

Abstract
Small cell lung cancer (SCLC) is a recalcitrant malignancy defined by subtypes based on differential expression of the ASCL1, NEUROD1 and POU2F3 transcription factors. The MUC1-C protein is activated in pulmonary epithelial cells by exposure to environmental carcinogens and promotes oncogenesis; however, there is no known association between MUC1-C and SCLC. We report that MUC1-C is expressed in classic neuroendocrine (NE) SCLC-A, variant NE SCLC-N and non-NE SCLC-P cells and activates the MYC pathway in these subtypes. In SCLC cells characterized by NE differentiation and DNA replication stress, we show that MUC1-C activates the MYC pathway in association with induction of E2F target genes and dysregulation of mitotic progression. Our studies further demonstrate that the MUC1-C→MYC pathway is necessary for induction of (i) NOTCH2, a marker of pulmonary NE stem cells that are the proposed cell of SCLC origin, and (ii) ASCL1 and NEUROD1. We also show that the MUC1-C→MYC→NOTCH2 network is necessary for self-renewal capacity and tumorigenicity of NE and non-NE SCLC cells. Analyses of datasets from SCLC tumors confirmed that MUC1 expression in single SCLC cells significantly associates with activation of the MYC pathway. These findings demonstrate that SCLC cells are addicted to MUC1-C and identify a potential new target for SCLC treatment.
Keywords: MUC1-C, SCL C, MYC, NOTCH2, dedifferentiation
Introduction
Small cell lung cancer (SCLC) is a highly aggressive malignancy that is associated with exposure to tobacco carcinogens and is characterized by rapid growth, metastasis, and drug resistance (1). SCLC that occurs de novo represents about 15% of lung cancers (1). Resistance of non-small cell lung adenocarcinomas (NSCLCs) to EGFR inhibitors is also associated with transdifferentiation to SCLCs at rates of 5–14% (2). Patients with SCLC have had limited therapeutic options other than standard chemotherapy (1, 3); however, treatment with immune checkpoint inhibitors (ICIs) has been associated with response rates of 10–14% and increases in survival when combined with chemotherapy (3). Despite these advances, patients diagnosed with SCLC have a median overall survival of ~1 year, emphasizing the critical need for identifying other effectors that contribute to SCLC progression. SCLCs arise from pulmonary neuroendocrine (NE) stem cells that are activated by lung injury and are regulated by p53, RB and NOTCH in the wound repair response (4). SCLCs exhibit widespread functional loss of p53 and RB, and dysregulation of the MYC and E2F pathways (1, 5–7). As a result, a notable characteristic of NE SCLC cells is activation of the replication stress response (RSR), which is dependent on DNA repair and cell cycle checkpoints for proliferation (8–10). NE SCLC subtypes have been characterized by differential expression of the ASCL1 and NEUROD1 transcription factors (TFs) and non-NE SCLCs by POU2F3 and YAP1 (1, 11). Otherwise, the identification of potential targets for SCLC treatment has remained a major unmet medical need (6).
MUC1-C appeared in mammals to provide protection of pulmonary and other types of epithelial cells from loss of homeostasis associated with exposure to the external environment (12). MUC1-C induces inflammatory, proliferative and remodeling signaling pathways that contribute to the wound healing response (12). In addition and in settings of chronic inflammation with repetitive cycles of damage and repair, prolonged MUC1-C activation promotes cancer progression (12). In concert with this capacity for driving oncogenesis, MUC1-C is aberrantly overexpressed in diverse carcinomas and is associated with poor clinical outcomes (12). MUC1-C contributes to multiple hallmarks of the cancer cell, including the epithelial-mesenchymal transition (EMT), cancer stem cell (CSC) state, epigenetic reprogramming and lineage plasticity (12, 13), which are associated with drug resistance and immune evasion (14). Along these lines, MUC1-C confers resistance of CSCs to genotoxic anticancer agents (12), at least in part, by (i) binding directly to ATM and γH2AX and promoting repair of DNA double strand breaks (DSBs) (15), and (ii) integrating epigenetic reprogramming with activation of poly-ADP-ribose polymerase (PARP) in the DNA damage response (DDR) (16). MUC1-C also promotes immune evasion by intrinsic production of effectors that suppress the tumor microenvironment (12, 17).
There is no known involvement of MUC1-C in SCLC. The present studies demonstrate that MUC1-C is of functional importance in SCLC by driving activation of the MYC pathway. We show that MUC1-C→MYC signaling is integrated with induction of E2F target genes and dysregulation of cell cycle progression. We also show that the MUC1-C→MYC pathway induces (i) NOTCH2, (ii) NE dedifferentiation, and (iii) self-renewal and tumorigenicity. These findings reveal a previously unrecognized addiction to MUC1-C, as defined by dependence for survival (18), in classic, variant and non-NE SCLC subtypes.
Material and Methods
Cell culture.
NCI-H69 cells (ATCC; nonsense mutations in TP53 and RB1) and COR-L279 cells (Sigma-Aldrich, St. Louis, MO; deletion mutation in TP53) were cultured in RPMI1640 medium (Corning, Corning, NY, USA) supplemented with 10% fetal bovine serum (FBS) and 2 mM glutamine. DMS53 cells (ATCC; missense mutation in TP53) were grown in Weymouth’s medium (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% FBS. NCI-H526 cells (ATCC; splice acceptor mutation in TP53) were cultured in RPMI1640 medium with 10% FBS. Authentication of the cells was performed by short tandem repeat (STR) analysis. Cells were monitored for mycoplasma contamination using the MycoAlert Mycoplasma Detection Kit (Lonza, Rockland, ME, USA). Cells were maintained for 3–4 months for performing experiments.
Gene silencing.
MUC1shRNA (MISSION shRNA TRCN0000122938; Sigma), MYCshRNA (MISSION shRNA TRCN0000039642; Sigma) or a control scrambled shRNA (CshRNA; Sigma) was inserted into the pLKO-tet-puro vector (Plasmid #21915; Addgene, Cambridge, MA, USA). Single guide RNAs targeting MUC1 exon 4 were inserted into the lentiCRISPR v2 (Plasmid #52961; Addgene). Single guide RNAs targeting NOTCH2 were inserted into the lentiCRISPR v2 hygro (Plasmid #98291; Addgene). The viral vectors were produced in 293T cells as described (19). Cells transduced with the vectors were selected for growth in 1-4 μg/ml puromycin or 100-400 μg/ml hygromycin. Single cell clones were isolated by the array dilution method. Cells were treated with 0.1% DMSO as the vehicle control or 500 ng/ml doxycycline (DOX; Millipore Sigma).
Quantitative reverse-transcription PCR (qRT-PCR).
Total cellular RNA was isolated for qRT-PCR as described (19) using primers listed in Supplementary Table S2.
Immunoblot analysis.
Total lysates prepared from subconfluent cells were subjected to immunoblot analysis using anti-ASCL1 (GTX129189; GeneTex, Irvine, CA, USA), anti-YAP1 (14074; Cell Signaling Technology (CST), Danvers, MA), anti-NEUROD1 (4373; CST), anti-POU2F3/SKN1 (ab101724; Abcam, Cambridge, MA, USA), anti-MUC1-C (HM-1630-P1ABX; Thermo Fisher Scientific), anti-GAPDH (5174; CST), anti-β-actin (A5441; Sigma-Aldrich), anti-CDK1 (ab131450; Abcam), anti-CCNA2/cyclin A2 (4656; CST), anti-PLK1 (4513, CST), anti-AURKA (ab1287, Abcam), anti-γH2AX (9718, CST), anti-NOTCH2 (5732; CST), anti-MDK (ab52637, Abcam), anti-BRN2 (12137; CST) and anti-MYC (ab32072; Abcam), anti-HES1 (11988, CST), anti-BMI1 (6964; CST), anti-CD133 (5860; CST) and anti-CD44 (KO601; TransGenic Inc., Tokyo, Japan).
RNA-seq analysis.
Total RNA from cells cultured in triplicates was isolated for generation of the RNA-seq datasets as described (19). Gene Set Enrichment Analysis (GSEA) was performed as described (19).
Tumorsphere formation assays.
Cells (1.5-3.0 x 104) were seeded per well in 6-well ultra-low attachment culture plates (Corning Life Sciences) in DMEM/F12 50/50 medium (Corning Life Sciences) with 20 ng/ml EGF (Millipore Sigma), 20 ng/ml bFGF (Millipore Sigma) and 1% B27 supplement (Gibco). In certain studies, cells were treated with (i) vehicle control or 500 ng/ml DOX and (ii) 5 μM GO-203 dissolved in PBS (19–23). Tumorspheres were counted under an inverted microscope in triplicate wells.
Colony formation assays.
Cells were treated with control vehicle or 5 μM GO-203 for 3 days, seeded at 1,500 cells per well in 24-well plates and then stained with 0.5% crystal violet in 25% methanol. Colonies >25 cells were counted in triplicate wells.
Mouse tumor model studies.
