

Abstract
Rhabdomyosarcoma (RMS) is the most common soft tissue cancer in children. Treatment outcomes, particularly for relapsed/refractory or metastatic disease, have not improved in decades. The current lack of novel therapies and low tumor mutational burden suggest that CAR T therapy could be a promising approach to treating RMS. Previous work identified Fibroblast Growth Factor Receptor 4 (FGFR4, CD334) as being specifically upregulated in RMS, making it a candidate target for CAR-T cells. We tested the feasibility of an FGFR4 targeted CAR for treating RMS using an NSG mouse with RH30 orthotopic (intramuscular) tumors. The first barrier we noted was that RMS tumors produce a collagen-rich stroma, replete with immunosuppressive myeloid cells, when T cell therapy is initiated. This stromal response is not seen in tumor-only xenografts. When scFV-based binders were selected from phage display, CARs targeting FGFR4 were not effective until our screening approach was refined to identify binders to the membrane-proximal domain of FGFR4. Having improved the CAR, we devised a pharmacologic strategy to augment CAR-T activity by inhibiting the myeloid component of the T cell-induced tumor stroma. The combined treatment of mice with anti-myeloid polypharmacy (targeting CSF1R, IDO1, iNOS, TGFbeta, PDL1, MIF and myeloid misdifferentiation) allowed FGFR4 CAR-T to successfully clear orthotopic RMS tumors, demonstrating that RMS tumors, even with very low copy number targets, can be targeted by CAR-T upon reversal of an immunosuppressive microenvironment.
Keywords: Rhabdomyosarcoma, CAR-T cells, FGFR4, myeloid cells, pediatric tumors, solid tumors, tumor microenvironment, tumor stroma
Introduction:
Rhabdomyosarcoma (RMS), a soft tissue cancer that arises in striated muscle, is the most common soft tissue cancer in children (1). Typical treatment of RMS patients is comprised of multimodal chemotherapy, with 70–80% of all RMS cases achieving remission-free survival, but not without significant toxicities. For patients with metastatic or relapsed/refractory RMS, survival is only 20–30% and has not improved in decades despite novel targeted therapies. Pediatric tumors, and especially the fusion-gene driven subsets, such as alveolar RMS (AMRS, FP-RMS), have among the lowest mutational burden of any tumor (2), suggesting that checkpoint therapies alone will be insufficient to control tumors. Indeed, trials of such inhibitors have been largely unsuccessful in pediatric patients (3). New therapies with improved specificity and reduced toxicities are needed.
Chimeric Antigen Receptors (CAR)-T cells are genetically engineered T lymphocytes expressing an extracellular binding domain and intracellular T cell signaling domains (4). Upon engagement of the binding domain (often antibody-derived) to the antigenic target on a tumor cell, activation and degranulation occurs resulting in cell-mediated toxicity induced death of the tumor cells. The key to CAR T cell therapy success is to guide engineered T cells to a molecular target that is tumor specific, expressed on the cell surface, and expressed at high enough levels for CAR-T activation (5). CAR T therapy has been successfully used to treat hematological cancers, but is currently far less effective against solid tumors, likely due to the uniquely immunosuppressive environment of solid tumors. Nonetheless, some successes have been seen clinically and suggest that T cell therapy can be adapted for targeting solid tumors (6–10) Previous work identified Fibroblast Growth Factor Receptor 4 (FGFR4, CD334) as being specifically upregulated in both major subtypes of RMS, a driver of metastasis and tumor survival, and aiding in resistance to chemotherapy, making it a promising candidate for targeting by CAR-T cells(11–15) ().
FGFR4 is a membrane tyrosine kinase expressed during embryonic muscle development and is largely absent in healthy tissues. FGFR4 was found to be upregulated in both FP- and FN-RMS, though the expression of FGFR4 is higher in FP-RMS where expression is driven by the PAX3-FOXO1 fusion protein (12,16,17). One report of FGFR4-targeted CAR demonstrated in vitro activity of a single-domain antibody binder used in a CAR format against RMS tumor lines, assessing the feasibility of such a CAR (18). In our own work, we reported that a CAR designed to target FGFR4 with an scFv derived from a human phage-display library against the extracellular domain of FGFR4 showed in vitro efficacy and some control of tumor in a metastatic (intravenous) RMS model, but failed to control orthotopic (intramuscular, i.m.) disease in vivo (19,20). We have developed a new generation of FGFR4-binding moieties to be tested as components of CARs and derived from both human F(ab)- and human VH-only (single chain domain antibodies, dAbs) phage display libraries. New binders were designed to target the Ig-III domain of FGFR4, as membrane proximal targeting has been shown to improve CAR and transgenic TCR function (21) and the Ig-III domain is not known to contain splice variants, making the target less amenable to antigen escape (22).In vitro screening identified top antigen-binding candidates active in vitro in CAR format, but when tested in vivo failed to control RH30 orthotopic tumors. Examination of the TME by immunohistochemistry demonstrated that in this model system, the infused T-cells induced a collagen-rich tumor stroma replete with suppressive myeloid cells that would have to be overcome for effective immunotherapy. Our findings are reminiscent of reports indicating that immunosuppressive macrophage populations (CD68+, CD163+) present in RMS are associated with poor prognosis (23) and that improving T cell’s ability to migrate through stromal matrix improves T cell therapies (24).
Immunohistochemical analysis of orthotopic (intramuscular) tumors clearly demonstrated the presence of immunosuppressive myeloid populations. mRNA expression analysis also revealed the presence of multiple soluble factors that play a role preventing effective CAR-T therapy. We used these findings to design an anti-myeloid polypharmacy approach specific to RMS intramuscular xenografts of the RH30 cell line in order to overcome the biology of the tumor lesion that inhibits T cell activity, and thereby allow FGFR4 CAR-T cells to clear orthotopic RMS tumors in an NSG model.
Methods and Materials:
Cell Lines and culture media
The aRMS RH30_19 used in these studies was produced by transducing RH30 cell line with truncated CD19 (CD19t) and firefly Luciferase (ffLuc). RH30_19 MIF KO was engineered by CRISPR-Cas9 editing of the MIF gene by transient transfection of the px330 plasmid (provided by Feng Zhang, Addgene #42230) containing MIF-targeted guide RNA (25). MIF guides were targeted to the sequences: CCTTTCCTCGCAGTACATCG, CAGTACATCGCGGTGCACG, and ACCGCGAAGGCCATGAGCTGGTCC. Single clones were cultured and MIF KO confirmed by ELISA (R&D Quantikine Kit). The eRMS cell line RD was transduced with GFP-ffLuc and CD19t. REH and NALM6 were transduced to express GFP:ffLuc and clonally selected for positive expression. Raji-Luc and K562-Luc were provided by Dr. Michael Jensen. STR fingerprinting was conducted to verify the identity of cell lines, and each cell line was validated to be Mycoplasma free by qPCR. RH30 and RD cell lines were grown in DMEM (Gibco) supplemented with 10% heat-inactivated FBS (VWR) and 2mM L-glutamine (Gibco). Raji, K562, NALM6, and REH lines were cultured in RPMI 1640 (Gibco) supplemented with 2 mM L-glutamine, 10 mM HEPES (Invitrogen), and 10% FBS. Healthy donor human PBMCs were obtained from Bloodworks Northwest and isolated with SepMate PBMC Isolation Tubes and Lymphoprep (Stemcell Technologies). T cells were cultured in TexMACS medium (Miltenyi Biotec) with recombinant IL-2 (premium grade, Miltenyi Biotec) as described below.
CAR design
Chimeric antigen receptors consisted of a GM-CSF receptor signal sequence, binder, CD8 hinge and transmembrane, 4–1BB (CD137) co-stimulation domain, and CD3ζ activation domain, as previously described (21,26–28). The LV sequence was designed with NheI and NotI restriction sites flanking binder domain sequences to facilitate screening of candidates. Additional CARs optimized for low density antigen recognition replaced CD8 H/TM with CD28 H/TM as in Majzner et al, 2020 (29).
Vector Production
Lentiviral vector was produced by transient transfection of HEK293T/17SF cells, seeded at 1×106/mL in 200mL of FreeStyle293 expression media (Gibco) in a 1L polycarbonate shaker flask (TriForest Labware). HEK293T/17SF cells were transfected the following day when cells reached 2x106/mL. PEIpro (Polyplus) was used to transfect cells with plasmids containing gag-pol, rev, VSV-G envelope protein and transfer plasmid containing the CAR. Sodium butyrate (MiiliporeSigma) was added 24 hour after transfection. Supernatant was collected on day 3 after transfection and filtered by 0.45uM filter (MilliporeSigma). LV was concentrated by centrifugation at 12,000xg for 4hr. Pelleted LV particles were resuspended in serum-free medium, aliquoted, and stored at −80°C.
Phage Panning and Binder Expression
To identify binders specifically targeting FGFR4 membrane proximal domain (IgIII), we developed a sequential phage panning strategy in which the full-length FGFR4 ectodomain (Sino Biological) was used in the first round of panning with human antigen binding fragment (Fab) and human Ig heavy chain variable domain (VH) phage libraries. For the 2nd and 3rd round, we used the recombinant IgIII domain, stably expressed as a human IgG1-Fc fusion protein in expi293 cells and purified by protein A resin. Panning enrichment was checked by the polyclonal phage ELISA binding to both full-length FGFR4 and the IgIII domain alone after three rounds of panning. After validating enrichment, monoclonal expression ELISA was used to screen positive monoclonal binders by using the full-length FGFR4 (Fc fusion protein) coated plates. During ELISA, we used an IgG1 m336 (non-specific binder), containing the Fc fragment, as the negative control. Positive clones were amplified and plasmids extracted, followed by Sanger sequencing. For expression of positive VH and Fab binders, phagemids were transfected into HB2151 E.coli bacteria, and proteins were purified from the periplasmic space by secretion mediated by ompA and pelB signal peptides. The protein purity was checked by SDS-PAGE and quantified by spectroscopy (Nanodrop).
CAR T cell preparation
PBMC were cultured at 2x106 cell per well in 24-well plates in TexMACS media supplemented with 40 U/mL rhIL-2 and 20ul/well TransAct (Miltenti Biotec) activation matrix. At 24 hours, PBMCs were transduced with LV in the presence of protamine sulfate (8 μg/mL). Media was replaced at 24 hours with fresh TexMACS, with 40 IU/mL rhIL-2. On day 4, media was replenished and rhIL-2 increased to 200 IU/mL. PBMC were expanded in TexMACS with rhIL-2 until day 9, at which point CAR surface expression and functional activity were assessed or T cells transferred into tumor-bearing mice.
