

SUMMARY
Master transcription factors (TFs) directly regulate present and future cell states by binding DNA regulatory elements and driving gene-expression programs. Their abundance influences epigenetic priming to different cell fates at the chromatin level, especially in the context of differentiation. In order to link TF protein abundance to changes in TF motif accessibility and open chromatin, we developed InTAC-seq, a method for simultaneous quantification of genome-wide chromatin accessibility and intracellular protein abundance in fixed cells. Our method produces high-quality data and is a cost-effective alternative to single-cell techniques. We showcase our method by purifying bone marrow (BM) progenitor cells based on GATA-1 protein levels and establish high GATA-1-expressing BM cells as both epigenetically and functionally similar to erythroid-committed progenitors.
In brief
Baskar et al. present a multi-omic epigenetics approach to assay chromatin accessibility of fixed cells defined by intracellular regulators (e.g., transcription factors). Their method, InTAC-seq, generates high-quality data with low cell numbers and links chromatin-binding protein levels to epigenetic landscapes within rare and transient cell states in human tissue.
Graphical Abstract
INTRODUCTION
Transcription factors (TFs) are the master drivers of cell identity and differentiation, and their binding to specific regulatory sequences across the genome controls gene networks conferring cell phenotype and function. The exact binding sites of a TF can depend on various factors including the pre-existing chromatin landscape, the presence of cooperative and antagonistic TFs, and the concentration of the TF in the cell (Spitz and Furlong, 2012). It has been shown that subtle differences in TF levels can have significant effects on cell determination, as evidenced by classical studies on TF gradients in defining anterior-posterior axis patterning in Drosophila embryogenesis (Courey and Huang, 1995; Rivera-Pomar and Jãckle, 1996). Similarly, studies of hematopoiesis in mice have shown that different levels of the master TF PU.1 can drive myeloid versus lymphoid fate, while insufficient levels of PAX5, a driver of the B cell lineage, can result in cells adopting an abnormal bipotent myeloid/lymphoid phenotype (DeKoter and Singh, 2000; Simmons et al., 2012). These examples highlight the importance of interrogating the molecular mechanisms underlying the consequences of TF levels on cellular phenotype. However, the effects of endogenous TF abundance on its set of target DNA-binding sites are difficult to interrogate in unmanipulated primary human tissue despite improvements in inducible gene-expression systems in precisely controlling expression and protein abundance (Pedone et al., 2019).
The identification of regulatory regions in a cell has been facilitated by techniques that map regions of open chromatin, such as the assay for transposase-accessible chromatin by sequencing (ATAC-seq) (Buenrostro et al., 2013; Corces et al., 2017). Measuring the association between TF abundance and chromatin accessibility in cells can link master TFs that drive cell-fate decisions to the epigenetic events that influence these decisions. Still, methods to directly measure regulatory protein abundance in a cell and link changes in their levels to genome-wide changes in chromatin accessibility are lacking. The challenge of revealing these relationships is exacerbated when cells of interest are rare and regulatory proteins are transiently expressed in primary human tissue, such as in hematopoietic lineage commitment in the bone marrow. However, by overcoming these challenges, measurements of protein abundance for lineage-defining TFs in minimally manipulated primary tissues could be integrated with epigenetic priming information through chromatin accessibility, thereby providing unprecedented granularity into cell-fate decision mechanisms.
There have been a number of attempts to link gene expression to chromatin-accessibility profiles employing single-cell ATAC-seq (scATAC-seq) alone or in combination with RNA sequencing (RNA-seq) in the same cell (Cao et al., 2018; Granja et al., 2019; Ma et al., 2020; Satpathy et al., 2019). While RNA levels are informative, they do not always reflect existing protein levels in the nucleus or account for post-translational regulation, which can dictate the functional state of a cell (Schwanhäusser et al., 2011; Vogel and Marcotte, 2012; Vogel et al., 2010). Protein expression has also been previously linked to chromatin accessibility in single cells (protein-indexed ATAC [piATAC]) (Chen et al., 2018). However, transcription start site (TSS) enrichment scores were considerably lower than those from live-cell ATAC-seq libraries, and piATAC datasets had comparatively few unique reads per cell. The lower enrichment in signal over background and decreased library complexity resulted in difficulty associating differences in TF protein abundance with significant changes in chromatin accessibility at predicted binding sites. Therefore, improvements in data quality are needed to quantitatively assess how protein regulators (i.e., TFs, chromatin modifiers, upstream signaling molecules) influence epigenetic states. Such methods will enable the robust, combinatorial measurement of specific chromatin-binding proteins, such as TFs, that affect gene expression and link their protein abundance to chromatin changes, thereby improving our understanding of the complex interplay between TF abundance and gene regulatory programs involved in cell fate.
Here, we present InTAC-seq, a method that profiles chromatin accessibility from cells isolated based on abundance of cell surface and intracellular proteins-of-interest, that has data quality and cost comparable to standard ATAC-seq on live cells. In-TAC-seq can be applied to chromatin-binding protein factors such as TFs and generates high-quality, quantitative data from primary tissue to robustly measure genome-wide chromatin accessibility associated with TF levels. We first benchmarked In-TAC-seq with the GM12878 lymphoblastoid cell line. We then demonstrated that variation in GATA-1 protein abundance was associated with altered chromatin accessibility at sites across the spectrum of GATA-1 binding affinity in the K562 erythroleukemic cell line. We further applied InTAC-seq to bone marrow progenitor cells isolated based on GATA-1 expression to profile GATA-1-associated chromatin accessibility in the context of human hematopoietic differentiation. The high-quality profiles produced by InTAC-seq enabled us to integrate our results with previous scATAC- and RNA-seq bone marrow datasets. Thus, we could position the isolated GATA-1 cells within this multi-omic landscape of human hematopoietic lineage commitment and link high GATA-1 expression to erythroid priming. By using surrogate surface markers, we show clonally homogeneous erythroid differentiation from GATA-1-high progenitor cells. Taken together, our method enabled measurement of chromatin accessibility associated with endogenous GATA-1 TF abundance in human bone marrow, thereby characterizing high-GATA-1-expressing progenitors as epigenetically and functionally similar to erythroid progenitors in human red blood cell (RBC) development.
Design
The main challenge with combining ATAC-seq with intracellular protein quantification lies in the fixation and permeabilization of cells required for direct measurement of intracellular proteins using affinity reagents, such as antibodies, and subsequent paraformaldehyde crosslinking reversal at high temperatures prior to library amplification. The previously applied 65°C crosslink reversal step results in dissociation and loss of shorter fragments that contribute to a large fraction of the final ATAC-seq library, resulting in reduced library complexity and lower TSS enrichment scores (Chen et al., 2018). To improve data quality, the InTAC-seq protocol uses a shorter fixation time with mild permeabilization and reversal of formaldehyde crosslinks with a catalyst to allow crosslink reversal to occur at 37°C (Karmakar et al., 2015; Figure 1A).
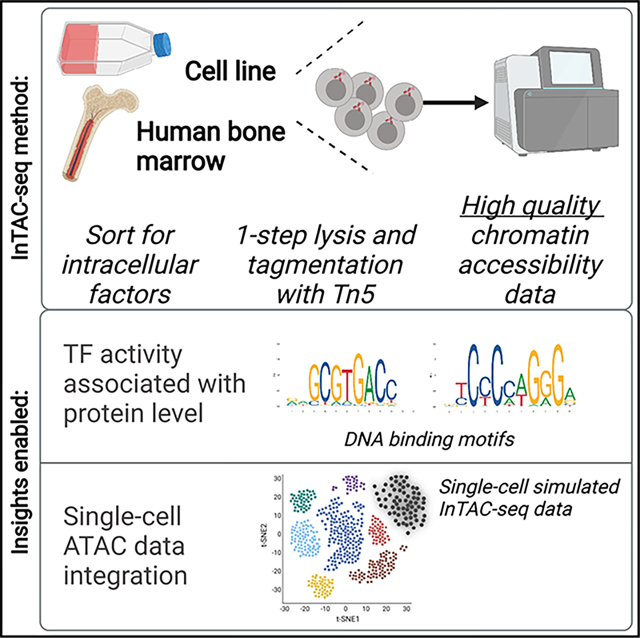
Figure 1. InTAC-seq data from fixed cells is of comparable quality to ATAC-seq data from live cells and allows interrogation of chromatin accessibility associated with TF protein abundance.
(A) Overview of InTAC-seq experimental protocol.
(B) Genome coverage of ATAC-seq data generated from live cells, fixed cells using InTAC-seq, or fixed cells using piATAC at the EBF1 locus in GM12878 cells.
(C) Normalized Tn5 insertion profiles centered at transcription start sites (TSSs) for the indicated ATAC-seq libraries.
(D) Scatterplot of estimated library size versus normalized TSS insertion score across all replicates of compared protocols
(E) Scatterplot of reads in consensus peaks averaged across replicates between InTAC-seq and live ATAC samples, with calculated Spearman correlation coefficient as shown.
(F) FACS plot of forward scatter (linear scale) versus GATA-1 protein abundance (log10 scale) and the gating strategy to isolate the highest and lowest 15% of GATA-1-expressing K562 cells.
(G) MA plot of log2 fold change in accessibility between GATA-1-high and GATA-1-low K562 populations versus log2 mean number of reads at all consensus peaks. Peaks with significant changes in accessibility are highlighted in red or blue.
(H) Most significantly enriched TF motifs in differentially accessible peaks in GATA-1-high cells calculated using Fisher’s test.
(I) Average accessibility of GATA-1 motif sites across all consensus ATAC-seq peaks binned by GATA-1 motif score. Accessibility is defined here as the area under the curve of a plot of bias-corrected, normalized Tn5 insertions centered at GATA-1 motif sites (as in Figure S1G), integrated from −50 to +50 bp and excluding the TF footprint from −10 to +10 bp.
(J) Difference in GATA-1 motif accessibility between GATA-1 high and GATA-1 low samples normalized to the accessibility in the GATA-1 low population for each motif score bin.
RESULTS
InTAC-seq libraries are comparable in quality to ATAC-seq libraries generated from fresh cells
Libraries generated from fixed GM12878 cells using InTAC-seq exhibited enrichment in Tn5 insertions at TSSs comparable to TSS enrichment in ATAC-seq libraries from unfixed live cells and approximately 2-fold greater than that in published bulk piA-TAC libraries (Chen et al., 2018; Figure 1C). The fragment length distribution for InTAC-seq libraries was also similar to the distribution in libraries from live cells due to preservation of sub-nucleosomal fragments at 37°C (Figure S1A). The retention of sub-nucleosomal fragments translated into a larger estimated number of unique ATAC-seq fragments from InTAC-seq and live ATAC libraries relative to piATAC libraries (Figure 1D). To compare InTAC-seq libraries with unfixed libraries, we plotted the correlation of reads in consensus peaks and found a strong correlation between InTAC-seq data generated from fixed cells and ATAC-seq data generated from live cells (Figures 1E and S1B). Genome Browser tracks further illustrated the concordance between InTAC-seq data and live-cell ATAC-seq data (Figure 1B). We further demonstrated that InTAC-seq can be performed on as low as 100 cells, enabling profiling of rare cell types (Figure S1C). Together, these results show that the InTAC-seq protocol produces data on par with live ATAC-seq, with comparable signal-to-background (as defined by TSS enrichment scores) and library complexity.
InTAC-seq is sufficiently sensitive to detect differences in chromatin accessibility associated with varying TF protein abundance
We next aimed to use InTAC-seq to detect chromatin accessibility differences in cells endogenously expressing different levels of chromatin-binding protein such as TFs. We compared the chromatin-accessibility profiles in K562 cells expressing the highest or lowest 15% of GATA-1 levels using InTAC-seq (Figures 1F and 1G). With ChromVAR (Schep et al., 2017), we found that GATA-binding motifs were among the most variable in accessibility across samples (Figure S1E). Specifically, we observed increased accessibility in cells with high levels of GATA-1 at GATA-1 motif sites (Figures S1G, S1H, and 1H). Differentially accessible peaks (false discovery rate [FDR] <0.1) between cells with high versus low GATA-1 were almost exclusively more accessible in GATA-1-high cells, and they were most significantly enriched for GATA motifs relative to all other peaks present across samples (Figures 1H and S1F), suggesting that accessibility differences are likely due to differences in GATA-1 abundance. However, we observed slight increases in accessibility at motif sites for other erythroid TFs in GATA-1-high cells, which could indicate that GATA-1-high cells within a K562 culture are further along in erythroid differentiation. It therefore remains possible that the increased accessibility at GATA-1 sites could be due to factors other than GATA-1.
We next asked how increases in accessibility at GATA-1 motif sites can vary based on the predicted binding affinity of the putative GATA-1 binding site. To address this, we grouped all consensus peaks into 20 bins of equal size based on the quality of their GATA-1 motif score and measured the average accessibility across these bins in cells with high versus low GATA-1 abundance. We found that cells with high GATA-1 levels exhibit a general increase in accessibility at GATA-1 motif sites above a threshold motif score that likely represents the minimum score for a true GATA-1 binding site (Figure 1I). However, we observed that the greatest change in accessibility between cells with high versus lower GATA-1 occurred at sites of moderate predicted affinity (i.e., bin 18), suggesting that the highest-affinity GATA-1 binding sites may be saturated even at lower GATA-1 levels (Figure 1J). These results showcase the ability of InTAC to measure subtle differences in chromatin accessibility among populations, allowing us to link natural variation in TF abundance to accessibility differences at specific genomic loci such as putative TF-binding motif sites.
