

Abstract
Non-genetic predisposition to colorectal cancer (CRC) continues to be difficult to measure precisely, hampering efforts in targeted prevention and screening. Epigenetic changes in the normal mucosa of CRC patients can serve as a tool in predicting CRC outcomes. We identified epigenetic changes affecting the normal mucosa of CRC patients. DNA methylation profiling on normal colon mucosa from 77 CRC patients and 68 controls identified a distinct subgroup of normally-appearing mucosa with markedly disrupted DNA methylation at a large number of CpGs, termed as “Outlier Methylation Phenotype” (OMP) and are present in 15/77 patients with cancer vs. 0/68 controls (p<0.001). Similar findings were also seen in publicly available datasets. Comparison of normal colon mucosa transcription profiles of OMP cancer patients with those of non-OMP cancer patients indicates genes whose promoters are hypermethylated in the OMP patients are also transcriptionally down-regulated, and that many of the genes most affected are involved in interactions between epithelial cells, the mucus layer, and the microbiome. Analysis of 16S rRNA profiles suggests that normal colon mucosa of OMPs are enriched in bacterial genera associated with CRC risk, advanced tumor stage, chronic intestinal inflammation, malignant transformation, nosocomial infections and KRAS mutations. In conclusion, our study identifies an epigenetically distinct OMP group in the normal mucosa of patients with CRC that is characterized by a disrupted methylome, altered gene expression and microbial dysbiosis. Prospective studies are needed to determine whether OMP could serve as a biomarker for an elevated epigenetic risk for CRC development.
Keywords: Colorectal cancer, Methylation, Outlier, Racial disparity, Expression, Microbiome, African Americans
Prevention Relevance Statement:
Our study identifies an epigenetically distinct OMP group in the normal mucosa of patients with CRC that is characterized by a disrupted methylome, altered gene expression and microbial dysbiosis. Identification of OMPs in healthy controls and CRC patients will lead to prevention and better prognosis, respectively.
INTRODUCTION
Despite the availability of an effective screening test, colorectal cancer (CRC) remains the third-leading cause of cancer deaths in men and women in the United States(1).
Over the years, scientists have discovered various molecular markers like gene mutations (KRAS, BRAF and APC genes); CpG island methylator phenotype (CIMP), microsatellite instability and so on to better understand the heterogeneous outcomes of colorectal cancer (2). However, it is noteworthy that all of these molecular subtypes are based on investigating the tumor tissues. We, on the other hand, study the normal tissues of colorectal cancer patients, which could harbor biomarkers to better understand CRC outcomes.
We have identified site-specific DNA methylation differences in normal colon mucosa that distinguish cancer patients from patients without cancer with high sensitivity and specificity(3). This observation, validated in both an independent population(4) and an animal model(5), suggests that these cancer patient “signature” methylation differences in normal tissues accumulate over time as a result of aging, environmental exposures and, perhaps, genetic influences. Our earlier observation(3) that the largest category of genes affected by differential methylation were those involved in carbohydrate and lipid metabolism is consistent with long-standing epidemiological evidence(6) that dietary factors affect CRC risk.
Colorectal cancer risk is not distributed uniformly across the population but is higher in patients of African descent than Caucasians, Hispanics or Asians(7). African American (AA) CRC patients also appear less likely to develop microsatellite-instable cancers (a form of colorectal cancer with improved outcome) than their Caucasian counterparts(7). In addition, AA patients who are asymptomatic are more likely to have proximal, large, pre-cancerous adenomatous polyps present on colonoscopy screening(8). While there are likely to be socioeconomic factors involved in disparities in cancer incidence and outcomes, it is also possible that race-associated differences in biology contribute(9).
Our current study was designed to investigate differences in the normal colon epigenome of CRC patients by performing genome-wide DNA methylation profiling on 77 CRC patients (42 AA and 35 Caucasians) and age-, sex- and race-matched controls (34 AA and 34 Caucasian). We also performed normal colon transcription profiling on selected cancer patients, as well as microbiome analysis via 16S rRNA sequencing. Our hypothesis is that environmental factors interact with the normal colon epigenome to engender epigenetic changes that predispose to cancer, and that these changes are greater and/or more frequent in AA than in Caucasians. We hypothesize, further, that environmental factors, principally diet, exert much of their effect on the normal colon epigenome through interactions with the microbiome.
MATERIALS AND METHODS
Samples
Normal colon tissues (fresh frozen) of 77 CRC patients were purchased from Fox Chase Cancer Centre biobank. These normal colon tissues adjacent (~10cm away) to tumors were collected from CRC patients as described previously(3,4). Similarly, normal colon tissues (fresh frozen) from age, sex, location and race matched healthy controls (n=70) were collected during routine screening colonoscopies after informed consent. Controls with previous colonoscopic finding of polyps were excluded.
Written informed consent from the patients were obtained and the study was conducted in accordance with Declaration of Helsinki ethical guidelines and the study was approved by Temple University’s institutional review board.
Sample processing
DNA extraction:
Genomic DNA was extracted from colon tissue samples using Invitrogen PureLink genomic DNA kit as per the manufacturer’s protocol.
RNA extraction:
RNA was extracted using Qiagen’s RNeasy Plus mini kit. Briefly, nearly 30mg of colon tissue was homogenized followed by isolation and purification using standard manufacturer’s protocol.
Quantification and Quality check:
Extracted DNAs and RNAs were quantified using ThermoFisher’s NanoDrop. RNA integrity was checked on Agilent 2100 Bioanalyzer.
Statistical Analyses
All the statistical analyses were done using different packages in R. Plots were made using both R and GraphPad Prism version8.
DNA methylation
Illumina EPIC array:
Extracted DNA was sent to external Genomic Facility at Penn State University to be run on Illumina’s EPIC array. Prior to array run, extracted DNA was treated with bisulfite using Zymo EZ DNA methylation kit. Bisulfite treated DNA is processed further to run Illumina’s EPIC array as described previously(10). The output data is generated in the .idat files. Two healthy samples failed to hybridize during the initial array processing.
Data processing:
Raw data files from 77 CRC patients and 68 healthy controls were preprocessed using minfi’s preprocessIllumina function to mimic Genome Studio’s background correction and normalization steps in the R environment. Probe normalization was also done via the preprocessIllumina function which equally recreates Genome Studio’s method of normalizing variability in red/green signal using paired red/green control probes in a reference sample. Beta values obtained after these preprocessing steps were used for all the subsequent analyses.
Quality control:
The quality of the samples was checked using the minfi getQC test.
Batch effect:
As the samples were run in batches, batch effect was checked using correlation and Bland Altmann analyses for the replicate samples (both intraplate or interwell replicates (same samples in different wells of the same plate) as well as interplate replicates (same samples in different plates)).
Cell composition/purity:
Epithelial cell purity between the tissues from healthy controls and CRC patients was estimated by leukocyte unmethylation for purity (LUMP) as described previously (11).
CpG selection for methylation analyses:
SNP associated and cross-reactive CpGs (12,13) and 59 SNP CpGs were excluded from analysis. Poor performing probes (missing values in >=20% of the samples) were also excluded This resulted in 819,239 CpGs which were included for the analyses.
