

Abstract
A limited number of cell lines have fueled the majority of preclinical Prostate Cancer (PCa) research, but their genomes remain incompletely characterized. Here, we utilized whole genome linked-read sequencing for comprehensive characterization of phased mutations and rearrangements in the most commonly used cell lines in PCa research including PC3, LNCaP, DU145, CWR22Rv1, VCaP, LAPC4, MDA-PCa-2b, RWPE-1, and four derivative castrate resistant (CR) cell lines LNCaP_Abl, LNCaP_C42b, VCaP-CR, and LAPC4-CR. Phasing of mutations allowed determination of “Gene-level Haplotype” to assess whether genes harbored heterozygous mutations in one or both alleles. Phased structural variant analysis allowed identification of complex rearrangement chains consistent with chromothripsis and chromoplexy. Additionally, comparison of parental and derivative CR lines revealed previously known and novel genomic alterations associated with the CR phenotype.
Implications:
This study therefore comprehensively characterized phased genomic alterations in the commonly used PCa cell lines, providing a useful resource for future prostate cancer research.
Keywords: Prostate Cancer Genome, Haplotype, Chromoplexy
INTRODUCTION
Cancer cell line models are vital resources in cancer research. For the prostate cancer (PCa) field, due to difficulties in establishing cell lines from cancer tissues, there are only about 20 unique parental cell lines established in the past half-century of research (1, 2). A query of PubMed revealed >23,000 publications with these cell lines, with seven of them (PC3, LNCaP, DU145, CWR22Rv1, VCaP, LAPC4, MDA-PCa-2b) accounting for >99% of the citations (Supplementary Methods). Early landmark efforts for molecular characterization of the PCa cell lines revealed both chromosomal instability and gene mutations as key oncogenic drivers (2, 3).
Advances in next generation sequencing (NGS) have fueled large scale genomic studies of >1,000 PCa genomes (4, 5). These studies identified recurrently mutated PCa driver genes even when they were rare across individuals (4). Whole genome sequencing (WGS) studies also identified key patterns of structural variants (SV), including complex chained and clustered rearrangement breakpoints termed chromoplexy and chromothripsis, as drivers of PCa (6, 7, 8, 9). NGS has also allowed ever more comprehensive and deep characterizations of the ways in which the PCa cell lines resemble or differ from primary human cancer tissues in their genetic alterations (10). However, it is still unclear to what extent the common PCa cell lines recapitulate the genomic mutational and structural alterations found in patient samples.
Long-read sequencing and linked-read sequencing have allowed comprehensive assessment of haplotypes and even end-to-end assembly of human genomes (8, 11, 12). Combining the power of sequencing fidelity and throughput of conventional Illumina sequencing with emulsion based barcoding of smaller fragments derived from very high molecular weight DNA, linked-read sequencing allows phasing of variants, and greater power for detecting structural alterations.
Here we report a comprehensive analysis of phased mutations and SVs determined through linked-read sequencing of the commonly-used PCa cell lines, including castrate resistant (CR) subclones of 3 of those cell lines. These analyses revealed that this collection of cell lines harbored a significant fraction of recurrent putative driver mutations and complex structural variants observed in human PCa.
MATERIALS AND METHODS
Cell lines and culturing methods
Cell lines were obtained from ATCC (CWR22Rv1, DU145, LNCaP, MDA-PCa-2b, RWPE-1, VCaP), Johns Hopkins Biorepository (LAPC4-CR, VCaP-CR), NCI (PC3), UroCor, OK (LNCaP_C42b), Charles Sawyer’s lab, UCLA (LAPC4), and Zoran Culig’s lab, Innsbruck Medical University (LNCaP_Abl). All were cultured in media as instructed by providers and used at passages less than 10 (Supplementary Table S1). Frozen pellets were used for mycoplasma testing (MycoDtect kit, Greiner Bio-One) and STR genotyping (GenePrint 10 System, Promega) performed by the Johns Hopkins Genetic Resource Core Facility in July 2019 right before sequencing was performed. All cell lines were mycoplasma free and passed the a priori defined STR genotype threshold of 70%. Sources, RRIDs, culture conditions, passage number, and STR genotype results are reported in Supplementary Table S1.
High Molecular weight (HMW) DNA Extraction
HMW DNA was extracted following the “salting out” protocol described by 10X Genomics (Pleasanton, CA). Briefly, two million cells were lysed overnight at 37°C in Lysis buffer containing 2mM EDTA, 300mM NaCl, 8mM Tris-HCl, 125ug/mL Proteinase K, and .5% SDS. HMW DNA was precipitated using 1M NaCl, washed in absolute ethanol, and resuspended overnight at 4 °C in TE buffer. The TapeStation 4200 system (Agilent) was used to assess DNA quality. All samples contained more than 80% of DNA having greater than 60kb in length (Supplementary Table S2).
Barcoding, library preparation, and sequencing
1 (+/−0.2) ng of DNA was quantified by Qubit Broad Range (Thermo Q32850) and used for GEM creation and barcoded library generation using the 10X Chromium Genome protocol. Resulting library fragment sizes were determined using the DNA 1000 and 2100 Kit BioAnalyzer (Agilent Technologies), and indexed with Chromium i7 Sample Index Plate (Cat 220103, 10X Genomics). The indexed libraries were sequenced to 30X- 50X average coverage on an Illumina HiSeqX platform using paired-end 150 bp × 150 bp reads. The resulting sequencing BCL files were processed by the Long Ranger Pipeline (2.2)(13) (10X Genomics) for alignment, variant discovery, and phasing.
