

Abstract
Endogenous replication stress is a major driver of genomic instability. Current assessments of replication stress are low throughput precluding its comprehensive assessment across tumors. Here we develop and validate a transcriptional profile of replication stress by leveraging established cellular characteristics that portend replication stress. The repstress gene signature defines a subset of tumors across lineages characterized by activated oncogenes, aneuploidy, extrachromosomal DNA amplification, immune evasion, high genomic instability, and poor survival, and importantly predicts response to agents targeting replication stress more robustly than previously reported transcriptomic measures of replication stress. Repstress score profiles the dual roles of replication stress during tumorigenesis and in established cancers and defines distinct molecular subtypes within cancers that may be more vulnerable to drugs targeting this dependency. Altogether, our study provides a molecular profile of replication stress, providing novel biological insights of the replication stress phenotype, with clinical implications.
Introduction
Genomic instability is an enabling characteristic of cancer, which by generating genetic diversity expedites the acquisition of multiple hallmark capabilities (1). DNA damage resulting from unabated replication – referred to as replication stress – is a major driver of genomic instability (2). Cells have evolved multiple mechanisms to sense and respond to replication stress, together referred to as the replication stress-response (3). When replication fork stalls, the exposed single-stranded DNA (ssDNA) is rapidly coated by ssDNA-binding proteins such as replication protein-A (RPA), leading to activation of ataxia telangiectasia and Rad3-related kinase (ATR), which subsequently phosphorylates downstream kinases including CHK1 (4). ATR and CHK1 negatively regulate cyclin dependent kinase (CDK) activity through phosphorylation of WEE1 and other substrates. ATR also delays exhaustion of RPA and global breakage of active forks by limiting origin firing (5). Together, the replication stress-response cascade prevents stalling of replication forks, controls the initiation of DNA replication, ensures sufficient supply of nucleotides, and limits mitotic entry of cells that have not yet completed DNA replication. Failure to resolve replication stress can lead to collapse of replication forks, DNA double strand breaks, and acquisition of mutations that are deleterious to genome integrity (2).
Replication stress is a feature of pre-cancerous (6) and cancerous cells (7). Cancer cells exhibit heightened replication stress-response, for example through CHEK1 amplification, to support rapid proliferation and tolerate the higher levels of replication stress (8). Replication stress itself and the mechanisms that mitigate replication stress are increasingly recognized as cancer cell-specific vulnerabilities that could be exploited therapeutically (9–12). However, rational targeting of these dependencies requires reliable approaches to assess replication stress and its cellular response in patient tumors. Measures of replication stress – including single-stranded DNA (ssDNA) or ssDNA-bound RPA levels, phosphorylated form of histone H2AX (γH2AX) – are widely used in experimental settings (13,14), but are not optimized for use in large cohorts of clinical tumor samples. Here we develop and validate a transcriptional profiling-based approach – the repstress gene signature – that characterizes the cellular response to replication stress at a functional network level (Fig. S1).
Materials and Methods
Data acquisition
RNA sequencing (RNA-seq), mutations, copy number states, drug activity, and doubling time in National Cancer Institute Development Therapeutics Program small cell lung cancer (NCI-DTP SCLC), Cancer Cell Line Encyclopedia (CCLE), Genomics of Drug Sensitivity in Cancer (GDSC), Cancer Therapeutics Response Portal (CTRP), and NCI60 were downloaded from CellMiner CDB (15,16). Clinical, pathological, and molecular characteristics, survival, RNA-seq, expression of RPPA, genomic alteration, and copy number alteration for the Cancer Cell Genome Atlas (TCGA) samples were retrieved from data hub of Pan-Cancer TCGA dataset in University of California Santa Cruz Xena platform (17). For other dataset used in this study, please refer supplementary text in Supplementary Materials.
Development of repstress gene signature
To develop repstress gene signature, we focused on four biological characteristics associating with replication stress in SCLC cell lines: MYC-paralogue genes amplification, sensitivity to cell cycle checkpoint inhibitors, high expression of phosphorylated Chk1 (p-Chk1), and neuroendocrine differentiation. We defined MYC amplified SCLC cell lines using the cutoff of 0.7 or more of copy number score (the average log2-transformed probe intensity ratio of gene specific chromosomal segment DNA relative to normal DNA) in either of MYC family genes (MYC, MYCL, MYCN). Cell cycle checkpoint inhibitor-sensitive SCLC cell lines were defined as those with drug activity score (standardized, z-score normalized measurements provided from the mean and standard deviation of -log10[molar concentration causing 50% cell growth inhibition, GI50] values over NCI-DTP SCLC cell lines) of more than 6 with CHK1 inhibitor AZD-7762 (drug ID: 754352) or WEE1 inhibitor MK-1775 (drug ID: 757148). For details of these scores, please refer a previous report describing methods used in CellMiner CDB (16). High expression of p-Chk1 was defined as Chk1_pS345 RPPA expression of more than 0.15. We subsequently applied gene set enrichment analysis (GSEA) using Hallmark gene sets (18) comparing differentially regulated pathways between SCLC cell lines with one of these characteristics and those without. By using adjusted P value of < 0.05, we identified two shared hallmark gene sets (HALLMARK_E2F_TARGET and HALLMARK_G2M_CHECKPOINT) as commonly upregulated pathways in SCLC cell lines with one of the repstress characteristics across all of the hallmark genesets. During the GSEA, 11 genes (AURKB, CCNA2, GINS1, KPNA2, LIG3, MTF2, ORC6, PRPS1, SRSF1, SUV39H1, TNPO2) were found as shared leading-edge genes of the two gene sets. Neuroendocrine status of SCLC cell lines (19) and clinical tumors in an independent cohort (20) were assessed using single sample GSEA(21) of previously described 50 neuroendocrine differentiation gene set, containing 25 genes associated with high neuroendocrine and 25 genes associated with low neuroendocrine differentiation (22). High-neuroendocrine score and low-neuroendocrine score were calculated by single sample GSEA separately using each of the 25 high of low neuroendocrine differentiation genes and compared the two scores to define high vs. low neuroendocrine differentiated SCLC cell lines (15) and clinical tumors (20). Subsequently, differentially expressing genes were analyzed between high vs. low neuroendocrine differentiated SCLC cell lines or tumors in each cohort. Among identified highly expressing genes in neuroendocrine differentiated SCLC, by false discovery rate of < 10% by Mann-Whitney U test followed by adjusting multiple testing with Benjamini-Hochberg test, those identified in both two cohort and involved in DNA damage repair pathways (23) were defined as additional repstress signature genes (GADD45G, POLA1, POLD4, POLE4, RFC5, RMI1, and RRM1). We finally excluded the gene KPNA2 from the repstress gene signature because it did not frequently express in cell lines other than SCLC (Table S1).
Repstress score was calculated by applying principal component analysis-based weighting score. In detail, SCLC cell lines were projected onto principal component analysis plot using the scores for biological characteristics associated with replication stress described above and the 17 repstress gene expression were also projected onto the plot, which achieved variable loadings of first principal component dimension for each gene as gene weight (Fig. S2A, Table S1). We summed up the measurements of repstress signature gene expressions (Z score-normalized in each cell line across all of sequenced gene expressions) multiplied by each gene weight and defined as reptress score. Repstress scores were Z score-normalized among samples used in each analysis and shown in figures.
SCLC Cell lines
Nine SCLC cell lines (NCI-H1048; RRID: CVCL_1453, NCI-H1341; RRID: CVCL_1463, NCI-H841; RRID: CVCL_1595, DMS114; RRID: CVCL_1562, NCI-H211; RRID: CVCL_1529, NCI-H446 RRID: CVCL_1562, NCI-H889: RRID: CVCL_1598, NCI-H146; RRID: CVCL_1473, NCI-H524; RRID: CVCL_1568) were purchased from ATCC and maintained in cell culture. H211, H889, H1048, and H1341 cell lines are female and the rest are male. Cell lines were authenticated using short tandem repeat analysis, and were monthly tested for mycoplasma contamination. Cell media was RPMI-1640 supplemented with 10% FBS for all lines to maintain consistency. Cells were grown at 37˚C and 5% CO2. were used in subsequent experiments.
Western blot
Cells were lysed with RIPA buffer containing protease inhibitor cocktail (Thermo Fisher Scientific) and micrococcal nuclease (Thermo Fisher Scientific). The resulting mixtures were incubated on ice for 30 minutes, then centrifuged 20 minutes to get the supernatants. After adding Tris-Glycine SDS sample buffer including 5% of 2-Mercaptoethanol, the lysates were boiled for 10 min, analyzed by SDS–polyacrylamide gel electrophoresis (SDSPAGE), and immunoblotted with various antibodies as follows: RPA phosphorylation (pS4/8, from Bethyl; RRID: AB_2891810); total RPA (from Bethyl; RRID: AB_185548); pATR (T1989, from Cell signaling; RRID:AB_2722679); and pCHK1(S345, from cell signaling; RRID:AB_330023). To proceed Western blot, block nitrocellulose membrane with 5% nonfat milk, then incubate with primary antibodies at 1:1000 dilution in PBST buffer (PBS containing 0.1% Tween 20) containing 1% nonfat milk, at 4C for overnight. After 3 times washing with PBST, the membrane was incubated with 2nd antibody at 1:2000 dilution in PBST buffer containing 1% nonfat milk, at room temperature for 1h. Develop the western blot results by BIO-RAD ChemiDoc MP Imaging System.
Immunofluorescence assay
Cells were fixed with 2% paraformaldehyde (PFA), followed by the incubation with 70% cold ethanol. After block with 5% BSA. Primary antibody staining was performed as follows: anti γH2AX (1:500, Millipore, 05–636), anti-pRPA (1:500, Bethyl lab, A300–245A; RRID: AB_210547). Secondary antibody staining was performed as follows: Alexa 488 conjugated anti-mouse lgG and Alexa 594 conjugated anti-rabbit lgG (1:500, Cell signaling Technology, 4408 and 8889). DAPI staining was performed with VECTASHIELD mounting medium with DAPI (H-1200, VECTOR Laboratories). A Zeiss LSM780 confocal microscope was used to capture the fluorescence. The Colocalization Plugin of the FIJI-ImageJ software was used to calculate the fluorescence density.
EdU incorporation and γH2AX induction upon topotecan treatment
Cell lines were plated at 1 million cells per 10 cm plate. After 24 hours cells were treated for two hours with either DMSO control or 10 µM topotecan, and for 1 hour (the second hour of topotecan treatment) with 1 µM EdU. Cells were fixed in and stained for γH2AX as previously described (24), followed by click it chemistry per manufacturer instructions utilizing the Click-iT™ Plus EdU Alexa Fluor™ 647 Flow Cytometry Assay Kit C10634 (Thermo Fisher). Flow cytometry data was collected using a BD LSRFortesa and analyzed utilizing FlowJo V10.7.1.
