

Abstract
Synthesis of glycosaminoglycans such as heparan sulfate (HS) and chondroitin sulfate (CS) occurs in the lumen of the Golgi but the relationship between Golgi structural integrity and glycosaminoglycan synthesis is not clear. In this study, we disrupted the Golgi structure by knocking out GRASP55 and GRASP65 and determined its effect on the synthesis, sulfation, and secretion of HS and CS. We found that GRASP depletion increased HS synthesis while decreasing CS synthesis in cells, altered HS and CS sulfation, and reduced both HS and CS secretion. Using proteomics, RNA-seq and biochemical approaches, we identified EXTL3, a key enzyme in the HS synthesis pathway, whose level is upregulated in GRASP knockout cells; while GalNAcT1, an essential CS synthesis enzyme, is robustly reduced. In addition, we found that GRASP depletion decreased HS sulfation via the reduction of PAPSS2, a bifunctional enzyme in HS sulfation. Our study provides the first evidence that Golgi structural defect may significantly alter the synthesis and secretion of glycosaminoglycans.
Keywords: Golgi, GRASP55, GRASP65, heparan sulfate (HS), chondroitin sulfate (CS), synthesis, sulfation, RNAseq, proteomic
Introduction
The Golgi apparatus is a central station in the intracellular trafficking pathway and serves as the principal hub for sorting and post-translational modifications of proteins and lipids (1). The basic structure of the Golgi is a stack of flattened cisternae. In mammalian cells, multiple Golgi stacks are latterly linked into a ribbon-like structure located in the perinuclear region of the cell. It has been previously demonstrated that two Golgi peripheral membrane proteins, GRASP55 and GRASP65, function as the “glue” that links Golgi membranes together and facilitates Golgi stacking and ribbon formation. Knocking down or knocking out either one of these two GRASP proteins decreases the number of cisternae per stack, whereas depleting both GRASPs disrupts the entire Golgi structure (2–4). Most recently, it has been shown that acute degradation of GRASP55 and GRASP65 proteins causes defects in Golgi ribbon linking and cisternae alignment (5), confirming an important role of GRASP proteins in Golgi structure formation.
Functional studies revealed that destruction of the Golgi structure by GRASP-depletion accelerates protein trafficking in the Golgi, but impairs accurate N-glycosylation and protein sorting (6). At the cellular level, GRASP depletion reduces cancer cell attachment and invasion mainly through the reduction of α5β1 integrin synthesis (7), indicating a role of GRASPs and/or the Golgi structure in transcription regulation. In addition to Golgi structure formation, GRASPs are also involved in autophagy and unconventional secretion. Under starvation or stress conditions, GRASP55, but not GRASP65, translocates from the Golgi to other membrane structures such as autophagosomes and endoplasmic reticulum (ER) to regulate autophagy and unconventional secretion of certain cytosolic or transmembrane proteins (8–13).
Glycosaminoglycans (GAGs) are main components of the cell surface glycome and extracellular matrix (14). GAGs are long linear polysaccharides consisting of repeating disaccharide units. Based on the core disaccharide structures, GAGs are classified into three major forms, heparan sulfate (HS), chondroitin sulfate (CS), and hyaluronan (HA). While HA is synthesized at the plasma membrane and released, HS and CS are synthesized and attached to serine residues of cargo proteins in the Golgi, from where they are transported to the cell surface and secreted to the extracellular space. The biosynthesis of both HS and CS begins with the formation of a tetrasaccharide linker on a serine residue in a protein core, which is subsequently diversified to HS or CS depending on the subsequent enzymatic reactions (Figure 1A) (14).
Figure 1. Golgi structure disruption by GRASP KO increases GAG synthesis but reduces its secretion.
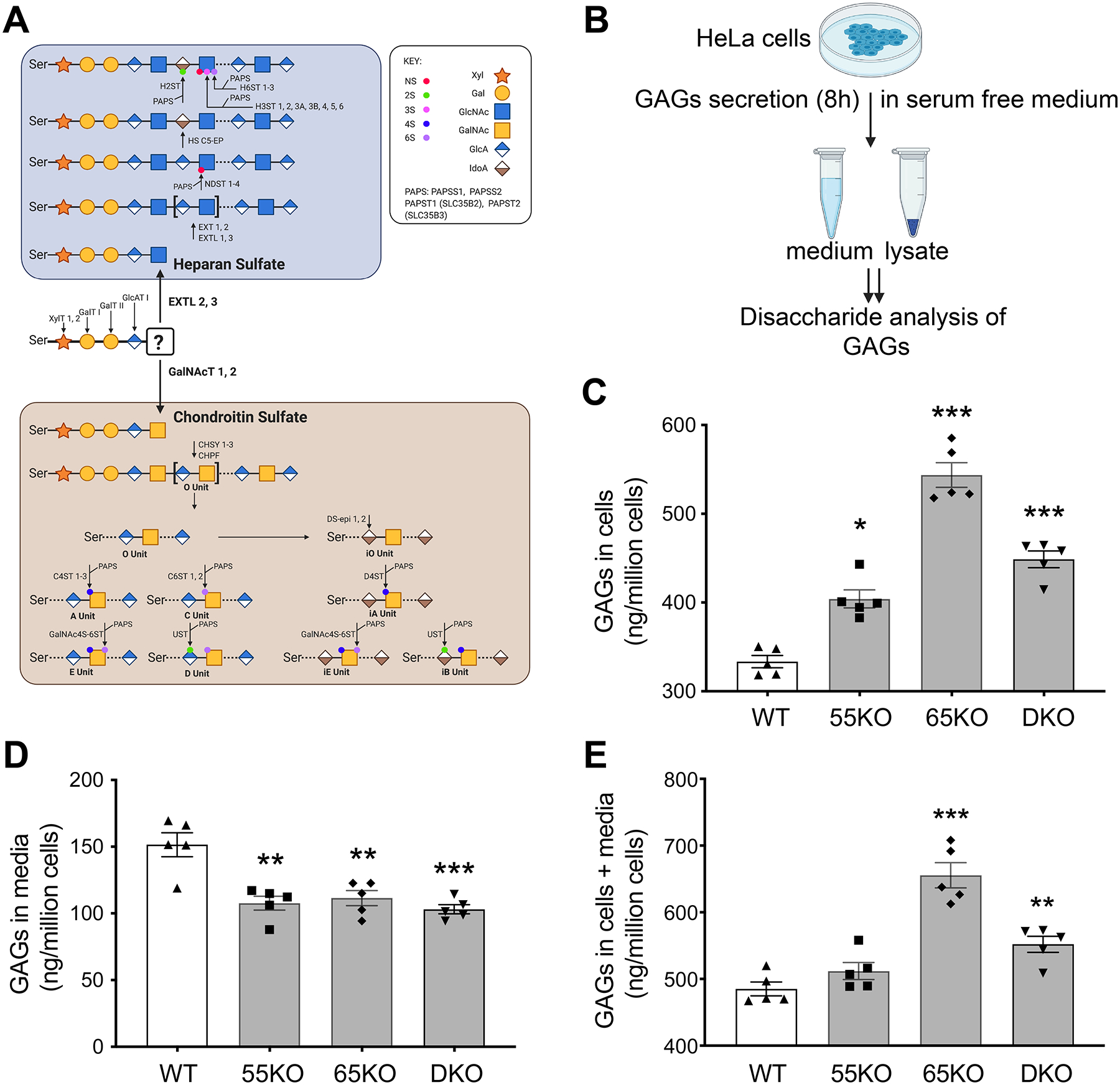
(A) Schematic diagram illustrating the HS and CS synthesis pathways; major enzymes in each pathway are indicated at their designated steps of reactions. (B) Schematic workflow for cell lysate and medium sample preparation to analyze GAGs by LC-MS. (C) GRASP KO increases the amount of GAGs in cells. Shown are the amounts of GAGs in the lysates of wildtype (WT), GRASP55 knockout (55KO), GRASP65 knockout (65KO), and GRASP55 and GRASP65 double knockout (DKO) HeLa cells. (D) GRASP KO decreases GAG secretion. WT and indicated GRASP KO cells were incubated in serum-free medium for 8 h and GAGs in the conditioned media were analyzed by LC-MS. (E) GRASP KO increases the total amount of GAGs in cells and media. Shown are the total amount of GAGs per million cells in both cell lysates and conditioned medium in each cell line. Results are presented as mean ± SEM, statistical analysis was assessed by comparing KO cells to WT cells by student’s t-test. *, p<0.05; **, p<0.01; ***, p<0.001.
For HS, the initiation of HS biosynthesis occurs as the transferases Exostosin-like 2 (EXTL2) and 3 (EXTL3) transfer an N-Acetylglucosamine (GlcNAc) to the initial linker chain (Figure 1A) (15, 16). Next, Exostosin-1 (EXT1) and −2 (EXT2), and Exostosin-like 1 (EXTL1) and EXTL3, extend the chain by alternatively transferring GlcNAc and D-Glucuronic acid (GlcA) residues to the sugar chain (17). This HS chain is then modified by the enzymes bifunctional heparan sulfate N-deacetylase/N-sulfotransferase 1–4 (NDST1–4) that have both N-deacetylase and N-sulfotransferase activities (18). Other enzymes involved in HS sulfation include heparan sulfate 2-O-sulfotransferase (H2ST), 6-O-sulfotransferases (H6ST1–3), and 3-O-sulfotransferases (H3ST) 1, 2, 3A, 3B, 4, 5, and 6 (19). These NDST enzymes and sulfotransferases rely on the sulfur donor 3’-phosphoadenosine 5’-phosphosulfate (PAPS), which is synthesized by 3’-phosphoadenosine 5’-phosphosulfate synthase-1 (PAPSS1) and −2 (PAPSS2) in the cytosol and transported by adenosine 3’-phospho 5’-phosphosulfate transporter 1 (PAPST1) and PAPST2 into the Golgi lumen (20–23). Most HS chains are sulfated, which significantly affects their activity and function. For example, it has been shown that 3-O-sulfation of HS increases its binding with Tau at the cell surface, which facilitates Tau internalization (24).
