

Abstract
Background:
The identification of patients with homologous recombination deficiency (HRD) beyond BRCA1/2 mutations is an urgent task, as they may benefit from PARP inhibitors. We have previously developed a method to detect mutational signature 3 (Sig3), termed SigMA, associated with HRD from clinical panel sequencing data, that is able to reliably detect HRD from the limited sequencing data derived from gene-focused panel sequencing.
Methods:
We apply this method to patients from two independent datasets: (1) high-grade serous ovarian cancer and triple-negative breast cancer (TNBC) from a Phase 1b trial of the PARP inhibitor olaparib in combination with the PI3K inhibitor buparlisib (BKM120) (NCT01623349), and (2) TNBC patients who received neoadjuvant olaparib in the Phase II PETREMAC trial (NCT02624973).
Results:
We find that Sig3 as detected by SigMA is positively associated with improved progression-free survival and objective responses. In addition, comparison of Sig3 detection in panel and exome sequencing data from the same patient samples demonstrated highly concordant results and superior performance in comparison with the genomic instability score.
Conclusion:
Our analyses demonstrate that HRD can be detected reliably from panel sequencing data that are obtained as part of routine clinical care, and that this approach can identify patients beyond those with germline BRCA1/2mut who might benefit from PARP inhibitors. Prospective clinical utility testing is warranted.
INTRODUCTION
Targeting DNA damage repair with PARP inhibitors (PARPi) has been approved in ovarian, breast, pancreatic, and prostate cancer.1–5 In breast cancer, patients are currently selected based on the presence of a germline BRCA1/2 pathogenic mutation. However, candidate lesions for PARP inhibitor sensitivity include loss-of-function mutations in a large number of other genes that result in homologous recombination deficiency (HRD), suggesting that testing based only on germline BRCA1/2mut may not identify the full set of patients who could benefit from PARPi. In addition, as loss-of-function mutations in these tumor suppressor genes frequently do not occur in hot spots, testing PARP inhibitor sensitivity based on the presence of individual mutations in clinical trials is not realistic for the less common HRD genes and may miss cases where HRD is the result of polygenic tumor development.5, 6
BRCA1/2mut and other HRD tumors have been shown to display a specific pattern of genome-wide somatic single nucleotide variations (SNVs) defined as “mutational signature 3” (Sig3) in the COSMIC signature catalog, which consists of several dozen “signatures” based on the base substitution types and the trinucleotide context in which the substitutions occur.7, 8 A pattern of insertions, deletions, and rearrangements have also been associated with BRCA1/2mut tumors.8 HRD status can also be inferred from the copy number variation (CNV) profile of the tumors. For example, a commercial platform for detecting HRD called MyChoice from Myriad Genetics calculates genomic instability score (GIS), comprised of loss-of-heterozygosity (LOH), telomeric allelic imbalance (TAI), and large-scale state transitions (LST).4 Both Sig3 and GIS are strongly associated with BRCA1/2 mutations and have been proposed as genomic markers of HRD, and their ability to predict sensitivity to PARPi has been shown in preclinical models.8–14 HRD is also associated with other specific alterations, such as excess of deletions with microhomology at the deletion junction and long tandem duplications. When whole-genome sequencing (WGS) data are available, one could also use the HRDetect diagnostic tool, which utilizes a weighted model that accounts for both substitution and rearrangement signatures to identify HRD cases, even when they are not BRCA1/2mut.11
Clinical validation of these genomic biomarkers of HRD has aimed at identifying patients who might benefit from platinum- or PARPi-based treatment. In breast cancers, LOH and LST scores were associated with platinum responses in the PrECOG 010515 and TBCRC 00916 trials. GIS predicted pathologic responses with neoadjuvant chemotherapy17 and improved disease-free survival with adjuvant epirubicin + cyclophosphamide chemotherapy in SWOG S9313.18 However, GIS has not been predictive of response in all studies — the Myriad HRD assay was not associated with responses to either neoadjuvant cisplatin or paclitaxel in TBCRC 03019 or to docetaxel or carboplatin in the metastatic setting in the TNT trial.20 In ovarian cancers, GIS was associated with progression-free survival (PFS) and overall survival.21 In terms of PARPi, several studies have shown that ovarian cancer patients with high GIS have longer PFS and higher response rates.1, 4, 22, 23 Instead of using GIS, the ARIEL3 investigators used genome-wide LOH (gLOH) as defined by Foundation Medicine’s T5 assay24 and showed that a gLOH > 16% was associated with longer PFS.25
Other methods for testing HRD status have practical limitations: standard Sig3 detection requires whole-exome sequencing (WES) or WGS data, GIS requires a proprietary assay for profiling BRCA1/2 mutations and copy number profiling, and HRDetect requires WGS. In particular, none of these methods could be applied to cancer gene panels (typically 100–400 genes) that are often employed in clinical settings. To overcome this limitation, we have previously developed the SigMA (Signature Multivariate Analysis) algorithm that can infer the presence of Sig3 even when the number of detected mutations is an order of magnitude smaller due to the narrow region of the genome targeted by the panel.14 We initially showed that Sig3 prediction from panels is a good proxy for Sig3 prediction from WES using several cancer cell lines that were treated with PARPi.14 Subsequently, we used SigMA to analyze data from the TOPACIO trial (combination of niraparib with pembrolizumab in ovarian cancer). We showed that Sig3-positivity inferred from panels was associated with response to combined PARP- and PD-1-inhibition in ovarian cancer.26
Here, we assess the validity of panel-based Sig3 (panel-Sig3) prediction by SigMA through a direct comparison with paired WES data as well as with GIS and BRCA1/2 status on the same tumors. The applicability of panel-Sig3 would be of great clinical importance because it would provide a quicker and much less expensive biomarker in PARPi and other HRD-related clinical studies. We utilized data from a study evaluating the combination of the PI3K inhibitor buparlisib (BKM-120) with olaparib in patients with advanced TNBC or high-grade serous ovarian cancer (OC) cancer,27 where the combined regimen yielded a response rate of 29%. We also externally validated panel-Sig3 using data from the Phase II PETREMAC trial, which investigated single-agent neoadjuvant olaparib in unselected and treatment-naïve triple-negative breast cancer (TNBC) and found an objective response rate of 56% not restricted to patients harboring germline BRCA1/2 mutations.28 In both studies, responses to PARP inhibition were not limited to carriers of a germline BRCA1/2mut, making these data ideal for our Sig3 validation study.
METHODS
Clinical data and patient selection
NCT01623349 is a multicenter phase 1b trial of escalating doses of olaparib and buparlisib.27 Clinical trial data was obtained from the study registered at ClinicalTrials.gov. This was a multicenter, open-label, phase 1b trial with a 3 + 3 dose-escalation design. Eligibility criteria included age of at least 18 years, diagnosis of recurrent TNBC, or diagnosis of high-grade serous ovarian cancer or recurrent breast cancer of any histology with germline BRCA mutations. We obtained whole-exome sequencing (WES) and panel sequencing from 37 participants (cohort 1) based on the availability of tissue. DNA extraction and construction of libraries for massively parallel sequencing were performed as previously described.29 For the analysis of the concordance between different HRD detection methods, all 37 patients were included. For the analysis of responses, there were 5 patients who went off study in the absence of progression (radiological or clinical) and thus were not analyzed for response rates and were censored in the time-to-event analyses. The PETREMAC trial (NCT02624973) is a multicenter Phase II study that included patients with stage II/III breast cancer that were stratified to eight different neoadjuvant treatment regimens. The cohort of 32 patients with TNBC (cohort 2) received initial olaparib monotherapy for up to 10 weeks, before assessment of tumor response. Clinical and radiological evaluation of tumor size was carried out by each local investigator. Tumor biopsies studies in this report were collected before olaparib treatment and underwent targeted DNA sequencing (360 genes), as previously described.28, 30 All study participants provided written informed consent; the studies were conducted in accordance with Declaration of Helsinki ethical guidelines and approved by an institutional review board (IRB).