InTAC-seq links high expression of GATA-1 protein to erythroid-committed bone marrow (BM) progenitors
While clinically significant for both cell-based therapies and hematopoietic dysplasia (Zivot et al., 2018), the regulatory landscape of erythropoietic cell homeostasis in the human bone marrow (BM) compartment is not well understood. BM progenitor populations are traditionally defined and isolated based on expression of cell-surface proteins, which viably preserves these cells for downstream functional assays (Akashi et al., 2000; Manz et al., 2002; Mori et al., 2015; Seita and Weissman, 2010). However, these surface molecules are not always functionally related to the cellular state that we associate them with, thereby resulting in oversimplification of the complex hematopoietic system (Paul et al., 2015). We therefore hypothesized that intracellular regulatory protein abundance would identify cellular states with higher fidelity and enable more accurate molecular characterization. To test this, we focused on human erythroid progenitor cells, which are regulated by GATA-1 (Gutiérrez et al., 2020; Stachura et al., 2006; Suzuki et al., 2003) but are generally defined by unrelated surface molecules, such as a CD45RA-negative population separated based on FLT3 (Doulatov et al., 2010) or IL3RA (CD123) (Manz et al., 2002), with the latter gating subsequently referred to as CD123− megakaryocyte-erythroid progenitor cells (MEPs; fully defined as CD34+/CD38+/CD10−/CD45RA−/CD123−). We applied InTAC-seq to interrogate the link between the abundance of the lineage-defining TF, GATA-1, and epigenetic changes related to GATA-1 abundance differences in RBC development.
First, we used InTAC-seq to profile the accessible chromatin landscape of GATA-1-high- (top 8%) and GATA-1-mid and low-expressing cells (referred to as GATA-1-mid/low cells in the lower 87%) within the landscape of the general BM progenitor compartment (i.e., CD34+CD38+) to determine if GATA-1-high cells were enriched for erythroid epigenetic signatures (Figure 2A). InTAC-seq libraries generated from these isolated subpopulations had TSS enrichment scores similar to ATAC-seq libraries from live GM12878 cells, indicating that InTAC-seq performs well on primary human samples (Figures S2A and S2B). We observed a marked increase in accessibility at regulatory regions surrounding the GATA-1 locus in GATA-1-high versus mid/low progenitors, consistent with GATA-1 expression levels, along with a decrease in accessibility at regulatory regions within the SPI1 locus, which encodes a TF known to antagonize GATA-1 activity and repress erythroid commitment (Arinobu et al., 2007; Nerlov et al., 2000; Rekhtman et al., 1999; Zhang et al., 2000) (Figure 2B). The binding motifs for these two TFs also exhibited strong differences in accessibility between GATA-1-high and GATA-1-mid/low progenitors (Figure 2C). We further observed a broader trend of increased accessibility surrounding the motifs for TFs that drive erythroid fates (e.g., GATA-1, Mecom [Shimizu et al., 2002]) and decreased accessibility at motifs for TFs that drive myeloid and lymphoid lineages (e.g., SPI1 [Hromas et al., 1993; Klemsz et al., 1990], EBF1 [Nechanitzky et al., 2013]) in GATA-1-high progenitors (Figure 2C). Analysis of differentially accessible peaks showed enrichment of these erythroid TF motifs in sites more accessible in GATA-1-high progenitors and enrichment of a variety of myeloid and lymphoid TFs in sites more accessible in GATA-1-mid/low-expressing progenitors (Figures 2D, 2E, and S2C). Deeper analysis into accessibility at GATA-1 motif sites of varying binding affinities across the genome showed that high GATA-1 abundance in progenitors is associated with marked increase in accessibility above a threshold motif score (~12; Figure 2F). The greatest increases were observed at sites with the highest predicted GATA-1 affinity, suggesting the preferential binding of GATA-1 to the genome at these sites. Altogether, these data suggest that high GATA-1 expression in human hematopoietic progenitors is likely linked to an erythroid epigenetic program while repressing alternative lineage fates.
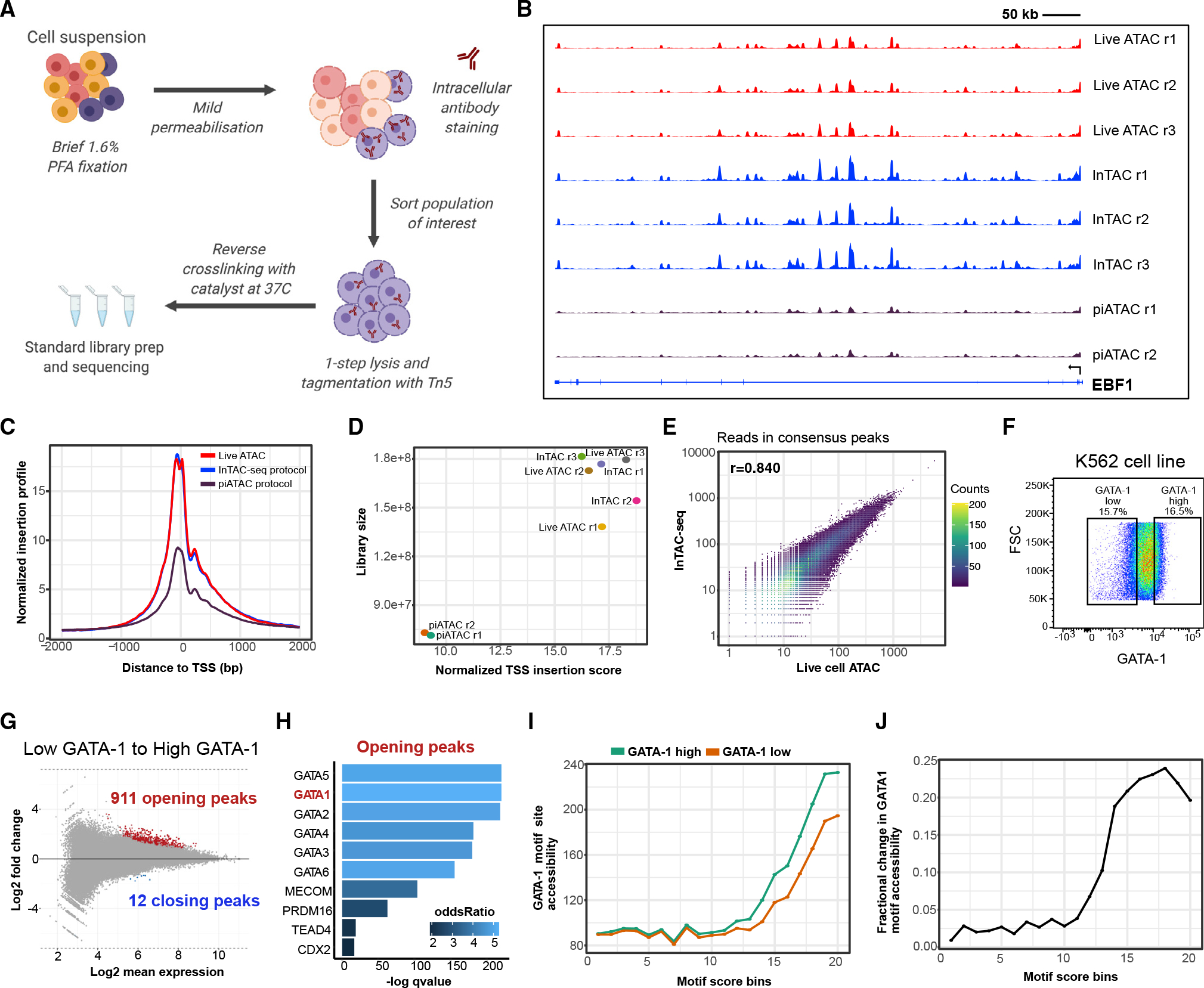
Figure 2. GATA-1-high BM progenitors show strong priming for erythroid lineage.
(A) BM aspirate is ficolled and enriched for CD34+ cells before gating for CD34+/CD38+ cells and selecting GATA-1-high population (top ~8%) and remaining GATA-1-expressing cells (denoted as GATA-1 mid/low) (bottom ~87%).
(B) InTAC-seq genome coverage plots at GATA1 and SPI1 loci for GATA-1-high and -mid/low BM progenitors.
(C) Heatmap of chromVAR deviation scores across GATA-1 high and mid/low BM progenitors for top 50 most variable motifs.
(D) MA plot of log2 fold change in accessibility between GATA-1 high and mid/low BM progenitors versus log2 mean number of reads in consensus peaks. Peaks with significant changes in accessibility are highlighted in red or blue.
(E) Most significantly enriched TF motifs in differentially accessible peaks between GATA-1-high and -mid/low BM progenitors calculated using Fisher’s test.
(F) (Top) Average accessibility of GATA-1 motif sites across all consensus ATAC-seq peaks binned by GATA-1 motif score. Accessibility is defined here as the area under the curve of a plot of bias-corrected, normalized Tn5 insertions centered at GATA-1 motif sites, integrated from −50 to +50 bp and excluding the TF footprint from −10 to +10 bp. (Bottom) Difference in GATA-1 motif accessibility between GATA-1 high and mid/low samples normalized to the accessibility in the GATA-1 mid/low population for each motif score bin.
(G) UMAP of previously published and annotated BM scATAC dataset with Seurat clusters manually annotated as key BM populations.
(H) Normalized GATA1 gene expression across BM progenitors in UMAP space (expression derived from scRNA-seq data integrated with scATAC-seq data).
(I) Bulk BM progenitor InTAC-seq data simulated as scATAC counts and projected onto scATAC UMAP space.
To further assess the position of these progenitors within the hematopoietic hierarchy, we projected our InTAC-seq samples onto a scATAC-seq uniform manifold approximation and projection (UMAP) of filtered BM mononuclear cells (BMMCs) from published datasets to identify their closest hematopoietic cell type using ArchR (Granja et al., 2019, 2020). Given our focus on erythropoiesis, we performed a more fine-grained analysis of the erythroid cluster and separated it into early, mid, and late erythroid progenitor populations based on differences in gene accessibility and chromVAR deviation scores of key TFs (Figures 2G, S2D, and S2F). Despite diffuse GATA1 expression across hematopoietic populations, we see highest enrichment of GATA1 gene expression at the erythroid arm of UMAP (Figure 2H). Likewise, when projected, we found that progenitors with high GATA-1 protein abundance, represented by the top 8% of GATA-1-expressing BM progenitor cells, were positioned within the mid erythroid progenitor branch while mid/low-GATA-1-abundance progenitors spanned scRNA-annotated granulocyte/monocyte progenitor (GMP), lymphomyeloid primed progenitor (LMPP), and common myeloid progenitor (CMP) clusters (Figure 2I). Strikingly, the position of GATA-1-high progenitors at the mid erythroid cluster (Figure 2I) combined with the overall enrichment of erythropoietic programs (Figures 2C–2E) suggests that the GATA-1 highest expressing cells are most similar to progenitors in the erythroid fate arm of the BM map.
Single-cell proteomic map of erythroid development complements InTAC-seq to reveal GATA-1 co-expression patterns
We next sought to better understand GATA-1 protein abundance dynamics in relation to other key TFs in erythroid progenitor development. We curated a 35-plex antibody panel comprised of key TFs implicated in hematopoiesis, such as GATA-1, GATA-2, BMI-1, and PBX-1 (Figure 3A). The panel also included surface markers for BM progenitor gating strategies and markers previously shown to be involved in erythropoiesis to better understand their dynamics relative to GATA-1 in erythroid commitment (Figure 3A). In order to construct a map of the BM progenitor space, we embedded our density-downsampled dataset from ~1 million BM progenitors into a force-directed layout (Jacomy et al., 2014) and visualized the topology of cell-state distributions in high-dimensional space (Traag et al., 2019; Wolf et al., 2018) (Figures 3B and S3B). We then overlaid manually gated, previously defined progenitor population labels (Manz et al., 2002) (Figures 3B, left, and S3A, progenitor gating). Expectedly, reference populations do not project in the previously established hierarchical manner and instead present as a continuum of states from early progenitor to distinct endpoints characterized by an erythroid-primed branch (indicated by GATA-1), CMPs or GMPs (indicated by CD123), common lymphoid progenitors (CLPs; indicated by CD10), and myeloid cells (immature myeloid indicated by CD117 and mature myeloid by CD33) (Paul et al., 2015) (Figures 3B, right, 3C, and S3C, surface marker distributions).
