

Abstract
Epidermal growth factor receptor exon 20 insertion mutations (EGFR exon20ins) are detected in approximately 2% of patients with non-small cell lung cancer (NSCLC). Due to lack of effective therapy, the prognosis of these patients was poor. Sunvozertinib (DZD9008) was designed as an oral, potent, irreversible and selective EGFR tyrosine kinase inhibitor, showing activity against EGFR exon20ins and other mutations. In both cell lines and xenograft models, sunvozertinib shows potent antitumor activity. In the two ongoing phase 1 clinical studies, sunvozertinib was tolerated up to 400 mg once daily. The most common drug-related adverse events included diarrhea and skin rash. Antitumor efficacy was observed at the doses of 100 mg and above in patients with EGFR exon20ins NSCLC across different subtypes, with prior amivantamab treatment as well as with baseline brain metastasis. The median duration of response (DoR) has not been reached.
Statement of Significance:
We report the discovery and early clinical development of sunvozertinib, a potential treatment option for unmet medical need of EGFR exon20ins NSCLC.
Keywords: CLINICAL TRIAL RESULTS/Phase I clinical trials, LUNG CANCER/LUNG CANCER, SMALL MOLECULE AGENTS/Kinase inhibitors
Introduction
NSCLC remains one of the leading causes of cancer death worldwide. Genetic mutations, such as EGFR mutations, have been reported in NSCLC (1-4) and drugs have been approved for the treatment of NSCLC with EGFR sensitizing, T790M and some types of uncommon mutations (5). For NSCLC with EGFR exon20ins, which comprise about 2% of NSCLC (6), the development of targeted therapies has been more challenging. With available therapy, such as platinum-based chemotherapy, patients have poor clinical prognosis with median progression free survival (PFS) and overall survival (OS) of only around 6 months and 24 months, respectively (7-9). Recently, an EGFR and MET bispecific antibody amivantamab received accelerated approval from the US FDA for treating this disease. However, the objective response rate (ORR) with amivantamab treatment was only around 40% (10). Mobocertinib, an EGFR TKI, has also recently received accelerated approval, while its ORR was only around 28% in a pivotal phase 2 study (11). Also, about 40% of patients experienced ≥ grade 3 treatment-related adverse events (TEAEs), and 21% of patients experienced ≥ grade 3 diarrhea (11, 12), which required dose reduction or even discontinuation. Therefore, there is still room for improvement to treat EGFR exon20ins mutations.
Engineering one human EGFR exon20ins subtype 770_NPG into transgenic mice induced tumor formation in the lung (13), which suggests potential oncogenic driver role of EGFR exon20ins. So far more than 39 distinct subtypes of EGFR exon20ins have been reported in NSCLC (9). Each subtype might have different sensitivity to a particular EGFR TKI. Therefore, a compound with good tolerability which allows it to reach high enough doses to cover a wide spectrum of mutation subtypes is highly desirable.
Sunvozertinib (DZD9008) is an oral, potent, irreversible and selective EGFR TKI targeting EGFR exon20ins as well as EGFR sensitizing, T790M and uncommon mutations with weak activity against wild-type EGFR. In addition, sunvozertinib exhibits desirable drug metabolism and pharmacokinetics (DMPK) properties as an oral drug in both preclinical and clinical settings. Moreover, nonclinical toxicity studies demonstrated a favorable safety margin of sunvozertinib to support its clinical development. Currently, sunvozertinib is under phase 1 clinical development in the USA, Australia, South Korea, Taiwan (ClinicalTrials.gov: NCT03974022) and China (Chinadrugtrial: CTR20192097).
Here we present the in vitro and in vivo activities of sunvozertinib on inhibiting EGFR exon20ins at the enzymatic and cellular levels, and in xenograft and transgenic animal models. In addition, we also illustrated the mechanism of sunvozertinib on suppressing EGFR exon20ins through structure-activity relationship. Finally, safety, pharmacokinetics (PK) and antitumor efficacy results from the two ongoing phase 1 clinical studies were also reported.
Results
Design and structure of sunvozertinib which can selectively bind to EGFR exon20ins protein
We focused on designing inhibitors with irreversible (covalent) binding mode with C797 similar to that of osimertinib. Without any clear structural rationale for attaining wild-type selectivity, we opt for using the osimertinib scaffold as the starting point for optimization for EGFR exon20ins potency and monitoring selectivity against wild-type EGFR using cellular assays. In addition, maintaining potent activity against EGFR sensitizing mutations and double mutations of T790M are also key attributes in our inhibitors design strategy. Based on the available structural information of EGFR exon20ins, we decided to replace the rotationally less flexible methylindole on osimertinib by a more flexible anilinophenyl moiety on C-4 of the pyrimidine hinge binding motif. Different substitution patterns on the phenyl ring were extensively investigated. Screening of the newly designed compounds began with the direct testing in EGFR cellular target engagement and anti-proliferation assays, bypassing the unknown effect of ATP concentration on compounds' activity in biochemical assays with different EGFR exon20ins. A brief discussion for the structure-activity-relationship (SAR) cumulated to the discovery of sunvozertinib (Figure 1) is as follows. Initial screening campaign using cellular pEGFR assays (L858R/T790M, ASV, NPH, and wild-type) on our irreversible EGFR inhibitors library identified both triazine and pyrimidine hinge binding scaffolds as hits with EGFR exon20ins activities that were more potent than that of osimertinib (Supplementary Table S1). Initial lead optimization on the novel and more potent triazine series led to the identification of ortho-substituted anilinophenyl head-groups as the preferred substructure for potent EGFR exon20ins potency. The enhanced potency of the inhibitors with EGFR exon20ins mutants can be envisioned as the ortho-substituent on the anilinophenyl group occupying part of the back pocket on the ATP binding site and interacting favorably with the α-C helix. Among all the ortho-substituents investigated, we settled with the 2-hydroxypropan-2-yl moiety as in compound Cpd-5 for further optimization based on its potency and in vitro DMPK parameters. Incubation of compound Cpd-5 with human hepatocytes indicated oxidative liability that occurred predominantly on the head-group phenyl ring and the dimethylaminoethyl side chain. To this end, introducing both 4-fluoro and 5-chloro substitutions as in compound Cpd-6 successfully reduced human hepatocytes clearance from 34 to 7.1 (μL/min)/(106 cells). Additional modification of the flexible dimethylaminoethyl side chain in Cpd-6 into a conformational more rigid (dimethylamino) pyrrolidin-1-yl in compound Cpd-8 further decrease the human hepatocytes clearance to 2.8 (μL/min)/(106 cells), while maintaining the required EGFR mutants activity. Towards the end game, a match-pair direct comparison between compounds in the triazine and pyrimidine series was performed. Although the triazine series consistently showed better EGFR exon20ins potency, the pyrimidine core offered a more desirable selectivity against the wild-type EGFR. Finally, DZD9008 in the pyrimidine series was selected for further profiling in numerous in vivo efficacy and safety experiments, and was eventually recommended as the development candidate of sunvozertinib. Nonclinical DMPK properties of sunvozertinib was summarized in Supplementary Table S2. Sunvozertinib is highly permeable with reasonable bioavailability in both rat and dog. At least 2-fold cellular selectivity between cells expressing major EGFR exon20ins subtypes and wild type EGFR was observed. This is probably due to different binding affinity of sunvozertinib against mutants and wild-type EGFR protein in cytoplasmic environment with presence of variable and high concentrations of ATP. The synthesis of sunvozertinib was described in Supplementary Methods and the 1H and 13C nuclear magnetic resonance spectra of sunvozertinib were illustrated in Supplementary Figure S1.
Figure 1. Modeling of sunvozertinib with EGFR exon20ins 770_NPG (PDB code: 4LRM).

Key interactions of sunvozertinib with EGFR proteins include (i) bidentate interactions of aminopyrimidine with hinge (Met796); (ii) acrylamide group forms irreversible covalent bond with Cys797; (iii) 2-hydroxypropan-2-yl group occupy space next to c-helix; (iv) polar interaction of dimethylaminopyrrolidine with solvent channel residues. Green: carbon; Purple: nitrogen; Red: oxygen; Dark green: chloride; Light blue: fluorine. Colors on protein surface represent the ATP binding pocket and are for clarity only.