For CS, chain formation initiates with N-acetylgalactosaminyltransferases, GalNAcT1–2, and is then elongated by chondroitin sulfate synthase 1–3 (CHSY1–3) and chondroitin sulfate glucuronyltransferase (CHPF) that alternatively transfer GalNAc and GlcA residues to the sugar chain (Figure 1A) (19). CS is sulfated by sulfotransferases chondroitin 4-sulfotransferase 1–3 (C4ST1–3), chondroitin 6-sulfotransferase 1–2 (C6ST1–2) and N-acetylgalactosamine 4-sulfate 6-O-sulfotransferase (GalNAc4S-6ST), or by dermatan sulfate epimerases 1–2 (DS-epi 1–2) if the 2-O-sulfated D-glucuronic acid (GlcA) residues are C5-epimerized to L-iduronic acid (IdoA). Subsequently, when the chain is further sulfated by dermatan sulfotransferase D4ST, uronyl 2-O-sulfotransferase (UST), or GalNAc4S-6ST, it is referred to as dermatan sulfate (DS) (19). As with HS, the sulfotransferases for CS also rely on the sulfur donor PAPS.
While the synthesis of the tetrasaccharide linker of HS and CS is initiated in the ER or ERGIC, the bulk parts of HS and CS are synthesized in the Golgi lumen (25), similar to that of N-linked glycans. Consistently, most enzymes involved in HS and CS synthesis, such as EXT1 and EXT2 in HS synthesis and GalNAcT-1 in CS synthesis, reside in the Golgi (26, 27). Therefore, it is reasonable to speculate that Golgi structural defect may significantly impact the synthesis of HS and CS as N-glycans as we previously showed (6). Indeed, depletion of certain subunits of the Conserved Oligomeric Golgi (COG) complex, which targets Golgi enzymes to their proper locations within the Golgi, reduces GAG modification (28); while depletion of giantin, a membrane tether in the Golgi, reduces the mRNA level of polypeptide N-acetylgalactosaminyltransferase 3 (GALNT3/GalNAcT3) (29). However, the relationship between Golgi structural integrity and GAG synthesis has not been systematically explored.
In this study, we disrupted the Golgi structure by knocking out GRASP55 and GRASP65 and determined the effect on GAG synthesis, sulfation, and secretion. We also performed proteomic and RNA-seq analysis to identify the enzymes whose alternation is responsible for the defects in HS and CS synthesis in GRASP knockout (KO) cells.
Results
GRASP KO increases GAG synthesis but decreases their secretion.
Given that GRASP55 and GRASP65 are major regulators of Golgi stack formation, we knocked them out, single or in combination, in HeLa cells to disrupt the Golgi structure (3), and thereby determined the effect on GAG synthesis. As shown in Figure 1B, we cultured wildtype (WT), GRASP55 knockout (55KO), GRASP65 knockout (65KO), and GRASP55 and GRASP65 double knockout (DKO) cells in serum-free medium for 8 h, collected cell lysate and conditioned media, and performed GAG analysis by liquid chromatography-mass spectrometry (LC-MS) (Supplemental Tables 1 and 2). The amount of GAGs, including (HS, CS and HA) in the cell lysate was increased in GRASP KO cells, with its level in 65KO the highest (Figure 1C). In contrast to the cell lysate, the amount of GAGs in the conditioned media was reduced (Figure 1D). The total amount of GAGs (cells + media) was higher in GRASP KO cells compared to WT, again with 65KO to be the highest (Figure 1E). In summary, disruption of the Golgi structure by GRASP depletion increases GAG synthesis while reducing its secretion.
GRASP KO increases HS synthesis but decreases its sulfation and secretion.
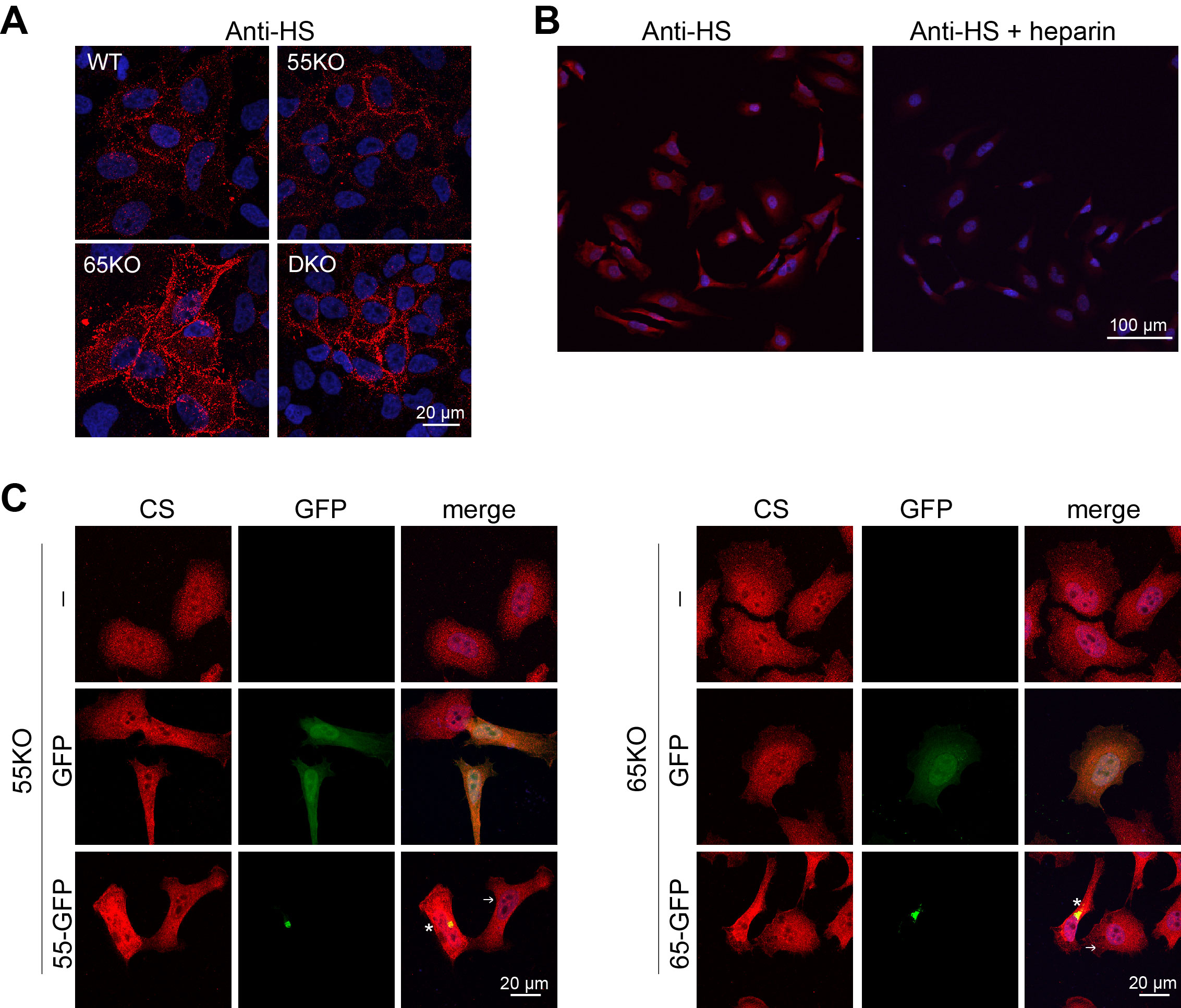
Given that HS and CS but not HA are synthesized in the Golgi, we further characterized HS and CS synthesis and sulfation in GRASP KO cells. When the Golgi structure was disrupted by GRASP KO, HS synthesis was significantly increased compared to WT cells as analyzed by LC-MS (Figure 2A; Supplemental Table 1–2). The increase of HS in GRASP KO cells or at the cell surface was confirmed by immunostaining of HS with an anti-HS antibody 10E4 followed by immunofluorescence microscopy with or without permeabilization (Figure 2B; Supplemental Figure 1A). These results were confirmed in three different clones of 55KO, 65KO and DKO cell lines (Supplemental Figure 1C). The HS signal by this antibody was specific as it was largely quenched by the preincubation of the antibody with heparin (Supplemental Figure 1B). The increased level of HS in GRASP KO cells was further validated by flow cytometry (Figure 2C).
Figure 2. GRASP KO increases HS synthesis but reduces its sulfation and secretion.