Cluster analysis:
Unsupervised clustering using bootstrap method was performed using the pvclust package in R.
Principal component analysis:
Principal component analysis was done by using prcomp function in R.
Outlier analysis:
Outliers or individuals with Outlier Methylation Phenotype (OMPs) were identified by following a two- step procedure(14,15). In the first step, each of the 819,239 CpG sites was analyzed for the presence of outliers (methylation levels beyond 1.5 times the interquartile range below the first quartile (“hypomethylated outliers”) or above the third quartile (“hypermethylated outliers”) of the distribution). In the second step, the distribution of outlier CpGs was plotted for each sample and similar outlier calculations as in Step 1 were done, to identify individuals with extremely large number of outlier CpGs compared to rest of the population. Outliers of Step 2 were considered as the individuals with OMP.
Differential methylation analysis:
Between group comparisons were done using two-sided t-test for methylation values. Bonferroni’s correction was used to correct for multiple testing. We checked the location/feature of each of the 819239 CpGs and corrected for 108,498 features (because methylation levels are highly correlated at CpGs within the same feature, and are, thus, not independent) resulting in p values less than 4.6E-07 as the cut off for significance. A cut off (0.05) for magnitude of difference in beta values was also introduced. Hence, CpGs with p value less than 4.6E-07 and magnitude of difference >0.05 were considered to be significant. Differential methylation analyses were done using two-sided t test in R. Differential methylated regions (DMRs) were identified using “DMRcate” package in R.
Gene expression
RNAseq:
RNAseq libraries were prepared using Illumina’s TruSeq stranded mRNA kit by following the standard manufacturer’s protocol. Libraries were sequenced in Illumina HiSeq 4000 at GENEWIZ.
Data processing:
Sequencing data quality was assessed by FastQC. Sequencing reads were trimmed using Trim Galore and aligned using mapping software STAR (16). Transcripts were counted using HTSeq.
Differential gene expression analysis:
We used R package DESeq2, version 3.13(17) for differential gene expression analyses.
Gene ontology (GO) analysis:
We used the R package ReportingTools (https://bioconductor.org/packages/release/bioc/html/ReportingTools.html) to generate GO pathways (18).
Microbiome
16S rRNA sequencing:
16S rRNA libraries were generated using a modified Illumina 16S protocol that increases input DNA to 62.5ng. Barcoded libraries were generated with Nextera XT adapters per Illumina’s 16S protocol. Purified libraries were quantified via Qubit and analyzed on the Agilent DNA Bioanalyzer in order to generate 10mM pooled libraries to be sequenced on the MiSeq platform.
Amplicon sequence variants:
We pre-processed raw 16S rRNA sequences generated for 70 colon tissue samples collected from African American patients using QIIME2, version 2019.1(19). We obtained a total of 3,845,964 quality-screened DNA sequences, with an average count of 54,942 sequence reads per sample. We applied the DADA2 algorithm(20) via the q2-dada2 plugin to denoise the sequence data and generate unique amplicon sequence variants (ASVs). Taxonomic classification of representative ASVs was conducted using the classify-sklearn naïve Bayes classifier(21) against the Greengenes, version 13_8 99% reference database(22).
Taxonomic composition and differential abundance:
We used R package phyloseq, version 1.24.2(23) to describe the taxonomic composition of each cohort at the phylum and genus level. In addition, differential abundance analysis using R package DESeq2, version 3.13(17) was applied to identify bacterial taxa that were significantly different between the cohorts studied. Differential abundances in bacterial species were assessed using a log2foldchange value, and cohort comparisons were conducted applying the Wald test with the Benjamini-Hochberg correction.
Microbiome diversity:
A rarefied sampling depth of 14,214 DNA reads per sample and R package phyloseq, version 1.24.2(23) were further used to assess microbiome diversity across sampling cohorts. Diversity within samples (alpha diversity) was estimated as observed number of ASVs and Shannon diversity index and significance of differences was tested using non-parametric Wilcoxon rank sum-tests. Rarified samples were also used to calculate Bray-Curtis beta diversity (dissimilarity between samples), and non-metric multidimensional scaling (NMDS) was performed. Significance of differences in beta diversity between cohorts was assessed by permutational analysis of variance (PERMANOVA) and permutation tests for homogeneity in multivariate dispersion (PERMDISP) in R package vegan, version 2.5–6(24) with 999 permutations.
Data Availability Statement
The datasets generated during the current study are available in the GEO repository (GSE 199057).
RESULTS
Quality control (QC) of methylation dataset
All the samples passed the QC test on minfi (Supplementary Figure 1A). No batch effects were observed for the processed methylation data. All the replicates (irrespective of intraplate or interplate) were strongly correlated (R2=0.99). Similarly, all the replicates (Supplementary Figures 1B–E) showed similar results on Bland Altmann analyses wherein nearly 45K CpGs (5%) were outside agreement boundary irrespective of whether those were intraplate (Supplementary Figures 1B–C) or interplate (Supplementary Figures 1D–E) replicates. Furthermore, there was no difference (p=0.4626) in the cell purity of normal tissues from healthy controls and CRC patients on LUMP analysis (Supplementary Figure 1F).
Identification of an outlier methylation group in normal tissues of cancer patients
We performed unsupervised hierarchical cluster analysis, using methylation data from 819,239 CpGs to determine whether our study population could be subdivided on the basis of the normal colon epigenome. Interestingly, we observed a group of 14 CRC individuals (11 African American and 3 Caucasians) and a Caucasian CRC patient clustering separately (highlighted in yellow in Figure 1A) from rest of the dataset. We also performed principal component analysis to determine whether quantitative variation at multiple sites might distinguish the study groups (Figure 1B). Patients without cancer were less variable compared to the colon cancer groups of both races. The Caucasian healthy (CH) group had the least variability followed by the African American healthy group (AH) group. The African American cancer (AC) group had the highest variability followed by the Caucasian cancer group (CC). Very high variability in the cancer groups was exacerbated by the samples (11AC, 4CC) at the right side of the PCA plot (values >590 in PC1, samples within the black ellipse). It is noteworthy, that these 15 individuals are the ones that clusters separately in Figure 1A.
Figure 1: Analysis of methylation data.