Six- to 8-week old nude mice (Taconic Farms, Germantown, NY, USA) were injected subcutaneously in the flank with 3-5 x 106 cells in 100 μl of a 1:1 solution of medium and Matrigel (BD Biosciences). When the mean tumor volume reached 100–150 mm3, mice were pair-matched into groups. In studies of H69/tet-MUC1shRNA and COR-L279/tet-MUC1shRNA tumors, mice were fed without or with DOX (625 ppm, daily). Mice bearing established COR-L279 tumors were treated with 5 mg/kg GO-203 administered intraperitoneally (ip) each day. Tumor measurements and body weights were recorded twice each week. Mice were sacrificed when tumors reached >2000 mm3 as calculated by the formula: (width)2 x length/2.
Immunohistochemistry (IHC).
Formalin-fixed, paraffin-embedded (FFPE) sections of SCLC tissue samples were deparaffinized in xylene. Antigen retrieval was performed at 95-99°C for 40 min and at room temperature for 20 min in pH 8.5 EDTA buffer (E1161, Sigma-Aldrich). Peroxidase blocking (Peroxidazed 1, Biocare Medical, Pacheco, CA, USA) was performed for 5 min followed by universal blocking (Background sniper, Biocare Medical) for 10 min. Sections were incubated with anti-MUC1-C (dilution 1:100, MA5-11202; Thermo Fisher Scientific) for 2 h at room temperature, anti-Armenian hamster secondary antibody (dilution 1:200, ab5745; Abcam) for 30 min at room temperature and diaminobenzidine tetrahydrochloride chromagen reagents for 5 min. Immunostained sections were counterstained with hematoxylin.
Statistical analysis.
Data obtained from experiments performed at least three times are expressed as the mean±SD. Data was analyzed using the unpaired Mann-Whitney U test with pvalues of <0.05 (*) considered statistically significant.
Analysis of human SCLC tumor datasets.
Data analysis was performed using the cBioPortal Cancer Genomic and Oncomine websites (24, 25). GSE60052 was downloaded from Gene Expression Omnibus (GEO)(https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE60052). GSE138267 is the accession number of the scRNA-seq SCLC dataset (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE138474). Processed scRNA-seq data comprising untreated xenograft tissue derived from circulating tumor cells captured from 7 SCLC patients (25) were obtained from the Gene Expression Omnibus (GSE138474). SCLC cell data was re-analyzed via Seurat (26) for variable feature selection, dimensionality reduction (PCA), and uniform manifold approximation and projection (UMAP) low-dimensional representation using a workflow and parameters described by Stewart et al. SCLC cell gene expression was imputed using MAGIC (27), implemented via the Rmagic package. Single-cell pathway enrichment was performed on imputed expression by AUCell (28), using select HALLMARK pathways. Associations between MUC1 expression and select genes or signatures within SCLC cells were examined by Pearson correlation analysis.
Data Availability.
The accession numbers for the RNA-seq data are GEO Submissions GSE159801 and GSE192475.
Results
MUC1-C regulates the transcriptomes of SCLC-A cell lines.
The human NCI-H69 SCLC cell line is characteristic of the SCLC-A classic subtype that expresses ASCL1 and predominantly grows as aggregates of floating cells (Fig. 1A) (1). Human DMS53 SCLC cells grow largely as adherent monolayers and express ASCL1, consistent with the NE SCLC-A subtype (Fig. 1A) (1). DMS53 cells also express YAP1, which has been questioned as another marker of SCLC subtypes (1). In H69 cells, MUC1-C is expressed as N-glycosylated 25-20 kDa and unglycosylated ~17 kDa MUC1-C proteins (Fig. 1A). Expression of MUC1-C in DMS53 cells is predominantly the 25-20 kDa form (Fig. 1A). To assess potential MUC1-C functions, we established H69 cells stably expressing a tet-inducible control shRNA (tet-CshRNA) or a tet-MUC1shRNA. Treatment with doxycycline (DOX) was associated with downregulation of MUC1-C in H69/tet-MUC1shRNA, and not H69/tet-CshRNA, cells (Fig. 1B, left and right). Similar results were obtained from DOX-treated DMS53/tet-MUC1shRNA cells (Fig. 1C, left and right), supporting potential models for RNA-seq studies to assess MUC1-C-driven changes in gene expression. Along these lines, volcano plots of the RNA-seq datasets from H69 and DMS53 cells with MUC1-C silencing revealed substantial effects on repressed and activated genes (Fig. 1D, left and right), among which we identified 1634 downregulated and 1410 upregulated genes that are common to both cell lines (Fig. 1E). Analysis by GSEA using the BENPORATH collection identified MUC1-C-regulated signatures associated with proliferation, cell cycle progression and the embryonic stem cell (ESC) state (Fig. 1F). We also found that silencing MUC1-C in H69 and DMS53 cells associates with downregulation of the BENPORATH MYC TARGETS WITH EBOX gene signature (Figs. 1F and 1G, left and right; Supplemental Fig. S1A) and MYC target genes that are expressed in both cell lines (Supplemental Fig. S1B).
Figure 1. MUC1-C regulates common transcriptomes in SCLC-A cells.
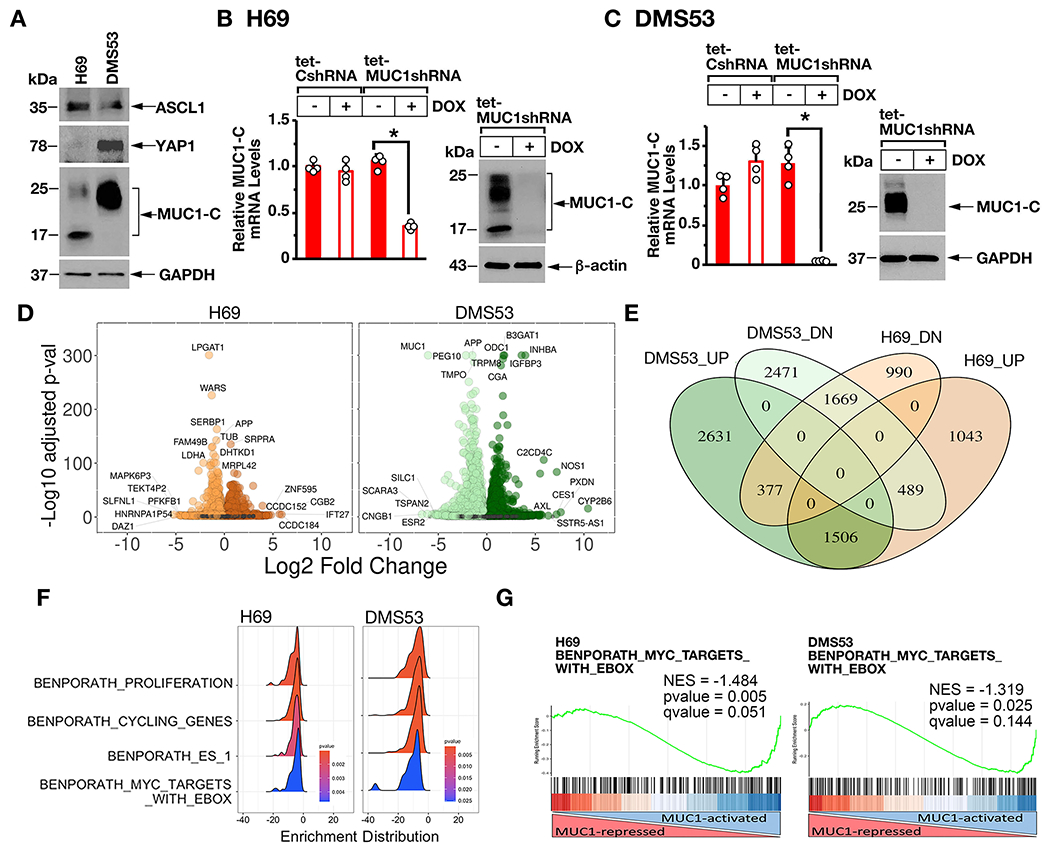
A. Lysates from H69 and DMS53 cells were immunoblotted with antibodies against the indicated proteins. B and C. H69 (B) and DMS53 (C) cells expressing a tet-CshRNA or a tet-MUC1shRNA were treated with vehicle or 500 ng/ml DOX for 7 days. The cells were analyzed for MUC1-C mRNA levels by qRT-PCR using primers listed in Supplemental Table S1. The results (mean±SD of four determinations) are expressed as relative mRNA levels compared to that obtained for vehicle-treated cells (assigned a value of 1) (left). Lysates were immunoblotted with antibodies against the indicated proteins (right). D. RNA-seq was performed in triplicate on H69/tet-MUC1shRNA and DMS53/tet-MUC1shRNA cells treated with vehicle or DOX for 7 days. The datasets were analyzed for effects of MUC1-C silencing on repressed and activated genes as depicted by the Volcano plots. E. Overlap of down- and up-regulated genes in H69 and DMS53 cells with MUC1-C silencing. F. RNA-seq datasets from H69 (left) and DMS53 (right) cells were analyzed with GSEA for enrichment distribution using the indicated BENPORATH collection gene signatures. G. RNA-seq datasets from H69 (left) and DMS53 (right) cells were analyzed with GSEA using the BENPORATH MYC TARGETS WITH EBOX gene signatures.
MUC1-C regulates common MYC target genes in classic, variant and non-NE SCLC cells.
