

Abstract
Microscaled proteogenomics was deployed to probe the molecular basis for differential response to neoadjuvant carboplatin and docetaxel combination chemotherapy for triple-negative breast cancer (TNBC). Proteomic analyses of pre-treatment patient biopsies uniquely revealed metabolic pathways, including oxidative phosphorylation, adipogenesis and fatty acid metabolism, that were resistance-associated. Both proteomics and transcriptomics revealed sensitivity was marked by elevation of DNA repair, E2F targets, G2M checkpoint, interferon-gamma signaling and immune checkpoint components. Proteogenomic analyses of somatic copy number aberrations identified a resistance-associated 19q13.31–33 deletion where LIG1, POLD1 and XRCC1 are located. In orthogonal datasets, LIG1 (DNA ligase I) gene deletion and/or low mRNA expression levels were associated with lack of pathological complete response, higher chromosomal instability (CIN) and poor prognosis in TNBC, as well as carboplatin-selective resistance in TNBC pre-clinical models. Hemizygous loss of LIG1 was also associated with higher CIN and poor prognosis in other cancer types, demonstrating broader clinical implications.
Introduction
Ten to 15% of breast cancers are designated triple negative (TNBC) because of low expression of HER2, the estrogen receptor (ER) and the progesterone receptor. TNBC exhibits high mortality and frequent chemotherapy resistance (1). A minority of TNBC cases are linked to hereditary homologous recombination defects (HRD), most commonly in the BRCA1 gene, and are treatable with poly ADP ribose polymerase (PARP) inhibitors (2). However, the majority of TNBC cases do not have an obvious hereditary explanation, and therefore the underlying DNA repair defects are more obscure (3). Cytotoxic chemotherapy is standard of care but is only partially effective; hence, lack of pathological complete response (pCR) after neoadjuvant chemotherapy is frequent and associated with poor survival (4). Post non-pCR, salvage therapy with adjuvant capecitabine has modest efficacy (5). The programmed cell death receptor (PD1)-targeting antibody pembrolizumab is also approved for neoadjuvant TNBC treatment based the results of the KEYNOTE-522 trial (6). In combination with neoadjuvant chemotherapy, pembrolizumab significantly prolongs event-free survival versus neoadjuvant chemotherapy alone (7). In contrast to metastatic TNBC, however, outcome improvements are not predicted by PD-L1 immunohistochemistry (IHC) (8). Carboplatin also has efficacy in TNBC. The BrighTNess trial enrolled patients with stage II or III operable TNBC and randomized patient treatment to one of three arms prior to doxorubicin and cyclophosphamide: paclitaxel/carboplatin/veliparib (Arm A), paclitaxel/carboplatin (Arm B), or paclitaxel alone (Arm C). Carboplatin-containing arms A and B showed significantly improved pCR compared with paclitaxel alone (53% and 58%, respectively, vs 31%) (9). The efficacy of carboplatin addition is supported by two other randomized neoadjuvant trials CALGB 40603 (Alliance) (10) and GeparSixto (11). Thus, in the absence of predictive markers for individual components of each regimen, the neoadjuvant treatment for TNBC involves up to seven different drugs.
Herein we describe the first study to deploy microscaled proteogenomics (12) to discover neoadjuvant chemotherapy response biomarkers in TNBC. Snap-frozen, optimal cutting temperature compound (OCT)-embedded core needle biopsies were accrued from patients enrolled into two clinical trials that investigated a simplified carboplatin and docetaxel regimen designed to be less toxic by omitting doxorubicin and cyclophosphamide (NCT02547987 and NCT02124902) (13). This discovery dataset included germline-matched tumor whole exome DNA sequencing (WES), RNA-seq, and tandem mass tag (TMT)-based proteomics and phosphoproteomics. Analyses focused on the identification of biomarker associations with pCR, with the goal of identifying patients who would be better served with investigational drugs at diagnosis rather than suffer an ineffective standard of care. Multiple independent data sets were used to validate findings in the discovery analysis, including mRNA profiles of other TNBC clinical trials, immunohistochemistry (IHC), preclinical therapeutic studies in patient-derived TNBC xenografts (PDX) and pan-cancer analysis using data from The Cancer Genome Atlas (TCGA).
Results:
Overview of the Proteogenomic analysis approach
OCT-embedded snap-frozen core needle biopsies were accrued from consented patients with clinical stage 2 or 3 TNBC (70% Caucasian, 27% African American, and 3% other racial categories). Patients were subsequently treated with six cycles of neoadjuvant carboplatin and docetaxel combination chemotherapy (NCT02547987 and NCT02124902). Pre-treatment samples from 59 patients had >25% tumor content and were ultimately analyzed. For 16 patients, an additional sample was obtained 48 to 72 hours after initiating chemotherapy. A REMARK (Reporting Recommendations for Tumor Marker Prognostic Studies) diagram demonstrates sample flow into different analytical pipelines (Fig 1A). Using previously described BioTEXT sample processing and microscaled proteogenomics methods (12), frozen core biopsies were processed on a cryotome to produce 50um sections for analyte extraction interspersed with 5um sections to document tumor content. Alternating 50um sections were distributed into three different analyte preparation approaches to ensure even representation of analytes from different layers in the biopsy. Multianalyte extraction allowed for paired normal/tumor DNA exome sequencing (100X), RNA sequencing and quantitative, multiplexed (TMT) mass spectrometry (MS)-based proteomics and phosphoproteomics (12) (Fig 1B, Supplementary Table S1-3).
Fig 1: TNBC patient sample overview.
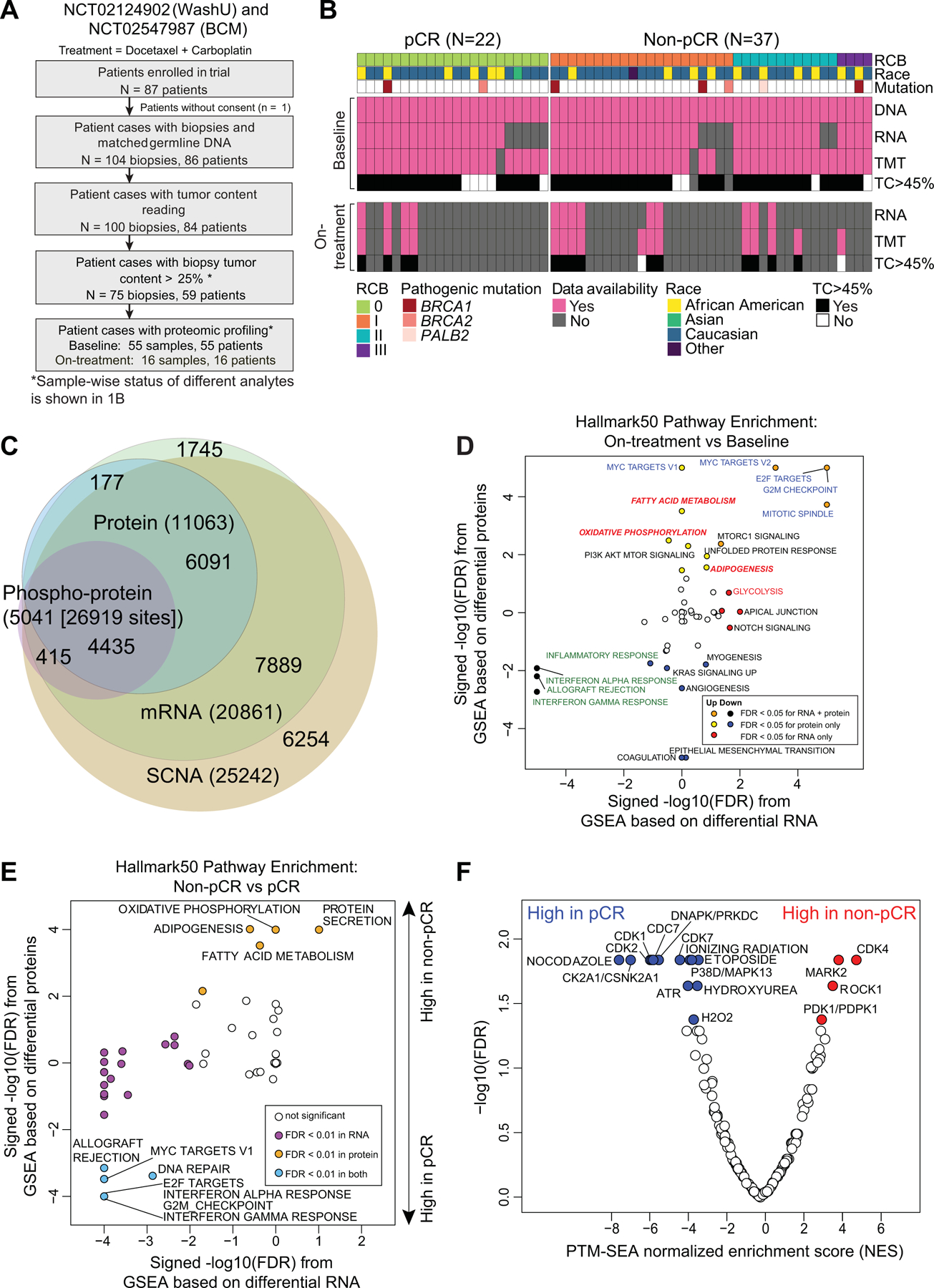
A) REMARK diagram showing pre- and on-treatment sample accrual schema from TNBC patients enrolled in two clinical trials (NCT02544987 (BCM), NCT201404107 (WashU)) and treated with carboplatin and docetaxel in the neoadjuvant setting. * <45% samples were later excluded from analysis based on evidence from data QC.
B) Overview of available omics datasets from 59 patients (22 tumors with pCR and 37 tumors without pCR). Pathogenic BRCA1/2 and PALB2 mutation status, residual cancer burden (RCB) and patient race are indicated via color-coded annotation tracks.
C) Venn-diagram showing overlap of gene IDs detected across multiple analytes and omics data profiled.
D) Hallmark metabolism pathways are induced by chemotherapy exclusively at the protein level. Scatter plot shows signed -log10 FDR from GSEA using the signed (by direction of change) -log10 p-values from paired Wilcoxon signed rank tests comparing RNA (x-axis) and protein levels (y-axis) for on-treatment (cycle 1 day 3) samples to matching baseline samples (n = 14).
E) MSigDB Hallmark metabolism pathways are elevated in baseline non-pCR tumors at the protein level while immune and cell cycle pathways are elevated in baseline pCR tumors at both RNA and protein levels. Scatter plot shows the signed -log10 FDR values from Gene Set Enrichment Analysis (GSEA) using ranked lists of signed (by direction of change) -log10 p-values from Wilcoxon rank sum tests comparing RNA (x-axis) and protein (y-axis) levels in non-pCR tumors to pCR tumors.
F) Cell cycle kinase targets and PTM-SigDB phosphosites associated with genotoxic stress are enriched in pCR tumors relative to non-pCR tumors at baseline. Volcano plot shows results from Post-Translational Modification-Set Enrichment Analysis (PTM-SEA) using the signed -log10 p-values from Wilcoxon rank sum tests comparing phosphosite levels in non-pCR tumors to pCR tumors. Red and blue dots indicate significant (FDR <0.05) PTM signatures.
Sample-level mRNA to protein correlations deteriorated in seven samples with an average tumor content (TC) below 45% (Supplementary Fig S1A). Based on this cutoff, a total of nine samples with proteomics data (including 1 sample that lacked RNA and 1 sample that lacked both RNA and protein) were therefore excluded from further bioinformatic analyses. TMT11 multiplexes were linked using a pooled sample common reference to serve as a denominator for calculating peptide and phosphosite ratios (12). The common reference samples showed very strong correlations across multiplexes, indicating consistent data quality (Supplementary Fig S1B). For each qualified sample, DNA, RNA and protein level information was available for an average of 10,500 genes (Fig 1C) and phosphoproteomic analysis quantified ~27,000 phosphorylation sites in ~5,000 distinct phosphoproteins (Fig 1C). Comparable to previous CPTAC proteogenomic analyses, median per gene mRNA to protein correlation was 0.37 (14) (Supplementary Fig S1C). Genes with significant positive RNA-protein correlations were enriched for KEGG pathways involved in cellular respiration, and amino acid and lipid metabolism. Genes with lower correlations were enriched in pathways containing large protein complexes serving the spliceosome, replication, transcription, and pyrimidine metabolism (Supplementary Fig S1C). Consistent with previous observations, protein data significantly outperformed RNA data for co-expression-based gene function predictions (Supplementary Fig S1D) (12,15–17).