DNA combing analysis
As previously described (25), asynchronous DMS114 and H524 cells were sequentially labelled with 20 μM IdU for 20 min and 50 μM CldU for 20 min. To preserve long genomic DNA fibers, cells were embedded in low melting point agarose plugs and incubated in cell lysis buffer with proteinase K at 50°C for overnight. Washed plugs with TE buffer, and then melted plugs in 0.1M MES (pH 6.5) at 70°C for 20 min. Agarose was subsequently degraded by adding 2 μl of β-agarase (New England Biolabs). DNA fibers were then stretched onto salinized coverslips (Genomic Vision, cov-002-RUO) using an in-house combing machine. Combed DNA on coverslips was then baked at 60 °C for 2 hours and denatured in 0.5 N NaOH for 20 min. IdU, CldU and single-strand DNA were detected using a mouse antibody directed against BrdU (IgG1, Becton Dickinson, 347580, 1:25 dilution), a rat antibody directed against BrdU (Accurate chemical, OBT0030, 1:200 dilution) and a mouse antibody directed against single-stranded DNA (ssDNA) (IgG 2a, Millipore, MAB3034, 1:100), respectively. The secondary antibodies used were goat anti-mouse Cy3 (Abcam ab6946), goat anti-rat Cy5 (Abcam, ab6565), and goat anti-mouse BV480 (Jackson ImmunoResearch, 115–685-166) for ssDNA. Slides were scanned with a FiberVision Automated Scanner (Genomic Vision). Replication signals on single DNA fibers were analyzed using FiberStudio (Genomic Vision).
Graph generation and statistical analysis
All figures were generated using CellMiner CDB (16), GraphPad PRISM software version 8.1.2 (GraphPad Software), R version 1.2.135 (R Foundation for Statistical Computing), and STATA software version 16.0 (Stata-Corp). Box plots in this manuscript were shown by box and Tukey whiskey appearances, unless specifically indicated in figure legends. Methods for statistical analyses were indicated in the manuscript and figure legends and were performed using softwares described above. Overall survival (OS) curves were created by the Kaplan-Meier method and compared by log-rank test. All statistical tests were two-sided.
Data Availability
The data analyzed in this study were obtained from public database. The experimental data generated in this study are available upon request from the corresponding author.
Results
Development and validation of a replication stress-response signature
While replication stress is widely prevalent across cancers, it is more central to the tumorigenesis of some cancers than others (7). We chose to develop a replication stress-response signature in SCLC, a fast-growing and deadly cancer with molecular and clinical features distinct from other lung cancers. We reasoned that signatures that report replication stress-response in SCLC could then be extended to other tumors that also exhibit this phenotype.
SCLCs are characterized by high degree of genomic instability, an important consequence of replication stress (26). Nearly all SCLCs have loss-of-function alterations in tumor suppressors RB1 and TP53, and frequently exhibit amplification and overexpression of oncogenes such as MYC (20). SCLCs also exhibit sustained high expression of lineage transcription factors, which contribute to replication stress (27), and are highly vulnerable to perturbation of the transcriptional state (28,29). Not surprisingly, the standard treatment of SCLC consists mostly of DNA damaging agents such as platinum compounds, topoisomerase I and II inhibitors, and an alkylating agent temozolomide.
To obtain a comprehensive molecular understanding of the replication stress-response, we examined a panel of 67 SCLC cell lines characterized by microarray-based gene expression, representing the molecular diversity of the disease (15,19). We reasoned that SCLC cells under high replication stress might be characterized by amplification of MYC and its paralogs MYCN and MYCL (30,31); expression of p-Chk1 (32); sensitivity to inhibitors of cell cycle checkpoints CHK1 and WEE1 (33); and neuroendocrine differentiation (12,29,34). GSEA was performed to define differentially regulated biological processes between SCLCs with and without these features, revealing cell-cycle related targets of E2F transcription factors and genes involved in the G2/M checkpoint (AURKB, CCNA2, GINS1, LIG3, MTF2, ORC6, PRPS1, SRSF1, SUV39H1, TNPO2) and DNA replication and repair genes associated with neuroendocrine differentiation (GADD45G, POLA1, POLD4, POLE4, RFC5, RMI1, and RRM1), together designated as the repstress gene signature (Fig. 1A, Table S1). Repstress signature score was calculated using weighted principal component analysis (Fig. S2A, Table S1), with most genes providing positive signature weightings except POLD4 and POLE4.
Fig. 1. Generation and in vitro functional validation of a repstress gene signature in small cell lung cancer cell lines.
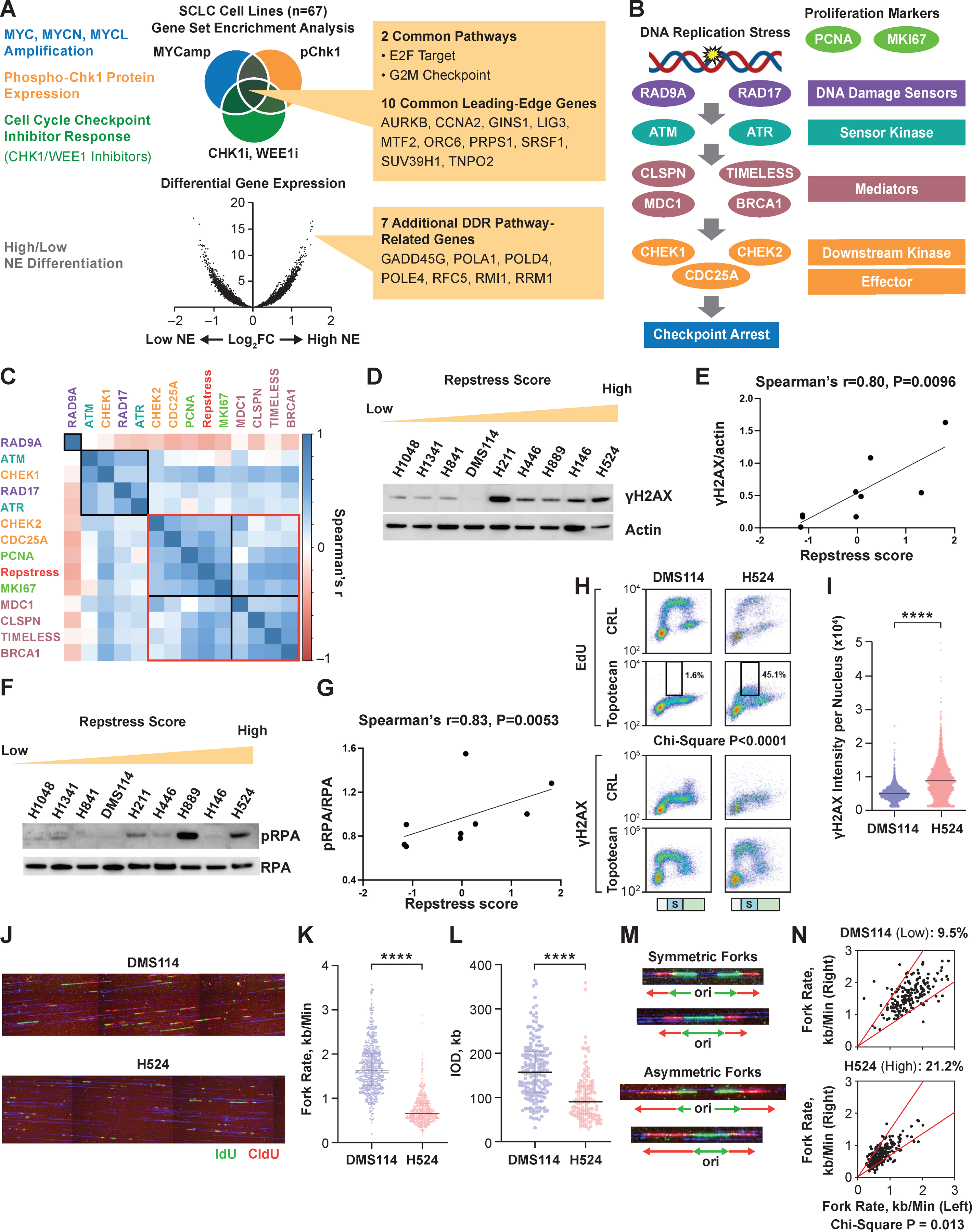
(A), Schematic representation of the repstress gene signature derivation, which is based on four key characteristics associated with replication stress: (i) amplification of MYC paralogs; (ii) expression of p-Chk1; (iii) sensitivity with CHK1 and WEE1 inhibitors; and (iv) neuroendocrine differentiation.
(B), Schematic representation highlighting key components of the replication stress-response pathway
The DNA damage sensors recruit kinases ATM and ATR that in turn phosphorylate mediators such as MDC1 and BRCA1 which sustain the DNA damage response signaling. DNA damage response signaling then engages downstream kinases CHK1 and CHK2 and eventually activates downstream effectors such as CDC25A phosphatases triggering transient cell cycle arrest.
(C), Pairwise correlations between expression of DNA damage response genes, proliferation markers PCNA and MKI67, and repstress score in 67 SCLC cell lines
Colors of gene name labels denote replication stress-response functions indicated in panel B. Genes are clustered by Euclidean distance, using the complete-linkage clustering method, indicated with squares with black and red lines.
(D), (E), Western blot (D) and correlations (E) of γH2AX signal with repstress score in SCLC cell lines
SCLC cell lines are ordered from low to high repstress score (range: −1.2 – 1.8) from left to right in panel D.
(F), (G), Western blot (F) and correlations (G) of pRPA signal with repstress score in SCLC cell lines
SCLC cell lines are ordered from low to high repstress score (range: −1.2 – 1.8) from left to right in panel F.
(H), (I), S phase arrest and induction of γH2AX by exogenous replication stress by topotecan treatment in S phase SCLC cell lines
EdU incorporation (top) and γH2AX induction (bottom) in SCLC cell lines with low (DMS114) and high repstress score (H524) are shown in panelH. Cell cycle effects are defined by PI staining (Fig. S5) and G1, S, G2/M phases are indicated on the bottom of the panels with light green, light blue with the letter of S, and light orange bars, respectively. Black squares indicate proportion of EdU incorporating S phase cells, gated by cutoff of EdU signal intensity > 1.0 × 103. A comparison of quantified γH2AX signal intensity per nucleus with topotecan treatment in S phase cells is shown in panel I. ****: P < 0.0001 by unpaired Student t test.
(J), (K), (L), DNA combing analysis of SCLC cell lines with low (DMS114) and high (H524) repstress scores
Representative images (J) and quantifications of replication fork speed (K) and inter-origin distance (L) are shown. Green and red lines in panel J indicate IdU and CIdU, respectively. ****: P < 0.0001 by Mann-Whitney U test.
(M), (N), Representative images (M) and quantification (N) of fork asymmetry in DNA combing analysis of SCLC cell lines with low (DMS114) and high (H524) repstress score Fork asymmetry was defined by > 30% difference of fork speed between one direction with the other as previously described (25), indicating with a redline in panel N. The proportions of DNAs with fork asymmetry in each cell line were indicated on top of panel N.