To extend these results to other SCLC subtypes, we studied (i) variant SCLC-N COR-L279 cells that express NEUROD1, and exhibit characteristics of NE differentiation, and (ii) SCLC-P H526 cells that express POU2F3, but not the NE phenotype (Fig. 2A) (1). MUC1-C was detectable in COR-L279 cells and at higher levels in H526 cells (Fig. 2A). Accordingly, we silenced MUC1-C in these cells (Figs. 2B and 2C) and by RNA-seq analysis (Supplemental Fig. S2A) identified 488 downregulated and 411 upregulated differentially expressed genes (DEGs) common to H69, DMS53, COR-L279 and H526 cells (Figs. 2D and 2E). Consistent with findings in H69 and DMS53 cells, RNA-seq analysis of COR-L279 and H526 cells further demonstrated that MUC1-C is necessary for activation of the BENPORATH MYC TARGETS WITH EBOX signature (Fig. 2F, left and right). Shared sets of MUC1-C-regulated MYC target genes were identified COR-L279 and H526 cells (Supplemental Fig. S2B). Moreover, MUC1-C was necessary for the upregulation and downregulation of common sets of MYC target genes in H69, DMS53, COR-L279 and H526 cells (Supplemental Figs. S2C and S2D). MUC1-C activates MYC expression by a WNT pathway-mediated mechanism that is dependent on cancer cell context (12). MUC1-C also binds directly to the MYC HLH-LZ domain to drive MYC target genes (29). MYC, MYCL and MYCN are expressed in the major SCLC subtypes (1). We found that silencing MUC1-C results in the downregulation of MYC in H69, DMS53 and COR-L279 cells (Fig. 2G). MUC1-C was also necessary for expression of MYCL and MYCN in H69 cells, but not in DMS53, COR-L279 and H526 cells (Fig. 2G). Accordingly, we focused our studies on MUC1-C-induced regulation of the MYC pathway.
Figure 2. MUC1-C regulates common and distinct transcriptomes in NE and non-NE SCLC subtypes.
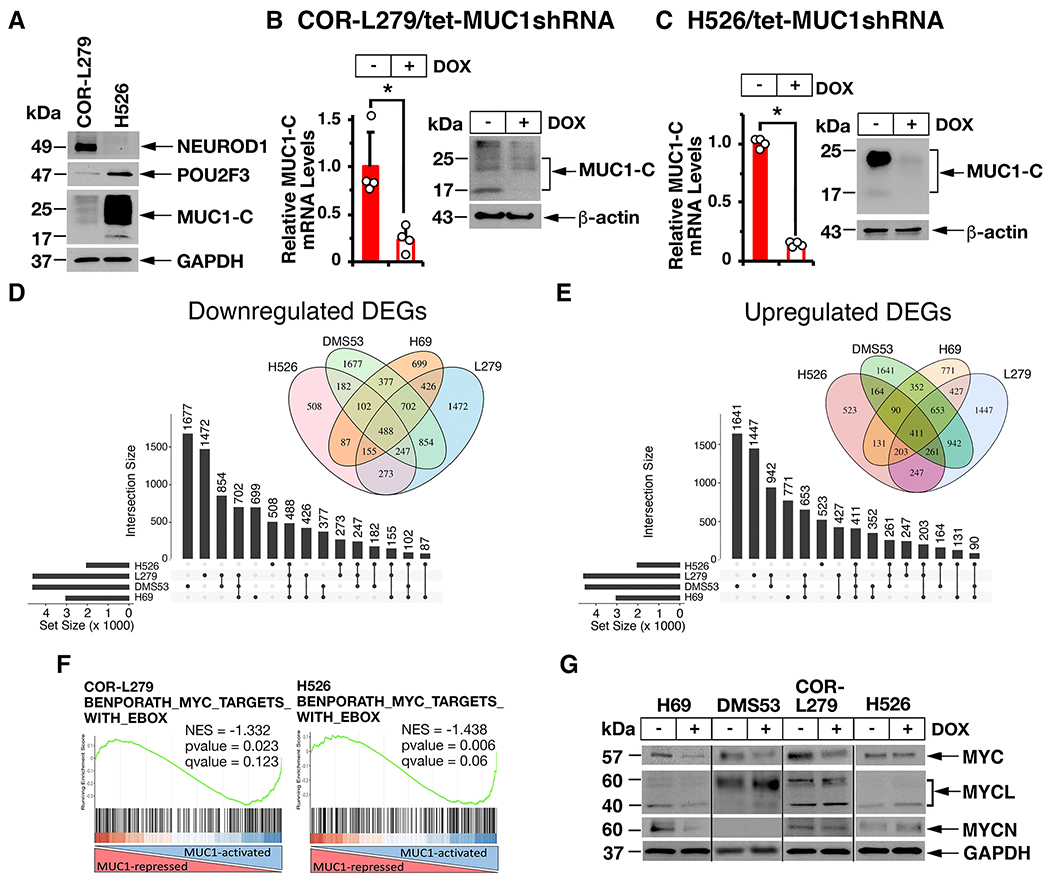
A. Lysates from COR-L279 and H526 cells were immunoblotted with antibodies against the indicated proteins. B and C. COR-L279 (B) and H526 (C) cells expressing a tet-CshRNA or a tet-MUC1shRNA were treated with vehicle or 500 ng/ml DOX for 7 days. The cells were analyzed for MUC1-C mRNA levels by qRT-PCR. The results (mean±SD of four determinations) are expressed as relative mRNA levels compared to that obtained for vehicle-treated cells (assigned a value of 1) (left). Lysates were immunoblotted with antibodies against the indicated proteins (right). D and E. RNA-seq analysis was performed in triplicate on COR-L279/tet-MUC1shRNA and H526/tet-MUC1shRNA cells treated with vehicle or DOX for 7 days. Common differentially expressed genes (DEGs) downregulated (D) and upregulated (E) by MUC1-C silencing in the indicated SCLC cells. The lollipop plots show common numbers of DEGs in the 4 different SCLC cells. F. RNA-seq datasets from the indicated cells were analyzed with GSEA using the BENPORATH MYC TARGETS WITH EBOX. G. The designated SCLC cells expressing a tet-MUC1shRNA were treated with vehicle of DOX for 7 days. Lysates were immunoblotted with antibodies against the indicated proteins.
MUC1-C drives cell cycle progression and DNA replication stress response in SCLC cells.
MYC cooperates with loss of RB in driving SCLC proliferation (5). Analysis of the H69, DMS53 and COR-L279 cell RNA-seq datasets revealed that MUC1-C significantly associates with downregulation of the BENPORATH CYCLING GENES (Supplemental Fig. S3A) and GO CELL DIVISION gene signatures (Supplemental Fig. S3B). In contrast, analysis of the RNA-seq datasets from non-NE H526 cells revealed upregulation of these cell cycle-related gene signatures (Supplemental Figs. S3A and S3B). To extend the potential involvement of MUC1-C in driving SCLC cell cycle progression, we focused on H69 and DMS53 cells and found that MUC1-C induces expression of CCNA2 (cyclin A2) and CDK1, which in concert regulate entry into mitosis (30) (Supplemental Fig. S3C). To confirm these results, we targeted MUC1-C with CRISPR/Cas9 editing using two different MUC1sgRNAs and found that MUC1-C drives CCNA2 and CDK1 expression (Supplemental Fig. S3D). Analysis of MUC1-C-driven genes also uncovered PLK1 and AURKA (Supplemental Fig. S3E), which are of significance in that their encoded proteins have been identified as potential targets for SCLC treatment (9). PLK1 plays a role in the DNA replication stress response (RSR) and regulation of the G2/M checkpoint (30). Aurora kinase A activates PLK1 and protects DNA forks during replicative stress (30). In addition, PLK1 and aurora kinase A function with CDK1 in regulating mitotic progression (30). Consistent with these functions, silencing MUC1-C with downregulation of the cyclin A2, CDK1, PLK1 and aurora kinase A proteins was associated with induction of DNA replication stress as evidenced by increases in γH2AX (Supplemental Fig. S3F, left and right) and delays in G2/M phase progression (Supplemental Fig. S3G). GSEA analysis further demonstrated that MUC1-C significantly associates with activation of the RSR DEFECT GENE SIGNATURE (Supplemental Fig. S3H) and suppression of the GO REGULATION OF RESPONSE TO DNA DAMAGE STIMULUS gene signature (Supplemental Fig. S3I). These findings collectively indicated that MUC1-C regulates (i) MYC signaling in both NE and non-NE SCLC subtypes, and (ii) cell cycle pathways in SCLC cells that have been associated with NE differentiation and replication stress (10).
MUC1-C→MYC signaling is necessary for NOTCH2 expression.