Cytotoxicity Assays
1 x 104 tumor cells (target) were co-cultured with CAR-T cells (effector) at the indicated effector to target (E:T) ratios in a 96-well, flat-bottom plate, in triplicate wells. Cells were co-incubated for 20 hours in 200 μl TexMACS media without added cytokines. 100 μl of media was removed for cytokine analysis and 100 μl SteadyGlo reagent (Promega) was added to each well. After 10 minutes at room temperature, luminescence was quantified using an Espire plate reader (Perkin Elmer). Target cell only wells (positive, 100% viable) and tumor cells with 1% Tween-20 (negative, non-viable) were included. Percent specific lysis was calculated as (sample-negative)/(positive-negative)*100%. For multiple-day challenge cytotoxicity assays, cells were cultured in a 96-well plate with an E:T ratio of either 1:1 or 1:4 with CAR T effector cells and RH30 target cells. At 48 hours, cells were analyzed for the fraction of effector and target cells remaining in culture using flow cytometry, and original ratios were re-established. This was repeated for 3 consecutive challenges over 6 days.
Cytokine Assay
Cytokine production was measured from the supernatants of cytotoxicity assays (described above). Supernatants were centrifuged at 1000xg for 5 minutes to clear cells and debris, then analyzed using bead-based multiplex cytokine assays (LengendPlex, Biolegend) for INF-γ, TNFα, and IL-2 per manufacturer’s protocol. Samples were acquired on a LSR Fortessa cell analyzer (BD Biosciences) and analyzed using LEGEND Plex software (v8.0, Biolegend).
Cell staining and Flow Cytometry
Samples were processed into single cell suspensions and stained in FACS buffer (PBS + 2% FBS + 0.1% sodium Azide) with antibody for 45 minutes on ice followed by secondary (APC- or PE-conjugated streptavidin) binding for 20 minutes. Antibodies used were CD8-BUV395 (RPA-T8, BD Biosciences), CD4-Bv605 (RPA-T4, BD Biosciences), CD3-APC (SP34–2, BD Biosciences), FGFR4-PE (4FR6D3, Biolegend), CD19-PE (HIB19, Biolegend), PDL1-APC (MIH1, BD Bioscience), PDL2-Bv421 (MIH18, BD Biosciences). Surface CAR was detected using biotinylated CD19 with Fc-tag (Acro Bio) or biotinylated FGFR4-Fc (Sino Bio). For evaluation of FGFR4 binders identified from phage screening, 500 nM VH or Fab proteins were incubated with cells at 4 °C for 30 minutes followed by PE conjugated anti-human Fc antibody (Miltenyi Biotec, 130–101-576) for 30 minutes at 4 °C. Cells were then analyzed by flow cytometry using BD LSR II (San Jose, CA). Tumor cell surface antigen was quantified using Quanti-Brite beads (BD Biosciences) according to the manufacturer’s instructions. Quantibrite beads and corresponding tumor cells stained for FGFR4 antigen were acquired on a BD LSRII and analysed using Graphpad Prism. Data were analyzed by FlowJo V10.
ELISA
Supernatants from tumor cell cultures were collected after treatment with IFN-γ for 24 or 48 hours. TGFβ1, MIF, LIF, and CCL24 protein was quantified by ELISA using manufacturer’s protocol (R&D Biosystems, Quantikine). Plates were read on an Espire plate reader (Perkin Elmer), analyzed in Microsoft Excel, and graphed in Prism (v. 9.2.0, Graphpad). For evaluation of FGFR4 binders identified from phage screening, recombinant antigen (full-length FGFR4 ectodomain or IgIII alone) was coated into Corning® 96-well Half Area Clear Flat Bottom Polystyrene High Bind Microplate (Cat.no. 3690) at 4C overnight. Plates were blocked using PBS buffer containing 3% non-fat milk (PBSM) for one hour. For detection antibody binding, the VH and Fab binders were 5-fold serially diluted with starting concentration of 1 μM and incubated for two hours at room temperature. After incubation, the plates were washed using PBS containing 0.05% Tween-20 (PBST). Horseradish peroxidase (HRP) conjugated anti-FLAG tag antibody (Sigma, A8592) diluted in the PBSM buffer (1:1000), were added into plate and incubated for one hour at room temperature, followed by washing with PBST. The binding signals were developed by adding HRP substrate TMB (Sigma, T0440) and stopped by using 1M H2SO4. The plate was read using Biorad iMark™ Microplate Absorbance Reader.
Nanostring Profiling
RNA was extracted from in vitro cultures of RH30_19 cells or from single cell suspensions of tumor tissue. RNA was isolated from 3000 cells/ul per sample. RH30_19 tumor cells were isolated from whole tumor samples using anti-CD19 magentic beads (Miltenyi Biotec). RNA expression profiling was performed with the Nanostring nCounter using the Human Immunology V2 panel (Nanostring Technologies). Normalization and analysis was conducted in Rosalind (Nanostring Technologies). Gene categories were selected in Microsoft Excel using the Human Immunology V2 Panel Annotations summary.
IHC and immunofluorescence
Immunofluorescence was performed on paraffin fixed paraffin embedded tissues sectioned 5µm thick. Sections were deparaffinized and pretreated with Diva decloaking buffer (Biocare Medical) at 120°C for 30 seconds. Sections were first stained for rabbit anti-CD4 (Abcam #243872) and rabbit anti-CD8 (Abcam #4055) each diluted 1:200 together using the goat anti-rabbit tyrimide amplification kit (Thermo #B40943) according to manufacturer instructions. Slides were then reheated in Diva buffer to 120°C and then stained for goat anti firefly luciferase (Novus #NB100–1677) followed by rabbit anti-CD11b (1:500, Abcam #33357), rat-anti F4/80 (1:500, Thermo #MF48000), and CD206 (1:500, R&D #AF2535-SP) stained with Alexa Fluor labeled secondaries (Thermo #A-11056, #A-11036, #A-21247) each diluted 1:500 in blocking buffer. Ten non-overlapping fields of each slide were imaged using a Nuance multispectral camera (Akoya Bio) on a Nikon Eclipse Ci microscope with both 10x (NA 0.45) and 40x (NA 0.95) objectives. Images were analyzed using InForm Tissue analysis software (v2.4.1, Akoya Bio). Sections were also stained with H&E or TriChrome (Sigma Aldrich, HT15).
Mice and Tumor Model
NOD-scid IL2Rgammanull (NSG) mice, used at 8–12 weeks old, were purchased from Jax Labs or bred in-house. RH30_19 tumor cells were prepared at 20x106/mL in PBS. 50 μL (1x106 RH30_19 or RH30_19 MIF KO) of the single cell suspension were injected intramuscularly into the right hind leg of NSG mice. T cells (CAR T or UTD) were injected intravenously or intratumorally, as per procedure. Mice were imaged weekly after tumor inoculation using IVIS Spectrum in vivo Imaging system (Perkin Elmer) 15 minutes after subcutaneous D-luciferase (Xenolight D-luciferin K+ salt, Perkin Elmer) injection. Endpoint was determined by tumor volume (1000 mm3) as measured by digital caliper using the formula: volume = (width2 * length)/2. All-trans retinoic acid (ATRA) was delivered by timed release subcutaneous pellet (5mg, 21-day release, Innovative Research of America). PLX3397 (50mg/kg/day, MedChemExpress), Epacadostat (75mg/kg/day, MedChemExpress)(30,31), and SD-208 (25 mg/kg/day, MedChemExpress)(32) were delivered by oral gavage using plastic feeding tubes (Instech, FTP-18–30). L-NAME (50 mg/kg/day, Cayman Chemical) and anti-PD1 antibody (200 μg/dose, BioXCell, BE0188) were delivered by I.P. injection. Doses were 5 days on/2 days off unless otherwise noted.
ALT assay
Serum was collected from mice at euthanization, based on disease-burden. Whole blood was collected, allowed to sit at room temperature for 15 minutes, then centrifuged for 20 minutes at 4C. The resulting serum was collected and stored at −80C until use. ALT was measured using a commercially available ALT Activity Assay (Sigma Aldrich), following the manufacturer’s protocol.
Statistical Methods
Plots and statistical analyses were analyzed using Prism software (v. 9.2.0, GraphPad Software, LLC). Experimental replicates are noted in each figure legend. Statistical significance is indicated as p<0.05 (*), p<0.01 (**), p<0.001 (***). Animal survival was plotted as a Kaplan-Meier curve and analyzed using a log-rank (Mantel-Cox) test.
Approvals
Animal experiments were conducted in an AAALAC accredited facility, following Seattle Children’s Research Institute institutional animal care and use committee (IACUC) approval of procedures (IACUC00417)
Data Availability Statement:
Data were generated by the authors and available on request (permitted only for data types for which a community-recognized, structured repository does not exist)
Results
In Vitro characterization of FGFR binding domains on chimeric antigen receptors
To improve FGFR4 CAR-T performance, we screened a new generation of binding moieties. Previous binders were generated against the entire extracellular domain of FGFR4, consisting of three Ig-like domains and an acid box domain (Figure 1A). To target new CARs to the membrane proximal Ig-III domain of FGFR4, we screened for FGFR4 binders (single domain VH and Fab) by panning against the full extracellular domain of FGFR4, followed by two rounds of panning against the Ig-III domain alone (Figure 1B). Binding was confirmed by ELISA against the full extracellular domain and the Ig-III domain and with binding to cell surface associated FGFR4 binding in cell culture (Figure 1C and Figure S1A-C). We developed eight VH binders and seven Fab binders from this screening process. The binders were cloned into a second generation chimeric antigen receptor featuring a GM-CSF signal sequence, CD8 hinge/transmembrane domain, 4–1BB costimulatory domain, and CD3ζ activation domain (Figure 1D). scFv-based binders were cloned in both the VL-VH and VH-VL orientation (Figure 1D). FGFR4 CARs were screened in vitro for surface expression, cytotoxicity, and cytokine production in primary human T cells. We confirmed the expression of FGFR4 CARs nine days after activation. CAR expression varied greatly between binders, with some CARs being undetectable despite previous validation as effective binders by ELISA (Figure 1E). All 22 CARs generated were then cloned in LV-expression vectors, tested for cytotoxicity against the aRMS cell line, RH30 (transduced to co-express truncated CD19 as a model antigen (RH30_19) and luciferase to facilitate analysis, and a control (FGFR4 negative) cell line, Raji. Day nine FGFR4 CAR-T were co-incubated with target cells for 20 hours at the indicated effector:target (E:T) ratios. Nearly all new FGFR4 binders demonstrated in vitro cytotoxicity specific for RMS cell line without cytotoxicity against controls (Figure 1F). The FGFR4 CARs were also tested for cytokine production during co-incubation with RH30_19 target cells. Supernatants from CAR-T-tumor target co-incubation assays were also tested for the presence of IFN-γ and TNFα. Again, depending on the binder incorporated into the CAR-T construct, variable cytokine production was seen (Figure S1D). Based on surface detection of CAR, high cytokine production, and high specific cytotoxicity, we selected the most active FGFR4 binders, with ready detection by flow cytometry, to continue in vitro screening.