Abbreviations: SCLC: small cell lung cancer; DDR: DNA damage response; NE: neuroendocrine differentiation; MYCamp: MYC amplification; WEEi1: WEE1 inhibitor; CHK1i: CHK1 inhibitor; p-Chk1: phosphorylated Chk1; pRPA: phosphorylated replication protein A; EdU: 5-ethynyl-2’-deoxyuridine; Cont: control; PI: propidium iodide; DAPI: 4′,6-diamidino-2-phenylindole; IdU: iododeoxyuridine; CIdU: chlorodeoxyuridine; kb: kilobase; IOD: inter-origin distance; ori: origin.
Repstress signature included genes involved in mitosis (AURKB), cell cycle progression (CCNA2), initiation of replication and replisome progression (GINS1, ORC6, RFC5), nuclear transport (TNPO2), DNA and RNA metabolism (LIG3, PRPS1, RMI1, RRM1), transcriptional regulation (MTF2, SUV39H1), RNA splicing (SRSF1), and DNA polymerases (POLA1, POLD4, POLE4). High repstress cells had elevated expression of MDC1, CLSPN, and TIMELESS, genes involved in replication stress tolerance by protecting the replication fork, downstream effectors CHEK2 and CDC25A, and genes associated with proliferation PCNA and MKI67 (ranges of Spearman’s correlation coefficient and multiple testing adjusted P value: 0.22 to 0.61 and 5.5 × 10−7 to 7.7 × 10−4, respectively). In contrast, DNA damage sensors RAD9A and RAD17 and sensor kinases ATM and ATR were less correlated with repstress score (Fig. 1B, C, S2B–D). Repstress score correlated positively with the expression of genes involved in solving topological problems during replication (TOP2A), facilitating the repair and restart of stalled replication forks (FANCD2), resolving barriers to replication fork progression (RNASEH2A), and DNA repair (POLQ and PARP1) (Fig. S2E–I).
Stalled replication forks require the surrounding chromatin to be compacted for their stabilization (35); the expansion of heterochromatic regions is mediated by histone modifications and attenuates replication stress signaling. We reasoned that if repstress score captures replication stress-response at a functional network level, it may be able to predict the heterochromatin response as well. To test this possibility, we examined pairwise correlations between the repstress score and expression of chromatin remodelers and histone modifiers. Repstress score correlated positively with the expressions of heterochromatin proteins HP1α, HP1β and HP1γ that associate with methylated histone H3 on nucleosomes and mediate heterochromatin formation (ranges of Spearman’s correlation coefficients and multiple testing adjusted P values: 0.44 to 0.56 and 1.4 × 10−5 to 2.8 × 10−3, respectively). In contrast, genes involved in INO80 chromatin remodeling complex (INO80 and ARP8) were less correlated with repstress signature and clustered separately (0.11 to 0.25 and 0.6 to 1.0, respectively, Fig. S2J).
Stressed DNA replication results in DNA double-strand breaks, which induce rapid phosphorylation of H2AX on Ser139, termed as γH2AX. γH2AX is a sensitive albeit indirect indicator of replication stress (36). We detected higher basal endogenous expression of γH2AX by Western blot in SCLC cells with high repstress score compared to cells with low repstress score (Spearman’s correlation coefficient and P value: 0.80 and 0.0096, respectively, Fig. 1D, E). Other replication-stress associated proteins such as phosphorylated RPA, Chk1, and ATR also had positive correlations with repstress score (Fig. 1F, G, S3). Higher basal levels of γH2AX and phosphorylated RPA were also detected by fluorescence microscopy in repstress-high H524 cell line compared with repstress-low DMS114 (Fig. S4).
We then assessed whether cells with variable repstress scores responded differentially to exogenous replication stress, using topotecan which produces replication blocks by generating topoisomerase I−DNA cleavage complexes, in two representative cell lines H524 and DMS114 with high and low repstress scores, respectively. At basal levels without drug treatment, H524 cells exhibited lower DNA synthesis and more DNA damage during S-phase, as indicated by the proportion of cells labelled with 5-ethynyl-2’-deoxyuridine (EdU) and γH2AX respectively, compared with DMS114 cells. Upon treatment with topotecan, DNA synthesis and cell proliferation were inhibited to a much lesser extent in H524 cells compared with DMS114 (Fig. 1H, S5), resulting in higher induction of γH2AX in H524 (Fig. 1H, I). The γH2AX induction by topotecan treatment correlated with the repstress score in a larger panel of SCLC cell lines (Fig. S6). To further elucidate the dynamics of DNA replication, we performed DNA combing assay. H524 cells had markedly lower fork velocities and inter-origin distances compared with DMS114 (Fig. 1J–L). Shorter inter-origin distances can result from activation of dormant origins due to oncogene-induced replication stress which slows or stalls replication forks (37). Further, the patterns of bidirectional fork movement were more asymmetric in H524 cells compared with DMS114 (Fig. 1M, N), indicating that higher repstress gene expression associates with replication fork stalling.
Together, we find that the molecular components involved in replication stress-response are interconnected. Repstress score captures the coordinate expression of key components of this cascade downstream of checkpoint sensors and kinases with the associated chromatin changes.
Even in an unchallenged S phase, high repstress score cells exhibit more endogenous replication stress and robust activation of DNA damage response than low repstress cells. However, they are hypersensitive to exogenous replicative stress likely because further recruitment of replication stress-response is less effective. Thus, the repstress gene signature could allow for interrogation of endogenous replication stress and efficiency of the replication stress-response in SCLC cell lines.
Repstress score captures transcriptional responses to replication stress across cancer types
To determine whether the repstress gene signature was generalizable and able to predict replication stress-response signaling in cancers beyond SCLC, we queried RNA-seq and Reverse Phase Protein Array (RPPA) data from the CCLE of 937 cell lines across 20 cancer types (Fig. 2A) (38). Highest repstress scores were found in SCLC (the number and proportion of SCLC cells with repstress score ≥ 95% confidence interval of repstress score across all CCLE cell lines: 48/50, 96.0%), hematopoietic malignancies (non-Hodgkin’s lymphoma [43/49, 87.8%] and leukemia [57/78, 73.1%]) and sarcoma (55/87, 63.2%), consistent with previous reports of these malignancies exhibiting high replication stress phenotype (39,40). Low repstress scores were observed in renal cell carcinoma (the number and proportion of cells with repstress score <95% confidence interval of repstress score across all CCLE cell lines: 22/31, 71.0%), pancreatic cancer (15/23, 65.2%), ovarian cancer (30/46, 65.2%), melanoma (35/56, 62.5%), and thyroid cancer (6/11, 54.5%). The distribution of repstress score across cancer types was overall similar when DNA repair genes associated with neuroendocrine differentiation were excluded from the signature, with SCLC and hematopoietic malignancies exhibiting the highest scores (Fig. S7), suggesting that the high repstress score in SCLC is not confounded by neuroendocrine differentiation, a pathophysiological characteristic of this cancer.
Fig. 2. Across cancer cell lines, repstress score profiles replication stress at a functional network level.
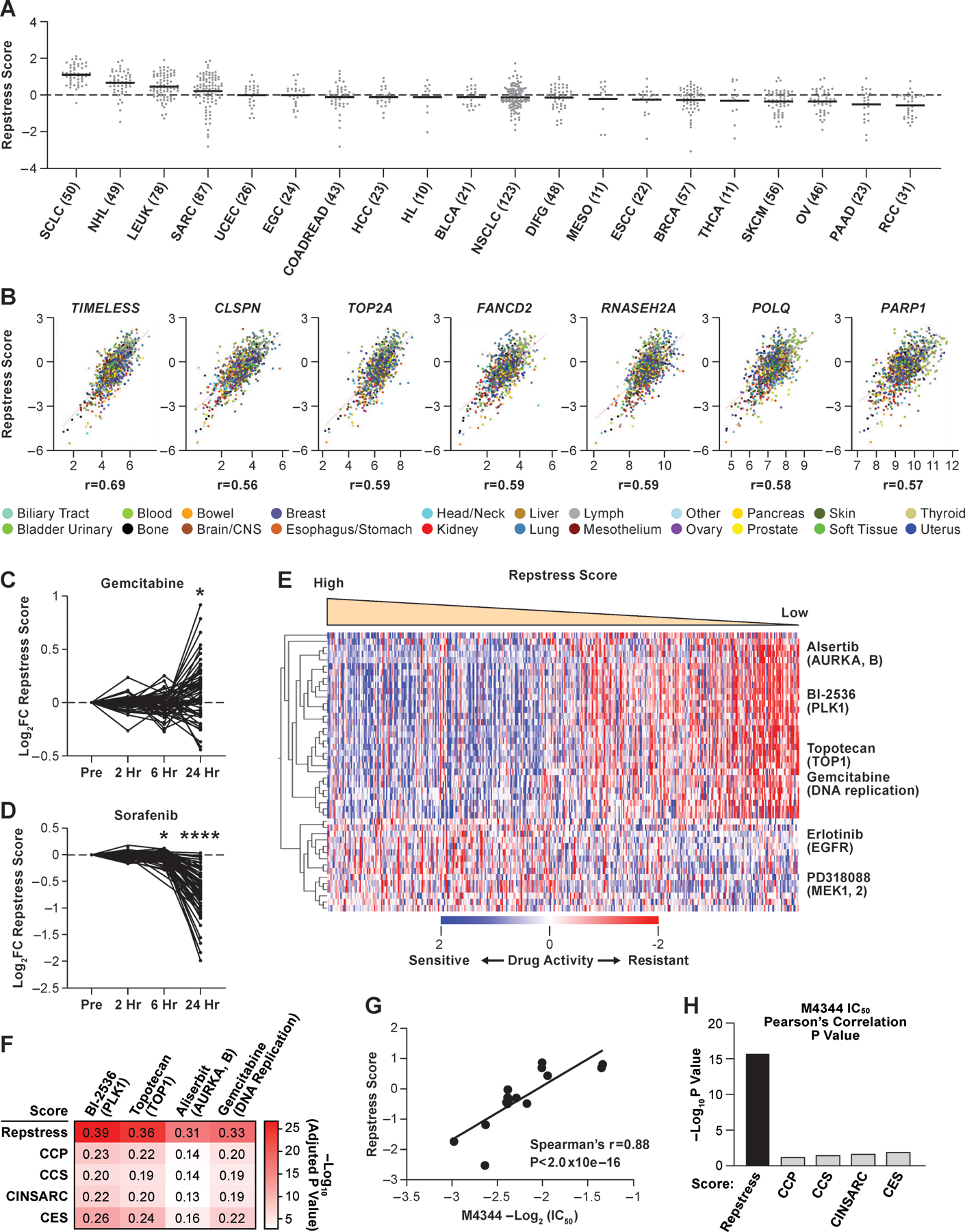
(A), Dot plot showing distribution of repstress score across 839 cancer cell lines from 20 cancer types represented in the CCLE
A black bar in each cancer type indicates the mean repstress score within each cancer type. Dash line indicates zero of Z-normalized repstress score across all of cancer cell lines in CCLE. The numbers with cancer type labels on x-axis indicate the numbers of cell lines included.