Analysis for phased mutations and SVs
Long Ranger pipeline was used for phasing of small variants (SNP and micro-indels), phased SVs, and unphased copy number variants (CNV). Passed variants from the Long Ranger phased_variants.vcf output files were annotated using the vcf2maf (14) pipeline and the Uniprot canonical isoform reference. Gene-level haplotype was inferred through a custom R (version 4.0.0) script that used the haplotype (column GT) and phase set (column PS) information provided by Long Ranger in phase_variants.vcf files. Any gene with at least one mutation with GT=”1|1” was categorized as “Hemi/homozygous”, and any gene with only one heterozygous (GT=”0|1” or “1|0”) mutation was categorized as “Monoallelic”. For any gene with multiple heterozygous mutations, if all mutations were on the same phase set and belonged to the same haplotype, it was categorized as “Multi-monoallelic”. If all mutations were on the same phase set and there was at least one mutation belonging to the opposite allele, it was categorized as “Biallelic heterozygous”. In cases where multiple heterozygous mutations did not belong to the same phase set or were unphased (GT=”0/1”), the genes were categorized as “no info”.
Phased structural variants (SV) were defined as large SV calls in large_sv_calls.bedpe file that had phased set information (PS1/PS2). A custom R script, termed ChainLink, allowed inference of phased structural variant breakpoints as follows: SVs were chained together based on both shared phased set and shared haplotype for each breakpoint. Each SV chain contained at least 2 SVs.
ChainFinder (9) was used in parallel using phased SVs from all 12 cell lines to determine background breakpoint frequency. “Copy number type” was set as “Seq” and Deletion_threshold was set at 2. Other parameters were left as default. SnpEff 4.5.1 (RRID:SCR_005191) was used to annotate the closest gene to each breakpoint coordinate.
Graphs were prepared using 10x Genomics Loupe Browser 4.1.0, the R packages Rcircos (1.2.1) (RRID:SCR_003310), and ggplot2 (3.3.5) (RRID:SCR_014601), and assembled on Adobe Illustrator (24.1.1) (RRID:SCR_010279).
RESULTS
Linked-read sequencing of human PCa cell lines
We performed linked-read sequencing of high molecular weight DNA from 11 PCa cell lines and one immortalized non-malignant prostatic epithelial cell line RWPE-1 (Supplementary Table S1, S2; Workflow: Supplementary Figure S1A). The majority of cell lines were of low passage from the original source, and shared a majority of mutations found in driver genes from the Cancer Cell Line Encyclopedia (CCLE) project (Supplementary Table S3 and S4). Whole genome copy number and gene level copy number analysis also showed a high degree of correlation between related cell lines when comparing the current data to that from cells of later passages or with CCLE data, while unrelated cell lines showed lower correlation (Supplementary Figure S1B; Supplementary Table S3, S4). As expected, we confirmed that cell lines with known microsatellite instability (MSI) and mismatch repair deficiency exhibited high mutation rates, and microsatellite stable (MSS) VCaP and PC3 cells exhibited lower mutational burdens (Supplementary Figure S1C, Supplementary Table S5) (15). In addition to MMR gene defects in some cell lines, many cell lines showed mutations in HDR genes, as indicated in Supplementary Table S6.
Gene-level phasing of somatic mutations to distinguish between mono- and bi-allelic gene mutations
Linked-read sequencing allowed phasing of heterozygous mutations, thus revealing allelic mutation status at the gene level. Using this phasing information, we developed the concept “Gene-level haplotype” for each gene to determine whether a given gene is altered at a single or both alleles (Figure 1A). We focused our analysis on 97 putative driver genes recurrently mutated in PCa cataloged by Armenia et al (4) that we called “Longtail” genes. Among these, we found 87 genes to be mutated in our panel of 12 cell lines (Figure 1B and Supplementary Figure S1D). MSI cell lines showed higher percentage of genes inactivated by biallelic heterozygous mutations, whereas MSS cell lines showed mostly mono-allelic or hemi/homozygous mutations (Figure 1B). To enrich missense mutations among potential cancer drivers, we filtered to those specific missense mutations documented in the Cancer Mutation Census (CMC) project under COSMIC (16), yielding 58 Longtail genes with putative driver mutations found in our panel of cell lines (Figure 1C).
Figure 1: Phasing of somatic mutations to determine Gene-level Haplotype.