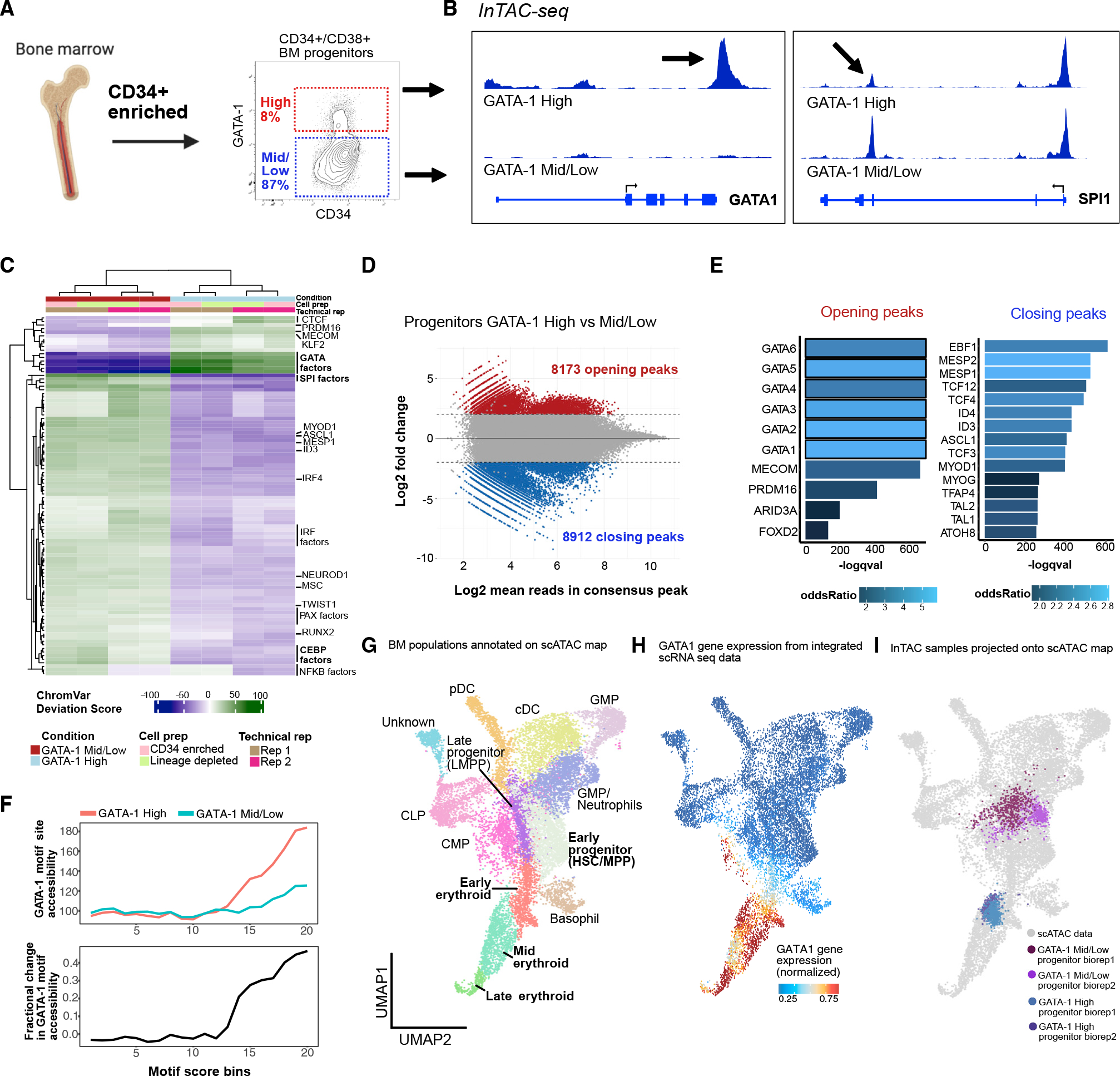
Figure 3. High-dimensional, single-cell proteomic analysis of BM progenitors identifies TF and surface-marker trends in erythroid commitment.
(A) BM aspirate is ficolled and enriched for CD34+ cells before staining with antibodies to surface marker panel and key TF to capture single-cell protein abundance using mass cytometry. High-dimensional data with over 1 million cells were used to delineate heterogeneity in BM progenitors and find surface-marker surrogates for GATA-1 TF abundance for further functional validation.
(B) Force-directed layout (ForceAtlas2) of density downsampled (to 250,000 cells) CD45+-gated BM progenitors colored by manually gated populations.
(C) Normalized marker expression of key surface and TF proteins (in orange) across force-directed layout. Orange arrow denotes GATA-1-high region in single-cell map.
(D) Leiden clusters of BM progenitors (resolution = 1) visualized on force-directed layout.
(E) Barplot of frequency of manually gated BM progenitor populations across 11 Leiden clusters.
(F) Normalized diffusion pseudotime calculation visualized on force-directed layout with trajectory from HSCs to erythroid-primed progenitors across Leiden clusters 3, 1, and 11 (in red).
(G) Row-normalized heatmap of median marker abundance at 100 bins across diffusion pseudotime-aligned trajectory. Orange font and arrows indicate TF protein trends, and bold font with black arrows indicate key surface marker trends in trajectory.
(H) Column-normalized heatmap of mutual information scores calculated on cells in Leiden clusters 2, 7, and 8 and normalized across key TF and surface-marker pairs.
The CD123− MEP population (Manz et al., 2002) occupies a large portion of the map indicating heterogeneity of states within this population, which is also exemplified by numerous Leiden clusters in this space that show enrichment of distinct cell states (Figures 3B, bottom right, 3D, 3E, S3E, and S3F). Previous work has shown myeloid-primed states existing within CD123− MEPs (Manz et al., 2002), and we also observe these cells in clusters 3 and 4, marked by high CD117 and CD33 (Psaila et al., 2016) (Figure S3F). The large number of BM cells assayed (about 1 million) also enabled us to detect a small frequency of lymphoid-primed CD123− MEPs marked by mid to high CD45RA and high CD10 expression in cluster 5 (Figure S3F). In contrast, cells expressing the highest GATA-1 are primarily localized within the erythroid arm (Figure 3C, orange arrow). Together, these results confirm the lineage-priming heterogeneity present in the CD123− MEP population and localize cells with high expression of GATA-1 to erythroid-primed progenitor space.
Next, we aimed to better understand changes in levels of and relationships between functional markers in erythroid progenitor development with respect to GATA-1. We constructed a differentiation trajectory from hematopoietic stem cells (HSCs) to high GATA-1 cells (i.e., putative erythroid progenitors) using computed diffusion pseudotime (DPT) (Haghverdi et al., 2016) (Figures 3F, red arrow, and 3G). Notably, the erythroid arm is adjacent to early progenitors (both in single-cell proteomic and transcriptomic space), corroborating previous claims of erythroid progenitors arising directly from HSCs without intermediate states (Grinenko et al., 2018) (Figures 3F and S3G). When binned across normalized DPT, the ordering of cells enabled us to quantify key TF and surface marker trends through erythroid commitment (Figure 3G). We observe that CD71 and CD84, both known markers of erythroid commitment (Psaila et al., 2016; Sanada et al., 2016; Zaiss et al., 2003), increase during erythroid differentiation (Figures 3G and S3D). Our trajectory also showed expected trends in erythroid lineage-priming TFs, with GATA-2 being expressed earlier than GATA-1 in early progenitors (clusters 2 and 7, respectively) (Suzuki et al., 2013) (Figures 3G and S3F). Interestingly, stemness maintenance TFs, PBX-1 and BMI-1, that have previously been implicated in mouse RBC development (Kim et al., 2015; Manavathi et al., 2012) showed expression in erythroid progenitor clusters, indicating a potential role in human erythroid development (Figures 3G, 3C, and S3D). PBX-1 is likely acting to promote self-renewal in lineage-primed progenitor pools as indicated by its expression in early myeloid and early erythroid progenitor clusters (clusters 3 and 7) (Figure S3F). PBX-1 and GATA-1 show the highest mutual information only in cluster 8 despite lower PBX-1 abundance, supporting their known co-regulation through human PBX-1-interacting protein (PBXIP1/HPIP) (Manavathi et al., 2012) (Figure 3H). Additionally, we observe co-expression of BMI-1 with GATA-1, which is supported by previous work implicating BMI-1 in regulating self-renewal and ribosome biogenesis in erythroid progenitors at late-stage erythroid commitment (Gao et al., 2015; Kim et al., 2015; Liu et al., 2021) (Figure S3D). BMI-1 abundance increases with GATA-1 expression only toward later-stage erythroid progenitor states (cluster 8), hinting at unipotency at this stage (Figure 3G). Our single-cell, high-dimensional proteomic map complements our InTAC-seq assay in revealing trends of relevant surface markers and TFs in GATA-1 high cells within the context of erythroid progenitor development in human BM.
GATA-1-high BM progenitors exhibit significant clonal enrichment for erythroid-committed cells
To functionally assess erythroid progenitor commitment in high-GATA-1-expressing cells, we sought to benchmark them against CD123− MEPs (fully gated as CD34+CD38+CD10−CD123−CD45RA−) using a colony-forming assay for clonal hematopoietic lineage potential. In order to isolate viable cells with high GATA-1 abundance for the assay, we first identified surface-protein surrogates for GATA-1 using our BM cytometry by time-of-flight (CyTOF) data. We found that CD71 and CD84, both known to be enriched in erythroid cells (Psaila et al., 2016; Sanada et al., 2016; Zaiss et al., 2003), best represented high GATA-1 expression (Pearson correlation = 0.372 and 0.378, respectively; Figure 4A). The myeloid-enriched surface protein CD33 was also mildly anti-correlated with GATA-1 (Pearson correlation = −0.079). Additionally, when we selected the highest expressing GATA-1-high progenitors (Figure 4B) at a frequency similar to that used to sort for GATA-1-high progenitors in the In-TAC-seq experiment (~8%), we observed an enrichment for CD71 and CD84 and a depletion for CD33 in the high-GATA-1-expressing cells relative to mid/low-GATA-1 progenitors (Figures 4B and 4C). A data-driven, back-gating approach (Aghaeepour et al., 2018) was employed, and CD33, CD84, and CD71 were predicted to best gate for the high-GATA-1-expressing target population with an F score of 0.99 (Figures S4A and S4B, red cells). By selecting for CD71+CD84+CD33− cells within the CD34+CD38+ compartment, we confirmed that these cells have higher expression of GATA-1 relative to cells in the CD123− MEP gate (Figures S4C and S4D).
Figure 4. Surface-marker-defined BM population surrogate for high GATA-1 protein abundance clonally enriches for erythroid lineage.
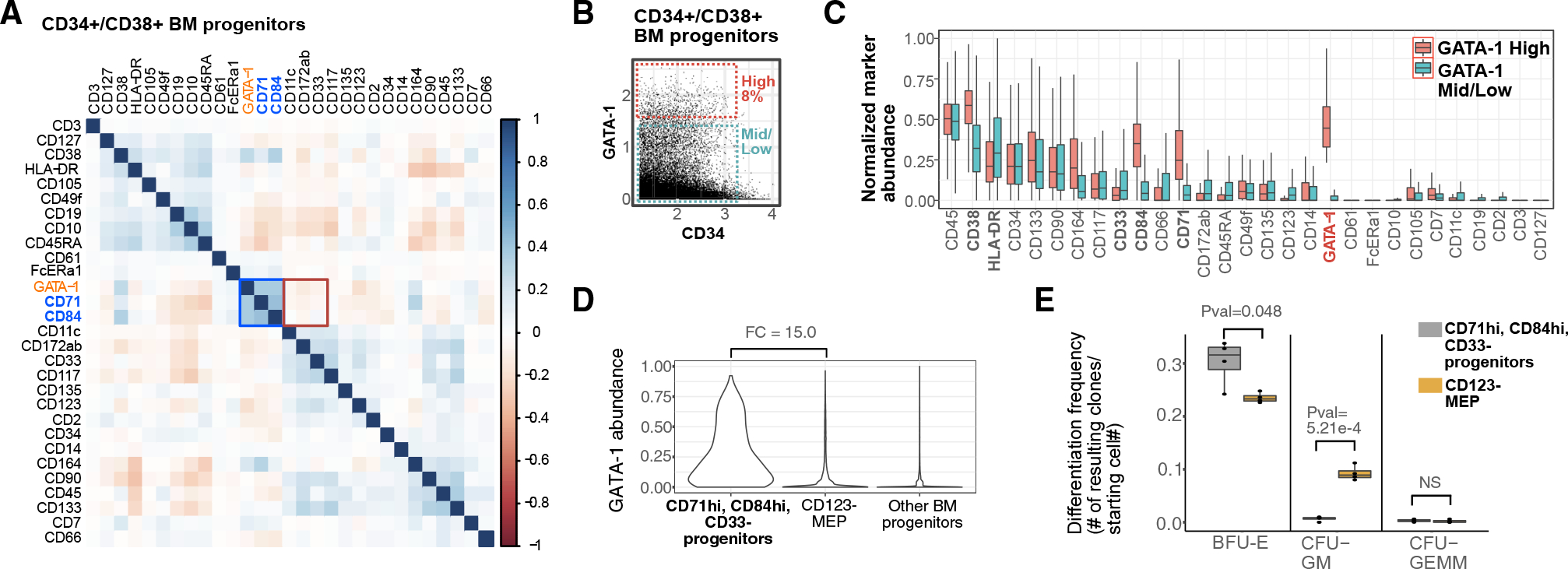
(A) Spearman correlation plot of surface markers and GATA-1 abundance in BM progenitors as measured by mass cytometry.
(B) Top 8% of GATA-1-expressing cells in CD34+/CD38+ BM progenitors gated as GATA-1-high BM cells, and remaining GATA-1-mid/low cells used for subsequent analysis.