Figure 1. Isolation of new FGFR4 binding moieties and Screening anti-FGFR4 CAR-T.
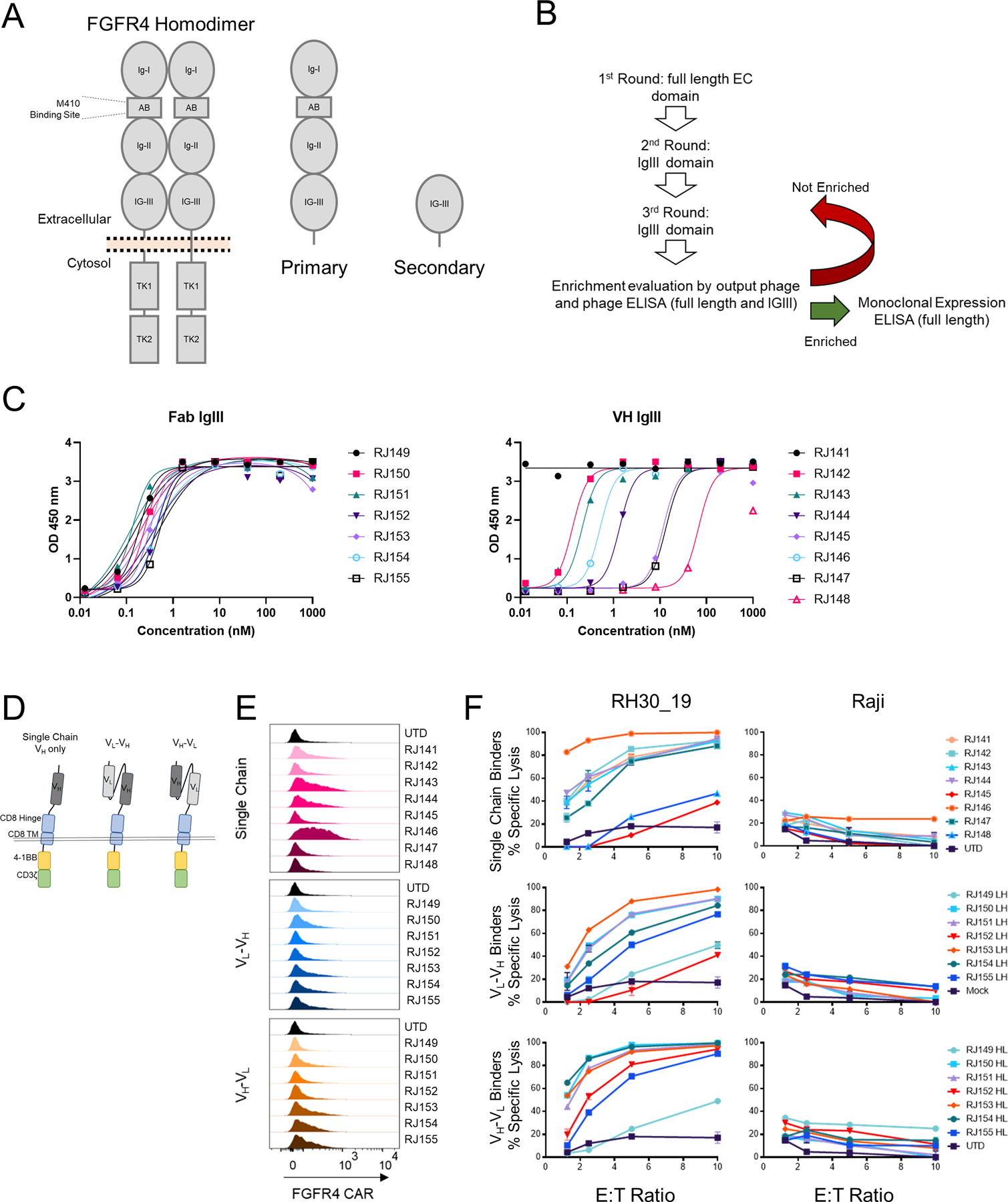
A. Schematic of FGFR4 homodimer and the FGFR4 protein fragments used for panning new binders. Full length antigen (Primary) was used for the first round of panning FGFR4 binders, followed by an IgIII-domain only fragment (Secondary) in subsequent rounds. B. Panning workflow included three rounds of panning, phage enrichment and binding evaluation. C. ELISA targeting FGFR4 IgIII domain only with either Fab binders (left panel) or VH-only binders (rightpanel). Increasing amounts of soluble binder (x-axis) were added to antigen coated plates, and specific binding quantified by ELISA (y-axis, as in Methods).D. Schematic of CAR structure. The thin double line represents the plasma membrane, which is transited by the CD8 transmembrane (TM) domain and linked to intracellular signaling domains derived from 4–1BB and the CD3-ζchain. The extracellular aspect of the CAR contains a hinge domain derived from CD8 which links to single chain (dark gray) or scFv-based (light gray and dark gray) antigen binding domains derived from phage display, Figure 1. E. Surface expression of anti-FGFR4 CARs was tested in healthy donor PBMCs day 9 post activation. CAR was detected using recombinant FGFR4-Fc-Biotin and SA-PE to directly bind CAR on the cell surface. Surface-stained cells were visualized using flow cytometry. F. Cytotoxicity of CAR T cells was assessed using CTL assays against FGFR4-expressing RH30_19 RMS cell line and FGFR4-negative Raji-ffLuc cells. Target cells were plated at 10,000/well with CAR T cells added at the indicated E:T ratios, and co-incubated for 20 hours. Percent specific lysis was determined by luminescent signal from surviving tumor cells. D. IFN-γ and TNFα cytokines released during 20-hour co-incubation with FGFR4-expressing RH30_19 target cells were quantified using LegendPlex bead-based cytokine assay. Controls include unstimulated T cells (Unstim) and T cells activated for transduction but not exposed to LV (UTD). All assays were conducted in triplicate and independently repeated three times.
Five FGFR4 binders, two VH-only and three scFv-based (RJ144, RJ146, RJ151-LH, RJ150-HL, and RJ154-HL) were selected for additional in vitro vetting. These were compared to two previous FGFR4 binders generated during previous studies, m410 and m412 (20). Surface detection of FGFR4 CAR varied between constructs (Figure S2A) and CAR expression did not change the in vitro expansion kinetics of transduced T cells (Figure S2B). As sarcoma lines are sensitive to NK-like activity (33–35), and CAR-T cells can exhibit non-specific NK-like killing (36) which can complicate the interpretation of CAR-T based killing, the cytotoxicity of these CARs was also tested, with or without cold target inhibition (CTI). CTI with K562 cells serves to blunt NK-like killing activity associated with both NK cells and activated T cells and was accomplished by adding a 30-fold excess of unmodified K562 to the cytotoxicity, effectively redirecting CAR non-specific, NK-like cytolysis. We also confirmed that NK cells were not present in CAR-T cell populations (Figure S2C).With the addition of CTI, only two binder candidates maintained strong cytotoxicity against RH30_19, with the remaining three CARs showing greatly diminished cytotoxicity (Figure 2A). To better understand the non-specific cytotoxicity of these CARs, we also tested them against K562, with and without CTI. All five CARs had cytotoxicity against the FGFR4-negative targets that was blocked with CTI, suggesting NK-like cytotoxicity was present in our cytotoxicity assays (Figure 2B). FGFR4-CAR cells had variable non-specific cytotoxicity against REH and NALM6 cells lines, which was also reduced by cold target inhibition (Figure S2C). We next tested the durability of these selected CARs in a multiple-challenge cytotoxicity assay. FGFR4 CAR-T cells were plated with tumor targets at a 1:1 or 1:4 ratio for 48 hours. The remaining cell populations were then harvested from wells, and assessed for total number and the ratio of persisting tumor and FGFR4 CAR-T. The original ratio was then re-established by adding target cells and the process was repeated for a total of three challenge cycles. FGFR4-CARs demonstrated varied responses after the initial challenge, with RJ144 decreasing in effectiveness after the initial challenge, RJ146 retaining the strongest cytotoxicity throughout the three challenges, and the remaining three CARs falling between these responses (Figure 2C). Cytokine production was also assessed. All newer generation FGFR4 binders targeting the membrane proximal domain of FGFR4 outperformed previously generated CARs (m410 and m412) (Figure 2D). From this data, we selected RJ150-HL and RJ154-HL based on their specific and strong cytotoxicity, cytokine expression, and in vitro persistence based on tumor-clearance activity against multiple rounds of tumor challenge.
Figure 2. Screening anti-FGFR4 CAR T cells for specificity and function.