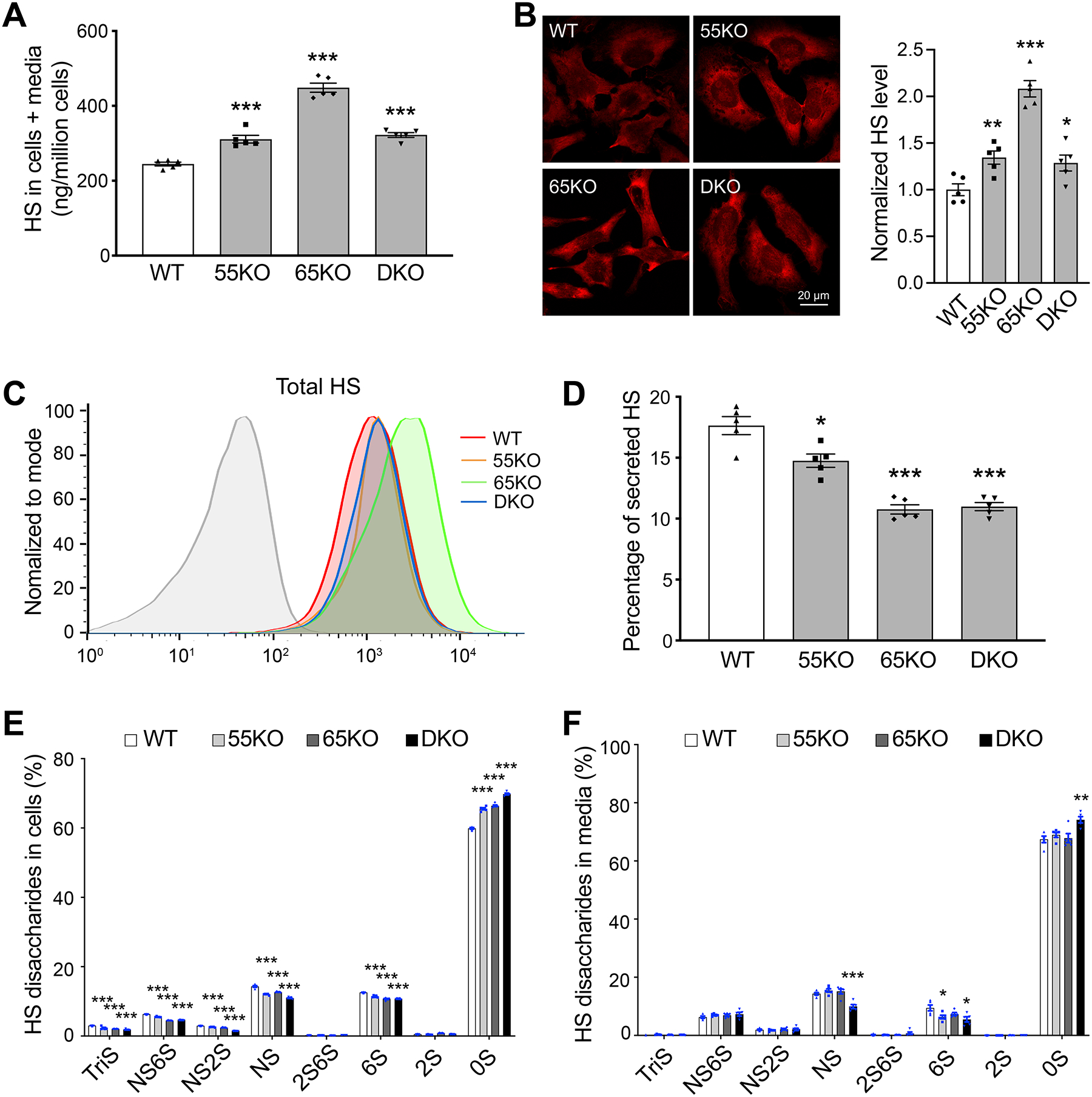
(A) GRASP KO increases HS synthesis analyzed by LC-MS. Shown are the total amount of HS in both cells and medium of indicated cell lines. (B) GRASP KO increases HS synthesis analyzed by immunofluorescence microscopy. Indicated cells were permeabilized and stained for HS with an HS antibody 10E4. Shown are microscopic images (left) and quantitation (right). (C) GRASP KO increases HS synthesis analyzed by flow cytometry. Indicated cells were permeabilized, stained for HS with an HS antibody and analyzed by flow cytometry. (D) GRASP KO decreases HS secretion. The percentage of secreted HS (HS in conditioned media/HS in both cell lysate and conditioned media) was analyzed by LC-MS. (E) GRASP KO reduces HS sulfation in the cell lysate. Shown are the percentage of each sulfated form of HS in cells. (F) GRASP KO reduces HS sulfation in the conditioned media. Shown are the percentage of each sulfated form of HS in the conditioned media.
HS is covalently linked to core proteins that are secreted by cells, so we analyzed the level of HS in the conditioned media. GRASP KO largely reduced HS secretion compared to that of WT cells (Figure 2D). Although the absolute amount of HS in the media was not reduced by GRASP KO, the percentage of HS in the media was significantly lower in the KOs than WT due to the increased HS synthesis (Supplemental Tables 1–2). Given the importance of HS sulfation, we also quantified the different sulfated forms of HS in cells and conditioned media by 2-aminoacridone (AMAC) labeling and LC-MS (Supplemental Tables 3 and 4). In contrast to the increased level of HS synthesis, the overall sulfation of HS was significantly reduced by GRASP KO in both cells and conditioned media (Figure 2E–F). This indicates that Golgi destruction via GRASP depletion negatively regulates the sulfation pathway of HS. Taken together, disruption of the Golgi structure by GRASP KO increases HS synthesis but decreases its sulfation and secretion.
GRASP KO decreases CS synthesis and secretion.
Like HS, CS is synthesized in the Golgi and sulfated. HS and CS share the same tetrasaccharide precursor, which branches into either the HS pathway via the action of the EXTL enzymes, or the CS pathway via the reaction of the GalNAcT enzymes (Figure 1A). Therefore, it is reasonable to speculate that increased branching into the HS pathway may lead to reduced branching into the CS pathway. Indeed, GRASP KO reduced CS synthesis as analyzed by LC-MS, the opposite to HS (Figure 3A; Supplemental Tables 1 and 2). To confirm this result by an alternative approach, we stained WT and GRASP KO cells with an CS antibody (CS-56) and analyzed the levels of CS by fluorescence microscopy and flow cytometry. Consistent with the LC-MS results, GRASP KO reduced the level of CS in cells compared to WT (Figure 3B–C). Here, the degree of CS reduction examined by microscopy and flow cytometry was more dramatic than by LC-MS. The cause of this difference could be that LC-MS includes both CS and DS in the results, while the CS antibody only recognizes CS but not DS (30), which more accurately reflects the CS level in cells.
Figure 3. GRASP KO reduces CS synthesis and secretion.
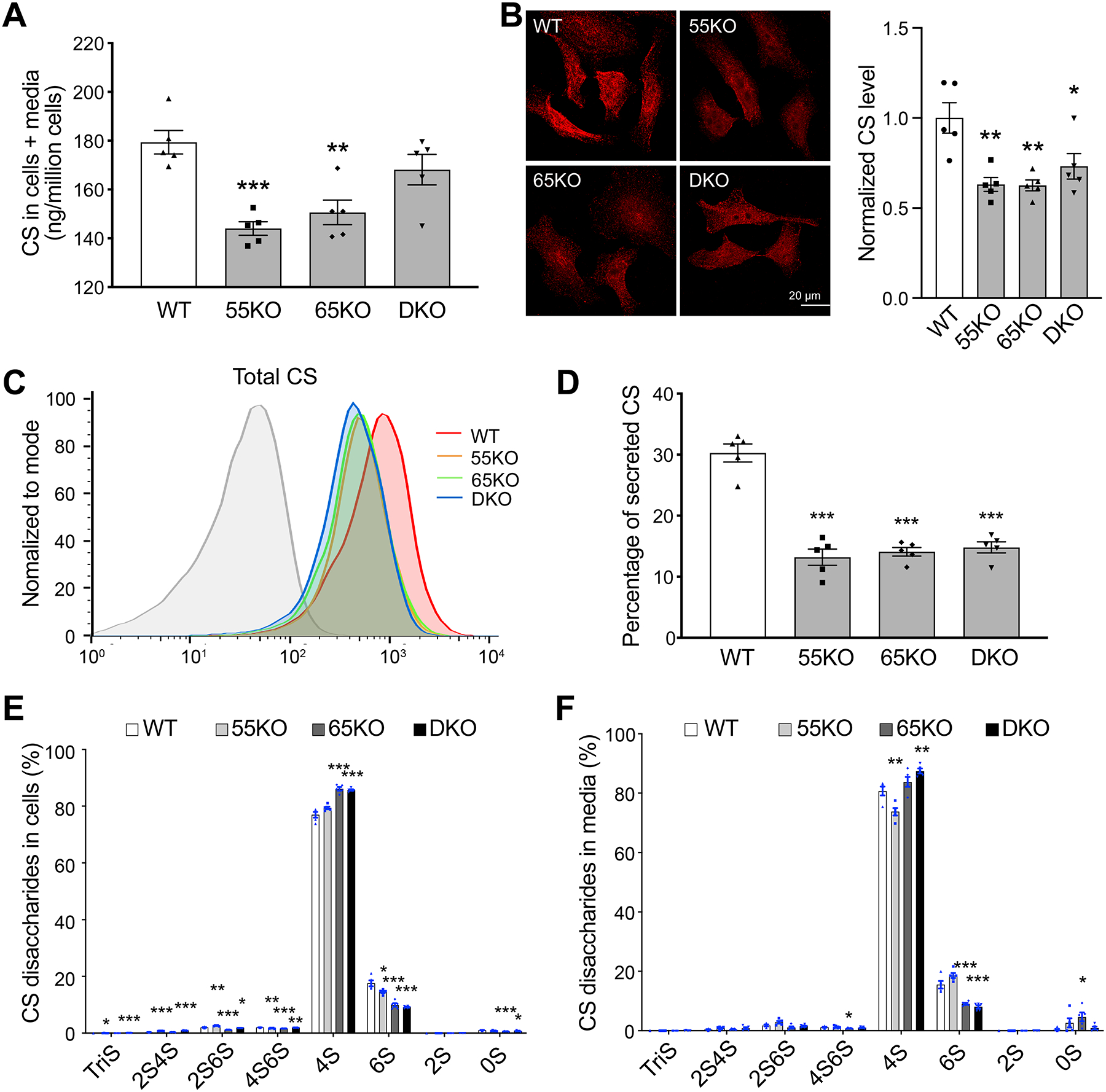
(A) GRASP KO reduces CS synthesis analyzed by LC-MS. Shown are the total amount of CS in both cells and medium of indicated cell lines. (B) GRASP KO decreases CS synthesis analyzed by immunofluorescence microscopy. Indicated cells were permeabilized and stained for CS with a CS antibody (CS-56). Shown are microscopic images (left) and quantitation (right). (C) GRASP KO decreases CS synthesis shown by flow cytometry. Indicated cells were permeabilized, stained for CS with a CS antibody and analyzed by flow cytometry. (D) GRASP KO decreases CS secretion. The percentage of secreted CS (CS in conditioned media/CS in both cell lysate and conditioned media) was analyzed by LC-MS. (E) GRASP KO alters CS sulfation in the cell lysate. Shown are the percentage of each sulfated form of CS in cells. (F) GRASP KO alters CS sulfation in the conditioned media. Shown are the percentage of each sulfated form of CS in the conditioned media. Note that GRASP depletion increased 4-sulfation while decreases 6-sulfation in both the cell lysate (E) and conditioned media (F).
