RNF43 non-G569fs variants and RNF43/RSPO WT cases is displayed in Supplementary Figure 2.
DISCUSSION
Inappropriate activation of Wnt/β - catenin signaling is a key oncogenic event in a significant subset of CRCs (18) and is associated with tumor cell proliferation and drug resistance (19, 20). While the most frequent LOF mutation in the Wnt/β-catenin pathway, namely APC, has been very well studied (21), genetic alterations in the Wnt receptor complex emerged only recently as a potential new therapeutic target (21, 22). LOF mutations in RNF43 and RSPO fusion proteins were described previously to occur in a small subgroup of CRCs (15, 23, 24). However, the molecular landscape of these genetic alterations in CRC remains previously unexplored. Herein, we studied the molecular profile of CRC patients harboring a RNF43 mutation or a RSPOfp. Our study revealed that the molecular landscape of RNF43-mut CRC substantially differs from the genetic portrait of RSPOfp CRC. In fact, a higher rate of MSI-H/dMMR was observed in RNF43-mut compared to RSPOfp-positive tumors. This is in line with findings previously reported in the literature, that RNF43 mutations are more frequently encountered in patients with MSI-H/dMMR cancers (15), both in sporadic cases, and, to a lesser extent, in patients with a Lynch syndrome (25). However, when focusing on the subgroup of MSS/RNF43-mut tumors, the genetic profile exhibited greater similarity to that observed in RSPOfp-positive tumors. From this first perspective this finding might indicate that a part of RNF43 mutations might be a secondary mutation effect triggered by MSI. However, when analyzing the subset of MSI-H patients, distinct differences of the molecular landscape according to RNF43 status were observed. Hence, it remains elusive to which extent the genomic landscape is altered either due to MSI-H/dMMR status or RNF43 mutations.
Up to now, conflicting data exists regarding the pathogenicity of specific RNF43 mutations. In 2019, Tu and colleagues reported that the G659fs mutation does not seem to have an impact on carcinogenesis and seems to be fully functional (26). In contrast, two studies published in 2020 were not able to corroborate this finding (27). In particular, Yu and colleagues could show that the G659fs mutation induces LOF (16). The current uncertainty whether the G659fs mutation represents a LOF is also reflected in our analysed cohorts. In the discovery cohort the G659fs mutational variant was considered pathogenic whereas this specific mutation was excluded in the analyses of the validation cohort. Of note, we observed that virtually all patients harboring a RNF43 G659fs mutation were characterized by a MSI-H/dMMR and a TMB-high phenotype. Up to now, the impact of the G659fs mutation on WNT activation remains elusive. Therefore, further mechanistic studies are highly desirable to unravel the pathogenic interplay between MSI-H/dMMR and different RNF43 mutations.
To date, only limited data is available regarding prognostic significance of the respective alterations. Matsumoto and colleagues reported that RNF43 mutations are associated with an aggressive phenotype in BRAF-mut CRC leading to poor outcome (28). In line with this finding, survival analysis of the discovery cohort showed that patients harboring RNF43 mutations are characterized by inferior overall survival. Additionally, for the first time we observed that RSPO fusions represent a poor prognostic factor.
Anatomic location, or ‘sidedness’, of CRC has emerged over the past several years as an important predictive and prognostic biomarker in this cancer entity (29, 30). In particular, enrichment of mutations in BRAF, and also co-association with MSI-H/dMMR in right-sided CRC tumors is associated with a worse prognosis (31, 32), and many of the additional molecular factors associated with this worse outcome are under active investigation. Thus, we hypothesized that RNF43 and/or RSPOfp are associated with this genomic signature. In pursing this hypothesis, we detected a higher prevalence of RNF43 mutations in right-sided CRC primaries compared to tumors originating in left-sided locations irrespective of MSI status. This observation opens further options to combinational treatment approaches in this subset of patients with CRC. Indeed, in patients with MSI-H/dMMR tumors immune checkpoint inhibition has been proven to be efficacious (33, 34). To date, the reasons why patients with MSS cancers do not respond to immunotherapy have not been fully elucidated at the cellular and molecular levels, so far. Besides the hypothesis of reduced neoantigen formation in MSS tumors (35), other authors have reported that T-cells are actively excluded from the tumor (36). One possible pathway that modulating T-cell activity is the Wnt pathway, whose activation has been shown to prevent anti-tumor response in melanoma (37). Hence, inhibition of Wnt signaling seems to activate the immune system by activating dendritic cells as well as T-cells (38, 39).