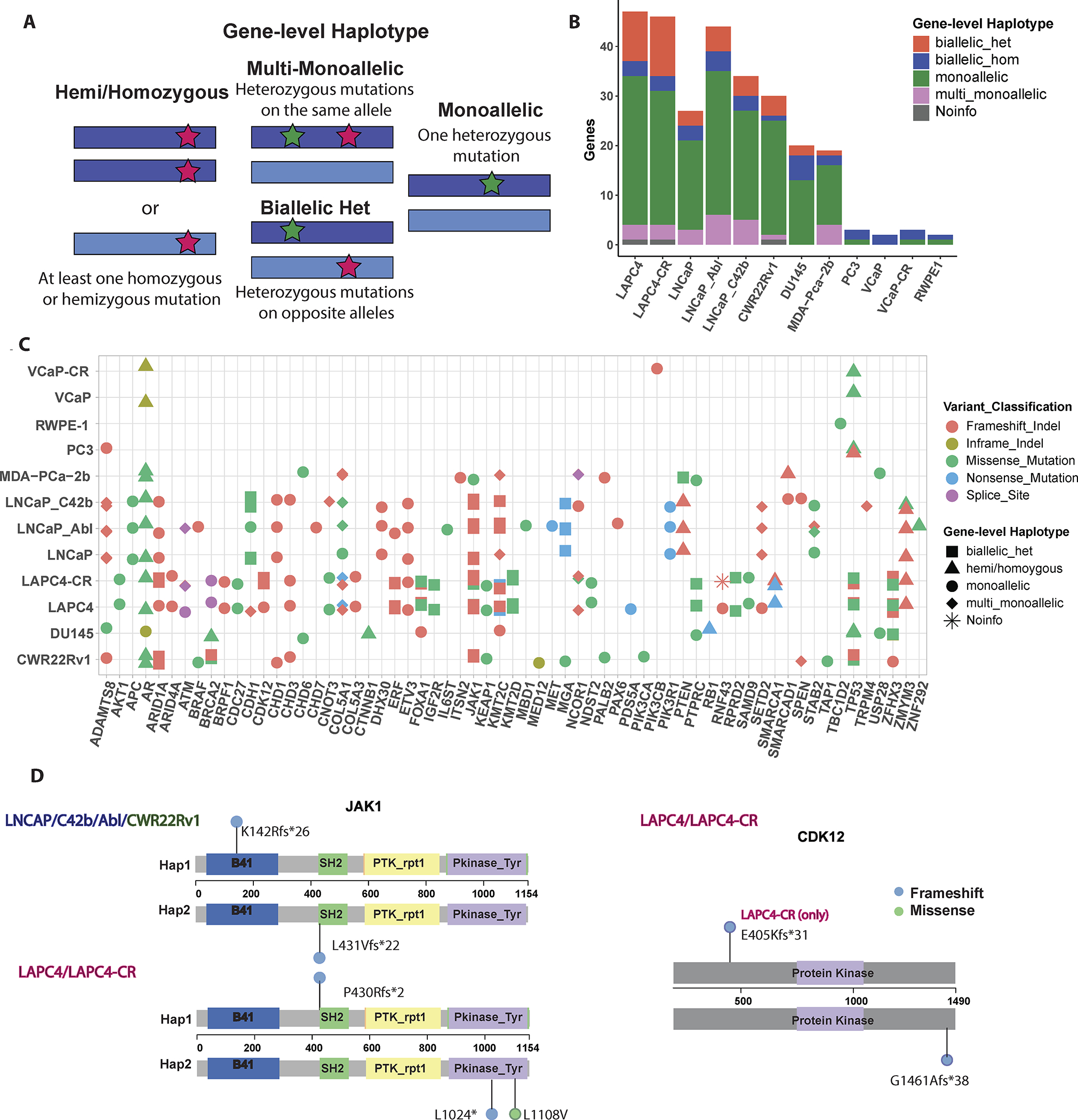
(A) Schematic of gene-level Haplotype, derived from phased linked read sequencing data, distinguishes between genes with multiple heterozygous mutations on the same or different alleles. Dark blue or light blue bars represent each allele for a given gene. Red and green stars indicate examples for the positions of mutations. (B) Somatic mutations on “Longtail” genes previously found to be recurrently mutated in PCa (4) stratified by Gene-level haplotype. (C) Cell line mutations in the Longtail panel of cancer driver genes that are recurrently mutated in human PCa, with variant classification for each mutation, and Gene-level haplotype for each gene annotated for each cell line. (D) Lollipop plots showing locations of JAK1 and CDK12 biallelic heterozygous mutations relative to positions of Pfam domains. Colors of the lollipop heads indicate the variant classification for each mutation (blue= frameshift; green = missense).
Gene-level haplotypes allowed identification of several genes inactivated in a mono- vs. bi-allelic manner (Supplementary Table S7). Illustrative examples of genes with biallelic alterations are shown for CDK12 and JAK1 (Figure 1D). CDK12 mutation denotes a distinct subclass of PCa characterized by tandem duplication and high neoantigen burden (6). In our cell lines, LAPC4 and its CR clone both had CDK12 G1461Afs*38 (COSM2837928) mutation. This somatic mutation is the most common frameshift CDK12 mutation documented in the CMC, recurrently found in human cancers (16). It is interesting to note that this frameshift mutation led to alteration of the last 30 amino acids of the protein, along with lengthening the protein by 9 amino acids. It is thus intriguing to speculate that it may have led to dominant negative function rather than just loss of function. LAPC4-CR lost the other copy of CDK12 from an E405Kfs*31 mutation, which truncated the remaining two thirds of the protein, and therefore had mutations in both alleles (Figure 1D, Supplementary Table S7).
Another example to note was JAK1, a tyrosine kinase implicated in immune and apoptosis evasion (17). LNCaP, its CR clones, and CWR22Rv1 all had p.L431Vfs*22 (COSM41842) and p.K142Rfs*26 (COSM1639943), both of which are recurrent JAK1 mutations in human cancer (Figure 1D). LAPC4 and LAPC4-CR shared p.P430Rfs*2 (COSM1560531) and LAPC4-CR had additional mutations at p.L1108V and p.L1024* on the kinase domain. Loss of function mutations in JAK1 is common among MSI cell lines and associated with reduced interferon response (17). Among the cell lines examined here, biallelic heterozygous mutations on JAK1 were observed in three MSI cell lines. We can speculate that these mutations may have been involved in evasion of apoptosis and immune surveillance in the original cancers from which these cell lines were derived (17).