The Cancer Genome Atlas (TCGA) data
The germline mutation calls by TCGA were also downloaded from GDC data portal;31 the overall pathogenicity determined in that publication allowed us to select samples with germline mutations. Four samples with Prioritized VUS were not considered as germline BRCA1/2mut. The consensus somatic single base substitution and indel calls32 were also downloaded from the GDC data portal.31 The samples with somatic BRCA1/2 mutations in the TCGA BRCA and OV cohorts were selected based on the presence of pathogenic annotation ClinVar database, or a damaging/deleterious effect prediction by SIFT or PolyPhen as long as these alterations were not annotated to be benign or likely benign in ClinVar.
Mutation calls
Single base substitutions:
Somatic single base substitutions were calculated using MuTect2 and germline SNPs were called with HaplotypeCaller, according to the GATK best practices.33 Somatic single base substitutions with supporting read count less than 8 and 2, total depth less than 20 and 5, allelic fraction less than 0.1 and 0.05, for MSK-IMPACT panels and WES data, respectively, were filtered out.
Copy number alterations:
Segment level copy number alterations (CNAs) were calculated using Sequenza34 and gene-level CNAs using GISTIC35 from the segment-level Sequenza results.
HRD detection methods
GIS calculation:
Using Sequenza34 copy number calls and scarHRD algorithm,36 we calculated LOH (median and standard deviation, SD, in BRCA1/2mut tumors were 16.5 and 6.8, respectively), LST (median and SD of 25.5 and 10.3, respectively) and TAI (median and SD of 24.0 and 8.0, respectively). The sum of these quantities yields GIS. We compared the distribution of GIS values in our cohort with those from TCGA for BRCA1/2mut tumors (Supplementary Figure 1). The GIS values for the TCGA were calculated from SNP arrays by the TCGA Pan-Cancer Atlas project.37, 38
Sig3 calculation:
The Sig3 calculation was carried out with SigMA algorithm.14 For analysis of TCGA tumors the in-built multivariate classifiers with data parameter ‘tcga_mc3’ were used. Similarly, for MSK-IMPACT panels and WES data in cohort 1, Sig3 predictions were performed with the in-built multivariate classifiers with data parameter ‘msk’ and ‘seqcap’, respectively. The predictions were done for breast cancers and ovarian cancers separately using the corresponding tumor type settings (tumor_type parameter ‘breast’ and ‘ovary’). For MSK-IMPACT panels samples with less than five SNVs are classified as Sig3−. The 77% of panels in cohort 1 has ≥ 5 SNVs. In simulations, 91% of Sig3+ and 66% of samples have ≥ 5 SNVs. The samples that have < 5 SNVs are accounted for in the calculation of 74% sensitivity at 10% FPR. For cohort 2 a new classifier was trained using simulations that are generated following the same procedure as previously described.14 Due to the smaller library size of panels in cohort 2 and the lower counts of SNVs, Sig3 detection was performed for all samples with ≥ 4 SNVs, and 75% of samples satisfied this criterion. In simulations 90% of Sig3+ and 65% of Sig3− samples have ≥ 4 SNVs and samples that have < 4 SNVs are accounted for in the calculation of 70% sensitivity at 10% FPR. In this study, we used the threshold in WES data that corresponds to 5% FPR and 90% sensitivity, and the one that corresponds to 10% FPR and 75% sensitivity in panel data to define Sig3+ tumors. The sensitivity and FPR are determined using panel and WES simulations generated by down-sampling WGS data with respect to the ground truth defined based on the Sig3 calculation in WGS data.
SigProfiler calculation:
To compare SigMA based Sig3 calculation to other popular signature analysis methods, we used SigProfiler.39 The de novo signature analysis with SigProfiler found 3 signatures in our cohort. The signatures were matched to the COSMIC catalog v3 and those with the highest cosine similarity were identified. Signature A was most similar to Sig3 (Supplementary Figure 2).
Sig3 selection thresholds for SigMA:
To define Sig3+ samples from panel and WES data we used thresholds that correspond to 10% and 5% FPR, respectively. The 10% FPR threshold was found to capture more true positives and had an overall better predictive value of response, while in WES data the 5% threshold selects samples with higher Sig3 exposure (Supplementary Figure 3) and allows better separation in PFS. These results are summarized in the comparison of:
Comparison of GIS and Sig3 exposures in WES data between samples that have SigMA scores above the 5% and 10% FPR thresholds (Supplementary Figure 3).
Comparison of PFS between Sig3+ and Sig3− samples with different thresholds. In panel data, the PFS was also better associated with the 10% FPR (reported in results section) compared to 5% FPR (p = 0.57, median 5.7 and 7.7). Instead, in WES data, the 10% threshold provided worse PFS separation (p = 0.19, median 5.63 and 6.97) compared to 5% threshold (reported in the results section).
The number of patients that are BRCA1/2mut in different Sig3 categories (Supplementary Figure 4).
Comparison of Sig3 detection with SigMA in WES data compared to other methods:
The detection of Sig3 from WES with SigMA and 5% FPR threshold compared to alternative signature detection approaches. We compared PFS of WES-Sig3+ classification with SigMA to PFS of samples with positive Sig3 exposure calculated by NNLS (Supplementary Figure 5) as well as with non-negative matrix factorization (NMF, as implemented in SigProfiler algorithm; Supplementary Figure 2). A significantly longer PFS is observed in samples that had a Signature A exposure greater than 25, but the association was still inferior in comparison to SigMA classification (lower significance, smaller PFS increase 2.2 months versus 4.24 months with SigProfiler and SigMA, respectively). Confirming our observation of lower Sig3 exposure in Sig3− samples with NNLS, the NMF analysis yielded a similarly low exposure distribution in the WES-Sig3− group (Supplementary Figure 2) even if there were samples with positive exposures. The Sig3+ samples selected with SigMA associate better to the PFS compared to other signature analysis methods.
Survival analysis
Survival analyses were performed with the Kaplan-Meier method using survival R package. Patients stratified according to Sig3 status and tumor type. Curves were compared using the log-rank method. A Cox regression analysis was performed to estimate the effect size of each feature. The p-values for Cox-regression were calculated by Wald test.
Pathway analysis
The difference in copy number gain and loss frequencies of genes is used to calculate geneset enrichment scores with ReactomePA package (Supplementary Table 1). The gene level differences can be found in Supplementary Table 2.
Code availability
The SigMA algorithm is available on GitHub (https://github.com/parklab/SigMA). The code used in the analysis will be provided upon request from the authors.
Data availability
The authors support the dissemination of research data that has been generated, and increased cooperation between investigators. The data that support the findings of this study are available at doi:10.17632/zg5xvm3f3w.1 Raw data for this study were generated at the Dana-Farber Cancer Center (Boston, MA) and Memorial Sloan Kettering Cancer Center (New York, NY). Derived data supporting the findings of this study are available at the database of Genotypes and Phenotypes (dbGaP) via study accession: phs003019.v1.p1. Further de-identified individual participant data will be provided according to institutional procedures. Requests addressed to G.M. Wulf must include a description of the nature of the proposed research and extent of data requirements. Data recipients are required to enter a formal data sharing agreement that describes the conditions for release and requirements for data transfer, storage, archiving, publication and intellectual property. Requests are reviewed by the study team in terms of scientific merit and ethical considerations, including patient consent.
