

Summary
Background
Enteropathy is prevalent in tuberculosis-endemic areas, and it has been shown to impair intestinal absorptive function; therefore, enteropathogen burden might negatively affect antimycobacterial pharmacokinetics, particularly among malnourished children. We sought to quantify enteropathogen burden among children initiating tuberculosis treatment in rural Tanzania and determine the effect of enteropathogen burden on serum antimycobacterial pharmacokinetics.
Methods
We performed a prospective cohort study at one site in rural Tanzania as an exploratory substudy of a large multicountry cohort study. We included children younger than 15 years of age with confirmed or probable tuberculosis undergoing treatment with first-line tuberculosis therapy; children were excluded from the study if they were unable to undergo sample collection. Participants were consecutively recruited from the inpatient paediatric wards or the outpatient tuberculosis clinic at Haydom Lutheran Hospital, Tanzania. The main outcome was to quantify symptomatic enteropathogen burden and the effect on serum antimycobacterial pharmacokinetics. We quantified enteropathogen burden (defined as the sum of distinct enteropathogens detected in stool) using a multipathogen PCR capable of simultaneous detection of 37 bacterial, viral, and parasitic species or species groups from stool collected within 72 h of treatment initiation. Comprehensive clinical assessment, including presence of gastrointestinal symptoms, was performed at baseline, and serum was collected approximately 2 weeks after treatment initiation at steady state and throughout the dosing interval with concentrations of isoniazid, rifampicin, pyrazinamide, and ethambutol measured by liquid chromatography with a tandem mass spectrometry assay to quantify peak (Cmax) and total area under the concentration curve (AUC0–24), as determined by non-compartmental analysis. Enteropathogen burden was compared with pharmacokinetic measurements using bivariable and multivariable linear regression.
Findings
58 children were assessed for eligibilty and enrolled between June 25, 2016, and Feb 6, 2018; 44 had complete stool testing and serum pharmacokinetic data, and they were included in the analyses. 20 (45%) were female, and 24 (55%) were male. 37 (84%) had moderate or severe malnutrition. A mean of 2·1 (SD 1·3) enteropathogens were detected per participant. Target peak concentrations of rifampicin were reached in eight (18%) of 44 participants, isoniazid in 24 (54%) of 44 participants, pyrazinamide in 28 (74%) of 38 participants, and ethambutol in six (15%) of 39 participants. Compared with controlled comparisons, each summative additional bacterial enteropathogen detected was associated with a 40% lower rifampicin Cmax (95% CI −62 to −5) and a 36% lower ethambutol Cmax (−52 to −14), while viral pathogens were associated with a 51% lower isoniazid Cmax (−75 to −7). The combination of gastrointestinal symptoms and detection of an additional enteropathogen was associated with a 27% reduction in rifampicin AUC0–24 (95% CI −47 to −1).
Interpretation
Tanzanian children undergoing tuberculosis treatment rarely attained pharmacokinetic targets; enteropathogen carriage was common and enteropathogen burden was associated with significant reductions in the concentrations of some antimycobacterial drugs. Further research should explore mechanistic relationships of individual pathogens and antimycobacterial pharmacokinetics in larger cohorts, or determine if screening for and treating enteropathogens at tuberculosis treatment initiation improves pharmacokinetic target attainment.
Funding
National Institute of Allergy and Infectious Diseases, National Institutes of Health.
Introduction
Before the start of the COVID-19 pandemic in 2020, tuberculosis was the leading cause of death by a single infectious disease worldwide.1 In 2018, 10 million people were diagnosed with tuberculosis, with over 1 million cases in children younger than 15 years; despite the availability of a first-line treatment regimen with more than 90% efficacy in controlled settings, tuberculosis was the cause of death in over 200 000 children. Pharmacokinetic variability is an important driver of treatment failure; and low serum drug concentrations have been associated with a delayed response to treatment, poor outcomes, and acquired drug resistance.2,3 Most first-line tuberculosis medications are concentration dependent in their activity, with peak (Cmax) and total area under the concentration time curve (AUC) predictive of microbial kill rate and prevention of acquired resistance.4 Although in 2014 the WHO updated their guidelines for weight-based dosing in paediatric tuberculosis treatment, there is continued evidence of poor attainment of target Cmax and AUC values in children undergoing tuberculosis treatment.5–7 We previously showed that among 28 children undergoing tuberculosis treatment in rural Tanzania, none achieved target peak serum Cmax for rifampicin, isoniazid, pyrazinamide, and ethambutol simultaneously, and 54% did not reach target Cmax for any drug.7 In addition to drug dose, malnutrition was associated with lower drug concentration in multivariable models. In the past decade, multicountry cohorts have associated specific enteropathogen frequency with childhood malnutrition and growth trajectories,8 but enteropathogen burden has not been investigated in children treated for tuberculosis.
97% of tuberculosis cases occur in low-income and middle-income countries, where enteropathy is an important contributor to childhood morbidity.1,9 Enteropathogen burden—ie, the number of concurrent pathogens detected in stool—is known to adversely impact small bowel absorptive capacity, which has been measured by functional mannitol excretion tests in children without tuberculosis.10 It has also been shown that adult patients with tuberculosis (without known enteropathogen infection) also have impaired small bowel absorptive function, and they have lower serum rifampicin and isoniazid levels compared with healthy controls.11 Importantly, orally administered isoniazid, pyrazinamide, and ethambutol are preferentially absorbed in the small bowel, whereas oral rifampicin is also absorbed in the stomach.12 Thus, pathogens leading to inflammation along the upper gastrointestinal tract can affect antimycobacterial drug absorption and subsequent pharmacokinetic parameters; the impact might also be driven by individual drug-pathogen effects.
To quantify enteropathogen burden and determine the association between this burden and antimycobacterial pharmacokinetics, we used a multipathogen PCR array card for simultaneous detection of 37 bacterial, viral, and parasitic species or species groups among children undergoing tuberculosis treatment in rural Tanzania as part of a prospective pharmacokinetic study.13 We hypothesised that those with evidence of symptomatic enteropathogen infection (ie, gastrointestinal symptoms) and high enteropathogen burden would have a lower serum Cmax and AUC of first-line tuberculosis medication than those without evidence of enteropathogen infection.