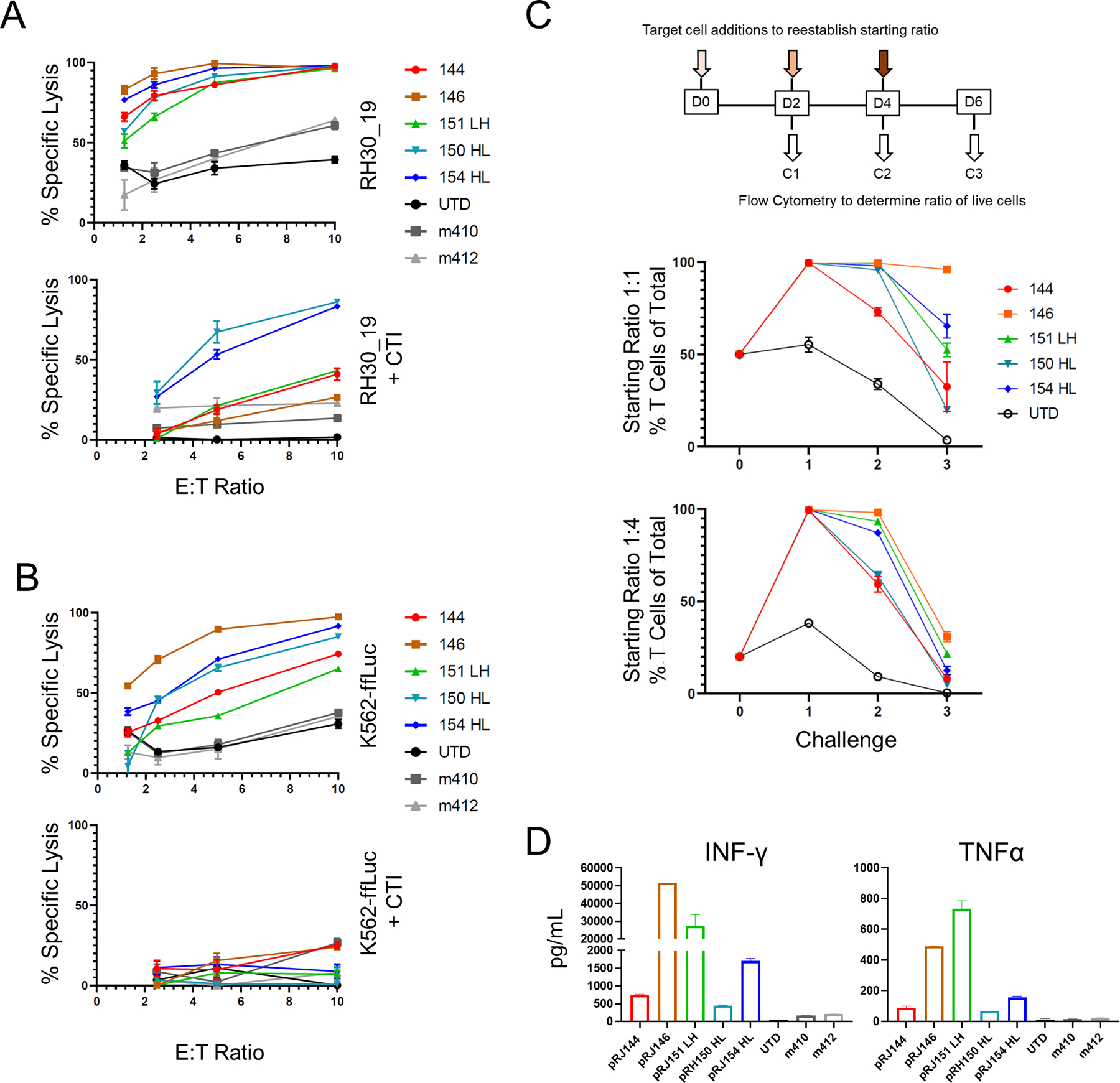
A, B Top candidate FGFR4 binders were tested for cytotoxicity against RH30_19 (A) or K562-ffLuc (B) with or without cold target inhibition (+CTI) using unlabeled K562 cells at 30-fold excess. C. Multiple challenge cytotoxicity assay was conducted by plating anti-FGFR4 CAR T cells with RH30_19 or Raji cells at a starting E:T ratio of 1:1 or 1:4. Cell populations were assessed by counting and flow cytometry at 48 hours, then original ratios re-established. This was repeated for three tumor-cell challenge cycles and the percentage effector T cells of the total population graphed at the end of each cycle (%T cells of total). Thus, 100% (y-axis) indicates no tumor detectable. D. Cytokine production at 20 hours of co-culture with RH30_19 target cells. All assays were conducted in triplicate and independently repeated at least twice.
Low antigen density limits FGFR4 CAR effectiveness against orthotopic RMS tumors
We tested two FGFR4 CARs, RJ150-HL and RJ154-HL, and a CD19 CAR (FMC63 binder) against RH30_19 intramuscular tumors in NSG mice. CARs were delivered either by intravenous delivery (IV) on day 3 post tumor inoculation or intratumor delivery (IT) on day 7 post tumor inoculation, the earliest time point at which tumor could be reliably palpated (Figure 3A). Tumor growth was assessed weekly by luciferase activity (IVIS) and caliper measurement. Mice with FGFR4 CARs did not show any difference from UTD or untreated mice, whereas CD19 CAR had a transient effect, as evidenced by slowing or reducing tumor growth when delivered by IV (Figure 3B, C). None of these CARs were able to significantly extend survival (Figure 3D). Furthermore, delivering CAR T cells by IT had no benefit over IV, despite directly delivering the effector cells into the tumor and bypassing obstacles to T cell trafficking to the tumor. We propose that CAR-T either do not persist in the tumor interior, or that migration to tumor stoma is an active process, whether CAR-T start inside the tumor or arrive through the vasculature. CD19 CAR-T cells were able to delay tumor growth when delivered by IV, possibly by reducing the accumulation of immunosuppressive TAMs in the TME (Figure S3A).
Figure 3. CAR-T cell therapy alone fail to control I.M. RMS tumors.
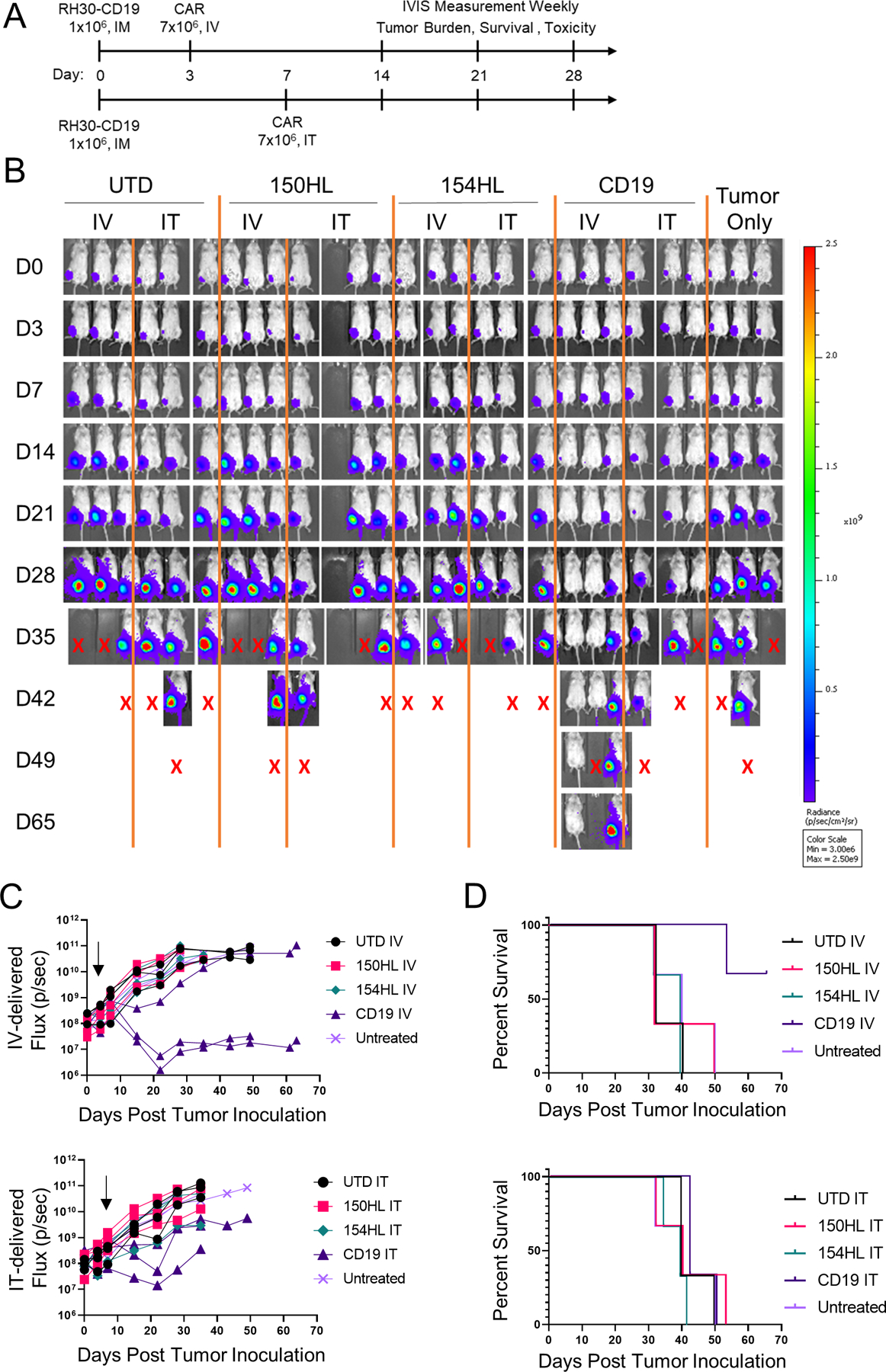
A. Schematic experimental timeline. Tumors were injected intramuscularly on day 0. CAR-T cells were injected I.V. on day 3 for one group (top line) or injected intratumorally (IT) on day 7 in a separate group of tumor-bearing mice, 7x106 per mouse. The top two anti-FGFR4 CAR candidates, RJ150HL and RJ154HL, as well as CD19 were used for treatment. B. Weekly IVIS measurements was used to determine tumor growth until mice reached endpoint (tumor volume >1000 mm3). Groups of three mice received CAR-T cells IV, IT, or control (UTD). Three mice received tumor only. X indicates sacrifice as per protocol. C. Flux (photons/sec) of individual mice treated with either IV-delivered CAR-T cells (left) or IT-delivered CAR-T cells (right) is plotted. Arrows indicated the time of CAR-T injection. D. Kaplan-Meier survival curves for mice receiving IV- (left) or IT- (right) delivered CAR-T cells. Two mice treated with IV CD19 CAR-T survived to day 65.
To better understand why FGFR4 CARs showed no effect against RH30_19 tumors whereas CD19 CAR showed a transient effect, we quantified the target surface molecules per cell. CD19t was expressed 35-fold higher than FGFR4 (95,000 CD19t molecules/cell compared to 2,700 FGFR4) on the RH30_19 cell line in culture (Figure 4A). Furthermore, when FGFR4 surface expression was tested on tumor cells that were excised from tumor-bearing animals, we observed a further decrease in expression (700 FGFR4 molecules/cell) which is similar to the low levels of expression seen on the eRMS cell line RD (400 FGFR4 molecules/cell) (Figure 4B). To overcome low antigen density, we tested additional CAR designs that have previously been shown to improve CAR function against a low-density antigen (29). We tested the RJ154-HL binder (hereafter referred to as FGFR4 CAR) with our standard CAR design (standard), a CAR with two CD3ζ domains (double zeta), and a CAR with a CD28 hinge/transmembrane domain (CD28 H/TM) and CD137 (4–1BB) and a single CD3ζ domain (Figure 4C). The double zeta FGFR4 CAR reduced CAR surface expression and function compared to our standard CAR design, as determined by cytotoxicity and cytokine production in response to the RD cell line. In contrast, the CD28 H/TM CAR maintained surface expression and cytokine production while improving cytotoxicity (Figure 4D, E, F). A similar pattern was observed with our CD19 CAR construct (Figure S4A, B). Based on the improved function of the CD28 H/TM CAR, this format was used in subsequent experiments.
Figure 4. Tuning anti-FGFR4 CARs for low-density antigens.