(C) Boxplots of normalized surface-marker abundance of GATA-1-high BM progenitors (top 8% of expression) and GATA-1-mid/low BM progenitors (bottom 87% of expression) from mass cytometry.
(D) Violin plots of GATA-1 protein abundance in manually gated target populations (as defined by CD71+, CD84+, CD33−), CD123− MEP populations, and in other BM progenitor populations.
(E) Boxplot of clonal differentiation frequency of target population and CD123− MEP population to different lineages/population types across 4 biological replicates. (p values calculated using Student’s t test)
We then compared the hematopoietic colony-forming potential of the CD71+CD84+CD33− (i.e., GATA-1-high) population with CD123− MEPs using a methylcellulose assay. While it is known that CD123− MEPs are heterogeneous (Mori et al., 2015; Sanada et al., 2016) and contain erythroid progenitors as well as other lineage-primed progenitors (Figures 4E and S4D), in comparison, our GATA-1-high surrogate cell population (CD71+CD84+CD33−) was significantly (p = 0.048) enriched for erythroid (BFU-E) colonies (Figures 4E and S4D). These data confirm that the highest GATA-1 expressing cells within human CD34+CD38+ progenitors have erythroid potential by functional colony-forming assay and are consistent with the epigenetic programs we observed in our InTAC-seq data.
Consistent with their mixed-lineage potential demonstrated by colony-forming unit (CFU) analysis, the CD123− MEPs have a spread of expression for GATA-1 (Figure 5A). To examine the nature of high-GATA-1 expressing cells from within CD123− MEPs and how they compare to the high GATA-1 CD34+CD38+ progenitors, we isolated them and carried out In-TAC-seq (Figure 5A). Despite the low cell numbers obtained from these rare primary BM populations (down to ~250 cells), the resulting data were of high quality (Figures S5A and S5B). As expected, projection of high GATA-1-expressing cells isolated from CD34+CD38+ progenitors and CD123− MEP compartments onto the scATAC UMAP of BMMCs demonstrated similar embedding within the mid-erythroid population (Figure 5B). These data suggest that they are indeed similar cells, further confirming that high GATA-1 expression in CD34+CD38+ BM progenitors is sufficient to enrich for erythroid-committed progenitors without further gating.
Figure 5. High GATA-1 protein abundance delineates epigenetic program for erythroid commitment in RBC developmental trajectory.
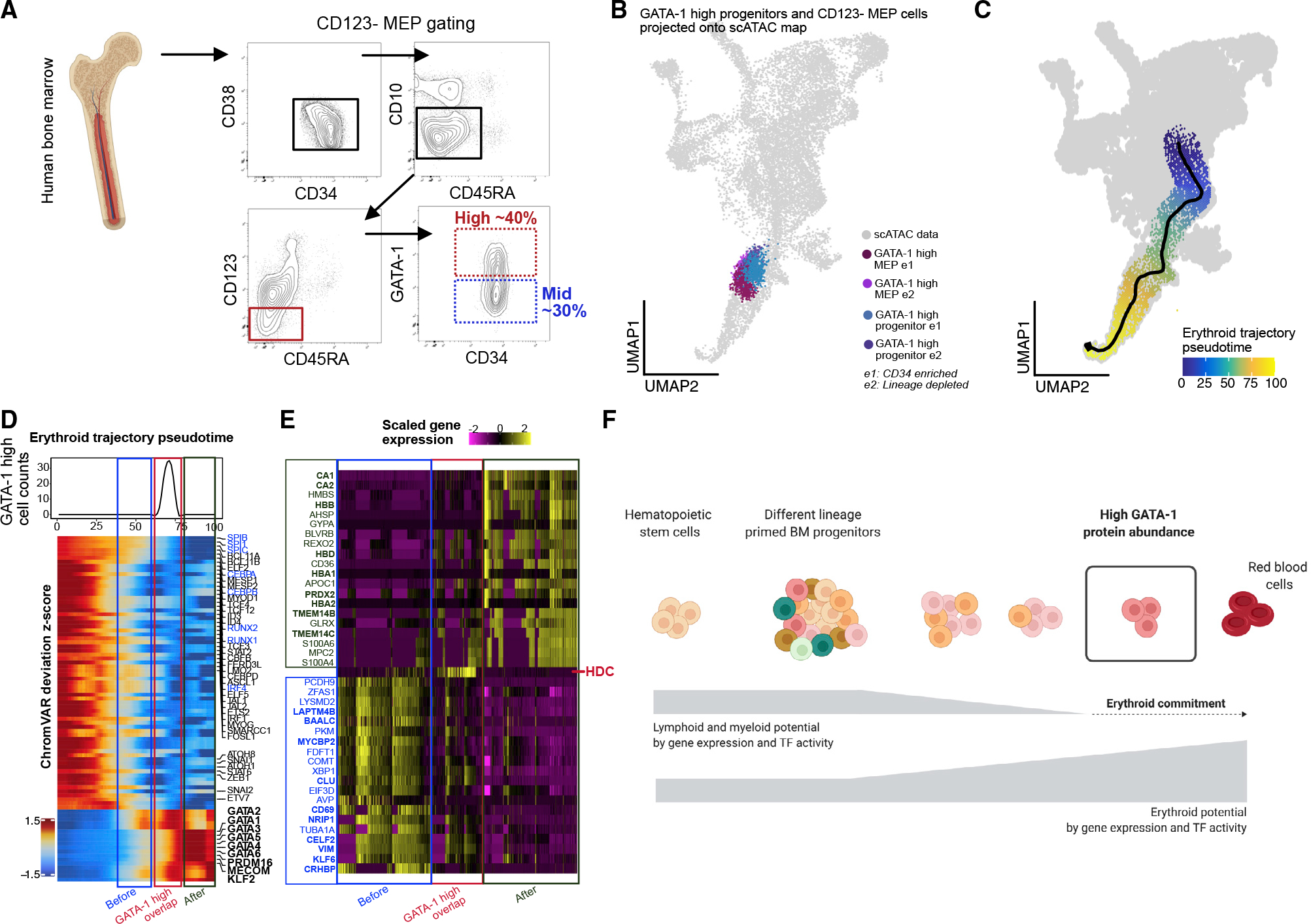
(A) BM aspirate is ficolled and enriched for CD34+ cells before gating for CD123− MEP population (CD34+/CD38+/CD10−/CD45RA−/CD123−) and selecting high-(25%–40%) and mid- (~lower 30%) GATA-1-expressing cells within each compartment.
(B) GATA-1-high cells from CD34+/CD38+ and CD123− MEP compartments InTAC-seq data were simulated as scATAC counts and projected on scATAC UMAP space.
(C) Putative erythropoiesis trajectory constructed from HSCs to late erythoid populations and overlaid on scATAC UMAP.
(D) Heatmap of top variable TFs by ChromVAR deviation scores across constructed erythroid trajectory with the projected position of GATA-1-high InTAC-seq samples indicated in red as the point of GATA-1-high overlap, in blue as the before point, and in green as the after point. Top: line plot of InTAC-seq-denoted GATA-1-high-simulated scATAC cells as binned across pseudotime.
(E) Top 20 genes significantly enriched (of fold change 2 and above) in integrated scRNA-seq data between the 3 bins, before, at, and after GATA-1-high overlap points in trajectory.
(F) Summary schematic of continuous differentiation to erythrocytes in BM with downregulation of lymphoid/myeloid TF activity and gene expression programs and upregulation of erythroid TF activity and gene expression programs. High GATA-1 protein abundance overlaps epigenetic program shift to erythroid lineage commitment in human BM.
High GATA-1 protein expression overlaps with epigenetic switch in BM progenitors to erythroid lineage
During erythropoiesis, BM progenitors undergo global changes in TF activity and chromatin accessibility in order to restrict non-erythroid lineage potential and drive the erythroid program. In order to model this process and understand the epigenetic transitions associated with GATA-1 acquisition, and erythroid commitment, we calculated a pseudotime ordering of cells from HSCs to the late erythroid cluster from BM scATAC-seq data (Granja et al., 2019) (Figure 5C). Using our InTAC-seq data from high GATA-1-expressing cells as a landmark, we identified the positions of their closest scATAC BM cells within the erythroid trajectory (Figure S5H). To assess the chromatin accessibility changes across erythroid differentiation, we then plotted chromVAR deviations for variable TF motifs along this derived erythroid trajectory. We observed a coordinated decrease in accessibility at motif sites for TFs that drive alternative lineages and an increase in accessibility at erythroid TF motif sites precisely where our high GATA-1-expressing progenitors are positioned within the trajectory (Figure 5D). Interestingly, there is a global increase in accessibility of the GATA TF family motifs during erythroid differentiation coinciding with high GATA-1 protein expression (Figure 5D). The preceding stage of erythroid differentiation still shows persistence of non-erythroid TF motif accessibility, suggesting that functional lineage restriction and erythroid fate determination is tied to GATA-1 protein abundance.
Using existing scRNA-seq data from the same BM samples, we next integrated measured TF protein abundance data with gene expression, gene accessibility (gene score), and motif accessibility along a differentiation trajectory. We observe that gene accessibility changes before and after our experimentally determined GATA-1-high trajectory point concur with expected trends for lymphoid/myeloid and erythroid genes, respectively (Figure S5J). Differential analysis of integrated scRNA-seq data at these 3 stages of erythroid differentiation reveal key hemoglobin subunit genes (e.g., HBD, HBA1, HBB) and heme metabolism (TMEM14B/C) after our determined GATA-1 high point (Figure 5E, green and red). We also observe differential upregulation of histidine decarboxylase (HDC), an enzyme in the histamine-synthesis pathways, in cells at the GATA-1 high point, with decreasing HDC expression as cells move into the final erythroid stage. HDC has been implicated in RBC development in mice (Otsuka et al., 2021), and its expression in adult BM coincides with our InTAC-seq-identified GATA-1 high point in the erythroid developmental trajectory. In contrast, preceding the GATA-1 high point, we see enrichment for genes involved in various non-erythroid lineages, including NRIP1 for hematopoietic stem cell quiescence (Forsberg et al., 2010; Huang et al., 2008) (Figure 5E). Our integrated plots further reveal the discord between the different regulatory layers for GATA1 and GATA2 genes and highlight the importance of directly measuring protein levels of key TFs and associated chromatin accessibility (Figure S5I).
Given that CD123− MEPs have mixed GATA-1 expression (Figure 5A) and correspond to mixed commitment to the erythroid lineage (Figure 4E), we also examined how the high and mid GATA-1-expressing CD123− MEPs differed. For example, we observed differences in accessibility at the gene locus for the erythropoiesis-enhancing TF MYB, a known target of GATA-1 binding and repression, between these two populations, consistent with their GATA-1 abundance levels (Figure S5C). Globally, we found that GATA-1 expression marked distinct cellular subsets with thousands of differentially accessible sites between GATA-1 high and mid GATA-1-expressing CD123− MEP populations (Figures S5D and S5E). While megakaryocyte and erythroid TF motifs were enriched in GATA-1 high cells, motifs for TFs involved in myeloid and B cell development were enriched in other subsets (Figures S5F and S5G). These results suggest that mid GATA-1-expressing cells within the CD123− MEP population retain some myeloid and/or lymphoid potential, likely resulting in their mixed lineage functional output in clonal assays. These observations are consistent with a previous study that described a subpopulation of cells within the CD123− MEP compartment exhibiting both erythroid and myeloid potential based on functional assays, although lymphoid potential was not tested (Psaila et al., 2016).
DISCUSSION
In summary, we have developed a robust protocol for profiling chromatin accessibility in fixed cells that enables staining and isolation of populations based on protein levels of intracellular regulators prior to ATAC-seq. This protocol, which we termed In-TAC-seq, enables us to integrate endogenous differences in key TFs with associated chromatin accessibility profiles to probe the link between their protein level abundance and accessibility across TF-binding motif sites. We use our approach to reveal GATA-1-associated epigenetic profiles and infer motif binding stoichiometry.
Importantly, our protocol captures high-quality data from primary human tissue and can be used with low (~100 cells) inputs, which allows robust integrated profiling of intracellular functional drivers and associated genome-wide chromatin accessibility in rare populations such as progenitors in human BM. We identified high GATA-1 protein expression as associated with epigenetic priming to erythropoiesis in human BM (Figure 5F) and believe current methods do not enable an equivalent analysis of such rare cells in unmanipulated primary human tissue. We further demonstrated that, as predicted based on their epigenetic profiles, these rare cells within the BM compartment that are highest in GATA-1 expression are clonally enriched for RBC differentiation. In addition, our single-cell proteomic map captured the dynamics of relevant TFs and surface markers in GATA-1-high progenitor cells within the trajectory of erythroid development. InTAC-seq enables biologists to connect levels of functionally pertinent intracellular proteins such as TFs and other chromatin-binding proteins to chromatin accessibility profiles in order to answer gene-regulatory questions and assess transient cellular molecular states previously difficult to access. By measuring endogenous intracellular regulators in complex biological contexts, InTAC-seq delineates epigenetic landscapes associated with master TFs or chromatin remodelers in primary human tissue. This allows us to interrogate how levels of these key proteins and their cooperative and antagonistic partners could influence global chromatin accessibility and local binding to drive cellular state and function. This technique provides information complementary to that provided by more targeted chromatin-binding assays such as CUT&Tag (Kaya-Okur et al., 2019), which can directly assay chromatin binding by a regulatory protein but cannot provide information on how changes in the protein’s abundance influences its target-site accessibility or the overall epigenetic landscape.