Methods
Study design and participants
We performed a pharmacokinetic cohort study at one hospital in rural Haydom, Tanzania, that included children and enteropathogen burden as an exploratory analysis of a larger multicountry pharmacokinetic trial regarding pharmacokinetics of tuberculosis treatment, with prespecified pharmacokinetic parameters. The larger trial was not assessing enteropathogen burden. Haydom was selected due to the known coburden of malnutrition. Haydom was also the only paediatric site. Participants were consecutively recruited from the inpatient paediatric wards or the outpatient tuberculosis clinic at Haydom Lutheran Hospital from June 25, 2016, to Feb 6, 2018. Children were included if they were younger than age 15 years with confirmed or probable tuberculosis (defined by the National Institutes of Health consensus case definitions for tuberculosis research in children) and within 72 h of starting first-line tuberculosis treatment.14 Children were excluded from the study if they were unable to undergo sample collection of blood or stool by a treating physician, if their parent or guardian was unable to consent, and if they were unable to attend for follow-up. Follow-up for the primary outcome occurred at 2 weeks. Additional follow-up for clinical outcomes occurred at 4, 8, 24, 48, 72, and 96 weeks after treatment initiation; these outcomes are not described here as they do not relate to this study. All parents or guardians gave written informed consent and children older than 7 years provided assent. All study patients were enrolled in Haydom, with a protocol approved by the institutional review boards for human subjects research at the University of Virginia and the Tanzania National Institute for Medical Research. Reporting is in accordance with STROBE guidelines for cohort studies where applicable.
The study protocol is available at ClinicalTrials.gov, NCT03559582.
Procedures
Data on demographics and anthropometrics, including age, sex, height, weight, presence of abdominal pain or diarrhoea, presence and duration of fever, and previous medical history were collected by trained study staff at baseline. The primary outcomes were to quantify the symptomatic enteropathogen burden and determine the effect of this burden on the pharmacokinetic parameters of serum Cmax and AUC over 24 h (AUC0–24) for isoniazid, rifampicin, pyrazinamide, and ethambutol. The secondary outcomes were to determine the effect of individual enteropathogens on rifampicin, isoniazid, pyrazinamide, and ethambutol Cmax and AUC0–24. To quantify the enteropathogen burden stool samples from participants were collected at baseline by study nurses in sterile containers, stored at −80°C, and shipped to the Kilimanjaro Clinical Research Institute, Tanzania, where entero_pathogen burden was assessed by real-time PCR using a TaqMan Array Card (TAC) (Thermo Fisher, Carlsbad, CA, USA) as described elsewhere13 (appendix pp 3–4). JG, JL, and ERH assessed the data.
QIAamp Fast Stool DNA Mini Kit (Valencia, CA, USA) was used to extract nucleic acids, which were mixed with AgPath-ID One-Step RT-PCR buffer (Thermo Fisher, CA, USA) and nuclease-free water, and were run on a custom-designed TAC. Organisms included on the TAC were Campylobacter spp, Shigella spp, enteroaggregative Escherichia coli, typical and atypical enteropathogenic E coli, enterotoxigenic E coli, shiga-toxin-producing E coli, Salmonella enterica, Aeromonas spp, Plesiomonas shigelloides, Helicobacter pylori, Clostridioides difficile, Mycobacterium tuberculosis, rotavirus, norovirus, adenovirus 40 and adenovirus 41, astrovirus, sapovirus, Cryptosporidium, Giardia, Enterocytozoon bieneusi, Trichuris, Encephalitozoon intestinalis, Cyclospora, Isospora, Entamoeba histolytica, Ancyclostoma, Ascaris, Necator, and Strongyloides. A measure of stool pathogen burden was calculated for each participant, defined as the sum of all enteropathogens detected in stool. M tuberculosis was excluded in the summation, as detection in stool was thought to represent sputum ingestion. Summative measures of bacterial, viral, and parasitic enteropathogen burden were also calculated. Testing was performed in batch, and stool results were not available to treating clinicians.
To quantify the pharmacokinetic parameters procedures were completed as inpatients, or for those patients previously discharged or enrolled as outpatients, procedures were completed at the research facility of the hospital. Each patient received weight-based dosing of oral antimicrobials: rifampicin, isoniazid, pyrazinamide, and ethambutol while fasted, as per Tanzanian national guidelines.15 Pyrazinamide and ethambutol were not administered in some instances at the discretion of the treating physicians. If paediatric dispersible tablets were not available, adult formulations were substituted (appendix pp 2–3).
Blood samples were collected 14 days after the tuberculosis medication was started; this was to allow for rifampicin autoinduction of clearance and steady-state kinetics. The study team, including a study nurse and doctor, observed drug dosing and administration the day before blood samples were collected to ensure consumption of the medicine. The following day, blood samples were collected at 1, 2, and 6 h after observed morning medication administration, to collect capture peak and elimination phase for the oral tuberculosis medication, and serum was stored at −80°C until batch shipment to the University of Florida Infectious Diseases Pharmacokinetics Laboratory, USA, where drug concentrations were measured by a validated liquid chromatography with a tandem mass spectrometry assay.7
Additional outcomes included assessing physiological perturbance in addition to pathogen presence, two biomarkers to determine the magnitude of gut inflammation and permeability were tested on baseline stool. Concentrations of stool myeloperoxidase (Alpco, Salem, NH, USA), a marker of intestinal inflammation,9 and α1-antitrypsin (Biovendor, Candler, NC, USA), a marker of intestinal permeability, were determined by ELISA. Assays were performed as described in the package insert, except initial dilutions for the myeloperoxidase assay were performed at 1:500.