(B), Across cancers, repstress score correlates with expression of representative genes involved in: (i) increasing replication stress tolerance by protecting replication forks (TIMELESS, CLSPN), (ii) solving topological problems during replication (TOP2A), (iii) facilitating the repair and restart of stalled replication forks (FANCD2), (iv) resolving barriers to replication fork progression (RNASEH2A), and (v) DNA damage repair factors (POLQ and PARP1)
Correlations are analyzed in CellMiner CDB (16). Spearman’s correlation coefficients (r) are indicated. All of P values by Spearman’s correlation test are < 0.0001.
(C), (D), Dynamics of normalized repstress score with treatment of gemcitabine (C) and sorafenib (D) in NCI60 cell lines
Dynamics of gene expression pre- and post-treatment are retrieved from The NCI Transcriptional Pharmacodynamics Workbench (42). *: P < 0.05; ****: P < 0.0001 by Wilcoxon signed rank test. For detailed method, please refer supplementary text in Supplementary Materials.
(E), Heatmap of sensitive or resistant agents in cell lines with high vs. low repstress score in the CTRP
Drug activity scores indicate calculated area under the curve over a 16-point concentration range using an automated, high-throughput workflow fitting concentration-response curves (43). The drug activity score are retrieved from CellMiner CDB (16) and z score-normalized in the heatmap. Cell lines are sorted by repstress score from high (left) to low (right). The heatmap shows 30 mostly sensitive compounds in high repstress score cell lines, and all of sensitive compounds in low repstress score cell lines with false discovery rate of < 5%. For detailed method, please refer supplementary text in Supplementary Materials.
(F), Heatmap of Pearson’s correlations between gene signature scores and activities of drugs targeting replication stress
The color in each column indicates log-transformed P value of Pearson’s correlation between annotated gene signature score and drug activity score. The number in each column shows Pearson’s correlation coefficient between them. CCP, CCS, CINSARC, and CES scores are calculated as previously reported (39,45–47).
(G), Correlations between IC50 of M4344 (an ataxia telangiectasia and Rad3-related inhibitor) and repstress score in different cancer type cell lines
The IC50 of M4344 in different cancer type cell lines was examined in a previous report (48).
(H), Comparison of Spearman’s correlations between M4344 IC50 and scores of repstress and other cell proliferation gene signatures
Each bar represents log-transformed P value of Spearman’s correlation between annotated gene signature and M4344 IC50. The IC50 of M4344 in different cancer type cell lines is examined in a previous report (48). CCP, CCS, CINSARC, and CES scores are calculated as previously reported (39,45–47).
Abbreviations: CCLE; Cancer Cell Line Encyclopedia; SCLC: small cell lung cancer; NHL: non-Hodgkin’s lymphoma; LEUK: leukemia; SARC: sarcoma; UCEC: uterine endometrioid cancer; EGC: esophagogastric adenocarcinoma; COADREAD: colorectal adenocarcinoma; HCC: hepatocellular carcinoma; HL: Hodgkin’s lymphoma; BLCA: bladder urothelial carcinoma; NSCLC: non-small cell lung cancer; DIFG: diffuse glioma; MESO: mesothelioma; ESCC: Esophageal squamous cell carcinoma; BRCA: breast carcinoma; THCA: Thyroid cancer; SKCM: skin melanoma; OV: ovarian cancer; PAAD: pancreatic adenocarcinoma; RCC: renal cell carcinoma; FC: fold change; hr: hour; CTRP: Cancer Therapeutics Response Portal; AURKA, B: aurora kinase A and B; PLK1: polo-like kinase-1; TOP1: topoisomerase I; EGFR: Epidermal growth factor receptor; MEK1, 2: mitogen-activated protein kinase kinase 1 and 2. IC50: half maximal inhibitory concentration; CCP: cell cycle progression; CCS: cell cycle score; CINSARC: complexity index in sarcomas; CES: Centromere and kinetochore gene Expression Score; IC50: half maximal inhibitory concentration.
Similar to SCLC cell lines, the repstress score was positively correlated with expression of key genes involved in increasing replication stress tolerance across cancer types (Fig. 2B). Pairwise correlations recapitulated the correlation of repstress score with expression of DNA damage response mediators, effectors, and heterochromatin, in contrast to sensors and sensor kinases at the mRNA and protein levels (Fig. S8).
Genotoxic agents currently used for cancer therapy include many potent inducers of replication stress, such as platinum derivatives, topoisomerase inhibitors, and nucleotide analogues (41). We hypothesized that repstress gene signature may profile these changes in diverse cancers types. To investigate this possibility, we examined repstress score dynamics pre-and post-treatment with 15 anticancer agents across a panel of 60 human cancer cell lines of different lineages (42). Cells were exposed to these agents at concentrations below the human peak plasma concentration and the average concentration resulting in 50% cell growth inhibition. In a group of cell lines, we identified similar transcriptional responses to gemcitabine, cisplatin, and topotecan, which resulted in notable induction of repstress gene expression after treatment (Fig. 2C, S9A–C). Topotecan and cisplatin induce replication blocks respectively by generating topoisomerase I−DNA cleavage complexes and platinum–DNA adducts, whereas gemcitabine stalls replication through its integration into DNA and depletion of the deoxyribonucleotide pool. In contrast, treatment with tyrosine kinase inhibitors sorafenib and dasatinib, and the histone deacetylase inhibitor vorinostat resulted in uniformly decreased repstress gene expression (Fig. 2D, S9A, D, E).
Together, repstress gene signature stratifies cancer cell lines across tumor types based on their adaptability to replication stress and profiles transcriptional responses to drug-induced modulation of replication stress. Molecular features that contribute to the replication stress phenotype including drug responses across cancer cell line databases may be explored at this web-based resource: https://discover.nci.nih.gov/cellminercdb/ (15,16).
Repstress score predicts sensitivity to replication stress targeted therapies including novel ATR inhibitors
Cancers with heightened replication stress-response may be particularly vulnerable to drugs that target this dependency. We investigated whether the repstress score predicts drug sensitivity using 481 anticancer drugs across 823 cell lines of the Cancer Therapeutics Response Portal (CTRP) (43). Drug sensitivities were compared between cell lines defined by the lowest (<25th) and highest (≥75th) repstress score percentiles. With false discovery rate of 5%, 280 compounds were identified as significantly more or less active in repstress-high compared with repstress-low cell lines (Fig. S10A). High repstress score cells were more sensitive to inhibitors of polo-like kinase-1 (BI-2536: adjusted P value = 2.4 × 10−28), topoisomerase I (topotecan: adjusted P value = 1.1 × 10−21), aurora kinase A and B (alisertib: adjusted P value = 2.0 × 10−20), and regulators of cell cycle progression and DNA replication (gemcitabine: adjusted P value = 9.4 × 10−17) (Fig. 2E, S10). In contrast, low repstress score cells were more sensitive to compounds targeting pathways such as mitogen-activated protein kinase (MEK) and epidermal growth factor receptor (Fig. 2E, S10A). This observation is consistent with a recent study in isogenic cell lines which reported MEK signaling-dependence in replication stress-response defective cells (44). Repstress score exhibited a higher positive correlation with response to agents that induce replication stress, including alisertib, BI-2536, topotecan, and gemcitabine, than the currently available cell cycle proliferation genes (39,45–47) (Fig. 2F, S11).
Because of the critical functions of ATR in protecting cells under replication stress, small-molecule ATR inhibitors are being explored as cancer therapeutic agents to selectively kill cancer cells under replication stress (9). A reliable method to measure replication stress levels could in principle enable patient stratification for ATR inhibitor therapies. We examined whether the repstress signature predicted sensitivity to ATR inhibitors (48). Across 16 cancer cell lines from different histologies, cells with high repstress score showed higher sensitivity to ATR inhibitor M4344 than cells with low repstress score (Spearman’s r = 0.88, P < 2.0 × 10−16, Fig. 2G). Repstress score better predicted ATR inhibitor response than the previously described signatures of replication stress and proliferative gene expression signatures (Fig. 2H, S12) (39,45–47).
Repstress score defines subsets of cancers characterized by genomic instability, immune evasion, and poor prognosis across tumor types
Replication stress is a driver for cancer progression and is linked to genomic instability in precancerous lesions and cancers (7). In precancerous lesions, the replication stress-response provides a barrier to delay or prevent tumorigenesis (6,8,49). Using repstress score, we assessed replication stress along the continuum of cancer development (50). Repstress scores were higher in bronchial precancerous lesions which eventually regressed and those that progressed to become cancers, compared with lesions that maintained stable precancerous characteristics (Fig. 3A), supporting the dual roles of replication stress in promoting genomic instability, and in slowing down cell proliferation and activating anticancer barriers (8).
Fig. 3. Across cancer types, repstress score defines cancers characterized by genomic instability, immune evasion, and poor prognosis.