Sunvozertinib is an EGFR exon20ins inhibitor showing good pharmacokinetic (PK)/pharmacodynamic (PD) correlation in xenograft models and weak activity against wild-type EGFR
To test the enzymatic activity of sunvozertinib against EGFR exon20ins mutations, we utilized the commerically available recombinant kinase domain of EGFR exon20ins NPG enzyme to establish the enzymatic assay. Sunvozertnib showed potent inhibition with IC50 of 2.1 nM against EGFR exon20ins NPG and slightly weaker inhibition against wild-type EGFR with IC50 of 2.4 nM in an enzymatic assay performed with compound in pre-incubation condition (Supplementary Figure S2A).
To explore a broader kinome selectivity profile, sunvozertinib was evaluated by in vitro kinase assay of 117 recombinant human kinases performed without compound pre-incubation condition (Supplementary Figure S2B). At 1 μM, sunvozertinib inhibited 15 of 117 kinases by > 50%, including EGFR, HER2, HER4, and EGFR resistance mutation (Supplementary Table S3). Dose-response curves were established in these kinases whose activity were inhibited by > 50% in the single-point screening to determine IC50 (Supplementary Table S4). Of these 15 kinases, only one kinase (EGFR T790M mutation) was inhibited with IC50 < 150 nM and two additional kinases (BTK, wild-type EGFR) were inhibited with IC50s < 250 nM. The IC50 of EGFR T790M is about 10-fold potent than that of wild-type EGFR. Overall, sunvozertinib only displayed potent kinase inhibitory activity against the mutant EGFR, with limited off-target activity against the rest of the kinome.
To test cellular activity of sunvozertinib, 14 different subtypes of EGFR exon20ins were engineered into Ba/F3 cell lines. In these cell lines, sunvozertinib showed potent activity in downregulating pEGFR with IC50 of 6 nM to 40 nM (Figure 2A). In other cell lines expressing EGFR sensitizing mutations, T790M resistant mutations and uncommon mutations, sunvozertinib more potently downregulated pEGFR, with IC50 ranging from 1.1 nM to 12 nM (Figure 2A). The effect of sunvozertinib on wild-type EGFR was assessed by measuring pEGFR expression in A431 cell line that over-expressed wild-type EGFR, which was used by other clinically approved EGFR TKIs (14-15). In this cell line, sunvozertinib was less potent in downregulating pEGFR with IC50 of 58 nM (Figure 2A). Thus, sunvozertinib has 1.4~9.6-fold, 4.8-fold and 52-fold selectivity on the EGFR exon20ins, uncommon and sensitizing/resistant mutations, respectively, over wild-type EGFR. Especially it has 2.5~3.1-fold selectivity on the most common subtypes of EGFR exon20ins (insASV, insSVD and insNPH) vs. wild-type EGFR. The anti-proliferation potency IC50s in cells carrying different EGFR mutant variants or wild-type EGFR was shown in Supplementary Figure S2C. Sunvozertinib potently suppresses cell proliferation with GI50 of 6 nM to 88 nM in these cells. The wild-type pEGFR IC50 and anti-proliferation GI50 was 52 nM and 113 nM, in Ba/F3 cell clones, respectively, which was comparable to the data on A431 cell line.
Figure 2. In vitro and in vivo antitumor activity of sunvozertinib in EGFR exon20ins, sensitizing mutation or resistant mutation cell lines and animal models.

A. The cellular activity of sunvozertinib on EGFR exon20ins, sensitizing mutation or resistant mutation vs wild-type EGFR, shown as pEGFR IC50. Cell lines carrying EGFR exon20ins were treated with sunvozertinib at a series of concentrations for 4 hours, and then pEGFR (Tyr1068) was measured with MSD SECTOR® Imager. In A431 cell line carrying wild-type EGFR, after compound treatment for 4 hours, cells were stimulated with 100 ng/ml of recombinant human EGF for 10 minutes before lysis. The potency in each cell line was the average values from 3 biologically independent experiments. Data were presented as mean±standard error of the mean. One-way ANOVA test was used for comparison with wild-type EGFR. **P < 0.01, ****P < 0.0001. B. Antitumor activity of sunvozertinib in PDX model LU0387 carrying EGFR exon20ins insNPH. C. Antitumor activity of sunvozertinib in PDX model LU3075 carrying EGFR exon20ins insDNP. D. Antitumor activity of sunvozertinib in A431 xenograft model expressing wild-type EGFR. Tumor volume in different treatment group at the end point was performed by two-way ANOVA. * p < 0.05, *** P < 0.001, **** P < 0.0001. bid: twice daily. E. PK/PD relationship of sunvozertinib in PDX LU3075 model. The pEGFR (Tyr1068) and pERK (Thr202/Tyr204) expression in tumor tissues was detected by immunohistochemistry and normalized to the vehicle control group. Each time point had tumor tissues from 3 mice to detect pEGFR or pERK signal, except time point of 24 hours in 50 mg/kg group, which included only one mouse due to complete remission of tumor nodules in some mice. pEGFR: phosphorylated EGFR; pERK: phosphorylated ERK. DZD9008 = sunvozertinib.
In EGFR exon20ins patient derived xenograft (PDX) model LU0387 (EGFR exon20ins 773_NPH) or LU3075 (EGFR exon20ins 772_DNP), oral administration of sunvozertinib demonstrated profound antitumor efficacy in a dose-dependent manner. At the doses of ≥ 25 mg/kg, tumor regression was observed (Figure 2B and 2C). In contrast, no tumor regression was observed at 25 mg/kg in xenograft model carrying wild-type EGFR (Figure 2D), suggesting its selectivity between mutant and wild-type EGFR. The waterfall plots of the anti-tumor activity of sunvozertinib in xenograft models were shown in Supplementary Figure S3A and S3B to visually reflect such difference. In addition, antitumor activity of sunvozertinib was also demonstrated in a transgenic mice model carrying one of the most prevalent EGFR exon20ins mutation subtypes insASV (Supplementary Figure S4A and S4B), which further confirmed its antitumor activity against EGFR exon20ins. Moreover, antitumor activity of sunvozertinib was also assessed in a brain metastasis model carrying T790M mutation. As shown in Supplementary Figure S5, sunvozertinib induced profound tumor regression in luci-H1975 BM models at the tested doses of 25 mg/kg bid or 50 mg/kg bid. The antitumor effect of sunvozertinib was comparable to the effect of osimertinib at its clinically equivalent dose.
At the end of efficacy study, blood samples and tumor tissues were collected to analyze the relationship between drug concentrations and pEGFR or pERK inhibition in tumor tissues. As shown in Figure 2E, increased dose of sunvozertinib led to higher drug concentration in the plasma and more profound pEGFR inhibition. Its plasma concentrations at the dose of ≥ 25 mg/kg covered in vitro pEGFR IC50 of DNP engineered Ba/F3 cells for more than 16 hours. At ≥ 25 mg/kg, sunvozertinib led to more than 50% pEGFR inhibition from 2 hours post dosing and the effect maintained for about 24 hours. In addition, dose-dependent modulation of pERK (Thr202/Tyr204) was also detected in this model. Sunvozertinib at ≥ 25 mg/kg led to more than 70% pERK inhibition, and the effect lasted for ≥ 8 hours. This data suggested good in vivo PK and PD relationship by sunvozertinib.
Phase 1 clinical studies of sunvozertinib in NSCLC patients with EGFR or HER2 mutations
There are two ongoing phase 1 clinical studies for sunvozertinib (WU-KONG1, ClinicalTrials.gov identifier: NCT03974022 and WU-KONG2, Chinadrugtrial identifier: CTR20192097). WU-KONG1 is being conducted in the USA, Australia, South Korea and Taiwan, and WU-KONG2 is being conducted in China. Patients were eligible for the studies if they had locally advanced or metastatic NSCLC harboring EGFR or HER2 mutations (including EGFR exon20 insertion) and relapsed from prior stand of treatment, with radiologically measurable disease. Patients with brain metastasis could be enrolled under stable and asymptomatic condition. As the key study design of these two studies is similar, pooled analysis was performed to assess safety, PK and antitumor efficacy.