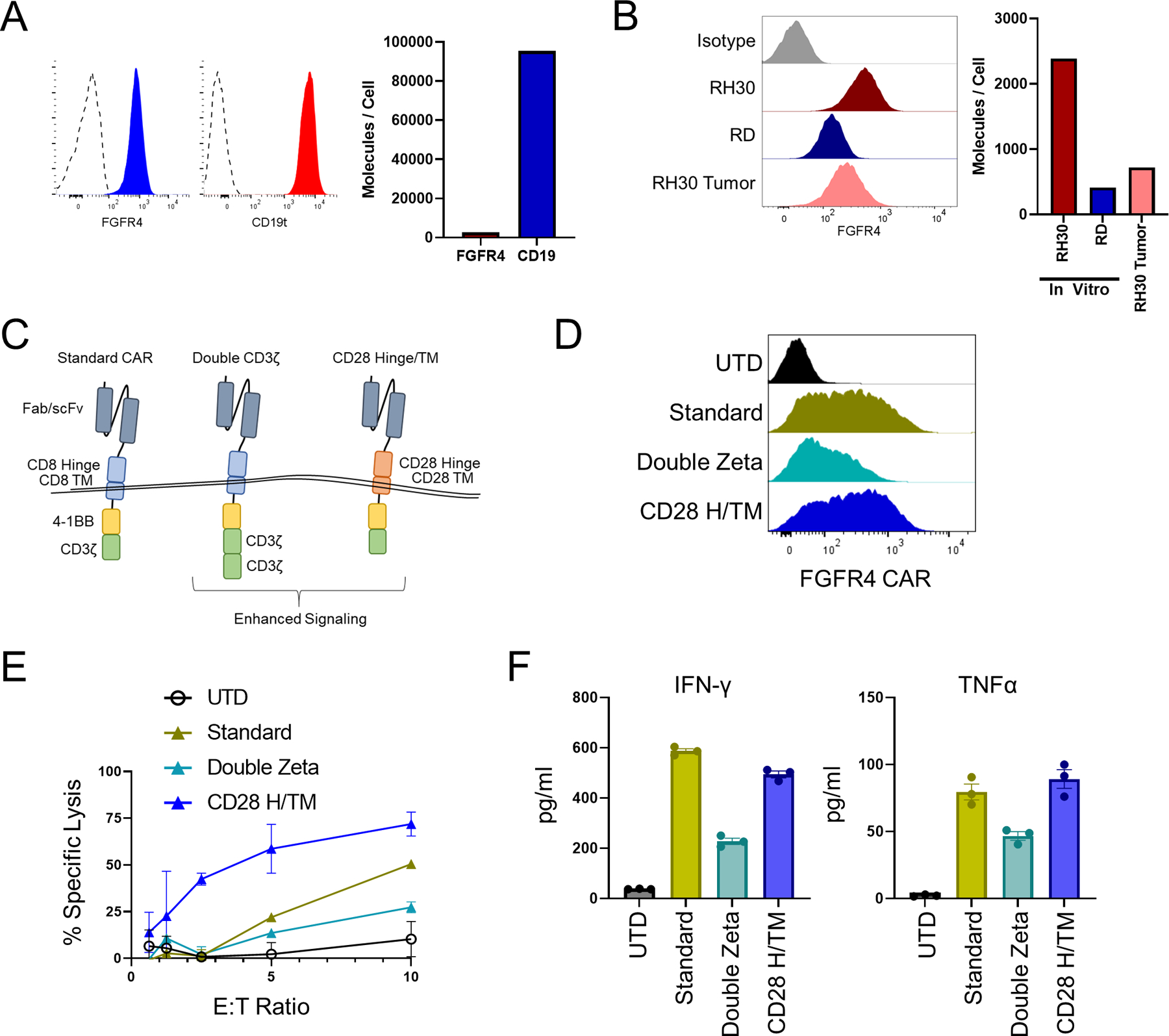
Expression of RH30_19 antigen surface expression of FGFR4 and CD19 as assessed by flow cytometry (A) and quantified with QuantiBrite beads (B). Quantification of surface FGFR4 expression of cultured RH30_19 and RD cell lines or RH30_19 cells derived from excised tumors. Both flow analysis (left panel) and quantification (right panel) are shown. C. New CAR structural and signaling formats evaluated: double CD3ζ CAR, and CD28 H/TM CAR. D. Surface expression of RJ154HL CAR (anti-FGFR4 CAR) with indicated new signaling domains assessed by flow cytometry. E. Cytotoxicity of anti-FGFR4 CAR-T cells against the RD-ffLuc RMS cell line after 5 hours of co-culture at indicated E:T ratios. F. Cytokine production by anti-FGFR4 CAR after 20 hours of co-culture with RH30_19 target cells. Average of three wells and SEM are indicated for each new CAR format and control UTD.
The TME of RMS remodels in response to T cells
RH30_19 tumors were excised from NSG mice that had either received no treatment, or received activated, untransduced T cells. Histochemical analysis, H&E stained FFPE tumor sections, of these tumors showed that the tumors exposed to activated T cells developed a thick collagen-rich stroma (Figure 5A). H&E showed minimal stromal deposition in untreated orthotopic tumors growing IM in NSG mice, (top panels, black arrows, Fig. 5A), and a thick stroma in T cell treated mice (bottom panels, yellow arrows). The collagen deposition attending this stromal reaction was highlighted by Trichrome staining (Figure 5B). As this reaction was seen with UTD T cells and CAR-T cells, we ascribe it to the production of factors (such as IFN-γ) known to activate murine myeloid cells (37), as seen in mouse models of sarcoma. We confirmed that UTD T cells do produce IFN-γ in response to mouse cells by co-incubating T cells with single cell suspensions of mouse spleen, bone marrow (BM), and excised RH30 tumor. We observed low levels of IFN-γ production when the T cells were in the presence of mouse cells, but not when co-incubated with cultured RH30 cells (Figure S5A). To determine the cellular composition of the tumor and stroma, we used immunofluorescent staining for myeloid cell markers (CD11b, F4/80) and ffLuc antibody staining for tumor cells (ffLuc) to produce a phenotypic map of the tumor lesion, and imaging analysis to create masks that segregate tumor and tumor-excluded (stomal) regions (Figure 5C, top panel). Cellular masks allowed for individual cells to be counted (Hoechst nuclei) and phenotype ascribed to each. We found that the total number of myeloid cells in the tumor increased, and that this increase was primarily in the tumor excluded, stromal regions (Figure 5C). Furthermore, the presence of M2-like tumor associated macrophages (TAM) was largely absent in the untreated tumors but became more abundant when treated with either untransduced T cells or FGFR4 CAR T cells (Figure 5D). Finally, untransduced, CD19 CAR, and FGFR4 CAR T cells all localized to the stromal regions of the tumor in close association with myeloid cells (Figure 5E), consistent with a immune-excluded tumor phenotype.
Figure 5. T cell therapy induces tumor stroma formation.
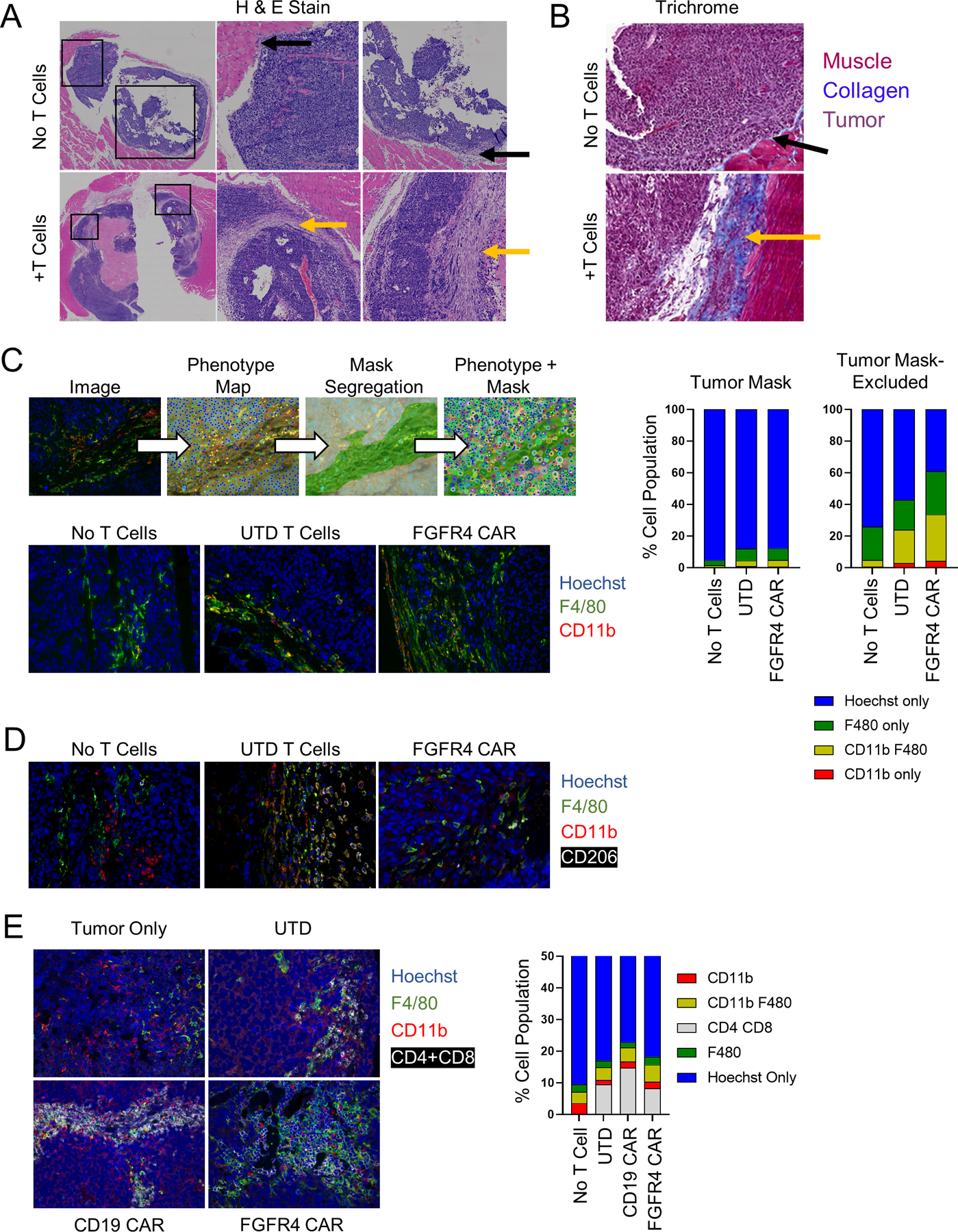
A, B. Untreated or T cell treated intramuscular RH30_19 tumors were excised and stained with H&E, with central and right panels showing enlargements of the indicated boxed areas (A) or TriChrome stained to highlight collagen deposition (blue) (B). Yellow arrows indicate regions of stroma/collagen deposition as opposed to the tumor-stromal interface in untreated mice (black arrows). C. Untreated or T cell treated RH30_19 tumors were stained with α-Luciferase (tumor), mouse α-CD11b, and α-F4/80. A phenotype map was created to indicate the identity of each cell within the field of view. Luciferase staining was used to determine tumor cells and create a mask of tumor and non-tumor regions. Myeloid cells were counted within both regions (phenotype + mask) as demonstrated. The proportion of myeloid cells in untreated, T cell treated, or anti-FGFR4 CAR treated mice is shown in the representative images and plotted to the right. D. The presence of tumor-associated macrophages was determined with immunofluorescence staining of CD206 along with CD11b and F4/80 in RH30_19 tumors excised from untreated, untransduced T cell, or FGFR4 CAR-T cell treated mice. E. CD4 and CD8 T cells localized to myeloid-rich regions as shown by immunofluorescent staining with CD4+CD8, CD11b, and F4/80 in RH30_19 tumor samples with the indicated treatments.