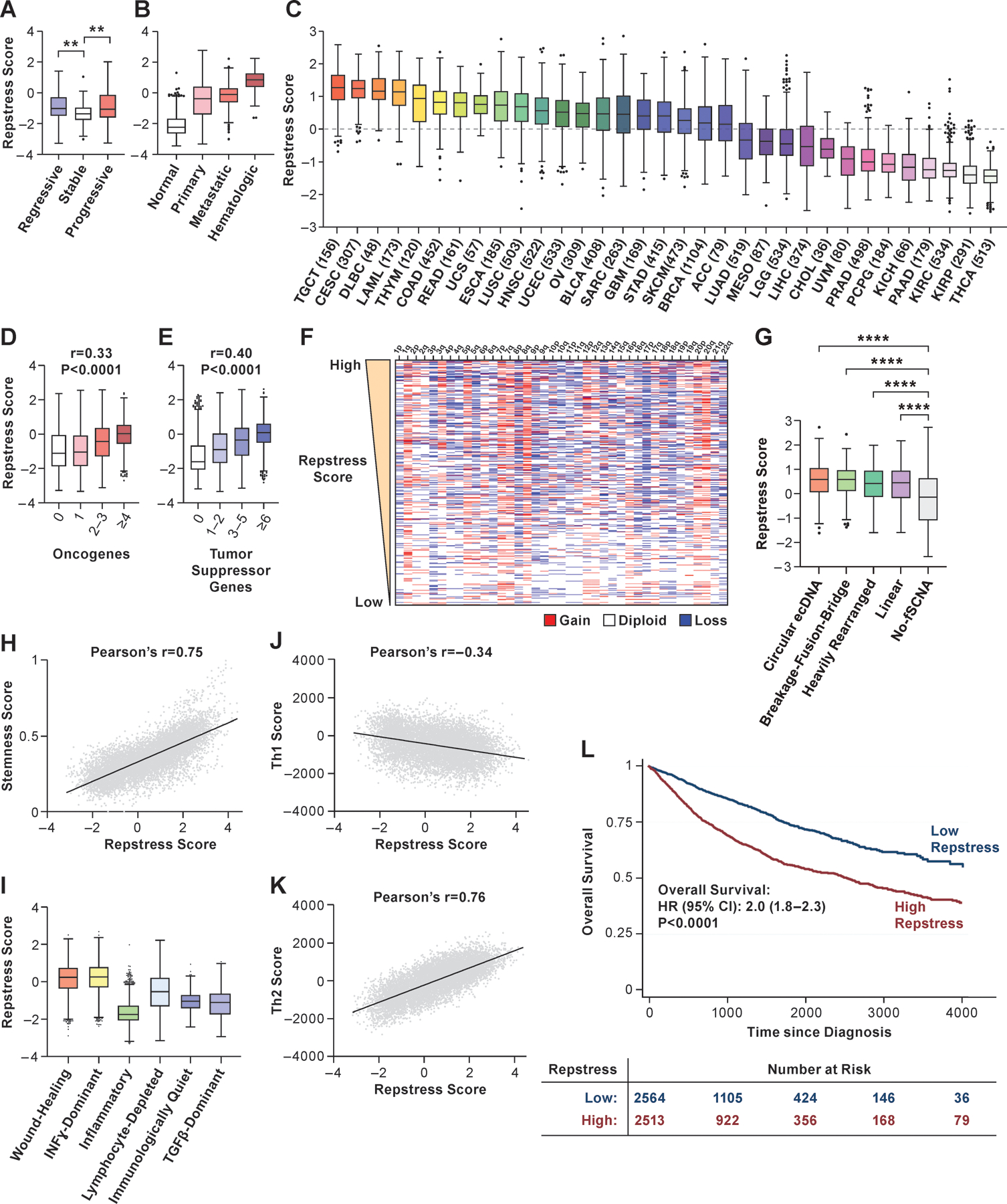
(A), Comparison of repstress score among bronchial premalignant lesions which regressed to normal tissue (regressive), did not change the premalignant histology (stable), and progressed to invasive malignancy (progressive) after biopsy
Gene expression data are obtained from a previous report (50). **: P < 0.01 by one-way ANOVA followed by Tukey’s multiple comparison test.
(B), Comparison of repstress score among TCGA normal tissue, primary and metastatic epithelial cancers, and hematopoietic malignancies
P < 0.0001 by comparing repstress scores in normal tissues vs. primary and metastatic epithelial cancers, and hematologic malignancies; and comparing those in hematologic malignancies vs. primary cancer and metastatic cancers; whereas P > 0.05 comparing those in primary and metastatic epithelial cancers. P values are analyzed by One-way ANOVA followed by Tukey’s multiple comparison test.
(C), Distribution of repstress scores across 33 cancer types in TCGA
The number in the x-axis label indicates the number of tumors included in each cancer type. A dash line indicates zero of Z-normalized repstress score across all of tumors in TCGA.
(D), (E), Pan-cancer analysis showing the relationship between repstress score with the number of mutated oncogenes (D) and tumor suppressor genes (E) (54)
Spearman’s correlation coefficient (r) and p values are indicated on top of each panel. Hypermutated tumors (i.e. mutational burden of ≥ 50 mutations per megabase) are excluded.
(F), Copy number alteration heatmap sorted by high (top) to low (bottom) repstress score Chromosome with copy number deletion or gain are indicated with blue and red, respectively. Copy number alteration data in TCGA tumors are retrieved from a previous report (81).
(G), Comparison of repstress scores among tumors with amplicons of circular ecDNA, breakage-fusion-bridge, heavily rearranged, linear, and no focal somatic copy number amplification
Annotations of amplification for each tumor in TCGA are reported previously (55). ****: P < 0.0001 by One-way ANOVA followed by Tukey’s multiple comparison test.
(H), Correlation between cancer stemness score and repstress score
Cancer stemness score is derived by integrative transcriptome- and methylation-based analysis (57). The P value of Pearson’s correlation is < 0.0001.
(I), Comparison of repstress score across six distinct TCGA immune subtypes, derived by gene signature-based clustering approach
Immune subtypes are previously described (58). P < 0.0001 by comparing repstress score in Wound Healing group vs. the others; IFN-gamma Dominant group vs. the others; and Inflammatory vs. the others, respectively. P values are analyzed by One-way ANOVA followed by Tukey’s multiple comparison test.
(J), (K), Correlations between Th1 (J) and Th2 (K) scores, and repstress score across cancer types
Th1 and Th2 scores are available in a previous report (58). The P values of Pearson’s correlation are < 0.0001 in panel J and K.
(L), Overall survival in cancer patients with high vs. low repstress score
High vs. low repstress scores are defined as patients whose cancers have repstress score ≥ 75th or < 25th percentiles across TCGA tumors. P value is derived from the log-rank test.
Abbreviations: TCGA: The Cancer Genome Atlas; ecDNA: extrachromosomal DNA; fSCNA: focal somatic copy number alteration; Th1: type 1 helper T cell; Th2: type 2 helper T cell; HR: hazard ratio; CI: confidence interval; Abbreviations for cancer types in TCGA are available in https://gdc.cancer.gov/resources-tcga-users/tcga-code-tables/tcga-study-abbreviations.
To explore the replication stress-response profiles of cancers, we analyzed over 10,000 tumors of 33 cancer types from TCGA. As with cell lines, expressions of genes required for survival of replication stress and DNA damage repair (TIMELESS, CLSPN, TOP2A, FANCD2, RNASEH2A, POLQ, and PARP1) positively correlated with repstress scores (Fig. S13A–G). These associations were also maintained at the protein level across tumor types; expression of proteins that most highly correlated with repstress score included CYCLINB1, CYCLINE1, CHK2, 4EBP1, phosphorylated CDK1 and PCNA (Fig. S13H). We next assessed repstress scores across normal tissue, localized, and metastatic cancers. Normal tissue had the lowest repstress score compared with cancers, and hematologic malignancies had higher repstress score than epithelial cancers (Fig. 3B).
We observed large variance in repstress scores across cancer types, implying significant differences in replication stress-response proficiency among different cancers (Fig. 3C). High repstress gene expression was observed in testicular germ cell tumors (TCGT, the number and proportion of TCGT with repstress scores ≥ 95% confidence interval of repstress score across TCGA: 148/156, 94.9%), cervical squamous cell carcinoma (CESC: 302/307, 98.4%), and hematologic malignancies (diffuse large B cell lymphoma, DLBCL: 46/48, 95.8%; and acute myeloid leukemia, LAML: 161/173, 93.1%). In general, tumors with high repstress scores were highly proliferative tumors typically treated with DNA damaging therapies such as platinum and topoisomerase inhibitors. A notable exception was thymoma which had high repstress scores (THYM: 96/120: 80.0%) despite a relatively indolent growth pattern. This may be explained by the prominent role of E2F2 in promoting unscheduled cell division and oncogenic transformation of thymic epithelial cells (51). Cancer types with lower repstress scores included thyroid cancers (THCA: the number and proportion of THCA with repstress scores <95% confidence interval of repstress score across TCGA: 513/513, 100%), kidney cancers (renal papillary cell carcinoma [KIRP]: 284/291, 97.6%; renal clear cell carcinoma [KIRC]: 521/534, 97.6%; kidney chromophobe [KICH]: 63/66, 95.5%), and pancreatic adenocarcinoma (PAAD: 172/179, 96.1%). The distribution of repstress score across cancers was overall similar even we excluded the seven genes associated with neuroendocrine differentiation (Fig. S14).
Since replication stress is driven by activation of oncogenes and absence of tumor suppressor genes (52), we examined the association between repstress score and mutations or copy number states in these genes. Tumors with mutated oncogenes (Fig. 3D) and tumor suppressor genes (Fig. 3E) had higher repstress scores compared with tumors with no mutations affecting these genes. In most cancer types, repstress score was significantly higher in tumors harboring mutations in DNA repair and cell cycle-related genes (Fig. S15A), suggesting deregulation of these pathways underlying increased replication stress. Tumors with TP53 or RB1 mutations had significantly higher repstress score compared with those without (Fig. S15B, C) and a loss of Rb1 function score (53) positively correlated with repstress score (Fig. S15D). Notably, there was no association between repstress score and the number of point mutations (Fig. S15E). In contrast, somatic copy number alterations (54) at chromosome, arm, and focal levels (Fig. 3F, S15F) and whole genome doubling (Fig. S15G) were positively correlated with repstress score. Extrachromosomal DNA (ecDNA) amplification has recently been reported to promote aneuploidy and genomic instability (55). Tumors with ecDNA amplification had higher repstress scores compared with those without (Fig. 3G), with increasing number of ecDNA amplicons associated with higher repstress scores (Fig. S16). Consistent with cancer stem cells displaying robust replication stress-response to prevent the accumulation of genetic lesions (56), a cancer stemness gene signature score (57) positively correlated with repstress score (Fig. 3H).
Next, we examined repstress score among previously defined cancer immune subtypes (58). The wound healing and interferon-gamma dominant subtypes had higher repstress scores compared with the other immune subtypes, including notably the inflammatory subtype which had lower repstress scores (Fig. 3I). The association of wound healing and repstress score (Pearson’s r = 0.81, P < 0.0001, Fig. S17A) (58), consistently observed across nearly all cancer types (Fig. S17B), is supported by previous work showing the similarities in cellular responses to cancer progression and wound healing (59). Helper T (Th) cells play a key role in the adaptive immune system by coordinating effector functions leading to destructive responses, including pathogen clearance and autoimmunity. A proinflammatory Th1 subtype response score was negatively correlated with repstress score (Pearson’s r = −0.34, P < 0.0001), whereas immunosuppressive Th2 subtype response score correlated positively (Pearson’s r = 0.76, P < 0.0001) (Fig. 3J, K). Accordingly, high repstress score was associated with poor survival in an independent cohort of melanoma patients treated with immune checkpoint inhibitor nivolumab (60) (Fig. S18).
Finally, we analyzed the impact of repstress score on patient outcomes. Patients with high repstress tumors had poorer OS compared to patients with low repstress tumors (hazard ratio [95% confidence interval]: 2.0 [1.8–2.3], P < 0.0001 by log-rank test, Fig. 3L). Multivariate Cox regression analysis revealed that the repstress score independently contributed to poor survival after adjusting known variables associated with survival including age at diagnosis, sex, pathological/clinical stage, and cancer type (Table S2, Fig. S19). Together, these analyses functionally link replication stress and its cellular response as measured by the repstress score with oncogene alterations, tumor aneuploidy, ecDNA amplification, cancer stemness, immunosuppressive T cell responses, and inferior survival across cancers.