GSEA of the RNA-seq datasets using the BENPORATH_ES1 signature, which is derived from genes enriched in ESCs and CSC-like phenotypes (Ben-Porath I, Nat. Genet. 2008], revealed that MUC1-C associates with the regulation of NOTCH signaling. Along these lines, we found that MUC1-C drives expression of (i) NOTCH2 in H69, DMS53, COR-L279 and H526 cells, (ii) NOTCH1 in DMS53 and H526 cells, and (iii) NOTCH3 in DMS53 and COR-L279 cells (Supplemental Figs. S4A-4D). NOTCH2 is a marker of NE stem cells, which initiate NE reprogramming after injury and are the proposed origin of SCLC (4). Based on MUC1-C-induced regulation of MYC target genes in H69, DMS53, COR-L279 and H526 cells, we analyzed NOTCH2 for potential MYC binding motifs and identified E-boxes in the promoter and proximal enhancer regions (Fig. 3A). Consistent with MUC1-C binding directly to the MYC HLH-LZ domain (29), we performed ChIP studies which demonstrated occupancy of MUC1-C and MYC on the NOTCH2 promoter region (Fig. 3B, left). Similar results were obtained from ChIP analysis of the NOTCH2 proximal enhancer region (Fig. 3B, right). Re-ChIP analysis further demonstrated that MUC1-C forms a complex with MYC on both regions (Fig. 3C, left and right). Silencing MUC1-C was associated with decreases in MYC occupancy (Fig. 3D) and downregulation of NOTCH2 expression (Fig. 3E, left and right). Moreover, silencing MYC decreased NOTCH2 mRNA levels (Fig. 3F, left and right). In concert with the demonstration that MUC1-C is necessary for activation of MYC target genes in COR-L279 and H526 cells, silencing MUC1-C in these models also resulted in suppression of NOTCH2 transcripts (Fig. 3G, left and right), in support of a MUC1-C→MYC→NOTCH2 pathway in NE and non-NE SCLC cells.
Figure 3. MUC1-C activates NOTCH2 expression by a MYC-mediated mechanism.
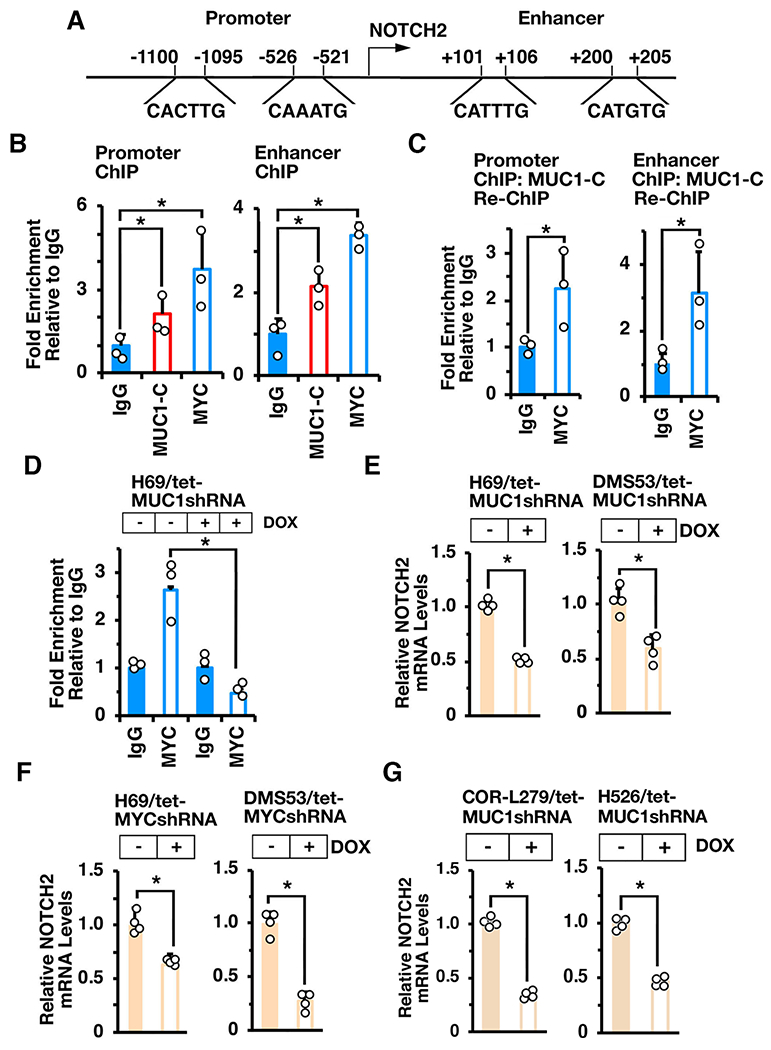
A. Schema of the NOTCH2 promoter and enhancer regions with locations of potential E-boxes. B and C. Soluble chromatin from H69 cells was precipitated with anti-MUC1-C, anti-MYC or a control IgG (B). Soluble chromatin was precipitated with anti-MUC1-C (ChIP) and then reprecipitated with anti-MYC or a control IgG (re-ChIP) (C). The DNA samples were amplified by qPCR with primers for the NOTCH2 promoter and enhancer regions. D. Soluble chromatin from H69/tet-MUC1shRNA cells treated with vehicle or DOX for 7 days was precipitated with anti-MYC or a control IgG. The DNA samples were amplified by qPCR with primers for the NOTCH2 promoter region. The results (mean±SD of 3 determinations) are expressed as the relative fold enrichment compared to that obtained with the IgG control (assigned a value of 1). E. H69/tet-MUC1shRNA (left) and DMS53/tet-MUC1shRNA (right) cells treated with vehicle or DOX for 7 days were analyzed for NOTCH2 mRNA levels by qRT-PCR. F. H69/tet-MYCshRNA (left) and DMS53/tet-MYCshRNA (right) cells treated with vehicle or DOX for 7 days were analyzed for NOTCH2 mRNA levels by qRT-PCR. G. COR-L279/tet-MUC1shRNA (left) and H526/tet-MUC1shRNA (right) cells treated with vehicle or DOX for 7 days were analyzed for NOTCH2 mRNA levels by qRT-PCR. The results (mean±SD of four determinations) are expressed as relative mRNA levels compared to that obtained for vehicle-treated cells (assigned a value of 1).
MUC1-C→MYC signaling integrates induction of NOTCH2 and NE dedifferentiation.
SCLC subtypes have been defined by differential expression of the ASCL1, NEUROD1, POU2F3 and YAP1 TFs (1, 11). In classic SCLC-A cells, ASCL1 is a key determinant of NE cell differentiation (1). In addition to NOTCH2, silencing MUC1-C in H69 cells resulted in suppression of ASCL1, as well as POU3F2/BRN2, another neural TF that promotes NE differentiation (Fig. 4A) (31). These findings were extended with similar results in H69/MUC1sgRNA cells; that is, targeting MUC1-C decreases NOTCH2, ASCL1 and BRN2 (Fig. 4B). DMS53 cells express ASCL1, as well as the YAP1 TF, which is activated by the HIPPO pathway (1). In DMS53 cells, we found that silencing MUC1-C is associated with decreases in NOTCH2, ASCL1, YAP1 and BRN2 (Fig. 4C). The importance of the MUC1-C→MYC pathway was further supported by the demonstration that silencing MYC in H69 (Fig. 4D) and DMS53 (Fig. 4E) cells also results in suppression of NOTCH2, ASCL1, YAP1 and BRN2. In extending this analysis to SCLC-N COR-L279 cells, we found that MUC1-C is necessary for expression of NOTCH2 and NEUROD1 (Fig. 4F). In H526 cells, silencing MUC1-C decreased NOTCH2 levels, but had little if any effect on POU2F3 expression (Fig. 4G). These findings supported involvement of MUC1-C→MYC signaling in integrating activation of the NOTCH2 pathway and dedifferentiation with induction of the ASCL1 and NEUROD1 TFs.
Figure 4. Targeting MUC1-C suppresses ASCL1, NEUROD1 and YAP1 expression.
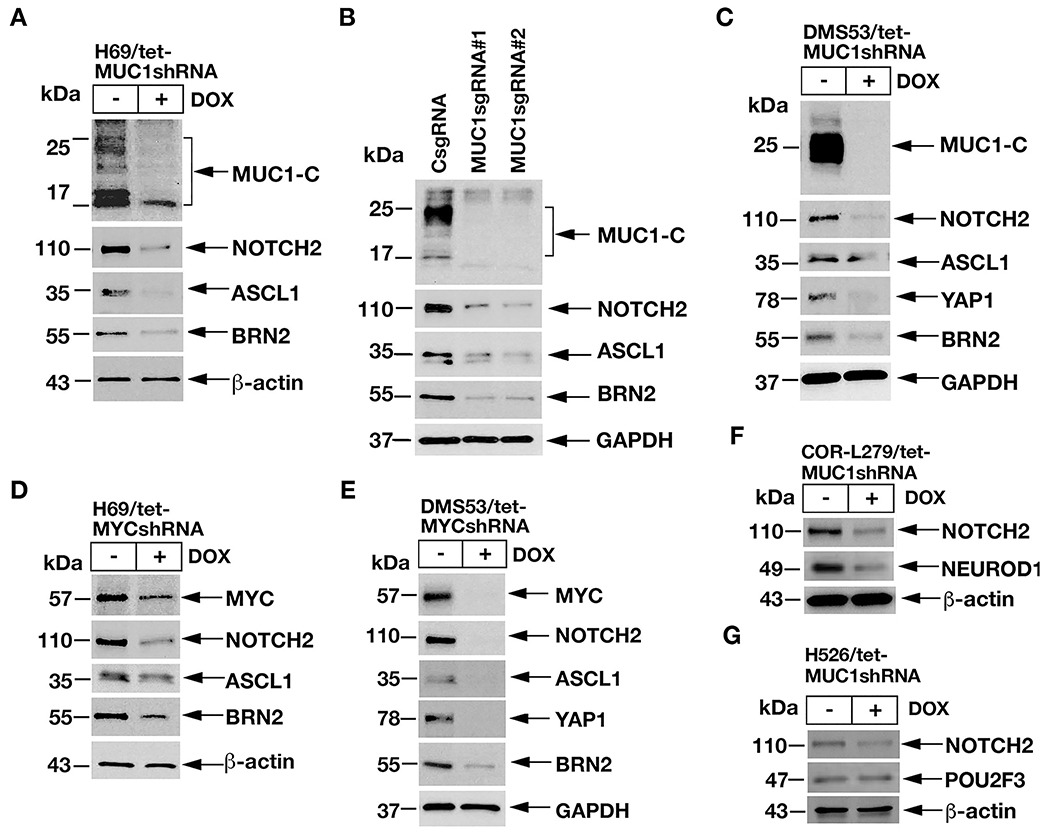
A. Lysates from H69/tet-MUC1shRNA cells treated with vehicle or DOX for 7 days were immunoblotted with antibodies against the indicated proteins. B. Lysates from H69/CsgRNA, H69/MUC1sgRNA#1 and H69/MUC1sgRNA#2 cells were immunoblotted with antibodies against the indicated proteins. C. Lysates from DMS53/tet-MUC1shRNA cells treated with vehicle or DOX for 7 days were immunoblotted with antibodies against the indicated proteins. D and E. Lysates from H69/tet-MYCshRNA (D) and DMS53/tet-MYCshRNA (E) cells treated with vehicle or DOX for 7 days were immunoblotted with antibodies against the indicated proteins. F and G. Lysates from COR-L279/tet-MUC1shRNA (F) and H526/tet-MUC1shRNA (G) cells treated with vehicle or DOX for 7 days were immunoblotted with antibodies against the indicated proteins.