A pairwise analysis was also conducted using 14 cases with baseline high tumor content (out of 16 pairs) matched to a second high tumor content specimen collected 48–72 hours after treatment (only 13 pairs had RNA data, Fig1B). Whereas immune-related pathways were downregulated upon treatment at both the RNA and protein level, cell cycle and metabolic pathways (except glycolysis) were significantly upregulated specifically at the protein level (Fig. 1D, Supplementary Table S4). Induction of DNA replication and repair pathways linked to the cell cycle was observed, likely in response to genotoxic stress triggered by chemotherapy exposure (18). This observation was also present in the phosphorylation site data (Supplementary Fig S1E). Sets of phosphosites induced by treatment had correlation with those established to be induced by nocodazole and ionizing radiation treatment, which is logical in the setting of taxotere and carboplatin exposure. Increases in phosphorylation were also detected for targets of the cell cycle and DNA damage kinases CDK1, CDK2, and ATM (Supplementary Fig S1E).
Exploration of proteogenomic pathway signatures and response to chemotherapy.
Primary study endpoints were pCR and residual cancer burden (RCB) in the surgical specimen where 0 indicates pCR and I-III indicate increasing levels of residual disease (19). PAM50 intrinsic subtype (20), TNBCtype (21), and racial categories lacked association with pCR, as did other cohort-specific clinical metadata (Supplementary Fig S1F). Expected associations for pCR with germline mutations in the homologous recombination (HR) genes BRCA1/2 or PALB2 (22) or with HR deficiency-associated COSMIC signature 3 were also not observed (23,24). These negative findings emphasize the limitations of our study in terms of sample size. However, an elevated COSMIC Signature 6 score, indicating a mismatch repair defect, (23,24) was associated with high RCB (II or III) (p=0.03, Supplementary Fig S1G). Gene set enrichment analysis of proteogenomic features (Supplementary Table S5) that differed by pCR status indicated upregulation of MSigDB Hallmark metabolic pathways including oxidative phosphorylation, fatty-acid metabolism, and adipogenesis in samples without pCR. These associations were observed in the proteomic data but not at the mRNA level (Fig 1E, Supplementary Table S6). In contrast, immune signaling (interferon alpha and gamma response) and cell cycle (G2M checkpoint and E2F and MYC target) pathways were elevated in pCR cases in both the proteomic and transcriptomic datasets (Fig 1E). Enrichment analysis of differential phosphorylation sites (PTM-SEA) (25) logically demonstrated elevated phosphoproteome-driven signatures in samples from pCR cases for treatment with inhibitors that generate DNA damage (etoposide, hydroxyurea and ionizing radiation) (Fig 1F). Elevated MARK2 target sites were enriched in non-pCR tumors (Fig 1F), corroborating prior evidence for higher MARK2 levels in cisplatin resistance in other cancer types (26,27). Consistent with significantly elevated cell cycle pathways observed in pCR samples in the RNA and protein data, CDK1, 2, and 7 and CDC7 target phosphosites were also significantly higher in pCR samples (Fig 1F). Further sample-wise investigation of cell-cycle proteogenomic features revealed that multi-gene proliferation scores (MGPS), single-sample Gene Set Enrichment Analysis (ssGSEA) and PTM-SEA scores for cell cycle-related pathways and cyclin dependent kinases were higher in pCR but were variable in non-pCR (Fig. S2A). Of note, a subset of non-pCR samples had elevated CDK4 activity and Rb phosphorylation (highlighted in box in Fig. S2A), and Rb phosphorylation was marginally higher in non-pCR tumors (Fig. S2B). To study the therapeutic significance of these findings, TNBC cell lines from the DepMap resource were explored (www.depmap.org). In this database higher Rb protein was associated with reduced carboplatin response but enhanced CDK4/6 inhibitor response (Supplementary Fig S2C).
Immune pathways and response to chemotherapy.
Since interferon alpha and gamma response signatures were elevated in samples from pCR cases, signals from the immune microenvironment were further explored (Fig 2A). Protein-derived immune stimulatory scores, previously found to be well-correlated with immune infiltration (14), as well as PD-L1 RNA, protein, and phosphorylation levels, were significantly higher in pCR-associated samples (Fig. 2B). Non-synonymous mutation load was associated with neither pCR (Wilcoxon rank sum p=0.57, median for pCR=77, median for non-pCR=78) nor immune scores (Spearman rho=−0.17, p=0.25), suggesting increased mutation burden was not a strong determinant of immune infiltration in this TNBC data set. Rather, immune scores were significantly anti-correlated with chromosomal instability score (CIN) (Spearman Rho=−0.61, p=6.2e-6; Fig 2C). Both PD-L1 protein and phosphoprotein levels significantly correlated with PD-L1 IHC (Fig. 2D-E). Similar correlations were also observed between PD-L1 RNA and IHC (Supplementary Fig S2D). Representative IHC images for high and low PD-L1 staining are shown in Supplementary Fig S2E and F, respectively.
Fig 2: Proteogenomic features associated with pCR in TNBC tumors.
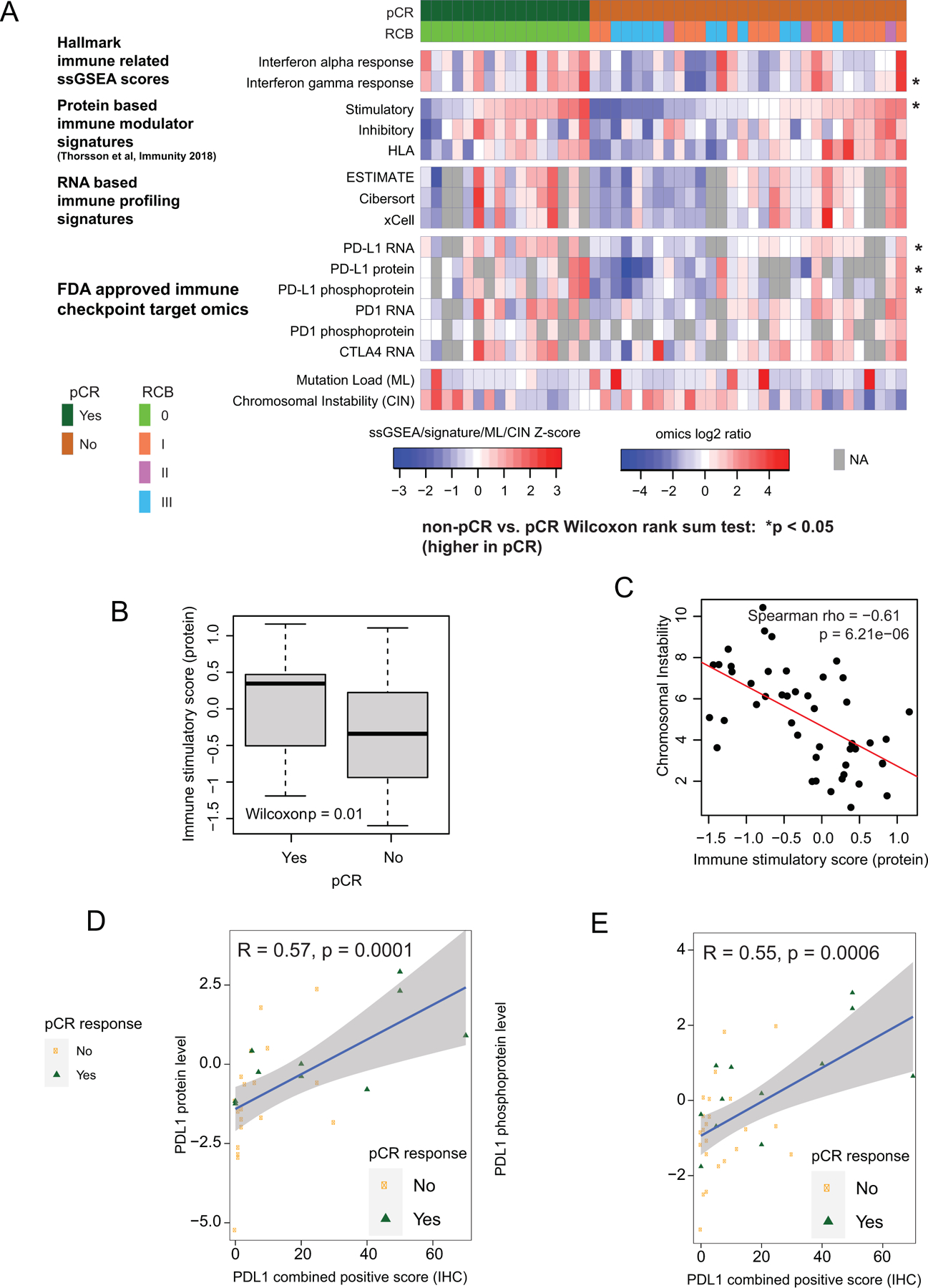
A) Proteogenomic features associated with the immune microenvironment are elevated in pCR tumors relative to non-pCR tumors. Heatmap shows protein-based Hallmark single sample GSEA (ssGSEA) scores, protein-based immune modulator (IM) scores, RNA-based immune profiles, and proteogenomic features for immune checkpoint genes that are targets of FDA-approved inhibitors. Within each group (pCR and non-pCR), samples are ordered by increasing immune stimulatory score. Asterisks indicate p<0.05 by Wilcoxon rank sum test comparing non-pCR to pCR tumors.
B) The protein based immune stimulatory score is significantly higher in pCR tumors than non-pCR tumors (p=0.01, Wilcoxon rank sum test). Boxplots show interquartile range (IQR) with median marked in center. Whiskers indicate 1.5x IQR.
C) The immune stimulatory score is negatively correlated to chromosomal instability index (CIN; Spearman Rho = −0.612, p = 6.2e-6). Scatter plot shows immune stimulatory score on the x-axis and CIN on the y-axis.
D-E) Scatter plots showing correlation between PDL1 IHC levels with PDL1 protein (D) and phosphoprotein levels (E). pCR cases are shown in green and non-pCR in orange.
Metabolic pathway analysis and response to chemotherapy.
As noted above, (Fig 1D) metabolic pathway enrichment appeared specific to proteomic data (with false discovery correction). Both GSEA (Fig 1E) and ssGSEA analyses showed differential metabolic pathways including oxidative phosphorylation, adipogenesis, fatty acid metabolism, as well as glycolysis were significantly higher in pre-treatment tumors without subsequent pCR (Fig 3A). Further analyses at the individual protein level identified many chemotherapy-resistance associated metabolic proteins, such as those directly involved in the tricarboxylic acid (TCA) cycle (ACO2, FH, MDH2, SUCLG1, SUCLG2, PDP1, DLAT), the electron transport chain (SDHC, UQCR10), fatty acid metabolism (CRAT, ACADS, ACAT1, DECR1, ECHS1, HADHB), and amino acid catabolism (ALDH6A1, HMGCL, DBT, BCKDHB) (Fig 3B). While pCR-associated metabolic pathway scores were more robust at the proteomic data than transcriptomic data, this did not equate to lack of mRNA and protein correlation for all metabolism gene products associated with non-pCR. A subset (29 out of 43) from the relevant Hallmark metabolic pathways showed sufficient protein/mRNA correlation to allow independent validation of metabolic gene expression associations with pCR at the mRNA level (Fig 3B) in the BrighTNess trial dataset (9). In this study, patients on arms A and B received combination treatment with carboplatin and paclitaxel plus/minus veliparib (addition of which did not affect outcomes), as well as subsequent treatment with doxorubicin and cyclophosphamide (9). Baseline RNA expression data for the subset of metabolism-associated resistance genes with high mRNA-protein corrrelation were for association with pCR status on these two arms combined. Geometric mean metabolic scores were significantly higher for non-pCR cases as compared to pCR cases (Wilcoxon rank sum test, p=0.003; N=359 Supplementary Fig S3). Additionally, increasing metabolic scores were observed as the RCB category increased (Kruskal-Wallis test, p= 0.0024; Fig 3C).
Fig 3: Proteogenomic features associated with lack of pCR and that are altered upon treatment in TNBC tumors.