Phasing of structural alterations in PCa cell lines
We next examined structural variants in the PCa cell lines. At a glance, in contrast with the high number of mutations (Supplementary Figure S1C), MSI cell lines exhibit less SVs compared to MS stable cell lines (Supplementary Figure S2A). By combining rearrangement breakpoint information with the associated haplotype information of each segment of the breakpoint, we phased multiple breakpoints to the same allele to identify complex rearrangements consistent with chromoplexy or chromothripsis. We developed a pipeline called ChainLink to infer clustered SVs by combining phase information and SV identification (Figure 2A). We identified complex rearrangements in all PCa cell lines, ranging from 3 chains in LNCaP to 11 chains in VCaP-CR (Figures 2B, 2C, Supplementary Table S8). The modal number of SVs in one chain was 3. The non-cancer line RWPE-1 notably did not have any complex SVs (Figures 2B, 2C, Supplementary Table S8), but did have evidence of having subclonal population(s) with potential aneuploidy (Supplementary Figures S3A and S3B). Many of these complex rearrangements found in PCa were consistent with chromoplexy and chromothripsis (Figures 2C, 2D; Supplementary Figures S2D, S2E).
Figure 2: Identification and phasing of structural alterations.
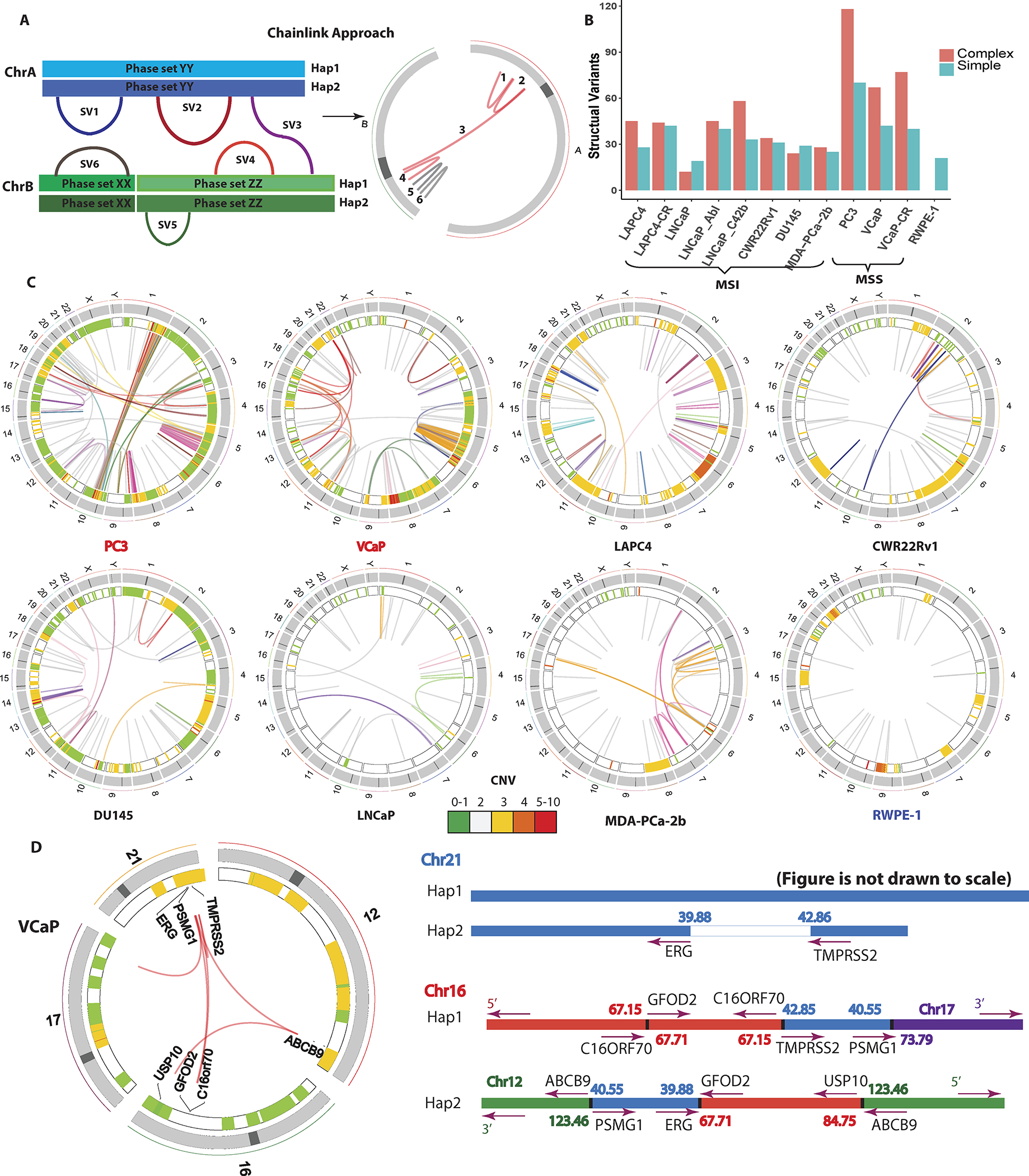
(A) The ChainLink approach uses phase information for each breakpoint to chain together complex SVs. In this schematic, SV 1, 2, 3, 4 on two different chromosomes are chained together into a complex SV cluster based on phase information on each breakpoint; SV 5 and 6 are considered simple SVs. (B) Number of SVs classified as simple or complex in each cell line. (C) Circos plots representing CNV and SVs on the 8 parental cell lines in this study: MSI stable (red), MSI high (black) and non-malignant immortalized prostatic epithelium (navy). Heatmap track beneath chromosome ideograms represents CNV with colors representing copy number. Innermost link track represents large SVs: complex SVs are colored by chain; simple SVs are gray. (D) Left: Circos plot showing the chromoplexy event associated with the TMPRSS2-ERG fusion gene formation in VCaP. Genes interrupted by these SVs are labeled on the innermost track. Right: genomic anatomy of the breakpoints involved in the chromoplexy event leading to the TMPRSS2-ERG fusion gene in VCaP and detailed rearrangement configuration of the 3Mb sequence between the TMPRSS2-ERG genomic breakpoint. Genomic coordinates are in Mb per reference genome hg19. Arrows represent gene direction.