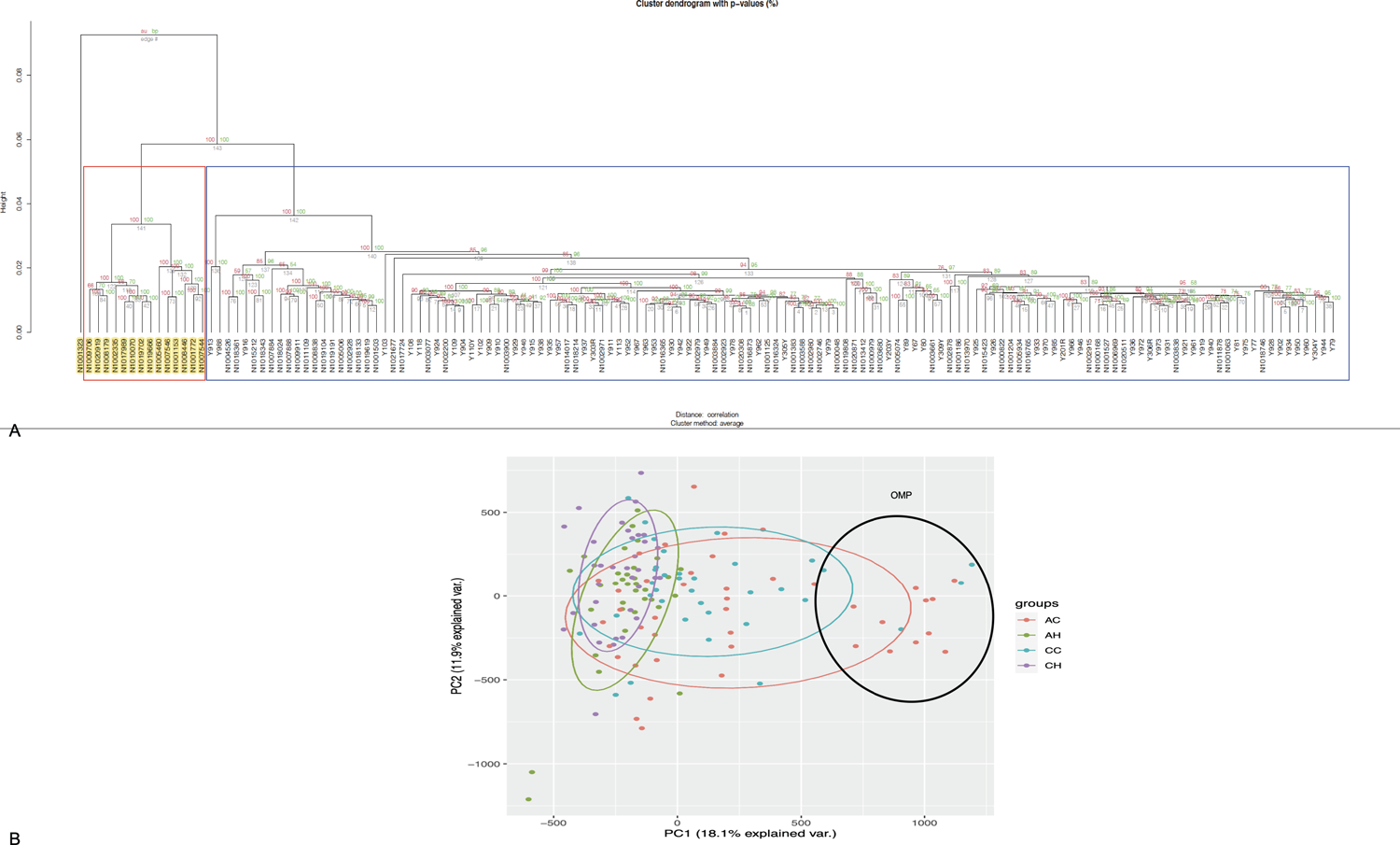
(A) Unsupervised cluster analysis of study samples. Hierarchial cluster plots using unsupervised cluster analysis showing separate cluster for the OMPs. (B) Principal component analyses. Principal component analyses of study groups using 819,239 CpGs. AC African American Cancer; AH African American Healthy; CC Caucasian Cancer; CH Cancer Healthy.
Definition of an Outlier Methylation Phenotype (OMP) group
Because both PCA and cluster analysis suggested the existence of a group with dramatically disrupted normal tissue methylomes, we applied the same metric we have used previously(14,15) to identify individuals with “Outlier Methylation Phenotype” (OMP) (Figure 2). Although this method (see Materials and Methods) transforms a fundamentally quantitative trait (methylation values) into a discrete classifier (OMP status), it simplifies further analysis of factors that may contribute to this phenotype. In other words, converting a quantitative variable to a categorical variable simplifies the downstream analysis for better characterization of this group (OMP). We plotted the number of CpGs in which an individual was hyper- (Figure 2A) or hypo-methylated (Figure 2B) at greater than 1.5-times the interquartile range to identify those individuals who were OMPs.
Figure 2: Identification of samples with Outlier Methylation Phenotype (OMP).
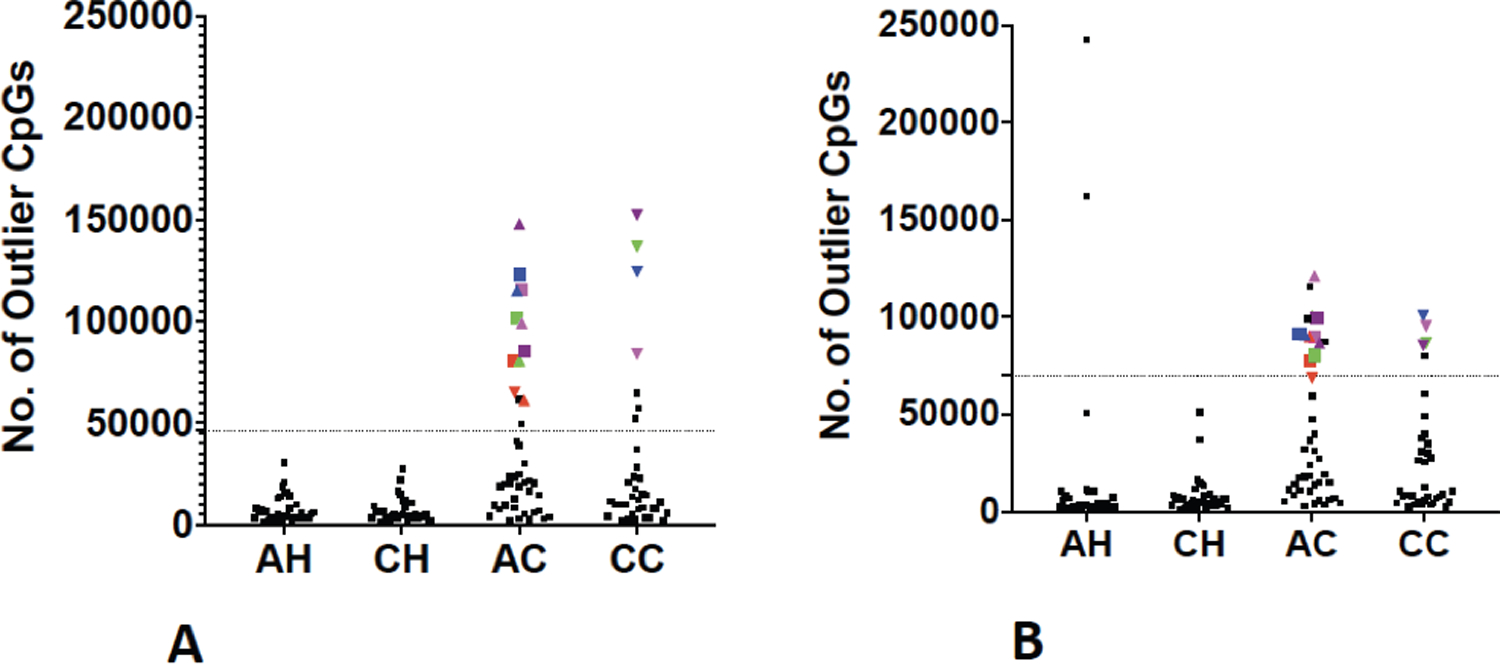
Number of CpGs in which a sample is (A) Hypermethylated outlier (B) Hypomethylated outlier. Dotted line indicates outlier boundary. Each symbol is a sample. Symbols above the dotted lines are outliers in respective plots. Colored symbols indicate samples who are outliers in both the plots and are termed as “OMPs”. Samples represented by colored symbols are OMPs. Same color and shape show the same individuals in both the plots. AH African American Healthy; CH Caucasian Healthy; AC African American Cancer; CC Caucasian Cancer.