MUC1-C→MYC→NOTCH2 signaling drives SCLC cell self-renewal capacity.
In extending involvement of the MUC1-C→MYC→NOTCH2 pathway in SCLC dedifferentiation, we found that silencing MUC1-C in H69 (Fig. 5A, left and right) and DMS53 (Fig. 5B, left and right) cells suppresses self-renewal capacity as assessed by decreases in tumorsphere formation. We also found that silencing NOTCH2 in H69 and DMS53 cells is associated with downregulation of (i) the NOTCH target gene, HES1, which like NOTCH2 contributes to stemness (32), and (ii) the CD133, CD44 and BMI1 stemness factors (Fig. 5C, left and right). In addition, silencing NOTCH2 resulted in induction of γH2AX expression (Figs. 5C, left and right), in concert with intersection of the RSR and CSC state (33, 34) and suppressed self-renewal capacity (Fig. 5D, left and right). In extending this work to variant COR-L279 cells, we found that silencing MUC1-C similarly inhibits self-renewal capacity (Fig. 5E). As an additional approach, we treated SCLC cells with the GO-203 inhibitor, which is a cell-penetrating peptide that blocks MUC1-C homodimerization, nuclear localization and function (12, 20–23). Treatment of H69, DMS53, COR-L279 and H526 cells with GO-203 demonstrated similar effects on (i) viability with IC50 values of ~2-4 μM and (ii) proliferation and (iii) induction of cell death (Supplemental Figs. S5A-S5H). Moreover and of importance, GO-203 was effective in suppressing SCLC cell capacity for colony formation and self-renewal (Figs. 5F–5H, left and right).
Figure 5. MUC1-C is necessary for SCLC self-renewal.
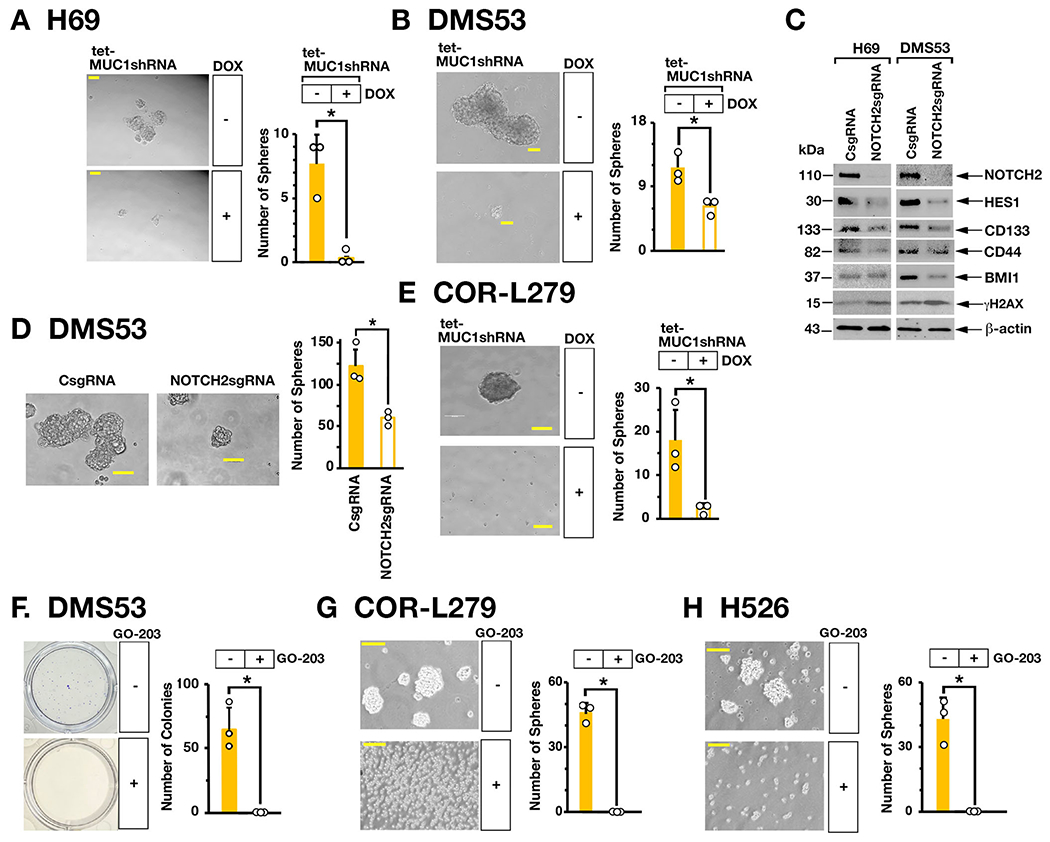
A and B. Representative images of tumorspheres derived from H69/tet-MUC1shRNA (A) and DMS53/tet-MUC1shRNA (B) cells treated with control vehicle or DOX for 7 days (left). Bar represents 50 microns. The number of tumorspheres is expressed as the mean±SD of three determinations (right). C. Lysates from H69 (left) and DMS53 (right) cells expressing a CsgRNA or NOTCH2sgRNA were immunoblotted with antibodies against the indicated proteins. D. Representative images of tumorspheres derived from DMS53/CshRNA and DMS53/NOTCH2shRNA cells (left). Bar represents 50 microns. The number of tumorspheres is expressed as the mean±SD of three determinations (right). E. Representative images of tumorspheres derived from COR-L279/tet-MUC1shRNA cells treated with control vehicle or DOX for 7 days (left). Bar represents 50 microns. The number of tumorspheres is expressed as the mean±SD of three determinations (right). F. DMS53 cells treated with control vehicle or 5 μM GO-203 for 3 days were seeded in dishes for 12 days. Representative images are shown for colonies stained with crystal violet (left). The number of colonies is expressed as the mean±SD of three determinations (right). G and H. Representative images of tumorspheres derived from COR-L279 (G) and H526 (H) cells treated with control vehicle or 5 μM GO-203 for 7 days (left). Bar represents 50 microns. The number of tumorspheres is expressed as the mean±SD of three determinations (right).
MUC1-C→MYC→NOTCH2 pathway is necessary for SCLC tumorigenicity.
To determine if MUC1-C→MYC→NOTCH2 pathway is also of importance in promoting tumorigenicity, we fed mice bearing established H69/tet-MUC1shRNA tumors with DOX to silence MUC1-C expression. DOX treatment was associated with significant inhibition of tumor growth (Fig. 6A). Analysis of tumors from control and DOX-treated mice demonstrated downregulation of MUC1-C, as well as MYC and NOTCH2, expression (Fig. 6B). In addition, targeting MUC1-C with a MUC1sgRNA, which suppressed MYC and NOTCH2 in vitro, resulted in complete inhibition of tumor growth (Fig. 6C). By extension, silencing NOTCH2 also decreased H69 tumor growth (Figs. 6D and 6E). As a second model, we confirmed that DOX treatment of mice with COR-L279/tet-MUC1shRNA tumors results in suppression of MUC1-C signaling and tumor growth (Supplemental Figs. S6A and S6B). Moreover, for potential translational relevance, we treated mice bearing established COR-L279 tumors with GO-203 and found inhibition of tumor growth in association with downregulation of MUC1-C, MYC, NOTCH2 and NEUROD1 (Figs. 6F and 6G), indicating that the MUC1-C→MYC→NOTCH2 pathway is necessary for conferring SCLC tumorigenicity.
Figure 6. Targeting MUC1-C suppresses SCLC tumorigenicity.