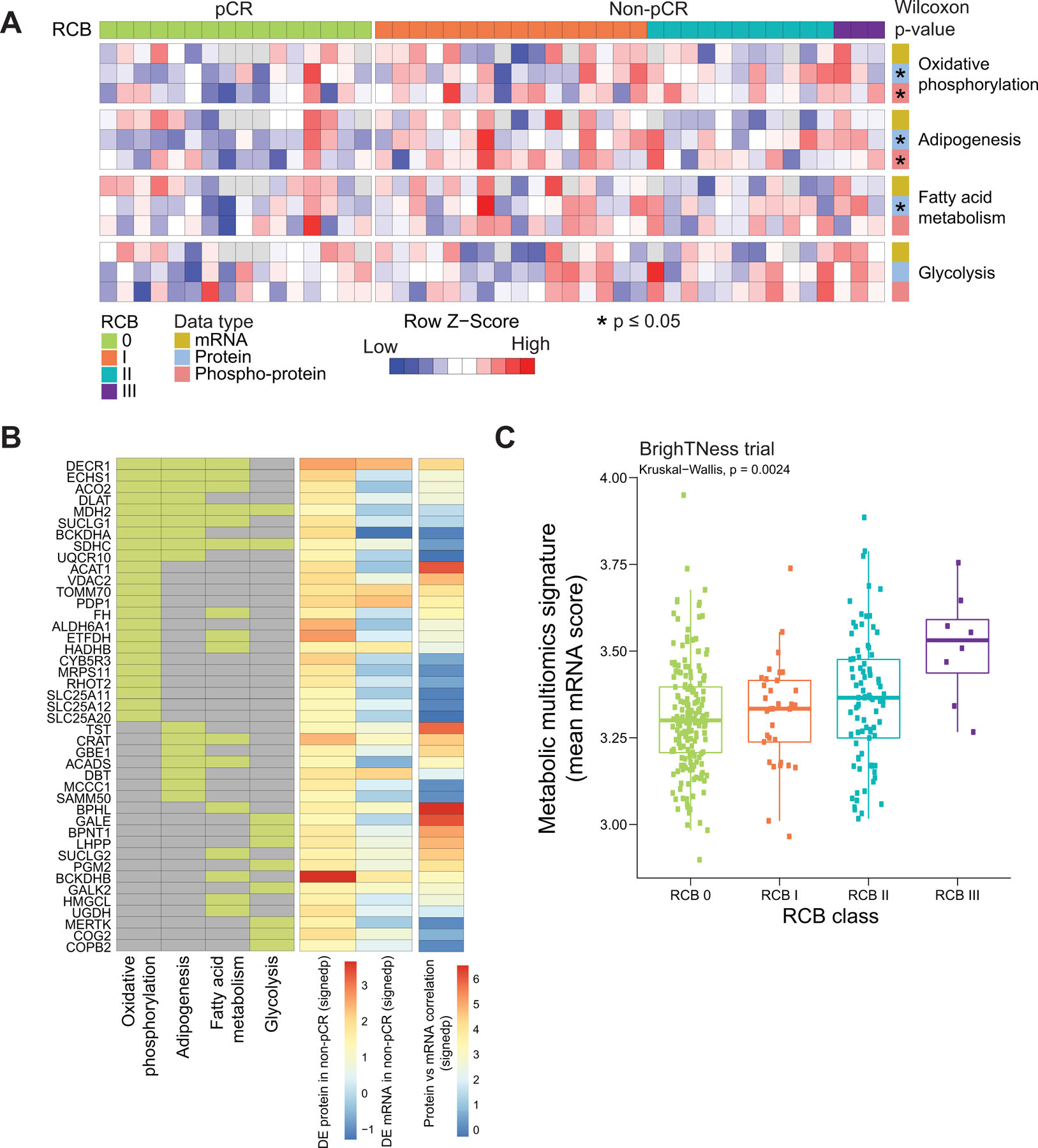
A) Heatmap showing single-sample GSEA NES for metabolic Hallmark pathways that are significantly higher in non-pCR cases, arranged by RCB0 (pCR) and RCBI/II/III (non-pCR). Shown are the 4 pathways that showed significant enrichment at either the RNA or protein level in Fig. 1E. Single sample pathway enrichment scores were assessed at the level of mRNA (yellow), protein (blue) and phospho-protein (red). The Wilcoxon rank sum test was used to compare scores for non-pCR vs pCR scores, * p<0.05.
B) Membership of differentially regulated proteins to pathways highlighted in A. Proteins (rows) belonging to a given pathway (columns) are shown in light green. The differential expression at protein and mRNA levels for each gene along with mRNA-protein correlation scores are shown as signed -log10 p-value (signedp).
C) A multi-omics metabolic gene signature derived for genes that are correlated at mRNA and protein level was further investigated in patients treated with carboplatin and paclitaxel in the BrighTNess clinical trial (Treatment arms A and B) for which RNA-seq data was available. The mean mRNA expression score for this signature was significantly higher in higher RCB tumors.
Proteogenomic analyses of copy number alteration reveals novel chemotherapy response biomarkers.
The somatic landscape of TNBC is dominated by recurrent copy number alterations (CNA) (28), however the significance of many recurrent CNA events remains unclear, because typically many genes are involved in larger scale chromosomal deletions and rearrangements (29). A typical pattern of CNA for TNBC was observed in the discovery data (Supplementary Fig S4A). To explore whether chemotherapy response correlates with the expression of genes within specific chromosomal locations involved in recurrent can gains or losses, GSEA was utilized to statistically evaluate relationships between cytoband location and upregulated or downregulated gene expression at the mRNA or protein level (Fig 4A). Individual gene expression ranks was derived from the non-pCR versus pCR dichotomy using a signed -log 10 p-value derived from the Wilcoxon test were used as the input for this analysis. This unbiased prioritization demonstrated that expression of gene products from the 8q21.3 (amplified) and 19q13.31–33 (deleted) cytobands were elevated and suppressed, respectively, in non-pCR versus pCR tumors (Fig 4B). Four genes located at 8q21.3, RMDN1, CPNE3, DECR1, and OTUD6B, showed higher mRNA and protein expression in non-pCR tumors (Supplementary Fig S4B). In addition, RIPK2, which may mediate metastasis in advanced breast cancer (28), also located on 8q21.3, was significantly higher in non-pCR tumors, but only at the protein level. Similarly, four genes located on 19q13.31–33, LIG1, PPP5C, BCL3, and NOSIP, showed lower mRNA and protein expression in non-pCR tumors (Fig 4C). Both mRNA and protein level expression from these coordinately downregulated genes were confirmed to be suppressed in association with single copy LIG1 loss (GISTIC = −1) status in a subset of non-pCR-associated samples (Supplementary Fig S4C). Hallmark pathway GSEA analysis of the genes on cytoband 19q13.31–33 showed enrichment in the DNA damage repair (DDR) pathway with LIG1, XRCC1, POLD1 and ERCC1 comprising the leading-edge genes (Fig 4D). LIG1 showed the strongest association with treatment response at the protein level, followed by POLD1 (Fig 4D).
Fig 4: Discovery of DNA repair and replication components enriched in non-pCR TNBC tumors.
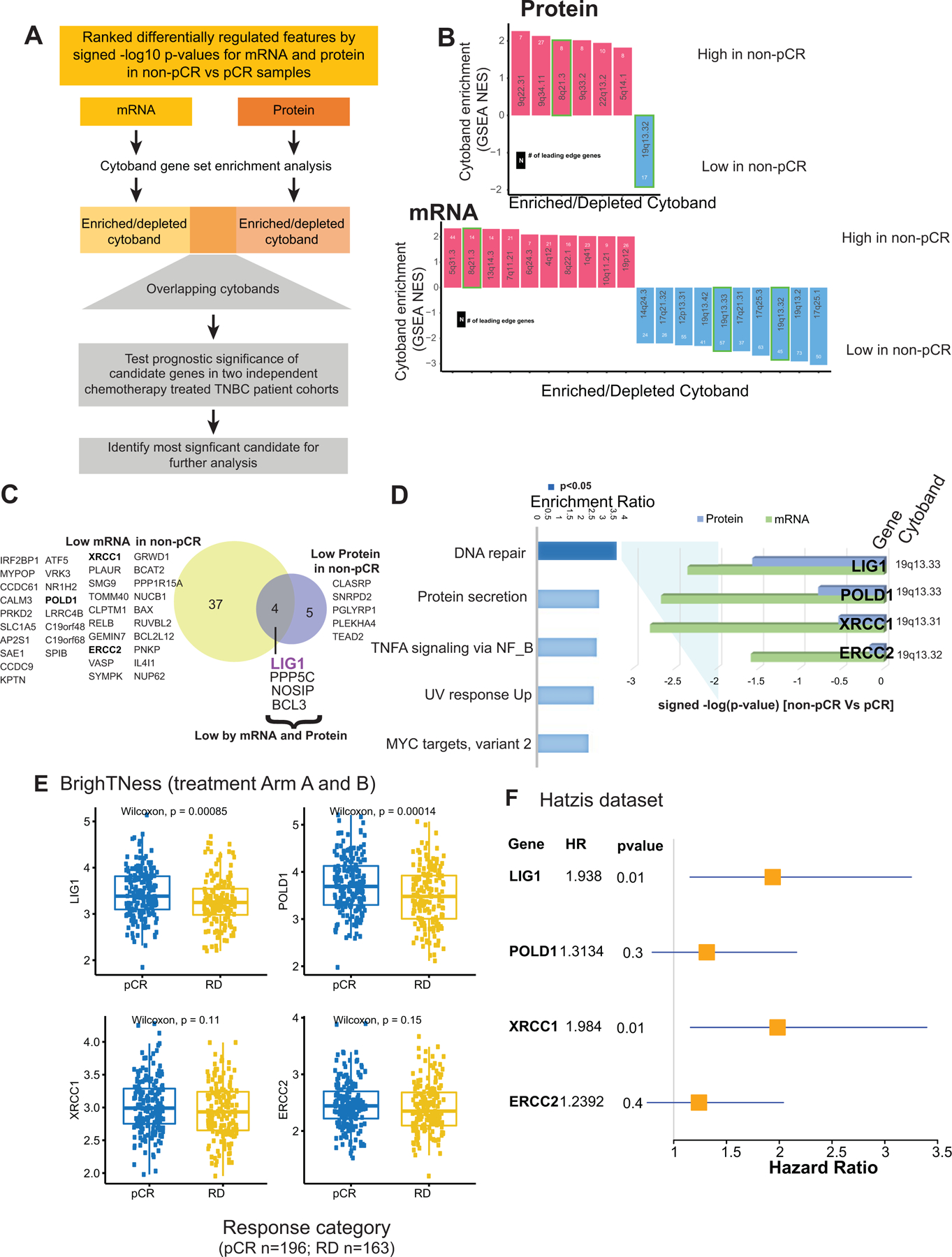
A) p=Cytobands enriched in genes differentially expressed between non-pCR and pCR for both mRNA and protein. To identify up or downregulated features over-represented in certain cytobands within the chromosome, GSEA was used to identify regions from chromosomal location databases enriched with differential genes (GSEA input was ranked expression list (signed -log10 pvalue) from Wilcoxon rank sum tests). Overrepresented cytobands that were either enriched or depleted using differentially expressed mRNA and protein are indicated in B, and the overlapping sets were used for further analysis. Genes downregulated in non-pCR samples correspond to cytoband 19q13.31–33 is indicated in C.
B) Plot showing significantly enriched or depleted cytobands obtained by running differential mRNA and protein ranked lists through GSEA.
C) Venn-diagram showing differential (non-pCR vs. pCR) mRNA and proteins located on cytoband 19q13.3.
D) Over-representation analysis (ORA) shows that differential 19q13.31–33 genes are enriched with Hallmark DNA repair pathway genes. Downregulation of these DNA repair genes at mRNA and protein level in non-pCR cases is shown in the bar chart on the right as signed -log10 p-values from Wilcoxon rank sum tests.
E) Boxplot showing comparing RNA expression of DNA-repair genes located on 19q13.31–33 in the previously published BrighTNess clinical trial (Treatment Arm A and B), where patients were treated with carboplatin and paclitaxel. The Wilcoxon rank sum test was used to compare residual disease (RD) cases to pCR cases.
F) Forest plot showing hazard ratios (HR) and p-values for metastasis free survival associated with LIG1, POLD1, XRCC1 and ERCC2. HR is based on categorizing samples using median expression cutoff for each gene in the Hatzis dataset.