None of the CH individuals were hyper- or hypo-methylated outliers (Figure 2A, B), whereas two of the AH individuals were hypo-methylated outliers. Fifteen AC patients were hyper- or hypo-methylated outliers and 11 were bidirectional (both hyper- and hypo-methylated) outliers. Among CC patients, seven samples were hyper-methylated and five samples were hypo-methylated outliers, of which only four patients were bidirectional outliers. Individuals who were outliers in both hyper- and hypo-methylated plots were classified as Outlier Methylation Phenotype (OMP)(14,15). Further justification for classifying only bidirectional outliers as OMPs (11 AC and 4 CC) is that these individuals are the same patients who form separate groups in the cluster analyses (Figure 1A) and are furthest from the other CRC patients in the PCA analysis (Figure 1B).
We also analyzed whether the OMP (red box) and non-OMP (blue box) clusters (Figure 1A) were based on any particular feature (like age, sex). As shown in Supplementary Table 1, these two clusters showed significant differences in cancer status. All other variables (age, sex, location) were not significantly different. Furthermore, we did see a borderline association (p=0.05) for race but the significance was lost after correcting for multiple (four) tests.
Validation of OMP group in publicly available colorectal cancer datasets
We selected three colorectal cancer datasets from Gene Expression Omnibus (GEO) which had 450K methylation array data for both healthy controls and normal tissues from CRC patients. We performed outlier analysis in each of the datasets and identified individuals with OMPs (or bidirectional outliers) as described above. As shown in Supplementary Table 2A, all of the datasets show higher frequency of OMPs in the CRC group compared to healthy controls. Additionally, the largest dataset (GSE132804) had significantly higher frequency of OMPs in CRC patients compared to controls. Furthermore, we also analyzed if any cofounding variables in dataset GSE132804 influenced the OMP output. Supplementary Table 2B clearly indicates that the two groups (cancer and controls) were matched for age, sex and location, justifying that OMP is not an outcome of unbalanced co-variables. This validates our finding that normal tissues of CRC patients are more prone to have disrupted epigenome or OMP characteristic compared to healthy controls.
Effect of OMPs on Differential methylation in Normal Colon Mucosa of CRC Patients
African American and Caucasian CRC patients, combined, showed significantly different methylation at 85,178 CpGs (10.40%) compared with healthy controls (Figure 3A). On race-stratified subgroup analysis, the AC patients had 26,803 differentially methylated CpGs compared with the AH controls (Figure 3B), whereas the CC patients had 12,016 (Figure 3C) differentially methylated CpGs compared with the (CH) controls. More than 60% of the differentially methylated CpGs (7,341 CpGs) in the Caucasian CRC patients were also differentially methylated in African American CRC patients (Figure 3B–C), suggesting that many of the cancer-associated methylation alterations were common to both AC and CC patients. However, African American CRC patients had a much larger number of abnormally methylated CpG sites compared with their healthy controls (an additional >14,000 CpG sites), than did their Caucasian counterparts.
Figure 3: Volcano plots showing differential methylation analyses.
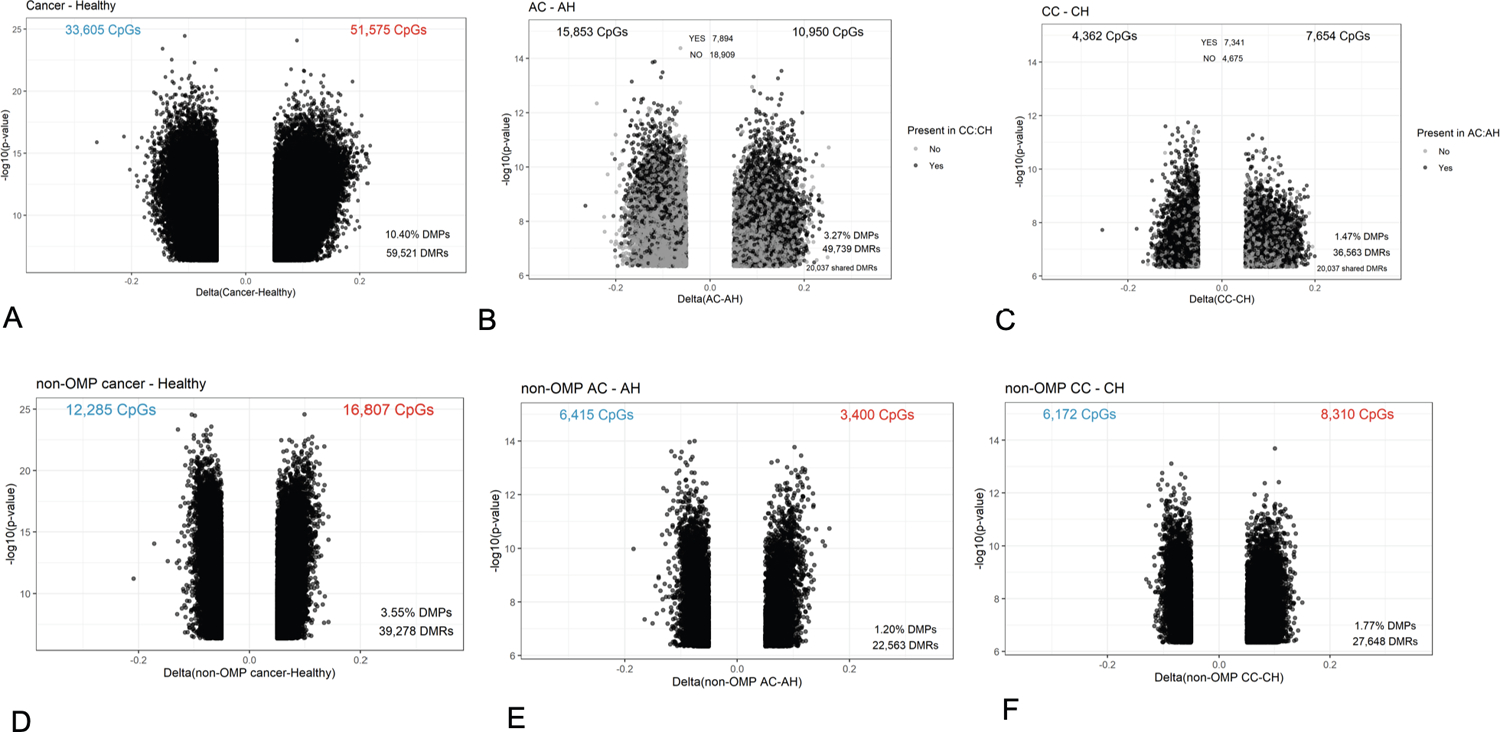
(A-C) Colon cancer vs Healthy in (A) All samples (B) African Americans (C) Caucasians.