Using InTAC-seq, one could also devise more complex isolation schemes based on the expression of a multitude of intracellular regulators rather than those which could be accomplished with CUT&Tag (Kaya-Okur et al., 2019). Such an approach could identify cooperative or antagonistic functions with the key proteins-of-interest and associate their expression levels with specific cell states. Alternatively, we can design experiments that introduce exogenous factors into biological systems and use In-TAC-seq to isolate and profile populations with a range of expression of the exogenous proteins to understand the relationship between their abundance and chromatin architecture. In-TAC-seq is also complementary to single-cell genomic techniques such as scATAC-seq and scRNA-seq as it can be integrated with existing single-cell data to study populations-of-interest within the context of the larger biological system. Overall, InTAC-seq enables the profiling of chromatin accessibility of cell populations defined by abundance of intracellular proteins such as TFs with a cost-effective alternative and with ease and robustness that is on par with standard ATAC-seq.
Limitations of study
Our method combines fluorescence-activated cell sorting (FACS) with a modified ATAC-seq protocol that can robustly capture chromatin-accessibility profiles of low numbers of fixed cells. Thereby, we note assay limitations shared by chromatin accessibility profiling methods such as ATAC-seq (Buenrostro et al., 2013; Corces et al., 2017) and by antibody-based quantification methods such as flow cytometry (McKinnon, 2018). As we measure genome-wide chromatin accessibility, we infer activity of multiple TFs through changes in accessibility of DNA regions flanking putative TF motif sites. Our method cannot determine definitive TF occupancy on chromatin unlike chromatin immunoprecipitation sequencing (ChIP-seq) or CUT&Tag assays (Kaya-Okur et al., 2019; Park, 2009) that directly measure TF binding. As a result, this method cannot resolve or detect the previously characterized GATA switch phenomenon in erythroid development (Suzuki et al., 2013). Additionally, our method relies on the validated affinity and binding specificity of the FACS antibody to transcription or chromatin binding factor-of-interest under staining conditions. We also note that our method captures chromatin accessibility in bulk populations and, while being able to profile down to a few (~100) cells, is not a single-cell method. Other methods, namely piATAC (Chen et al., 2018), have linked single-cell chromatin accessibility to protein abundance; however, the complexity and high costs of such methods are not practical for most labs to apply at scale. Thus, InTAC-seq fills that niche by providing similar information at significantly lower cost and higher data quality.
Though we are able to measure genome-wide chromatin profiles associated with specific TF levels such as GATA-1, we cannot assert direct interaction with chromatin for driving accessibility, including TF motif changes. In complex developmental systems such as the human BM, there are multiple TFs and gene regulatory and chromatin binding proteins affecting chromatin structure and accessibility for expression and subsequent cell-fate determination (Wang et al., 2021). We cannot rule out that the above-mentioned epigenetic profiles measured, while associated with GATA-1 protein abundance, could be a result of differentiation and/or other TF activity. Further experimentation with overexpression of TF, for example, would have to be carried out to parse out direct causal links. Instead, our study uses endogenous levels of GATA-1 as a “lineage reporter” in human BM to understand the epigenetic and functional state of high GATA-1-expressing BM progenitors. Finally, though we show significant erythroid commitment through differentiation in clonal assay, we do not contend that this cell population represents true MEPs or that they are the definitive erythroid progenitor population in BM. Further work is needed to assess our GATA-1-high population in comparison to other MEP and erythroid progenitor populations as defined in the literature (Doulatov et al., 2010; Edvardsson et al., 2006; Mori et al., 2015; Notta et al., 2016; Psaila et al., 2016; Sanada et al., 2016).
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to the lead contact, Sean Bendall (bendall@stanford.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
InTAC and ATAC-seq data from study have been deposited at GEO and are publicly available as of the date of publication at GEO:-GSE167934. Accession numbers are additionally listed in the key resources table. Fluorescence-activated cell sorting (FACS) data have been deposited at FlowRepository and are publically available at FlowRepository:FR-FCM-Z539. Mass cytometry data have been deposited at FlowRepository and are publicly available as of the date of publication at FlowRepository:FR-FCM-Z5ZA.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| CD45–89Y | Fluidigm | Cat# 3089003, RRID:AB_2661851 |
| CD235–113ln | Biolegend | Cat# 349102, RRID:AB_10612565 |
| CD71–115 | Biolegend | Cat# 334102, RRID:AB_1134247 |
| CD61–139La | Biolegend | Cat# 336402, RRID:AB_1227584 |
| CD3–141Pr | BD Biosciences | Cat# 561416, RRID:AB_10612021 |
| CD19–142Nd | Biolegend | Cat# 302202, RRID:AB_314232 |
| CD90–143Nd | BD Biosciences | Cat# 555594, RRID:AB_395968 |
| CD14–144Nd | Biolegend | Cat# 301802, RRID:AB_314184 |
| CD164–145Nd | Biolegend | Cat# 324802, RRID:AB_756020 |
| CD34–148Nd | Biolegend | Cat# 343502, RRID:AB_1731898 |
| CD105–150Nd | Biolegend | Cat# 323202, RRID:AB_755954 |
| CD123–151Eu | Biolegend | Cat# 306002, RRID:AB_314576 |
| CD10–152Sm | Biolegend | Cat# 312202, RRID:AB_314913 |
| FcER1–153Eu | Biolegend | Cat# 334602, RRID:AB_1227649 |
| CD84–154Sm | Biolegend | Cat# 326002, RRID:AB_830813 |
| CD33–158Gd | Biolegend | Cat# 303402, RRID:AB_314346 |
| CD11c-159Tb | Biolegend | Cat# 301602, RRID:AB_314172 |
| GATA-1–160Gd | Cell Signalling | Cat# 3535, RRID:AB_2108288 |
| CD7–162Dy | BD Biosciences | Cat# 555359, RRID:AB_395762 |
| CD49f-164Dy | Biolegend | Cat# 555734, RRID:AB_2296273 |
| CD127–165Ho | Biolegend | Cat# 351302, RRID:AB_10718513 |
| CD66–167Er | BD Biosciences | Cat# 551354, RRID:AB_394166 |
| CD38–168Er | Biolegend | Cat# 303502, RRID:AB_314354 |
| CD45RA-169Tm | Biolegend | Cat# 304102, RRID:AB_314406 |
| CD135–170Er | Biolegend | Cat# 313302, RRID:AB_314987 |
| CD117–171Yb | Biolegend | Cat# 313202, RRID:AB_314981 |
| CD133–172Yb | Miltenyi Biotec | Cat# 130–108-062 |
| CD172ab-173Yb | Biolegend | Cat# 323802, RRID:AB_830701 |
| CD2–174Yb | Biolegend | Cat# 309202, RRID:AB_314752 |
| HLA-DR-209Bi | Biolegend | Cat# 307602, RRID:AB_314680 |
| CD15-biotinylated | Biolegend | Cat# 301914, RRID:AB_2561326 |
| CD3-biotinylated | Biolegend | Cat# 300404, RRID:AB_314058 |
| CD7-biotinylated | Thermo Fisher Scientific | Cat# 13–0079-82, RRID:AB_891490 |
| CD56-biotinylated | Biolegend | Cat# 362536, RRID:AB_2565653 |
| CD34-FITC | Myltenyi | Cat# 130–113-178, RRID:AB_2726005 |
| CD38-BV421 | Biolegend | Cat# 303526, RRID:AB_10983072 |
| CD45RA-AF700 | Biolegend | Cat# 304120, RRID:AB_493763 |
| CD10-BV650 | BD Biosciences | Cat# 563734, RRID:AB_2738393 |
| CD123-PECy7 | Biolegend | Cat# 306010, RRID:AB_493576 |
| GATA-1-PE | Cell Signalling | Cat# 13353, RRID:AB_2798187 |
| CD38-APC/Cy7 | Biolegend | Cat# 303534, RRID:AB_2561605 |
| CD71-PE | Biolegend | Cat# 334108, RRID:AB_10915138 |
| CD33-PE/Cy7 | Biolegend | Cat# 303434, RRID:AB_2734265 |
| CD84-APC | Biolegend | Cat# 326010, RRID:AB_2814188 |
| GATA1 | Abcam | Cat# ab181544 |
| Cleaved caspase 3-PE | BD Biosciences | Cat# 550821, RRID:AB_393906 |
|
| ||
| Biological samples | ||
|
| ||
| Adult bone marrow | All Cells Inc | https://allcells.com/research-grade-tissue-products/bone-marrow/ |
|
| ||
| Deposited data | ||
|
| ||
| ATAC-seq data | This paper | GEO:GSE167934 |
| FACS data | This paper | FlowRepository:FR-FCM-Z539 |
| Mass cytometry data | This paper | FlowRepository:FR-FCM-Z5ZA |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| K562 | ATCC | Cat# CCL-243, RRID:CVCL_0004 |
| GM12878 | Coriell Institute | Cat# GM12878, RRID:CVCL_7526 |
|
| ||
| Software and algorithms | ||
|
| ||
| Cutadapt | Martin, 2011 | https://cutadapt.readthedocs.io/en/stable/ |
| Bowtie2 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Picard tools | Broad Institute | https://broadinstitute.github.io/picard/ |
| ChrAccR | Fabian Mueller, https://doi.org/10.5281/zenodo.6091218 | https://github.com/GreenleafLab/ChrAccR, https://zenodo.org/record/6091218 |
| DESeq2 | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| SPADE | Qiu et al., 2011 | https://github.com/nolanlab/spade |
| Scanpy | Wolf et al., 2018 | https://scanpy.readthedocs.io/en/stable/ |
| GateFinder | Aghaeepour et al., 2018 | https://www.bioconductor.org/packages/release/bioc/html/GateFinder.html |
| ArchR | Granja et al., 2020 | https://www.archrproject.com/ |
| Harmony | Korsunsky et al., 2019 | https://portals.broadinstitute.org/harmony/index.html |
| MAGIC | Dijk et al., 2018 | https://github.com/KrishnaswamyLab/MAGIC |
| Seurat | Hao et al., 2021 | https://github.com/satijalab/seurat/ |
This paper does not report original code.
Any additional information required to reanalyse the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Cell lines and primary cultures
The human female chronic myeloid leukemia cell line, K562, were obtained from the American Type Culture Collection (Manassas, VA, USA). The human female lymphoblastic cell line GM12878 were obtained from Coriell Institute. Cells were cultured in RPMI 1640 medium containing 15% fetal bovine serum (FBS) and penicillin/streptomycin, and maintained at 37 C, 5% CO2.
All fresh human adult whole BM used in this study was collected in heparin sulfate anticoagulant and purchased from All Cells, Inc. BM mononuclear cells (BMMNC) were separated using Ficoll-Paque Plus (Amersham Biosciences). Next, BMMNC were either cryopreserved in FBS with 10% of DMSO or previously enriched for CD34+ (CD34 MicroBead Kit, Miltenyi Biotec) before cryopreservation.
METHOD DETAILS
K562 GATA-1 staining and sorting
Cells were washed once with PBS, then incubated in LIVE/DEAD™ Fixable Aqua Dead Cell Stain (Invitrogen L34965) diluted in PBS for 30 mins on ice. After a PBS wash, cells were fixed with 1.6% formaldehyde in PBS for 1 min, then quenched with an equal volume of 1X eBioscience permeabilization buffer (Thermofisher Scientific 00-8333-56). Cells were immediately centrifuged for 5 min at 600g and washed once with permeabilization buffer. Cells were then stained with anti-cleaved caspase 3-PE (BD #550821) and anti-GATA-1 (Abcam ab181544) for 30 mins at room temperature and FITC anti-rabbit secondary (Cell Signaling Technologies #4412S) for 30 mins. Washes were performed using permeabilization buffer and cells were resuspended in PBS for FACS and sorted using a BD FACSAria II. Cells positive for Aqua live/dead stain or cleaved caspase 3 were gated out and a narrow FSC gate was used to control for cell size. Cells in the lowest and highest 15% of GATA-1 expression were then sorted into PBS containing 30% FBS for InTAC-seq.
Bone marrow processing and GATA-1 sorting
On the day of the sorting, BM enriched CD34+ cells or BMMNC were thawed into cell culture medium supplemented with 25 U/mL benzonase (Sigma-Aldrich). For BMMNC, cells underwent magnetic lineage depletion according to the manufacturer’s instructions using BD Streptavidin Particles Plus (BD Biosciences #557812) and the BD IMag Cell Separation Magnet (BD Biosciences) with biotinylated anti-CD3, CD15, CD7, and CD56. Next, cells were incubated with LIVE/DEAD fixable Aqua dead cell stain (Invitrogen #L34957) for 30 minutes at room temperature (RTP) in dark, followed by a wash with PBS, before Fc receptor blocking (Human TruStain FcX, Biolegend #422302) for 10 minutes. Surface staining was carried out with CD34-FITC, CD38-BV421, CD45RA-Alexa-Fluor700, CD10-BV650, and CD123-PECy7 at 4°C in dark. Cells were then washed with cell staining media (PBS + 0.5% BSA). Fixation was carried out with 1.6% paraformaldehyde for 5 min at RTP before quenching with 1X eBioscience permeabilization buffer (Thermo Fisher Scientific 00-8333-56). Two washes were carried out with permeabilization buffer at 600G for 5 minutes each. Cells were incubated with GATA-1-PE in permeabilization buffer for 45 minutes at RTP, followed by a wash with CSM and sorting using a BD FACS Aria II (BD Biosciences). GATA-1 high and mid/low gates were applied on BM progenitors (gated as singlet, viable CD34+, CD38+ cells) and on CD123- MEP population (gated as singlet, viable, CD34+, CD38+, CD10−, CD123−, CD45RA− cells; antibody panels in Table S2).