Between 9 July 2019 and 3 April 2021, sunvozertinib was administered to 102 patients with EGFR or HER2 mutant NSCLC, among whom 54 and 48 patients were from WU-KONG1 and WU-KONG2 studies, respectively (Figure 3). Forty-three, 54 and 5 patients were enrolled into dose escalation, expansion or food effect cohorts, respectively. Sixty-two out of 102 patients (60.8%) carried EGFR exon20ins. As of 3 April 2021, enrollment of dose escalation cohorts has been completed, food effect and dose expansion cohorts are still open. In the dose escalation cohorts, five dose levels of sunvozertinib were explored, 50, 100, 200, 300 and 400 mg once daily. Based on safety, tolerability, PK and efficacy signals in dose escalation cohorts, three dose levels (200 mg, 300 mg and 400 mg) were selected for expansion. At the data cut-off date, 6, 28 and 3 patients in dose escalation, expansion or food effect cohorts, respectively, are still receiving sunvozertinib treatment.
Figure 3. Trial profiles.
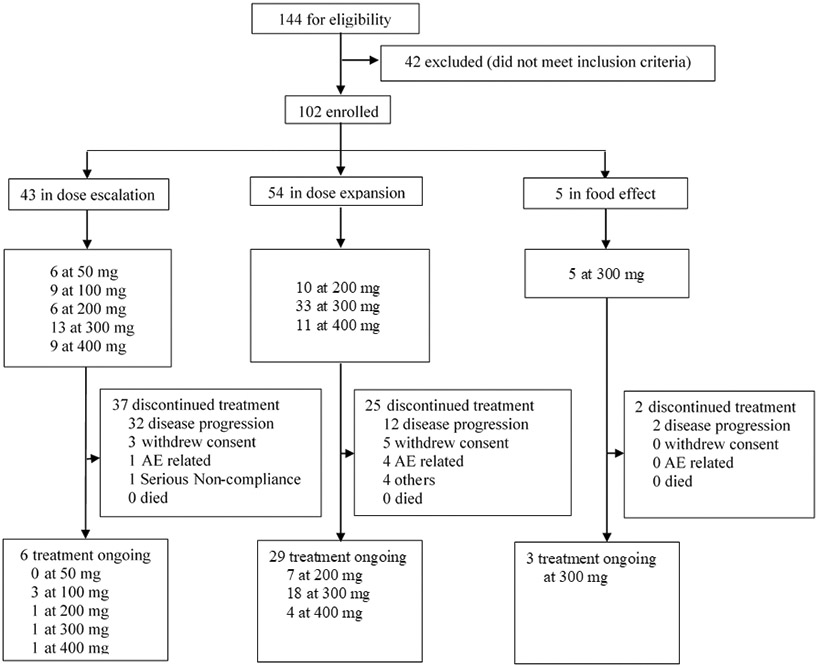
Pooled summary of WU-KONG1 and WU-KONG2 studies. Data cut-off date: 3 April 2021. DZD9008 was dosed once daily.
Patient demographics were shown in Table 1. Median age was 59 years, 57 of 102 patients (55.9%) were women and 101 patients (99%) had ECOG score ≤ 1. The median lines of prior therapy were 3 (1 – 10), and 93 patients (91.2%) had received at least one line of prior chemotherapy. Mutation types of the 102 patients include EGFR exon20ins (61%, 62/102), EGFR sensitizing mutation (4%, 4/102), EGFR T790M (1%, 1/102), EGFR sensitizing/T790M double mutation (6%, 6/102), EGFR uncommon point mutation (1%, 1/102) and HER2 exon20ins (28%, 28/102).
Table 1.
Patient demographics and baseline characteristics
| Characteristics | 50 mg (n = 6) |
100 mg (n = 9) |
200 mg (n = 16) |
300 mg (n = 51) |
400 mg (n = 20) |
Total (n = 102) |
|---|---|---|---|---|---|---|
| Age | ||||||
| Median | 48.5 | 60.0 | 60.5 | 62.0 | 53.5 | 59.0 |
| Min, Max | 36, 72 | 48, 83 | 36, 83 | 32, 82 | 47, 85 | 32, 85 |
| Gender, n (%) | ||||||
| Female | 5 (83.3) | 5 (55.6) | 11 (68.8) | 26 (51.0) | 10 (50.0) | 57 (55.9) |
| Male | 1 (16.7) | 4 (44.4) | 5 (31.3) | 25 (49.0) | 10 (50.0) | 45 (44.1) |
| Race, n (%) | ||||||
| Asian | 5 (83.3) | 8 (88.9) | 15 (93.8) | 40 (78.4) | 17 (85.0) | 85 (83.3) |
| White | 1 (16.7) | 1 (11.1) | 1 (6.3) | 11 (21.6) | 3 (15.0) | 17 (16.7) |
| Baseline ECOG, n (%) | ||||||
| 0 | 5 (83.3) | 2 (22.2) | 11 (68.8) | 15 (29.4) | 5 (25.0) | 38 (37.3) |
| 1 | 1 (16.7) | 6 (66.7) | 5 (31.3) | 36 (70.6) | 15 (75.0) | 63 (61.8) |
| 2 | 0 (0.0) | 1 (11.1) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (1.0) |
| Baseline brain metastasis, n (%) | ||||||
| Yes | 2 (33.3) | 4 (44.4) | 4 (25.0) | 25 (49.0) | 9 (45.0) | 44 (43.1) |
| No | 4 (66.7) | 5 (55.6) | 12 (75.0) | 26 (51.0) | 11 (55.0) | 58 (56.9) |
| Number of prior cancer therapy | ||||||
| Median | 3.0 | 3.0 | 1.5 | 2.0 | 2.5 | 3.0 |
| Min, Max | 1, 5 | 2, 6 | 1, 6 | 1, 10 | 1, 8 | 1, 10 |
| Mutation Types, n (%) | ||||||
| EGFR exon20ins | 3 (50.0) | 2 (22.2) | 11 (68.8) | 36 (70.6) | 10 (50.0) | 62 (60.8) |
| EGFR sensitizing mutation | 0 (0.0) | 1 (11.1) | 1 (6.3) | 0 (0.0) | 2 (10.0) | 4 (3.9) |
| EGFR T790M | 0 (0.0) | 0 (0.0) | 1 (6.3) | 0 (0.0) | 0 (0.0) | 1 (1.0) |
| EGFR double mutation | 2 (33.3) | 2 (22.2) | 0 (0.0) | 2 (3.9) | 0 (0.0) | 6 (5.9) |
| HER2 exon20ins | 1 (16.7) | 4 (44.4) | 3 (18.8) | 12 (23.5) | 8 (40.0) | 28 (27.5) |
| EGFR uncommon mutation | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (2.0) | 0 (0.0) | 1 (1.0) |
Pooled analysis from WU-KONG1 and WU-KONG2 studies. Data cut-off date: 3 April 2021. ECOG: Eastern Cooperative Oncology Group
In the dose escalation cohorts, sunvozertinib was tolerated up to 400 mg. Two patients experienced dose limiting toxicities (DLTs), including one from 300 mg cohort who experienced grade 3 diarrhea and later respiratory distress syndrome, and another one from 400 mg cohort who experienced grade 3 cardiac arrythmia. Across all dose levels, 34 (33.3%) experienced ≥ grade 3 drug-related AEs assessed by investigators. Twenty-four (23.5%), 16 (15.7%), and 6 (5.9%) had dose interruption, reduction and discontinuation due to drug-related TEAEs (Table 2). The most common TEAEs (> 10%) were listed in Supplementary Table S5. All grade diarrhea and rash occurred in 53.9% (55/102) and 40.2% (41/102) patients, respectively, while incidence of ≥ grade 3 diarrhea was only 4.9% (5/102), and no patients experienced ≥ grade 3 rash. Based on safety and tolerability data in dose escalation cohorts, 400 mg was defined as maximum tolerated dose (MTD), and 200 mg to 400 mg were selected for dose expansion.
Table 2.