(D-F) non-Outlier Colon cancer vs Healthy in (D) All samples (E) African Americans (F) Caucasians. AH African American Healthy; CH Caucasian Healthy; AC African American Cancer; CC Caucasian Cancer; DMPs Differentially Methylated Positions; DMRs Differentially Methylated Regions.
We also analyzed whether any confounding effects between the control and CRC groups account for these differences. Table 1 shows the demographic profile of the analyzed samples. None of the variables were significantly different between cancer and control groups. Hence, all the differentially methylated probes (overall or race-specific) are associated with CRC.
Table 1:
Demographic profile of analyzed samples
| All samples | ||||
|---|---|---|---|---|
| Cancer (n=77) | Control (n=68) | P value* | ||
| Age (Mean± Standard deviation) | 57.67± 9.68 | 56.81± 8.81 | 0.5757 | |
| Sex | Males | 38 | 33 | 1.0000 |
| Females | 39 | 35 | ||
| Race | Caucasian | 35 | 34 | 0.6198 |
| African American | 42 | 34 | ||
| Location | Distal | 42 | 37 | 1.0000 |
| Proximal | 35 | 31 | ||
| Caucasians | ||||
| Cancer (n=35) | Control (n=34) | P value* | ||
| Age (Mean± Standard deviation) | 56.80±9.33 | 56.06±9.61 | 0.6905 | |
| Sex | Males | 15 | 15 | 1.0000 |
| Females | 20 | 19 | ||
| Location | Distal | 19 | 19 | 1.0000 |
| Proximal | 16 | 15 | ||
| African Americans | ||||
| Cancer (n=42) | Control (n=34) | P value* | ||
| Age (Mean± Standard deviation) | 58.40±10.02 | 57.56±8.00 | 0.7461 | |
| Sex | Males | 23 | 18 | 1.0000 |
| Females | 19 | 16 | ||
| Location | Distal | 23 | 18 | 1.0000 |
| Proximal | 19 | 16 | ||
ttest for Age and Fisher’s exact test for other variables
Because we had identified a group of patients with dramatically disrupted normal colon methylation profiles, and the groups was composed of largely AA patients, we asked whether the increased number of differences between AC and AH groups compared with the CC and CH groups were driven by the OMPs by excluding them from the analysis. When this was done, the number of differentially methylated CpGs was reduced by more than 50% in overall cancer vs healthy comparison (Figure 3D). A similar trend was observed in AC vs AH (Figure 3E). However, we did not observe a reduction in abnormally methylated CpGs between the CC vs CH groups (Figure 3F), suggesting that OMPs in the AC group contributed much more variance than in the CC group.
It is noteworthy that DNA methylation profiles of normal colon mucosa between the controls and CRC patients of African American and Caucasian races are mostly similar. We observed a very small fraction of race-associated differences in site-specific CpG methylation between either healthy controls (0.10%, or 794 sites) or between cancer patients (0.02%, or 193 sites) (Supplementary Figure 2). These observations suggest that racial disparities in colon cancer incidence and outcome are not a result of large numbers of methylation differences at different CpG sites, with the caveat that not all CpG sites are interrogated by the Illumina platform used.
Each of the above analyses (cluster, PCA, outlier, differential methylation) indicates the presence of a highly epigenetically disrupted group of CRC patients, of which the majority are African American. We examined this OMP group of patients, further, to determine what factors might influence this phenotype, and whether it might contribute to observed racial disparity in CRC incidence and outcome.
Differential expression of genes with differentially methylated promoter CpGs in African American OMPs
A working hypothesis on racial disparities in colon cancer developed from our analysis of normal tissue DNA methylation is that OMPs, although not unique to African Americans, are more prevalent among African Americans and OMPs may be at higher risk of cancer. It is noteworthy that of the 178,469 CpGs that were differentially methylated between OMP cancer patients and non-OMP cancer patients (Supplementary Figure 3), 40,961 CpGs were present in the promoter regions of 11,357 genes.
Again, because the majority (~75%) of OMPs were African American and we wished to characterize this group further, we compared gene expression levels between OMPs (AO) and non-OMPs (AC) among African American CRC patients for whom we were able to obtain normal colon RNA samples by bulk RNAseq (3 OMPs vs 5 non-OMPs). More than 17% (1,964) of the promoter differentially methylated genes also exhibited differential expression levels (Supplementary Figure 4). The majority (1,151 genes) of the differentially expressed genes were hypermethylated in the promoters of OMPs. As expected, most of these hypermethylated genes (1,021 or 88.7%) were downregulated in the OMPs compared to non OMPs (Supplementary Figure 4).
Supplementary Table 3 lists the differentially expressed genes. Multiple genes linked to mucins (MUC17, MUC3A, MUC12, MUC4, MUC5B, MUC20, MUC2, MUC13, MUC1); claudins (CLDN8, CLDN3, CLDN4, CLDN7, CLDN12, CLDN9), cadherins (CDHR2, CDHR5, CDH1, CDH17, CDHR1) and other transmembrane junction proteins (DSC2, CGN, CAPN13, CDHR2, TMPRSS2, AMN) were differentially expressed (down regulated) in OMPs. In addition, among those genes that were hypo-methylated (Supplementary Table 3), the proinflammatory cytokine genes IL6 and IL11 were both up-regulated. The top significant biological processes (Supplementary Table 4) associated with the differentially expressed genes included xenobiotic processes (response to xenobiotic stimulus, xenobiotic metabolic process), leading us to perform an analysis of gut microbiome components.
Differential microbiome in OMPs (AO) compared to non-OMPs (AC) in African American CRC patients
Similar to expression analysis, additional microbiome analysis was restricted to African American patients and included 35 AH, 25 AC and 10 AO patients. In total, we identified 18,522 amplicon sequence variants (ASVs) across all samples analyzed. At the phylum level and across all cohorts, the microbiota was dominated by ASVs assigned to the phyla Firmicutes, Bacteroidetes, Proteobacteria, Actinobacteria, Fusobacteria, and Verrumicrobia (Figure 4A). At the genus level, ASVs were assigned primarily to the genera Bacteroides, Oscillospira, Clostridium, Coprococcus, Previotella and Ruminococcus (Figure 4B).
Figure 4: Analysis of the microbiome from African American samples.