Statistical analysis
A formal sample size calculation was not performed for this analysis given the exploratory nature of this study as part of a larger pharmacokinetic trial. Demographic characteristics were determined by simple frequencies. Mann-Whitney U test was used for continuous variables and Fisher’s exact test was used for categorical variables. Cmax and estimated AUC0–24 were determined by non-compartmental analysis using Phoenix WinNonlin version 8.0 (Certara USA, Princeton, NJ, USA); and AUC0–24 was calculated using the trapezoidal rule with at least two samples, with one in the elimination phase. Pharmacokinetic parameters were descriptively compared with targets from previous studies, which determined drug concentrations predictive of long-term treatment outcomes (isoniazid minimum targets of Cmax ≥3 mg/L, AUC0–24 ≥52 mg × h/L; rifampicin minimum targets of Cmax ≥8 mg/L, AUC0–24 ≥35 mg × h/L; pyrazinamide minimum targets of Cmax ≥20 mg/L, AUC0–24 ≥363 mg × h/L; ethambutol minimum targets of Cmax ≥2 mg/L, AUC0–24 target not defined).4,16
For the primary comparison of the drug exposure with the number of stool pathogens identified and the outcome of serum pharmacokinetic measurements, log-Cmax and log-AUC0–24 values for each drug were modelled as functions of enteropathogen burden using multivariable linear regression with percentage change in Cmax or AUC0–24 per each pathogen detected (95% CI) as the estimated effect size. To account for asymptomatic carriage of pathogens, and to estimate associations among individuals with gastrointestinal symptoms, we included a binary term indicating the presence of gastrointestinal symptoms (ie, diarrhoea or abdominal pain at the time of stool sample collection). Models were further adjusted for mg/kg drug dose, sex, age, and body-mass index (BMI) Z score. All participants were kept in the models. Models that used the presence of an abnormal faecal myeloperoxidase or α1-antitrypsin rather than gastrointestinal symptoms were also constructed. An exploratory analysis evaluating associations between individual pathogens and serum drug exposure was performed using the same framework as described above; whereas log-Cmax and log-AUC0–24 were modelled as functions of the presence of individual enteropathogens, with percentage change in Cmax or AUC0–24 if the pathogen was detected (95% CI) as the estimated effect size. Data were analysed in R (version 3.6.1). Statistical controls were included in the models to control for potential confounding variables including age, sex, drug dose, and malnutrition. Partipants lost to follow-up were not included in analysis. Not all children had ethambutol and pyrazinamide pharmackinetics if they were not treated with these medicines (table 2); these children were not included in models in which ethambutol or pyrazinamide was the dependent variable.
Table 2:
Summary of pharmacokinetic measurements
| Rifampicin (n=44) | Isoniazid (n=44) | Pyrazinamide (n=38)* | Ethambutol (n=39)* | |
|---|---|---|---|---|
| C max , mg/L | ||||
| Concentration | 4·5 (2·6–7·4) | 3·2 (2·1–5·5) | 30·2 (20·2–40·5) | 0·9 (0·7–1·7) |
| Number at target† | 8 (18%) | 24 (54%) | 28 (74%) | 6 (15%) |
| AUC 0–24 , mg×hr/L | ||||
| Concentration | 22·7 (12·6–39·0) | 13·0 (7·1–21·3) | 276·0 (181·8–391·4) | 9·4 (5·1–13·7) |
| Number at target‡ | 13/37 (35%) | 2/40 (5%) | 11/34 (32%) | .. |
| Dose, mg/kg | 11·8 (9·2–13·9) | 6·0 (4·6–7·0) | 30·4 (23·5–36·5) | 21·2 (16·6–25·6) |
Data are n (%) or median (IQR) unless otherwise specified. Cmax=peak serum concentration. AUC0–24=area under the concentration curve from 0–24 hours.
Pyrazinamide and ethambutol were not administered in some instances at the discretion of the treating physicians.
Target Cmax defined as ≥8 mg/L for rifampicin, ≥3 mg/L for isoniazid, ≥20 mg/L for pyrazinamide, ≥2 mg/L for ethambutol.16
Target AUC0–24 defined as ≥35 mg × h/L for rifampicin, ≥52 mg × h/L for isoniazid, ≥363 mg × h/L for pyrazinamide.4
Role of the funding source
The funder had no role in study design, data collection, data analysis, data interpretation, or writing of the report.
Results
58 children undergoing tuberculosis treatment with first-line therapy were enrolled between June 25, 2016, and Feb 6, 2018. Stool samples were provided by 51 participants at the time of enrolment. Pharmacokinetic data (blood samples) were collected in 44 participants (four were lost to follow-up, two died before pharmacokinetic sampling, and we were unable to obtain adequate serum sampling in one); these 44 were included in the final analysis. The median age of participants was 2·25 years (IQR 1·28–5·23), 20 (45%) were female, 24 (55%) were male (gender was reported by the child’s guardian), and 37 (84%) had moderate or severe malnutrition, with median weight-for-age Z scores of −3·42, median height-for-age Z scores of−2·37, and median BMI Z scores of −1·98 (table 1). 32 (73%) had gastrointestinal symptoms and 41 (93%) received non-dispersible drug formulations. All children were negative for HIV.
Table 1:
Baseline characteristics and enteropathogen detection
| Participants (n=44) | |
|---|---|
| Age, years | 2·25 (1·28–5·23) |
| Sex | |
| Female | 20 (45%) |
| Male | 24 (55%) |
| Moderate or severe malnutrition* | 37 (84%) |
| Weight-for-age Z score | −3·42 (−4·38 to −2·01) |
| Height-for-age Z score | −2·73 (−4·17 to −1·47) |
| BMI Z score | −1·98 (−3·06 to −0·73) |
| Haemoglobin, mg/dL | 9·6 (8·15–11·05) |
| Previous history of tuberculosis | 3 (7%) |
| Microbiological confirmation† | 29 (66%) |
| HIV positive | 0 |
| Fever for 1 week | 40 (91%) |
| Gastrointestinal symptoms‡ | 32 (73%) |
| Abdominal pain | 1 (25%) |
| Diarrhoea | 27 (61%) |
| Antibiotic use before stool collection | 40 (91%) |
| Non-dispersible (crushed) tuberculosis drug preparation | 41 (93%) |
| Number of enteropathogens detected per participant | |
| 0 | 6 (14%) |
| 1 | 7 (16%) |
| 2 | 13 (30%) |
| 3 | 14 (32%) |
| 4 | 2 (5%) |
| 5 | 1 (2%) |
| 6 | 1 (2%) |
| Enteropathogens detected | 2·1 (1·3) |
| Bacterial enteropathogens | 1·3 (1·0) |
| Enteroaggregative Escherichia coli | 28 (64%) |
| Clostridioides difficile | 23 (52%) |
| Campylobacter | 9 (20%) |
| Typical enteropathogenic E coli | 9 (20%) |
| Enterotoxigenic E coli | 8 (18%) |
| Shigella | 3 (7%) |
| Atypical enteropathogenic E coli | 3 (7%) |
| Helicobacter pylori | 2 (5%) |
| Shiga-toxin-producing E coli | 1 (2%) |
| Salmonella | 1 (2%) |
| Viral enteropathogens | 0·4 (0·5) |
| Sapovirus | 9 (20%) |
| Norovirus | 8 (18%) |
| Adenovirus | 2 (5%) |
| Astrovirus | 2 (5%) |
| Parasitic enteropathogens | 0·5 (0·6) |
| Giardia | 16 (34%) |
| Cryptosporidium | 4 (9%) |
| Enterocytozoon bieneusi | 3 (7%) |
Data are n (%), mean (SD), or median (IQR) unless otherwise specified. Enteropathogens not depicted were not detected. BMI=body mass index.