Summary of safety profiles of sunvozertinib
| AE category | 50 mg (n = 6) n (%) |
100 mg (n = 9) n (%) |
200 mg (n = 16) n (%) |
300 mg (n = 51) n (%) |
400 mg (n = 20) n (%) |
All (n = 102) n (%) |
|---|---|---|---|---|---|---|
| Patients with Any TEAE | 6 (100.0) | 9 (100.0) | 16 (100.0) | 51 (100.0) | 20 (100.0) | 102 (100.0) |
| Patients with Any TEAE with CTCAE Grade ≥ 3 | 2 (33.3) | 2 (22.2) | 2 (12.5) | 20 (39.2) | 14 (70.0) | 40 (39.2) |
| Patients with Any Drug-related AE | 6 (100.0) | 8 (88.9) | 16 (100.0) | 49 (96.1) | 20 (100.0) | 99 (97.1) |
| Patients with Any Drug-related AE with CTCAE Grade ≥ 3 | 1 (16.7) | 1 (11.1) | 1 (6.3) | 17 (33.3) | 14 (70.0) | 34 (33.3) |
| Patients with Any Treatment-emergent SAE | 0 (0.0) | 2 (22.2) | 3 (18.8) | 14 (27.5) | 7 (35.0) | 26 (25.5) |
| Patients with Any Drug-related SAE | 0 (0.0) | 0 (0.0) | 1 (6.3) | 9 (17.6) | 6 (30.0) | 16 (15.7) |
| Patients with Any TEAE Leading to Death | 0 (0.0) | 0 (0.0) | 0 (0.0) | 2 (3.9) | 1 (5.0) | 3 (2.9) |
| Patients with Any Drug-related AE Leading to Death | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (2.0) | 0 (0.0) | 1 (1.0) |
| Patients with Any TEAE Leading to Treatment Discontinuation | 0 (0.0) | 1 (11.1) | 0 (0.0) | 4 (7.8) | 2 (10.0) | 7 (6.9) |
| Patients with Any Drug-related AE Leading to Treatment Discontinuation | 0 (0.0) | 0 (0.0) | 0 (0.0) | 4 (7.8) | 2 (10.0) | 6 (5.9) |
| Patients with Any TEAE Leading to Dose Reduction | 0 (0.0) | 0 (0.0) | 0 (0.0) | 6 (11.8) | 10 (50.0) | 16 (15.7) |
| Patients with Any Drug-related AE Leading to Dose Reduction | 0 (0.0) | 0 (0.0) | 0 (0.0) | 6 (11.8) | 10 (50.0) | 16 (15.7) |
| Patients with Any TEAE Leading to Drug Interruption | 0 (0.0) | 2 (22.2) | 2 (12.5) | 16 (31.4) | 8 (40.0) | 28 (27.5) |
| Patients with Any Drug-related AE Leading to Drug Interruption | 0 (0.0) | 1 (11.1) | 1 (6.3) | 14 (27.5) | 8 (40.0) | 24 (23.5) |
Pooled analysis of WU-KONG1 and WU-KONG2 studies. Data cut-off date: 3 April 2021. Causality assessed by investigators. AE: adverse event; CTCAE: Common Terminology Criteria for Adverse Events version 5.0; SAE: serious adverse event.
Plasma samples were obtained from patients in dose escalation (n = 40) and expansion cohorts (n = 24) for PK analysis. Across all dose levels (50 mg - 400 mg), sunvozertinib was absorbed with a median tmax of 4 to 7 hours after single oral dosing and tss,max of 5 to 6 hours after repeat dosing. With once daily dosing, plasma concentrations of sunvozertinib appears to approach steady state within 15 days. At the steady state, sunvozertinib exhibited approximately dose-proportional increases in exposures (Css,max and AUCss) across the dose range investigated. Comparing repeat dosing with single dosing, there was around 3-fold accumulation of drug exposure, probably due to its half-life of around 50 hours. At the doses of ≥ 200 mg, the geometric mean of Css,max and Css,min (Supplementary Figure S6, Supplementary Table S6) were above pEGFR IC50 (Figure 2A) of majority of EGFR exon20ins subtypes.
In 56 patients with EGFR exon20ins who were evaluable for efficacy assessment, partial response (PR) was observed at the doses of ≥ 100 mg. Across all dose levels, the best objective response rate (ORR) was 41.1%, and confirmed ORR was 37.5%. At the recommended phase 2 doses (PR2Ds), 200 mg and 300 mg, the ORRs were 45.5% (confirmed, 45.5%) and 48.4% (confirmed, 41.9%), respectively (Table 3). In dose expansion cohorts, the best and confirmed ORRs were 47.4% and 44.7%, respectively, across 200 mg to 400 mg. Antitumor efficacy was observed in different EGFR exon20ins subtypes (Supplementary Table S7). PR was observed in patients with baseline brain metastasis or those with prior amivantamab treatment (Figure 4A). By the data cut-off, with a median follow-up duration of 4.2 months, median duration of response was > 3.5 months and had not been reached, the longest duration of response was > 8 months, and 15 out of 23 patients (65.2%) were ongoing treatment and still responding (Figure 4B). With a median follow-up duration of 5.6 months, the median progression free survival was > 4 months and had not been reached.
Table 3.
Antitumor activity of sunvozertinib in patients with previously treated EGFR exon20ins NSCLC
| 50 mg (n = 3) |
100 mg (n = 2) |
200 mg (n = 11) |
300 mg (n = 31) |
400 mg (n = 9) |
Total (n = 56) |
|
|---|---|---|---|---|---|---|
| Best response, n (%) | ||||||
| Partial response (unconfirmed) | 0 (0.0) | 1 (50.0) | 5 (45.5) | 15 (48.4) | 2 (22.2) | 23 (41.1) |
| Partial response (confirmed) | 0 (0.0) | 1 (50.0) | 5 (45.5) | 13 (41.9) | 2 (22.2) | 21 (37.5) |
| Stable disease | 2 (66.7) | 1 (50.0) | 4 (36.4) | 15 (48.4) | 5 (55.5) | 27 (48.2) |
| Progressive disease | 1 (33.3) | 0 (0.0) | 2 (18.2) | 3 (9.7) | 2 (22.2) | 8 (14.3) |
| Confirmed ORR, n (%) | 0 (0.0) | 1 (50.0) | 5 (45.5) | 13 (41.9) | 2 (22.2) | 21 (37.5) |
| Disease control rate, n (%) | 2 (66.7) | 2 (100.0) | 9 (81.8) | 28 (90.3) | 7 (77.7) | 48 (85.7) |
Pooled data from WU-KONG1 and WU-KONG2 studies. Data cut-off date: 3 April 2021. Tumor response was assessed by investigators. ORR: objective response rate.
Figure 4. Clinical activity of sunvozertinib in EGFR exon20ins NSCLC patients with post-baseline target lesion assessments.

A. Best percentage change from baseline in target lesions by dose level, molecular subtype, prior amivantamab or poziotinib treatment status and baseline brain metastasis. B. Plot showing percentage change from baseline in target lesion by time on treatment and dose level. Pooled analysis of WU-KONG1 and WU-KONG2 studies was performed. Data cut-off date: 3 April 2021. Tumor response was assessed by investigators according to RECIST1.1 (Response Evaluation Criteria In Solid Tumors, version 1.1). BM: brain metastasis; PR: partial response; SD: stable disease; PD: progressive disease. *: confirmed response. EGFR exon20ins subtypes were confirmed by next-generation sequencing using tumor tissue or/and plasma ctDNA. DZD9008 = sunvozertinib. JNJ-61186372 = amivantamab.
Note: Among the 56 patients, a total of 45 subjects had tumor tissue and/or plasma samples tested by central laboratory using NGS (next generation of sequencing), 41 of 45 were confirmed as EGFR Exon 20 insertion positive, the overall concordance rate was 91%. However, 31 subjects had only tumor tissue tested by central laboratory, the concordance rate between local and central laboratory testing was 97% (30/31).
In addition, preliminary antitumor activity of sunvozertinib was also observed in patients with EGFR sensitizing mutation, EGFR sensitizing/T790M double mutation and HER2 exon20ins (Supplementary Table S8).