We compared the performance of ChainLink to a previous method called ChainFinder, which uses rearrangement breakpoints identified by conventional paired-end sequencing data to infer chained rearrangements based on comparing breakpoint locations to an expected distribution of breakpoints as if they had arisen independently (9). ChainLink identified all chains called by ChainFinder (Supplementary Figures S2B, S2C). Moreover, ChainFinder identified fewer chains, and fewer structural variants per chain, relative to ChainLink (Supplementary Figures S2B, S2C). These analyses suggested that the use of phasing information from linked-read sequencing can increase the sensitivity of identifying rearrangements within complex chained rearrangements compared to more indirect statistical methods.
Using ChainLink, we were able to decipher the complete genomic anatomy of the complex SV underlying the TMPRSS2-ERG fusion gene in VCaP cells (Figure 2D; Supplementary Table S9). It was previously known that the 3Mbp sequence between the TMPRSS2 and ERG genes was rearranged to other genomic segments in this cell line, rather than being deleted (18); however, the precise genomic anatomy of rearrangements involving this 3Mbp stretch was not well understood. Here we show that this 3Mb piece was broken up into two parts, with one rearranged with sequences on chromosomes 16 and 17, and the other rearranged with sequences on chromosomes 16 and 12. Altogether, this rearrangement consisted of 7 structural variant breakpoints in a highly complex genomic anatomy (Figure 2D). Parallel ChainFinder analysis only called three of these structural variants and could not fully decipher the complex anatomy of the rearrangements associated with this fusion gene (Supplementary Table S9).
In addition to the aforementioned chromoplexy event, VCaP also harbored a highly complex chromothripsis event on chromosome 5 characterized by clustered SVs with random orientation and alternating copy number changes (Supplementary Figure S2E). PC3 also showed evidence of chromothripsis, occurring on multiple chromosomes. In addition to two striking chromothripsis rearrangement clusters on chromosome 5q and 8q, we noted an unusual interchromosomal chromothripsis event bridging chromosomes 1 and 10 (Supplementary Figure S2D, Supplementary Table S9, S10). This was suggestive of an inter-chromosomal rearrangement between chromosomes 1 and 10 in the PC3 cells, with subsequent catastrophic chromothripsis of the newly formed fused chromosome.
Somatic variants associated with resistance to androgen deprivation in PCa cell lines
Understanding mechanisms of resistance to androgen receptor targeted therapy remains an important area of PCa research. Among the cell lines with AR expression, it was interesting to note that there were several alterations in the AR gene itself, many of which have been previously documented (1, 2). CWR22Rv1, MDA-PCa-2b, VCaP, LAPC4, and LNCaP all have mutations in AR; CWR22Rv1 has duplication around the third exon of AR, and VCaP showed amplification of the entire AR locus (19) (Supplementary Figs. S3C, S3D, Supplementary Table S11). While some of these alterations, such as the AR T878A mutation in LNCaP, have well-documented functional implications, the functional significance of other mutations are not well understood (20). To understand whether there are acquired mutations associated with progression to resistance to androgen deprivation, we next compared the somatic mutational and structural variant landscape in the parental LNCaP, LAPC4, and VCaP cell lines versus their previously established androgen-deprivation resistant subclones (2, 20, 21) (Figure 3). While the CR cell lines largely retained the mutations present in their parental cell line, confirming that they are truly subclones of those parental lines, they gained many additional mutations not present in the parental cell lines (Figures 3A, 3B). LNCaP_Abl, LNCaP_C42b, and LAPC4-CR all gained mutations in genes encoding epigenetic machinery, including members of the SWI/SNF chromatin remodeling complex (ARID1A, ARID1B, SMARCA4 and SS18) (Figures 3A, 3B). Interrogating the cBioPortal database for mutations in 28 SWI/SNF subunits across 5 PCa genomic studies (13, 22, 23, 24, 25) found that eleven percent of. PCa samples (170/1578 total patients) showed mutations in at least one SWI/SNF subunit (Supplementary Figures S4A, S4B), with more mutations found in castrate resistant PCa than castrate sensitive PCa (Supplementary Figures S4B, 4C). These preliminary observations suggest SWI/SNF mutations potentially play a role in driving castrate resistant PCa. Other epigenetic machinery proteins that gained mutations in the CR cell lines include BRD3, SMARCAD1, MBD1, KMT2C, and NCOR2 (Figures 3A, B).
Figure 3: Comparison of LNCaP, LAPC4 parental cell lines with their respective CR derivatives.