Many studies reported, that RSPOfp alterations do not occur in tumors with APC mutations (6, 15), although it is not clear if RSPOfp mutations have a functional redundancy with APC mutations. However, in both (experimental and validation) cohorts, we observed that some RNF43-mut/RSPOfp-positive tumors harbor co-mutations in APC, which represents a novel finding.
Furthermore, despite the observation that Wnt/β-catenin activation is one of the key drivers of tumorigenesis in CRC, inhibition of the Wnt/β-catenin pathway has not been proven to be an efficacious therapeutic strategy to date (40). However, new attempts are being made to efficiently target the Wnt/β-catenin signaling pathway. One strategy could consist of inhibiting ligand-mediated activation of the Wnt/β-catenin cascade by PORCN inhibitors in CRC patients carrying RSPO rearrangements (17). For the RNF43 G659fs mutation as a predictive marker for a Wnt/β-catenin inhibiting treatment is still inconclusive, as some authors suggest that this mutation does not alter the protein’s function (26, 41). However, others provide evidence that this frameshift mutation leads to a responsiveness to PORCN inhibition (16). Other strategies may include to target the DKK-1, a modulator of Wnt/β-catenin activity (42), for which the monoclonal antibody DKN-01 is currently under clinical investigation in several gastrointestinal malignancies (e.g. NCT04057365 or NCT04166721) or targeting the Wnt co-receptor LRP5/6 for which the inhibitor BI905677 is currently under early clinical investigation (NCT03604445). Moreover, drugs directly blocking the interaction of β-catenin and CREB are currently being investigated in clinical trials (43). Taken together, it is tempting to speculate that the emergence of effective Wnt/β-catenin inhibitors, such as the PORCN inhibitors LGK974 (44), ETC-159 (45), or CGX1321 (41) might reshape the immunologic sensitivity of a subset of CRCs overcoming resistance to immunotherapeutical approaches, especially in the MSS subcohorts.
Several limitations apply to our study: 1.) Validation of our findings regarding RSPO rearrangements was not feasible, since in the validation cohort no fusion panel analysis was performed. 2.) Due to the retrospective study design a potential selection bias might have existed. 3.) Lacking the option of prospective longitudinal analyses we were not able to account for the possibility of sub-clonal RNF43 mutations. 4.) Due to limited availability of tissue and specific restrictions, no additional IHC stainings, depicting a variety of immunogenic markers and immune cell infiltration, and respective correlation with RNF43/RSPO status, could be conducted. Future prospective trials using sequential analyses during the molecular patient journey and further techniques (i.e. liquid biopsy, single-cell analysis) are desirable to dismantle the above mentioned limitations.
Taken together, in this large cohort of CRC patients whose tumors underwent molecular profiling we have identified a significant subset of CRCs harboring a RNF43 mutation or a RSPO fusion protein which are characterized by a distinct genetic landscape. Thus, these detectable gene alterations represent a potential new therapeutic target and several clinical trials are currently ongoing to prove the efficacy of different Wnt/β-catenin signaling inhibitors in RNF43/RSPO-positive tumors. Furthermore, MSI-H/dMMR were observed in a subgroup of RNF43-mutated tumors suggesting that immune checkpoint inhibition with and without Wnt/β-catenin signaling inhibitors may be a reasonable combinational therapeutic approach that should be tested in prospective trials.