To determine whether these observations were reproducible in other data sets, the association of LIG1, XRCC1, POLD1 and ERCC1 with pCR and RCB was evaluated at the mRNA level in the BrighTNess trial. For this analysis the two carboplatin- and paclitaxel-containing arms were combined to parallel the docetaxel and carboplatin treatment in the discovery data set (9). LIG1 and POLD1 were confirmed to be significantly downregulated in baseline tumor samples from patients who experienced residual disease (Fig 4E). Similar differences were not observed in the paclitaxel-only arm, although the sample size was smaller (treatment arm C, p>0.05) (Supplementary Fig S4D). Low RNA expression levels for LIG1 and XRCC1 were also significantly associated with poor metastasis-free survival in the TNBC subset of another neoadjuvant chemotherapy-treated patient cohort (30) (Fig 4F and S4E). Finally, a trial where a modest number of patients were treated with single-agent cisplatin neoadjuvant therapy was interrogated (31). Consistent with the other data sets, LIG1 mRNA levels were significantly lower in samples associated with stable or progressive disease (SD+PD) as opposed to samples associated with a complete or partial response (CR+PR) (Supplementary Fig S4F). Of the four DDR genes located within 19q13.31–33, LIG1 expression was the most consistently associated with chemotherapy resistance and poor metastasis-free survival across datasets (Fig 4E, F, S4E-F).
Molecular features of TNBCs harboring LIG1 deletion
The associations between LIG1 deletion and/or reduced expression with tumor pathophysiological features were further investigated in the discovery set. Low LIG1 copy number level (GISTIC = −1) were observed in eight of the thirty-one (~26%) tumors without pCR (Fig 5). LIG1 copy number log ratios were strongly and positively correlated with the level of both LIG1 mRNA (Pearson, R = 0.67, p = 2.8e−06) and LIG1 protein (R = 0.55, p = 8.2e−05) (Supplementary Fig S5A-B). At the genomic level, COSMIC HRD Signature 3 was lower in tumors with LIG1 loss (T test, p=0.01) (Fig 5). In contrast, tumors harboring LIG1 loss exhibited significantly higher chromosomal instability (CIN) scores (T test, p=0.0003, Fig 5). While no significant differences were observed in immune stimulatory scores when LIG1 loss tumors were compared other tumors, tumors with LIG1 loss had lower immune stimulatory (IM) scores when compared to tumors that were associated with pCR (p=0.01) (Supplementary Fig S5C). At the level of phosphosite expression-based PTM-SEA (25) analysis, the IL33 pathway was significantly down-regulated in LIG1 loss tumors (Supplementary Fig S5D-E). Tumors with LIG1 loss also had significantly higher protein-based proliferation scores (p-MGPS), Wilcoxon p=0.004, Fig 5) as well as upregulation of CDK1/2 activity (Supplementary Fig S5D) in PTM-SEA analysis of differential phosphosites (25), supporting increased cell cycle activity (FDR p<0.05). Collectively these results suggest that loss of LIG1 is associated with a constellation of poor prognosis features including higher proliferation rates, a less active immune microenvironment and higher copy number instability. Furthermore, when the phosphoproteomic data was examined, signatures of EGFR (gefitinib) and PI3K (wortmannin) perturbations were significantly enriched in LIG1 loss tumors but in a negative direction (Supplementary Fig S5D-E). Since LIG1 loss tumors have suppressed EGFR and PI3K signaling they may be less responsive to EGFR, PI3K or AKT inhibition.
Fig 5: Proteogenomic features associated with LIG1.
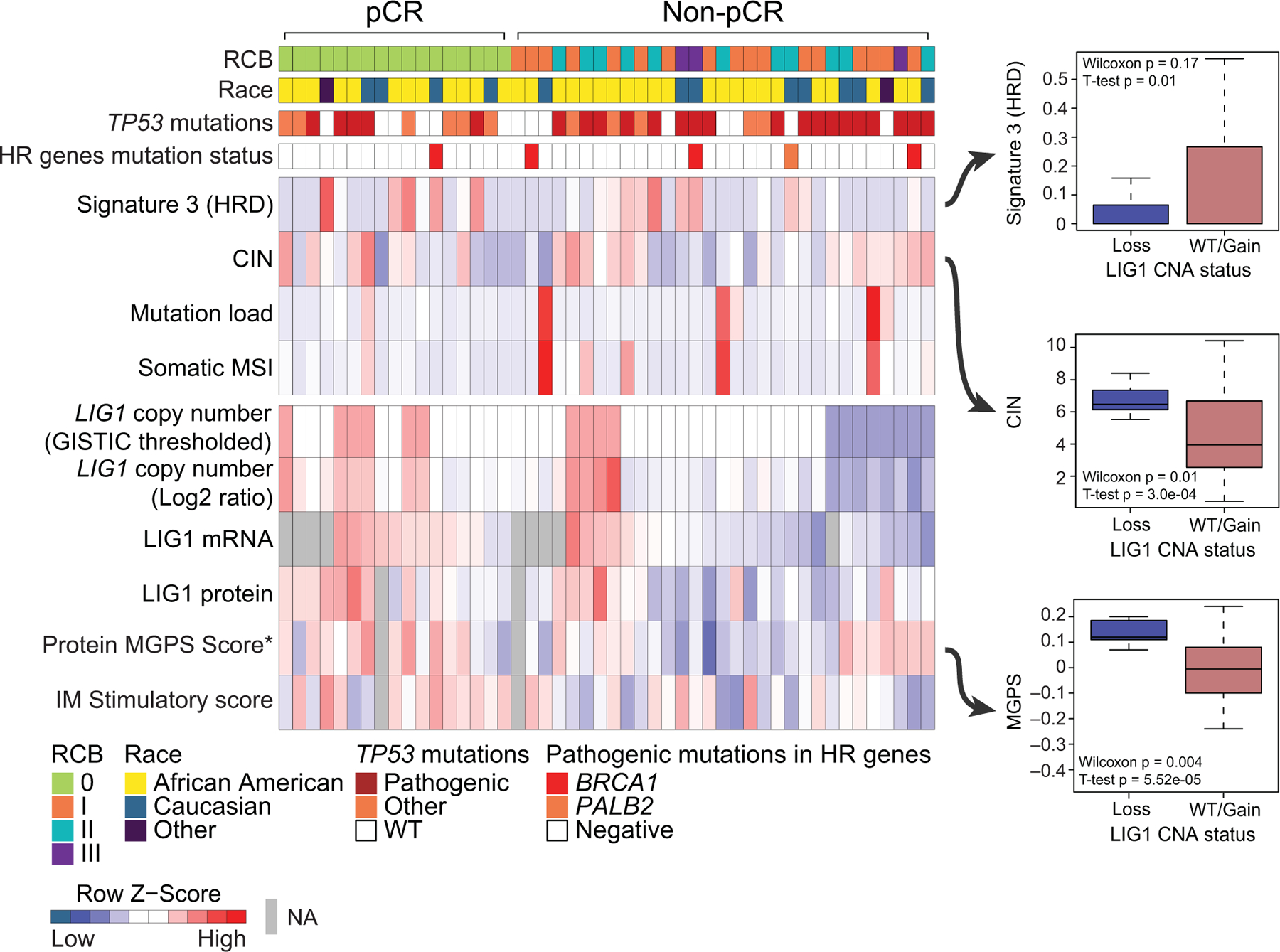
Heatmap showing copy number, mRNA and protein levels of LIG1, which are significantly (Wilcoxon rank sum test) lower (blue) in non-pCR tumors. Corresponding boxplots show that tumors with low level copy loss of LIG1 (GISTIC = −1, likely single copy number loss) display significantly higher chromosomal instability and multi-gene proliferation scores and significantly lower Signature 3 (COSMIC mutational signature associated with homologous recombination defect) than tumors that that are WT for or show gain of CNA (GISTIC >= 0). T-tests and wilcoxon rank sum tests were used to compare LIG1 loss cases to LIG1 intact (WT/Gain) cases.
LIG1 and chemotherapy response in model systems.
When chemotherapy resistance biomarkers are identified, the question arises as to whether the biomarker relationship is drug selective. Model systems can be useful in this regard because patients almost always receive multiple drugs. Another concern is whether a biomarker is associated with intrinsic resistance, acquired resistance or both. Patient data suggested higher frequency of LIG1 copy loss in metastatic disease (Supplementary Fig S6A). Patterns of 19q13.31–33 loss during malignant progression were therefore explored using three orthotopic PDX models generated from a single patient on discovery trial NCT02544987. WHIM68 grew from the pretreated breast primary, WHIM74 from a surgical sample accrued after five months of neoadjuvant carboplatin and docetaxel, and WHIM75 from a liver metastasis that appeared one year after treatment initiation. Proteogenomic analysis revealed progressive loss of LIG1 at the copy number, mRNA, and protein levels as the tumor progressed to a chemotherapy resistant state (Fig 6A). Progressive loss of LIG1 protein was confirmed by western blotting (Fig 6A and Supplementary Fig S6B) along with similar reductions of POLD1 and XRCC1 protein expression. Consistent with the progressive loss of chemotherapy sensitivity observed clinically, WHIM68, which expressed the highest LIG1 level, was sensitive to carboplatin, while WHIM74 and 75 were progressively and remarkably less sensitive (Fig 6B, Supplementary Table S7). Interestingly, this relationship was not as marked with docetaxel treatment (Supplementary Fig S6C, Supplementary Table S7). Of note, a BRCA2 loss of function somatic mutation was present in the baseline PDX (WHIM68) but was undetectable in the two PDX derived from after treatment biopsy, suggesting treatment-induced clonal selection, i.e., as the patient was treated, the BRCA2 mutant clone regressed, and a LIG1-deleted clone expanded. To further assess the potential association between LIG1 loss and selective carboplatin insensitivity, a large TNBC PDX cohort from the NCI PDXnet program was examined (32). LIG1 mRNA levels were significantly lower in PDX that failed to demonstrate a complete response to carboplatin (Fig 6C), and this relationship was not significant for docetaxel treatment (Supplementary Fig S6D). A second independent TNBC PDX samples with short-term in vitro treatment with multiple different oncology drugs was also examined (33, BCaPE database). This dataset demonstrated that LIG1 copy number loss was uniquely correlated with carboplatin resistance among over 100 drugs tested (Fig 6D).
Fig 6: LIG1 association with advanced TNBC disease in preclinical models.