Repstress score defines distinct molecular subtypes within cancer types
Given the wide range of repstress scores in individual cancers (Fig. 3C), we hypothesized that the repstress score can identify distinct molecular subtypes within cancer types. Among breast cancers, the basal subtype, characterized by expression of markers such as cytokeratins 5 and 6 (61), had significantly higher repstress score compared with the luminal A, luminal B, and HER2-enriched subtypes (Fig. 4A). Triple-negative breast cancers, which share similarities to the basal subtype, were also characterized by higher repstress score gene expression than tumors that expressed estrogen, progesterone, or HER2 receptors (Fig. S20A). Pancreatic cancers with transcriptionally defined basal characteristics and squamous features on histology harbored higher repstress score than those without these features in TCGA and an independent cohort (Fig. 4B, S20B–F) (62). Malignant mesothelioma with sarcomatoid histology, defined by infiltrative spindle or mesenchymal appearing cells and poor prognosis, were characterized by higher repstress score than epithelioid mesothelioma (Fig. 4C). Among prostate cancers, repstress score showed a positive correlation with Gleason score (Fig. 4D), an indicator of prostate cancer differentiation, with the highest Gleason score associated with the most poorly differentiated and aggressive subtype (63). Additionally, prostate cancers with higher copy number alterations (64) had higher repstress scores compared to those with less frequent copy number alterations (Fig. 4E). Similarly, uterine corpus endometrial carcinoma with genomic instability defined by high copy number alterations, POLE mutations, and microsatellite instability (65) had higher repstress score compared with low copy number altered tumors (Fig. 4F). Repstress score also identified a proliferative subtype of ovarian cancer (66) (Fig. 4G), and aggressive subtypes of hepatocellular carcinoma (iCluster 3) (67) with higher degree of chromosomal instability and TP53 mutations (Fig. 4H).
Fig. 4. Resptress score identifies distinct molecular subtypes among various cancer types.
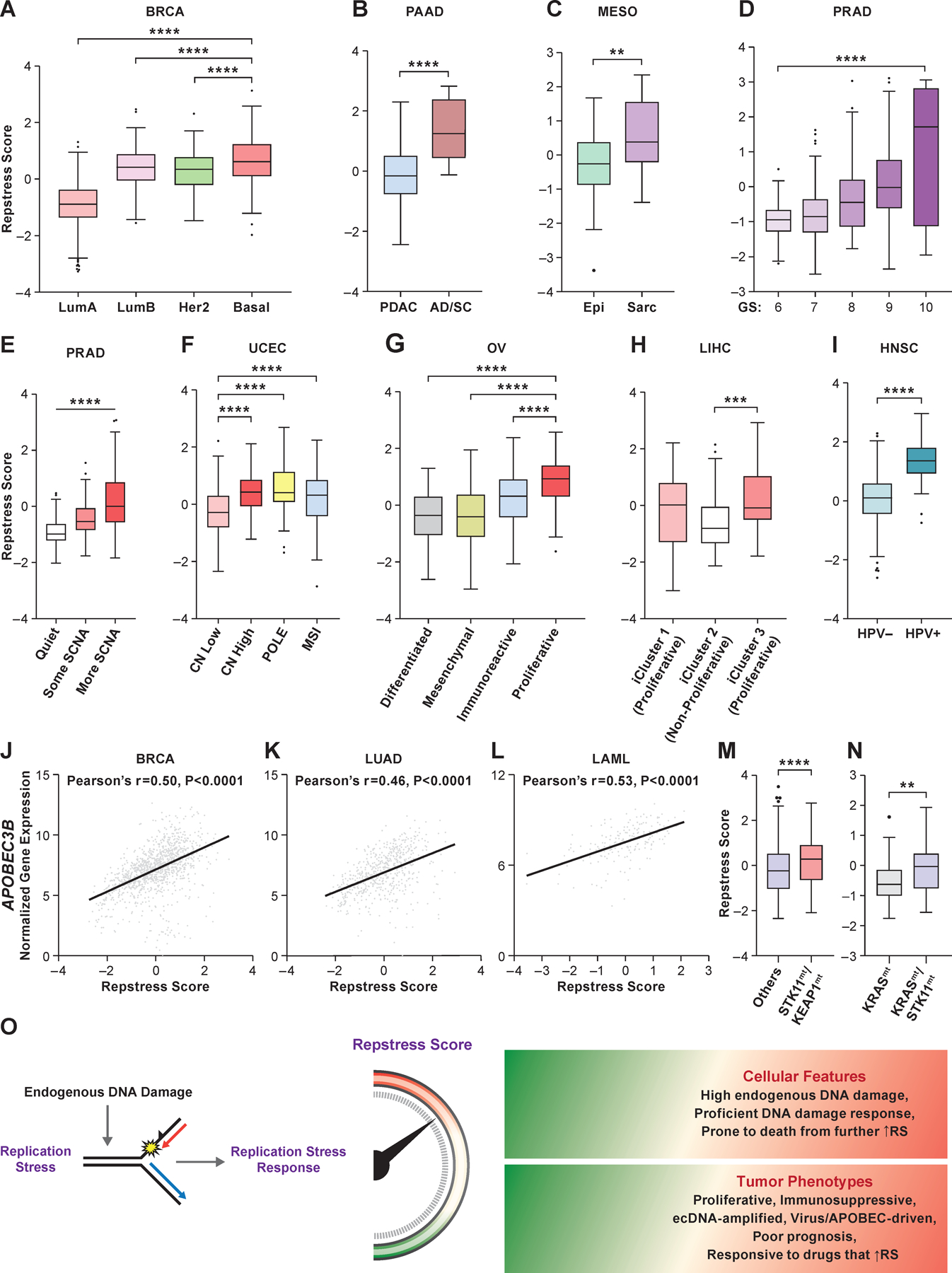
(A), Repstress scores among different breast cancer molecular subtypes
****: P < 0.0001 by one-way ANOVA followed by Tukey’s multiple comparison test.
(B), Repstress scores in pancreatic cancers with adenocarcinoma (PDAC) vs. adenosquamous (AD/SC) histology
****: P < 0.0001 by Mann-Whitney U test
(C), Repstress scores in malignant mesothelioma with epithelioid (Epi) vs. sarcomatoid or mixed epithelioid and sarcomatoid (Sarc) histology
**: P < 0.01 by Mann-Whitney U test
(D), Repstress scores among prostate cancers with different Gleason scores
****: P < 0.0001 by linear trend test from left to right.
(E), Repstress scores and somatic copy number alterations (SCNA) of TCGA prostate cancers SCNA subtype are defined by copy number-based clustering in a previous report (64). ****: P < 0.0001 by linear trend test from left to right.
(F), Repstress scores among uterine corpus endometrial carcinomas with different somatic copy number alteration (SCNA) subtypes
SCNA subtypes are defined by copy number-based clustering in a previous report (65). ****: P < 0.0001 by one-way ANOVA followed by Tukey’s multiple comparison test.
(G), Repstress scores among transcriptomic subtypes in ovarian carcinoma
The molecular subtypes are defined based on transcriptome-based clustering in a previous report (66). ****: P < 0.0001 by one-way ANOVA followed by Tukey’s multiple comparison test.
(H), Repstress scores among genomic subtypes in hepatocellular carcinoma The molecular subtypes (iCluster) are defined based on an integrative analysis of DNA copy number, DNA methylation, mRNA expression, microRNA expression, and reverse phase protein array in a previous report (67). ***: P < 0.001 by one-way ANOVA followed by Tukey’s multiple comparison test.
(I), Repstress scores between patients with HPV-null (HPV-) and HPV-driven (HPV+) head and neck cancers
****: P < 0.0001 by unpaired Student t test.
(J), (K), (L), Correlations between gene expression of APOBEC3B and repstress score in breast cancer (J), lung adenocarcinoma (K), and acute myeloid leukemia (L)
(M), (N), Repstress score comparison between tumors with KEAP1/STK11 co-alterations compared with those without (M), and tumors with KRAS/STK11 co-alterations compared with KRAS single-altered tumors (N) in lung adenocarcinoma
Gene alterations or copy number deletion (either heterozygous or homozygous) are considered as genetically alteration in KRAS, KEAP1, and STK11. Lung adenocarcinoma with KRAS/TP53 or KRAS/CDKN2A co-mutations are excluded from the analysis in panel (N) given a previous study reporting that non-small cell lung cancer with these co-mutations is different subtype from KRAS/STK11 co-mutated subtype (71,72). ****: P < 0.0001; **: P < 0.01 by Mann-Whitney U test.
(O), A schema of repstress gene signature characterizing replication stress and its response
Abbreviations: TCGA: The Cancer Genomic Atlas; LumA: luminal A; LumB: luminal B; PDAC: pancreatic adenocarcinoma; AD/SC: adenosquamous; Epi: epithelioid; Sarc: sarcomatoid; GS: Gleason score; SCNA: somatic copy number alteration; CN: copy number; POLE: DNA polymerase epsilon, catalytic subunit; MSI: microsatellite instable; HPV: human papilloma virus; APOBEC: apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like; STK11: Serine/threonine kinase 11; KEAP1: Kelch-like ECH-associated protein 1; RS: replication stress; Abbreviations for cancer types in TCGA are available in https://gdc.cancer.gov/resources-tcga-users/tcga-code-tables/tcga-study-abbreviations.
Given recent studies linking oncoviruses with genomic instability and replication stress (68), we examined repstress score in oncovirus-derived cancers. Human papilloma virus (HPV)-associated head and neck cancers had significantly higher repstress scores compared with non-HPV-associated cancers (Fig. 4I). A similar trend was also observed in cervical cancer, another HPV-related cancer (Fig. S20G). Replication stress exposes tracts of ssDNA that form substrates for APOBEC3-deaminase-mediated mutagenesis (69). Accordingly, repstress score positively correlated with APOBEC3B expression in breast cancer, lung adenocarcinoma, and acute myeloid leukemia, malignancies wherein APOBEC3B is upregulated and plays a key role in mutagenesis (70) (Fig. 4J-L). STK11 and KEAP1 co-mutated lung adenocarcinoma that are associated with aggressive tumor growth and immunotherapy resistance (71) had higher repstress scores compared with lung adenocarcinoma without concomitant loss of these genes (Fig. 4M). Among KRAS-mutant lung adenocarcinoma, a particularly aggressive subset with STK11 co-mutations (72) had higher repstress scores compared to tumors without co-mutations (Fig. 4N). Non-small cell lung cancer cell lines with KRAS/STK11 co-mutations were more sensitive to a CHK1/2 inhibitor than cell lines without STK11 co-mutations (Fig. S21). Together, our analysis brings to light the dependence of certain tumor types and subtypes of tumors on replication stress-response, potentially representing important therapeutic opportunities.