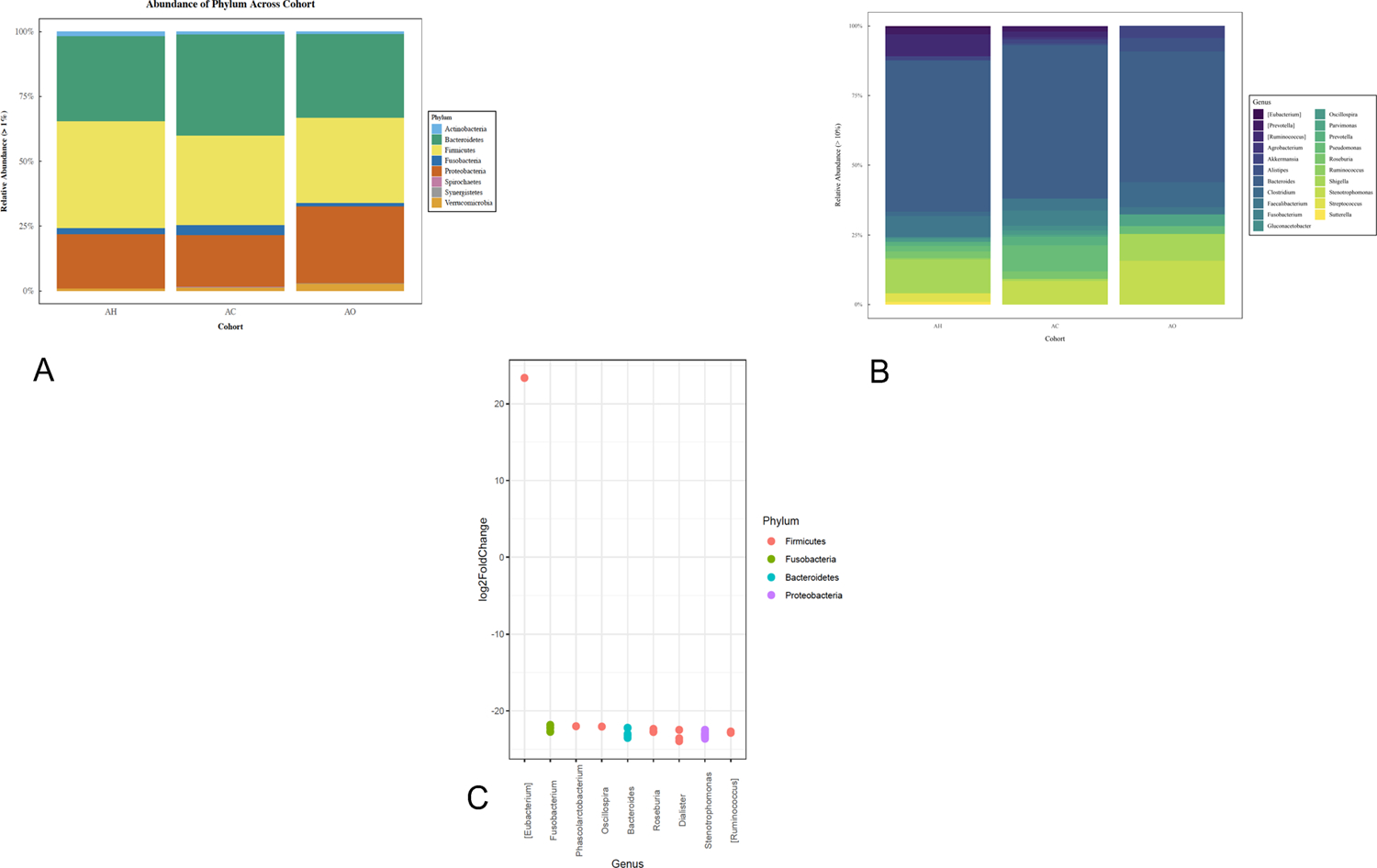
(A)Taxonomic composition of colon tissue microbiomes at the phylum level. (B) Taxonomic composition of colon tissue microbiomes at the genus level. (C) Differential abundance analysis between microbiome samples of AC and AO cohorts. Each point represents ASV belonging to respective bacteria species. ASVs were considered significant if their false discovery rate-corrected P-value was < 0.05. Multiple points visualized under the same genus represent ASVs that are classified within the same genus but differ by one or more nucleotides. Taxa in square brackets are annotations for proposed taxonomy supplied by the Greengenes database. AH African American Healthy; AC African American Cancer non-OMPs; AO African American Cancer OMPs.
Although neither alpha nor beta diversity estimates were significantly different between AH, AC and AO cohorts (Wilcoxon rank sum tests P>0.05, Supplementary Figure 5A; PERMANOVA, P = 0.084, Supplementary Figure 5B), differential abundance analysis (Supplementary Figure 5C) revealed that significant differences among cohorts were detected in the phyla Actinobacteria, Bacteroidetes, Firmicutes and Proteobacteria. More specifically, we detected an increased abundance of the Eubacterium genus in AC tissues when directly compared to taxonomic profiles of the AO cohort, whereas the genera Fusobacterium, Phascolarctobacterium, Bacteroides, Roseburia, Dialister, Stenotrophomonas and Ruminococcus were more prevalent in the AO cohort (Figure 4C).
DISCUSSION
We performed genome-wide DNA methylation profiling on normal colon mucosa from African American and Caucasian CRC patients and age-, sex-, and race-matched controls. Our hypothesis was that CRC incidence and outcome were associated with underlying differences in the normal tissue epigenome.
Unsupervised hierarchical cluster analysis (Figure 1A) and principal component analysis (Figure 1B) both suggested the existence of a separate group of CRC patients with dramatically disrupted normal tissue methylomes. Interestingly, we were also able to identify this same group of epigenetically disrupted individuals by using a simple metric of outlier determination as used previously by our group(14,15). We have termed this group of CRC patients as “Outlier Methylation Phenotype” (OMP) (see also Figure 2). We also identified OMPs in publicly available colorectal cancer datasets. OMP frequencies varied from 1 to 2% in controls and 8 to 30% in the CRC patients. This clearly suggests that CRC patients are more prone to develop OMPs compared to controls. Furthermore, the varying percentage of OMPs among CRC patients (<10% in GSE48684 and GSE131013; and ~30% in GSE 132804) in these datasets could be explained by smaller sample size (24 CRC patients in GSE48684) or difference in ethnicity (Spanish population in GSE131013). Unfortunately, these datasets do not have any African American samples, so we could not perform race-specific analyses.
While we identified many differences in average site-specific methylation between CRC patients and controls, confirming and extending our previous studies(3,4), the major difference we identified between African American and Caucasian CRC patients was in the number of patients with OMP(15). African Americans (26%) were more than twice as likely to be OMPs compared to Caucasian (11%) CRC patients. Furthermore, African Americans CRC patients displayed higher abnormality in methylation profiles (AC vs AH) than their Caucasian counterparts (CC vs CH). However, methylation differences between the AC and AH groups were greatly reduced on excluding the OMPs, suggesting a substantial role for OMPs in causing epigenetic disbalances in African American CRC patients.
Because the frequency of OMPs appears higher among African Americans (Figure 2), and OMPs have sometimes been associated with undesirable outcomes in other diseases(14), as well as cancer(25,26), a greater frequency of OMPs among African American CRC patients could be associated with racial disparities in CRC incidence and outcome. However, too few OMPs have been identified to determine whether this unusual molecular phenotype is associated with any clinical outcome or any established molecular subtype in CRC patients. However, it is noteworthy, that our previous study on OMP in TCGA data showed that OMP is independent of CIMP (15). Another important aspect of cancer including CRC is the significance of epigenetic aging in tumorigenesis, and its potential use for cancer risk prediction (27). It would be interesting to further evaluate if OMPs have epigenetic age drift in normal tissues, which could be used as a predictive and prognostic tool. Nevertheless, determining the cause of OMP in normal tissues is of interest because of its potential to affect gene expression in normal colon mucosa, as well as the potential for environmental factors to influence this phenotype.