Moderate or severe malnutrition defined as weight-for-age, height-for-age or BMI-for-age Z score less than −2.
Microbiological confirmation by positive sputum GeneXpert.
Gastrointestinal symptoms defined as presence of abdominal pain or diarrhoea at the time of stool collection.
The prevalence of individual enteropathogens as determined by stool PCR is shown in table 1. Of note, C difficile was detected in 23 (52%) participants; rates of other enteropathogens detected were otherwise consistent with studies from the past decade of children 2 years or younger without tuberculosis, conducted at the same location.8 After excluding enteroaggregative E coli—which was detected in the majority of participants, can frequently be asymptomatic,17,18 and nearly always co-occurred with a second enteropathogen that could better explain gastrointestinal symptoms—38 (86%) participants had one or more enteropathogens detected in stool, with a mean of 2·1 pathogens (SD 1·3) per participant. Bacterial enteropathogens were most commonly detected, with a mean of 1·3 pathogens (1·0) per participant, followed by parasitic pathogens with a mean of 0·5 (0·6), and viral pathogens with a mean of 0·4 (0·5).
A summary of serum pharmacokinetic measurements is shown in table 2. Target peak concentrations of rifampicin were reached in eight (18%) of 44 participants, isoniazid in 24 (53%) of 44 participants, pyrazinamide in 28 (62%) of 38 participants, and ethambutol in six (15%) of 39 participants. Antimycobacterial Cmax and AUC0–24 values were modelled as functions of enteropathogen burden and shown in figure 1, including all 44 children in the model. There were a few significant results, including decreased rifampicin AUC0–24 associated with detection of any additional enteropathogen, rifampicin Cmax and ethambutol Cmax with bacterial pathogens, and isoniazid Cmax with viral pathogens. Detection of each additional enteropathogen of any type among individuals with gastrointestinal symptoms was associated with a 27% reduction in rifampicin AUC0–24 (95% CI −47 to −1) (figure 1). Each additional bacterial pathogen among individuals with gastrointestinal symptoms was associated with a 40% lower rifampicin Cmax (95% CI −62 to −5) and a 36% lower ethambutol Cmax (95% CI −52 to −14), while each additional viral pathogen was associated with a 51% lower isoniazid Cmax (95% CI −75 to −7).
Figure 1: Combined effect of enteropathogen burden and gastrointestinal symptoms on Cmax and AUC0–24.
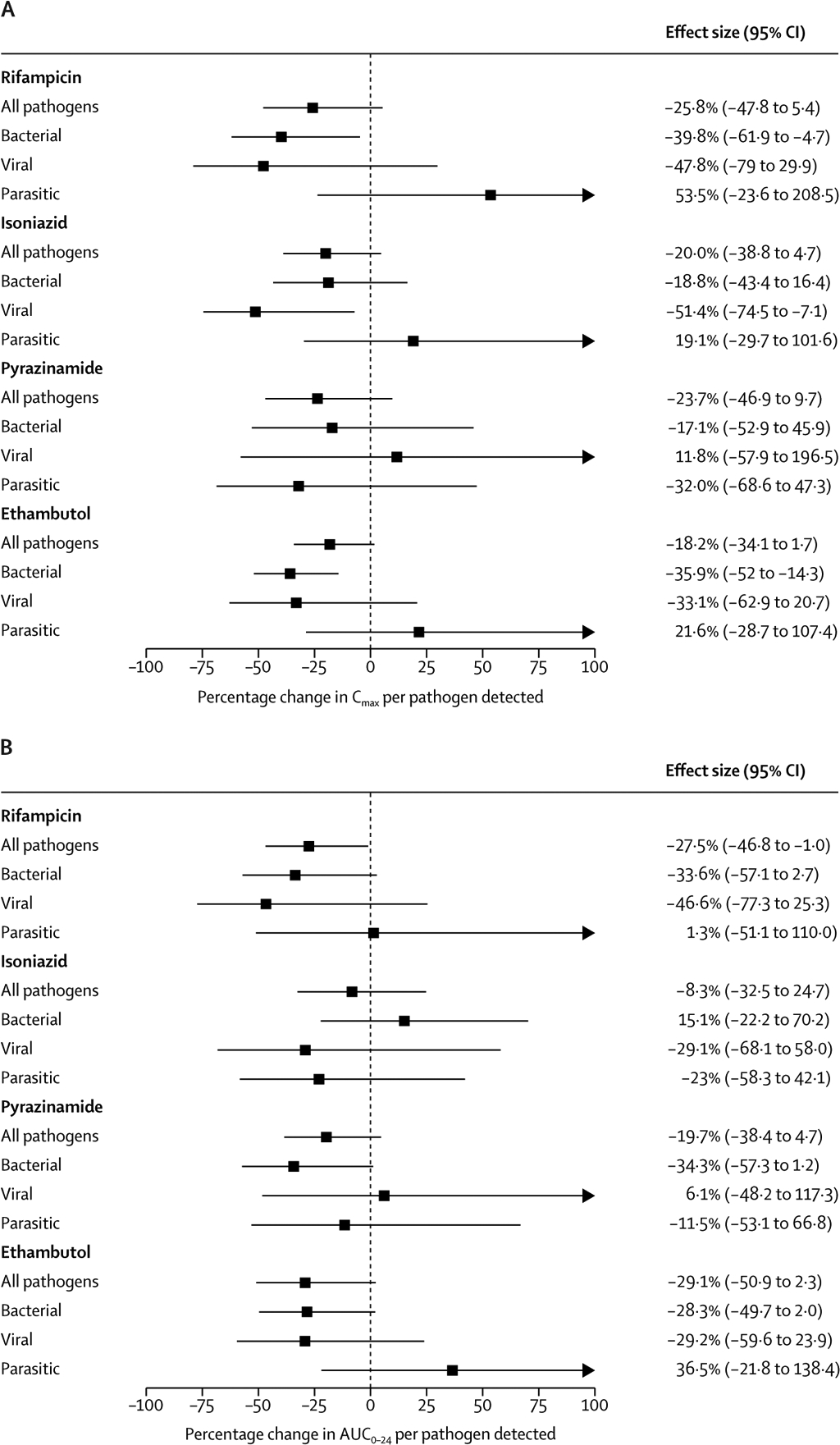
Cmax=peak drug concentration. AUC0–24=total exposure area under the concentration curve over 24 hs (A) Percentage change in Cmax per pathogen detected. (B) Percentage change in AUC0–24 per pathogen detected. Log-Cmax and log-AUC0–24 were modelled as functions of enteropathogen burden and the presence of gastrointestinal symptoms. Estimates were back-transformed to calculate the relative change in Cmax or AUC0–24. Estimates were adjusted for mg/kg drug dose, age, sex, and body-mass index Z score. 95% CIs are shown and are truncated above 100%. Results from bivariable analysis without adjustment for covariates are available in the appendix pp 5–6.