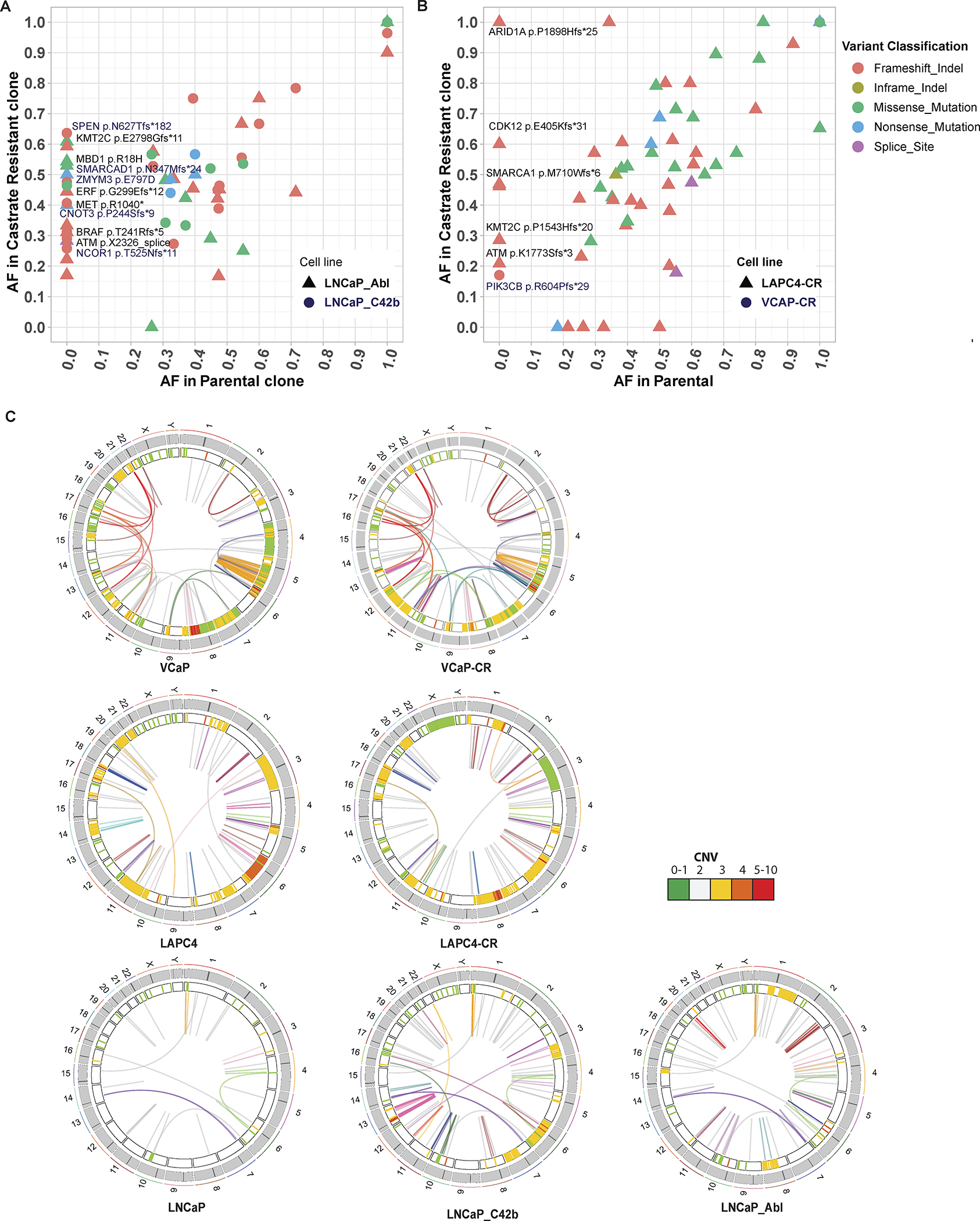
(A, B) Allele frequencies for each mutation in CR clones LNCaP_Abl, LNCaP_C42b, LAPC4-CR and their associated parental lines (LNCaP and LAPC4). Mutations seen along the y-axis represent new mutations arising in the CR lines not found in the parental cells. (C) Juxtaposition of SVs in parental and CR clones. Heatmap track beneath chromosome band represents CNV with colors representing copy number. Innermost link track represents large SVs as defined by barcode overlap: complex SVs are colored by chain; simple SVs are gray.
CR clones also accumulated more SVs and copy number variants (CNVs), many of which may have contributed to their CR progression (Figure S5). Most notably, 8q24 amplification was found in both LAPC4-CR and LNCaP_Abl (Supplementary Figures S5A, S5B). In LNCaP_Abl, there were three copy number gains at 8q24 with one spanning the MYC enhancer (Supplementary Figure S5A). The LAPC4-CR line, which harbored biallelic mutation of CDK12, showed multiple tandem duplications around both the MYC gene and its enhancer (Supplementary Figures S5B, S5E); such an association between CDK12 mutation and tandem duplications around the MYC gene and its enhancer was recently reported in human PCa (7). VCaP-CR has an AR enhancer amplification, a well-studied recently discovered SV driving castrate resistant phenotype (Supplementary Figure S5C) (8). The AR enhancer duplication in VCaP-CR occurred in addition to the AR amplification already present in VCaP. Lastly, C42b gained a series of complex SVs on chromosome 10, 11, 12, 13, and 14 (Figure 3C; Supplementary Figure S5D; Supplementary Table S10). Most notable was a chromothripsis-like complex rearrangement on chromosome 13 (Supplementary Figure S5D and Supplementary Table S10). These complex rearrangements involved multiple amplifications and translocations within chromosome 13, dysregulating genes within 13q12.11 to 13q13.2 and 13q31.3. Amongst these, LATS2 encodes a tumor suppressor protein kinase that functions as a positive regulator of TP53 and as a corepressor of androgen-responsive gene expression. Future mechanistic studies can examine the role of these mutations in driving resistance to androgen deprivation or PCa progression.
DISCUSSION
Linked-read sequencing allowed allelic phasing of both mutations and structural variants in the commonly used prostate cancer cell lines. The phasing information of somatic mutations was used to derive “Gene level haplotypes”, which distinguished between multiple heterozygous mutations on the same or different copies of each gene. This allowed for the first time a genome-wide analysis of mono- vs. biallelic driver gene alterations in the PCa cell lines, providing a valuable resource for future studies (Supplementary Table S6–S13). While it is certainly possible and likely that the cell lines can be prone to cell line drift, the high degree of similarity, with respect to mutations and copy number alterations, between the cell lines used in this study and those reported in the CCLE project and in batches of cell lines from independent lab and across passage numbers, suggests that many of the alterations and findings reported here should be generalizable (Supplementary Figure S1, Supplementary Tables S3, S4).