Our analysis of gene expression, comparing normal colon mucosa of OMP cancer patients with non-OMP cancer patients, indicated that the major pathways differentially affected in OMP patients were involved in repression of genes mediating the interaction between the intestinal epithelium/mucus barrier and the microbiome. For instance, a number of genes from the cadherin superfamily, claudins and other transmembrane junction proteins were downregulated in the OMP group. Cadherins and claudins are integral parts of adherens and tight junctions, respectively. Cadherins are important cell adhesion molecules and loss of cell adhesion, specifically by downregulation of E-cadherin (CDH1) has been associated with malignant characteristics including tumor progression, loss of differentiation, invasion and metastasis(28). On the other hand, claudins are transmembrane proteins that maintain the barrier functioning of tight junctions(29). Clearly, loss of expression of these and other transmembrane junction proteins leads to deregulation of normal tissue function and development of epithelium related diseases, including cancer(30). Furthermore, genes belonging to the mucin family were downregulated in OMP cancer patients. Aberrant mucin expression is linked to chronic inflammation and CRC, as mucus functions as a physical barrier and influences microbial composition by providing nutrients and attachment sites for the microbial community(31).
Analysis of the microbiome further showed differential abundance of several genera between OMPs vs non-OMP CRC patients. The genus Eubacterium was found to be in lower abundance in OMPs in our study. Interestingly, the abundance of Eubacterium hallii, and Eubacterium ventriosum were found to be significantly higher in healthy samples than in CRC samples(32). E. hallii utilizes glucose and the fermentation intermediates acetate and lactate to form butyrate and hydrogen, which are important in maintaining intestinal metabolic balance(33).
Fusobacterium and Bacteroides, which are among the most prominent CRC associated bacteria, were highly abundant in OMPs compared to non-OMPs (34). Fusobacterium is also known to be associated with microsatellite instability (MSI), hypermethylation and malignant transformation of epithelial cells (35). On the other hand, Bacteroides fragilis cause a series of inflammatory reactions due to B. fragilis toxin (BFT), which leads to chronic intestinal inflammation and tissue injury and plays a crucial role leading to CRC(36).
Other genera found to be in higher abundance in OMPs, such as Phascolarctobacterium, Roseburia, Ruminococcus, Diallister and Stenotrophomonas have also been reported to be in higher abundance in CRC patients in other studies(37–40). Furthermore, Ruminococcus gnavus has been positively associated with KRAS mutations (a known CRC mutation)(41). Recent studies have also highlighted the role of Dialister pneumosintes in advanced CRC patients(42). Stenotrophomonas maltophilia is a nosocomial pathogen which is found in higher abundance in CRC patients after radio or chemotherapy(43).
A recent study (44) showed that the overall microbial composition in normal adjacent tissues is relatively similar to their tumor tissues, with the exceptions of some bacteria which show different prevalence between these two tissue types. This suggests that some of the microbiome changes that we observe may be affected by the presence of an adjacent neoplasm.
African American race is widely understudied and underrepresented in both publicly available datasets (like TCGA) and tissue biobanks. We were limited by the number of African American biospecimens available in the biobank. It is to be noted that some of the largest CRC biobanks and Consortiums have negligible representation of African Americans.
Although our sample size was insufficient to clinically characterize (like tumor grade, side of tumor, age, sex) the OMP group, analysis of the microbiome clearly reflected that normal colon mucosa of OMPs are enriched in bacterial genera associated with CRC risk, advanced tumor stage, chronic intestinal inflammation, malignant transformation, nosocomial infections and KRAS mutations. These observations suggest that OMP patients may have microbial dysbiosis that is distinct from that of non-OMP patients.
In conclusion, we identified a distinct group of highly abnormally methylated CRC patients, termed “OMPs”, and validated their existence using multiple statistical approaches and in multiple datasets. This epigenetically disrupted OMP group was more prevalent among African American CRC patients than Caucasian CRC patients. Furthermore, we showed that the vast majority of methylation differences between African Americans CRC patients and healthy controls are driven by this OMP group. We were also able to demonstrate downregulation of crucial genes in the OMP group, especially mucins and transmembrane junction genes. Finally, microbiome analysis showed higher abundance of microbial genera that are associated with CRC risk, malignancy and advanced tumor stage in OMP cancer patients compared to non-OMP cancer patients.
Whether these differences might be a cause or effect of normal colon OMP is unclear. Such questions are only likely to be answered by examination of a much larger number of OMP patients. In this regard, a major consideration for future studies is the relative rarity of OMP individuals, and a major weakness of the present study is the small number of OMP individuals examined. If OMPs are, in fact, more prevalent among patients of African ancestry, examination of a much larger number of such patients might shed additional light on the significance of this phenotype, as well as whether it might be associated with observed racial disparities in colon cancer incidence and outcome.
Supplementary Material
Financial Support:
The project described was supported by TUFCCC/HC Regional Comprehensive Cancer Health Disparity Partnership, Award Number U54 CA221704(5) from the National Cancer Institute (to C. Sapienza, J.P.J. Issa, F.E. Kleiman, K. Krampis). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute or the National Institutes of Health. Work in the Issa laboratory is supported by National Institutes of Health grants CA214005 (to J.P.J. Issa). Work in the Sapienza laboratory is also supported by two PA Cure grant 914103047 (to C. Sapienza) from Pennsylvania Department of Health and R21 CA264213 (to C. Sapienza and J. Ghosh) from National Institute of Health.
Footnotes
Conflict of Interest: The authors declare no potential conflicts of interest.