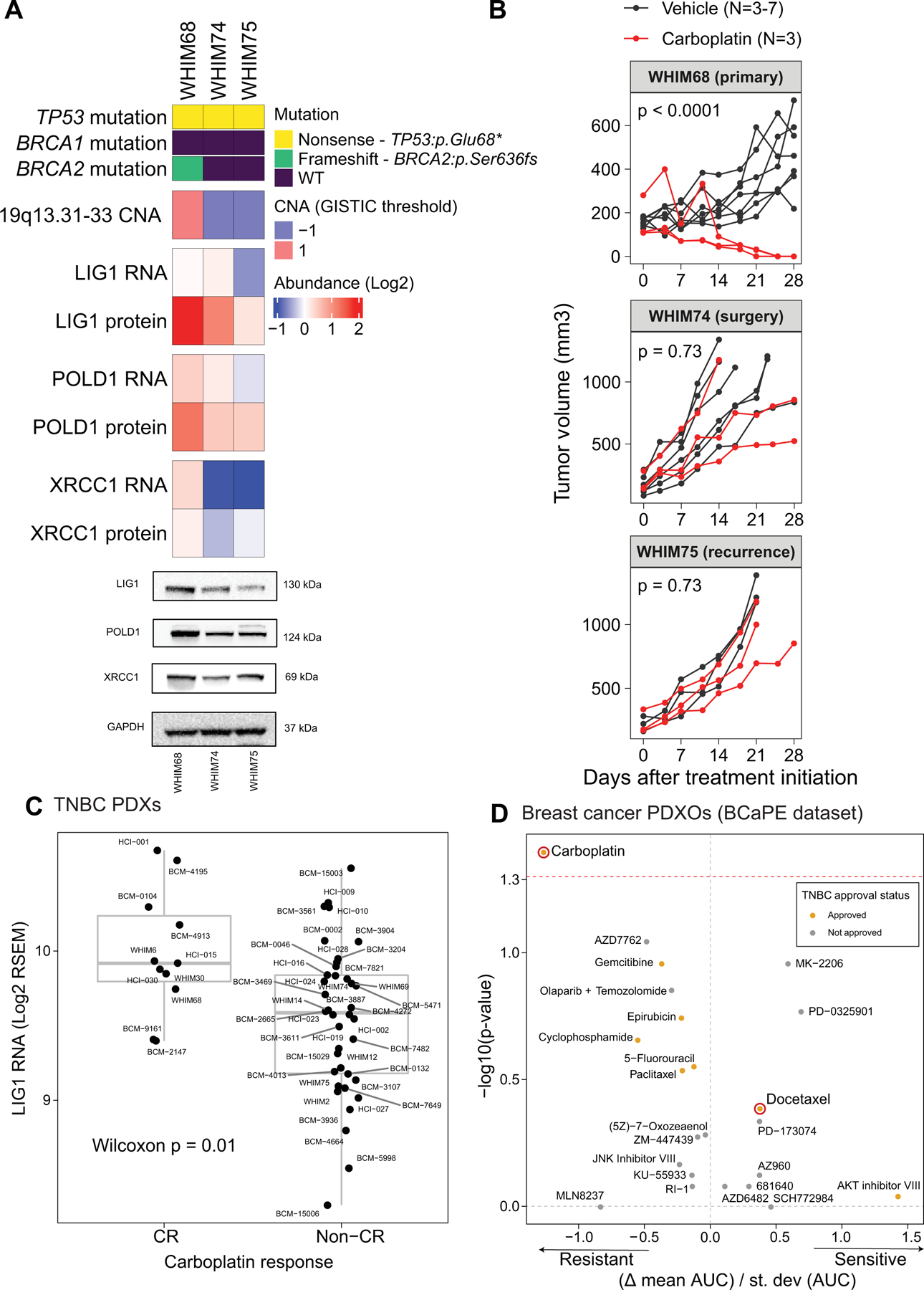
A) Proteogenomic status of LIG1, POLD1 and XRCC1 in three PDX models derived from longitudinal biopsies from the same TNBC patient prior to any treatment (WHIM68), at the time of surgery after completing 5 months of neoadjuvant carboplatin and docetaxel (WHIM74), and from a liver metastasis one year after treatment initiation (WHIM75). Mutation and copy number data were derived from WES and RNA from RNAseq, and protein data was obtained from TMT proteomics generated by this current study. Bottom panels show representative western blots from 3 biological repeats for LIG1, POLD1 and XRCC1 protein levels. GAPDH is used as loading control.
B) Tumor volume was measured in three PDX models. Black and red lines indicate changes in tumor volume in PDXs treated with vehicle and carboplatin respectively. WHIM68, with highest LIG1 protein levels, was most sensitive to carboplatin while WHIM74 and 75, which displayed progressive LIG1 loss at the copy number, mRNA, and protein levels, were insensitive to carboplatin treatment. P-values derived from a general linear model within each PDX were computed using estimated mean log2 fold changes in tumor volume at Day 28 vs Day 0 for each treatment arm.
C) Boxplots showing LIG1 mRNA levels in TNBCs PDXs categorized into complete response (CR) and non-CR groups. After 4 weeks of carboplatin treatment, CR was defined as PDXs with non-palpable tumors, and non-CR was defined as PDXs with residual tumors with measurable dimensions. The Wilcoxon rank sum test was used to compare the two groups.
D) Association between LIG1 copy number loss and treatment response in patient-derived xenograft organoids (PDXOs) obtained from the BCaPE database. Carboplatin and docetaxel are highlighted in red.
LIG1 copy number loss is associated with poor progression-free survival and CIN across multiple cancer types
Gene copy number analysis of tumors characterized by TCGA demonstrated that LIG1 single copy loss is present in other cancer types. In the TCGA “pan-cancer” data set, LIG1 heterozygous loss was associated with poor progression-free survival (PFS) (Fig 7A, p<0.0001), significantly higher CIN (Fraction genome altered, Fig 7B), and lower signature 3 scores (suggesting proficient homologous recombination, Fig 7C). Cancer types driving these relationships include endometrial carcinoma (HR=2.23, p=0.02), head & neck squamous cell carcinoma (HR=1.46, p=0.03), prostate adenocarcinoma (HR=2.07, p=0.02), colon adenocarcinoma (HR=1.75, p=0.03) and most convincingly renal papillary cell carcinoma (HR=4, p= 0.0001) (Fig 7D). Despite a marginal association between PFS and LIG1 loss in testicular germ cell tumors (TGCT), the seminoma subtype, which demonstrates exquisite sensitivity to carboplatin (34), displayed no cases of LIG1 loss (Supplementary Fig S7A). Higher CIN (fraction genome altered) was observed in association with LIG1 loss in several other cancers (Fig 7E (TCGA cohorts) and Supplementary Fig S7B (CPTAC cohorts)).
Fig 7: Pan-cancer analysis of LIG1 loss.
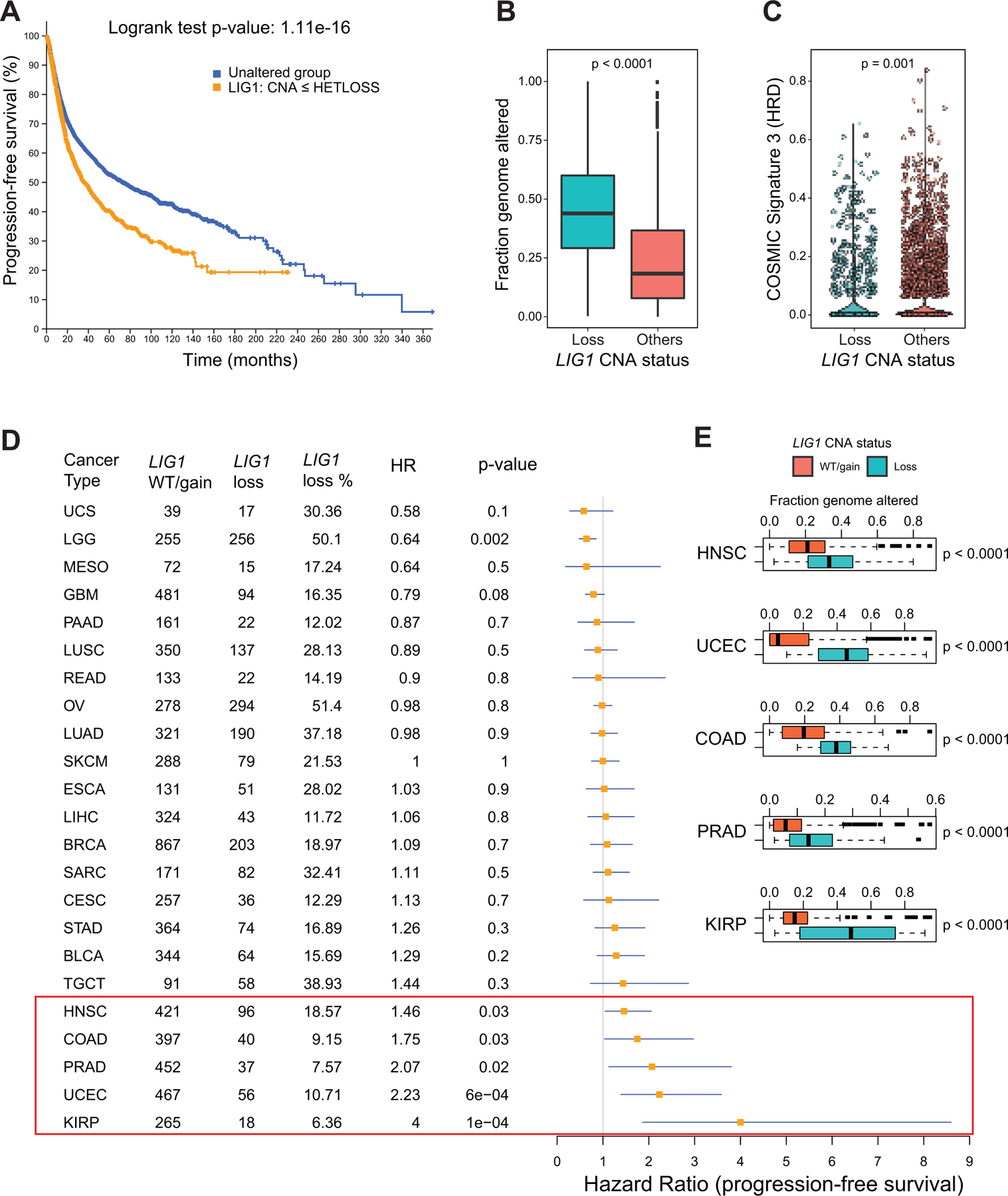
A) Kaplan-Meier curve showing significantly reduced (Log-rank P value) progression free survival (PFS) for tumors with single copy loss of LIG1 (HETLOSS, GISTIC <=−1, indicated in orange) in the TCGA pan-cancer cohort.
B) Boxplot showing higher fraction genome altered (FGA) in tumors with LIG1 copy number loss tumors (shown in teal) relative to tumors that were LIG1 wild-type or displayed LIG1 gain (shown in orange).
C) Violin plot showing significantly lower (Wilcoxon rank sum test) COSMIC Signature 3 scores in LIG1 loss tumors (shown in teal).
D) Forest plot showing impact of LIG1 copy number loss on PFS by cancer type along with LIG1 WT/gain/loss frequency, HR and corresponding p-value.
E) Boxplot showing significantly higher (by Wilcoxon rank sum test) FGA (representing chromosomal instability) in tumors that had LIG1 copy number loss versus tumors with either wild-type LIG1 or with LIG1 copy number gain. Shown are the only 5 cancers (HNSCC, UCEC, COAD, PRAD and KIRP) that displayed significant association between LIG1 loss and adverse prognosis
Discussion
The absence of a baseline pCR predictor is a persistent unmet need for the precision treatment of breast cancer. Patients without pCR suffer prolonged exposures to toxic and ineffective treatment and therefore do not receive alternative treatment soon enough. Additionally, PD-L1 IHC assays have failed to predict the benefit of immune checkpoint blockade in TNBC (35). Thus, alternative biomarkers for antitumor immunity are required. The data presented suggest that integrated proteogenomic characterization provides more extensive information on the immune microenvironment that could be used to complement PDL1 IHC. While a TMT-based proteomic assay for PD-L1 would not be practical, targeted proteomic assays optimized for quantitative measurement using heavy isotope labelled peptides for multiple immune response components is an efficient and low-cost approach that could complement IHC (36). We also observed a novel association for baseline oxidative phosphorylation and fatty acid metabolism gene products with chemo-resistance in TNBC. These findings are supported by functional studies in TNBC model systems demonstrating a role for oxidative phosphorylation and fatty acid metabolism as drivers of TNBC chemo-resistance (37,38). In fact, fatty acid synthase inhibition using the proton pump inhibitor omeprazole in combination with neoadjuvant chemotherapy in TNBC patients is currently being evaluated in a phase II trial (NCT02595372). pCR prediction models could be therefore strengthened by the inclusion of protein level analysis of these pathways. The cellular origin of these resistance-associated metabolic signals is unresolved. An additional possibility is immunosuppressive tumor associated macrophages with high lipid content (39). A third class of potential pCR predictors are G2M checkpoint components, E2F regulators and MYC target pathways. For example, TNBC tumors with high/intact Rb protein and phosphorylation levels have lower pCR rates and lower levels of proliferation and E2F target gene expression than tumors with loss of Rb protein (14). CDK4/6 or CDK2 inhibitors could therefore be an alternative treatment for RB intact TNBC. Finally, proteomic analysis clearly assists in the prioritization of genomic chromosomal alterations associated with pCR status, exemplified herein by the identification of LIG1 as a TNBC chemotherapy resistance and multi-cancer type poor prognosis marker. The finding from preclinical models that LIG1 loss is a selective biomarker for carboplatin resistance is provocative. The use of carboplatin adds toxicity to an already toxic anthracycline-based regimen and could potentially be avoided in LIG1-depleted tumors.