Next, we analyzed the secretion of CS in WT and GRASP KO cells. Both the amount and percentage of CS in the conditioned media were largely reduced by GRASP KO (Figure 3D; Supplemental Tables 1–2). Lastly, we analyzed the different subtypes of CS sulfation and found that GRASP KO increased 4-sulfation but decreased 6-sulfation in both cell lysate and conditioned media (Figure 3E–F; Supplemental Tables 5 and 6). Taken together, disruption of the Golgi structure by GRASP KO decreases CS synthesis, alters its sulfation, and decreases its secretion.
GRASP KO regulates key enzymes in HS and CS synthesis and sulfation.
The regulation of HS and CS synthesis and sulfation is complex and involves numerous enzymes (Figure 1A). A majority of these enzymes are localized in the Golgi and thus their level and localization could be regulated by Golgi structural changes. Therefore, we performed systematic RNA-seq and proteomic analysis of WT and GRASP KO cells to identify genes related to the observed alterations in HS and CS synthesis and sulfation. As expected, many enzymes involved in HS and CS synthesis were affected by GRASP KO (Table 1). Consistent with the increased HS level in cells, the mRNA levels of several HS synthesis enzymes, such as EXTL2, EXTL3, EXT1, NDST1 and PAPST2 (SLC35B3), were increased in GRASP KO compared to WT cells. Similar to the trend of CS reduction in GRASP KO cells, the mRNA levels of many CS synthesis enzymes, including GalNAcT1, CHSY1, C4ST2, GalNAc4S-6ST, C6ST1 and DS-epi1, were all decreased upon GRASP KO (Figure 4A). To analyze the alterations in HS and CS synthesis more systematically, we applied the RNAseq data to the “GlycoMaple” program for prediction of the changes in specific steps of HS and CS synthesis upon GRASP KO. The results revealed an increase in multiple steps of HS synthesis while reduction in multiple steps of CS synthesis (Supplemental Figure 1D).
Table 1. GRASP KO alters HS and CS synthesis enzymes at both the mRNA and protein levels.
WT and GRASP KO cells were analyzed by proteomic and RNA-seq analyses. Shown are the fold changes of the indicated GAG synthesis enzymes in GRASP KO cells compared to that in WT cells. Enzymes in bold were further tested by Western blot.
| Enzymes | Proteomics | RNA-seq | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Common Enzymes | p-value | p-value | p-value | p-value | p-value | |||||
| XylT1 | NA | NA | NA | |||||||
| XylT2 | 0.770 | 0.290 | 0.740 | |||||||
| GalT1 (B4GALT7) | 0.440 | 0.500 | 0.590 | |||||||
| GalT2 | 0.020 | 0.340 | 0.840 | |||||||
| GlcAT1 (B3GAT3) | 0.210 | 0.750 | 0.960 | |||||||
| PAPSS 1 | 0.411 | 0.082 | 0.990 | 0.790 | 0.460 | |||||
| PAPSS 2 | 1.854E-05 | 2.289E-06 | 7.920E-10 | 2.269E-06 | 3.599E-06 | |||||
| PAPST 1 (SLC35B2) | 0.034 | 0.256 | 0.750 | 0.080 | 0.060 | |||||
| PAPST 2 (SLC35B3) | 0.190 | 0.800 | 0.050 | |||||||
| Enzymes in HS synthesis | ||||||||||
| EXTL2 | 0.420 | 0.030 | 0.670 | |||||||
| EXTL3 | 0.120 | 0.620 | 0.160 | |||||||
| EXTL1 | NA | NA | NA | |||||||
| EXT1 | 0.420 | 3.090E-05 | 0.430 | |||||||
| EXT2 | 0.790 | 0.690 | 0.240 | |||||||
| NDST 1 | 0.260 | 0.600 | 0.400 | |||||||
| NDST 2 | 0.620 | 0.960 | 0.940 | |||||||
| NDST 3 | 0.700 | 0.200 | 0.850 | |||||||
| NDST 4 | ||||||||||
| HS C5-EP (HSGLCE) | ||||||||||
| H2ST | ||||||||||
| H3ST 1 | ||||||||||
| H3ST 2 | ||||||||||
| H3ST 3A | ||||||||||
| H3ST 3B | ||||||||||
| H3ST 4 | ||||||||||
| H3ST 5 | ||||||||||
| H3ST 6 | ||||||||||
| H6ST 1 | ||||||||||
| H6ST 2 | ||||||||||
| H6ST 3 | ||||||||||
| Enzymes in CS synthesis | ||||||||||
| GalNAcT1 (GALNT1) | 8.206E-05 | 1.34308E-05 | 2.856E-09 | 3.668E-26 | 7.254E-42 | |||||
| GalNAcT2 (GALNT2) | 0.094 | 0.111 | 0.480 | 0.040 | 0.040 | |||||
| CHPF2 | 0.368 | 0.227 | 0.950 | 0.910 | 0.540 | |||||
| CHSY1 | 8.023E-07 | 9.417E-05 | 1.442E-03 | |||||||
| CHSY2 | ||||||||||
| CHSY3 | 0.790 | 0.900 | 0.740 | |||||||
| C4ST 1 (CHST11) | 0.472 | 0.461 | 0.424 | |||||||
| C4ST 2 (CHST12) | 0.732 | 0.935 | 0.925 | |||||||
| C4ST 3 (CHST13) | 0.635 | 0.240 | 0.008 | |||||||
| GalNAc4S-6ST (CHST15) | 0.866 | 0.003 | 0.007 | |||||||
| C6ST 1 (CHST3) | 0.471 | 0.005 | 1.600E-04 | |||||||
| C6ST 2 (CHST4) | NA | NA | NA | |||||||
| UST | ||||||||||
| DS-epi 1 (DSE) | 6.146E-10 | 3.265E-28 | 7.330E-12 | |||||||
| DS-epi 2 (DSEL) | 8.811E-05 | 8.939E-03 | 9.967E-01 | |||||||
| D4ST (CHST14) | ||||||||||
| Enzymes in HA synthesis | ||||||||||
| HAS1 | NA | NA | NA | |||||||
| HAS2 | 0.850 | 0.888 | 0.356 | |||||||
| HAS3 | 0.992 | 0.819 | 0.915 | |||||||
NA, data not available; ND, not detected.
Figure 4. GRASP KO alters the expression level of GAG synthesis and sulfation enzymes.

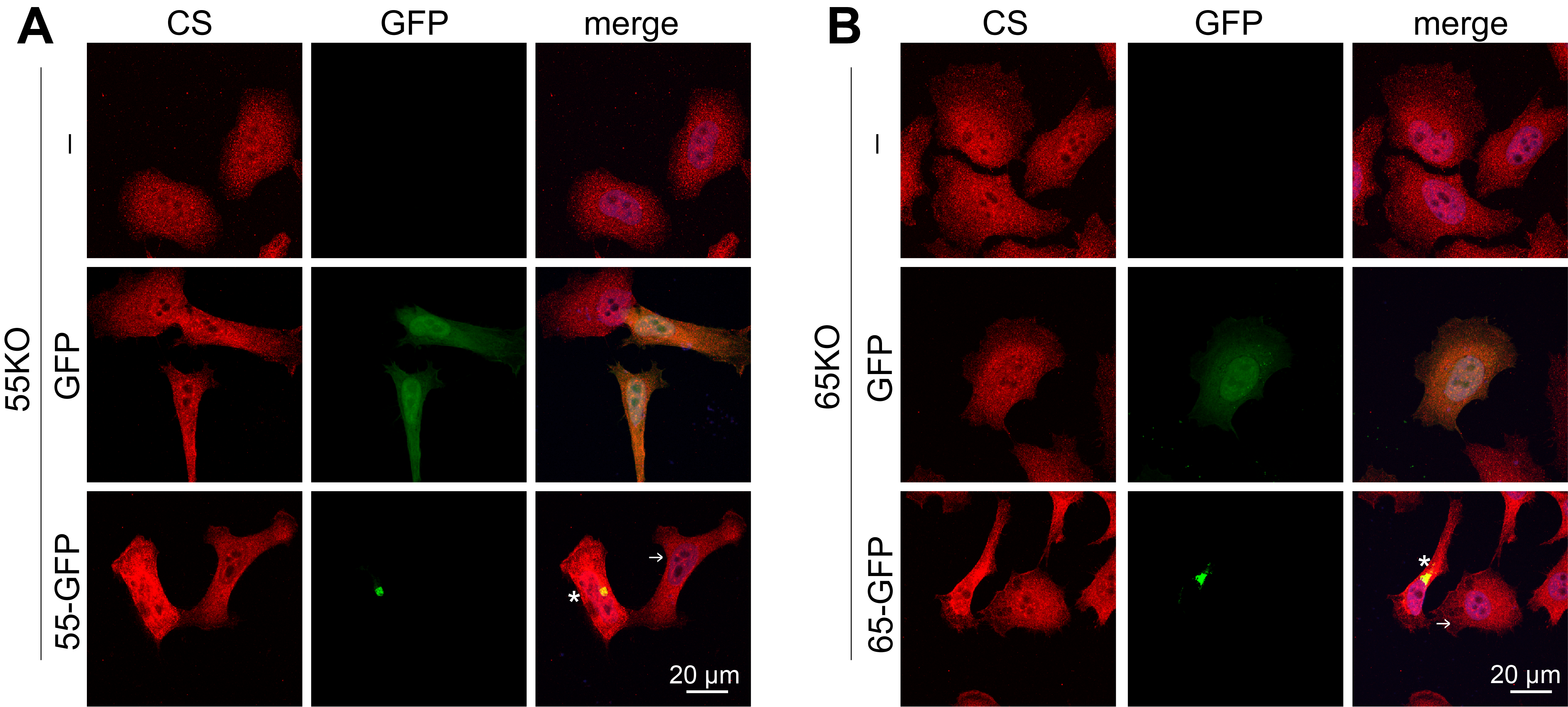
(A) GRASP KO increases the expression of HS synthesis enzymes while decreases CS synthesis enzymes. Results are based on RNA-Seq analysis of each cell line for indicated genes. (B) GRASP KO increases the protein level of HS synthesis enzymes while decreases that of CS synthesis enzymes. Cell lysate of indicated cells were analyzed for three HS synthesis enzymes EXTL3, EXT1 and EXT2, and a key CS synthesis enzyme GalNAcT1. Note the increased level of EXTL3 and decreased GalNAcT1 level in GRASP KO cells. Results are representative of three independent experiments. (C) Quantification of B. (D) GRASP KO decreases the protein level of the HS sulfation enzyme PAPSS2. Shown are representative Western blots of indicated proteins in the four cell lines from three independent experiments. (E) Quantitation of D. (F) Re-expression of GRASP proteins in GRASP KO cells corrects the expression level of HS and CS synthesis enzymes. Indicated cell lines were transfected with GRASP constructs and probed for EXTL3, GalNAcT1, GRASP65, GRASP55, GFP, and actin. The major enzymes EXTL3 and GalNAcT1 in 55KO and 65KO cells were rescued by expressing GRASP55-GFP or GRASP65-GFP, respectively, but not by GFP alone (lanes 4 & 7 vs. 3 & 6). (G) Re-expression of GRASP proteins corrects the HS and CS defects in GRASP KO cells. Confocal images of WT HeLa cells and GRASP KO cell lines transfected with indicated constructs followed by HS staining. The level of HS in 55KO and 65KO cells was decreased by the expression of GRASP55-GFP or GRASP65-GFP, respectively, but not by GFP alone. Note the different HS signals in cells expressing GRASP55- or GRASP65-GFP (asterisks) vs. non-transfected cells (arrows). (H-I) Quantification of G. Results are presented as mean ± SEM, statistical analysis was assessed by comparing KO cells to WT cells by student’s t-test. *, p<0.05; **, p<0.01; ***, p<0.001.