Discussion
DNA replication is a tightly regulated process. Replication stress and DNA damage ensue when these regulatory mechanisms fail. Causes of replication stress are diverse. Even single oncogene can induce replication stress by different mechanisms depending on the context (73). In fact, the causes of replicative stress might be quite dynamic during tumorigenesis. Independent of the causes of replication stress, cells have evolved a complex mechanism which ensures that the genome is accurately duplicated in each cell cycle. Despite its critical role in tumorigenesis and emerging importance as a potential therapeutic target, replication stress and its phenotypic characteristics have not been explored in high-throughput sequencing studies of human cancers. Many available studies examining replication stress to date have focused on individual tumor types, for example in ovarian cancer (74), pancreatic cancer (75,76), or selected features that drive replication stress, for example overexpression of oncogenes (via overexpression of CDC25A, CCNE1 or MYC) (77) or replication stress response defects (via depletion of ATR, ATM, CHEK1, or CHEK2) (44). Here we describe a gene expression signature, capturing broad measures of replication stress-related gene expression using an approach compatible with formalin-fixed paraffin-embedded clinical samples, allowing interrogation of replication stress at a functional network level across cancers, independent of the underlying mechanisms. The global view of replication stress provided by the repstress signature reveals heightened genomic instability, immune evasion, and poor survival in subsets of tumors across lineages, and enabled identification of cancer subtypes that may be more vulnerable to replication and replication stress-response inhibitors including the novel ATR inhibitors (Fig. 4O, S1).
Repstress score provides a framework to investigate the link between replication stress and its functional consequences. Our analyses implicate copy number alterations rather than base-pair mutations as a key consequence of genomic instability linked to DNA replication stress. These results support the oncogene-induced DNA replication stress model for cancer development wherein chromosomal instability in sporadic cancers results from oncogene-induced collapse of DNA replication forks, which in turn leads to DNA double-strand breaks and genomic instability (78). Another consequence of replication stress is abnormal chromosome segregation which may result in formation of micronuclei (79) and non-chromosomal DNA elements (55). Indeed, we find a positive correlation between repstress gene expression and ecDNA amplification, suggesting that oncogene-induced replication, abnormal chromosome segregation, and chromosome instability may be driving ecDNA formation.
Repstress gene signature reveals the dynamic nature of the replication stress-response during tumorigenesis and following drug treatment. Bronchial precancerous lesions that eventually regress and those that progress to become cancers are characterized by high repstress score compared with lesions that maintain stable precancerous characteristics. These results are consistent with the fundamental role of replication stress-response in early stages of cancer development maintaining genomic integrity and preventing tumorigenesis (6,8) while generating DNA damage and contributing to rapid evolution and genetic heterogeneity in established cancers (52). Whether these insights could enable the currently sparse toolset to identify and treat premalignant lesions at risk for progression to cancer needs further study (80). Modulation of repstress score following treatment suggests the utility of the signature to profile to study agents in terms of their impact on replication stress.
Repstress score provides insights into tumor phenotypes associated with high replication stress. Across multiple datasets, repstress score was an independent predictor of poor survival after adjusting known variables associated with survival. Notably, we find substantial enrichment of TCGA wound healing and interferon-gamma dominant phenotypes among high repstress tumors. The dominant anti-inflammatory Th2 response and rapid tumor growth that preclude immune control may explain the notably less favorable outcomes in high repstress score tumors despite a substantial immune component. It is also likely that these tumors have already been remodeled by the existing robust Th1 infiltrate and have escaped immune recognition. Further, the repstress score enabled delineation of several prognostically relevant subtypes within diverse cancer types, including high Gleason score prostate cancer, basal subtype of breast cancer, sarcomatoid mesothelioma, proliferative subtypes of ovarian cancer and hepatocellular carcinoma, and pancreatic cancer with squamous differentiation.
Additional studies are warranted to define clinically relevant and tumor-type specific repstress score thresholds, but it is notable, and probably the singular strength of the study, that repstress gene signature stratifies tumors across and within cancer types beyond SCLC based on the likelihood of drug response and prognosis. The generalizability of repstress score beyond SCLC suggests that while the causes of replication stress are varied, the replication stress-response pathways are conserved across cancers, and thus may represent a shared therapeutic vulnerability. Up-regulation of cell cycle genes is a common denominator between highly proliferative cells and cells under high replication stress, and further studies are needed to understand the contribution of individual repstress genes to these characteristics. It is notable that repstress signature better predicted response to ATR inhibitors than previously described gene signatures of proliferation (39,45–47), suggesting that repstress signature captures transcriptional changes of replication stress in addition to proliferation. In conclusion, gene expression profiling-based assessment of replication stress using the repstress signature represents a powerful approach to dissect the replication stress-response. We anticipate the repstress score to have therapeutic implications, enabling stratification of patients for therapies that modulate replication stress.
Supplementary Material
Significance.
We develop a transcriptional profile of replication stress which characterizes replication stress and its cellular response, revealing phenotypes of replication stress across cancer types. We envision the repstress score to serve as an effective discovery platform to predict efficacies of targeting replication stress and clinical outcomes.
Acknowledgements
Funding: This study was supported by the Center for Cancer Research, the Intramural Program of the National Cancer Institute (ZIA BC 011793). AT and YP report research funding from AstraZeneca, Tarveda, EMD Serono, and Prolynx.
Footnotes
Conflict of interest disclosure statement: The authors have declared that no conflict of interest exists.
References
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144:646–74 [DOI] [PubMed] [Google Scholar]
- 2.Zeman MK, Cimprich KA. Causes and consequences of replication stress. Nature cell biology 2014;16:2–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Osborn AJ, Elledge SJ, Zou L. Checking on the fork: the DNA-replication stress-response pathway. Trends in cell biology 2002;12:509–16 [DOI] [PubMed] [Google Scholar]
- 4.Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Molecular cell 2010;40:179–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Toledo LI, Altmeyer M, Rask MB, Lukas C, Larsen DH, Povlsen LK, et al. ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell 2013;155:1088–103 [DOI] [PubMed] [Google Scholar]
- 6.Gorgoulis VG, Vassiliou LV, Karakaidos P, Zacharatos P, Kotsinas A, Liloglou T, et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 2005;434:907–13 [DOI] [PubMed] [Google Scholar]
- 7.Macheret M, Halazonetis TD. DNA replication stress as a hallmark of cancer. Annual review of pathology 2015;10:425–48 [DOI] [PubMed] [Google Scholar]
- 8.Bartkova J, Horejsí Z, Koed K, Krämer A, Tort F, Zieger K, et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 2005;434:864–70 [DOI] [PubMed] [Google Scholar]
- 9.Lecona E, Fernandez-Capetillo O. Targeting ATR in cancer. Nature reviews Cancer 2018;18:586–95 [DOI] [PubMed] [Google Scholar]
- 10.Fang Y, McGrail DJ, Sun C, Labrie M, Chen X, Zhang D, et al. Sequential Therapy with PARP and WEE1 Inhibitors Minimizes Toxicity while Maintaining Efficacy. Cancer cell 2019;35:851–67.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thomas A, Redon CE, Sciuto L, Padiernos E, Ji J, Lee MJ, et al. Phase I Study of ATR Inhibitor M6620 in Combination With Topotecan in Patients With Advanced Solid Tumors. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2018;36:1594–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thomas A, Takahashi N, Rajapakse VN, Zhang X, Sun Y, Ceribelli M, et al. Therapeutic targeting of ATR yields durable regressions in small cell lung cancers with high replication stress. Cancer cell 2021;39:566–79.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Buisson R, Boisvert JL, Benes CH, Zou L. Distinct but Concerted Roles of ATR, DNA-PK, and Chk1 in Countering Replication Stress during S Phase. Molecular cell 2015;59:1011–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bensimon A, Simon A, Chiffaudel A, Croquette V, Heslot F, Bensimon D. Alignment and sensitive detection of DNA by a moving interface. Science (New York, NY) 1994;265:2096–8 [DOI] [PubMed] [Google Scholar]
- 15.Tlemsani C, Pongor L, Elloumi F, Girard L, Huffman KE, Roper N, et al. SCLC-CellMiner: A Resource for Small Cell Lung Cancer Cell Line Genomics and Pharmacology Based on Genomic Signatures. Cell reports 2020;33:108296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rajapakse VN, Luna A, Yamade M, Loman L, Varma S, Sunshine M, et al. CellMinerCDB for Integrative Cross-Database Genomics and Pharmacogenomics Analyses of Cancer Cell Lines. iScience 2018;10:247–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goldman MJ, Craft B, Hastie M, Repečka K, McDade F, Kamath A, et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nature biotechnology 2020;38:675–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liberzon A, Subramanian A, Pinchback R, Thorvaldsdóttir H, Tamayo P, Mesirov JP. Molecular signatures database (MSigDB) 3.0. Bioinformatics (Oxford, England) 2011;27:1739–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Polley E, Kunkel M, Evans D, Silvers T, Delosh R, Laudeman J, et al. Small Cell Lung Cancer Screen of Oncology Drugs, Investigational Agents, and Gene and microRNA Expression. Journal of the National Cancer Institute 2016;108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.George J, Lim JS, Jang SJ, Cun Y, Ozretić L, Kong G, et al. Comprehensive genomic profiles of small cell lung cancer. Nature 2015;524:47–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hänzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC bioinformatics 2013;14:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang W, Girard L, Zhang YA, Haruki T, Papari-Zareei M, Stastny V, et al. Small cell lung cancer tumors and preclinical models display heterogeneity of neuroendocrine phenotypes. Translational lung cancer research 2018;7:32–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Knijnenburg TA, Wang L, Zimmermann MT, Chambwe N, Gao GF, Cherniack AD, et al. Genomic and Molecular Landscape of DNA Damage Repair Deficiency across The Cancer Genome Atlas. Cell reports 2018;23:239–54.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tanaka T, Halicka D, Traganos F, Darzynkiewicz Z. Cytometric analysis of DNA damage: phosphorylation of histone H2AX as a marker of DNA double-strand breaks (DSBs). Methods in molecular biology (Clifton, NJ) 2009;523:161–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fu H, Martin MM, Regairaz M, Huang L, You Y, Lin CM, et al. The DNA repair endonuclease Mus81 facilitates fast DNA replication in the absence of exogenous damage. Nature communications 2015;6:6746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thomas A, Pommier Y. Small cell lung cancer: Time to revisit DNA-damaging chemotherapy. Science translational medicine 2016;8:346fs12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kotsantis P, Silva LM, Irmscher S, Jones RM, Folkes L, Gromak N, et al. Increased global transcription activity as a mechanism of replication stress in cancer. Nature communications 2016;7:13087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rudin CM, Poirier JT, Byers LA, Dive C, Dowlati A, George J, et al. Molecular subtypes of small cell lung cancer: a synthesis of human and mouse model data. Nature reviews Cancer 2019;19:289–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Balanis NG, Sheu KM, Esedebe FN, Patel SJ, Smith BA, Park JW, et al. Pan-cancer Convergence to a Small-Cell Neuroendocrine Phenotype that Shares Susceptibilities with Hematological Malignancies. Cancer cell 2019;36:17–34.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dominguez-Sola D, Gautier J. MYC and the control of DNA replication. Cold Spring Harbor perspectives in medicine 2014;4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Petroni M, Sardina F, Heil C, Sahún-Roncero M, Colicchia V, Veschi V, et al. The MRN complex is transcriptionally regulated by MYCN during neural cell proliferation to control replication stress. Cell death and differentiation 2016;23:197–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dai Y, Grant S. New insights into checkpoint kinase 1 in the DNA damage response signaling network. Clinical cancer research : an official journal of the American Association for Cancer Research 2010;16:376–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang J, Dai Q, Park D, Deng X. Targeting DNA Replication Stress for Cancer Therapy. Genes 2016;7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang L, Smith BA, Balanis NG, Tsai BL, Nguyen K, Cheng MW, et al. A genetically defined disease model reveals that urothelial cells can initiate divergent bladder cancer phenotypes. Proceedings of the National Academy of Sciences of the United States of America 2020;117:563–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sulli G, Di Micco R, d’Adda di Fagagna F. Crosstalk between chromatin state and DNA damage response in cellular senescence and cancer. Nature reviews Cancer 2012;12:709–20 [DOI] [PubMed] [Google Scholar]