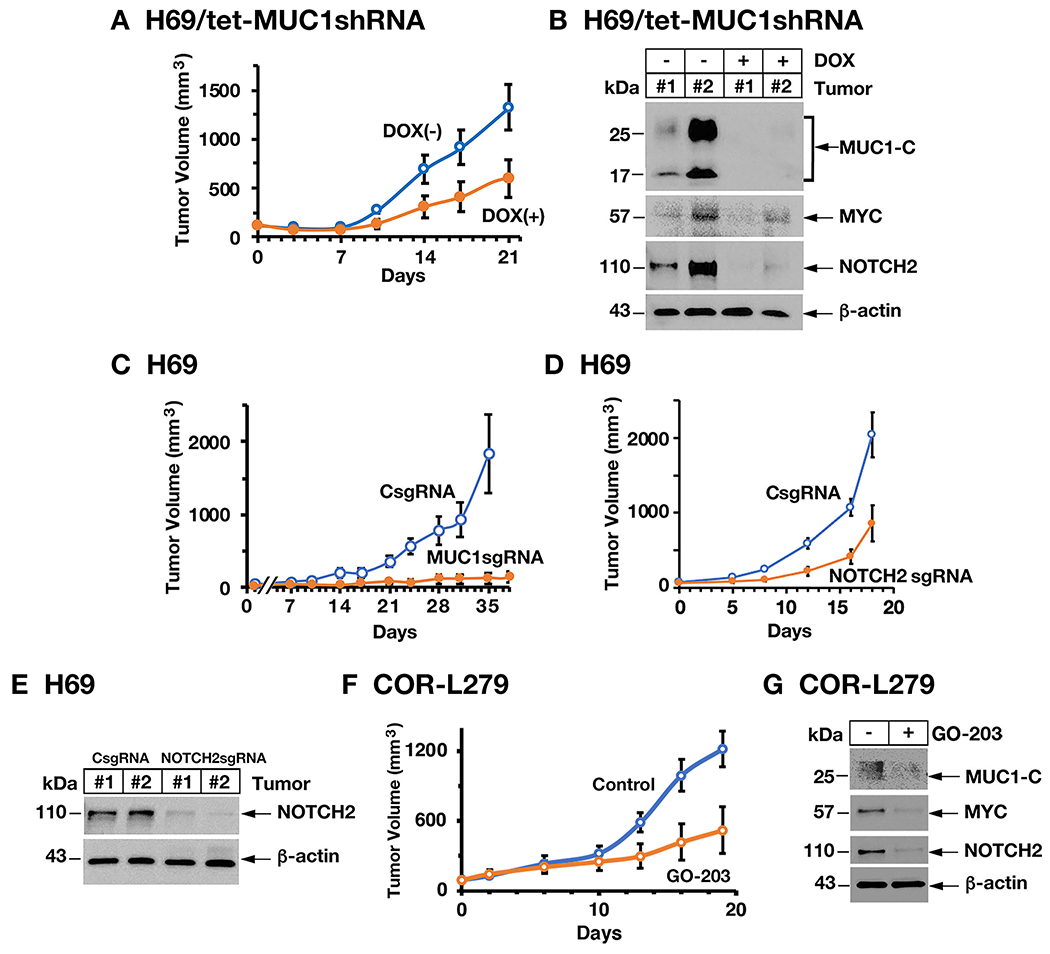
A. Six-week old nude mice were injected subcutaneously in the flank with 1 × 106 H69/tet-MUC1shRNA cells. Mice were pair-matched into two groups when tumors reached 100–150 mm3 and were fed without and with DOX. Tumor volumes are expressed as the mean±SD for six mice. B. Lysates from two untreated and two DOX-treated H69/tet-MUC1shRNA tumors obtained on day 21 were immunoblotted with antibodies against the indicated proteins. C. Six-week old nude mice were injected subcutaneously in the flank with 1 × 106 H69/CsgRNA and H69/MUC1sgRNA cells. Tumor volumes are expressed as the mean±SD for six mice. D. Six-week old nude mice were injected subcutaneously in the flank with 1 × 106 H69/CsgRNA and H69/NOTCH2sgRNA cells. Tumor volumes are expressed as the mean±SD for six mice. E. Lysates from two H69/CsgRNA and two H69/NOTCH2sgRNA tumors obtained on day 18 were immunoblotted with antibodies against the indicated proteins. F and G. Six-week old nude mice were injected subcutaneously in the flank with 3 × 106 COR-L279 cells. Mice pair-matched into two groups when tumors reached 100-150 mm3 were treated intraperitoneally each day with PBS or GO-203 for 19 days. Tumor volumes are expressed as the mean±SEM for six mice (F). Lysates from tumors harvested on day 19 were immunoblotted with antibodies against the indicated proteins (G).
Expression of MUC1 in individual SCLC tumor cells.
In extending these findings to SCLC tumor samples, we first analyzed the GSE60052 bulk RNA-seq dataset derived from 79 previously untreated patients (24). In support of our in vitro studies, we found that expression of MUC1 in these tumors significantly correlates with MYC and NOTCH2 (Fig. 7A). Further analysis was performed using the scRNA-seq dataset generated from SCLC circulating tumor cells obtained from 7 patients and grown as xenografts (CDXs) (25). The datasets were filtered, normalized and clustered by UMAP (Fig. 7B, left). MUC1 expression was detectable in each of the 7 tumor clusters (Fig. 7B, right). By extension, we found that normalized MUC1 expression was uniform within tumors with SC55 exhibiting higher and SC39 lower MUC1 levels (Fig. 7C). In concordance with our in vitro findings, MUC1 expression in single SCLC tumor cells was significantly associated with activation of the HALLMARK MYC TARGETS V1 gene signature (Fig. 7D). Consistent with these results, MUC1-C expression was detectable in individual SCLC cells by IHC staining of tumor tissue (Fig. 7E).
Figure 7. Expression of MUC1 in SCLC tumor tissues.
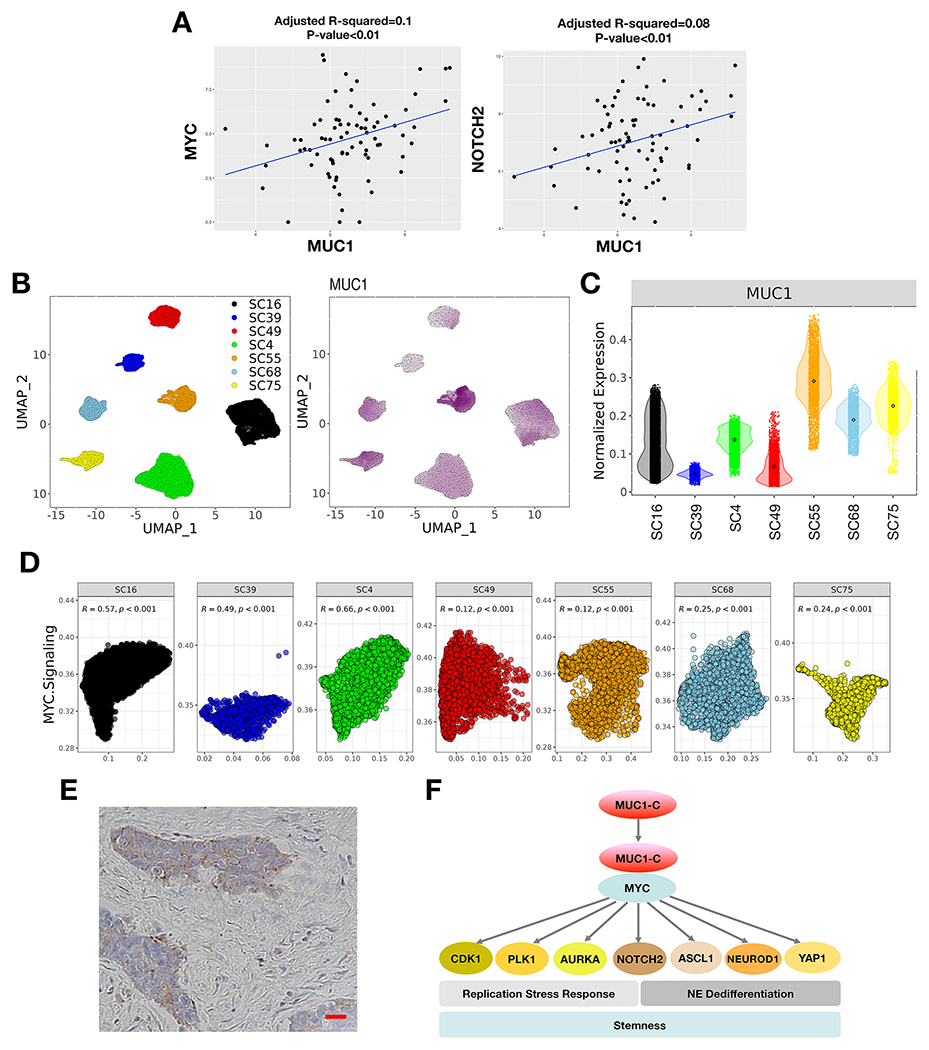
A. Analysis of the GSE60052 SCLC bulk RNA-seq dataset demonstrating significant correlations of MUC1 with MYC and NOTCH2 expression in SCLC tumor tissues. B. Analysis of the GSE138267 SCLC scRNA-seq dataset demonstrating MUC1 expression in clusters of SCLC cells obtained from 7 patient-derived CDX models. C. Distribution of MUC1 expression in the 7 CDX models. D. Significant associations of MUC1 expression with activation of the HALLMARK MYC TARGETS V1 pathway in individual tumors. E. Representative IHC staining of MUC1-C expression in SCLC tumor tissue. Bar represents 50 microns. F. Proposed model for MUC1-C in driving SCLC progression. MUC1-C activates MYC and E2F target genes that contribute to the RSR and mitotic progression. MUC1-C→MYC signaling also drives expression of NOTCH2 and NE dedifferentiation. MUC1-C thereby integrates activation of the dysregulated MYC and E2F pathways and RSR with NE dedifferentiation, stemness and self-renewal in SCLC cells.