Regarding LIG1 loss as a potential pathogenetic event in TNBC, there are already mechanistic studies of LIG1 loss that support this hypothesis. LIG1 encodes an ATP-dependent DNA ligase that seals DNA nicks during replication, recombination, and a variety of DNA damage responses (40). Of the three DNA ligases in the human genome (LIG1, 3 and 4), LIG1 is the main enzyme responsible for ligating Okazaki fragments during lagging-strand synthesis at the replication fork during S-phase (41–43). LIG1 also ligates single-stranded or double-stranded DNA breaks in various DNA damage repair pathways including long-patch base-excision repair, nucleotide-excision repair, and alternative non-homologous end-joining repair (44,45). A phenotype for LIG1 deficiency in humans was first identified in an immunodeficient patient with homozygous germline hypomorphic LIG1 alleles causing impaired Okazaki fragment ligation (46). Insufficient LIG1 activity results in the accumulation of replication intermediates that cause single-stranded and double-stranded breaks (DSB) (47,48), ultimately leading to reduced genome integrity. In transgenic mice hypomorphic LIG1 alleles were associated with high susceptibility to cancer formation (49). However, the relevance of these observations can be challenged in the setting of TNBC, because single copy LIG1 loss observed in our studies may not produce sufficient functional deficiency to generate a phenotype. However, co-deletion of LIG1, POLD1 and XRCC1 on 19q13.31–33 may produce a hemizygous compound deficiency phenotype since all three genes serve lagging strand synthesis. XRCC1 is particularly noteworthy because LIG3/XRCC1 provides a backup pathway for LIG1 mediated DNA ligation during DNA repair and lagging strand DNA synthesis (50).
The presence of LIG1 loss was found to be orthogonal to the HRD mutational signature 3. Consequently, LIG1 cells may be required to be proficient in DSB repair, i.e. HRD and LIG1 loss are orthogonal routes to TNBC pathogenesis and this potentially could explain the correlation with carboplatin insensitivity. The PDX study (Fig 6) hints at this, as the model derived from the pretreatment sample (WHIM68) had a BRCA2 frameshift mutation and no LIG1 loss and the subsequent carboplatin resistant lines (WHIM74 and 75) had lost the BRCA2 mutation and gained a LIG1 hemizygous deletion. It remains unclear why LIG1 loss is so strongly associated with chromosomal instability across cancer types and mechanistic studies connecting these events are an important next step. However, cells that enter mitosis with unrepaired lagging strands are at risk for chromosomal breakage, illicit chromosomal fusion events and aneuploidy.
In conclusion, our findings emphasize the potential of microscaled proteogenomic approaches for the investigation of cancer treatment resistance. Follow-up mechanistic studies are clearly warranted, not just for LIG1-related biology but also, for example, the role of lipid-related metabolic signatures in chemotherapy resistance. However, lack of complete mechanistic insight does not diminish the clinical importance of novel chemotherapy drug-selective predictive biomarkers in a setting where a genomic approach or transcriptomic analyses have yet to produce actionable models.
Methods
Clinical sample collection:
Eligible patients for the two clinical trials (NCT02547987 and NCT02124902) included pre or post-menopausal women at least 18 years old with clinical stages II/III ER negative and HER2 negative (0 or 1 + by IHC or FISH negative) invasive breast cancer. The study was approved by the IRB at both participating sites, WashU and BCM, and written informed consent from the patients was obtained. The studies were conducted in accordance with recognized ethical guidelines and followed the Declaration of Helsinki and Good Clinical Practice guidelines. All patients were uniformly treated (without randomization or blinding) with neoadjuvant intravenous docetaxel 75 mg/m2 and carboplatin every 21 days for 6 cycles with granulocyte colony-stimulating factor support (13). Research tumor biopsies for correlative studies were obtained at baseline prior to chemotherapy and on cycle 1 day 3 (C1D3). On-treatment biopsy on C1D3 and biopsy at time of relapse were optional. Details of the clinical cohort have been recently published (13). Treatment response information was provided by clinical teams associated with these trials and residual cancer burden was calculated using RCB calculator (http://www3.mdanderson.org/app/medcalc/index.cfm?pagename=jsconvert3).
Immunohistochemistry:
For immunohistochemistry (IHC), cut tissue sections (5mm) on charged glass slides were baked for 10–12 hours at 58oC in a dry slide incubator, deparaffinized in xylene and rehydrated via an ethanol step gradient. The IHC slides were stained for CD3 and PDL1. Pathology slide scoring was performed using established professional guidelines for TNBC, when appropriate. All immunohistochemistry results were evaluated against positive and negative tissue controls. See Supplementary Data and Methods for more details.
Genomic analysis:
Whole exome sequencing (WES):
Tumor DNA was extracted from fresh-frozen biopsies and matched leukocyte germline DNA from blood samples. WES data was generated for 59 unique baseline DNA samples using the Illumina platform. For this, paired-end libraries were constructed as described previously(51) with the modifications described in Supplementary Data and Methods Whole exome sequencing (WES) section.
RNA-Seq data:
Transcriptome data was generated for 60 samples in this study. For this, strand-specific, poly-A+ RNA-seq libraries for sequencing on the Illumina platform were prepared as previously described (52). See Supplementary Data and Methods RNA-Seq data section for additional details. Between 59.96 and 112.62M total reads were generated for these 60 samples. The average strand-specificity and rRNA rate was 97.04% and 1.79%, respectively. The transcripts for 22868 to 27856 genes were detected in these samples.
The paired-end reads were mapped to the human genome version GRCh38.d1.vd1 (From GDC) using STAR-2.7.1a. Gene expression estimation was performed using RSEM-1.3.1, and RSEM and FPKM values were upper-quartile normalized. Unless otherwise noted, gene median-centered log2-transformed RSEM values were used for the analyses presented here.
Somatic and copy number variant calling:
Somatic variants were called using paired tumor and blood normal from WES data. Tools used for somatic variant calling were Strelka2, Mutect2, CARNAC, and Pindel (v 0.2.5b9). Filtering steps are described in Supplementary Data and Methods Somatic and copy number variant calling section. Similarly, germline mutations were called by comparing normal WES against the reference genome. Hg19.UCSC.add_miR.140312.refgene was used to map the copy number information to genes. COSMIC mutational signature scores for every sample were estimated using deconstructSigs (53).
For somatic copy number alteration analysis, bam files were processed by the CopywriteR package (54) to derive log2 tumor-to-normal copy number ratios, and the circular binary segmentation (CBS) algorithm (55) implemented in the CopywriteR package was used for the copy number segmentation, with the default parameters.
Chromosomal instability for each chromosome in each sample was inferred from the segmentation data using a weighted-sum approach in which the absolute values of the log2 ratios of all segments within a chromosome were weighted by the segment length and summed up (16). The genome-wide chromosome instability index (CIN) was derived by adding up the instability scores for all 22 autosomes in each sample. MSIsensor (56) was used to calculate somatic MSI counts.
GISTIC2 (57) was used to retrieve gene-level copy number values and call significant copy number alterations in the cohort. A threshold of +/−0.3 was applied to log2 copy number ratio to identify gene-wise gain or loss of copy number, respectively. Each gene of every sample was assigned a thresholded copy number level that reflects the magnitude of its deletion or amplification. These are integer values ranging from −2 to 2, where 0 means no amplification or deletion of magnitude greater than the threshold parameters described above. Amplifications are represented by positive numbers: 1 means amplification above the amplification threshold; 2 means amplification larger than the arm level amplifications observed in the sample. Deletions are represented by negative numbers: −1 means deletion beyond the threshold; −2 means deletions greater than the minimum arm-level copy number observed in the sample.
For the pancancer analysis, GISTIC value +/− 2 exceed the high-level thresholds for amplifications/deep deletions, and those with +/− 1 exceed the low-level thresholds but not the high-level thresholds. The low-level thresholds are just the ‘ampthresh’ and ‘delthresh’ noise threshold input values to GISTIC (typically 0.1 or 0.3) and are the same for every threshold.
Proteomics data generation and analysis:
Proteomic sample preparation:
Samples were prepared for proteomic analysis as described in a previous microscaled proteogenomic study (12) with minimal alterations. The details are described in Supplementary Data and Methods proteomic sample preparation section. TMT labeling: A total of 30 ug peptides in 100 uL 50 mM HEPES, pH 8.5, were labeled with 240 ug TMT reagent for an 8:1 TMT:peptide ratio and incubated at 25C for 1 hour. Excess TMT reagent was quenched by incubating with 5 uL 5% hydroxylamine (Sigma) for 15 min. Samples within each plex were combined according to the ratios determined to achieve sample representation within +/− 15% error margin to all other samples. The combined peptides were desalted on a 100 mg tC18 Sep-Pak (Waters), eluted with 50% acetonitrile/0.1% FA, and dried in a vacuum centrifuge.
Experimental design for proteomics and phospho-proteomics:
Samples were analyzed in a TMT11 format as described above. To measure relative protein and phosphosite expression, common references were constructed. The first core common reference consisted of peptide material from all clinical core samples, such that an even proportion was contributed for each of the 60 patients. The second common reference (“prospective BRCA CR”) was from a previous large cohort breast cancer proteomics study (14). Protein and phosphosite expression were reported as the TMT intensity ratio between each sample and the core common references within each plex. For analysis of clinical core samples, eight TMT 11-plexes each contained peptides from 9 core needle biopsies in the first 9 channels. If available, paired pre- and post-treatment tumor samples from a patient were grouped within the same 11-plex. As a quality control measure, we obtained protein and phosphopeptide ratios between prospective BRCA common reference and the core common reference, and the results are shown in Supplementary Fig S1C
Basic reverse phase fractionation and phosphoenrichment
For basic reverse phase fractionation, ~330 ug of peptides were dissolved in 500 uL of 5 mM ammonium formate and 5% acetonitrile using an offline Agilent 1260 LC with a 30 cm long, 2.1 mm inner diameter C18 column, running at 200 uL per minute in a total of 72 fractions, and further concatenated into 18 fractions for proteome analysis and 6 fractions for Fe3+ immobilized metal affinity chromatography (IMAC) based phosphoproteomics analysis. The details of this method are described (12) and Supplementary Data and Methods Basic reverse phase fractionation and phospho-enrichment section.
Proteomic data acquisition and processing
Proteome and phosphoproteome data acquisition was performed with a Proxeon nLC-1200 coupled to Thermo Lumos instrumentation with parameters described in Supplementary Data and Methods Proteomic data acquisition and processing section.
Raw files were searched against the human (clinical cores) or humanRefSeq protein databases complemented with 553 small-open reading frames (smORFs) and common contaminants (Human: RefSeq.20171003_Human_ucsc_hg38_cpdb_mito_259contamsnr_553smORFS.fasta), using Spectrum Mill (Broad Institute) using parameters described in Supplementary Data and Methods Proteomic data acquisition and processing section.
Quantification, normalization and filtering of proteomics data
Before calculation of protein and phosphopeptide ratios, reporter ion signals were corrected for isotopic impurities. Relative abundances of proteins and phosphosites were selected as the median of TMT reporter ion intensity ratios from all PSMs matching to the protein or phosphosite. PSMs were excluded if they lacked a TMT label, had a precursor ion purity < 50%, or had a negative delta forward-reverse score. To normalize across 11-plex experiments, TMT intensities were divided by the common reference for each protein and phosphosite. Log2 TMT ratios were further normalized by median centering and median absolute deviation scaling. Proteins and phosphosites quantified in fewer than 30% of samples (i.e., missing in > 70% of samples) were removed from the respective datasets.
PDX proteomics data generation and analysis
For the PDX experiment, cryopulverized PDX tumor tissues were lysed and digested as described above. 50ug peptides were dissolved in 200ul 50 mM HEPES, pH 8.5 and labeled with 400ug of TMT reagent. TMT sample generation, basic reverse fractionation and proteomic analysis was performed identical to that of clinical core biopsies. Raw files were searched against the human and mouse (PDX samples) UniProt protein databases complemented with 553 small-open reading frames (smORFs) and common contaminants (Human and mouse: UniProt.human.mouse.20171228.RIsnrNF.553smORFs.264contams.fasta) using Spectrum Mill subgroup-specific (SGS) option described in Supplementary Data and Methods PDX proteomics data generation and analysis section.