The inability of intratumorally-delivered CAR T cells to control tumor growth (Figure 3B, C) and the immune-excluded phenotype observed by immunofluorescence (Figure 5E), indicates that delivery of CAR T cells is unlikely to have a durable effect on tumor growth unless the stromal formation which serves as a cellular sink for introduced T cells can be overcome in parallel. To identify molecular targets for improving CAR T cell therapy, we first used quantitative RNA-expression profiling panels (Nanostring analysis) of tumor cells in culture. RH30_19 cells were left untreated or treated with IFN-γ for 24 hours to mimic T cell effects on tumors in vivo. Using the nCounter Human Immunology V2 panel, we identified the most highly expressed genes after IFN-γ treatment and curated the list to highlight the top soluble factors (Figure S6A) and immune cell receptor transcripts (Figure S6B). Among the highest expressed soluble factors were macrophage migration inhibitory factor (MIF), Transforming Growth Factor β1 (TGFB1), and Leukemia inhibitory factor (LIF). The presence of MIF, LIF, and TGFB1 are all associated with poor prognosis in multiple cancer types. Indoleamine 2,3-Dioxygenase 1 (IDO1) was also included, as expression of IDO1 is well-established as an immunosuppressive marker. Additionally, the chemokines CXCR4 and CCL24 decreased with IFN-γ treatment (Figure 6A). In vivo tumor samples from untreated or T cells treated RH30 tumor-bearing mice were also analyzed for gene expression, with similar candidates identified (Figure S6C, D). Select soluble factors were confirmed by ELISA in multiple RMS tumor cell lines, including RH30_19, RH30 parental line (FP-RMS), RD (FN-RMS), and RMS13 (FP-RMS) after 24 or 48 hours of of IFN-γ treatment. To verify RNA-expression results at the protein level, ELISA assays of tumor culture supernatants were tested for MIF (Figure S6E), TGFB1 (Figure S6F), CCL24 (Figure S6G) and LIF (Figure S6H). Differences were noted between RMS cell lines, indicating that the presence of some soluble factors may be subtype specific. PDL1 and PDL2 surface expression was analyzed by flow cytometry in untreated or IFN-γ treated RH30_19 cultures (Figure S6I). PDL1 expression increased with IFN-γ treatment, mirroring the gene expression data (Figure S6B, I). PDL2 is expressed on RH30_19 cells, but does not increase with IFN-γ, despite minimal gene expression (Figure S6B, I). These results suggest several well-described tumor-produced immune defense mechanisms that could be targeted to improve FGFR4-targeted CAR T cells control of RMS tumors.
Figure 6. Anti-myeloid poly-pharmacy allows CAR-T cells to clear orthotopic RMS tumors.
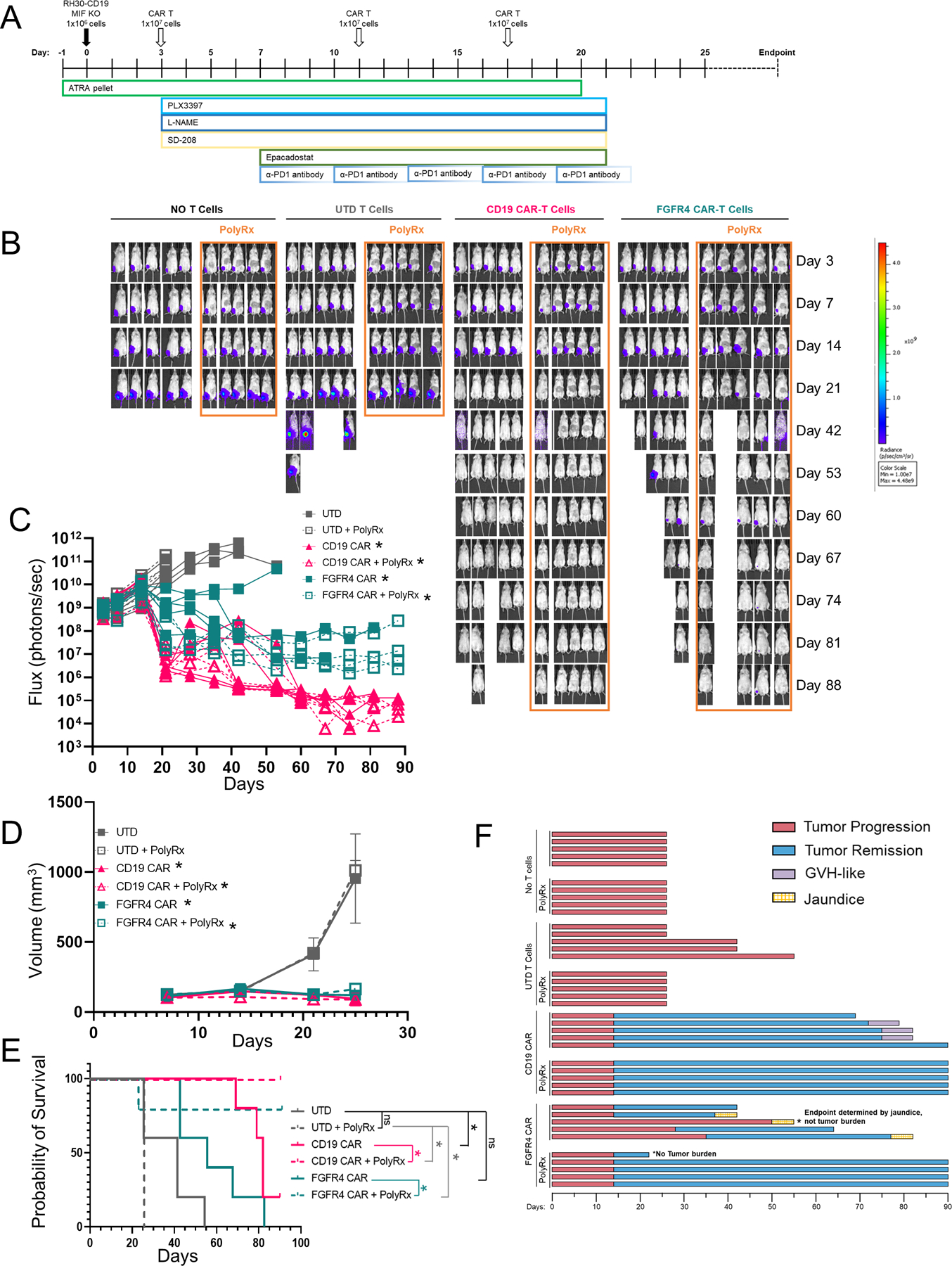
A. Schematic timeline of anti-myeloid PolyRx treatments and CAR-T treatment. Closed boxes around each treatment indicate its duration. B. Mice were imaged weekly by IVIS to assess tumor growth. C. Flux measured by IVIS was plotted for individual mice treated with UTD T cells, anti-CD19 CAR, or anti-FGFR4 CAR (PS525) with and without PolyRx. D. Tumor volume as measured by caliper measurement, averaged for each treatment group. E. Kaplan-Meier survival curve of treated groups with (dotted lines) or without (solid lines) PolyRx. F. Swimmer plot of mouse survival (length of bar), tumor progression/regression (red/blue, respectively), and toxicities (graft versus host, purple; jaundice, yellow), to the end of the assay (day 90).
Anti-myeloid polypharmacy allows CAR T cells to clear orthotopic RMS tumors
With a deeper characterization of the TME of RMS tumors, we developed a series of small molecule-based interventions aimed at disrupting the myeloid cells that were invoked in the region due to our T cell intervention. We hypothesized that a poly-pharmacy approach would be required to overcome the multiple tumor defenses described in order to allow CAR T cells to engage the tumor cells and clear the lesion. We targeted myeloid-derived suppressor cells that are known to be present in NSG tumor models with all-trans retinoic acid (ATRA), M2-like tumor-associated macrophages (TAM) with the CSF1R inhibitor Pexidartinib, TGFβ signaling with a TGFBRI inhibitor, SD-208, IDO1 with Epacadostat, iNOS with L-NAME, and α-PD1 antibody to prevent T cell exhaustion (Figure 6A). Additionally, we used an MIF knockout line of RH30_19 (RH30_19 MIF KO). We termed this approach PolyRx. When PolyRx was combined with CD19 or FGFR4 CAR T cells, but not untransduced T cells, the T cells were able to clear i.m. RH30_19 tumors (Figure 6B-D) and extend the survival of CD19 or FGFR4 CAR treated mice, whereas FGFR4 CAR alone controlled tumors to a lesser extent and did not significantly extend survival (Figure 6E). Mice receiving PolyRx in the No T Cell and UTD T Cells groups had fewer total macrophages and CD206+ macrophages residing in their tumors (Figure S7A, B). Interestingly, mice that received PolyRx plus CAR T cells demonstrated fewer signs of autoreactive T cells – graph versus host (GVH) disease with CD19 CAR or jaundice associated with on target/off tumor targeting of the FGFR4 CAR (Figure 6F). PolyRx did not show any obvious signs of toxicity, evidenced by equal weight gain throughout treatment (Figure S8A). Jaundice was clearly evident in FGFR4 CAR-T treated tumor bearing mice that did not receive PolyRx as evidenced by yellowed eyes, skin, and mesenteric tissues upon necropsy. GvHD was noted by thin and matted fur, hunched posture, and mice were sacrificed according to protocol due to weight loss. Liver toxicity was assessed by ALT activity assay. Increased serum ALT was observed only in FGFR4 CAR-T treated mice, suggesting that the resulting jaundice was due to FGFR4 CAR-T activityrather than PolyRx treatment (Figure S8B). We propose in further work to determine the causation of these specific CAR-T product associated toxicities and why they are not apparent in PolyRx treated tumor-bearing mice. Further studies will help eludcidate the individual and combined impact of PolyRx targets in RMS and other pediatric solid tumors. We have observed that MIF knockout slows RH30 tumor growth in NSG mice, similar to previous studies of MIF knockout in other tumor lines (Figure S9A-C) (38). Preliminary data also suggests that reducing PolyRx can provide a similar level of efficacy in combination with FGFR4 CAR in WT RH30_19 tumors (Figure S10A-D).These data demonstrate that orthotopic RMS tumors can be targeted by CAR T cells, even against low density target molecules like FGFR4, by reversing the immunosuppressive features of the tumor microenvironment.