To further characterise enteropathy, faecal myeloperoxidase and α1-antitrypsin were quantified in 43 (98%) participants (table 3). Myeloperoxiase and α1-antitrypsin was not quantified in one participant due to assay failure. Median stool myeloperoxidase concentration was 1574 ng/mL (IQR 597–3806; normal range <2000 ng/mL) and median α1-antitrypsin was 217 μg/mL (146–438; normal range <270 μg/ml). 19 (44%) had elevated myeloperoxidase and 15 (35%) participants had elevated α1-antitrypsin concentrations. Of 31 participants with gastrointestinal symptoms, 19 (61%) had at least one abnormal biomarker, compared with four (33%) of 12 without gastrointestinal symptoms (p=0·10). Relationships of stool pathogen burden and either myeloperoxidase or α1-antitrypsin concentrations were not observed in bivariable regression (appendix p 7).
Table 3:
Stool myeloperoxidase and α1-antitrypsin concentrations
| All participants (n=43) | Gastrointestinal* symptoms (n=31) | No gastrontestinal symptoms (n=12) | p-value† | |
|---|---|---|---|---|
| Myeloperoxidase | ||||
| Concentration, ng/mL | 1574 (597–3806) | 2225 (812–4033) | 663 (295–2335) | 0·088 |
| Number elevated‡ | 19 (44%) | 16 (52%) | 3 (25%) | 0·11 |
| α1-antitrypsin | ||||
| Concentration, μg/mL | 217 (146–438) | 257 (142–475) | 211 (186–261) | 0·80 |
| Number elevated§ | 15 (35%) | 13 (42%) | 2 (17%) | 0·12 |
| Total number of elevated myeloperoxidase or α1-antitrypsin | 23 (53%) | 19 (61%) | 4 (33%) | 0·10 |
Data are n (%) or median (IQR) unless otherwise specified.
Gastrointestinal symptoms of diarrhoea or abdominal pain.
Comparisons between results in those with gastrointestinal symptoms and those without gastrointestinal symptoms were calculated by Mann-Whitney U test for continuous variables and by Fisher’s exact test for categorical variables.
Defined as myeloperoxidase >2000 ng/mL.
Defined as α1-antitrypsin >270 μg/mL.
Detection of each additional enteropathogen among children with an abnormal myeloperoxidase or α1-antitrypsin (rather than gastrointestinal symptoms) demonstrated similar inverse associations with Cmax and AUC0–24, although most did not reach statistical significance (supplemental figure 3; appendix p 8). Additionally, gastrointestinal symptoms or pathogen burden alone did not significantly affect pharmacokinetic measurements independently of each other in controlled comparisons (appendix pp 9–10), suggesting pathogen burden has a negative association with drug concentrations but only in the setting of clinical findings consistent with enteropathy.
Given the variety of individual pathogens detected, relationships of individual pathogens with pharmacokinetic measurements were considered exploratory but were modelled using the same approach (figure 2). The detection of typical enteropathogenic E coli among individuals with gastrointestinal symptoms was associated with a 79% reduction in rifampicin Cmax (95% CI −93 to −36), a 63% lower isoniazid Cmax (−84 to −13), a 68% lower ethambutol Cmax (−85 to −30), and a 71% lower rifampicin AUC0–24 (95% CI −91 to −4) compared with patients with gastrointestinal symptoms but without typical enteropathogenic E coli. Enterotoxigenic E coli was associated with a 70% lower rifampicin Cmax (−91 to −4), sapovirus with a 69% lower rifampicin Cmax (−90 to −5), norovirus with a 72% lower isoniazid AUC0–24 (−90 to −24·4), and Giardia was associated with a 65% lower pyrazinamide Cmax (−86 to −12).
Figure 2: Combined effect of individual enteropathogens and gastrointestinal symptoms on Cmax and AUC0–24.
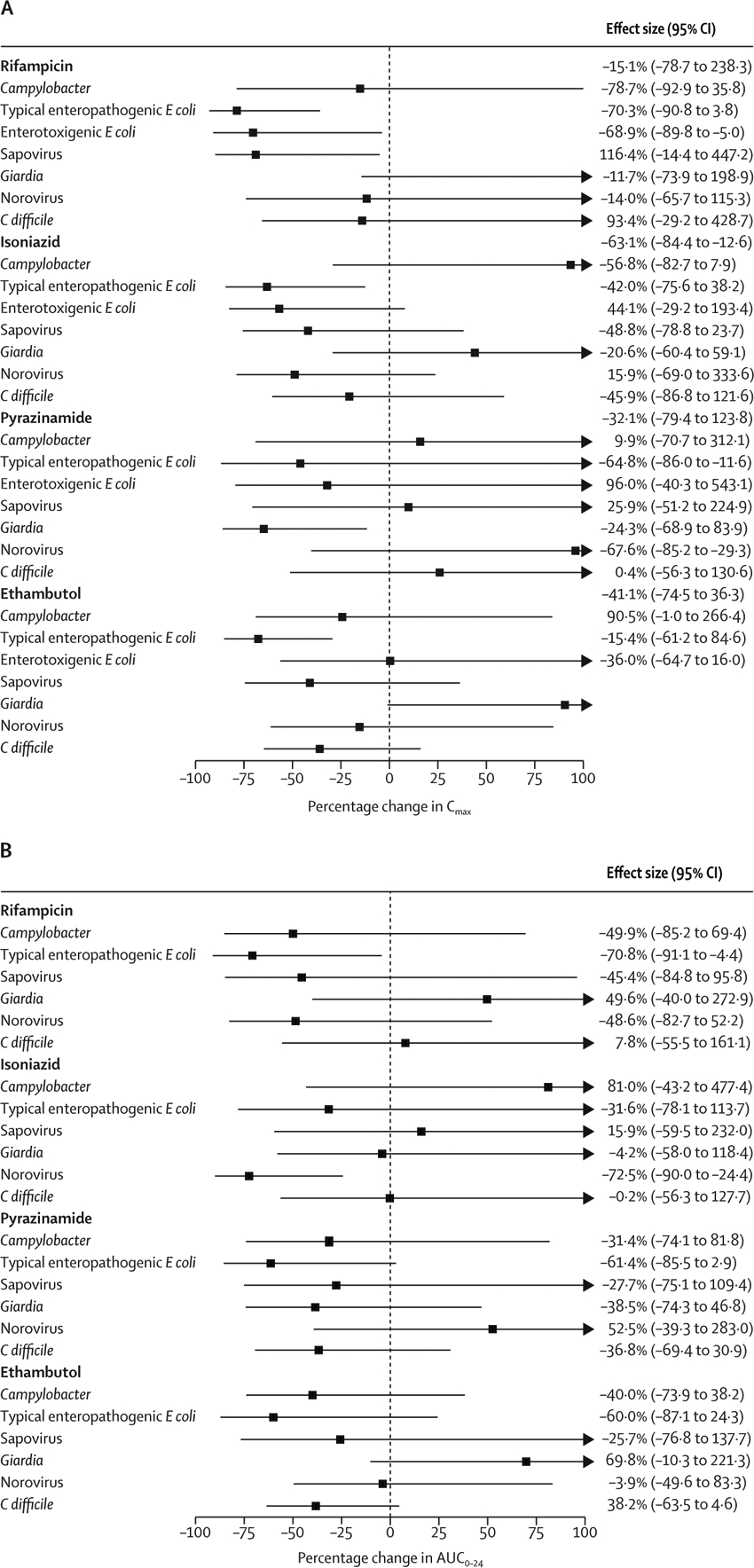
E Coli=Escherichia coli. C difficile=Clostridiodies difficile. Cmax=peak drug concentration. AUC0–24=total exposure area under the concentration curve over 24 hs. (A) Percentage change in Cmax associated with pathogen detection. (B) Percentage change AUC0–24 associated with pathogen detection. Log-Cmax and log-AUC0–24 were modelled as functions of enteropathogen burden and the presence of gastrointestinal symptoms. Estimates were back-transformed to calculate the relative change in Cmax or AUC0–24. Estimates were adjusted for mg/kg drug dose, age, sex, and body-mass index Z score. 95% CIs are shown and are truncated above 100%.
Discussion
Although a growing evidence base suggests that optimal pharmacokinetic exposure is critical for successful treatment of tuberculosis, our understanding of the determinants of pharmacokinetic variability remains incomplete, particularly among paediatric populations. In this study of children undergoing first-line tuberculosis treatment in rural Tanzania, we found that the majority of participants had antimycobacterial drug concentrations below target and that intestinal pathogens were common. Enteropathogen burden was identified as a significant risk factor for decreased serum antimycobacterial drug concentrations among participants with symptoms consistent with enteropathy, these participants represented the majority of the study population.