Haukeland University Hospital and the University of Bergen support the dissemination of research data and cooperation between investigators nationally and internationally. Based on current Norwegian laws and regulations, and the biobank approval for the PETREMAC trial given by the Regional Ethics Committee Western Norway before the study commenced, genomic data are not to be made openly available. After publication and upon formal request, raw sequencing data, including de-identified individual participant data and a data dictionary defining each field in the data set, may be shared according to institutional guidelines, pending project-specific approvals from the Regional Ethics Committee in Norway. Requests are via a standard pro forma describing the nature of the proposed research and extent of data requirements. Data recipients are required to enter a formal data sharing agreement that describes the conditions for release and requirements for data transfer, storage, archiving, publication and intellectual property. Requests are reviewed by the PETREMAC study team in terms of scientific merit and ethical considerations, including patient consent. An evaluation as described above will typically take three months. Requests may be directed to H.P. Eikesdal.
RESULTS
Cohorts and high throughput sequencing
NCT01623349 was a multicenter phase 1b trial of escalating doses of olaparib and buparlisib.27 Archival tumor material was collected prior to enrollment, and WES, and panel sequencing was obtained from the 37 patients (cohort 1) based on the availability of tissue. These patients include 26 with high-grade serous ovarian cancer (including four fallopian tube and two primary peritoneal tumors) and 11 with triple-negative breast cancer (Figure 1A). Panel sequencing was performed using the MSK-IMPACT (Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets) panel, which covers 341 genes (2.1 Mb). WES was performed using solution hybrid selection (73.7 Mb). We determined the Sig3 status from both panel and WES data using SigMA. We also calculated the GIS from WES data with the scarHRD algorithm.36 We compared the GIS in BRCA1/2 mutant tumors in our cohort to the calculation by the PanCanAtlas consortium in the TCGA data. See methods for detailed information. The PETREMAC study was a Phase II trial evaluating olaparib monotherapy in the neoadjuvant setting for 32 patients with untreated TNBC (cohort 2).28 Pre-treatment biopsies were extracted and submitted for targeted DNA sequencing applying a 360-gene panel (1.5 Mb).30 Cohort 1 had on average 9.9 mutations (median = 7, standard deviation 9.5) per sample and cohort 2 had 6.96 (median = 5, standard deviation = 5.0) mutations. It is important to note that using synonymous and non-coding region mutations increases the counts of mutations available for signature analysis and improves the detection accuracy. The 77% and 57% of mutations used in the signature analysis for cohorts 1 and 2 were either non-coding or synonymous.
Figure 1.
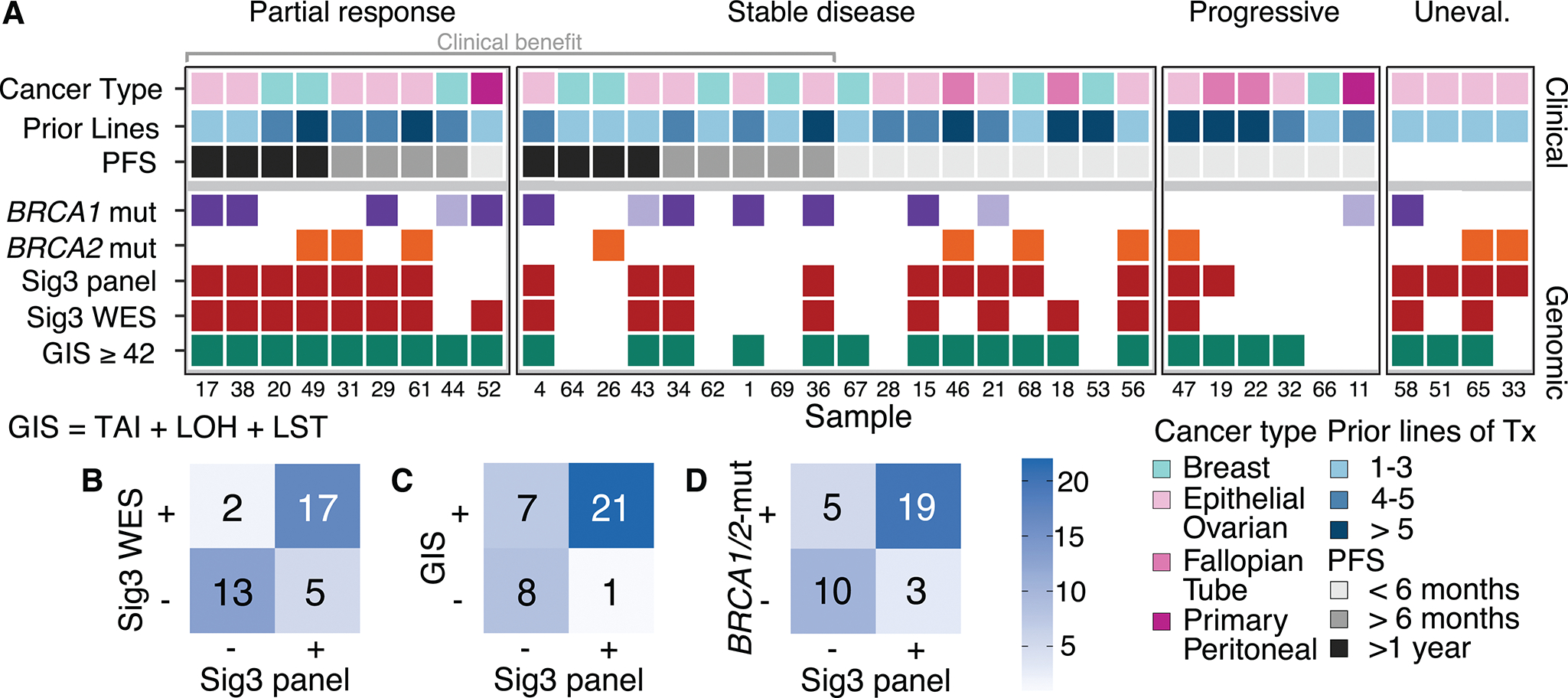
Cohort 1 details and comparison of different HRD detection methods. A, Clinical and genomic characteristics for each sample (small numbers at the bottom represent the individual study ID of each participant), grouping based on RECIST (Response Evaluation Criteria in Solid Tumors) best overall response criteria. Clinical characteristics include the different cancer types and number of prior treatments. Different intervals of progression-free survival are shown in the third row. Genomic characteristics include BRCA1/2 mutation status, Sig3+ classification by SigMA from panel and WES data, and the GIS calculated from WES. The lighter fill color of BRCA1/2m indicates lack of LOH. B, 2×2 table of Sig3 status identified from WES and panel concordance is 81%.C, 2×2 table of GIS calculated with scarHRD versus Sig3-status from panel data. D, Mutations of BRCA1/2 versus Sig3 status. One patient had both a BRCA1 and BRCA2 mutation. WT: wild type. Tx: therapy. GIS: Genomic Instability Score. TAI: Telomeric Allelic Imbalance. LOH: Loss of Heterozygosity. LST: Large Scale Transitions.
Sig3 predictions with SigMA algorithm
The SigMA algorithm infers the HRD status from panel data by estimating the likelihood that the observed mutational spectrum results from Sig3.14 A compendium of WGS cases collected by the International Cancer Genome Consortium (ICGC) provides the resource for determining which signatures tend to co-occur and their frequencies in different tumor tissue types.40 The likelihood measure indicates the compatibility of mutations identified in a patient to the expected mutational spectra of Sig3+ tumors in the ICGC data. By combining these likelihoods with other signature-related features in a multivariate machine-learning classifier, SigMA robustly predicts the presence of mutations from Sig3. Classifiers are trained using realistic simulations of panels or WES obtained by down-sampling whole genomes (WGS) that provide the ‘true’ Sig3 tags. The sensitivity and false-positive rate (FPR) are calculated by comparing the Sig3 classification in simulated panels or WES to the categories in WGS data. For MSK-IMPACT panels used to sequence cohort 1, Sig3 detection was shown to have a sensitivity of 74% at 10% FPR.14 In cohort1, the previously described14 classifiers and thresholds (< 5% and < 10% FPR) were employed directly without any retraining for Sig3 classification from panels and WES. For cohort2, following the procedure described above, we simulated panels according to the specific library coverage and used the simulations to train a new classifier yielding a sensitivity of 70% at 10% FPR. For panel data in cohorts 1 and 2, we defined samples with a SigMA score larger than that corresponding to 10% FPR as Sig3. We adopted a more stringent selection criterion corresponding to a < 5% FPR to define Sig3+ tumors in WES. Higher counts of mutations in WES compared to panels allow a reduction in FPR without leading to a dramatic decrease in sensitivity (see Methods for more information).