Discussion:
Our novel anti-FGFR4 CAR paired with anti-myeloid poly-pharmacy reversed the immunosuppressive myeloid compartment of solid RMS tumors, allowing for successful clearance of orthotopic (IM) tumors. These results, while striking on their own, also provide insight into targeting low-density antigens on other solid tumors. We clearly demonstrate that the anti-FGFR4 CAR is able to clear solid tumors to the same efficiency as anti-CD19 CARs, despite FGFR4 molecules being 40 times less frequent on the cell surface. Our new generation of FGFR4 binders demonstrates the role of target selection in CAR T cell testing. While the original FGFR4 binders targeted the more membrane distal acidic box region of FGFR4, the new binders selected for binding to the membrane proximal Ig-III domain of FGFR4 improved CAR performance (Figure 1A, Figure 2A, B, D), in line with data from previous CAR T cell optimization reports (21,39). The improvement reported here by targeting a membrane proximal domain rather than distal domains may be one of the few rules governing CAR design, whereas signaling domains (CD28, 4–1BB, CD3ζ) and structural domains (spacers, hinge, transmembrane, linkers) require empirical optimization that will be unique to the CAR based on receptor affinity and target density among other under-recognized variables. The need for empirical testing of new CARs has been demonstrated by other groups (29,40). The density of the target on the tumor surface is one important factor to consider. We found that modifying the intracellular signaling domains of our CARs (both CD19 and FGFR4) was able to improve the cytotoxicity of our CAR, similar to a recent study demonstrating CAR tuning specifically to low density antigens (29). While swapping out a CD8 H/TM for a CD28 H/TM improved the CD19 and RJ154HL FGFR4 CAR, we did not see any improvement in the RJ150HL FGFR4 CAR (Figure 4E, F; Figure S4A, B; and data not included), highlighting lack of standardized rules in CAR tuning.
Despite the improvements in FGFR4 CAR cytotoxicity and cytokine production seen with our new generation of binders, the FGFR4 CAR alone was unable to control orthotopic RMS tumors (Figure 3). Orthotopic tumors are more challenging to control than metastatic and subcutaneous tumor models but better model the TME. By using intramuscular tumors in our model, we can better understand the complex interactions of the tumor and CAR T cells in a more native environment. We found that RMS tumors respond to the presence of activated T cells, likely due to IFN-γ release, by producing a thick, collagen-rich stroma populated by anti-inflammatory TAMs (Figure 5A-D). CD206 positive myeloid cells in the tumor microenvironment have been found to suppress T cells (41). Specifically in RMS, immunosuppressive TAMs infiltrate the tumor, reside proximal to vasculature, and correlate with poor overall and event-free survival (23,42). These studies did not see other common sources of immunosuppression, such as FoxP3+ Tregs or PD-L1 expression. Similar to these reported studies of RMS patient samples, we observed myeloid infiltration into orthotopic RMS tumors, which polarized into a more suppressive state upon T cell treatment, replicating the immunosuppressive TAMs observed in patients. NSG mice to not have Tregs, but the absence of Tregs in clinical samples suggests that these immunosuppressive cells do not play a major role in RMS. Interestingly, PD-L1 is low in both patient samples and in our untreated animals but increases upon treatment with T cells. Whether RMS tumors will respond to immunotherapies by upregulating PD-L1 remains untested but understanding this response will be important for the success of CAR-T therapies in treating pediatric solid tumors.
Furthermore, the stroma sequestered both suppressive myeloid cells and T cells in the same compartment, effectively protecting the tumor from anti-tumor cellular therapy and maintaining an immune-excluded TME phenotype (Figure 5E). This data is supportive of the current models of an immune excluded phenotype (43,44), however we add the novel observation that this stroma can be due to the response of the tumor to immune surveillance and thus also represents a model of immune editing (45,46). While immune editing often refers to antigen recognition by T cells, in our system immune editing represents the innate immune response and tissue remodeling mechanisms invoked by the presence of activated T cells.
Identifying key components and pathways of tumor defense proved to be instrumental in allowing CAR T therapy to be effective. We modeled tumor response to T cells in culture by treating RMS cell lines with IFN-γ, then analyzed gene expression. We focused on soluble factors, surface receptors, and several known tumor defense mechanisms. This approach identified key targets in RMS tumors while showcasing several differences between RMS lines that are important to account for in targeted therapy (Figure S6C-F). Armed with a clearer understanding of our RMS model, we have shown for the first time the ability to treat orthotopic RMS with CAR-T in an NSG model. Highly active CAR-T and myeloid cell inhibition are both required for tumor control (Figure 6, Figure S11). Unexpectedly, we also found that PolyRx treatment decreased the propensity for autoreactivity of CAR T cells in our model (GVH with CD19 CAR and jaundice with FGFR4 CAR), although the mechanism preventing these effects remains unclear. The focusing of a polyclonal immune response has been noted in the creation of Epstein-Barr virus-specific T cell lines for the treatment of EBV-associated malignancies such as post-transplant lymphoproliferative disease (47) These lines could be used in a haplo-identical manner, once expanded against EBV antigens. Similarly, the use of NY-ESO-1 recombinant TCRs, when introduced into autologous activated T cells does not create self-reactive cells (48). Here we have established a unique model system in which off-target CAR-T effects are modulated by increased anti-tumor effectiveness.
Given the low expression in vivo (700 molecules/cell), our model suggests that even low expression or aberrant modification of surface proteins can be targeted for CAR T therapy when immunosuppression from the TME is also resolved. Furthermore, our model provides the framework for multi-modal interventions paired with CAR T therapy. The complex cellular make-up of solid tumor provides multiple mechanisms for tumors to suppress T cell therapies. Different tumors may favor various immunosuppressive signaling mechanisms, of which several may need to be overcome for effective T cell therapy. Therefore, targeting multiple tumor-specific immune evasion pathways, as with our anti-myeloid polypharmacy, or additional armoring of CAR T cells may be a common requirement for targeting solid tumors, such as RMS (Figure S11). Thus we will both develop protocols featuring our signaling optimized FGFR4 CAR-T, PS525, in the context of PolyRx, as well as incorporate armoring mechanisms into the LV itself such as decoy receptors for TGFβ and PD-L1(49–52).
Supplementary Material
Acknowledgements:
This work was funded by NIH grant 1R21CA241023–01, and by St. Baldrick’s-Stand Up to Cancer Dream Team Translational Research Grant (SU2C-AACR-DT-27–17). The St. Baldrick’s Foundation collaborates with Stand Up To Cancer. Research Grants are administered by the American Association for Cancer Research, the Scientific Partner of SU2C. This work was also supported by the Seattle Children’s Foundation, Seattle, WA, including support from The Rachel Lynn Henley Foundation, and Make Some Noise: Cure Kids' Cancer Foundation Inc. (in memory of Timothy Gearty)
Financial Support
This work was supported by NIH Grant 1R21CA241023–01, a St. Baldrick's Foundation - Stand Up To Cancer Pediatric Dream Team Translational Research Grant (SU2C-AACR-DT-27–17), and the Seattle Children’s Foundation. Stand Up To Cancer is a division of the Entertainment Industry Foundation. The indicated Stand Up To Cancer grant is administered by the American Association for Cancer Research, the Scientific Partner of SU2C
Footnotes
Conflict of Interest Disclosure statement:
Rimas J. Orentas: Research support, Miltenyi Biotec. Board and Consulting: Umoja Biopharma and Abound Bio, Inc. R.J.O. has filed patents on some of the FGFR4 binders and CARs described herein.
Dimiter S. Dimitrov: Employment and Research Support: Abound Bio, Inc. D.S.D. has filed patents on some of the FGFR4 binders and derived CARs described herein.
Wei Li: W.L. has filed patents on some of the FGFR4 CARs described herein.
Peter M Sullivan: has filed patents on use of FGFR4 CARs.
Michael C. Jensen: has interests in Umoja Biopharma and Juno Therapeutics, a Bristol-Myers Squibb company. He is a seed investor and holds ownership equity in Umoja, serves as a member of the Umoja Joint Steering Committee, and is a Board Observer of the Umoja Board of Directors. M.C.J. also holds patents, some of which are licensed to Umoja Biopharma and Juno Therapeutics.