Discussion
The MUC1 gene evolved in mammals to protect epithelial cells, including those lining the respiratory tract, against loss of homeostasis resulting from exposure to the external environment (12). The MUC1-C subunit contributes to the repair of epithelial cell damage by activating signals associated with inflammatory, proliferative and reprogramming phases of the wound healing response, which if prolonged can promote oncogenesis (12). MUC1 is upregulated in pulmonary epithelial cells exposed to tobacco carcinogens (35). Smoking is a major risk factor for the development of SCLC (1, 3); however, there has been no known involvement of MUC1-C in SCLC progression. Our studies demonstrate that expression of MUC1-C in classic NE SCLC-A, variant NE SCLC-N and non-NE SCLC-P cells activates the MYC pathway, which is dysregulated in SCLC cells and promotes tumor progression, drug resistance and poor clinical outcomes (1). In NE SCLC cells, we also found that MUC1-C drives the E2F pathway, which is aberrantly activated by mutational loss of the RB1 tumor suppressor gene and thereby derepression of E2F-driven gene transcription (1). These findings uncover a previously unrecognized role for MUC1-C in integrating dysregulation of the (i) MYC pathway in NE and non-NE SCLC cells and (ii) E2F pathway in NE SCLC cells (Fig. 7F). MUC1-C has also been linked to integrating the induction of MYC and E2F1 target genes that encode the EZH2/PRC2, BMI1/PRC1, BAF and PBAF complexes and contribute to MUC1-C-induced epigenetic reprogramming in the DDR and CSC state (13, 16, 36, 37).
Functional loss of p53 and RB with dysregulation of the MYC and E2F pathways and unrestrained proliferation of NE SCLC cells promotes stalled replication forks and activation of the RSR (8–10). NE SCLC cells are dependent on the RSR to protect against genome instability before completion of mitosis (8–10). MYC functionally promotes E2F activity by inducing the expression of cyclins and CDKs that contribute to DNA damage and cell cycle checkpoints (38–40). We identified CCNA2 and CDK1 as MUC1-C-driven MYC and E2F target genes in SCLC cells (Fig. 7F). Cyclin A2 activates CDK1, which promotes progression through G2/M phase (30). We also found that MUC1-C induces (i) PLK1, which is dysregulated in SCLC and is a target for SCLC treatment (41) and (ii) aurora kinase A, which has been targeted in MYC amplified SCLC cells with alisertib and other aurora kinase inhibitors (5)(Fig. 7F). PLK1 is activated in the G2/M DNA damage repair pathway and in conjunction with CDK1 is necessary for triggering mitosis (30). Aurora kinase A also contributes to the G2/M transition by promoting centrosome maturation and mitotic spindle assembly (30). Taken together, these results support involvement of MUC1-C in activating MYC and E2F target genes that promote dysregulation of mitotic progression. In concert with these findings and evidence that SCLC NE differentiation associates with increases in replication stress (10), we found that silencing MUC1-C in NE SCLC cells (i) promotes DSB formation as evidenced by increases in γH2AX, (ii) delays G2/M phase progression, and (iii) downregulates G2/M and mitotic spindle checkpoint gene signatures.
Epithelial wound healing includes phases of inflammation, proliferation and remodeling (12, 42). In this regard, pulmonary NE stem cells, which proliferate in the response to injury to promote repair, have been linked to the development of SCLC by constitutive activation of self-renewal (4). In addition, damage to the pulmonary epithelium is associated with reprogramming of NE stem cells and the induction of NOTCH2 as a marker of stemness in the repair response (4). We found that MUC1-C→MYC signaling induces NOTCH2 in NE and non-NE SCLC subtypes (Fig. 7F). MUC1-C/MYC complexes were detectable on the NOTCH2 promoter and proximal enhancer regions and MUC1-C was necessary for MYC occupancy, consistent with MUC1-C functioning in the activation of MYC target genes (29). We also found that MUC1-C→MYC signaling is associated with induction of ASCL1 and NEUROD1, as well as YAP1, which regulates expression of NE-associated genes (1, 11)(Fig. 7F). These findings support an essential role for MUC1-C in MYC driven SCLC NE dedifferentiation (43)(Fig. 7F). The activities of MYC family members have been ascribed to the regulation of (i) specific subsets of genes, and (ii) the majority of gene promoters in licensing elongation of all RNA PolII complexes, which can pose challenges in the analysis of RNA-seq data (44, 45). Along these lines, cell number-normalized RNA-seq, Ser2PolII immunoblots and nascent transcription techniques can be employed to address these challenges. In the present work, we confirmed our RNA-seq data by demonstrating that MUC1-C and MYC are indeed necessary for activation of the NOTCH2 stemness-associated gene. We found that targeting MUC1-C genetically as well as pharmacologically with the GO-203 inhibitor suppresses NE and non-NE SCLC cell self-renewal and tumorigenicity. Targeting MYC and NOTCH2 also decreased the capacity for self-renewal and tumor formation corroborating the importance of the MUC1-C→MYC→NOTCH2 pathway in driving the SCLC CSC state. Of note, MUC1 is not designated in DepMap as a common essential gene in SCLC cells. Our results show that targeting MUC1 genetically slows G2/M cell cycle progression, which apparently is insufficient to significantly score in DepMap. Nonetheless and importantly, targeting MUC1 with different MUC1shRNAs and MUC1sgRNAs inhibits self-renewal capacity as determined by tumorsphere formation and tumorigenicity that are not scored by DepMap.
Our findings that MUC1 significantly associates with MYC and NOTCH2 in SCLC tumors further indicate that MUC1-C may be a potential target for inhibiting CSC self-renewal in the treatment of NE and non-NE SCLCs. Of translational relevance, antibodies generated against the MUC1-C extracellular domain have been advanced for clinical evaluation as CAR-T cells and are being developed preclinically as antibody-drug conjugates (46). In dose escalation clinical trials, the MUC1-C GO-203 inhibitor was well-tolerated at low μM plasma levels and, because of a short circulating half-life, is being reformulated in nanoparticles for sustained administration (47). Additional studies will now be needed that determine the effectiveness of targeting MUC1-C in settings of SCLC cells that are resistant to treatment with cytotoxic agents, such as cisplatin and etoposide. Nonetheless, the present findings and the availability of anti-MUC1-C agents could provide new therapeutic options for SCLC patients.
Supplementary Material
Implications.
The present work uncovers addiction of SCLC cells to MUC1-C, which is a druggable target that could provide new opportunities for advancing SCLC treatment.
Financial support:
Research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health under grant numbers CA97098, CA166480 awarded to D. Kufe and CA233084 awarded to D. Kufe and K-K. Wong and CA232979 awarded to S. Liu.
Footnotes
Conflict of Interest Disclosure: DK has equity interests in Genus Oncology, Reata Pharmaceuticals and Hillstream BioPharma and is a paid consultant to Reata and CanBas. KKW has equity interests in G1 Therapeutics, Zentalis Therapeutics, Epiphanies Therapeutics and Hillstream BioPharma. The other authors declared no competing interests.