Validation of Sig3 predictions from panels using exomes
The Sig3 classification was in agreement between WES and panel data for 30 out of 37 patients (81% concordance). For the remaining seven patients, five tested Sig3+ only for their panel data, and two tested Sig3+ only for their WES data (Figure 1B). More stringent selection criterion in WES data preferentially selects samples with a higher burden of Sig3, and the Sig3− category may include tumors with a non-negligible but low burden of Sig3, partly explaining why there are more panel-Sig3+/WES-Sig3− samples compared to panel-Sig3−/WES-Sig3+ cases (Methods; Supplementary Figure 1–2).
HRD predictions using Sig3 from panel and GIS have a significant overlap
We then compared the level of concordance with GIS for panel-Sig3 and WES-Sig3. For GIS, we imposed a threshold of 42 to define HRD based on prior work.7 As shown in Figure 1C, between panel-Sig3 and GIS, there was an agreement for 29 out of 37 cases (78% concordance); between WES-Sig3 and GIS, there was an agreement for 28 out of 37 samples (76% concordance). This suggests that panel-Sig3 provides an adequate proxy for WES-Sig3 in comparing substitution-based signature with genome instability as measured by GIS. There are seven cases with GIS+ / panel-Sig3−, but five of these were also WES-Sig3−, suggesting that the difference is between substitution-based vs. CNV-based signature, rather than a panel vs. exome difference. Two tumors were GIS− / panel-Sig3+ / WES-Sig3−, potentially indicating a false positive HRD assignment from panel sequencing. In addition, four out of seven with GIS+ / Sig3− (panel) had borderline GIS values. Although a very good correlation has been reported between WES and SNP-array dependent GIS values,36 minor adjustments may be necessary for selecting the optimal threshold. A higher threshold of GIS ≥ 49 provided the highest concordance of 86% with panel-Sig3 and 84% with WES-Sig3 (Supplementary Figure 3).
Overall, the high concordance between Sig3 calls from panel and exomes as well as between GIS score and Sig3 positivity provides orthogonal validation that panel-Sig3 can be used to identify HRD tumors. The HRD tumors defined by Sig3-positivity have a higher response rate to PARPi. The small number of discordant cases between Sig3 and GIS may reflect either the differences in the accuracies of the methods in identifying HRD, differences in underlying mechanisms that cause copy number variations and single-base substitutions in HRD tumors, or a combination of both. The impact of these different HRD biomarkers on clinical outcomes following treatment with PARPi is discussed in a later section.
Sig3 and GIS detect HRD in most BRCA1/2mut tumors
Cohort 1 had 24 BRCA1/2mut tumors of germline origin with 14 BRCA1mut and 10 BRCA2mut cases (Figure 1A). As shown in Figure 1D, there was a significant association of BRCA1 or BRCA2 mutations (p = 0.0049, Fisher’s exact test) with Sig3-positivity; 79% (19/24) of BRCA1/2mut were panel-Sig3+, 71% (17/24) were WES-Sig3+, and 87% (21/24) were GIS+. Five patients – four BRCA1mut and one BRCA2mut – had panel-Sig3−, and four of these patients were also WES-Sig3−, indicating that a fraction of BRCA1/2mut cancers arises through mechanisms unrelated to the development of Sig3. These Sig3− BRCA1/2mut samples also exhibited a numerically lower GIS than Sig3+ BRCA1/2mut cases (Supplementary Figure 3). Among the BRCA1/2 WT samples, 23% (3/13) were identified as panel-Sig3+. For 6 of the 24 BRCA mutations, the pathogenicity of the mutation according to ClinVar was uncertain (n = 2), or loss-of-heterozygosity of the second BRCA1/2 allele with a copy number loss or somatic mutations could not be established (n = 4). The two samples with BRCA1/2 alterations that qualify as variants of uncertain significance (VUS) were also Sig3+, supporting their classification as HRD-positive. Two of the four samples without biallelic loss were Sig3+, and the remaining two samples were Sig3−. Overall, we find that 20/24 (83%) BRCA1/2mut patients had a biallelic loss, mostly due to LOH.
It is important to highlight that the response rate among BRCA1/2mut tumors to PARPi therapy is approximately 50%. Thus discordance – or imperfect overlap – between these HRD detection approaches and BRCA1/2 mutations does not necessarily suggest weakness of the method. Instead, it may indicate the potential for further refinement of patient selection. Hence, comparing different methods in terms of patient outcomes is a critical step in assessing these biomarkers.
Sig3+ predicts longer PFS better than GIS+ and BRCA1/2mut in ovarian or breast cancer treated with PARPi
We evaluated the relationship of Sig3-positivity with PFS in this cohort of 37 patients with advanced and heavily pre-treated ovarian cancer (n = 26) and TNBC (n = 11) who were uniformly treated with a PARPi-containing regimen in a clinical trial setting (see Methods for details).27 PFS was significantly longer (log-rank test; p = 0.036) in the panel-Sig3+ group, with a median of 7.7 months, compared to the Sig3− group, with a median of 5.6 months (Figure 2A). Similarly, median PFS in the Sig3+ group identified with SigMA on WES data was 8.5 compared to 5.0 in the Sig3− group, (log-rank test; p=0.0057) (Figure 2B).
Figure 2.