The altered expression of HS and CS synthesis enzymes in GRASP KO cells was further confirmed at the protein level by proteomic analysis (Table 1) and Western blots (Figure 4B). GRASP KO increased the protein level of EXTL3, a key enzyme in HS synthesis, while decreased the level of GalNAcT1 in the CS synthesis pathway (Figure 4B–C). The upregulation of EXTL3 in GRASP KO cells was further confirmed by immunofluorescence microscopy, although its Golgi localization was unaffected by GRASP KO (Supplemental Figure 1E). Furthermore, the upregulation of EXTL3 and downregulation of GalNAcT1 protein levels were confirmed in three different clones of 55KO, 65KO, and DKO cell lines (Supplemental Figure 1F). Interestingly, the increase of HS level in 65KO was more dramatic than that in 55KO and DKO cells. Correlatively, the protein levels of EXT1 and EXT2 were slightly higher in 65KO cells compared to ctrl, 55KO or DKO (Figure 4B), which might explain why 65KO cells have the highest HS level in all cell lines. It is unclear, however, how EXT2 is more upregulated in 65KO but not 55KO and DKO cells.
To decipher the underlying mechanism of the reduced HS sulfation in GRASP KO cells, we analyzed the levels of multiple HS sulfation enzymes in the RNA-seq data and found that GRASP KO reduced the expression of the sulfur synthase PAPSS2 and the major PAPS transporter PAPST1 (Table 1). Consistently, the protein level of PAPPS2 was also significantly lower in GRASP KOs than WT cells as shown by proteomic analysis (Table 1) and western blot (Figure 4D–E). Unlike a global reduction in HS sulfation, we only observed a significant shift from 6-sulfation (6S) to 4-sulfation (4S) in CS (Figure 3E–F). GRASP depletion largely increased the ratio of 4S/6S compared to WT. Repeating disaccharide units of CS are sulfated at C4 and C6 by C4ST-1 and C6ST-1, respectively. In our RNA-seq analysis, the mRNA levels of C6ST1 and C6ST2 were significantly decreased in GRASP KO cells, especially 65KO and DKO (Table 1), which may explain the reduction of 6S in GRASP KO cells.
To confirm that the observed effects in HS and CS were caused by GRASP KO, we transfected 55KO and 65KO cells with GRASP55-GFP and GRASP65-GFP, respectively, which has previously been shown to rescue the Golgi structure and correct the defects in N-glycosylation and cell attachment (3, 6, 7). Indeed, re-expression of GRASP55 and GRASP65 in the corresponding KO cells not only restored the levels of major enzymes such as EXTL3 and GalNAcT1 (Figure 4F), but also normalized the levels of HS (Figure 4G–I) and CS (Supplemental Figure 2). Taken together, these results revealed that Golgi structure formation and defect regulate HS and CS synthesis and sulfation through modulating the expression of key enzymes.
Discussion
In this study we found that GRASP depletion and subsequent disruption of the Golgi structure increased overall GAG synthesis but decreased their secretion. As HS and CS are the two main types of GAGs synthesized in the Golgi, we further analyzed their levels and sulfation in WT and GRASP KO cells. Our results revealed that GRASP depletion increased HS synthesis but reduced its sulfation mainly through the upregulation of EXTL3 and downregulation of PAPSS2, respectively. GRASP depletion, however, reduced CS synthesis by decreasing GalNAcT1 expression; GRASP KO also altered the balance between 4-sulfation and 6-sulfation of CS, two main forms of CS detected in LC-MS. These effects were due to GRASP depletion as re-expression of GRASPs corrected the observed defects in HS and CS synthesis.
HS and CS levels and sulfation are tightly related to their functions in various biological processes including extracellular matrix (ECM) assembly, cell adhesion, coagulation and immune response (31). It has been shown that abnormal sulfation of HS causes defects in FGF-2–induced proliferation and survival of multipotent progenitor cells via reducing FGF-2 and FGFR1 interaction, which contribute to Hurler syndrome (32). Similarly, in Drosophila melanogaster and Caenorhabditis elegans, reduced HS sulfation results in a delay in wound closure and defects in actin stress fiber formation (33). In another example, reduction of functional heparan sulfate proteoglycan (HSPG) has been shown to increase pericyte number while reducing its adhesion to nascent sprouts via the regulation of transforming growth factor β signal transduction (34). Similar to HS, CS and its synthesis enzymes are also altered in multiple disease conditions. Deficiency of an essential chondroitin synthase CHSY1 causes Temtamy preaxial brachydactyly syndrome (TPBS) (35). GalNAcT1 deficient mice showed defects in heart valve development and cardiac function via the remodulation of ECM and mitogen-activated protein kinase (MAPK) signaling pathway (36). In developing mouse brain, the 6S level of CS is gradually decreased while 4S is gradually increased, resulting in a progressive increase of 4S/6S ratio during brain development. This change in CS sulfation was shown to reduce cortex plasticity (37). Interestingly, in neurodegenerative diseases such as Alzheimer’s, the Golgi is fragmented possibly due to the loss of function of the GRASP proteins (38), indicating a potential link between Golgi fragmentation, increased 4S/6S ratio, and reduced cortex plasticity in aging and neurodegenerative diseases.
How Golgi structural defect and/or GRASP depletion affect the expression of HS and CS synthesis and sulfation enzymes is an interesting but unanswered question. This not only includes EXTL3 and GalNAcT1 that reside in the Golgi, but also PAPSS2 that is localized in the cytosol. It has been shown that many signaling molecules including mTOR, KRAS and some transcription factors such HIF1a are localized on the Golgi (39–41). Golgi unstacking may affect these signaling pathways or the activity of the transcription factor, which in turn may regulate the expression of HS and CS enzymes. Most recently, it has been shown that a transcription regulator, NFIL3 (nuclear factor, interleukin 3 regulated), binds to the promoter region of EXTL3 and suppresses its transcription (42). Interestingly, our RNA-seq analysis revealed that NFIL3 is downregulated in GRASP KOs compared to WT cells. It is possible that GRASP depletion reduces EXTL3 expression via the downregulation of NFIL3. Similar to GAG synthesis enzymes, it has been shown that GRASP depletion reduces the synthesis of α5β1 integrins, major cell adhesion molecules at the cell surface, which subsequently decreases cell adhesion but increases cell growth (7). This indicates an exciting possibility that cells may possess a sensing mechanism for Golgi structural changes, which when activated, may regulate the expression of multiple proteins to control different cellular activities. This will be tested in our future studies.
The disruption of the Golgi stacks by GRASP-depletion used in our study is fundamentally different from the block of ER-to-Golgi trafficking by brefeldin A (BFA) treatment used in previous studies. It has been reported that disruption of the Golgi structure by BFA treatment affects HS and CS biosynthesis (43, 44). BFA blocks ER-to-Golgi trafficking and causes a merge of the Golgi stack (not the TGN) to the ER. Because HS and CS synthesis enzymes are localized in distinct sub-compartments of the Golgi, BFA treatment affects HS and CS synthesis differently. Unlike BFA treatment, GRASP depletion disrupts the Golgi stack structure (2) but does not block membrane trafficking (3, 6). In addition, our systematic RNA-seq and proteomic studies identified critical genes whose alterations are responsible for the changes in HS and CS synthesis and sulfation. To our knowledge, this is the first study that links Golgi structure formation to the transcriptional regulation of O-glycosylation enzymes.
It was surprising to see that the secretion of both HS and CS was reduced in GRASP KO cells compared to WT as it has previously been shown that GRASP depletion accelerated protein trafficking through the Golgi membranes (6, 45, 46). This result can be explained in several ways. First, given that all marker proteins used in the previous studies, including the vesicular stomatitis virus G (VSV-G) protein, CD8, and α5β1 integrins, are mainly modified by N-glycosylation, it is possible that GRASP depletion and/or Golgi structural disruption may affect the trafficking and secretion of different cargo molecules differently. Similarly, it has been shown that GRASP depletion alters the level of glycolipids by decreasing the level of globotriaosylceramide (Gb3) and increasing the level of monosialotetrahexosylganglioside (GM1) (3). Second, altered sulfation of HS and CS may affect the secretion and stability of the core proteins. As sugar modifications affect protein stability and activity (47), reduced sulfation may lead to certain core proteins to be sent for degradation instead of secretion. Third, there is a possibility that more HS and CS are degraded in the conditioned media of GRASP KO cells. It has previously been shown that GRASP depletion causes mis-sorting of lysosomal enzymes and results in their secretion (6). It is possible that GRASP KO cells may secrete HS and CS degrading enzymes such as endoglycosidases and exohydrolases that normally reside in the lysosomes (48). Nevertheless, the molecular mechanism that reduces HS and CS secretion in GRASP depleted cells requires further investigation.