Despite a relatively small number of participants, we found that each additional enteropathogen was negatively associated with rifampicin AUC0–24, that bacterial enteropathogens were associated with decreased rifampicin and ethambutol peak concentrations, and that viral enteropathogens were associated with lower isoniazid Cmax peak concentrations among participants with gastrointestinal symptoms. Suboptimal serum isoniazid concentrations in this study can partly be explained by the low median dose of 6 mg/kg despite a recommended dose of 10 mg/kg,5 which was a consequence of the intermittent availability of paediatric fixed-dose combinations that matched the updated guidelines at the time of study. However, rifampicin, pyrazinamide, and ethambutol were adequately dosed, yet target Cmax and AUC0–24 were still infrequently achieved.
The identification of enteropathogen burden as a risk factor for suboptimal antimycobacterial pharmacokinetics has far-reaching implications. Diarrhoeal disease is globally recognised as a major cause of childhood illness, and despite improvements in global mortality within the past decade, it continues to be a major cause of childhood morbidity, particularly in low-income and middle-income countries where the prevalence of paediatric tuberculosis is greatest.17 The low attainment of target drug concentrations in our work is consistent with a growing number of pharmacokinetic studies performed among children with tuberculosis in endemic areas.6,7 Our findings suggest that enteropathogen burden might play a previously under recognised, antagonistic role in the pharmacotherapy of paediatric patients with tuberculosis, and warrants further study in larger and more diverse paediatric settings in tuberculosis endemic regions.
Our hypothesis that enteropathogen burden would affect serum drug concentrations assumed a primary mechanism of malabsorption due to intestinal infection. Both enteropathogen burden and tuberculosis disease are independently associated with impaired small bowel absorptive function,10,11 and malabsorption has been implicated in causing low concentrations of rifampicin and isoniazid among patients with HIV, tuberculosis disease, and diarrohea.18 We found that faecal myeloperoxidase (a marker of intestinal inflammation) and α1-antitrypsin (a marker of intestinal permeability) were commonly above the normal range in this study;19,20 although, we did not find these markers to be directly associated with pharmacokinetic measurements.
Several of the individual pathogens identified as associated with poor pharmacokinetics are also notable causes of intestinal inflammation or permeability. Typical enteropathogenic E coli was associated with decreased rifampicin, isoniazid, and ethambutol peak concentrations, as well as a reduction in rifampicin AUC0–24, and particularly among young children it is a cause of gastroenteritis and malabsorption.21 Sapovirus and norovirus were associated with significant reductions in isoniazid exposure and rifampicin Cmax, respectively, and additional presence of any viral species significantly reduced isoniazid Cmax. Peak concentrations of isoniazid might be specifically blunted with viral pathogens as isoniazid is more diffusely absorbed throughout the small bowel and not in the stomach, which is the primary site for rifampicin absorption.12 Commonly detected viral pathogens in this study, norovirus and the related sapovirus of the Caliciviridae family, lead to primary inflammation in the jejunum but notably spare the stomach.22 That inflammatory process can lead to gastric emptying, which would more severely affect isoniazid absorption compared with pyrazinamide (a drug that is exceedingly well absorbed) or rifampicin.23,24
Giardia was the most frequently detected parasitic pathogen; yet, unexpectedly we observed a negative association between Giardia carriage and pyrazinamide Cmax levels, but not with rifampicin, isoniazid, or ethambutol concentrations. It would be unusual for malabsorption of any cause to affect pyrazinamide but not the other first-line drugs. Yet, one possible explanation is increased metabolism of pyrazinamide in children with giardiasis. Xanthine oxidase, which converts pyrazinamide into 5-hydroxypyrazinamide, is expressed in the liver and is also found in high concentrations in the gut.25 The immunopathogenesis of Giardia infection is complex and differs from other pathogens detected in this cohort; paradoxically, it has been associated with a decreased risk for diarrhoea as well as reductions in faecal myeloperoxidase and systemic C-reactive protein.26 Importantly, Giardia induces a lymphocytic inflammation with a prominent increase in interferon-γ,27 which is among the strongest inducers of xanthine oxidase.28 Giardiasis might therefore alter pyrazinamide metabolism via these immunomodulatory changes but would require a dedicated experimental design to best investigate.