Data QC and differential expression and pathway enrichment analysis
Samples with estimated tumor content below 45% were entirely removed from the dataset due to lack of RNA to protein correlation in these samples (Supplementary Fig S1B). The Wilcoxon rank sum test in R was used to identify genes (RNA), proteins, phosphosites, and phosphoproteins (mean of all sites on a given protein) that were differential between samples from pCR and non-pCR cases (Supplementary Table S5) and between samples with LIG1 loss (GISTIC = −1) and those without loss (LIG1 WT/Gain, GISTIC >= 0). WebGestaltR (58) and PTM-SEA(25) were used to identify MSigDB Hallmark pathways (gene level data) and PTM signature sets (phosphosite level data), respectively, that show enrichment in pCR or non-pCR tumors by applying the GSEA/PTM-SEA algorithms to signed (by direction of change) log10 p-values from the differential expression analysis (Supplementary Table S6). Additionally, the ssGSEA R package (59,60) was applied to data from three “omes”, and scores for Hallmark pathways were obtained for individual samples (Supplementary Table S6). Normalized enrichment scores (NES) were utilized for visualization purposes. The Wilcoxon signed rank test in R was used for paired differential analysis of on-treatment to baseline measurements for RNA, protein, phosphosite, and phosphoprotein data for 14 patients with matched on-treatment and baseline biopsies (only 13 had matched RNA data). GSEA using WebGestaltR and PTM-SEA were applied to signed log10 transformed p=s from this analysis. PTM-SEA was also applied to phosphosite log2 TMT ratios for each baseline sample to obtain single sample kinase activity scores (normalized enrichment scores for kinase target PTM sets).
Functional prediction based on gene co-expression
Co-expression network construction using mRNA and protein expression data and network-based gene function prediction for KEGG pathways were performed as previously described in Wang et al, 2017 (15) using OmicsEV (https://github.com/bzhanglab/OmicsEV).
Multi-gene proliferation and immune profiling scores
RNA-based multi-gene proliferation scores (MGPS) were calculated as described previously (14,61) by averaging the gene-centered log2 RSEM data for all genes previously characterized as cycle-regulated (62) in each sample. Protein-based MGPS were generated for each sample by averaging log2 TMT ratios for all proteins that showed significant correlation with the RNA-based MGPS (Pearson correlation, p<0.01 after Benjamini-Hochberg fdr correction). Immune profile and microenvironment scores were inferred from the FPKM version of the RNA-seq data using ESTIMATE (63), Cibersort (64) (run in absolute mode), and xCell (65). Protein-based immune modulator scores were calculated as described previously (14) by averaging log2 TMT ratios for expert curated sets of immune modulators belonging to three categories: immune stimulatory, immune inhibitory, and human leukocyte antigen (HLA) (66).
Immunoblotting
Fresh frozen WHIM68, WHIM74, and WHIM75 tumors were cryopulverized (Covaris CP02) then lysed in RIPA buffer. Lysates were blotted for LIG1 (cat# 18051–1-AP, ProteinTech, 1:1000), POLD1 (cat# 15656–1-AP, ProteinTeech, 1:1000), or XRCC1 (cat# ab134056, Abcam, 1:1000). GAPDH (cat# sc-47724, Santa Cruz Biotechnology, 1:4000) was used as a loading control. Details described in the Supplementary Data and Methods Immunoblotting section)
Validation using DepMap
Global-TMT measurements for RB1 and response profiles to approved drugs from the Cancer Response Therapeutics Response Portal (CTRP), Genomics of Drug Sensitivity in Cancer (GDSC), and Profiling Relative Inhibition Simultaneously in Mixtures (PRISM) drug response datasets for cancer cell lines were retrieved from the DepMap resource (www.depmap.org). TNBC cell lines were selected based on ERneg_HER2neg lineage_sub_subtype for breast lineages from sample information provided by DepMap. For TNBC cell lines, Pearson’s correlation was calculated between RB1 protein abundance (log2 TMT ratio) and drug responses (AUC). P-values < 0.05 were considered significant.
Data availability
The genomics and transcriptomics data has been deposited in the dbGAP database under the accession code phs002505.v1, and the proteomics data is accessible through NCI Proteomics Data Commons (PDC: https://pdc.cancer.gov/pdc/) with accessing identifiers PDC00040 (TNBC biopsies proteome raw files), PDC000409 (TNBC biopsies phosphoproteome raw files), and PDC000410 (TNBC PDX proteome raw files). Mass Spectrometry raw files can also be accessed via MASSIVE (https://massive.ucsd.edu/) with accession identifier MSV000089758.
Supplementary Material
Statement of significance.
Proteogenomic analysis of triple negative breast tumors reveal a complex landscape of chemotherapy response associations including a 19q13.31–33 somatic deletion encoding genes serving lagging-strand DNA synthesis (LIG1, POLD1 and XRCC1) that correlate with lack of pathological response, carboplatin-selective resistance and, in pan cancer studies, poor prognosis and chromosomal instability.
Acknowledgments:
Authors would like to acknowledge funding support U24CA160034 (SAC), U24CA210986 (SAC, MAG), U01CA214125 (MJE/MA, SAC), U24CA210979 (DRM), R03OD032626 (DRM), U24CA210954 (BZ), P50 CA186784–06 (MJE), U54CA224083 (SL), U54 CA224076 (MTL), U24 CA226110 (MTL), P30 NCI-CA125123 (CKO), RR160027 (BZ CPRIT Scholar in Cancer Research), RR RR140027 (MJE CPRIT Scholar in Cancer Research), RR200009 (GVE); CPRIT Scholar in Cancer Research), RP170691 (CPRIT Core Facilities Support Grant), GH0005083 Breast Cancer Research (SL). NCI-SPORE Career enhancement award to MA (part of P50 CA186784–06); T32CA203690 support to JTL; 1S10OD028671–01 to GM, K12 CA167540 to F.O.A are also acknowledged. MJE. is a Susan G. Komen Foundation Scholar. MJE and BZ are McNair Scholars supported by the McNair Medical Institute at The Robert and Janice McNair Foundation. This work was also supported generous gifts from the Korell family, Lisa and Ralph Eads and Washington University School of Medicine, Department of Oncology (Dr John Dipersio). Authors are most grateful to the National Cancer Institute (NCI) Clinical Proteomic Tumor Analysis Consortium along with patients and caregivers participating in this study. Authors thank the patient advocates (GRASP Huddle) for their insightful comments and suggestions.
References
- 1.Dent R, Trudeau M, Pritchard KI, Hanna WM, Kahn HK, Sawka CA, et al. Triple-negative breast cancer: clinical features and patterns of recurrence. Clin Cancer Res 2007;13(15 Pt 1):4429–34 doi 10.1158/1078-0432.CCR-06-3045. [DOI] [PubMed] [Google Scholar]
- 2.Rottenberg S, Jaspers JE, Kersbergen A, van der Burg E, Nygren AO, Zander SA, et al. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc Natl Acad Sci U S A 2008;105(44):17079–84 doi 10.1073/pnas.0806092105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Staaf J, Glodzik D, Bosch A, Vallon-Christersson J, Reutersward C, Hakkinen J, et al. Whole-genome sequencing of triple-negative breast cancers in a population-based clinical study. Nat Med 2019;25(10):1526–33 doi 10.1038/s41591-019-0582-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Symmans WF, Wei C, Gould R, Yu X, Zhang Y, Liu M, et al. Long-Term Prognostic Risk After Neoadjuvant Chemotherapy Associated With Residual Cancer Burden and Breast Cancer Subtype. J Clin Oncol 2017;35(10):1049–60 doi 10.1200/JCO.2015.63.1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Masuda N, Lee SJ, Ohtani S, Im YH, Lee ES, Yokota I, et al. Adjuvant Capecitabine for Breast Cancer after Preoperative Chemotherapy. N Engl J Med 2017;376(22):2147–59 doi 10.1056/NEJMoa1612645. [DOI] [PubMed] [Google Scholar]
- 6.Schmid P, Cortes J, Pusztai L, McArthur H, Kummel S, Bergh J, et al. Pembrolizumab for Early Triple-Negative Breast Cancer. N Engl J Med 2020;382(9):810–21 doi 10.1056/NEJMoa1910549. [DOI] [PubMed] [Google Scholar]
- 7.Schmid P, Cortes J, Dent R, Pusztai L, McArthur H, Kummel S, et al. Event-free Survival with Pembrolizumab in Early Triple-Negative Breast Cancer. N Engl J Med 2022;386(6):556–67 doi 10.1056/NEJMoa2112651. [DOI] [PubMed] [Google Scholar]
- 8.Fountzila E, Ignatiadis M. Neoadjuvant immunotherapy in breast cancer: a paradigm shift? Ecancermedicalscience 2020;14:1147 doi 10.3332/ecancer.2020.1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Loibl S, O’Shaughnessy J, Untch M, Sikov WM, Rugo HS, McKee MD, et al. Addition of the PARP inhibitor veliparib plus carboplatin or carboplatin alone to standard neoadjuvant chemotherapy in triple-negative breast cancer (BrighTNess): a randomised, phase 3 trial. Lancet Oncol 2018;19(4):497–509 doi 10.1016/S1470-2045(18)30111-6. [DOI] [PubMed] [Google Scholar]
- 10.Sikov WM, Berry DA, Perou CM, Singh B, Cirrincione CT, Tolaney SM, et al. Impact of the addition of carboplatin and/or bevacizumab to neoadjuvant once-per-week paclitaxel followed by dose-dense doxorubicin and cyclophosphamide on pathologic complete response rates in stage II to III triple-negative breast cancer: CALGB 40603 (Alliance). J Clin Oncol 2015;33(1):13–21 doi 10.1200/JCO.2014.57.0572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.von Minckwitz G, Schneeweiss A, Loibl S, Salat C, Denkert C, Rezai M, et al. Neoadjuvant carboplatin in patients with triple-negative and HER2-positive early breast cancer (GeparSixto; GBG 66): a randomised phase 2 trial. Lancet Oncol 2014;15(7):747–56 doi 10.1016/S1470-2045(14)70160-3. [DOI] [PubMed] [Google Scholar]
- 12.Satpathy S, Jaehnig EJ, Krug K, Kim BJ, Saltzman AB, Chan DW, et al. Microscaled proteogenomic methods for precision oncology. Nat Commun 2020;11(1):532 doi 10.1038/s41467-020-14381-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ademuyiwa FO, Chen I, Luo J, Rimawi MF, Hagemann IS, Fisk B, et al. Immunogenomic profiling and pathological response results from a clinical trial of docetaxel and carboplatin in triple-negative breast cancer. Breast Cancer Res Treat 2021;189(1):187–202 doi 10.1007/s10549-021-06307-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krug K, Jaehnig EJ, Satpathy S, Blumenberg L, Karpova A, Anurag M, et al. Proteogenomic Landscape of Breast Cancer Tumorigenesis and Targeted Therapy. Cell 2020;183(5):1436–56 e31 doi 10.1016/j.cell.2020.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang J, Ma Z, Carr SA, Mertins P, Zhang H, Zhang Z, et al. Proteome Profiling Outperforms Transcriptome Profiling for Coexpression Based Gene Function Prediction. Mol Cell Proteomics 2017;16(1):121–34 doi 10.1074/mcp.M116.060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vasaikar S, Huang C, Wang X, Petyuk VA, Savage SR, Wen B, et al. Proteogenomic Analysis of Human Colon Cancer Reveals New Therapeutic Opportunities. Cell 2019;177(4):1035–49 e19 doi 10.1016/j.cell.2019.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang C, Chen L, Savage SR, Eguez RV, Dou Y, Li Y, et al. Proteogenomic insights into the biology and treatment of HPV-negative head and neck squamous cell carcinoma. Cancer Cell 2021;39(3):361–79 e16 doi 10.1016/j.ccell.2020.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Christmann M, Kaina B. Transcriptional regulation of human DNA repair genes following genotoxic stress: trigger mechanisms, inducible responses and genotoxic adaptation. Nucleic Acids Res 2013;41(18):8403–20 doi 10.1093/nar/gkt635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Symmans WF, Peintinger F, Hatzis C, Rajan R, Kuerer H, Valero V, et al. Measurement of residual breast cancer burden to predict survival after neoadjuvant chemotherapy. J Clin Oncol 2007;25(28):4414–22 doi 10.1200/JCO.2007.10.6823. [DOI] [PubMed] [Google Scholar]