Supplementary Material
Translational Relevance:
To provide meaningful rationales to develop new impactful targeted approaches for colorectal cancer (CRC) patients, we comprehensively described the mutational landscape of R-spondin fusion proteins (RSPOfp) and RNF43 mutations, which are known to induce Wnt signaling. Using a cohort of 7,245 CRC samples we could identify five new RSPO rearrangements and could describe the unique molecular portrait of RSPOfp and RNF43 mutations in CRCs. The genetic profile of RSPOfp positive tumors is similar to RNF43-mutated CRC and is characterized by a higher frequency of BRAF, SMAD4 and KMT2D mutations in comparison to RSPOfp/RNF43 negative cases. Of note, a subgroup of RNF43-mutated tumors is associated with microsatellite instability. Our data could support clinical and pre-clinical research developing treatments targeting the Wnt pathway and could also provide a rationale for combinational approaches to overcome primary resistance to immunotherapy in CRC.
Funding:
This work was supported by the National Cancer Institute (P30CA 014089).
Footnotes
Conflict of interests: FB, KZ, GS, FK, DR, AP, BAW, PES, KHL, AG, GST, HT, SHL, HLN, AA, ZT, MDB, and DW have no competing interests. AFS has received research funding, travel payments, and is on the speaker’s panel for Caris. AS, RMG, JLM, EL, HJL has received travel payments, and is on the speaker`s panel for Caris. YB, JX, JS, ME, CN, and WMK are employers of Caris. VND is employer of A*Star.
REFERENCES
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70(1):7–30. [DOI] [PubMed] [Google Scholar]
- 2.Malvezzi M, Carioli G, Bertuccio P, Boffetta P, Levi F, La Vecchia C, et al. European cancer mortality predictions for the year 2018 with focus on colorectal cancer. Ann Oncol. 2018;29(4):1016–22. [DOI] [PubMed] [Google Scholar]
- 3.Hinoi T, Akyol A, Theisen BK, Ferguson DO, Greenson JK, Williams BO, et al. Mouse model of colonic adenoma-carcinoma progression based on somatic Apc inactivation. Cancer Res. 2007;67(20):9721–30. [DOI] [PubMed] [Google Scholar]
- 4.Sansom OJ, Reed KR, Hayes AJ, Ireland H, Brinkmann H, Newton IP, et al. Loss of Apc in vivo immediately perturbs Wnt signaling, differentiation, and migration. Genes Dev. 2004;18(12):1385–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dow LE, O'Rourke KP, Simon J, Tschaharganeh DF, van Es JH, Clevers H, et al. Apc Restoration Promotes Cellular Differentiation and Reestablishes Crypt Homeostasis in Colorectal Cancer. Cell. 2015;161(7):1539–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Seshagiri S, Stawiski EW, Durinck S, Modrusan Z, Storm EE, Conboy CB, et al. Recurrent R-spondin fusions in colon cancer. Nature. 2012;488(7413):660–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Han T, Schatoff EM, Murphy C, Zafra MP, Wilkinson JE, Elemento O, et al. R-Spondin chromosome rearrangements drive Wnt-dependent tumour initiation and maintenance in the intestine. Nat Commun. 2017;8:15945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.de Lau W, Peng WC, Gros P, Clevers H. The R-spondin/Lgr5/Rnf43 module: regulator of Wnt signal strength. Genes Dev. 2014;28(4):305–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Min BH, Hwang J, Kim NK, Park G, Kang SY, Ahn S, et al. Dysregulated Wnt signalling and recurrent mutations of the tumour suppressor RNF43 in early gastric carcinogenesis. J Pathol. 2016;240(3):304–14. [DOI] [PubMed] [Google Scholar]
- 10.Ryland GL, Hunter SM, Doyle MA, Rowley SM, Christie M, Allan PE, et al. RNF43 is a tumour suppressor gene mutated in mucinous tumours of the ovary. J Pathol. 2013;229(3):469–76. [DOI] [PubMed] [Google Scholar]
- 11.Wang K, Yuen ST, Xu J, Lee SP, Yan HH, Shi ST, et al. Whole-genome sequencing and comprehensive molecular profiling identify new driver mutations in gastric cancer. Nat Genet. 2014;46(6):573–82. [DOI] [PubMed] [Google Scholar]