Taken together, our results showed that disruption of the Golgi stacked structure via GRASP depletion led to the increase of total GAG synthesis, where HS level was increased due to the upregulation of EXTL3 expression and CS level was reduced because of GalNAcT1 down-regulation. In addition, Golgi defect also reduced HS sulfation via the reduction of PAPSS2. In summary, this study revealed that Golgi structural integrity and GAG synthesis are tightly linked.
Materials and Methods
Cell culture and transfection
Wild type, GRASP55 knockout (55KO), GRASP65 knockout (65KO), GRASP55 and GRASP65 double knockout (DKO) HeLa cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM 4.5 g/l glucose) supplemented with 10% bovine calf serum (Hyclone, SH30072.03), 1% L-glutamine, 1% penicillin-streptomycin under 5% CO2 at 37°C as previously described (3). GRASP single and double knockout cell lines were previously established (3). In brief, multiple sg-RNAs targeting the coding regions of GRASP55 and GRASP65 were cloned into Cas9 vector pSpCas9(BB)-2A-Puro(PX459) for puromycin selection or pSpCas9(BB)-2A-GFP(PX458) vector for cell sorting to select positive cells. Single clones of CRISPR knockouts of both GRASP55 and GRASP65 were made by co-transfection of cells with two constructs and selected by puromycin and cell sorting. Knockout was confirmed by Western blotting, immunofluorescence and genomic sequencing.
To express exogenous GRASP proteins, HeLa cells of ~50% confluency were transfected with indicated GRASP constructs (2, 49). For a 6 cm plate, 6 μg of pEGFP-N1-GRASP65 or pEGFP-N1-GRASP55 (both wild type) construct was mixed with 18 μl polyethylenimine (PEI) and 0.5 ml serum-free medium for 15 min at room temperature and then added to the cells in 4 ml DMEM containing 10% bovine calf serum. For control transfection, 4 μg of pEGFP-N1 construct was mixed with 18 μl PEI and 0.5 ml serum-free medium for 15 min and then added to the cells in 4 ml DMEM containing 10% super calf serum. At 24 h post-transfection, cells were collected for western blotting or immunofluorescence microscopy. In the rescue experiments (Figure 4 F–I; Supplemental Figure 2), the transfection rates of GRASP65 and GRASP55 were above 70%.
Materials and sample preparation for LC-MS and PAGE analysis
Unsaturated disaccharide standards of HS (ΔUA-GlcNAc; ΔUA-GlcNS; ΔUA-GlcNAc6S; ΔUA2S-GlcNAc; ΔUA2S-GlcNS; ΔUA-GlcNS6S; ΔUA2S-GlcNAc6S; ΔUA2S-GlcNS6S), unsaturated disaccharide standards of CS (ΔUA-GalNAc; ΔUA-GalNAc4S; ΔUA-GalNAc6S; ΔUA2S-GalNAc; ΔUA2S-Gal-NAc4S; ΔUA2S-GalNAc6S; ΔUA-GalNAc4S6S; ΔUA2S-GalNAc4S6S), and unsaturated disaccharide standard of HA (ΔUA-GlcNAc), where ΔUA is 4-deoxy-α-L-threo-hex-4-enopyranosyluronic acid, were purchased from Iduron (UK). Actinase E was obtained from Kaken Biochemicals (Japan). Chondroitin lyase ABC from Proteus vulgaris was expressed in Linhardt’s laboratory. Recombinant Flavobacterial heparin lyases I, II, and III were expressed in Linhardt’s laboratory using Escherichia coli strains provided by Jian Liu (College of Pharmacy, University of North Carolina). 2-Aminoacridone (AMAC), sodium cyanoborohydride were obtained from Sigma-Aldrich (St. Louis, MO, USA). All solvents used in LC-MS were HPLC grade.
GAG preparation for disaccharide analysis
Cells were proteolyzed at 55°C with 500 μl of 10-mg/mL actinase E for 24 h and followed by actinase E deactivation at 100°C for 30 min. The volume of the above solution containing 2 million cells was transferred to a 3-kDa molecular weight cut off (MWCO) spin tube. The filter unit was washed three times with 400 μl distilled water and then added with 300-μl digestion buffer (50 mM ammonium acetate containing 2 mM calcium chloride adjusted to pH 7.0). Recombinant heparin lyase I, II, III (pH optima 7.0−7.5) and recombinant chondroitin lyase ABC (pH optimum 7.4, 10 mU each) were added to each filter unit containing sample and mixed well. The samples were all incubated at 37 °C for 24 h. The enzymatic digestion was terminated by ultrafiltration through the 3-kDa spin tube. The filtrate was collected, and the filter unit was washed twice with 200 μl distilled water. All the filtrates containing the disaccharide products were combined and dried via freeze dry. For the medium samples, 400-μl medium from each specimen was ultrafiltrated through a 3-kDa molecular weight cut off (MWCO) spin tube to remove small molecular compounds, and then went through the same digestion procedure as mentioned above.
AMAC labeling of disaccharides
The dried samples were AMAC-labeled by adding 10 μl of 0.1 M AMAC in DMSO/acetic acid (17/3, V/V) incubating at room temperature for 10 min, followed by adding 10 μl of 1 M aqueous sodium cyanoborohydride and incubating for 1 h at 45°C.A mixture containing all 17-disaccharide standards prepared at 0.5ng/μl was similarly AMAC-labeled and used for each run as an external standard. After the AMAC-labeling reaction, the samples were centrifuged, and each supernatant was recovered.
LC-MS GAG analysis
LC was performed on an Agilent 1200 LC system at 45 °C using an Agilent Poroshell 120 ECC18 (2.7 μm, 3.0 × 50 mm) column. Mobile phase A (MPA) was 50 mM ammonium acetate aqueous solution, and the mobile phase B (MPB) was methanol. The mobile phase passed through the column at a flow rate of 300 μl/min. The gradient was 0–10 min, 5–45% B; 10–10.2 min, 45–100%B; 10.2–14 min, 100%B; 14–22 min, 100–5%B. Injection volume is 5 μl.
A triple quadrupole mass spectrometry system equipped with an ESI source (Thermo Fisher Scientific, San Jose, CA) was used a detector. The online MS analysis was at the Multiple Reaction Monitoring (MRM) mode. MS parameters: negative ionization mode with a spray voltage of 3000 V, a vaporizer temperature of 300°C, and a capillary temperature of 270°C.
Western blot
Wild type and GRASP KO cells are lysed in 20 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1% Triton X-100 and protease inhibitors for 30 min on ice. Lysates were cleared by centrifugation (20,000 g for 20 min at 4°C). After electrophoresis and transfer, nitrocellulose membranes were incubated with antibodies to actin (Sigma, A2066), EXT1 (Santa Cruz, sc-515144), EXT2 (Santa Cruz, sc-514092), EXTL3 (Santa Cruz, sc-271986), GalNAcT1 (Novus, NBP1–81852), GFP (Proteintech, 66002–1-Ig), GRASP55 (Proteintech, 10598–1-AP), GRASP65 (Santa Cruz, sc-374423), or PAPSS2 (Santa Cruz, sc-271429) overnight at 4°C. The membranes were extensively washed and further incubated with HRP conjugated goat anti-Rabbit or goat anti-mouse secondary antibodies for 1 h at room temperature and exposed to a FluorChem M machine (Proteinsimple).
Immunofluorescence microscopy
Cells were grown on sterile glass coverslips and rinsed with phosphate buffered saline (PBS) before fixation. For total protein staining, cells were fixed in 4% paraformaldehyde for 10 min and permeabilized with 0.2% Triton X-100 in PBS for 10 min. For cell surface staining, cells were fixed in 1% paraformaldehyde for 10 min and not permeabilized. Cells were incubated with primary antibodies for HS (10E4, Amsbio 370255, 1:100) and CS (CS-56, Abcam ab11570, 1:50) overnight at 4°C, washed and probed with the appropriate secondary antibodies conjugated to TRITC for 45 min. To confirm that the detected 10E4 signal was specific to HS, the HS antibody (10E4 1:100) was incubated with or without 40 μg/ml heparin overnight at 4°C and then used for primary antibody staining of fixed cells. DNA was stained with Hoechst for 5 min. Coverslips were rinsed with PBS and mounted with Mowiol onto slides. Images were taken with a 20x air objective or a 63x oil objective on a Nikon ECLIPSE Ti2 Confocal microscope and shown as max projections. For quantification of HS and CS signals, five replicates of images with around 200 cells were quantified. The Average Normalized Intensity was used in the quantification results, with WT cells normalized to 1.
Flow cytometry
HeLa cells (WT and KO) were detached using 20 mM EDTA and resuspended in PBS with 0.5% BSA. The cells were fixed and permeabilized with 4% PFA for 10 min and 0.2% Triton X-100 for 10 min, respectively. After washing with PBS twice, the cells were incubated with the primary antibody for HS (10E4, 1:100) or CS (CS-56, 1:50) (or control without a primary antibody) with rotation for 1.5 h at room temperature. Both primary antibodies and the control group were incubated with goat anti mouse secondary antibodies (TRITC) for 1 h with rotation at room temperature. The cells were sorted with a Sony MA900 Multi-Application Cell Sorter with the following settings: forward scatter (FSC), and side scatter (SSC) signals were collected, and gates were set for single cells. TRITC signals from >4000 events were collected. The data was analyzed with FlowJo software.