This study had other limitations. First, although stool samples were collected and enteropathogen burden was quantified at the time of treatment initiation, serum drug concentrations were not measured until the second week of treatment. Bacterial pathogen carriage could have been altered with rifampicin administration, or new enteropathogens could have been acquired before pharmacokinetic measurement; in addition, some children had been on antimicrobials for up to 72 h before stool collection. Second, the high rates of C difficile found in this study must be interpreted cautiously; although the assay used a toxin-specific nucleic acid target, confirmatory assays were not performed. Children ultimately diagnosed with tuberculosis disease frequently undergo several rounds of antibiotic therapy before diagnosis, which likely contributes to gut microbiome dysbiosis and allows C difficile proliferation. While often overlooked in tuberculosis endemic areas, C difficile has been recognised as an important cause of hospital-acquired and nosocomial diarrhoea in resource-limited settings,29 and it has increasingly been recognised as a complication of tuberculosis therapy.30 Third, a functional investigation (such as lactulose mannitol excretion tests) would be better than myeloperoxidase and α1-antitrypsin to characterise gut disruption and absorptive capacity and establish a mechanistic link between enteropathogen burden and pharmacokinetics. Fourth, N-acetyltransferase 2 (NAT2) genotyping, which can be predictive of isoniazid metabolism was not included, but it might have less impact in younger children for whom NAT2 enzymatic maturation is incomplete.31 Fifth, the study analyses included 44 participants, although this was sufficient to describe several significant effects of enteropathogen burden on antimycobacterial drug concentrations, to determine generalisability these findings will require confirmation in other comparable cohorts; to determine the impact of individual pathogens these findings will require confirmation with larger cohorts; and to allow more complete assessment of total serum exposures these findings will require confirmation with more extensive sampling strategies and at different days within the total treatment duration. Finally, given the exploratory nature of the study, a large number of analyses were performed without adjustment for multiple comparisons, further necessitating confirmatory studies.
In summary, children treated for tuberculosis disease in rural Tanzania rarely attained pharmacokinetic targets. Enteropathogen burden was identified as a risk factor for subtarget serum concentrations of rifampicin, isoniazid, and ethambutol. Future work should involve further characterisation of the mechanistic explanations of these findings, confirmation within larger and more diverse cohorts of children from other tuberculosis endemic settings, and lastly testing of interventions to ameliorate enteropathogen burden to improve antimycobacterial pharmacokinetic target attainment or to personalise tuberculosis treatment based on point-of-care knowledge of an individual’s pharmacokinetics.
Supplementary Material
Research in context.
Evidence before this study
We searched PubMed from database inception, without language restrictions for studies of enteropathogen prevalence in tuberculosis patients using the keywords “enteropathogen” AND “tuberculosis” before June 28, 2020, which returned 16 results. Of these, only one study attempted to quantify prevalence of enteropathogens among people with tuberculosis; this study was performed in adults with HIV or tuberculosis, and it was only evaluated for parasitic pathogens. A second search using the keywords “enteropathogen” AND “tuberculosis” AND “pharmacokinetics” returned no results.
Added value of this study
Among a subset of children with tuberculosis participating in a prospective pharmacokinetic cohort study, we performed broad-spectrum stool polymerase chain reaction for enteropathogens, we obtained full pharmacokinetic profiles of each of the first-line antimycobacterial drugs, and we measured biomarkers of enteropathy. This is the first study to quantify enteropathogen burden and species composition among children with tuberculosis. These results add to a growing body of data showing suboptimal serum target attainment among children with tuberculosis in endemic areas; yet, they are the first to demonstrate a negative association between enteropathogen burden and antimycobacterial pharmacokinetic target.
Implications of all the available evidence
These findings indicate that enteropathogens are an important and overlooked aspect of clinical care in children with tuberculosis. Given the heavy enteropathogen burden seen in this cohort and the documented global prevalence of enteropathogen carriage in tuberculosis endemic areas, further resources should be devoted to the investigation of the pathogenesis and consequences of enteropathogen and tuberculosis coinfection. Moreover, these findings strongly suggest that enteropathogens play an antagonistic role in the pharmacotherapy of childhood tuberculosis; and these findings provide insight into enteropathogen species and individual drug relationships that can be targeted for further mechanistic or interventional studies.
Acknowledgements
This work was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health (NIH). SKH and ERH received grant U01AI115594 from the National Institute of Allergy and Infectious Diseases of the NIH. DVA received training grant, T32AI007046, from the National Institute of Allergy and Infectious Diseases of the NIH. We are grateful to the community of Haydom, Tanzania, and the Haydom Lutheran Hospital; and we are indebted to the parents, guardians, and participants for their generosity in participating in the study.
Footnotes
Declaration of interests
We declare no competing interests.
See Online for appendix
Data sharing
The primers list for multiplex PCR of enteropathogens are available in the appendix pp 3–4. Other metadata and standard operating procedures for PCR testing are available upon request to the corresponding author.
References
- 1.WHO. Global tuberculosis report 2019. 2019. https://www.who.int/publications/i/item/9789241565714 (accessed June 8, 2020).