The linked read sequencing data are well-suited to identifying rearrangement breakpoints as overlap of barcodes from non-contiguous genomic regions. Furthermore, by developing a simple approach termed ChainLink, we leveraged the phasing/haplotype information from the linked read data to phase SV breakpoints and identify chained complex rearrangements, including those consistent with chromoplexy and chromothripsis (Supplementary Table S10). First, we were able to decipher for the first time the precise genomic anatomy of several complex rearrangement events, including the highly recurrent TMPRSS2-ERG rearrangement present in VCaP cells (Figure 2D, Supplementary Table S9), as well as several chromoplexy and chromothripsis events in numerous cell lines that were previously unrecognized (Figure 2C; Supplementary Figures S2D, 2E). Second, by phasing all of the individual breakpoints that constituted these complex rearrangements, we showed that the breakpoints all occurred across a single phased allele, and did not represent rearrangements that were staggered independently in different alleles. This supports the model that chromoplexy and chromothripsis occur in a single concerted event, rather than through accumulation of multiple independent rearrangements.
These analyses also established the relevance of this set of PCa cell lines to human PCa. First, the cell lines captured a large fraction of recurrently mutated driver genes in human PCa as have been reported in large scale PCa genome sequencing studies (4). Additionally, integrating the phased mutation and SV information, we saw many associations between mutations and SV patterns that have been described in human cancers including: i) the association of CDK12 mutations with tandem duplications as seen in the LAPC4/LAPC4-CR cell lines; ii) the association of TP53 mutations with presence of chromothripsis (PC3 and VCaP cells); and iii) correlation between pathogenic BRCA2 mutations and genome wide deletion in DU145 (Figures 1C, 2C) (7). Finally, comparing the CR cell lines to the castration sensitive parental cell lines, we found that several mutations in epigenetic pathways occurred in the CR lines.
One limitation of this study is that the approaches used for phasing variants and rearrangement breakpoints would not have accounted sufficiently for aneuploidy. As a result of this, we would be limited in determining the phasing of variants that arose after a specific allele gained a copy. Nonetheless, since we had such a high rate of phasing of the identified somatic mutations, it is likely that most of these mutations arose prior to any copy number gains of those segments.
Taken together, this study serves as a comprehensive compilation of genomic features for the most commonly studied PCa cell lines, provides important insights into the pathogenesis of chromoplexy, represents a valuable resource for the field, and presents genomic analysis frameworks for future long-read sequencing studies.
Supplementary Material
ACKNOWLEDGEMENTS:
We would like to thank Jennifer Meyers from the SKCCC Experimental and Computational Genomics Core and Lisa Haley from the Pathology Molecular Diagnostics Lab for their help with sequencing runs and quality controls of DNA samples.
Financial support and funding:
This work was supported by NIH/NCI grants P50CA058236, U01CA196390, R01CA183965, DOD CDMRP grant W81XWH-21-1-0295, and by the Prostate Cancer Foundation, Commonwealth Foundation, Maryland Cigarette Restitution Fund, and the Irving Hansen Foundation. The Experimental and Computational Genomics Core at the SKCCC was supported by the NIH/NCI Cancer Center Support Grant P30CA006973, and MG was supported by the National Science Foundation Graduate Research Fellowship under grant number DGE-1746891.
Footnotes
Conflict of Interest Statement
S.Y. has received sponsored research support to his institution from Celgene/BMS, Janssen, and Cepheid/Danaher, has served as a paid consultant to Cepheid/Danaher, and is a co-founder and equity owner in Digital Harmonic LLC and Brahm Astra Therapeutics. W.G.N. is a cofounder of and equity owner in Digital Harmonic LLC and Brahm Astra Therapeutics, an equity owner in and member of the board of directors of Armis Biopharma, a stockholder in Becton, Dickinson and Company, and a member of the scientific advisory board for Cepheid, and has rights to earn royalties from patents licensed by Johns Hopkins to Exact Sciences. The remaining authors have no financial relationships to declare.
DATA AVAILABILITY STATEMENT
Raw data were made available at SRA with the following object ID: PRJNA751700.
SVs and CNVs data were made available at dbVar under the accession number nstd213.
Supplementary Data are included.