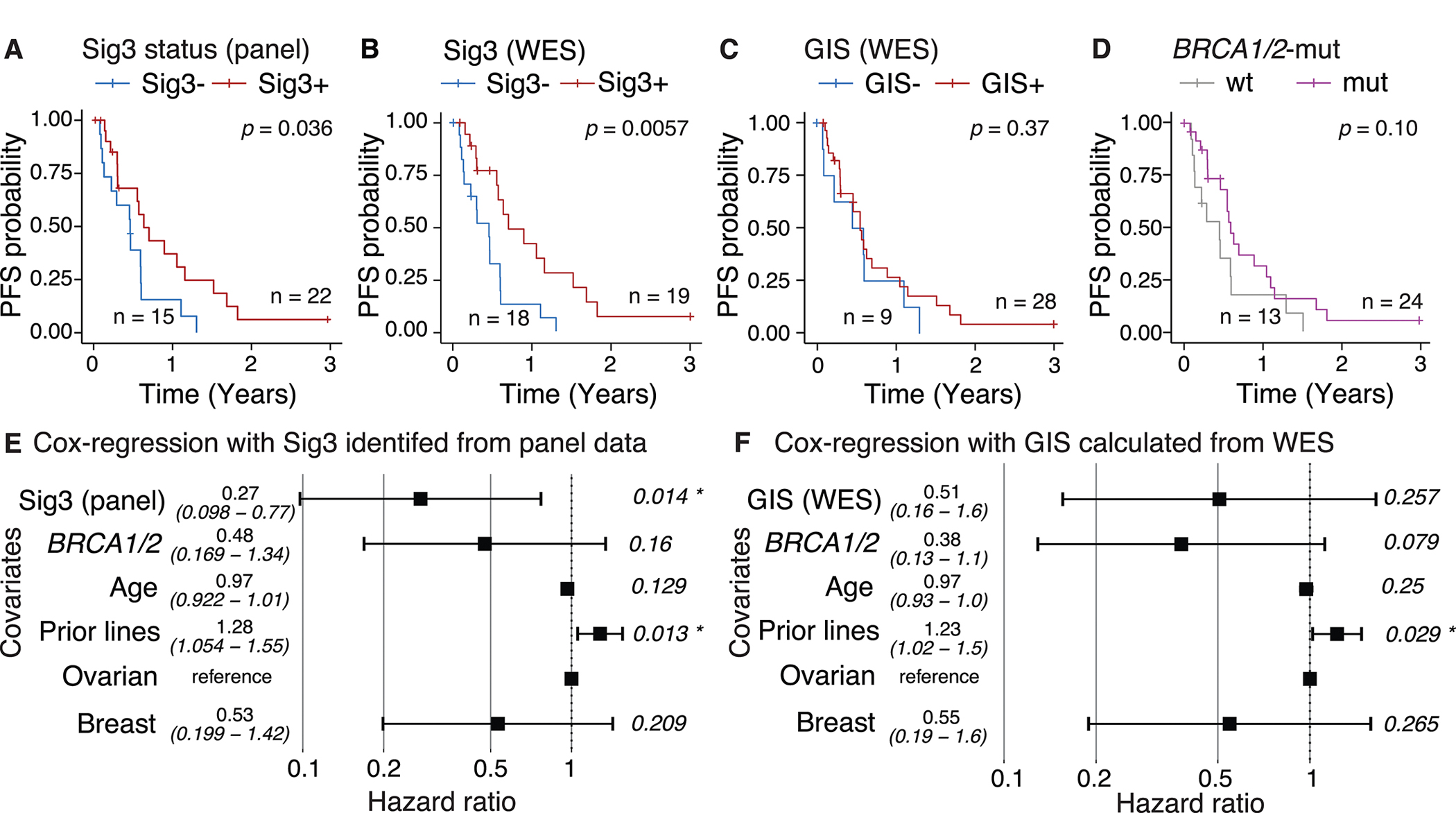
Survival analysis according to A-B Sig3-status using panel data (median survival: 5.6 versus 7.7 months in Sig3− and Sig3+ samples, respectively) and WES (median survival: 5.0 versus 8.5 in Sig3− and Sig3+ samples respectively), C, GIS (median survival: 5.6 versus 7.0 months in GIS+ and GIS− samples, respectively), D, BRCA1/2mut or WT, (median survival: 5.5 versus 7.3 months in WT versus BRCA1/2mut samples, respectively). Cox multivariate regression including E, Sig3-status, or F, GIS. Age, ECOG and stage as covariates did not have significant hazard ratio and were not included as covariates. “Prior lines” refers to the number of previous lines of therapy in the metastatic setting.
Whereas Sig3+ (WES or panel) was associated with a significantly longer PFS, GIS, and BRCA1/2mut were not observed to have any association with PFS (log-rank test: p = 0.37 and p = 0.10, respectively; Figure 2C, Figure 2D). The GIS+ / panel-Sig3− patients had a short PFS, similar to the GIS− / panel-Sig3− cases (Supplementary Figure 5). The selection with GIS ≥ 49 was somewhat better in predicting PFS than the GIS ≥ 42 selection but did not change the general conclusion (Supplementary Figure 1). Accounting for potential confounding factors such as prior lines of treatment, age, and cancer type in the Cox regression analysis decreased hazard ratios of progression or death for BRCA1/2mut and patients with GIS ≥ 49, although Sig3 panel or WES remained the most promising biomarker with the lowest hazard ratio and tighter confidence intervals (Supplementary Figure 1 and 6). Loss-of-function mutations in other genes potentially associated with homologous recombination that could explain the response to PARPi were not identified.
When combined in a Cox regression model, Sig3 status was significant (p = 0.03, hazard ratio = 0.34; Figure 2E) and BRCA1/2 status was not (p = 0.26). This combined analysis shows that Sig3 brings additional information that allows better stratification of HRD patients beyond BRCA1/2mut status. This finding suggests that there is a great deal of heterogeneity in PFS among the BRCA1/2mut cases and that BRCA1/2mut status is less informative than the Sig3 status. When GIS is used in place of Sig3, it was not statistically significant (p = 0.40 and p = 0.069 for 42 and 49 thresholds, respectively: Figure 2F–Supplementary Figure 1E). The number of previous lines of therapy was significantly associated with a higher risk of progression (p = 0.003, hazard ratio = 4.24 in the Sig3 model and p = 0.006, hazard ratio = 3.49 in the GIS model; Figure 2E and 2F), likely related to the acquisition of cross-resistance between platinum and other DNA damaging chemotherapies and PARPi. These results suggest that panel-Sig3 may serve as a biomarker to identify HRD cancers most likely to respond to PARPi independently of previous therapy.
PARPi have been approved for BRCA1/2 carriers in various cancer types. Therefore, an immediate step towards more comprehensive HRD detection would be to include patients based on their BRCA1/2mut as well as Sig3 status to identify a larger number of patients potentially responding to PARP inhibition. A similar approach has been adopted for GIS-based commercial tests. In later stages, it may be possible to explore the use of Sig3 to increase the specificity of HRD detection by differentiating between BRCA1/2mut tumors with and without HRD. Our current analysis does not allow a definitive conclusion in this respect. Defining panel-Sig3+ and/or BRCA1/2mut patients as HRD, we find a significantly higher response rate (per Response Evaluation Criteria in Solid Tumors [RECIST], version 1.1 criteria) to PARPi in HRD patients (9/23: 39%) compared to non-HRD patients (0/10, 0%) (Fisher’s Exact Test; p = 0.03).
ATR amplification confers resistance to PARPi
To identify additional genetic factors that may impact PFS, we examined the association of copy number alterations of single genes implicated in HRD with PFS. We focused on the role of copy number alterations of ATR, TP53BP1, EZH2, and POLQ, as these genes have been implicated in PARPi resistance through replication fork stabilization.41 Among these, ATR amplification was significantly associated with shorter PFS (log-rank test: p = 0.01; Figure 3A). Although other copy number alterations also had a trend towards shorter PFS, they were not statistically significant (Supplementary Figure 7). Upon inclusion into the Cox regression model, ATR amplification is associated with worse PFS with a hazard ratio of 4.48, whereas HRD is associated with better PFS with a hazard ratio of 0.16 (Figure 3B). When samples are stratified according to HRD and ATR amplification into four groups, the longest PFS is obtained for Sig3+ patients without ATR amplification (Supplementary Figure 7). Notably, seven patients with HRD tumors did not have clinical benefit – defined as a radiologic response or stable disease for at least six months – from the treatment. Five of these seven tumors were found to have ATR amplification, possibly explaining the lack of response. These exploratory and hypothesis-generating results suggest that adding ATR amplification status to Sig3 may be informative although the determination as to whether ATR amplification is associated with primary or acquired resistance will require analysis of larger datasets of tumors from patients treated with PARP-inhibitors.
Figure 3.