Discussion
The discovery and development of EGFR TKI, such as gefitinib and osimertinib, has substantially improved clinical outcome of EGFRm NSCLC. However, due to the unique structural features of EGFR exon20ins proteins, the current approved EGFR TKIs for sensitizing and T790M mutations were not active against them, and thus a potent and selective inhibitor is urgently needed. Exon20ins mutations occur mostly along a loop between αC and β4 on the kinase domain, with a noticeable exception occurring within the αC-helix (763_764insFQEA) (16). Currently, only one exon20ins mutant (770_771insNPG) crystal protein structure is available in the public domain (PDB codes: 4LRM, 7LGS) (15, 17). Due to the scarce of crystallographic data on different exon20ins mutants, computational modeling was used to study the conformation of various exon20ins mutants and tried to identify their differences from the wild-type protein (18-20). Most studies concluded that exon20ins mutations preferentially stabilize the αC-helix in the catalytic active conformation. Apart from the αC-helix orientation, it is also speculated that different exon20ins mutants may also have an indirect effect on the conformation of the P-loop, hinge, or other β-sheet structures around the ATP binding pocket, making the ATP binding pocket smaller for the mutants. This rationale was used to explain the potency of poziotinib against EGFR exon20ins by Jacqulyne et al (13). However, the modeling studies could not reveal any structural differentiation within the ATP binding pockets between the EGFR exon20ins mutants and the wild-type protein, partly because all amino acids sequence within the ATP binding pockets are the same and majority of the exon20ins mutations occur outside the ATP binding pocket. Nevertheless, the differences between the mutants and wild-type ATP binding pockets can have subtle effect on ATP and inhibitors binding. Similar to the advantage previously been taken for the development of first-generation EGFR TKIs for exon19 deletion and L858R substitution mutants, the decrease in ATP binding Km with the exon19del and L858R mutants compared with that of the wild type led to the successful identification of potent and selective EGFR inhibitors, such as gefitinib. Unfortunately, this ATP Km difference was decreased when T790M resistant mutation occurred after gefitinib treatment. The development of third-generation irreversible inhibitors such as osimertinib (21), alleviated this detrimental effect of high ATP concentration in cells. Yasuda et al. has shown that exon20ins mutant D770_N771insNPG having ATP Km = 36.8 μM that is in between L858R (ATP Km = 68.5 μM) and wild type (ATP Km = 4.98 μM) (17). The lack of clear structural and biophysical data for most of the exon20ins mutants makes designing pan-exon20ins inhibitors with wild-type protein selectivity challenging. Recently, Gonzalvez et al have reported the design of mobocertinib (TAK-788) targeting EGFR exon20ins (15). The research group focused on exploiting the unoccupied space around the gatekeeper Thr790 and osimertinib. The isopropyl ester substitution on C-5 of the pyrimidine core of osimertinib was found to be optimal. However, it is unclear how a larger molecule mobocertinib interacts with predicted smaller ATP binding pockets for EGFR exon20ins mutants. It is also interesting that mobocertinib can maintain its potency against EGFR T790M mutation. The added isopropyl ester on C-5 on the pyrimidine hinge moiety may interact unfavorably with the larger methionine. Furthermore, the aforementioned unoccupied pocket lined in the proximity with the amino acids sequence on the far-loop between αC-β4, we envisioned insertion mutations on exon20, especially on the far-loop, may have a profound effect on that region. Nevertheless, the data from mobocertinib collectively suggested that inhibitors with modified substitutions on the pyrimidine hinge binding core can gain potency against EGFR exon20ins with a certain level of wild-type selectivity. For a more thorough understanding on how EGFR exon20ins and wild-type proteins interact with inhibitors with different scaffolds, the availability of more crystallography data is essential.
In our EGFR exon20ins program, in contrast to mobocertinib, we decided to keep the C-5 position (which is near Thr790) on the pyrimidine open, and directly replace the rotationally less flexible methylindole on osimertinib with a more flexible anilinophenyl moiety on C-4 of pyrimidine, hoping for a flexible accommodation of the inhibitor among different mutants with slightly variable sizes of the ATP binding pocket. Together with the suggestion from modeling studies that the c-helix in EGFR exon20ins mutants are more stabilized, we focused on designing inhibitors with substituents on the anilinophenyl moiety that may provide specific interactions with the adjacent c-helix and under the P-loop. After several design-make-test-analyze cycles, our discovery effort cumulated the identification of the clinical candidate sunvozertinib by optimizing the head-group on C-4 of the pyrimidine hinge binder and the solvent-exposed amino side-chain for both on-target activities against EGFR mutations with desirable drug-like properties (Supplementary Table S2).
The two phase 1 studies aimed to address the safety, tolerability, PK and antitumor efficacy of sunvozertinib. The starting dose of 50 mg and dose escalation scheme were defined based on nonclinical toxicity studies and PK/PD modeling. Sunvozertinib was generally tolerated up to 400 mg, with 1 out of 13 subjects in 300 mg group and 1 out of 9 subjects in 400 mg group experienced dose limiting toxicity. There was a trend showing increased incidence and severity of adverse events (AEs) with dose increase. Given that 50% of subjects in 400 mg group need a dose reduction, this dose was not selected for future development. The most common drug-related AEs were diarrhea and skin rash, which were consistent with the mechanism of sunvozertinib through inhibiting EGFR pathway. Comparing with other approved EGFR TKIs, such as afatinib (3) and dacomitinib (22), and EGFR exon20ins inhibitor, such as mobocertinib (11, 12), sunvozertinib showed a more favorable safety profile, with a few patients of 200 mg and 300 mg experiencing ≥ grade 3 drug-related TEAEs which required dose reduction or discontinuation. These data aligned with the original design on the selectivity of this compound.
From 50 mg to 400 mg, sunvozertinib showed a dose-proportional PK profile in human, suggesting its good oral absorption. Due to its long half-life, sunvozertinib exhibited around 3-fold accumulation of drug exposure after repeat dosing versus single dosing. Similar as PK profile of osimertinib (23), sunvozertinib exhibited a flat PK curve with small fluctuation between Cmax and Cmin at steady state, which provides stable and continuous coverage of the target. This PK pattern could potentially minimize drug related AEs. These data suggest that sunvozertinib has a favorable PK profile in human as an oral agent and at ≥ 100 mg dose levels, free plasma concentrations of sunvozertinib were able to cover pEGFR IC50 of some subtypes of EGFR exon20ins, and the doses of ≥ 200 mg could cover majority of subtypes (Supplementary Figure S6). Indeed, at the doses of ≥ 100 mg, antitumor efficacy was observed in the phase 1 studies.
Noteworthily, the antitumor activity of sunvozertinib was observed in heavily pre-treated patients, with median 3 lines of prior systemic therapy. In addition, in 4 prior amivantamab treated patients, two showed PR. With these data, sunvozertinib was granted by US FDA Breakthrough Therapy designation for the treatment of patients with locally advanced or metastatic NSCLC with EGFR exon20ins whose disease has progressed on or after platinum-based chemotherapy. Future study is warranted to confirm antitumor activity of sunvozertinib in a larger cohort of patients with prior amivantamab treatment or prior EGFR exon20ins tyrosine kinase inhibitor treatment. Moreover, it is encouraging to see antitumor activity of sunvozertininb in patients with baseline brain metastasis, while as the intracranial lesions were not assessed as target lesion in this study, future studies are warranted to confirm this finding.
In conclusion, both preclinical and phase 1 clinical data suggest that sunvozertinib has a good safety/tolerability, PK and efficacy profile, which warrants its further clinical development for treating NSCLC with EGFR exon20ins.
Materials and Methods
Cell lines
A431 human epidermoid carcinoma cell line and Ba/F3 murine proB cell line were obtained from ATCC. Human cell identity was confirmed by short-tandem repeat (STR) analysis (GenePrint® 10 System, Promega). A431 cells were cultured in DMEM (Gibco) with 10% FBS. Ba/F3 cells were cultured in RPMI-1640 with 10% FBS and 10% conditioned medium of WEHI3B (ATCC) as a source of IL-3. Ba/F3 cells carrying EGFR exon20ins 773_NPH was purchased from Crown Bioscience, Inc. (Beijing, China). All cells were maintained and propagated as monolayer cultures in a humidified incubator with 5% CO2 at 37°C.