References
- 1.Rudin CM, Poirier JT, Byers LA, Dive C, Dowlati A, George J, et al. Molecular subtypes of small cell lung cancer: a synthesis of human and mouse model data. Nat Rev Cancer 2019;19(5):289–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marcoux N, Gettinger SN, O’Kane G, Arbour KC, Neal JW, Husain H, et al. EGFR-mutant adenocarcinomas that transform to small-cell lung cancer and other neuroendocrine carcinomas: clinical outcomes. J Clin Oncol 2019;37(4):278–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rudin CM, Brambilla E, Faivre-Finn C, Sage J. Small-cell lung cancer. Nat Rev Dis Primers 2021;7(1):3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ouadah Y, Rojas ER, Riordan DP, Capostagno S, Kuo CS, Krasnow MA. Rare pulmonary neuroendocrine cells are stem cells regulated by Rb, p53, and Notch. Cell 2019;179(2):403–16 e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mollaoglu G, Guthrie MR, Bohm S, Bragelmann J, Can I, Ballieu PM, et al. MYC drives progression of small cell lung cancer to a variant neuroendocrine subtype with vulnerability to aurora kinase inhibition. Cancer Cell 2017;31(2):270–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Drapkin BJ, Rudin CM. Advances in small-cell lung cancer (SCLC) translational research. Cold Spring Harb Perspect Med 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chan JM, Quintanal-Villalonga A, Gao VR, Xie Y, Allaj V, Chaudhary O, et al. Signatures of plasticity, metastasis, and immunosuppression in an atlas of human small cell lung cancer. Cancer Cell 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thomas A, Pommier Y. Small cell lung cancer: time to revisit DNA-damaging chemotherapy. Sci Transl Med 2016;8(346):346fs12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bian X, Lin W. Targeting DNA replication stress and DNA double-strand break repair for optimizing SCLC treatment. Cancers (Basel) 2019;11(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thomas A, Takahashi N, Rajapakse VN, Zhang X, Sun Y, Ceribelli M, et al. Therapeutic targeting of ATR yields durable regressions in small cell lung cancers with high replication stress. Cancer Cell 2021;39(4):566–79 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gay CM, Stewart CA, Park EM, Diao L, Groves SM, Heeke S, et al. Patterns of transcription factor programs and immune pathway activation define four major subtypes of SCLC with distinct therapeutic vulnerabilities. Cancer Cell 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kufe D. MUC1-C in chronic inflammation and carcinogenesis; emergence as a target for cancer treatment. Carcinogenesis 2020;41(9):1173–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rajabi H, Hiraki M, Kufe D. MUC1-C activates polycomb repressive complexes and downregulates tumor suppressor genes in human cancer cells. Oncogene 2018;37(16):2079–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Quintanal-Villalonga A, Chan JM, Yu HA, Pe’er D, Sawyers CL, Sen T, et al. Lineage plasticity in cancer: a shared pathway of therapeutic resistance. Nat Rev Clin Oncol 2020;17(6):360–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang L, Liao X, Beckett M, Li Y, Khanna KK, Wang Z, et al. MUC1-C oncoprotein interacts directly with ATM and promotes the DNA damage response to ionizing radiation. Genes & Cancer 2010;1(3):239–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yamamoto M, Jin C, Hata T, Yasumizu Y, Zhang Y, Hong D, et al. MUC1-C integrates chromatin remodeling and PARP1 activity in the DNA damage response of triple-negative breast cancer cells. Cancer Res 2019;79(8):2031–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yamashita N, Long M, Fushimi A, Yamamoto M, Hata T, Hagiwara M, et al. MUC1-C integrates activation of the IFN-gamma pathway with suppression of the tumor immune microenvironment in triple-negative breast cancer. J Immunother Cancer 2021;9(1):e002115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weinstein I, Joe A. Oncogene addiction. Cancer Res 2008;68(9):3077–80. [DOI] [PubMed] [Google Scholar]
- 19.Yasumizu Y, Rajabi H, Jin C, Hata T, Pitroda S, Long MD, et al. MUC1-C drives lineage plasticity in progression to neuroendocrine prostate cancer. Nat Commun 2020;11(1):338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Joshi MD, Ahmad R, Raina D, Rajabi H, Bubley G, Kharbanda S, et al. MUC1 oncoprotein is a druggable target in human prostate cancer cells. Mol Cancer Ther 2009;8(11):3056–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Raina D, Kosugi M, Ahmad R, Panchamoorthy G, Rajabi H, Alam M, et al. Dependence on the MUC1-C oncoprotein in non-small cell lung cancer cells. Mol Cancer Ther 2011;10(5):806–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Raina D, Ahmad R, Rajabi H, Panchamoorthy G, Kharbanda S, Kufe D. Targeting cysteine-mediated dimerization of the MUC1-C oncoprotein in human cancer cells. Int J Oncol 2012;40(5):1643–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Raina D, Agarwal P, Lee J, Bharti A, McKnight C, Sharma P, et al. Characterization of the MUC1-C cytoplasmic domain as a cancer target. PLoS One 2015;10(8):e0135156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jiang L, Huang J, Higgs BW, Hu Z, Xiao Z, Yao X, et al. Genomic landscape survey identifies SRSF1 as a key oncodriver in small cell lung cancer. PLoS genetics 2016;12(4):e1005895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stewart CA, Gay CM, Xi Y, Sivajothi S, Sivakamasundari V, Fujimoto J, et al. Single-cell analyses reveal increased intratumoral heterogeneity after the onset of therapy resistance in small-cell lung cancer Nat Cancer 2020;1:423–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Satija R, Farrell JA, Gennert D, Schier AF, Regev A. Spatial reconstruction of single-cell gene expression data. Nat Biotechnol 2015;33(5):495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van Dijk D, Sharma R, Nainys J, Yim K, Kathail P, Carr AJ, et al. Recovering gene interactions from single-cell data using data diffusion. Cell 2018;174(3):716–29 e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aibar S, Gonzalez-Blas CB, Moerman T, Huynh-Thu VA, Imrichova H, Hulselmans G, et al. SCENIC: single-cell regulatory network inference and clustering. Nature methods 2017;14(11):1083–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hata T, Rajabi H, Takahashi H, Yasumizu Y, Li W, Jin C, et al. MUC1-C activates the NuRD complex to drive dedifferentiation of triple-negative breast cancer cells. Cancer Res 2019;79(22):5711–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Crncec A, Hochegger H. Triggering mitosis. FEBS Lett 2019;593(20):2868–88. [DOI] [PubMed] [Google Scholar]
- 31.Bishop JL, Thaper D, Vahid S, Davies A, Ketola K, Kuruma H, et al. The master neural transcription factor BRN2 is an androgen receptor-suppressed driver of neuroendocrine differentiation in prostate cancer. Cancer Discov 2017;7(1):54–71. [DOI] [PubMed] [Google Scholar]
- 32.Liu Z, Sanders AJ, Liang G, Song E, Jiang WG, Gong C. HEY factors at the crossroad of tumorigenesis and clinical therapeutic modulation of HEY for anticancer treatment. Mol Cancer Ther 2017;16(5):775–86. [DOI] [PubMed] [Google Scholar]
- 33.Manic G, Sistigu A, Corradi F, Musella M, De Maria R, Vitale I. Replication stress response in cancer stem cells as a target for chemotherapy. Semin Cancer Biol 2018;53:31–41. [DOI] [PubMed] [Google Scholar]
- 34.McGrail DJ, Lin CC, Dai H, Mo W, Li Y, Stephan C, et al. Defective replication stress response is inherently linked to the cancer stem cell phenotype. Cell Rep 2018;23(7):2095–106. [DOI] [PubMed] [Google Scholar]
- 35.Merikallio H, Kaarteenaho R, Linden S, Padra M, Karimi R, Li CX, et al. Smoking-associated increase in mucins 1 and 4 in human airways. Respir Res 2020;21(1):239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hagiwara M, Yasumizu Y, Yamashita N, Rajabi H, Fushimi A, Long MD, et al. MUC1-C activates the BAF (mSWI/SNF) complex in prostate cancer stem cells. Cancer Res 2021;81(4):1111–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hagiwara M, Fushimi A, Bhattacharya A, Yamashita N, Morimoto Y, Oya M, et al. MUC1-C integrates type II interferon and chromatin remodeling pathways in immunosuppression of prostate cancer. J OncoImmunol 2022;11(1):e2029298 (15 pages). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu H, Tang X, Srivastava A, Pecot T, Daniel P, Hemmelgarn B, et al. Redeployment of Myc and E2f1–3 drives Rb-deficient cell cycles. Nat Cell Biol 2015;17(8):1036–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bretones G, Delgado MD, Leon J. Myc and cell cycle control. Biochim Biophys Acta 2015;1849(5):506–16. [DOI] [PubMed] [Google Scholar]
- 40.Kent LN, Leone G. The broken cycle: E2F dysfunction in cancer. Nat Rev Cancer 2019;19(6):326–38. [DOI] [PubMed] [Google Scholar]
- 41.Liao Y, Yin G, Wang X, Zhong P, Fan X, Huang C. Identification of candidate genes associated with the pathogenesis of small cell lung cancer via integrated bioinformatics analysis. Oncol Lett 2019;18(4):3723–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ge Y, Fuchs E. Stretching the limits: from homeostasis to stem cell plasticity in wound healing and cancer. Nat Rev Genet 2018;19(5):311–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ireland AS, Micinski AM, Kastner DW, Guo B, Wait SJ, Spainhower KB, et al. MYC drives temporal evolution of small cell lung cancer subtypes by reprogramming neuroendocrine fate. Cancer Cell 2020;38(1):60-78 e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Loven J, Orlando DA, Sigova AA, Lin CY, Rahl PB, Burge CB, et al. Revisiting global gene expression analysis. Cell 2012;151(3):476–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zeid R, Lawlor MA, Poon E, Reyes JM, Fulciniti M, Lopez MA, et al. Enhancer invasion shapes MYCN-dependent transcriptional amplification in neuroblastoma. Nat Genet 2018;50(4):515–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Panchamoorthy G, Jin C, Raina D, Bharti A, Yamamoto M, Adeebge D, et al. Targeting the human MUC1-C oncoprotein with an antibody-drug conjugate. JCI Insight 2018;3(12):e99880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hasegawa M, Sinha RK, Kumar M, Alam M, Yin L, Raina D, et al. Intracellular targeting of the oncogenic MUC1-C protein with a novel GO-203 nanoparticle formulation. Clin Cancer Res 2015;21(10):2338–23347. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession numbers for the RNA-seq data are GEO Submissions GSE159801 and GSE192475.