REFERENCES
- 1.Siegel RL, Miller KD, Goding Sauer A, Fedewa SA, Butterly LF, Anderson JC, et al. Colorectal cancer statistics, 2020. CA Cancer J Clin 2020;70:145–64 [DOI] [PubMed] [Google Scholar]
- 2.Guinney J, Dienstmann R, Wang X, de Reyniès A, Schlicker A, Soneson C, et al. The consensus molecular subtypes of colorectal cancer. Nat Med 2015;21:1350–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Silviera ML, Smith BP, Powell J, Sapienza C. Epigenetic differences in normal colon mucosa of cancer patients suggest altered dietary metabolic pathways. Cancer Prev Res (Phila) 2012;5:374–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cesaroni M, Powell J, Sapienza C. Validation of methylation biomarkers that distinguish normal colon mucosa of cancer patients from normal colon mucosa of patients without cancer. Cancer Prev Res (Phila) 2014;7:717–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Leclerc D, Pham DN, Lévesque N, Truongcao M, Foulkes WD, Sapienza C, et al. Oncogenic role of PDK4 in human colon cancer cells. Br J Cancer 2017;116:930–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Giovannucci E, Willett WC. Dietary factors and risk of colon cancer. Ann Med 1994;26:443–52 [DOI] [PubMed] [Google Scholar]
- 7.Carethers JM. Clinical and Genetic Factors to Inform Reducing Colorectal Cancer Disparitites in African Americans. Front Oncol 2018;8:531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lieberman DA, Williams JL, Holub JL, Morris CD, Logan JR, Eisen GM, et al. Race, ethnicity, and sex affect risk for polyps >9 mm in average-risk individuals. Gastroenterology 2014;147:351–8; quiz e14–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zavala VA, Bracci PM, Carethers JM, Carvajal-Carmona L, Coggins NB, Cruz-Correa MR, et al. Cancer health disparities in racial/ethnic minorities in the United States. Br J Cancer 2021;124:315–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mani S, Ghosh J, Lan Y, Senapati S, Ord T, Sapienza C, et al. Epigenetic changes in preterm birth placenta suggest a role for ADAMTS genes in spontaneous preterm birth. Hum Mol Genet 2019;28:84–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aran D, Sirota M, Butte AJ. Systematic pan-cancer analysis of tumour purity. Nat Commun 2015;6:8971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pidsley R, Zotenko E, Peters TJ, Lawrence MG, Risbridger GP, Molloy P, et al. Critical evaluation of the Illumina MethylationEPIC BeadChip microarray for whole-genome DNA methylation profiling. Genome Biol 2016;17:208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou W, Laird PW, Shen H. Comprehensive characterization, annotation and innovative use of Infinium DNA methylation BeadChip probes. Nucleic Acids Res 2017;45:e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ghosh J, Mainigi M, Coutifaris C, Sapienza C. Outlier DNA methylation levels as an indicator of environmental exposure and risk of undesirable birth outcome. Hum Mol Genet 2016;25:123–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ghosh J, Schultz B, Coutifaris C, Sapienza C. Highly variant DNA methylation in normal tissues identifies a distinct subclass of cancer patients. Adv Cancer Res 2019;142:1–22 [DOI] [PubMed] [Google Scholar]
- 16.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013;29:15–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huntley MA, Larson JL, Chaivorapol C, Becker G, Lawrence M, Hackney JA, et al. ReportingTools: an automated result processing and presentation toolkit for high-throughput genomic analyses. Bioinformatics 2013;29:3220–1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol 2019;37:852–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJ, Holmes SP. DADA2: High-resolution sample inference from Illumina amplicon data. Nat Methods 2016;13:581–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bokulich NA, Kaehler BD, Rideout JR, Dillon M, Bolyen E, Knight R, et al. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 2018;6:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McDonald D, Price MN, Goodrich J, Nawrocki EP, DeSantis TZ, Probst A, et al. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J 2012;6:610–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McMurdie PJ, Holmes S. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 2013;8:e61217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oksanen J, Blanchet FG, Kindt R, Legendre P, Minchin PR, O’hara R, et al. Package ‘vegan’. Volume 2(9): Community ecology package, version; 2013. p 1–295. [Google Scholar]
- 25.Teschendorff AE, Gao Y, Jones A, Ruebner M, Beckmann MW, Wachter DL, et al. DNA methylation outliers in normal breast tissue identify field defects that are enriched in cancer. Nat Commun 2016;7:10478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Panjarian S, Madzo J, Keith K, Slater CM, Sapienza C, Jelinek J, et al. Accelerated aging in normal breast tissue of women with breast cancer. Breast Cancer Res 2021;23:58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yu M, Hazelton WD, Luebeck GE, Grady WM. Epigenetic Aging: More Than Just a Clock When It Comes to Cancer. Cancer Res 2020;80:367–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Christou N, Perraud A, Blondy S, Jauberteau MO, Battu S, Mathonnet M. E-cadherin: A potential biomarker of colorectal cancer prognosis. Oncol Lett 2017;13:4571–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chiba H, Osanai M, Murata M, Kojima T, Sawada N. Transmembrane proteins of tight junctions. Biochim Biophys Acta 2008;1778:588–600 [DOI] [PubMed] [Google Scholar]
- 30.Bujko M, Kober P, Mikula M, Ligaj M, Ostrowski J, Siedlecki JA. Expression changes of cell-cell adhesion-related genes in colorectal tumors. Oncol Lett 2015;9:2463–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Coleman OI, Haller D. Microbe-Mucus Interface in the Pathogenesis of Colorectal Cancer. Cancers (Basel) 2021;13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ai D, Pan H, Li X, Gao Y, Liu G, Xia LC. Identifying Gut Microbiota Associated With Colorectal Cancer Using a Zero-Inflated Lognormal Model. Front Microbiol 2019;10:826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Engels C, Ruscheweyh HJ, Beerenwinkel N, Lacroix C, Schwab C. The Common Gut Microbe Eubacterium hallii also Contributes to Intestinal Propionate Formation. Front Microbiol 2016;7:713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ternes D, Karta J, Tsenkova M, Wilmes P, Haan S, Letellier E. Microbiome in Colorectal Cancer: How to Get from Meta-omics to Mechanism? Trends Microbiol 2020;28:401–23 [DOI] [PubMed] [Google Scholar]
- 35.Zhou Z, Chen J, Yao H, Hu H. Fusobacterium and Colorectal Cancer. Front Oncol 2018;8:371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cheng WT, Kantilal HK, Davamani F. The Mechanism of Bacteroides fragilis Toxin Contributes to Colon Cancer Formation. Malays J Med Sci 2020;27:9–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Flemer B, Lynch DB, Brown JM, Jeffery IB, Ryan FJ, Claesson MJ, et al. Tumour-associated and non-tumour-associated microbiota in colorectal cancer. Gut 2017;66:633–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Loftus M, Hassouneh SA, Yooseph S. Bacterial community structure alterations within the colorectal cancer gut microbiome. BMC Microbiol 2021;21:98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weir TL, Manter DK, Sheflin AM, Barnett BA, Heuberger AL, Ryan EP. Stool microbiome and metabolome differences between colorectal cancer patients and healthy adults. PLoS One 2013;8:e70803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Peters BA, Dominianni C, Shapiro JA, Church TR, Wu J, Miller G, et al. The gut microbiota in conventional and serrated precursors of colorectal cancer. Microbiome 2016;4:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hong BY, Ideta T, Lemos BS, Igarashi Y, Tan Y, DiSiena M, et al. Characterization of Mucosal Dysbiosis of Early Colonic Neoplasia. NPJ Precis Oncol 2019;3:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Osman MA, Neoh HM, Ab Mutalib NS, Chin SF, Mazlan L, Raja Ali RA, et al. Parvimonas micra, Peptostreptococcus stomatis, Fusobacterium nucleatum and Akkermansia muciniphila as a four-bacteria biomarker panel of colorectal cancer. Sci Rep 2021;11:2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mori G, Rampelli S, Orena BS, Rengucci C, De Maio G, Barbieri G, et al. Shifts of Faecal Microbiota During Sporadic Colorectal Carcinogenesis. Sci Rep 2018;8:10329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nejman D, Livyatan I, Fuks G, Gavert N, Zwang Y, Geller LT, et al. The human tumor microbiome is composed of tumor type-specific intracellular bacteria. Science 2020;368:973–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during the current study are available in the GEO repository (GSE 199057).