Expression vectors and transfection
The cDNA sequence of wild-type EGFR gene was obtained from GenBank (accession number NM005228.3). Full-length cDNAs of different types of human EGFR exon20ins were generated at Shanghai Sunbio Biotechnology Co., Ltd (Shanghai, China) and confirmed by Sanger sequencing. The cDNAs were then subcloned into pMT143 lentiviral vector (Shanghai Sunbio Biotechnology Co., Ltd). The lentivirus was packaged in 293T/17 cells (ATCC Cat# CRL-11268, RRID: CVCL_1926) by transfection of lentiviral constructs and packaging mix (Shanghai Sunbio Biotechnology Co., Ltd). Ba/F3 cells were infected by lentivirus with 5 μg/ml polybrene (Sigma-Aldrich, St. Louis, MO, USA), selected in 2 μg/mL puromycin (Invitrogen, Carlsbad, CA, USA) as single cell clones, and maintained in 1 μg/mL puromycin with IL-3 depletion. Expression of exogenous EGFR in Ba/F3 cells was confirmed by Sanger sequencing at mRNA level and western blot at protein level.
pEGFR MSD assay
All cells were seeded into 96-well plate in their corresponding culture medium containing 1% FBS. The seeding density for Ba/F3 cells and A431 was 50,000 cells/well and 20,000 cells/well, respectively. After overnight incubation, all cells were treated with compounds at a series of concentrations for 4 hours and then were lysis directly except A431 or wild-type EGFR engineered Ba/F3 cell clone was stimulated with 100 ng/ml of recombinant human EGF for 10 minutes before lysis. Phosphorylated EGFR (Tyr1068) and total EGFR levels in the cell lysates were measured by an electrochemiluminescent method (MULTI-SPOT®96 4-Spot HB Prototype EGFR Triplex ANALYTES: pEGFR (Tyr1068), pEGFR (Tyr1173), Total EGFR; MESO SCALE DISCOVERY, Cat. N45ZB-1) with MSD SECTOR® Imager, which outputs the ratio of pEGFR/total EGFR for each well. The percentage of inhibition was calculated as: % Inhibition = 100 × [1 - (ratio of sample well - ratio of Min control well)/ (ratio of Max control well - ratio of Min control well)]. The concentration of compound producing 50% inhibition of EGFR phosphorylation was calculated in best-fit curves with GraphPad Prism (RRID:SCR_002798, GraphPad Software Inc).
Cell proliferation assay
Ba/F3 cells expressing EGFR mutations were seeded in 384-well plates at 1,250 cells/well in RPMI1640 medium containing 10% FBS. A431 cells were seeded in 384-well plates at 1,000 cells/well in DMEM medium containing 10% FBS. At the same time a day 0 plate was prepared with duplicate rows of each cell line. After overnight incubation, the assay plates were dosed with compounds at a series of concentrations. Alongside dosing the assay plates, the day 0 plate was processed using MTS or CellTiter–Glo assay to measure the number of viable cells (G0). MTS assay is a colorimetric method for determining the number of viable cells using Spark plate reader (Tecan). The assay plates were further incubated for 72 hours and the number of viable cells (G3) was measured by MTS or CellTiter-Glo assay. The percentage of proliferation was calculated as: % Proliferation = 100 × (G3 value of sample well - G0 value)/(G3 value of DMSO control - G0 value). The concentration of compound producing 50% proliferation inhibition (GI50) was further calculated in best-fit curves using XLFit software.
Generation of PDX xenograft models in mice and compound treatment
All studies involving animals were conducted according to the guidelines approved by Institutional Animal Care and Use Committees (IACUC) and the standard and local regulatory requirements of Dizal Pharmaceuticals or Crown Bioscience Inc. Six to eight week-old specific-pathogen-free immunocompromised female nude mice were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd or Nanjing Biomedical Research Institute of Nanjing University.
PDX models (LU0387 and LU3075) were established subcutaneously in immunocompromised female nude mice by implanting patients’ surgical tumor tissues and serially re-engrafting in vivo. When tumor nodules reach 500-600 mm3, tumor tissues were sliced into 3 mm × 3 mm × 3 mm fragments and implanted subcutaneously in female immune-compromised nude mice for compound testing.
Tumor nodules were measured in two dimensions with caliper and the tumor volume was calculated using the following formula: tumor volume = (length × width2) ×0.52. When the mean tumor volume reached 150 to 250 mm3, tumor-bearing mice were randomized into different treatment groups. Mice were then treated orally with either vehicle or drug twice daily from the day post randomization. The tumor volume and body weight of the mice were measured twice weekly, and the raw data were recorded according to their study number and measurement date in the assigned lab notebook. Tumor growth inhibition from start of treatment was assessed by comparison of the mean change in tumor volume between the control and treatment groups and presented as tumor growth inhibition. The arithmetic mean tumor volume was used for efficacy calculation. The calculation was based on the arithmetic mean of relative tumor volume (RTV) in each group. RTV was calculated by dividing the tumor volume on the treatment day with the initial tumor volume. The efficacy of tumor growth inhibition on specific day, for each treated group, was calculated by formula: Inhibition % = (CG-TG) × 100 / (CG-1), among which “CG” means the arithmetic mean of RTV of the control group, and “TG” means the arithmetic mean of RTV of the treated group.
Determination of sunvozertinib plasma concentrations for PK/PD study in animal model
Blood samples from the LU3075 PDX mice model were collected at 0.5, 1, 2, 4, 8, 24 hours on day 28 post the last dosing of sunvozertinib at 12.5, 25 and 50 mg/kg twice daily, put into different K2-EDTA coagulated tubes, and then plasma was harvested post centrifugation. All plasma samples were stored at around −80°C prior to LC/MS/MS analysis.
Standards were prepared by spiking blank plasma with sunvozertinib covering 1.2 to 1200 ng/ml. Plasma samples were precipitated by adding 4-fold volume of acetonitrile containing internal standard [tolbutamide 30 ng/ml]. After 2 minutes vortex and 10 minutes centrifugation at 4000 rpm, supernatant was analyzed by LC/MS/MS (QTRAP5500, Applied Biosystems, Foster City). Two sets of standard curves were run at the beginning and end of each batch from plasma sample analysis along with two sets of QCs at different concentrations.
Immunohistochemical (IHC) staining for pEGFR expression in tumor tissue
Tumor tissues from the LU3075 PDX mice model were collected at 2, 8, 24 hours on day 28 post last dosing of sunvozertinib. Samples were harvested following formalin fixation and paraffin embedding (FFPE) for further study. IHC was performed on 3 μm FFPE sections using a Lab Vision autostainer (Thermo) for pERK (Thr202/Tyr204) staining, and Ventana discovery XT automation (Roche) for pEGFR (Tyr1068) staining. Monoclonal rabbit anti-pEGFR (Tyr1068) antibody (Cat.2234) and pERK (Thr202/Tyr204) antibody (Cat.4376) were purchased from Cell Signaling Technology. The stained IHC slides were firstly reviewed and interpreted by a qualified pathologist and then quantified by HALO system. “H” score was also performed when the cases were illegible on HALO platform. Statistical analysis was performed by t-test.
Phase 1 clinical studies
WU-KONG1 (ClinicalTrials.gov: NCT03974022) and WU-KONG2 (Chinadrugtrial: CTR20192097)
Study objectives
The primary objectives were to assess safety and tolerability of sunvozertinib, and define its maximum tolerated dose (MTD). Secondary objectives included the pharmacokinetics of sunvozertinib and its antitumor efficacy assessed by investigator according to RECIST1.1.