- 20.Parker JS, Mullins M, Cheang MC, Leung S, Voduc D, Vickery T, et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol 2009;27(8):1160–7 doi 10.1200/JCO.2008.18.1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest 2011;121(7):2750–67 doi 10.1172/JCI45014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Byrski T, Huzarski T, Dent R, Marczyk E, Jasiowka M, Gronwald J, et al. Pathologic complete response to neoadjuvant cisplatin in BRCA1-positive breast cancer patients. Breast Cancer Res Treat 2014;147(2):401–5 doi 10.1007/s10549-014-3100-x. [DOI] [PubMed] [Google Scholar]
- 23.Nik-Zainal S, Alexandrov LB, Wedge DC, Van Loo P, Greenman CD, Raine K, et al. Mutational processes molding the genomes of 21 breast cancers. Cell 2012;149(5):979–93 doi 10.1016/j.cell.2012.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alexandrov LB, Nik-Zainal S, Wedge DC, Campbell PJ, Stratton MR. Deciphering signatures of mutational processes operative in human cancer. Cell Rep 2013;3(1):246–59 doi 10.1016/j.celrep.2012.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Krug K, Mertins P, Zhang B, Hornbeck P, Raju R, Ahmad R, et al. A Curated Resource for Phosphosite-specific Signature Analysis. Mol Cell Proteomics 2019;18(3):576–93 doi 10.1074/mcp.TIR118.000943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hubaux R, Thu KL, Vucic EA, Pikor LA, Kung SH, Martinez VD, et al. Microtubule affinity-regulating kinase 2 is associated with DNA damage response and cisplatin resistance in non-small cell lung cancer. Int J Cancer 2015;137(9):2072–82 doi 10.1002/ijc.29577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wei X, Xu L, Jeddo SF, Li K, Li X, Li J. MARK2 enhances cisplatin resistance via PI3K/AKT/NF-kappaB signaling pathway in osteosarcoma cells. Am J Transl Res 2020;12(5):1807–23. [PMC free article] [PubMed] [Google Scholar]
- 28.Smid M, Hoes M, Sieuwerts AM, Sleijfer S, Zhang Y, Wang Y, et al. Patterns and incidence of chromosomal instability and their prognostic relevance in breast cancer subtypes. Breast Cancer Res Treat 2011;128(1):23–30 doi 10.1007/s10549-010-1026-5. [DOI] [PubMed] [Google Scholar]
- 29.Mertins P, Mani DR, Ruggles KV, Gillette MA, Clauser KR, Wang P, et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 2016;534(7605):55–62 doi 10.1038/nature18003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hatzis C, Pusztai L, Valero V, Booser DJ, Esserman L, Lluch A, et al. A genomic predictor of response and survival following taxane-anthracycline chemotherapy for invasive breast cancer. JAMA 2011;305(18):1873–81 doi 10.1001/jama.2011.593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Silver DP, Richardson AL, Eklund AC, Wang ZC, Szallasi Z, Li Q, et al. Efficacy of neoadjuvant Cisplatin in triple-negative breast cancer. J Clin Oncol 2010;28(7):1145–53 doi 10.1200/JCO.2009.22.4725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Petrosyan V, Dobrolecki LE, Thistlethwaite L, Lewis AN, Sallas C, Rajaram R, et al. A Network Approach to Identify Biomarkers of Differential Chemotherapy Response Using Patient-Derived Xenografts of Triple-Negative Breast Cancer. bioRxiv 2021:2021.08.20.457116 doi 10.1101/2021.08.20.457116. [DOI]
- 33.Bruna A, Rueda OM, Greenwood W, Batra AS, Callari M, Batra RN, et al. A Biobank of Breast Cancer Explants with Preserved Intra-tumor Heterogeneity to Screen Anticancer Compounds. Cell 2016;167(1):260–74 e22 doi 10.1016/j.cell.2016.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alifrangis C, Sharma A, Chowdhury S, Duncan S, Milic M, Gogbashian A, et al. Single-agent carboplatin AUC10 in metastatic seminoma: A multi-centre UK study of 216 patients. Eur J Cancer 2020; 164:105–113 doi 10.1016/j.ejca.2020.08.031. [DOI] [PubMed] [Google Scholar]
- 35.Badve SS, Penault-Llorca F, Reis-Filho JS, Deurloo R, Siziopikou KP, D’Arrigo C, et al. Determining PD-L1 Status in Patients With Triple-Negative Breast Cancer: Lessons Learned From IMpassion130. J Natl Cancer Inst 2022;114(5):664–75 doi 10.1093/jnci/djab121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morales-Betanzos CA, Lee H, Gonzalez Ericsson PI, Balko JM, Johnson DB, Zimmerman LJ, et al. Quantitative Mass Spectrometry Analysis of PD-L1 Protein Expression, N-glycosylation and Expression Stoichiometry with PD-1 and PD-L2 in Human Melanoma. Mol Cell Proteomics 2017;16(10):1705–17 doi 10.1074/mcp.RA117.000037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee KM, Giltnane JM, Balko JM, Schwarz LJ, Guerrero-Zotano AL, Hutchinson KE, et al. MYC and MCL1 Cooperatively Promote Chemotherapy-Resistant Breast Cancer Stem Cells via Regulation of Mitochondrial Oxidative Phosphorylation. Cell Metab 2017;26(4):633–47 e7 doi 10.1016/j.cmet.2017.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Echeverria GV, Ge Z, Seth S, Zhang X, Jeter-Jones S, Zhou X, et al. Resistance to neoadjuvant chemotherapy in triple-negative breast cancer mediated by a reversible drug-tolerant state. Sci Transl Med 2019;11(488) doi 10.1126/scitranslmed.aav0936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Luo Q, Zheng N, Jiang L, Wang T, Zhang P, Liu Y, et al. Lipid accumulation in macrophages confers protumorigenic polarization and immunity in gastric cancer. Cancer Sci 2020;111(11):4000–11 doi 10.1111/cas.14616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Howes TR, Tomkinson AE. DNA ligase I, the replicative DNA ligase. Subcell Biochem 2012;62:327–41 doi 10.1007/978-94-007-4572-8_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Waga S, Bauer G, Stillman B. Reconstitution of complete SV40 DNA replication with purified replication factors. J Biol Chem 1994;269(14):10923–34. [PubMed] [Google Scholar]
- 42.Soderhall S, Lindahl T. DNA ligases of eukaryotes. FEBS Lett 1976;67(1):1–8 doi 10.1016/0014-5793(76)80858-7. [DOI] [PubMed] [Google Scholar]
- 43.Petrini JH, Xiao Y, Weaver DT. DNA ligase I mediates essential functions in mammalian cells. Mol Cell Biol 1995;15(8):4303–8 doi 10.1128/MCB.15.8.4303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Paul-Konietzko K, Thomale J, Arakawa H, Iliakis G. DNA Ligases I and III Support Nucleotide Excision Repair in DT40 Cells with Similar Efficiency. Photochem Photobiol 2015;91(5):1173–80 doi 10.1111/php.12487. [DOI] [PubMed] [Google Scholar]
- 45.Pascucci B, Stucki M, Jonsson ZO, Dogliotti E, Hubscher U. Long patch base excision repair with purified human proteins. DNA ligase I as patch size mediator for DNA polymerases delta and epsilon. J Biol Chem 1999;274(47):33696–702 doi 10.1074/jbc.274.47.33696. [DOI] [PubMed] [Google Scholar]
- 46.Barnes DE, Tomkinson AE, Lehmann AR, Webster AD, Lindahl T. Mutations in the DNA ligase I gene of an individual with immunodeficiencies and cellular hypersensitivity to DNA-damaging agents. Cell 1992;69(3):495–503 doi 10.1016/0092-8674(92)90450-q. [DOI] [PubMed] [Google Scholar]
- 47.Soza S, Leva V, Vago R, Ferrari G, Mazzini G, Biamonti G, et al. DNA ligase I deficiency leads to replication-dependent DNA damage and impacts cell morphology without blocking cell cycle progression. Mol Cell Biol 2009;29(8):2032–41 doi 10.1128/MCB.01730-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Harrison C, Ketchen AM, Redhead NJ, O’Sullivan MJ, Melton DW. Replication failure, genome instability, and increased cancer susceptibility in mice with a point mutation in the DNA ligase I gene. Cancer Res 2002;62(14):4065–74. [PubMed] [Google Scholar]
- 49.Bentley D, Selfridge J, Millar JK, Samuel K, Hole N, Ansell JD, et al. DNA ligase I is required for fetal liver erythropoiesis but is not essential for mammalian cell viability. Nat Genet 1996;13(4):489–91 doi 10.1038/ng0896-489. [DOI] [PubMed] [Google Scholar]
- 50.Le Chalony C, Hoffschir F, Gauthier LR, Gross J, Biard DS, Boussin FD, et al. Partial complementation of a DNA ligase I deficiency by DNA ligase III and its impact on cell survival and telomere stability in mammalian cells. Cell Mol Life Sci 2012;69(17):2933–49 doi 10.1007/s00018-012-0975-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rokita JL, Rathi KS, Cardenas MF, Upton KA, Jayaseelan J, Cross KL, et al. Genomic Profiling of Childhood Tumor Patient-Derived Xenograft Models to Enable Rational Clinical Trial Design. Cell Rep 2019;29(6):1675–89 e9 doi 10.1016/j.celrep.2019.09.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Peters TL, Kumar V, Polikepahad S, Lin FY, Sarabia SF, Liang Y, et al. BCOR-CCNB3 fusions are frequent in undifferentiated sarcomas of male children. Mod Pathol 2015;28(4):575–86 doi 10.1038/modpathol.2014.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rosenthal R, McGranahan N, Herrero J, Taylor BS, Swanton C. DeconstructSigs: delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol 2016;17:31 doi 10.1186/s13059-016-0893-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kuilman T, Velds A, Kemper K, Ranzani M, Bombardelli L, Hoogstraat M, et al. CopywriteR: DNA copy number detection from off-target sequence data. Genome Biol 2015;16:49 doi 10.1186/s13059-015-0617-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Venkatraman ES, Olshen AB. A faster circular binary segmentation algorithm for the analysis of array CGH data. Bioinformatics 2007;23(6):657–63 doi 10.1093/bioinformatics/btl646. [DOI] [PubMed] [Google Scholar]
- 56.Niu B, Ye K, Zhang Q, Lu C, Xie M, McLellan MD, et al. MSIsensor: microsatellite instability detection using paired tumor-normal sequence data. Bioinformatics 2014;30(7):1015–6 doi 10.1093/bioinformatics/btt755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, Getz G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol 2011;12(4):R41 doi 10.1186/gb-2011-12-4-r41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang J, Vasaikar S, Shi Z, Greer M, Zhang B. WebGestalt 2017: a more comprehensive, powerful, flexible and interactive gene set enrichment analysis toolkit. Nucleic Acids Res 2017;45(W1):W130–W7 doi 10.1093/nar/gkx356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 2009;462(7269):108–12 doi 10.1038/nature08460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102(43):15545–50 doi 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ellis MJ, Suman VJ, Hoog J, Goncalves R, Sanati S, Creighton CJ, et al. Ki67 Proliferation Index as a Tool for Chemotherapy Decisions During and After Neoadjuvant Aromatase Inhibitor Treatment of Breast Cancer: Results From the American College of Surgeons Oncology Group Z1031 Trial (Alliance). J Clin Oncol 2017;35(10):1061–9 doi 10.1200/JCO.2016.69.4406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Whitfield ML, Sherlock G, Saldanha AJ, Murray JI, Ball CA, Alexander KE, et al. Identification of genes periodically expressed in the human cell cycle and their expression in tumors. Mol Biol Cell 2002;13(6):1977–2000 doi 10.1091/mbc.02-02-0030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yoshihara K, Shahmoradgoli M, Martinez E, Vegesna R, Kim H, Torres-Garcia W, et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun 2013;4:2612 doi 10.1038/ncomms3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chen B, Khodadoust MS, Liu CL, Newman AM, Alizadeh AA. Profiling Tumor Infiltrating Immune Cells with CIBERSORT. Methods Mol Biol 2018;1711:243–59 doi 10.1007/978-1-4939-7493-1_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Aran D, Hu Z, Butte AJ. xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol 2017;18(1):220 doi 10.1186/s13059-017-1349-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Thorsson V, Gibbs DL, Brown SD, Wolf D, Bortone DS, Ou Yang TH, et al. The Immune Landscape of Cancer. Immunity 2019;51(2):411–2 doi 10.1016/j.immuni.2019.08.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The genomics and transcriptomics data has been deposited in the dbGAP database under the accession code phs002505.v1, and the proteomics data is accessible through NCI Proteomics Data Commons (PDC: https://pdc.cancer.gov/pdc/) with accessing identifiers PDC00040 (TNBC biopsies proteome raw files), PDC000409 (TNBC biopsies phosphoproteome raw files), and PDC000410 (TNBC PDX proteome raw files). Mass Spectrometry raw files can also be accessed via MASSIVE (https://massive.ucsd.edu/) with accession identifier MSV000089758.