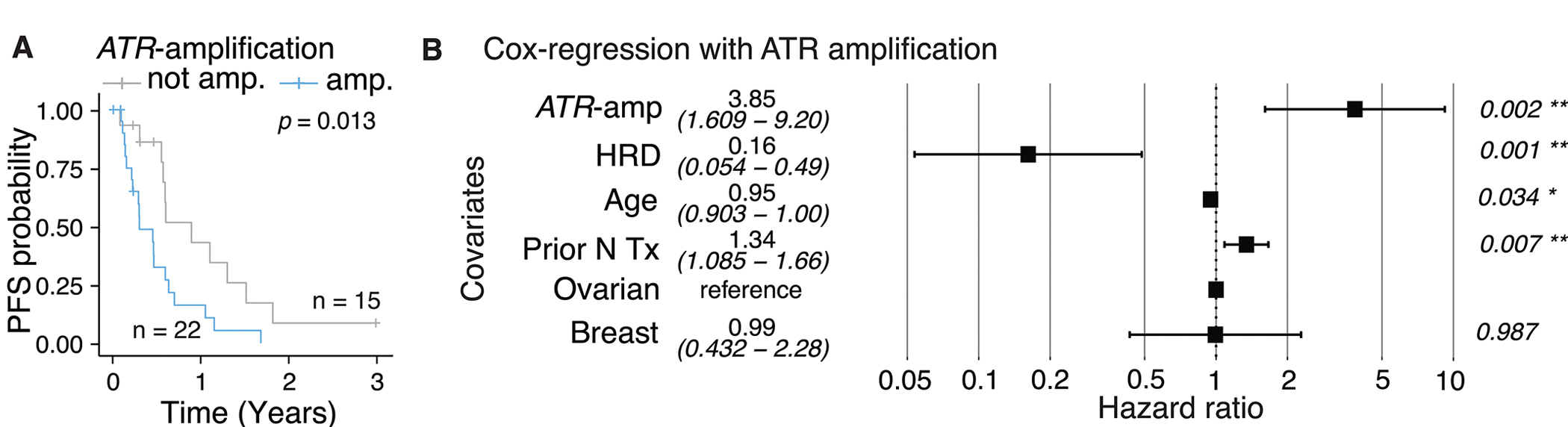
Survival analysis according to A, ATR amplification (median PFS 5.0 versus 10.9 months, respectively for samples with ATR-amplification and those without, respectively) B, Cox model same as in Figure 2E but with the additional covariate of ATR amplification. HRD is defined as panel-Sig3+ and/or BRCA1/2mut. “Prior N Tx” refers to number of prior lines of therapy.
External clinical validation of panel-Sig3 in TNBC using data from the Phase II PETREMAC trial
Cohort 2 (from the PETREMAC trial) and the study results were previously described in detail.28 In brief, the phase II PETREMAC trial included patients with primary TNBC >2 cm, and patients received olaparib for up to 10 weeks before response assessment. The calculation of panel-Sig3 using targeted DNA sequencing (360 genes) from tumor biopsies collected before olaparib (treatment-naïve tumors) revealed that most tumors (59%) are classified as Sig3+ (Figure 4A). For comparison, the frequency of Sig3+ tumors in TCGA (Figure 4B). Of the 19 patients classified as panel-Sig3+, only 4 patients were carriers of a germline BRCA1/2 mutation (Figure 4C), underscoring that Sig3 identifies patients with HRD beyond gBRCA1/2 mutations. We compared the ORR of patients classified as panel-Sig3+ with patients classified as panel-Sig3− and found that the ORR in the former group is higher than in the latter (74% vs. 31%, p = 0.029) (Figure 4D).
Figure 4.
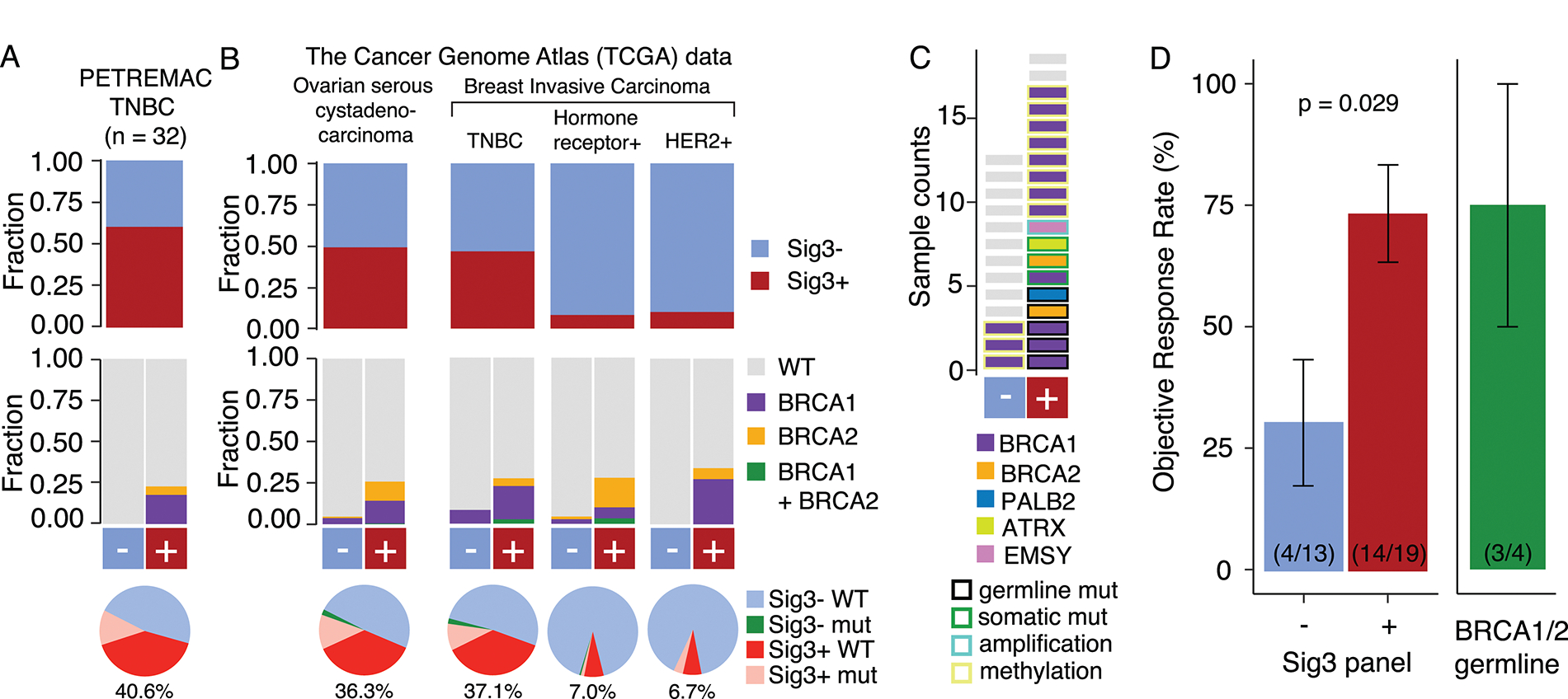
Fraction of samples that are Sig3+ (top), the proportion of samples with germline BRCA1/2mut for Sig3+ and Sig3− groups (middle) and pie charts showing the fraction of samples that are Sig3+ germline BRCA1/2 WT in TNBCs from PETREMAC trial (A) and TCGA ovarian cancers and the major clinical subtypes of breast cancer (B). The fraction of samples that are Sig3+ but not germline BRCA1/2mut are shown below the pie charts in red and the fractions are indicated below. Pink: Sig3+ and germline BRCA1/2mut, Green: Sig3− and germline BRCA1/2mut, and Blue Sig3− and germline BRCA1/2 WT. C, HR-gene alterations in patients from the PETREMAC trial. D, Objective response rate from patients from the PETREMAC trial according to panel-Sig3 classification and etiology of Sig3. Error bars denote the standard error. The number of samples and the responders are denoted in parenthesis.
TNBC: triple negative breast cancer, WT: wild type.
Analysis of TCGA data identifies many Sig3+ breast and ovarian cancers beyond BRCA carriers
Cohort 1 is enriched for BRCA1/2mut patients as opposed to an unbiased selection of TNBCs and HGSOCs. In order to provide estimates on the frequency of Sig3+ tumors in a patient population not enriched for tumors with deleterious BRCA1/2 mutation, we analyzed WES data from 901 unselected breast cancer samples and 454 unselected ovarian serous cystadenocarcinomas samples from The Cancer Genome Atlas (TCGA). We found that 47% and 50% of TNBC and ovarian cancers can be classified as Sig3+ while only 11% and 14% have a germline BRCA1/2 mutation, respectively (Figure 4B). Somatic BRCA1/2 mutations were found in only a minority of these tumors and constituted 8% and 5% of TNBCs and ovarian cancers, respectively. If we focus on TNBC, where the indication for PARPi is currently limited to carriers of a germline BRCA mutation, Sig3 could identify an additional 37% of patients who might be candidates for PARPi. Even though at a lower rate, hormone receptor or HER2-positive breast cancer were also classified as Sig3+ and hence these patients could potentially benefit from HRD-targeted therapy (i.e., PARPi). Thus, Sig3-based detection has the potential to substantially increase the number of patients who benefit from PARPi beyond those with germline BRCA1/2 mutations.