- 36.Gagou ME, Zuazua-Villar P, Meuth M. Enhanced H2AX phosphorylation, DNA replication fork arrest, and cell death in the absence of Chk1. Molecular biology of the cell 2010;21:739–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Courbet S, Gay S, Arnoult N, Wronka G, Anglana M, Brison O, et al. Replication fork movement sets chromatin loop size and origin choice in mammalian cells. Nature 2008;455:557–60 [DOI] [PubMed] [Google Scholar]
- 38.Ghandi M, Huang FW, Jané-Valbuena J, Kryukov GV, Lo CC, McDonald ER 3rd, et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 2019;569:503–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chibon F, Lagarde P, Salas S, Pérot G, Brouste V, Tirode F, et al. Validated prediction of clinical outcome in sarcomas and multiple types of cancer on the basis of a gene expression signature related to genome complexity. Nature medicine 2010;16:781–7 [DOI] [PubMed] [Google Scholar]
- 40.Walters DK, Wu X, Tschumper RC, Arendt BK, Huddleston PM, Henderson KJ, et al. Evidence for ongoing DNA damage in multiple myeloma cells as revealed by constitutive phosphorylation of H2AX. Leukemia 2011;25:1344–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Murai J, Thomas A, Miettinen M, Pommier Y. Schlafen 11 (SLFN11), a restriction factor for replicative stress induced by DNA-targeting anti-cancer therapies. Pharmacology & therapeutics 2019;201:94–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Monks A, Zhao Y, Hose C, Hamed H, Krushkal J, Fang J, et al. The NCI Transcriptional Pharmacodynamics Workbench: A Tool to Examine Dynamic Expression Profiling of Therapeutic Response in the NCI-60 Cell Line Panel. Cancer research 2018;78:6807–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rees MG, Seashore-Ludlow B, Cheah JH, Adams DJ, Price EV, Gill S, et al. Correlating chemical sensitivity and basal gene expression reveals mechanism of action. Nature chemical biology 2016;12:109–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McGrail DJ, Lin CC, Dai H, Mo W, Li Y, Stephan C, et al. Defective Replication Stress Response Is Inherently Linked to the Cancer Stem Cell Phenotype. Cell reports 2018;23:2095–106 [DOI] [PubMed] [Google Scholar]
- 45.Cuzick J, Swanson GP, Fisher G, Brothman AR, Berney DM, Reid JE, et al. Prognostic value of an RNA expression signature derived from cell cycle proliferation genes in patients with prostate cancer: a retrospective study. The Lancet Oncology 2011;12:245–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lundberg A, Lindström LS, Harrell JC, Falato C, Carlson JW, Wright PK, et al. Gene Expression Signatures and Immunohistochemical Subtypes Add Prognostic Value to Each Other in Breast Cancer Cohorts. Clinical cancer research : an official journal of the American Association for Cancer Research 2017;23:7512–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang W, Mao JH, Zhu W, Jain AK, Liu K, Brown JB, et al. Centromere and kinetochore gene misexpression predicts cancer patient survival and response to radiotherapy and chemotherapy. Nature communications 2016;7:12619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jo U, Senatorov IS, Zimmermann A, Saha LK, Murai Y, Kim SH, et al. Novel and Highly Potent ATR Inhibitor M4344 Kills Cancer Cells With Replication Stress, and Enhances the Chemotherapeutic Activity of Widely Used DNA Damaging Agents. Mol Cancer Ther 2021;20:1431–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 2006;444:633–7 [DOI] [PubMed] [Google Scholar]
- 50.Beane JE, Mazzilli SA, Campbell JD, Duclos G, Krysan K, Moy C, et al. Molecular subtyping reveals immune alterations associated with progression of bronchial premalignant lesions. Nature communications 2019;10:1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Scheijen B, Bronk M, van der Meer T, De Jong D, Bernards R. High incidence of thymic epithelial tumors in E2F2 transgenic mice. The Journal of biological chemistry 2004;279:10476–83 [DOI] [PubMed] [Google Scholar]
- 52.Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene-induced DNA damage model for cancer development. Science (New York, NY) 2008;319:1352–5 [DOI] [PubMed] [Google Scholar]
- 53.Ertel A, Dean JL, Rui H, Liu C, Witkiewicz AK, Knudsen KE, et al. RB-pathway disruption in breast cancer: differential association with disease subtypes, disease-specific prognosis and therapeutic response. Cell cycle (Georgetown, Tex) 2010;9:4153–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Davoli T, Uno H, Wooten EC, Elledge SJ. Tumor aneuploidy correlates with markers of immune evasion and with reduced response to immunotherapy. Science (New York, NY) 2017;355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kim H, Nguyen NP, Turner K, Wu S, Gujar AD, Luebeck J, et al. Extrachromosomal DNA is associated with oncogene amplification and poor outcome across multiple cancers. Nature genetics 2020;52:891–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vitale I, Manic G, De Maria R, Kroemer G, Galluzzi L. DNA Damage in Stem Cells. Molecular cell 2017;66:306–19 [DOI] [PubMed] [Google Scholar]
- 57.Malta TM, Sokolov A, Gentles AJ, Burzykowski T, Poisson L, Weinstein JN, et al. Machine Learning Identifies Stemness Features Associated with Oncogenic Dedifferentiation. Cell 2018;173:338–54.e15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Thorsson V, Gibbs DL, Brown SD, Wolf D, Bortone DS, Ou Yang TH, et al. The Immune Landscape of Cancer. Immunity 2018;48:812–30.e14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chang HY, Sneddon JB, Alizadeh AA, Sood R, West RB, Montgomery K, et al. Gene expression signature of fibroblast serum response predicts human cancer progression: similarities between tumors and wounds. PLoS biology 2004;2:E7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Riaz N, Havel JJ, Makarov V, Desrichard A, Urba WJ, Sims JS, et al. Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab. Cell 2017;171:934–49.e16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cancer_Genome_Atlas_Research_Network. Comprehensive molecular portraits of human breast tumours. Nature 2012;490:61–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hayashi A, Fan J, Chen R, Ho Y-j, Makohon-Moore AP, Lecomte N, et al. A unifying paradigm for transcriptional heterogeneity and squamous features in pancreatic ductal adenocarcinoma. Nature Cancer 2020;1:59–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Epstein JI, Egevad L, Amin MB, Delahunt B, Srigley JR, Humphrey PA. The 2014 International Society of Urological Pathology (ISUP) Consensus Conference on Gleason Grading of Prostatic Carcinoma: Definition of Grading Patterns and Proposal for a New Grading System. The American journal of surgical pathology 2016;40:244–52 [DOI] [PubMed] [Google Scholar]
- 64.Cancer_Genome_Atlas_Research_Network. The Molecular Taxonomy of Primary Prostate Cancer. Cell 2015;163:1011–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kandoth C, Schultz N, Cherniack AD, Akbani R, Liu Y, Shen H, et al. Integrated genomic characterization of endometrial carcinoma. Nature 2013;497:67–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cancer_Genome_Atlas_Research_Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011;474:609–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cancer_Genome_Atlas_Research_Network. Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 2017;169:1327–41.e23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Moody CA. Impact of Replication Stress in Human Papillomavirus Pathogenesis. Journal of virology 2019;93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kanu N, Cerone MA, Goh G, Zalmas LP, Bartkova J, Dietzen M, et al. DNA replication stress mediates APOBEC3 family mutagenesis in breast cancer. Genome biology 2016;17:185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Olson ME, Harris RS, Harki DA. APOBEC Enzymes as Targets for Virus and Cancer Therapy. Cell chemical biology 2018;25:36–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wohlhieter CA, Richards AL, Uddin F, Hulton CH, Quintanal-Villalonga À, Martin A, et al. Concurrent Mutations in STK11 and KEAP1 Promote Ferroptosis Protection and SCD1 Dependence in Lung Cancer. Cell reports 2020;33:108444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Skoulidis F, Goldberg ME, Greenawalt DM, Hellmann MD, Awad MM, Gainor JF, et al. STK11/LKB1 Mutations and PD-1 Inhibitor Resistance in KRAS-Mutant Lung Adenocarcinoma. Cancer discovery 2018;8:822–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hills SA, Diffley JF. DNA replication and oncogene-induced replicative stress. Current biology : CB 2014;24:R435–44 [DOI] [PubMed] [Google Scholar]
- 74.Konstantinopoulos PA, da Costa A, Gulhan D, Lee EK, Cheng SC, Hendrickson AEW, et al. A Replication stress biomarker is associated with response to gemcitabine versus combined gemcitabine and ATR inhibitor therapy in ovarian cancer. Nature communications 2021;12:5574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dreyer SB, Upstill-Goddard R, Paulus-Hock V, Paris C, Lampraki EM, Dray E, et al. Targeting DNA Damage Response and Replication Stress in Pancreatic Cancer. Gastroenterology 2021;160:362–77.e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Feng Z, Li K, Lou J, Ma M, Wu Y, Peng C. A Novel DNA Replication-Related Signature Predicting Recurrence After R0 Resection of Pancreatic Ductal Adenocarcinoma: Prognostic Value and Clinical Implications. Frontiers in cell and developmental biology 2021;9:619549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Guerrero Llobet S, Bhattacharya A, Everts M, Kok K, van der Vegt B, Fehrmann RSN, et al. An mRNA expression-based signature for oncogene-induced replication-stress. Oncogene 2022;41:1216–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Negrini S, Gorgoulis VG, Halazonetis TD. Genomic instability--an evolving hallmark of cancer. Nature reviews Molecular cell biology 2010;11:220–8 [DOI] [PubMed] [Google Scholar]
- 79.Sabatinos SA, Ranatunga NS, Yuan JP, Green MD, Forsburg SL. Replication stress in early S phase generates apparent micronuclei and chromosome rearrangement in fission yeast. Molecular biology of the cell 2015;26:3439–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Beane J, Campbell JD, Lel J, Vick J, Spira A. Genomic approaches to accelerate cancer interception. The Lancet Oncology 2017;18:e494–e502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Taylor AM, Shih J, Ha G, Gao GF, Zhang X, Berger AC, et al. Genomic and Functional Approaches to Understanding Cancer Aneuploidy. Cancer cell 2018;33:676–89.e3 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data analyzed in this study were obtained from public database. The experimental data generated in this study are available upon request from the corresponding author.