InTAC-seq protocol
Fixed, permeabilized cells were counted using a hemocytometer and up to 50,000 cells were used for ATAC-seq where possible. Cells were spun down at 600g for 5 mins and resuspended in transposition mix containing 1X TD buffer, 0.1% NP40, 0.01% digitonin, and Tn5. Cells were incubated at 37 degrees with 1200 rpm shaking for 1 hour. 2X reverse crosslinking buffer (2% SDS, 0.2mg/mL proteinase K, and 100mM N,N-Dimethylethylenediamine, pH 6.5 [Sigma Aldrich D158003]) was added at equal volume to transposed cells and reversal of crosslinks was performed at 37 degrees overnight with 600 rpm shaking. DNA was purified using Qiagen minelute PCR purification columns and ATAC-seq libraries were generated as previously described (Buenrostro et al., 2013). For preparation of live ATAC-seq samples from fresh cells as a comparison, samples were prepared as above, except DNA was purified immediately following transposition rather than performing crosslink reversal.
ATAC-seq data processing
Adapters were trimmed using cutadapt and reads were mapped using bowtie2 with max fragment length of 2000bp to hg19 (primary bone marrow samples) or hg38 (all cell lines). We then filtered for non-mitochondrial reads, mapq > 20, and properly paired reads. We then removed duplicates using Picard tools. Peaks were called using macs2 with the following parameters on Tn5 insertion sites: –shift −75 –extsize 150 –nomodel –call-summits –nolambda -p 0.01 -B –SPMR
ATAC-seq QC of live and fixed GM12878 samples
For estimating library complexity, libraries were downsampled to 13 million read pairs prior to deduplication and library size was estimated using Picard tools EstimateLibraryComplexity. For TSS enrichment, deduplicated libraries were down-sampled to 10 million read pairs except for cell titration experiments where libraries were down-sampled to 4 million read pairs. TSS enrichment was calculated using the getTssEnrichment function in the ChrAccR R package for gencode.v27 protein coding gene transcriptional start sites.
Differential accessibility analysis
Aligned, deduplicated bam files output from data processing pipeline were loaded into R in HDF5 format using DsATAC.bam function in the ChrAccR R package. Consensus peakset across technical and biological replicates was calculated using getPeakSet.snakeATAC function in the ChrAccR R package where peaks have to be consistently absent or present across replicates to be retained. Count matrix was calculated as insertion counts across samples at consensus peakset regions using ChrAccR region-Aggregation function. DESeq2 (Love et al., 2014) was used to calculate differentially accessible peaks and independent hypothesis weighting (Ignatiadis et al., 2016) was used to correct for multiple testing. ggmaplot package was used to visualize MA plot. Differentially accessible peaks for GATA-1 high or mid/low cells was used to calculate motif enrichment (getMotifEnrichment function in the ChrAccR R package) using the CIS-BP TF motif database (from chromVARmotifs package). Adjusted p value (q value) was converted to -log(q value) and top enriched motifs were plotted by -log(q value) and odds ratio.
ChromVAR analysis
Raw insertions counts at relevant consensus peakset regions were RPKM normalized, log2 transformed, and quantile normalized. ChromVAR deviation scores were calculated on the log transformed count matrix using getChromVarDev function in the ChrAccR R package. Top variable TF motifs’ deviation scores were plotted using ComplexHeatmap R package.
GATA-1 footprinting analysis
To calculate GATA-1 footprinting as a measure of GATA-1 occupancy, we calculated Tn5 bias-corrected, normalized insertions centered at GATA-1 motif sites across the GATA-1 consensus peak set using the aggregateRegionCounts in the ChrAccR R package using the following parameters: countAggrFun = “mean”, norm = “tailMean”, normTailW = 0.1, kmerBiasAdj = TRUE, k = 6. To compare accessibility in K562 cells with high vs low GATA-1 across GATA-1 sites of different binding affinity, we identified the highest scoring GATA-1 sequence motif within each consensus peak, binned all sites into 20 equal bins based on the GATA-1 motif score, and calculated the GATA-1 footprint. We then measured accessibility flanking the GATA-1 motif as the area under the GATA-1 footprint plot from −50bp to −10bp and from +10bp to +50bp. For each motif score bin, the fractional change in accessibility was calculated as the average difference in accessibility between GATA-1-high and GATA-1-low samples normalized to the accessibility in the GATA-1-low samples.
Mass cytometry experiment
BM enriched CD34 + cells were thawed and stained for viability using cisplatin protocol (Fienberg et al., 2012). After quenching and washing in CSM (at 250G for 5min), cells were stained for surface markers before fixing in 1.6% paraformaldehyde and permeabilizing in 100% methanol for 10min at 4C. GATA-1 and cleaved caspase antibodies were stained intracellularly before washing in CSM at 600G for 5min. Cells were then re-fixed in 1.6% paraformaldehyde and DNA intercalator44 before analyzing on Helios mass cytometer (Fluidigm). Table S1 details antibodies used, their clones and the metal isotope channels they were conjugated to. Resulting FCS files from Helios run contains single cell protein level abundance for ~1mil BM cells.
Mass cytometry analysis
FCS files were gated on the cytobank platform (Kotecha et al., 2010) for cisplatin low (viability) and then gated for the various BM progenitors as shown in Figures S3A–S3E. All CD45+, mature lineage depleted BM cells were exported into R, density downsampled to 250k cell events, and key population of GATA-1 high BM progenitors and candidate population (CD71hi, CD84hi, CD33−) was up-sampled. Density downsampling was carried out using function in SPADE R package (Gautreau, 2017; Qiu et al., 2011). Data was arcsinh transformed with a cofactor of 5 and normalized from 0–1 at 0.01–0.99th percentile. Data was annotated with manually gated population definitions and imported into python for processing in scanpy (Wolf et al., 2018), with kNN run at k = 20, leiden clustering (Traag et al., 2019) at resolution = 1 and subsequent PAGA graph construction (Wolf et al., 2019) on Leiden cluster nodes. Force-directed layout (Jacomy et al., 2014) was initialised on PAGA graph. Marker distribution across Leiden clusters was visualised in violin plots using scanpy.pl.stacked_violin function. Diffusion pseudotime (DPT) (Haghverdi et al., 2016) was calculated using 10 diffusion components and 0 branching using scanpy.tl.dpt function. Erythroid trajectory was defined as Leiden cluster 2 to 7 to 8 corresponding to a path from HSC to GATA+ high cells. DPT across trajectory was normalized 0–1 and cells aligned accordingly. Cells in trajectory were binned into 100 groups and median marker abundance across bins were normalized and plotted as a heatmap using ComplexHeatmap R package (Gu et al., 2016). Mutual information between key TFs and surface markers was calculated using knnmi.all function in parmigene R package at k=20 and normalized across clusters before plotting as a heatmap using pheatmap R package (visualised with column scaling).
CD34+/CD38+ gated BM progenitor mass cytometry data was then correlated across GATA-1 and all assayed surface markers using Spearman correlation and plotted using corrplot R package with hierarchal clustering. Boxplots of GATA-1 high and mid/ low BM progenitors were constructed using top 8% of GATA-1-expressing cells and remaining cells respectively in order to match frequency of GATA-1 positivity captured in sorting for InTAC-seq experiment. Manually gated CD123- MEP population, and deduced candidate populations were plotted for GATA-1 abundance using ggplot2 boxplot function. The data-driven, backgating algorithm GateFinder (Aghaeepour et al., 2018) was applied on all BM progenitors with candidate population as target with 2 gating step parameter and predicted gates were plotted as scatterplots using ggplot2.
Colony-forming unit (CFU) assay
BM enriched CD34+ cells were stained with CD34-FITC, CD38-APC/Cy7, CD71-PE, CD33-PE/Cy7, and CD84-APC and sorted for our putative GATA-1 high erythroid progenitor subpopulation. We also stained and sorted cells for the CD123- MEP population (CD34-FITC, CD38-BV421, CD45RA-AF700, CD10-BV650, CD123-PECy7) (Akashi et al., 2000). Antibody panels in Table S2. For viability, 7-amino actinomycin D (7-AAD) was used. Our putative population corresponding to GATA-1 high BM progenitors were gated as singlet, viable CD34+, CD38+, CD84hi, CD71hi, CD33− cells. Cells were sorting using a BD FACS Aria II (BD Biosciences) and collected in IMDM 2% FBS for further CFU assays.
CFU assays were performed using the MethoCult™ H4435 Enriched (STEMCELL Technologies). Briefly, progenitor BM sorted cells were seeded (250 or 500 cells/well) into 6 well SmartDish™ (STEMCELL Technologies). After incubation for 14 days, at 37°C in 5% CO2, hematopoietic colony-forming unit were automated counted and analyzed by STEMvision™ Human (STEMCELL Technologies). Differentiation frequency was calculated for each sorted population by number of resulting colonies/numbers of starting cells seeded.
scATAC processing and clustering
Raw data files were downloaded from published work which had scATAC and scRNA seq carried out in parallel on PBMC, BM and CD34-enriched BM (Granja et al., 2019). Processing was done using the ArchR package (Granja et al., 2020), where Harmony (Korsunsky et al., 2019) was used to batch correct and MAGIC (Dijk et al., 2018) was used to impute gene accessibility scores. Further processing including iterativeLSI and subsequent UMAP embedding was carried out using ArchR’s built-in functions of addIterativeLSI and addUMAP. Pre-determined population annotations (from scRNA seq) were integrated into the scATAC data using constrained integration of the scRNA seq data (addGeneIntegrationMatrix from ArchR which uses Seurat’s transferAnchor function) (Hao et al., 2021). Populations were filtered to exclude more differentiated PBMC and BM populations such as B cells, T cells and monocytes and focus the analysis on BM progenitors relevant to erythropoiesis. Seurat’s FindClusters approach was used on dimensionality reduced iterativeLSI embedding to cluster the scATAC data and clusters were labelled using predicted populations from annotated scRNA seq integration. MACS2 was run on the different scATAC clusters and reproducible peakset was curated using the addReproduciblePeakSet function with (n+1)/2 reproducibility with a maximum of 500 peaks per cell.
Populations were compared across accessibility in consensus peaks using binomial test after binarizing data and correcting for TSS enrichment and log10(nFrags) bias in getMarkerFeatures function of ArchR. Features differentially enriched across populations were plotted in a heatmap with a FDR<0.1 and Log2FC>0.5 cutoff. Key motifs such as GATA-1, CEBPA, GATA2, KLF1, KLF2, SPI1, RUNX1, IRF4 and IRF8 were plotted for chromVAR deviation scores as stacked histogram across populations using plotGroups function in ArchR.
Bulk sample projection onto scATAC space
Bulk ATAC count matrix calculated from relevant consensus peakset regions was converted into a summarizedExperiment data class. This was projected into the scATAC UMAP space after calculating iterativeLSI on bulk samples simulated as single cells using the projectBulkATAC function from ArchR. Resulting simulated single cell ATAC UMAP projection from bulk data (250 cells simulated per bulk sample) was plotted along with the scATAC data in the original UMAP embedding using ggplot. Closest 500 scATAC cells to simulated GATA-1 high samples’ bulk projection was quantified using mahanoblis distance to combined bulk sample centroid in projected UMAP space.
Erythroid trajectory analysis
Erythroid trajectory was quantified by fitting splines through the early progenitor, early erythroid, mid erythroid and late erythroid clusters using the addTrajectory function in ArchR and normalizing pseudotime between 0–100. Trajectory was plotted using the plotTrajectory function. The GATA-1 high sample demarcations on trajectory was found quantifying closest scATAC cells’ pseudotime values on trajectory.
Gene accessibility, expression and chromVAR motif deviation scores for cells across the trajectory were extracted using the get-Trajectory function from ArchR and heatmaps of top variable features were plotted. Normalized line plots were constructed by extracting GATA-1 high scATAC counts across 100 pseudotime bins and plotting using ggplot. GATA-1 high scATAC cells (by protein expression) was defined as GATA-1 high point on trajectory (65–75 on pseudotime scale) and subsequently before and after restriction point was binned (40–60 and 80–100 respectively) as per plotted gene expression inflection points.
Differential scRNA analysis
Integrated scRNA seq data was filtered for the abovementioned 3 InTAC-seq inferred bins (before, GATA-1 high point and after) and imported into Seurat R package. Data was log normalized with a scaling factor of 10000 and scaled after. FindAllMarkers function was used to detect only enriched markers in the 3 bins with genes detected in at least 10% of the total number of cells at a foldchange threshold of 2 using ROC analysis. Top 20 markers for each bin was plotted as a scaled heatmap using DoHeatmap function.