DISCUSSION
In this study, we show that the Sig3 classification from clinical panel sequencing could potentially serve as a surrogate for Sig3 from exomes and, more importantly, is predictive of response to a PARP-inhibitor combination, surpassing the predictive value of either BRCA1/2 status or GIS. We provide the first analytical validation that the Sig3 classification from gene panels has excellent concordance with that derived from WES data from patients with breast and ovarian cancers (cohort 1). This study also provides clinical validation of panel-Sig3 as a predictive biomarker in patients with breast and ovarian cancer receiving combined olaparib/buparlisib (cohort 1). In addition, for the first time, we clinically validate panel-Sig3 as predictive of olaparib response in a population with treatment-naïve and unselected TNBC (cohort 2). This means that the SigMA algorithm can be applied to panel sequencing data currently obtained for patients as part of their routine care for metastatic solid tumors, and thus, is an immediately available resource to contribute to HRD prediction. While panel sequencing is currently standard-of-care for most metastatic cancers in order to pinpoint therapeutic targets, the implementation of Sig3 calculation adds minimal compute cost and time. This contrasts with other methods such as GIS, which requires WES, or an additional commercial assay, or HRDetect, which requires WGS from fresh biopsies. Hence, Sig3 status extracted from panel sequencing commonly used in the clinic, has the potential to aid decision-making on the use of PARP inhibitors, and should be added as an exploratory biomarker in ongoing studies and as an integral marker in future clinical trials. It is important to note that all patients with partial response tested positive for Sig3 or a BRCA1/2 mutation, while three patients derived a clinical benefit who were neither Sig3-positive nor carried a BRCA1/2 mutation, implying that we still lack the tools to identify all patients who benefit from PARP inhibition, or that PARPi may have mechanisms of action independent of HRD. In cohort 1, two patients were panel-Sig3+ but did not carry a BRCA1/2 mutation, and one of these patients had a partial response and PFS longer than 1 year with the PARP-PI3K-inhibitor combination. Biologically, this finding raises the possibility that HRD is a polygenic phenotype that is only captured by a genomic signature rather than single mutational driver events in some tumors. If a patient’s tumor was panel-Sig3+ or BRCA1/2m, the objective response rate (ORR) was 39% and the clinical benefit rate was 65%. If neither was present, the ORR was 0%, and the clinical benefit rate was 30%. In cohort 2, the ORR was 74% for patients whose tumor was panel-Sig3+ or BRCA1/2m, while the group of patients with panel-Sig3− tumors had a significantly lower ORR of 31%.
It is noteworthy that in cohort 1, all patients who achieved an objective response tested positive for either Sig3 or BRCA1/2. Among patients who had progressive disease as their best response (primary resistance), there were two BRCA1/2mut patients, one of whom was Sig3+, but the patient had more than seven prior lines of treatment. Whether the absence of Sig3 can be used to predict primary resistance to PARP inhibition among BRCA1/2 carriers is unclear at this point. The overall response rate to olaparib in germline BRCA1/2 mutation carriers with metastatic breast cancer is 59.9 %, i.e. 40% of patients experience primary resistance,2 the mechanism of which is poorly understood. Secondary, acquired resistance to PARPi can occur through the reversal of HR gene mutations, restoration of HR through end-resection or replication fork stabilization.42 These secondary resistance mechanisms are thought to be facilitated by the high evolutionary plasticity of HRD tumors resulting in frequent copy number alterations that may lead to the recovery of some HR function during PARPi therapy. Observed secondary resistance mechanisms are the amplification of the ATR gene leading to upregulation of the ATR/CHK1 pathway improving fork stability43 or loss of TP53BP1 which partially restores HR.44 In cohort 1, we did find that ATR amplification was associated with shorter PFS. Ultimately, Sig3+ may be an indicator of the underlying HRD, but the clinical response to PARPi will also depend on the presence of mechanisms of resistance that are not captured with this signature.
Limitations of our study include the small numbers of patients in both cohorts, which limits our ability to explore the subgroups further, and the combination treatment in cohort 1. In addition, we were only able to analyze pre-treatment tumor tissue samples and no on- or post-treatment tissues were available to examine changes induced by PARP-inhibition. Among the 37 patients from cohort 1, there were only 13 patients who were BRCA1/2 WT, likely from ascertainment bias at enrollment favoring the enrollment of patients with BRCA1/2mut into a PARPi trial. Of note, BKM-120 was not considered to be efficacious at the studied doses as the maximum tolerated dose was only 60 mg/day, well below what was considered meaningful PI3K-inhibition based on pharmacokinetics and pharmacodynamics data.27 Therefore, we consider the responses seen in our cohort as mostly driven by olaparib, which was dosed at 200 mg twice a day in this study, a dose that to date would be considered below standard of care.
In summary, patients with Sig3+ tumors are more likely to respond to PARPi and have longer PFS. The implementation of Sig3 analysis with SigMA could be applied to clinical sequencing data without a considerable increase in cost and time. Prospective validation of the clinical utility is warranted in the setting of a biomarker-driven clinical trial.
Supplementary Material
Translational Relevance.
Among breast cancer patients, PARP inhibitors (PARPi) are approved for BRCA1/2 carriers, but there is growing evidence of benefit in additional patients. There is an immense clinical need for a more comprehensive biomarker of response to PARPi. We have previously developed a method to detect mutational signature 3 (Sig3) associated with homologous recombination deficiency from clinical panel sequencing data. The present work brings mutational signatures closer to the clinic by not only confirming our ability to reliably detect Sig3 from clinical sequencing but also relating these to meaningful clinical outcomes (progression-free survival and objective response rate). Our method is unique that it detects Sig3 from routine clinical sequencing (targeting a few hundred genes), whereas previous methods for Sig3 detection required whole-exome or whole-genome sequencing (not routine care). Sig3 could be used to design clinical trials to identify patients who will benefit from PARPi with high probability, without additional sequencing costs.
Acknowledgments
DG, GMW and PJP were supported by the Harvard Ludwig Center.
Institutional funding was provided to HPE, SK, PEL by The KG Jebsen Foundation (SKGJ-MED-020), AstraZeneca (ESR-14–10077) and Pfizer (GMGS #51752519). HPE was supported by Helse Vest Regionalt Helseføretak (#912252). SK was supported by the Norwegian Cancer Society (Kreftforeningen)(190281–2017). PEL was supported by the Norwegian Research Council (273354), Helse Vest Regionalt Helseføretak (912008) and the Norwegian Cancer Society (190275–2017).
GMW was supported by a Stand Up to Cancer-American Association for Cancer Research Dream Team Translational Cancer Research Grant SU2C-AACR-DT0209, and a National Institutes of Health (NIH) Research Project Grant 1R01CA226776–01. EPW, GMW, ELM are supported by the DF/HCC Specialized Program of Research Excellence (SPORE) in Breast Cancer, NIH Grant P50 CA168504. UAM is funded by the National Cancer Institute P50 CA240243–01A1 and the Breast Cancer Research Foundation. GMW reports grants from Mary Kay Ash Foundation, Ovarian Cancer Research Foundation, Breast Cancer Alliance, Breast Cancer Research Foundation.