- 2.Pasipanodya JG, McIlleron H, Burger A, Wash PA, Smith P, Gumbo T. Serum drug concentrations predictive of pulmonary tuberculosis outcomes. J Infect Dis 2013; 208: 1464–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Srivastava S, Pasipanodya JG, Meek C, Leff R, Gumbo T. Multeistant tuberculosis not due to noncompliance but to between-patient pharmacokinetic variability. J Infect Dis 2011; 204: 1951–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alffenaar JC, Gumbo T, Dooley KE, et al. Integrating pharmacokinetics and pharmacodynamics in operational research to end tuberculosis. Clin Infect Dis 2020; 70: 1774–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.WHO. Guidance for national tuberculosis programmes on the management of tuberculosis in children. 2014. https://apps.who.int/iris/bitstream/handle/10665/112360/9789241548748_eng.pdf;jsessionid=15F070C2FF9EEE9C1BE549B5ECDDA24F?sequence=1 (accessed June 8, 2020). [PubMed]
- 6.Kwara A, Enimil A, Gillani FS, et al. Pharmacokinetics of first-line antituberculosis drugs using WHO revised dosage in children with tuberculosis with and without HIV coinfection. J Pediatric Infect Dis Soc 2016; 5: 356–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Justine M, Yeconia A, Nicodemu I, et al. Pharmacokinetics of first-line drugs among children with tuberculosis in rural Tanzania. J Pediatric Infect Dis Soc 2020; 9: 14–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rogawski ET, Liu J, Platts-Mills JA, et al. Use of quantitative molecular diagnostic methods to investigate the effect of enteropathogen infections on linear growth in children in low-resource settings: longitudinal analysis of results from the MAL-ED cohort study. Lancet Glob Health 2018; 6: e1319–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kosek MN, Ahmed T, Bhutta Z, et al. Causal pathways from enteropathogens to environmental enteropathy: findings from the MAL-ED birth cohort study. EBioMedicine 2017; 18: 109–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee GO, McCormick BJJ, Seidman JC, et al. Infant nutritional status, feeding practices, enteropathogen exposure, socioeconomic status, and illness are associated with gut barrier function as assessed by the lactulose mannitol test in the MAL-ED birth cohort. Am J Trop Med Hyg 2017; 97: 281–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pinheiro VG, Ramos LM, Monteiro HS, et al. Intestinal permeability and malabsorption of rifampin and isoniazid in active pulmonary tuberculosis. Braz J Infect Dis 2006; 10: 374–79. [DOI] [PubMed] [Google Scholar]
- 12.Mariappan TT, Singh S. Regional gastrointestinal permeability of rifampicin and isoniazid (alone and their combination) in the rat. Int J Tuberc Lung Dis 2003; 7: 797–803. [PubMed] [Google Scholar]
- 13.Liu J, Gratz J, Amour C, et al. Optimization of quantitative PCR methods for enteropathogen detection. PLoS One 2016; 11: e0158199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Graham SM, Ahmed T, Amanullah F, et al. Evaluation of tuberculosis diagnostics in children: 1. Proposed clinical case definitions for classification of intrathoracic tuberculosis disease. Consensus from an expert panel. J Infect Dis 2012; 205: S199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.National Tuberculosis and Leprosy Programme. National guidelines for the management of tuberculosis in children, 3rd edn. Dar es Salaam, Tanzania: United Republic of Tanzania Ministry of Health and Social Welfare, 2016. [Google Scholar]
- 16.Alsultan A, Peloquin CA. Therapeutic drug monitoring in the treatment of tuberculosis: an update. Drugs 2014; 74: 839–54. [DOI] [PubMed] [Google Scholar]
- 17.Walker CLF, Rudan I, Liu L, et al. Global burden of childhood pneumonia and diarrhoea. Lancet 2013; 381: 1405–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gurumurthy P, Ramachandran G, Hemanth Kumar AK, et al. Malabsorption of rifampin and isoniazid in HIV-infected patients with and without tuberculosis. Clin Infect Dis 2004; 38: 280–83. [DOI] [PubMed] [Google Scholar]
- 19.Praharaj I, Revathy R, Bandyopadhyay R, et al. Enteropathogens and gut inflammation in asymptomatic infants and children in different environments in southern India. Am J Trop Med Hyg 2018; 98: 576–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Prata MM, Havt A, Bolick DT, Pinkerton R, Lima A, Guerrant RL. Comparisons between myeloperoxidase, lactoferrin, calprotectin and lipocalin-2, as fecal biomarkers of intestinal inflammation in malnourished children. J Transl Sci 2016; 2: 134–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hu J, Torres AG. Enteropathogenic Escherichia coli: foe or innocent bystander? Clin Microbiol Infect 2015; 21: 729–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schreiber DS, Blacklow NR, Trier JS. The mucosal lesion of the proximal small intestine in acute infectious nonbacterial gastroenteritis. N Engl J Med 1973; 288: 1318–23. [DOI] [PubMed] [Google Scholar]
- 23.Meeroff JC, Schreiber DS, Trier JS, Blacklow NR. Abnormal gastric motor function in viral gastroenteritis. Ann Intern Med 1980; 92: 370–73. [DOI] [PubMed] [Google Scholar]
- 24.Chirehwa MT, McIlleron H, Rustomjee R, et al. Pharmacokinetics of pyrazinamide and optimal dosing regimens for drug-sensitive and -resistant tuberculosis. Antimicrob Agents Chemother 2017; 61: e00490–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Auscher C, Amory N, van der Kemp P, Delbarre F. Xanthine oxidase activity in human intestines. Histochemical and radiochemical study. Adv Exp Med Biol 1979; 122B: 197–201. [DOI] [PubMed] [Google Scholar]
- 26.Rogawski ET, Bartelt LA, Platts-Mills JA, et al. Determinants and impact of Giardia infection in the first 2 years of life in the MAL-ED birth cohort. J Pediatric Infect Dis Soc 2017; 6: 153–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ebert EC. Giardia induces proliferation and interferon gamma production by intestinal lymphocytes. Gut 1999; 44: 342–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Page S, Powell D, Benboubetra M, et al. Xanthine oxidoreductase in human mammary epithelial cells: activation in response to inflammatory cytokines. Biochim Biophys Acta 1998; 1381: 191–202. [DOI] [PubMed] [Google Scholar]
- 29.Roldan GA, Cui AX, Pollock NR. Assessing the burden of Clostridium difficile infection in low- and middle-income countries. J Clin Microbiol 2018; 56: e01747–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Obuch-Woszczatyński P, Dubiel G, Harmanus C, et al. Emergence of Clostridium difficile infection in tuberculosis patients due to a highly rifampicin-resistant PCR ribotype 046 clone in Poland. Eur J Clin Microbiol Infect Dis 2013; 32: 1027–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rogers Z, Hiruy H, Pasipanodya JG, et al. The non-linear child: ontogeny, isoniazid concentration, and nat2 genotype modulate enzyme reaction kinetics and metabolism. EBioMedicine 2016; 11: 118–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The primers list for multiplex PCR of enteropathogens are available in the appendix pp 3–4. Other metadata and standard operating procedures for PCR testing are available upon request to the corresponding author.