References:
- 1.Shern JF, Yohe ME, Khan J. Pediatric Rhabdomyosarcoma. Crit Rev Oncog 2015;20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gröbner SN, Worst BC, Weischenfeldt J, Buchhalter I, Kleinheinz K, Rudneva VA, et al. The landscape of genomic alterations across childhood cancers. Nature 2018;555. [DOI] [PubMed]
- 3.Geoerger B, Kang HJ, Yalon-Oren M, Marshall LV, Vezina C, Pappo A, et al. Pembrolizumab in paediatric patients with advanced melanoma or a PD-L1-positive, advanced, relapsed, or refractory solid tumour or lymphoma (KEYNOTE-051): interim analysis of an open-label, single-arm, phase 1–2 trial. Lancet Oncol Lancet Publishing Group; 2020;21:121–33. [DOI] [PubMed] [Google Scholar]
- 4.Jackson HJ, Rafiq S, Brentjens RJ. Driving CAR T-cells forward. Nat Rev Clin Oncol 2016;13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Walker AJ, Majzner RG, Zhang L, Wanhainen K, Long AH, Nguyen SM, et al. Tumor Antigen and Receptor Densities Regulate Efficacy of a Chimeric Antigen Receptor Targeting Anaplastic Lymphoma Kinase. Mol Ther [Internet] Elsevier Ltd.; 2017;25:2189–201. Available from: 10.1016/j.ymthe.2017.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thomas S, Straathof K, Himoudi N, Anderson J, Pule M. An Optimized GD2-Targeting Retroviral Cassette for More Potent and Safer Cellular Therapy of Neuroblastoma and Other Cancers. PLoS One 2016;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pule MA, Savoldo B, Myers GD, Rossig C, Russell HV, Dotti G, et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med 2008;14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hegde M, Joseph SK, Pashankar F, DeRenzo C, Sanber K, Navai S, et al. Tumor response and endogenous immune reactivity after administration of HER2 CAR T cells in a child with metastatic rhabdomyosarcoma. Nat Commun 2020;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ahmed N, Brawley VS, Hegde M, Robertson C, Ghazi A, Gerken C, et al. Human Epidermal Growth Factor Receptor 2 (HER2) –Specific Chimeric Antigen Receptor–Modified T Cells for the Immunotherapy of HER2-Positive Sarcoma. J Clin Oncol 2015;33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Majzner RG, Mackall CL. Clinical lessons learned from the first leg of the CAR T cell journey. Nat Med 2019;25. [DOI] [PubMed] [Google Scholar]
- 11.Crose LES, Etheridge KT, Chen C, Belyea B, Talbot LJ, Bentley RC, et al. FGFR4 Blockade Exerts Distinct Antitumorigenic Effects in Human Embryonal versus Alveolar Rhabdomyosarcoma. Clin Cancer Res 2012;18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Khan J, Wei JS, Ringnér M, Saal LH, Ladanyi M, Westermann F, et al. Classification and diagnostic prediction of cancers using gene expression profiling and artificial neural networks. Nat Med 2001;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Orentas RJ, Sindiri S, Duris C, Wen X, He J, Wei JS, et al. Paired expression analysis of tumor cell surface antigens. Front Oncol 2017;7:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roidl A, Berger H-J, Kumar S, Bange J, Knyazev P, Ullrich A. Resistance to Chemotherapy Is Associated with Fibroblast Growth Factor Receptor 4 Up-Regulation. Clin Cancer Res 2009;15. [DOI] [PubMed] [Google Scholar]
- 15.Taylor VI JG, Cheuk AT, Tsang PS, Chung JY, Song YK, Desai K, et al. Identification of FGFR4-activating mutations in human rhabdomyosarcomas that promote metastasis in xenotransplanted models. J Clin Invest 2009;119:3395–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cao L, Yu Y, Bilke S, Walker RL, Mayeenuddin LH, Azorsa DO, et al. Genome-Wide Identification of PAX3-FKHR Binding Sites in Rhabdomyosarcoma Reveals Candidate Target Genes Important for Development and Cancer. Cancer Res 2010;70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gryder BE, Yohe ME, Chou H-C, Zhang X, Marques J, Wachtel M, et al. PAX3–FOXO1 Establishes Myogenic Super Enhancers and Confers BET Bromodomain Vulnerability. Cancer Discov 2017;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alijaj N, Moutel S, Gouveia ZL, Gray M, Roveri M, Dzhumashev D, et al. Novel FGFR4-Targeting Single-Domain Antibodies for Multiple Targeted Therapies against Rhabdomyosarcoma. Cancers (Basel) 2020;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shivaprasad N, Xiong Y, Yohe M, Schneider D, Shern J, Baskar S, et al. 649. Developing FGFR4 Chimeric Antigen Receptor CAR T Cell Therapy Against Rhabdomyosarcoma. Mol Ther [Internet]. The American Society of Gene & Cell Therapy; 2016;24:S257–8. Available from: 10.1016/S1525-0016(16)33457-8 [DOI] [Google Scholar]
- 20.Sullivan P, Kumar R, Li W, Wang L, Zhang Y, Cheuk A, et al. Abstract 1545: Development of FGFR4-targeted chimeric antigen receptors (CARs) for the treatment of rhabdomyosarcoma. Immunology American Association for Cancer Research; 2021. page 1545–1545.
- 21.Haso W, Lee DW, Shah NN, Stetler-Stevenson M, Yuan CM, Pastan IH, et al. Anti-CD22–chimeric antigen receptors targeting B-cell precursor acute lymphoblastic leukemia. Blood 2013;121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jackson HJ, Brentjens RJ. Overcoming Antigen Escape with CAR T-cell Therapy. Cancer Discov 2015;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kather JN, Hörner C, Weis C-A, Aung T, Vokuhl C, Weiss C, et al. CD163+ immune cell infiltrates and presence of CD54+ microvessels are prognostic markers for patients with embryonal rhabdomyosarcoma. Sci Rep 2019;9:9211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Caruana I, Savoldo B, Hoyos V, Weber G, Liu H, Kim ES, et al. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat Med 2015;21:524–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science (80- ) 2013;339:819–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schneider D, Xiong Y, Wu D, Nölle V, Schmitz S, Haso W, et al. A tandem CD19/CD20 CAR lentiviral vector drives on-target and off-target antigen modulation in leukemia cell lines. J Immunother Cancer 2017;5:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fry TJ, Shah NN, Orentas RJ, Stetler-Stevenson M, Yuan CM, Ramakrishna S, et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat Med 2018;24:20–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Long AH, Highfill SL, Cui Y, Smith JP, Walker AJ, Ramakrishna S, et al. Reduction of MDSCs with All-trans Retinoic Acid Improves CAR Therapy Efficacy for Sarcomas. Cancer Immunol Res 2016;4:869–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Majzner RG, Rietberg SP, Sotillo E, Dong R, Vachharajani VT, Labanieh L, et al. Tuning the Antigen Density Requirement for CAR T-cell Activity. Cancer Discov 2020;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yue EW, Douty B, Wayland B, Bower M, Liu X, Leffet L, et al. Discovery of Potent Competitive Inhibitors of Indoleamine 2,3-Dioxygenase with in Vivo Pharmacodynamic Activity and Efficacy in a Mouse Melanoma Model. J Med Chem 2009;52:7364–7. [DOI] [PubMed] [Google Scholar]
- 31.Liu X, Shin N, Koblish HK, Yang G, Wang Q, Wang K, et al. Selective inhibition of IDO1 effectively regulates mediators of antitumor immunity. Blood 2010;115:3520–30. [DOI] [PubMed] [Google Scholar]
- 32.Tandon M, Salamoun JM, Carder EJ, Farber E, Xu S, Deng F, et al. SD-208, a Novel Protein Kinase D Inhibitor, Blocks Prostate Cancer Cell Proliferation and Tumor Growth In Vivo by Inducing G2/M Cell Cycle Arrest. PLoS One 2015;10:e0119346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cho D, Shook DR, Shimasaki N, Chang Y-H, Fujisaki H, Campana D. Cytotoxicity of Activated Natural Killer Cells against Pediatric Solid Tumors. Clin Cancer Res 2010;16:3901–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vela M, Bueno D, González-Navarro P, Brito A, Fernández L, Escudero A, et al. Anti-CXCR4 Antibody Combined With Activated and Expanded Natural Killer Cells for Sarcoma Immunotherapy. Front Immunol 2019;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wagner J, Pfannenstiel V, Waldmann A, Bergs JWJ, Brill B, Huenecke S, et al. A Two-Phase Expansion Protocol Combining Interleukin (IL)-15 and IL-21 Improves Natural Killer Cell Proliferation and Cytotoxicity against Rhabdomyosarcoma. Front Immunol 2017;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang L, Zhang Y, Anderson E, Lamble A, Orentas RJ. Bryostatin Activates CAR T-Cell Antigen-Non-Specific Killing (CTAK), and CAR-T NK-Like Killing for Pre-B ALL, While Blocking Cytolysis of a Burkitt Lymphoma Cell Line. Front Immunol 2022;13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Highfill SL, Cui Y, Giles AJ, Smith JP, Zhang H, Morse E, et al. Disruption of CXCR2-Mediated MDSC Tumor Trafficking Enhances Anti-PD1 Efficacy. Sci Transl Med 2014;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Charan M, Das S, Mishra S, Chatterjee N, Varikuti S, Kaul K, et al. Macrophage migration inhibitory factor inhibition as a novel therapeutic approach against triple-negative breast cancer. Cell Death Dis 2020;11:774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.James SE, Greenberg PD, Jensen MC, Lin Y, Wang J, Till BG, et al. Antigen Sensitivity of CD22-Specific Chimeric TCR Is Modulated by Target Epitope Distance from the Cell Membrane. J Immunol 2008;180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Qin H, Yang L, Chukinas JA, Shah NN, Tarun S, Pouzolles M, et al. Systematic preclinical evaluation of CD33-directed chimeric antigen receptor T cell immunotherapy for acute myeloid leukemia defines optimized construct design. J Immunother Cancer 2021;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Peranzoni E, Lemoine J, Vimeux L, Feuillet V, Barrin S, Kantari-Mimoun C, et al. Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti-PD-1 treatment. Proc Natl Acad Sci U S A 2018;115:E4041–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen L, Oke T, Siegel N, Cojocaru G, Tam AJ, Blosser RL, et al. The Immunosuppressive Niche of Soft-Tissue Sarcomas is Sustained by Tumor-Associated Macrophages and Characterized by Intratumoral Tertiary Lymphoid Structures. Clin Cancer Res 2020;26:4018–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pagès C, et al. Type, Density, and Location of Immune Cells Within Human Colorectal Tumors Predict Clinical Outcome. Science (80- ) 2006;313:1960–4. [DOI] [PubMed] [Google Scholar]
- 44.Galon J, Mlecnik B, Bindea G, Angell HK, Berger A, Lagorce C, et al. Towards the introduction of the ‘Immunoscore’ in the classification of malignant tumours. J Pathol 2014;232:199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schreiber TH, Podack ER. A critical analysis of the tumour immunosurveillance controversy for 3-MCA-induced sarcomas. Br J Cancer 2009;101:381–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schreiber RD, Old LJ, Smyth MJ. Cancer Immunoediting: Integrating Immunity’s Roles in Cancer Suppression and Promotion. Science (80- ) 2011;331:1565–70. [DOI] [PubMed] [Google Scholar]
- 47.Orentas RJ, Lemas MV, Mullin MJ, Colombani PM, Schwarz K, Ambinder R. Feasibility of cellular adoptive immunotherapy for Epstein-Barr virus-associated lymphomas using haploidentical donors. J Hematother 1998;7:257–61. [DOI] [PubMed] [Google Scholar]
- 48.Ramachandran I, Lowther DE, Dryer-Minnerly R, Wang R, Fayngerts S, Nunez D, et al. Systemic and local immunity following adoptive transfer of NY-ESO-1 SPEAR T cells in synovial sarcoma. J Immunother cancer 2019;7:276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Oda SK, Anderson KG, Ravikumar P, Bonson P, Garcia NM, Jenkins CM, et al. A Fas-4–1BB fusion protein converts a death to a pro-survival signal and enhances T cell therapy. J Exp Med 2020;217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Oda SK, Daman AW, Garcia NM, Wagener F, Schmitt TM, Tan X, et al. A CD200R-CD28 fusion protein appropriates an inhibitory signal to enhance T-cell function and therapy of murine leukemia. Blood 2017;130:2410–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Burga RA, Yvon E, Chorvinsky E, Fernandes R, Cruz CRY, Bollard CM. Engineering the TGFβ Receptor to Enhance the Therapeutic Potential of Natural Killer Cells as an Immunotherapy for Neuroblastoma. Clin Cancer Res 2019;25:4400–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yvon ES, Burga R, Powell A, Cruz CR, Fernandes R, Barese C, et al. Cord blood natural killer cells expressing a dominant negative TGF-β receptor: Implications for adoptive immunotherapy for glioblastoma. Cytotherapy 2017;19:408–18. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data were generated by the authors and available on request (permitted only for data types for which a community-recognized, structured repository does not exist)