Study design and patients
WU-KONG1 is a phase 1/2, open-label, multinational study being conducted at 17 centers in the USA, Australia, Taiwan and South Korea. This study included two parts: part A (dose escalation, food effect and expansion cohorts), and part B (dose extension cohorts). This article reported the data from part A dose escalation, food effect and expansion cohorts. Dose escalation and food effect cohorts enrolled pre-treated NSCLC patients with EGFR or HER2 mutation, and dose expansion cohorts only enrolled pre-treated NSCLC patients with EGFR or HER2 exon20ins. Key inclusion criteria included: age 18 years or above; histologically or cytologically confirmed NSCLC with EGFR or HER2 mutation assessed in local laboratories; life expectancy of at least 3 months; an Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1; adequate organ system functions, patients with brain metastasis (BM) can only be enrolled under the condition that BM is stable. For dose expansion cohorts, patients should not have been previously treated with an EGFR exon20ins TKI. Key exclusion criteria included: unsolved ≥ grade 2 adverse events, according to Common Terminology Criteria for Adverse Events [CTCAE] version 5.0, from previous treatment; a medical history of interstitial lung disease; spinal cord compression or leptomeningeal metastasis; QTc > 470 msec.
WU-KONG2 is a phase 1, open-label, multicenter study being conducted at 8 centers in China. This study included two parts: part A dose escalation and part B dose expansion. A similar study design as that of WU-KONG1 dose escalation and expansion cohorts is applied to this study.
All patients provided written informed consent before participating into this study. Before site activation, the study protocol was approved by the institutional review board or ethics committee at every center participating in the study. The study was undertaken in accordance with the Declaration of Helsinki and Good Clinical Practice guidelines, as defined by the International Conference on Harmonization. A Safety Review Committee, of which investigators and study team core members employed by the sponsor, was formed for study monitoring, safety management, and decision making.
Procedures
In dose escalation cohorts, sunvozertinib was administered at 50, 100, 200, 300 and 400 mg once daily, respectively. In food effect cohort, sunvozertinib was given at 300 mg single dose, with cross-over of fed and high fat condition. After PK sampling, patients continued with repeat dosing of sunvozertinib. In dose expansion cohorts, sunvozertinib was administered at 200 or 300 mg once daily. Patients received sunvozertinib treatment until disease progression, intolerable AEs or withdrawal of consent. The investigators assessed safety at every scheduled visit. Categories of AEs were based on terms from the Medical Dictionary for Regulatory Activities (MedDRA), version 24.0. AEs were graded using CTCAE version 5.0, and investigators judged whether AEs were related to sunvozertinib. Tumor assessment was performed by investigators according to RECIST 1.1.
Plasma samples were collected at the scheduled visit and timepoints, after single or repeat dosing, to assess sunvozertinib PK profile. Plasma concentrations of sunvozertinib were determined by validated LC/MS/MS method following protein precipitation using acetonitrile with internal standard. The PK parameters were determined by noncompartmental analysis method.
Archived tumor tissues or/and plasma samples at baseline were collected for retrospective confirmation of EGFR exon20ins mutation status.
Statistical analysis
A Bayesian adaptive design was used to inform the dose escalation and MTD estimation (24). The MTD was defined as the highest dose at which the predicted probability of a dose-limiting toxicity was less than 30%. All patients who received at least one dose of sunvozertinib were included in the safety analysis, and patients with EGFR exon20ins NSCLC who had at least one post-treatment tumor assessment were included in the efficacy analysis.
Supplementary Material
Financial support:
This study was sponsored by Dizal Pharmaceuticals. Some of this work was supported in part, by grants (WX03-02B0105-062000-01) from Wuxi Municipal Bureau on Science and Technology (to Dr. L Zheng) on WU-KONG2 study.
Footnotes
Disclosure of Potential Conflicts of Interest
Mengzhao Wang reports the consultant role for Dizal. James Chih-Hsin Yang reports personal fees and other from Amgen, grants, personal fees and other from AstraZeneca, personal fees and other from Bayer, personal fees and other from Boehringer Ingelheim, personal fees and other from Bristol Myers Squibb, personal fees and other from Daiichi Sankyo, other from Eli Lilly, personal fees and other from Merck KGaA, Darmstadt, Germany, personal fees and other from Merck Sharp & Dohme, personal fees and other from Novartis, personal fees from Ono Pharmaceuticals, personal fees from Pfizer, personal fees and other from Roche/Genentech, personal fees and other from Takeda Oncology, personal fees and other from Yuhan Pharmaceuticals, other from JNJ, other from Puma Technology, other from Gilead, other from GSK, outside the submitted work. Paul L Mitchell reports having consultant role with AstraZeneca, Roche, Pfizer, Novartis, BMS, MSD and Amgen. Dr. Mitchell receives speaker fee from MSD. Jian Fang declares no conflict of interest. D. Ross Camidge receives research funding from Inivata. D. R. Camidge reports having an advisory role with Appolomics, AstraZeneca, Beigen, Medtronic, Mersana, Mirati, Onkure, Roche, Abbvie, Amgen, Anheart, Apollomics, Blueprint, Daiichi-Sankyo, Elevation, Eli Lilly, EMD Serono, Helsinn, Hengrui, Janssen, Kestrel, Mersana, Nuvalent, Puma, Ribon, Roche/Genentech, Sanofi, Seattle Genetics, Takeda, Turning Point, Amgen, Anchiarno, Bio-Thera (DSMB), BMS, GSK, Helssin, Janssen, Onkure, Mersana, Pfizer, Qilu, Roche, Sanofi, Seattle Genetics, Takeda. D. R. Camidge reports have clinical studies with Abbvie, AstraZeneca, Blueprint, Dizal, Inhibrx, Karyopharm, Pfizer, Phosplatin, Psioxus, Rain, Roche/Genentech, Seattle Genetics, Takeda, Turning Point, Verastem from 2020-2022. Weiqi Nian declares no conflict of interest. Chao-Hua Chiu receives honoraria from Amgen, AstraZeneca, Boehringer-Ingelheim, Bristol-Myers Squibb, Chugai Pharmaceutical, Eli Lilly, Janssen, Merck KGaA, Merck Sharp & Dohme, Novartis, Ono Pharmaceutical, Pfizer, Roche, and Takeda. Jianying Zhou declares no conflict of interest. Yanqiu Zhao declares no conflict of interest. Wu-Chou Su has consulting and advisory roles for Bayer, Bristol Myers Squibb, Eli Lily and Merck. Tsung-Ying Yang declares no conflict of interest. Viola Zhu has received honoraria from AstraZeneca, Blueprint, Roche-Foundation Medicine, Roche/Genentech, Takeda, Xcovery. Viola W Zhu had stock ownership of TP Therapeutics until May 2020, and currently is an employee of Nuvalent and holds stock ownership. Michael Millward reports grants from Bristol Myers Squibb, grants from Novartis, Roche, AstraZeneca, Takeda, GlaxoSmithKline, Beigene, Eli Lilly, Apollomics, Albion, AkesoBio, AbbVie, C-Stone Pharmaceuticals, Therapim, Five Prime Therapeutics, Dizal, Maxinovel, Amgen, Alpine Immune Sciences, Vivace Therapeutics, Turning Point Therapeutics, personal fees from Bristol Myers Squibb, personal fees from Roche, AstraZeneca, Merck Sharp & Dohme, Takeda, Pfizer, non-financial support from AstraZeneca and Bristol Myers Squibb, outside the submitted work. Yun Fan declares no conflict of interest. Wen-Tsung Huang declares no conflict of interest. Ying Cheng declares no conflict of interest. Liyan Jiang declares no conflict of interest. Daniel Brungs declares no conflict of interests. Lyudmila Bazhenova reports personal fees from ORIC, Turning point therapeutics, Neuvogen, Daichi, BMS, Janssen, Regeneron, Bayer, Takeda, Boehringer Ingleheim, Novartis, Genentech, Merck and Sanofi, grants from Beyondspring outside the submitted work. Chee Khoon Lee has received honoraria from Astrazeneca, Roche, Pfizer, Amgen, Takeda, Yuhan, Boehringer Ingelheim, MSD oncology, Novartis, GSK and Merck KGaA. C.K.L. reports the consulting or advisory role for Astrazeneca, Yuhan, Boehringer Ingelheim, Takeda, Novartis and Amgen. C.K.L. reports receiving the research funding from Astrazeneca, Roche and Merck KGaA. Bo Gao declares no conflict of interest. Yan Xu declares no conflict of interest. Wei-Hsun Hsu has received speaker fees/honoraria from AstraZeneca, Roche, Pfizer, Chugai, Takeda, Janssen, Ono, Norvatis, Boehringer Ingelheim, Guardant Health and ACT genomics outside the submitted work. Li Zheng is an employee and a shareholder of Dizal Pharmaceuticals. Pasi A. Jänne has received consultancy fees from and/or has had an advisory role with Pfizer, Boehringer Ingelheim, AstraZeneca, Merrimack, Chugai Pharma, Roche/Genentech, Loxo Oncology, Mirati Therapeutics, Araxes Pharma, Ignyta, Lilly, Takeda, Novartis, Biocartis, Voronoi Health Analytics, and SFJ Pharmaceuticals Group, Nuvalent, Esai, Accutar Biotech, Allorion Therapeutics and AbbVie; is the co-inventor on a Dana-Farber Cancer Institute–owned patent on EGFR mutations licensed to Labcorp, which P.A.J. received postmarketing royalties from; has stock/ownership in Gatekeeper Pharmaceuticals and Loxo Oncology; and has received research funding from AstraZeneca, Astellas Pharma, Daiichi Sankyo, Lilly, Boehringer Ingelheim, Puma Biotechnology, and Takeda.