Graphic design
All figures were constructed in Affinity Designer and schematics created with BioRender.com.
QUANTIFICATION AND STATISTICAL ANALYSIS
DESeq2 was used for identifying differentially accessible peaks as described in the Method Details section, with FDR cut-off of 0.05 (for all comparisons) and log2 fold change of 2 (for GATA-1 high vs mid/low progenitor comparison). For comparing differences in colony forming ability from different bone marrow populations, a student’s t-test was performed. The number of biological and technical replicates for each experiment are indicated in the figures and/or figure legends.
Supplementary Material
Highlights.
We developed InTAC-seq, a method to profile chromatin accessibility from fixed cells
InTAC-seq profiles rare cell populations defined by intracellular protein abundance
InTAC-seq matches live-cell ATAC in data quality, robustness, and cost of assay
GATA-1-high BM progenitors are epigenetically and functionally erythroid primed
MOTIVATION.
Cellular differentiation is a tightly regulated process where key lineage-determining transcription factors (TFs) play an important role in gene regulation. The abundance of these TFs influences epigenetic priming of cells toward different cell fates. In order to better understand this complex process, we developed InTAC-seq to capture epigenetic states of cells marked by particular TFs and to better associate TFs with differentiation and lineage restriction. We benchmark InTAC-seq in a homogeneous cell line, K562, in order to link GATA-1 protein abundance to chromatin changes in regular culture conditions and then extend it to the analysis of rare, primary human hematopoietic progenitor cells. Using InTAC-seq, we were able to profile chromatin accessibility landscapes associated with GATA-1 in bone marrow rare progenitor cells and characterize high GATA-1-expressing cells as erythroid-committed progenitors.
ACKNOWLEDGMENTS
We thank members of the S.C.B. and W.J.G. labs for their support and advice. In particular, we would like to thank Sizun Jiang for assistance at inception of the method and Fabian Mueller for computational advice. This study was supported by grants from the National Institutes of Health (1DP2OD022550-01, 1R01AG056287-01, 1R01AG057915-01, R01AG068279, and 1U24CA224309-01 to S.C.B.; RM1-HG007735, UM1-HG009442, UM1-HG009436, 1UM1-HG009442, U2CCA233311, U54-GH010426, and U19-AI057266 to W.J.G.); and GM110050 and GM127295 to E.T.K.). This work is also supported by the Defense Advanced Research Project Agency (W911NF1920185 to W.J.G.) and a Stanford Cancer Institute-Goldman Sachs Foundation Cancer Research Award (to W.J.G.). W.J.G. is a Chan Zuckerberg investigator. A.F.C. is supported by an NIH F32 postdoctoral fellowship (5F32GM135996-02). R.B. is supported by A*STAR National Science Scholarship (PHD) from A*STAR Graduate Academy, Singapore.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
INCLUSION AND DIVERSITY
One or more of the authors self-identifies as a member of the LGBTQ+ community and/or female ethnic minority in science.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.crmeth.2022.100188.
REFERENCES
- Aghaeepour N, Simonds EF, Knapp D, Bruggner RV, Sachs K, Culos A, Gherardini PF, Samusik N, Fragiadakis GK, Bendall SC, et al. (2018). GateFinder: projection-based gating strategy optimization for flow and mass cytometry. Bioinformatics 34, 4131–4133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akashi K, Traver D, Miyamoto T, and Weissman IL (2000). A clonogenic common myeloid progenitor that gives rise to all myeloid lineages. Nature 404, 193–197. [DOI] [PubMed] [Google Scholar]
- Arinobu Y, Mizuno S, Chong Y, Shigematsu H, Iino T, Iwasaki H, Graf T, Mayfield R, Chan S, Kastner P, et al. (2007). Reciprocal activation of GATA-1 and PU.1 marks initial specification of hematopoietic stem cells into myeloerythroid and myelolymphoid lineages. Cell Stem Cell 1, 416–427. [DOI] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, and Greenleaf WJ (2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao J, Cusanovich DA, Ramani V, Aghamirzaie D, Pliner HA, Hill AJ, Daza RM, McFaline-Figueroa JL, Packer JS, Christiansen L, et al. (2018). Joint profiling of chromatin accessibility and gene expression in thousands of single cells. Science 361, 1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Litzenburger UM, Wei Y, Schep AN, LaGory EL, Choudhry H, Giaccia AJ, Greenleaf WJ, and Chang HY (2018). Joint single-cell DNA accessibility and protein epitope profiling reveals environmental regulation of epigenomic heterogeneity. Nat. Commun 9, 4590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corces MR, Trevino AE, Hamilton EG, Greenside PG, Sinnott-Armstrong NA, Vesuna S, Satpathy AT, Rubin AJ, Montine KS, Wu B, et al. (2017). An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods 14, 959–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courey AJ, and Huang J-D (1995). The establishment and interpretation of transcription factor gradients in the Drosophila embryo. Biochim. Biophys. Acta 1261, 1–18. [DOI] [PubMed] [Google Scholar]
- DeKoter RP, and Singh H (2000). Regulation of B lymphocyte and macrophage development by graded expression of PU.1. Science 288, 1439–1441. [DOI] [PubMed] [Google Scholar]
- Dijk D. van, Sharma R, Nainys J, Yim K, Kathail P, Carr AJ, Burdziak C, Moon KR, Chaffer CL, Pattabiraman D, et al. (2018). Recovering gene interactions from single-cell data using data diffusion. Cell 174, 716–729.e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doulatov S, Notta F, Eppert K, Nguyen LT, Ohashi PS, and Dick JE (2010). Revised map of the human progenitor hierarchy shows the origin of macrophages and dendritic cells in early lymphoid development. Nat. Immunol 11, 585–593. [DOI] [PubMed] [Google Scholar]
- Edvardsson L, Dykes J, and Olofsson T (2006). Isolation and characterization of human myeloid progenitor populations—TpoR as discriminator between common myeloid and megakaryocyte/erythroid progenitors. Exp. Hematol 34, 599–609. [DOI] [PubMed] [Google Scholar]
- Fienberg HG, Simonds EF, Fantl WJ, Nolan GP, and Bodenmiller B (2012). A platinum-based covalent viability reagent for single-cell mass cytometry. Cytometry A 81, 467–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsberg EC, Passegué E, Prohaska SS, Wagers AJ, Koeva M, Stuart JM, and Weissman IL (2010). Molecular signatures of quiescent, mobilized and leukemia-initiating hematopoietic stem cells. Plos One 5, e8785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao R, Chen S, Kobayashi M, Yu H, Zhang Y, Wan Y, Young SK, Soltis A, Yu M, Vemula S, et al. (2015). Bmi1 promotes erythroid development through regulating ribosome biogenesis. Stem Cells 33, 925–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautreau G (2017). SPADEVizR: an R package for visualization, analysis and integration of SPADE results. Bioinformatics 33, 779–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granja JM, Klemm S, McGinnis LM, Kathiria AS, Mezger A, Corces MR, Parks B, Gars E, Liedtke M, Zheng GXY, et al. (2019). Single-cell multiomic analysis identifies regulatory programs in mixed-phenotype acute leukemia. Nat. Biotechnol 37, 1458–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granja JM, Corces MR, Pierce SE, Bagdatli ST, Choudhry H, Chang HY, and Greenleaf WJ (2020). ArchR: an integrative and scalable software package for single-cell chromatin accessibility analysis. Preprint at bioRxiv, 2020.04.28.066498. [Google Scholar]
- Grinenko T, Eugster A, Thielecke L, Ramasz B, Krüger A, Dietz S, Glauche I, Gerbaulet A, Bonin M, von, Basak O, et al. (2018). Hematopoietic stem cells can differentiate into restricted myeloid progenitors before cell division in mice. Nat. Commun 9, 1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z, Eils R, and Schlesner M (2016). Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849. [DOI] [PubMed] [Google Scholar]
- Gutiérrez L, Caballero N, Fernández-Calleja L, Karkoulia E, and Strouboulis J (2020). Regulation of GATA1 levels in erythropoiesis. Iubmb Life 72, 89–105. [DOI] [PubMed] [Google Scholar]
- Haghverdi L, Büttner M, Wolf FA, Buettner F, and Theis FJ (2016). Diffusion pseudotime robustly reconstructs lineage branching. Nat. Methods 13, 845–848. [DOI] [PubMed] [Google Scholar]
- Hao Y, Hao S, Andersen-Nissen E, Mauck WM, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zager M, et al. (2021). Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hromas R, Orazi A, Neiman RS, Maki R, Beveran CV, Moore J, and Klemsz M (1993). Hematopoietic lineage- and stage-restricted expression of the ETS oncogene family member PU.1. Blood 82, 2998–3004. [PubMed] [Google Scholar]
- Huang T-S, Hsieh J-Y, Wu Y-H, Jen C-H, Tsuang Y-H, Chiou S-H, Partanen J, Anderson H, Jaatinen T, Yu Y-H, et al. (2008). Functional network reconstruction reveals somatic stemness genetic maps and dedifferentiation-like transcriptome reprogramming induced by GATA2. Stem Cells 26, 1186–1201. [DOI] [PubMed] [Google Scholar]
- Ignatiadis N, Klaus B, Zaugg JB, and Huber W (2016). Data-driven hypothesis weighting increases detection power in genome-scale multiple testing. Nat. Methods 13, 577–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacomy M, Venturini T, Heymann S, and Bastian M (2014). ForceAtlas2, a continuous graph layout algorithm for handy network visualization designed for the gephi software. Plos One 9, e98679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karmakar S, Harcourt EM, Hewings DS, Scherer F, Lovejoy AF, Kurtz DM, Ehrenschwender T, Barandun LJ, Roost C, Alizadeh AA, et al. (2015). Organocatalytic removal of formaldehyde adducts from RNA and DNA bases. Nat. Chem 7, 752–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaya-Okur HS, Wu SJ, Codomo CA, Pledger ES, Bryson TD, Henikoff JG, Ahmad K, and Henikoff S (2019). CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat. Commun 10, 1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim AR, Olsen JL, England SJ, Huang Y-S, Fegan KH, Delgadillo LF, McGrath KE, Kingsley PD, Waugh RE, and Palis J (2015). Bmi-1 regulates extensive erythroid self-renewal. Stem Cell Rep 4, 995–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemsz MJ, McKercher SR, Celada A, Beveren CV, and Maki RA (1990). The macrophage and B cell-specific transcription factor PU.1 is related to the ets oncogene. Cell 61, 113–124. [DOI] [PubMed] [Google Scholar]
- Korsunsky I, Millard N, Fan J, Slowikowski K, Zhang F, Wei K, Baglaenko Y, Brenner M, Loh PR, and Raychaudhuri S (2019). Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotecha N, Krutzik PO, and Irish JM (2010). Web-based analysis and publication of flow cytometry experiments. Curr. Protoc. Cytom 10.1002/0471142956.cy1017s53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Wu M, Lancelot M, Deng J, Gao Y, Roback JD, Chen T, and Cheng L (2021). BMI1 enables extensive expansion of functional erythroblasts from human peripheral blood mononuclear cells. Mol. Ther 29, 1918–1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma S, Zhang B, LaFave LM, Earl AS, Chiang Z, Hu Y, Ding J, Brack A, Kartha VK, Tay T, et al. (2020). Chromatin potential identified by shared single-cell profiling of RNA and chromatin. Cell 183, 1103–1116.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manavathi B, Lo D, Bugide S, Dey O, Imren S, Weiss MJ, and Humphries RK (2012). Functional regulation of pre-B-cell leukemia homeobox interacting protein 1 (PBXIP1/HPIP) in erythroid differentiation. J. Biol. Chem 287, 5600–5614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manz MG, Miyamoto T, Akashi K, and Weissman IL (2002). Prospective isolation of human clonogenic common myeloid progenitors. Proc. Natl. Acad. Sci 99, 11872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17, 10–12. 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- McKinnon KM (2018). Flow cytometry: an overview. Curr. Protoc. Immunol 120, 5.1.1–5.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori Y, Chen JY, Pluvinage JV, Seita J, and Weissman IL (2015). Prospective isolation of human erythroid lineage-committed progenitors. Proc. Natl. Acad. Sci 112, 9638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nechanitzky R, Akbas D, Scherer S, Györy I, Hoyler T, Ramamoorthy S, Diefenbach A, and Grosschedl R (2013). Transcription factor EBF1 is essential for the maintenance of B cell identity and prevention of alternative fates in committed cells. Nat. Immunol 14, 867–875. [DOI] [PubMed] [Google Scholar]