- 12.Jiao Y, Yonescu R, Offerhaus GJ, Klimstra DS, Maitra A, Eshleman JR, et al. Whole-exome sequencing of pancreatic neoplasms with acinar differentiation. J Pathol. 2014;232(4):428–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Waddell N, Pajic M, Patch AM, Chang DK, Kassahn KS, Bailey P, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015;518(7540):495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eto T, Miyake K, Nosho K, Ohmuraya M, Imamura Y, Arima K, et al. Impact of loss-of-function mutations at the RNF43 locus on colorectal cancer development and progression. J Pathol. 2018;245(4):445–55. [DOI] [PubMed] [Google Scholar]
- 15.Giannakis M, Hodis E, Jasmine Mu X, Yamauchi M, Rosenbluh J, Cibulskis K, et al. RNF43 is frequently mutated in colorectal and endometrial cancers. Nat Genet. 2014;46(12):1264–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu J, Yusoff PAM, Woutersen DTJ, Goh P, Harmston N, Smits R, et al. The Functional Landscape of Patient-Derived RNF43 Mutations Predicts Sensitivity to Wnt Inhibition. Cancer Res. 2020;80(24):5619–32. [DOI] [PubMed] [Google Scholar]
- 17.Koo BK, van Es JH, van den Born M, Clevers H. Porcupine inhibitor suppresses paracrine Wnt-driven growth of Rnf43;Znrf3-mutant neoplasia. Proc Natl Acad Sci U S A. 2015;112(24):7548–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nusse R, Clevers H. Wnt/β-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell. 2017;169(6):985–99. [DOI] [PubMed] [Google Scholar]
- 19.Moradi A, Ghasemi F, Anvari K, Hassanian SM, Simab SA, Ebrahimi S, et al. The cross-regulation between SOX15 and Wnt signaling pathway. J Cell Physiol. 2017;232(12):3221–5. [DOI] [PubMed] [Google Scholar]
- 20.Yuan S, Tao F, Zhang X, Zhang Y, Sun X, Wu D. Role of Wnt/β-Catenin Signaling in the Chemoresistance Modulation of Colorectal Cancer. Biomed Res Int. 2020;2020:9390878. doi: 10.1155/2020/9390878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang L, Shay JW. Multiple Roles of APC and its Therapeutic Implications in Colorectal Cancer. J Natl Cancer Inst. 2017;109(8):djw332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jiang X, Cong F. Novel Regulation of Wnt Signaling at the Proximal Membrane Level. Trends Biochem Sci. 2016;41(9):773–83. [DOI] [PubMed] [Google Scholar]
- 23.Yan HHN, Lai JCW, Ho SL, Leung WK, Law WL, Lee JFY, et al. RNF43 germline and somatic mutation in serrated neoplasia pathway and its association with BRAF mutation. Gut. 2017;66(9):1645–56. [DOI] [PubMed] [Google Scholar]
- 24.Sekine S, Yamashita S, Tanabe T, Hashimoto T, Yoshida H, Taniguchi H, et al. Frequent PTPRK-RSPO3 fusions and RNF43 mutations in colorectal traditional serrated adenoma. J Pathol. 2016;239(2):133–8. [DOI] [PubMed] [Google Scholar]
- 25.Fennell LJ, Clendenning M, McKeone DM, Jamieson SH, Balachandran S, Borowsky J, et al. RNF43 is mutated less frequently in Lynch Syndrome compared with sporadic microsatellite unstable colorectal cancers. Fam Cancer. 2018;17(1):63–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tu J, Park S, Yu W, Zhang S, Wu L, Carmon K, et al. The most common RNF43 mutant G659Vfs*41 is fully functional in inhibiting Wnt signaling and unlikely to play a role in tumorigenesis. Sci Rep. 2019;9(1):18557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tsukiyama T, Zou J, Kim J, Ogamino S, Shino Y, Masuda T, et al. A phospho-switch controls RNF43-mediated degradation of Wnt receptors to suppress tumorigenesis. Nat Commun. 2020;11(1):4586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Matsumoto A, Shimada Y, Nakano M, Oyanagi H, Tajima Y, Kameyama H, et al. RNF43 mutation is associated with aggressive tumor biology along with BRAF V600E mutation in right-sided colorectal cancer. Oncol Rep. 2020;43(6):1853–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Snyder M, Bottiglieri S, Almhanna K. Impact of Primary Tumor Location on First-line Bevacizumab or Cetuximab in Metastatic Colorectal Cancer. Rev Recent Clin Trials. 2018;13(2):139–49. [DOI] [PubMed] [Google Scholar]