Proteomics analysis
Sample Preparation:
Three replicates of each WT, 55KO and 65KO cells were propagated as described above in 15 cm dishes. For cell lysates collection, after removing the media, cells were washed with PBS twice, collected in 10 ml PBS by scraping, and lysed in Pierce™ RIPA buffer (Thermo, 89900) with a protein inhibitor cocktail (Thermo). The protein concentration was tested with Bradford assay and normalized, 75 μg of each sample was provided to the Mass Spectrometry-Based Proteomics Resource Facility at Department of Pathology, University of Michigan for TMT labeling, LC-MS/MS and bioinformatics analysis.
Protein Digestion and TMT labeling:
Samples were proteolysed and labeled with TMT 10-plex essentially by following manufacturer’s protocol (ThermoFisher, Cat # 90110, Lot # VJ306782). Briefly, upon reduction and alkylation of cysteines, the proteins were precipitated by adding 6 volumes of ice-cold acetone followed by overnight incubation at −20°C. The precipitate was spun down, and the pellet was allowed to air dry. The pellet was resuspended in 0.1 M TEAB and overnight digestion with trypsin (1:50; enzyme:protein) at 37°C was performed with constant mixing using a thermomixer. The TMT 10-plex reagents were dissolved in 41 μl of anhydrous acetonitrile and labeling was performed by transferring the entire digest to TMT reagent vial and incubating at room temperature for 1 h. Reaction was quenched by adding 8 μl of 5% hydroxyl amine and further 15 min incubation. Labeled samples were mixed, and dried using a vacufuge. An offline fractionation of the combined sample (~200 μg) into 8 fractions was performed using high pH reversed-phase peptide fractionation kit according to the manufacturer’s protocol (Pierce; Cat # 84868). Fractions were dried and reconstituted in 9 μl of 0.1% formic acid/2% acetonitrile in preparation for LC-MS/MS analysis.
Liquid chromatography-mass spectrometry analysis (LC-multinotch MS3):
In order to obtain superior quantitation accuracy, we employed multinotch-MS3 (McAlister GC) which minimizes the reporter ion ratio distortion resulting from fragmentation of co-isolated peptides during MS analysis. Orbitrap Fusion (Thermo Fisher Scientific) and RSLC Ultimate 3000 nano-UPLC (Dionex) was used to acquire the data. 2 μl of the sample was resolved on a PepMap RSLC C18 column (75 μm i.d. × 50 cm; Thermo Scientific) at the flowrate of 300 nl/min using 0.1% formic acid/acetonitrile gradient system (2–22% acetonitrile in 150 min;22–32% acetonitrile in 40 min; 20 min wash at 90% followed by 50 min re-equilibration) and directly spray onto the mass spectrometer using EasySpray source (Thermo Fisher Scientific). Mass spectrometer was set to collect one MS1 scan (Orbitrap; 120K resolution; AGC target 2×105; max IT 100 ms) followed by data-dependent, “Top Speed” (3 seconds) MS2 scans (collision induced dissociation; ion trap; NCE 35; AGC 5×103; max IT 100 ms). For multinotch-MS3, top 10 precursors from each MS2 were fragmented by HCD followed by Orbitrap analysis (NCE 55; 60K resolution; AGC 5×104; max IT 120 ms, 100–500 m/z scan range).
Data analysis:
Proteome Discoverer (v2.4; Thermo Fisher) was used for data analysis. MS2 spectra were searched against SwissProt human protein database (20353 entries; downloaded on 06/20/2019) using the following search parameters: MS1 and MS2 tolerance were set to 10 ppm and 0.6 Da, respectively; carbamidomethylation of cysteines (57.02146 Da) and TMT labeling of lysine and N-termini of peptides (229.16293 Da) were considered static modifications; oxidation of methionine (15.9949 Da) and deamidation of asparagine and glutamine (0.98401 Da) were considered variable. Identified proteins and peptides were filtered to retain only those that passed ≤1% FDR threshold. Quantitation was performed using high-quality MS3 spectra (average signal-to-noise ratio of 10 and <50% isolation interference).
RNA-Seq analysis
RNA samples were collected from each of the four HeLa cell lines: WT, 55KO, 65KO, and DKO. Cells at an exponential growth phase (~ 80% confluency) in 6 well dishes were collected. Five replicates of each cell line were lysed using Trizol. RNA samples were prepared using the Direct-zol™ RNA Miniprep Plus kit and treated with DNase I provided in the same kit. The samples were sent to UMich Advanced Genomic Core for library creation and processing. At the core, after passing quality control for quantity and purity, RNA samples were used to create 3’ mRNA libraries using the QuantSeq 3’ mRNA-Seq Library Prep Kit FWD for Illumina kit with the UMI add-on kit (Lexogen, Cat # 081.96), which used oligoT priming to generate the first cDNA strand from RNAs with a poly-A tail. Single-read sequencing for the cDNA library was performed on Illumina NextSeq sequencer for 100 cycles. Sequencing results were trimmed using Trim Galore (v 0.5.0). Alignment of reads to human genome GRCh38 from ENSEMBL (https://useast.ensembl.org/index.html) was performed in house using STAR (v 2.6.0). Trim_Galore and STAR can be found on https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ and https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3530905/, respectively. Transcriptomic data was normalized using DESeq2 (50). Heatmaps were generated using matrix visualization software Morpheus by Broad Institute (RRID:SCR_017386) for genes detected in our data, which are involved in the HS and CS pathways. GlycoMaple (51) was used to visually compare and estimate glycosaminoglycan structures based on gene expression profiles of averaged GRASP KO samples and wildtype control, where fold change × > 1.2 and × < 0.8 were set as comparative thresholds.
Quantification and statistics
In all figures, the quantification results are expressed as the mean ± SEM (standard error of the mean) from 3–5 independent experiments, unless otherwise stated. The statistical significance of the results was assessed using Student’s t-test. *, p<0.05, **, p<0.01, ***, p<0.001.
Supplementary Material
{kind=link}
{kind=link}
Acknowledgments
We thank Ge Yu, Judith Meyers Opp and the University of Michigan Advanced Genomics Core for their contribution in the RNA-Seq experiment. We thank Drs. Venkatesha Basrur, Felipe da Veiga Leprevost, Alexey Nesvizhskii and the Mass Spectrometry-Based Proteomics Resource Facility in the Department of Pathology at the University of Michigan for their contribution in Proteomics analysis. We thank the members of the Wang lab and Linhardt lab for stimulating discussions and technical support. Y. W. was supported by the National Institutes of Health (Grant R35GM130331), Mizutani Foundation for Glycoscience, MCubed, and the Fast Forward Protein Folding Disease Initiative of the University of Michigan; E.A. was supported by a University of Michigan Rackham Predoctoral fellowship. R.J.L, F.Z. and Y.F. was supported by GlycoMIP a National Science Foundation Materials Innovation Platform funded through Cooperative Agreement DMR-1933525.
Footnotes
Supplemental Information
Supplemental Information includes two supplemental figures and six supplemental tables.
Competing Interest
The authors declare that no competing interests exist.
References
- 1.Li J, Ahat E, Wang Y. 2019. Golgi Structure and Function in Health, Stress, and Diseases. Results Probl Cell Differ 67:441–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xiang Y, Wang Y. 2010. GRASP55 and GRASP65 play complementary and essential roles in Golgi cisternal stacking. J Cell Biol 188:237–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bekier ME 2nd, Wang L, Li J, Huang H, Tang D, Zhang X, Wang Y. 2017. Knockout of the Golgi stacking proteins GRASP55 and GRASP65 impairs Golgi structure and function. Mol Biol Cell 28:2833–2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ahat E, Li J, Wang Y. 2019. New Insights Into the Golgi Stacking Proteins. Front Cell Dev Biol 7:131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang Y, Seemann J. 2021. Rapid degradation of GRASP55 and GRASP65 reveals their immediate impact on the Golgi structure. Journal of Cell Biology 220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xiang Y, Zhang X, Nix DB, Katoh T, Aoki K, Tiemeyer M, Wang Y. 2013. Regulation of protein glycosylation and sorting by the Golgi matrix proteins GRASP55/65. Nat Commun 4:1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ahat E, Xiang Y, Zhang X, Bekier ME, Wang Y. 2019. GRASP depletion-mediated Golgi destruction decreases cell adhesion and migration via the reduction of alpha5beta1 integrin. Mol Biol Cell 30:766–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang X, Wang L, Lak B, Li J, Jokitalo E, Wang Y. 2018. GRASP55 Senses Glucose Deprivation through O-GlcNAcylation to Promote Autophagosome-Lysosome Fusion. Dev Cell 45:245–261 e246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang X, Wang Y. 2018. GRASP55 facilitates autophagosome maturation under glucose deprivation. Mol Cell Oncol 5:e1494948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gee HY, Noh SH, Tang BL, Kim KH, Lee MG. 2011. Rescue of DeltaF508-CFTR Trafficking via a GRASP-Dependent Unconventional Secretion Pathway. Cell 146:746–760. [DOI] [PubMed] [Google Scholar]