REFERENCE
- 1.SOBEL RE and SADAR MD (2005) Cell lines used in prostate cancer research: A compendium of old and new lines—part 1. The Journal of Urology, 173, 342–359. [DOI] [PubMed] [Google Scholar]
- 2.van Bokhoven A, Varella-Garcia M, Korch C, Johannes WU, Smith EE, Miller HL, Nordeen SK, Miller GJ and Lucia MS (2003) Molecular characterization of human prostate carcinoma cell lines. The Prostate, 57, 205–225. [DOI] [PubMed] [Google Scholar]
- 3.van Bokhoven A, Caires A, Maria MD, Schulte AP, Lucia MS, Nordeen SK, Miller GJ and Varella-Garcia M (2003) Spectral karyotype (SKY) analysis of human prostate carcinoma cell lines. Prostate, 57, 226–244. [DOI] [PubMed] [Google Scholar]
- 4.Armenia J, Wankowicz SAM, Liu D, Gao J, Kundra R, Reznik E, Chatila WK, Chakravarty D, Han GC, Coleman I, et al. (2018) The long tail of oncogenic drivers in prostate cancer. Nat Genet, 50, 645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cancer Genome Atlas Research Network. (2015) The molecular taxonomy of primary prostate cancer. Cell, 163, 1011–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu Y, Cieślik M, Lonigro RJ, Vats P, Reimers MA, Cao X, Ning Y, Wang L, Kunju LP, de Sarkar N, et al. (2018) Inactivation of CDK12 delineates a distinct immunogenic class of advanced prostate cancer. Cell (Cambridge), 173, 1770–1782.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Quigley DA, Dang HX, Zhao SG, Lloyd P, Aggarwal R, Alumkal JJ, Foye A, Kothari V, Perry MD, Bailey AM, et al. (2018) Genomic hallmarks and structural variation in metastatic prostate cancer. Cell (Cambridge), 174, 758–769.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Viswanathan SR, Ha G, Hoff AM, Wala JA, Carrot-Zhang J, Whelan CW, Haradhvala NJ, Freeman SS, Reed SC, Rhoades J, et al. (2018) Structural alterations driving castration-resistant prostate cancer revealed by linked-read genome sequencing. Cell (Cambridge), 174, 433–447.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baca S, Prandi D, Lawrence M, Mosquera J, Romanel A, Drier Y, Park K, Kitabayashi N, MacDonald T, Ghandi M, et al. (2013) Punctuated evolution of prostate cancer genomes. Cell (Cambridge), 153, 666–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sienkiewicz K and Ratan A (2022) Protocol for integrative subtyping of lower-grade gliomas using the SUMO pipeline. STAR Protoc, 3, 101110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marks P, Garcia S, Barrio AM, Belhocine K, Bernate J, Bharadwaj R, Bjornson K, Catalanotti C, Delaney J, Fehr A, et al. (2019) Resolving the full spectrum of human genome variation using linked-reads. Genome Res, 29, 635–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zheng GXY, Lau BT, Schnall-Levin M, Jarosz M, Bell JM, Hindson CM, Kyriazopoulou-Panagiotopoulou S, Masquelier DA, Merrill L, Terry JM, et al. (2016) Haplotyping germline and cancer genomes with high-throughput linked-read sequencing. Nat Biotechnol, 34, 303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cancer Genome Atlas, Research Network, Weinstein JN, Collisson EA, Mills GB, Shaw KRM, Ozenberger BA, Ellrott K, Shmulevich I, Sander C and Stuart JM (2013) The cancer genome atlas pan-cancer analysis project. Nat. Genet, 45, 1113–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kandoth C (2020) Vcf2maf. 1.6.19,.
- 15.Sun X, Chen C, Vessella RL and Dong J (2006) Microsatellite instability and mismatch repair target gene mutations in cell lines and xenografts of prostate cancer. The Prostate, 66, 660–666. [DOI] [PubMed] [Google Scholar]
- 16.Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM, Bindal N, Boutselakis H, Cole CG, Creatore C, Dawson E, et al. (2018) COSMIC: The catalogue of somatic mutations in cancer. Nucleic Acids Res, 47, D941–D947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Albacker LA, Wu J, Smith P, Warmuth M, Stephens PJ, Zhu P, Yu L and Chmielecki J (2017) Loss of function JAK1 mutations occur at high frequency in cancers with microsatellite instability and are suggestive of immune evasion. Plos One, 12, e0176181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, Sun X, Varambally S, Cao X, Tchinda J, Kuefer R, et al. (2005) Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science, 310, 644. [DOI] [PubMed] [Google Scholar]
- 19.Li Y, Hwang TH, Oseth LA, Hauge A, Vessella RL, Schmechel SC, Hirsch B, Beckman KB, Silverstein KA and Dehm SM (2012) AR intragenic deletions linked to androgen receptor splice variant expression and activity in models of prostate cancer progression. Oncogene, 31, 4759–4767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Culig Z, Hoffmann J, Erdel M, Eder IE, Hobisch A, Hittmair A, Bartsch G, Utermann G, Schneider MR, Parczyk K, et al. (1999) Switch from antagonist to agonist of the androgen receptor bicalutamide is associated with prostate tumour progression in a new model system. Br. J. Cancer, 81, 242–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Haffner MC, Bhamidipati A, Tsai HK, Esopi DM, Vaghasia AM, Low JY, Patel RA, Guner G, Pham MT, Castagna N, et al. (2021) Phenotypic characterization of two novel cell line models of castration-resistant prostate cancer. Prostate, 81, 1159–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Abida W, Cyrta J, Heller G, Prandi D, Armenia J, Coleman I, Cieslik M, Benelli M, Robinson D, Van Allen EM, et al. (2019) Genomic correlates of clinical outcome in advanced prostate cancer. Proc. Natl. Acad. Sci. USA, 116, 11428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Robinson D, Van Allen E,M, Wu Y, Schultz N, Lonigro RJ, Mosquera J, Montgomery B, Taplin M, Pritchard CC, Attard G, et al. (2015) Integrative clinical genomics of advanced prostate cancer. Cell, 161, 1215–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Beltran H, Prandi D, Mosquera JM, Benelli M, Puca L, Cyrta J, Marotz C, Giannopoulou E, Chakravarthi, Balabhadrapatruni VSK, Varambally S, et al. (2016) Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat. Med, 22, 298–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stopsack KH, Nandakumar S, Wibmer AG, Haywood S, Weg ES, Barnett ES, Kim CJ, Carbone EA, Vasselman SE, Nguyen B, et al. (2020) Oncogenic genomic alterations, clinical phenotypes, and outcomes in metastatic castration-sensitive prostate cancer. Clin. Cancer Res, 26, 3230–3238. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw data were made available at SRA with the following object ID: PRJNA751700.
SVs and CNVs data were made available at dbVar under the accession number nstd213.
Supplementary Data are included.