- 30.Boeckx N, Koukakis R, Op de Beeck K, Rolfo C, Van Camp G, Siena S, et al. Primary tumor sidedness has an impact on prognosis and treatment outcome in metastatic colorectal cancer: results from two randomized first-line panitumumab studies. Ann Oncol. 2017;28(8):1862–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee MS, Menter DG, Kopetz S. Right Versus Left Colon Cancer Biology: Integrating the Consensus Molecular Subtypes. J Natl Compr Canc Netw. 2017;15(3):411–9. [DOI] [PubMed] [Google Scholar]
- 32.Gao XH, Yu GY, Gong HF, Liu LJ, Xu Y, Hao LQ, et al. Differences of protein expression profiles, KRAS and BRAF mutation, and prognosis in right-sided colon, left-sided colon and rectal cancer. Sci Rep. 2017;7(1):7882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med. 2015;372(26):2509–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.André T, Shiu KK, Kim TW, Jensen BV, Jensen LH, Punt C, et al. Pembrolizumab in Microsatellite-Instability-High Advanced Colorectal Cancer. N Engl J Med. 2020;383(23):2207–18. [DOI] [PubMed] [Google Scholar]
- 35.Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348(6230):124–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Feig C, Jones JO, Kraman M, Wells RJ, Deonarine A, Chan DS, et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci U S A. 2013;110(50):20212–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature. 2015;523(7559):231–5. [DOI] [PubMed] [Google Scholar]
- 38.Suryawanshi A, Manoharan I, Hong Y, Swafford D, Majumdar T, Taketo MM, et al. Canonical wnt signaling in dendritic cells regulates Th1/Th17 responses and suppresses autoimmune neuroinflammation. J Immunol. 2015;194(7):3295–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gattinoni L, Zhong XS, Palmer DC, Ji Y, Hinrichs CS, Yu Z, et al. Wnt signaling arrests effector T cell differentiation and generates CD8+ memory stem cells. Nat Med. 2009;15(7):808–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Martin-Orozco E, Sanchez-Fernandez A, Ortiz-Parra I, Ayala-San Nicolas M. WNT Signaling in Tumors: The Way to Evade Drugs and Immunity. Front Immunol. 2019;10:2854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li C, Cao J, Zhang N, Tu M, Xu F, Wei S, et al. Identification of RSPO2 Fusion Mutations and Target Therapy Using a Porcupine Inhibitor. Sci Rep. 2018;8(1):14244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Niehrs C Function and biological roles of the Dickkopf family of Wnt modulators. Oncogene. 2006;25(57):7469–81. [DOI] [PubMed] [Google Scholar]
- 43.Emami KH, Nguyen C, Ma H, Kim DH, Jeong KW, Eguchi M, et al. A small molecule inhibitor of beta-catenin/CREB-binding protein transcription [corrected]. Proc Natl Acad Sci U S A. 2004;101(34):12682–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li J, Wu G, Xu Y, Ruan N, Chen Y, Zhang Q, et al. Porcupine Inhibitor LGK974 Downregulates the Wnt Signaling Pathway and Inhibits Clear Cell Renal Cell Carcinoma. Biomed Res Int. 2020;2020:2527643. doi: 10.1155/2020/2527643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ng M, Tan DS, Subbiah V, Weekes CD, Teneggi V, Diermayr V, et al. First-in-human phase 1 study of ETC-159 an oral PORCN inhbitor in patients with advanced solid tumours. J Clin Oncol. 2017;35(15_suppl):2584 . [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The deidentified sequencing data are owned by Caris Life Sciences. The datasets generated during and analyzed during the current study are available from the authors upon reasonable request and with permission of Caris Life Sciences. Qualified researchers may contact the corresponding author with their request.