- Nerlov C, Querfurth E, Kulessa H, and Graf T (2000). GATA-1 interacts with the myeloid PU.1 transcription factor and represses PU.1-dependent transcription. Blood 95, 2543–2551. [PubMed] [Google Scholar]
- Notta F, Zandi S, Takayama N, Dobson S, Gan OI, Wilson G, Kaufmann KB, McLeod J, Laurenti E, Dunant CF, et al. (2016). Distinct routes of lineage development reshape the human blood hierarchy across ontogeny. Science 351, aab2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otsuka H, Endo Y, Ohtsu H, Inoue S, Noguchi S, Nakamura M, and Soeta S (2021). Histidine decarboxylase deficiency inhibits NBP-induced extramedullary hematopoiesis by modifying bone marrow and spleen microenvironments. Int. J. Hematol 113, 348–361. [DOI] [PubMed] [Google Scholar]
- Park PJ (2009). ChIP–seq: advantages and challenges of a maturing technology. Nat. Rev. Genet 10, 669–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul F, Arkin Y, Giladi A, Jaitin DA, Kenigsberg E, Keren-Shaul H, Winter D, Lara-Astiaso D, Gury M, Weiner A, et al. (2015). Transcriptional heterogeneity and lineage commitment in myeloid progenitors. Cell 163, 1663–1677. [DOI] [PubMed] [Google Scholar]
- Pedone E, Postiglione L, Aulicino F, Rocca DL, Montes-Olivas S, Khazim M, Bernardo D. di, Cosma MP, and Marucci L (2019). A tunable dual-input system for on-demand dynamic gene expression regulation. Nat. Commun 10, 4481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Psaila B, Barkas N, Iskander D, Roy A, Anderson S, Ashley N, Caputo VS, Lichtenberg J, Loaiza S, Bodine DM, et al. (2016). Single-cell profiling of human megakaryocyte-erythroid progenitors identifies distinct megakaryocyte and erythroid differentiation pathways. Genome Biol. 17, 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu P, Simonds EF, Bendall SC, Gibbs KD, Bruggner RV, Linderman MD, Sachs K, Nolan GP, and Plevritis SK (2011). Extracting a cellular hierarchy from high-dimensional cytometry data with SPADE. Nat. Biotechnol 29, 886–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rekhtman N, Radparvar F, Evans T, and Skoultchi AI (1999). Direct interaction of hematopoietic transcription factors PU.1 and GATA-1: functional antagonism in erythroid cells. Gene Dev 13, 1398–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera-Pomar R, and Jãckle H (1996). From gradients to stripes in Drosophila embryogenesis: filling in the gaps. Trends Genet 12, 478–483. [DOI] [PubMed] [Google Scholar]
- Sanada C, Xavier-Ferrucio J, Lu Y-C, Min E, Zhang P-X, Zou S, Kang E, Zhang M, Zerafati G, Gallagher PG, et al. (2016). Adult human megakaryocyte-erythroid progenitors are in the CD34+CD38mid fraction. Blood 128, 923–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satpathy AT, Granja JM, Yost KE, Qi Y, Meschi F, McDermott GP, Olsen BN, Mumbach MR, Pierce SE, Corces MR, et al. (2019). Massively parallel single-cell chromatin landscapes of human immune cell development and intratumoral T cell exhaustion. Nat. Biotechnol 37, 925–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schep AN, Wu B, Buenrostro JD, and Greenleaf WJ (2017). chromVAR: inferring transcription-factor-associated accessibility from single-cell epigenomic data. Nat. Methods 14, 975–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwanhäusser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, and Selbach M (2011). Global quantification of mammalian gene expression control. Nature 473, 337–342. [DOI] [PubMed] [Google Scholar]
- Seita J, and Weissman IL (2010). Hematopoietic stem cell: self-renewal versus differentiation. Wiley Interdiscip. Rev. Syst. Biol. Med 2, 640–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu S, Nagasawa T, Katoh O, Komatsu N, Yokota J, and Morishita K (2002). EVI1 is expressed in megakaryocyte cell lineage and enforced expression of EVI1 in UT-7/GM cells induces megakaryocyte differentiation. Biochem. Bioph Res. Commun 292, 609–616. [DOI] [PubMed] [Google Scholar]
- Simmons S, Knoll M, Drewell C, Wolf I, Mollenkopf HJ, Bouquet C, and Melchers F (2012). Biphenotypic B-lymphoid/myeloid cells expressing low levels of Pax5: potential targets of BAL development. Blood 120, 3688–3698. [DOI] [PubMed] [Google Scholar]
- Spitz F, and Furlong EEM (2012). Transcription factors: from enhancer binding to developmental control. Nat. Rev. Genet 13, 613–626. [DOI] [PubMed] [Google Scholar]
- Stachura DL, Chou ST, and Weiss MJ (2006). Early block to erythromegakaryocytic development conferred by loss of transcription factor GATA-1. Blood 107, 87–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki M, Kobayashi-Osaki M, Tsutsumi S, Pan X, Ohmori S, Takai J, Moriguchi T, Ohneda O, Ohneda K, Shimizu R, et al. (2013). GATA factor switching from GATA2 to GATA1 contributes to erythroid differentiation. Genes Cells 18, 921–933. [DOI] [PubMed] [Google Scholar]
- Suzuki N, Suwabe N, Ohneda O, Obara N, Imagawa S, Pan X, Motohashi H, and Yamamoto M (2003). Identification and characterization of 2 types of erythroid progenitors that express GATA-1 at distinct levels. Blood 102, 3575–3583. [DOI] [PubMed] [Google Scholar]
- Traag VA, Waltman L, and Eck N.J. van (2019). From Louvain to Leiden: guaranteeing well-connected communities. Sci. Rep 9, 5233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel C, and Marcotte EM (2012). Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet 13, 227–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel C, Abreu R. de S., Ko D, Le S-Y, Shapiro BA, Burns SC, Sandhu D, Boutz DR, Marcotte EM, and Penalva LO (2010). Sequence signatures and mRNA concentration can explain two-thirds of protein abundance variation in a human cell line. Mol. Syst. Biol 6, 400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Wang P, Li Y, Peng H, Zhu Y, Mohandas N, and Liu J (2021). Interplay between cofactors and transcription factors in hematopoiesis and hematological malignancies. Signal Transduct. Target Ther 6, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf FA, Angerer P, and Theis FJ (2018). SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 19, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf FA, Hamey FK, Plass M, Solana J, Dahlin JS, Goöttgens B, Rajewsky N, Simon L, and Theis FJ (2019). PAGA: graph abstraction reconciles clustering with trajectory inference through a topology preserving map of single cells. Genome Biol. 20, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaiss M, Hirtreiter C, Rehli M, Rehm A, Kunz-Schughart LA, Andreesen R, and Hennemann B (2003). CD84 expression on human hematopoietic progenitor cells. Exp. Hematol 31, 798–805. [DOI] [PubMed] [Google Scholar]
- Zhang P, Zhang X, Iwama A, Yu C, Smith KA, Mueller BU, Narravula S, Torbett BE, Orkin SH, and Tenen DG (2000). PU.1 inhibits GATA-1 function and erythroid differentiation by blocking GATA-1 DNA binding. Blood 96, 2641–2648. [PubMed] [Google Scholar]
- Zivot A, Lipton JM, Narla A, and Blanc L (2018). Erythropoiesis: insights into pathophysiology and treatments in 2017. Mol. Med 24, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
InTAC and ATAC-seq data from study have been deposited at GEO and are publicly available as of the date of publication at GEO:-GSE167934. Accession numbers are additionally listed in the key resources table. Fluorescence-activated cell sorting (FACS) data have been deposited at FlowRepository and are publically available at FlowRepository:FR-FCM-Z539. Mass cytometry data have been deposited at FlowRepository and are publicly available as of the date of publication at FlowRepository:FR-FCM-Z5ZA.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| CD45–89Y | Fluidigm | Cat# 3089003, RRID:AB_2661851 |
| CD235–113ln | Biolegend | Cat# 349102, RRID:AB_10612565 |
| CD71–115 | Biolegend | Cat# 334102, RRID:AB_1134247 |
| CD61–139La | Biolegend | Cat# 336402, RRID:AB_1227584 |
| CD3–141Pr | BD Biosciences | Cat# 561416, RRID:AB_10612021 |
| CD19–142Nd | Biolegend | Cat# 302202, RRID:AB_314232 |
| CD90–143Nd | BD Biosciences | Cat# 555594, RRID:AB_395968 |
| CD14–144Nd | Biolegend | Cat# 301802, RRID:AB_314184 |
| CD164–145Nd | Biolegend | Cat# 324802, RRID:AB_756020 |
| CD34–148Nd | Biolegend | Cat# 343502, RRID:AB_1731898 |
| CD105–150Nd | Biolegend | Cat# 323202, RRID:AB_755954 |
| CD123–151Eu | Biolegend | Cat# 306002, RRID:AB_314576 |
| CD10–152Sm | Biolegend | Cat# 312202, RRID:AB_314913 |
| FcER1–153Eu | Biolegend | Cat# 334602, RRID:AB_1227649 |
| CD84–154Sm | Biolegend | Cat# 326002, RRID:AB_830813 |
| CD33–158Gd | Biolegend | Cat# 303402, RRID:AB_314346 |
| CD11c-159Tb | Biolegend | Cat# 301602, RRID:AB_314172 |
| GATA-1–160Gd | Cell Signalling | Cat# 3535, RRID:AB_2108288 |
| CD7–162Dy | BD Biosciences | Cat# 555359, RRID:AB_395762 |
| CD49f-164Dy | Biolegend | Cat# 555734, RRID:AB_2296273 |
| CD127–165Ho | Biolegend | Cat# 351302, RRID:AB_10718513 |
| CD66–167Er | BD Biosciences | Cat# 551354, RRID:AB_394166 |
| CD38–168Er | Biolegend | Cat# 303502, RRID:AB_314354 |
| CD45RA-169Tm | Biolegend | Cat# 304102, RRID:AB_314406 |
| CD135–170Er | Biolegend | Cat# 313302, RRID:AB_314987 |
| CD117–171Yb | Biolegend | Cat# 313202, RRID:AB_314981 |
| CD133–172Yb | Miltenyi Biotec | Cat# 130–108-062 |
| CD172ab-173Yb | Biolegend | Cat# 323802, RRID:AB_830701 |
| CD2–174Yb | Biolegend | Cat# 309202, RRID:AB_314752 |
| HLA-DR-209Bi | Biolegend | Cat# 307602, RRID:AB_314680 |
| CD15-biotinylated | Biolegend | Cat# 301914, RRID:AB_2561326 |
| CD3-biotinylated | Biolegend | Cat# 300404, RRID:AB_314058 |
| CD7-biotinylated | Thermo Fisher Scientific | Cat# 13–0079-82, RRID:AB_891490 |
| CD56-biotinylated | Biolegend | Cat# 362536, RRID:AB_2565653 |
| CD34-FITC | Myltenyi | Cat# 130–113-178, RRID:AB_2726005 |
| CD38-BV421 | Biolegend | Cat# 303526, RRID:AB_10983072 |
| CD45RA-AF700 | Biolegend | Cat# 304120, RRID:AB_493763 |
| CD10-BV650 | BD Biosciences | Cat# 563734, RRID:AB_2738393 |
| CD123-PECy7 | Biolegend | Cat# 306010, RRID:AB_493576 |
| GATA-1-PE | Cell Signalling | Cat# 13353, RRID:AB_2798187 |
| CD38-APC/Cy7 | Biolegend | Cat# 303534, RRID:AB_2561605 |
| CD71-PE | Biolegend | Cat# 334108, RRID:AB_10915138 |
| CD33-PE/Cy7 | Biolegend | Cat# 303434, RRID:AB_2734265 |
| CD84-APC | Biolegend | Cat# 326010, RRID:AB_2814188 |
| GATA1 | Abcam | Cat# ab181544 |
| Cleaved caspase 3-PE | BD Biosciences | Cat# 550821, RRID:AB_393906 |
|
| ||
| Biological samples | ||
|
| ||
| Adult bone marrow | All Cells Inc | https://allcells.com/research-grade-tissue-products/bone-marrow/ |
|
| ||
| Deposited data | ||
|
| ||
| ATAC-seq data | This paper | GEO:GSE167934 |
| FACS data | This paper | FlowRepository:FR-FCM-Z539 |
| Mass cytometry data | This paper | FlowRepository:FR-FCM-Z5ZA |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| K562 | ATCC | Cat# CCL-243, RRID:CVCL_0004 |
| GM12878 | Coriell Institute | Cat# GM12878, RRID:CVCL_7526 |
|
| ||
| Software and algorithms | ||
|
| ||
| Cutadapt | Martin, 2011 | https://cutadapt.readthedocs.io/en/stable/ |
| Bowtie2 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Picard tools | Broad Institute | https://broadinstitute.github.io/picard/ |
| ChrAccR | Fabian Mueller, https://doi.org/10.5281/zenodo.6091218 | https://github.com/GreenleafLab/ChrAccR, https://zenodo.org/record/6091218 |
| DESeq2 | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| SPADE | Qiu et al., 2011 | https://github.com/nolanlab/spade |
| Scanpy | Wolf et al., 2018 | https://scanpy.readthedocs.io/en/stable/ |
| GateFinder | Aghaeepour et al., 2018 | https://www.bioconductor.org/packages/release/bioc/html/GateFinder.html |
| ArchR | Granja et al., 2020 | https://www.archrproject.com/ |
| Harmony | Korsunsky et al., 2019 | https://portals.broadinstitute.org/harmony/index.html |
| MAGIC | Dijk et al., 2018 | https://github.com/KrishnaswamyLab/MAGIC |
| Seurat | Hao et al., 2021 | https://github.com/satijalab/seurat/ |
This paper does not report original code.
Any additional information required to reanalyse the data reported in this paper is available from the lead contact upon request.