- 11.Cruz-Garcia D, Brouwers N, Duran JM, Mora G, Curwin AJ, Malhotra V. 2017. A diacidic motif determines unconventional secretion of wild-type and ALS-linked mutant SOD1. J Cell Biol doi: 10.1083/jcb.201704056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nüchel J, Tauber M, Nolte JL, Mörgelin M, Türk C, Eckes B, Demetriades C, Plomann M. 2021. An mTORC1-GRASP55 signaling axis controls unconventional secretion to reshape the extracellular proteome upon stress. Mol Cell doi: 10.1016/j.molcel.2021.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ahat E, Bui S, Zhang J, da Veiga Leprevost F, Sharkey L, Reid W, Nesvizhskii A, Paulson H, Wang Y. 2021. GRASP55 regulates mutant huntingtin unconventional secretion and aggregation. [DOI] [PMC free article] [PubMed]
- 14.Annaval T, Wild R, Cretinon Y, Sadir R, Vives RR, Lortat-Jacob H. 2020. Heparan Sulfate Proteoglycans Biosynthesis and Post Synthesis Mechanisms Combine Few Enzymes and Few Core Proteins to Generate Extensive Structural and Functional Diversity. Molecules 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim BT, Kitagawa H, Tamura J, Saito T, Kusche-Gullberg M, Lindahl U, Sugahara K. 2001. Human tumor suppressor EXT gene family members EXTL1 and EXTL3 encode alpha 1,4- N-acetylglucosaminyltransferases that likely are involved in heparan sulfate/heparin biosynthesis. Proc Natl Acad Sci U S A 98:7176–7181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kitagawa H, Shimakawa H, Sugahara K. 1999. The tumor suppressor EXT-like gene EXTL2 encodes an alpha1, 4-N-acetylhexosaminyltransferase that transfers N-acetylgalactosamine and N-acetylglucosamine to the common glycosaminoglycan-protein linkage region. The key enzyme for the chain initiation of heparan sulfate. J Biol Chem 274:13933–13937. [DOI] [PubMed] [Google Scholar]
- 17.Kreuger J, Kjellen L. 2012. Heparan sulfate biosynthesis: regulation and variability. J Histochem Cytochem 60:898–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carlsson P, Presto J, Spillmann D, Lindahl U, Kjellén L. 2008. Heparin/heparan sulfate biosynthesis: processive formation of N-sulfated domains. J Biol Chem 283:20008–20014. [DOI] [PubMed] [Google Scholar]
- 19.Maeda N 2015. Proteoglycans and neuronal migration in the cerebral cortex during development and disease. Front Neurosci 9:98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fuda H, Shimizu C, Lee YC, Akita H, Strott CA. 2002. Characterization and expression of human bifunctional 3’-phosphoadenosine 5’-phosphosulphate synthase isoforms. Biochem J 365:497–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stelzer C, Brimmer A, Hermanns P, Zabel B, Dietz UH. 2007. Expression profile of Papss2 (3’-phosphoadenosine 5’-phosphosulfate synthase 2) during cartilage formation and skeletal development in the mouse embryo. Dev Dyn 236:1313–1318. [DOI] [PubMed] [Google Scholar]
- 22.Kamiyama S, Sasaki N, Goda E, Ui-Tei K, Saigo K, Narimatsu H, Jigami Y, Kannagi R, Irimura T, Nishihara S. 2006. Molecular cloning and characterization of a novel 3’-phosphoadenosine 5’-phosphosulfate transporter, PAPST2. J Biol Chem 281:10945–10953. [DOI] [PubMed] [Google Scholar]
- 23.Dick G, Akslen-Hoel LK, Grondahl F, Kjos I, Maccarana M, Prydz K. 2015. PAPST1 regulates sulfation of heparan sulfate proteoglycans in epithelial MDCK II cells. Glycobiology 25:30–41. [DOI] [PubMed] [Google Scholar]
- 24.Zhao J, Zhu Y, Song X, Xiao Y, Su G, Liu X, Wang Z, Xu Y, Liu J, Eliezer D, Ramlall TF, Lippens G, Gibson J, Zhang F, Linhardt RJ, Wang L, Wang C. 2020. 3-O-Sulfation of Heparan Sulfate Enhances Tau Interaction and Cellular Uptake. Angew Chem Int Ed Engl 59:1818–1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Prydz K 2015. Determinants of Glycosaminoglycan (GAG) Structure. Biomolecules 5:2003–2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kobayashi S, Morimoto K, Shimizu T, Takahashi M, Kurosawa H, Shirasawa T. 2000. Association of EXT1 and EXT2, hereditary multiple exostoses gene products, in Golgi apparatus. Biochem Biophys Res Commun 268:860–867. [DOI] [PubMed] [Google Scholar]
- 27.Uliana AS, Giraudo CG, Maccioni HJ. 2006. Cytoplasmic tails of SialT2 and GalNAcT impose their respective proximal and distal Golgi localization. Traffic 7:604–612. [DOI] [PubMed] [Google Scholar]
- 28.Adusumalli R, Åsheim HC, Lupashin V, Blackburn JB, Prydz K. 2021. Proteoglycan synthesis in conserved oligomeric Golgi subunit deficient HEK293T cells is affected differently, depending on the lacking subunit. Traffic 22:230–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stevenson NL, Bergen DJM, Skinner REH, Kague E, Martin-Silverstone E, Robson Brown KA, Hammond CL, Stephens DJ. 2017. Giantin-knockout models reveal a feedback loop between Golgi function and glycosyltransferase expression. J Cell Sci 130:4132–4143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Avnur Z, Geiger B. 1984. Immunocytochemical localization of native chondroitin-sulfate in tissues and cultured cells using specific monoclonal antibody. Cell 38:811–822. [DOI] [PubMed] [Google Scholar]
- 31.Aquino RS, Lee ES, Park PW. 2010. Diverse functions of glycosaminoglycans in infectious diseases. Prog Mol Biol Transl Sci 93:373–394. [DOI] [PubMed] [Google Scholar]
- 32.Pan C, Nelson MS, Reyes M, Koodie L, Brazil JJ, Stephenson EJ, Zhao RC, Peters C, Selleck SB, Stringer SE, Gupta P. 2005. Functional abnormalities of heparan sulfate in mucopolysaccharidosis-I are associated with defective biologic activity of FGF-2 on human multipotent progenitor cells. Blood 106:1956–1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Götte M, Spillmann D, Yip GW, Versteeg E, Echtermeyer FG, van Kuppevelt TH, Kiesel L. 2008. Changes in heparan sulfate are associated with delayed wound repair, altered cell migration, adhesion and contractility in the galactosyltransferase I (beta4GalT-7) deficient form of Ehlers-Danlos syndrome. Hum Mol Genet 17:996–1009. [DOI] [PubMed] [Google Scholar]
- 34.Le Jan S, Hayashi M, Kasza Z, Eriksson I, Bishop JR, Weibrecht I, Heldin J, Holmborn K, Jakobsson L, Söderberg O, Spillmann D, Esko JD, Claesson-Welsh L, Kjellén L, Kreuger J. 2012. Functional overlap between chondroitin and heparan sulfate proteoglycans during VEGF-induced sprouting angiogenesis. Arterioscler Thromb Vasc Biol 32:1255–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sasarman F, Maftei C, Campeau PM, Brunel-Guitton C, Mitchell GA, Allard P. 2016. Biosynthesis of glycosaminoglycans: associated disorders and biochemical tests. J Inherit Metab Dis 39:173–188. [DOI] [PubMed] [Google Scholar]
- 36.Tian E, Stevens SR, Guan Y, Springer DA, Anderson SA, Starost MF, Patel V, Ten Hagen KG, Tabak LA. 2015. Galnt1 is required for normal heart valve development and cardiac function. PLoS One 10:e0115861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miyata S, Komatsu Y, Yoshimura Y, Taya C, Kitagawa H. 2012. Persistent cortical plasticity by upregulation of chondroitin 6-sulfation. Nat Neurosci 15:414–422, S411–412. [DOI] [PubMed] [Google Scholar]
- 38.Joshi G, Chi Y, Huang Z, Wang Y. 2014. Abeta-induced Golgi fragmentation in Alzheimer’s disease enhances Abeta production. Proc Natl Acad Sci U S A 111:E1230–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gosavi P, Houghton FJ, McMillan PJ, Hanssen E, Gleeson PA. 2018. The Golgi ribbon in mammalian cells negatively regulates autophagy by modulating mTOR activity. J Cell Sci 131. [DOI] [PubMed] [Google Scholar]
- 40.Philips MR. 2004. Sef: a MEK/ERK catcher on the Golgi. Mol Cell 15:168–169. [DOI] [PubMed] [Google Scholar]
- 41.Baumann J, Ignashkova TI, Chirasani SR, Ramirez-Peinado S, Alborzinia H, Gendarme M, Kuhnigk K, Kramer V, Lindemann RK, Reiling JH. 2018. Golgi stress-induced transcriptional changes mediated by MAPK signaling and three ETS transcription factors regulate MCL1 splicing. Mol Biol Cell 29:42–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weiss RJ, Spahn PN, Toledo AG, Chiang AW, Kellman BP, Li J, Benner C, Glass CK, Gordts PL, Lewis NE. 2020. ZNF263 is a transcriptional regulator of heparin and heparan sulfate biosynthesis. Proceedings of the National Academy of Sciences 117:9311–9317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Uhlin-Hansen L, Yanagishita M. 1993. Differential effect of brefeldin A on the biosynthesis of heparan sulfate and chondroitin/dermatan sulfate proteoglycans in rat ovarian granulosa cells in culture. J Biol Chem 268:17370–17376. [PubMed] [Google Scholar]
- 44.Meneghetti MCZ, Deboni P, Palomino CMV, Braga LP, Cavalheiro RP, Viana GM, Yates EA, Nader HB, Lima MA. 2021. ER-Golgi dynamics of HS-modifying enzymes via vesicular trafficking is a critical prerequisite for the delineation of HS biosynthesis. Carbohydr Polym 255:117477. [DOI] [PubMed] [Google Scholar]
- 45.Wang Y, Wei JH, Bisel B, Tang D, Seemann J. 2008. Golgi Cisternal Unstacking Stimulates COPI Vesicle Budding and Protein Transport. PLoS ONE 3:e1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee I, Tiwari N, Dunlop MH, Graham M, Liu X, Rothman JE. 2014. Membrane adhesion dictates Golgi stacking and cisternal morphology. Proc Natl Acad Sci U S A 111:1849–1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sola RJ, Griebenow K. 2009. Effects of glycosylation on the stability of protein pharmaceuticals. J Pharm Sci 98:1223–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Freeman C, Hopwood J. 1992. Lysosomal degradation of heparin and heparan sulphate. Adv Exp Med Biol 313:121–134. [DOI] [PubMed] [Google Scholar]
- 49.Tang D, Yuan H, Wang Y. 2010. The Role of GRASP65 in Golgi Cisternal Stacking and Cell Cycle Progression. Traffic 11:827–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huang YF, Aoki K, Akase S, Ishihara M, Liu YS, Yang G, Kizuka Y, Mizumoto S, Tiemeyer M, Gao XD, Aoki-Kinoshita KF, Fujita M. 2021. Global mapping of glycosylation pathways in human-derived cells. Dev Cell 56:1195–1209 e1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.