Data Availability
The data generated in this study are available within the article and its supplementary data files.
References
- 1.Maemondo M, Inoue A, Kobayashi K, Sugawara S, Oizumi S, Isobe H, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med 2010; 362:2380–8 [DOI] [PubMed] [Google Scholar]
- 2.Rosell R, Carcereny E, Gervais R, Vergnenegre A, Massuti B, Felip E, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol 2012; 13: 239–46 [DOI] [PubMed] [Google Scholar]
- 3.Wu YL, Zhou C, Hu CP, Feng J, Lu S, Huang Y, et al. Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): an open-label, randomised phase 3 trial. Lancet Oncol 2014; 15: 213–22 [DOI] [PubMed] [Google Scholar]
- 4.Moyer JD, Barbacci EG, Iwata KK, Arnold L, Boman B, Cunningham A, et al. Induction of apoptosis and cell cycle arrest by CP-358,774, an inhibitor of epidermal growth factor receptor tyrosine kinase. Cancer Res 1997; 57:4838–48 [PubMed] [Google Scholar]
- 5.Soria JC, Ohe Y, Vansteenkiste J, Reungwetwattana T, Chewaskulyong B, Lee KH, et al. Osimertinib in Untreated EGFR-Mutated Advanced Non-Small-Cell Lung Cancer. N Engl J Med 2018; 378:113–25 [DOI] [PubMed] [Google Scholar]
- 6.Arcila ME, Nafa K, Chaft JE, Rekhtman N, Lau C, Reva BA, et al. EGFR exon 20 insertion mutations in lung adenocarcinomas: prevalence, molecular heterogeneity, and clinicopathologic characteristics. Mol Cancer Ther 2013; 12: 220–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oxnard GR, Lo PC, Nishino M, Dahlberg SE, Lindeman NI, Butaney M, et al. Natural history and molecular characteristics of lung cancers harboring EGFR exon 20 insertions. J Thorac Oncol 2013; 8: 179–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Naidoo J, Sima CS, Rodriguez K, Busby N, Nafa K, Ladanyi M, et al. Epidermal growth factor receptor exon 20 insertions in advanced lung adenocarcinomas: Clinical outcomes and response to erlotinib. Cancer 2015; 121:3212–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang G, Li J, Xu H, Yang Y, Yang L, Xu F, et al. EGFR exon 20 insertion mutations in Chinese advanced non-small cell lung cancer patients: molecular heterogeneity and treatment outcome from nationwide real-world study. Lung Cancer 2020; 145: 186–94 [DOI] [PubMed] [Google Scholar]
- 10.Sabari JK, Shu CA, Park K, Leighl N, Mitchell P, Kim S, et al. Amivantamab in post-platinum EGFR Exon 20 insertion mutant non–small cell lung cancer [abstract OA04.04]. J Thorac Oncol 2021; 16: S108. [Google Scholar]
- 11.Zhou C, Ramalingam S, Li B, Fang J, Kim TM, Kim S, et al. Mobocertinib in NSCLC with EGFR exon 20 insertions: results from EXCLAIM and pooled platinum-pretreated patient populations [abstract OA04.03]. J Thorac Oncol 2021; 16: S108. [Google Scholar]
- 12.Riely GJ, Neal JW, Camidge DR, Spira AI, Piotrowska Z, Costa DB, et al. Activity and Safety of Mobocertinib (TAK-788) in Previously Treated Non–Small Cell Lung Cancer with EGFR Exon 20 Insertion Mutations from a Phase I/II Trial. Cancer Discov 2021; 11:1688–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Robichaux JP, Elamin YY, Tan Z, Carter BW, Zhang S, Liu S, et al. Mechanisms and Clinical Activity of an EGFR and HER2 Exon 20-selective Kinase Inhibitor in Non-small Cell Lung Cancer. Nat Med 2018; 24: 638–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cross DA, Ashton SE, Ghiorghiu S, Eberlein C, Nebhan CA, Spitzler PJ, et al. AZD9291, an Irreversible EGFR TKI, Overcomes T790M-Mediated Resistance to EGFR Inhibitors in Lung Cancer. Cancer Discov 2014; 4: 1046–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gonzalvez F, Vincent S, Baker TE, Gould AE, Li S, Wardwell SD, et al. Mobocertinib (TAK-788): A Targeted Inhibitor of EGFR Exon 20 Insertion Mutants in Non-Small Cell Lung Cancer. Cancer Discov 2021; 11:1672–87 [DOI] [PubMed] [Google Scholar]
- 16.Friedlaender A, Subbiah V, Russo A, Banna GL, Malapelle U, Rolfo C, et al. EGFR and HER2 exon 20 insertions in solid tumours: from biology to treatment. Nat Rev Clin Oncol 2022; 19 (1): 51–69. [DOI] [PubMed] [Google Scholar]
- 17.Yasuda H, Park E, Yun CH, Sng NJ, Lucena-Araujo AR, Yeo WL, et al. Structural, Biochemical, and Clinical Characterization of Epidermal Growth Factor Receptor (EGFR) Exon 20 Insertion Mutations in Lung Cancer. Sci Transl Med 2013; 5: 216ra177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ikemura S, Yasuda H, Matsumoto S, Kamada M, Hamamoto J, Masuzawa K, et al. Molecular dynamics simulation-guided drug sensitivity prediction for lung cancer with rare EGFR mutations. Proc Natl Acad Sci USA 2019; 116:10025–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koivu MKA, Chakroborty D, Tamirat MZ, Johnson MS, Kurppa KJ, Elenius K. Structural Basis for the Functional Changes by EGFR Exon 20 Insertion Mutations. Cancers 2021, 13:1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Di Gaetano S, Pirone L, Galdadas I, Traboni S, Iadonisi A, Pedone E, et al. Structural basis of the effect of activating mutations on the EGF receptor eLife 2021; 10: e65824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Finlay MR, Anderton M, Ashton S, Ballard P, Bethel PA, Box MR, et al. Discovery of a potent and selective EGFR inhibitor (AZD9291) of both sensitizing and T790M resistance mutations that spares the wild type form of the receptor. J Med Chem 2014; 57: 8249–67 [DOI] [PubMed] [Google Scholar]
- 22.Wu YL, Cheng Y, Zhou X, Lee KH, Nakagawa K, Niho S, et al. Dacomitinib versus gefitinib as first-line treatment for patients with EGFR-mutation-positive non-small-cell lung cancer (ARCHER 1050): a randomised, open-label, phase 3 trial. Lancet Oncol 2017; 18:1454–66. [DOI] [PubMed] [Google Scholar]
- 23.Jänne PA, Yang JC, Kim DW, Planchard D, Ohe Y, Ramalingam SS, et al. AZD9291 in EGFR Inhibitor–Resistant Non–Small-Cell Lung Cancer. N Engl J Med 2015; 372:1689–99. [DOI] [PubMed] [Google Scholar]
- 24.Babb J, Rogatko A, Zacks S. Cancer phase I clinical trials: efficient dose escalation with overdose control. Stat Med 1998; 17:1103–20. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data generated in this study are available within the article and its supplementary data files.