Footnotes
Disclosures
FB reports personal fees from Curio Science, outside of the submitted work. DG and PJP have a patent submitted for the SigMA algorithm and report license fees from Pfizer. EPW reports institutional research funding from Genentech/Roche, serving in advisory boards at Leap, and receiving personal fees from Athenex, Carrick Therapeutics, Genentech/Roche, Gilead, GSK, Jounce, Leap, and Syros. SK reports personal fess from AstraZeneca, Pfizer, and Pierre Fabre. SK reports the patents Patent EP2389450 A1, Patent WO 2012/01066, outside the current work. ELM reports serving as consultant at Lilly, Novartis, Pfizer, AstraZeneca, and Gilead. PEL reports personal fees (honoraria, consulting, or advisory role) from AstraZeneca, Abbvie, Pierre Fabre, Roche, Laboratorios Farmaceuticos Rovi, Akademikonferens, and Aptitude Health. PEL also reports a patent for Cytovation, outside the current work. UAM reports personal fees from Astrazeneca, Merck, Novartis, Blueprint Medicine, and NextCure. PAK reports institutional research funding from AstraZeneca and Novartis. PAK also reports serving in advisory boards at AstraZeneca, Pfizer, and Merck outside the submitted work. DBS has consulted for and received honoraria from Pfizer, Loxo/Lilly Oncology, Vividion Therapeutics, FORE Therapeutics, Scorpion Therapeutics and BridgeBio. HW is currently employed by Loxo Oncology. HPE reports personal fees (honoraria, consulting, or advisory role) from Amgen, AstraZeneca, Abbvie, Bristol-Myers-Squibb, Dagens Medisin, HAI Interaktiv AS, MSD, Novartis, Pfizer, Pierre Fabre, Roche, Seagen, Daiichi Sankyo, Eli Lilly, MSD, Novartis, and Sanofi. G.M. Wulf reports reports research funds from Merck outside of the current work and a patent for 8129131 Pin1 as a marker for abnormal cell growth issued, licensed, and with royalties paid from R&D Systems, Cell Signaling and 20140235630 Compositions and methods for the treatment of proliferative diseases pending. The other authors have nothing to disclose.
REFERENCES
- 1.Mirza MR, Monk BJ, Herrstedt J, et al. : Niraparib Maintenance Therapy in Platinum-Sensitive, Recurrent Ovarian Cancer. N Engl J Med 375:2154–2164, 2016 [DOI] [PubMed] [Google Scholar]
- 2.Robson M, Im S-A, Senkus E, et al. : Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. The New England Journal of Medicine 377:523–533, 2017 [DOI] [PubMed] [Google Scholar]
- 3.Golan T, Hammel P, Reni M, et al. : Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. New England Journal of Medicine 381:317–327, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.González-Martín A, Pothuri B, Vergote I, et al. : Niraparib in Patients with Newly Diagnosed Advanced Ovarian Cancer [Internet]. New England Journal of Medicine, 2019[cited 2020 Aug 13] Available from: 10.1056/NEJMoa1910962 [DOI] [PubMed]
- 5.de Bono J, Mateo J, Fizazi K, et al. : Olaparib for Metastatic Castration-Resistant Prostate Cancer. New England Journal of Medicine 382:2091–2102, 2020 [DOI] [PubMed] [Google Scholar]
- 6.Tung NM, Robson ME, Ventz S, et al. : TBCRC 048: A phase II study of olaparib monotherapy in metastatic breast cancer patients with germline or somatic mutations in DNA damage response (DDR) pathway genes (Olaparib Expanded). JCO 38:1002–1002, 2020 [Google Scholar]
- 7.Alexandrov LB, Nik-Zainal S, Wedge DC, et al. : Signatures of mutational processes in human cancer. Nature 500:415–421, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alexandrov LB, Kim J, Haradhvala NJ, et al. : The repertoire of mutational signatures in human cancer. Nature 578:94–101, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nik-Zainal S, Davies H, Staaf J, et al. : Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 534:47–54, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Polak P, Kim J, Braunstein LZ, et al. : A mutational signature reveals alterations underlying deficient homologous recombination repair in breast cancer. Nature Genetics 49:1476–1486, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Davies H, Glodzik D, Morganella S, et al. : HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nature Medicine 23:517–525, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Timms KM, Abkevich V, Hughes E, et al. : Association of BRCA1/2 defects with genomic scores predictive of DNA damage repair deficiency among breast cancer subtypes. Breast Cancer Res 16:475, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hoppe MM, Sundar R, Tan DSP, et al. : Biomarkers for Homologous Recombination Deficiency in Cancer. J Natl Cancer Inst 110:704–713, 2018 [DOI] [PubMed] [Google Scholar]
- 14.Gulhan DC, Lee JJ-K, Melloni GEM, et al. : Detecting the mutational signature of homologous recombination deficiency in clinical samples. Nat Genet 51:912–919, 2019 [DOI] [PubMed] [Google Scholar]
- 15.Telli ML, Jensen KC, Vinayak S, et al. : Phase II Study of Gemcitabine, Carboplatin, and Iniparib As Neoadjuvant Therapy for Triple-Negative and BRCA1/2 Mutation–Associated Breast Cancer With Assessment of a Tumor-Based Measure of Genomic Instability: PrECOG 0105. JCO 33:1895–1901, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Isakoff SJ, Mayer EL, He L, et al. : TBCRC009: A Multicenter Phase II Clinical Trial of Platinum Monotherapy With Biomarker Assessment in Metastatic Triple-Negative Breast Cancer. J Clin Oncol 33:1902–1909, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaklamani VG, Jeruss JS, Hughes E, et al. : Phase II neoadjuvant clinical trial of carboplatin and eribulin in women with triple negative early-stage breast cancer (NCT01372579). Breast Cancer Res Treat 151:629–638, 2015 [DOI] [PubMed] [Google Scholar]
- 18.Sharma P, Barlow WE, Godwin AK, et al. : Impact of homologous recombination deficiency biomarkers on outcomes in patients with triple-negative breast cancer treated with adjuvant doxorubicin and cyclophosphamide (SWOG S9313). Ann Oncol 29:654–660, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mayer EL, Abramson V, Jankowitz R, et al. : TBCRC 030: a phase II study of preoperative cisplatin versus paclitaxel in triple-negative breast cancer: evaluating the homologous recombination deficiency (HRD) biomarker. Ann Oncol 31:1518–1525, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tutt A, Tovey H, Cheang MCU, et al. : Carboplatin in BRCA1/2-mutated and triple-negative breast cancer BRCAness subgroups: the TNT Trial. Nat Med 24:628–637, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stronach EA, Paul J, Timms KM, et al. : Biomarker Assessment of HR Deficiency, Tumor BRCA1/2 Mutations, and CCNE1 Copy Number in Ovarian Cancer: Associations with Clinical Outcome Following Platinum Monotherapy. Mol Cancer Res 16:1103–1111, 2018 [DOI] [PubMed] [Google Scholar]
- 22.Hodgson DR, Dougherty BA, Lai Z, et al. : Candidate biomarkers of PARP inhibitor sensitivity in ovarian cancer beyond the BRCA genes. British Journal of Cancer 119:1401–1409, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ray-Coquard I, Pautier P, Pignata S, et al. : Olaparib plus Bevacizumab as First-Line Maintenance in Ovarian Cancer. New England Journal of Medicine 381:2416–2428, 2019 [DOI] [PubMed] [Google Scholar]
- 24.Sokol ES, Pavlick D, Khiabanian H, et al. : Pan-Cancer Analysis of BRCA1 and BRCA2 Genomic Alterations and Their Association With Genomic Instability as Measured by Genome-Wide Loss of Heterozygosity [Internet]. JCO Precision Oncology, 2020[cited 2020 Aug 21] Available from: 10.1200/PO.19.00345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Coleman RL, Oza AM, Lorusso D, et al. : Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 390:1949–1961, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Färkkilä A, Gulhan DC, Casado J, et al. : Immunogenomic profiling determines responses to combined PARP and PD-1 inhibition in ovarian cancer [Internet]. Nat Commun 11, 2020[cited 2021 May 2] Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7081234/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Matulonis UA, Wulf GM, Barry WT, et al. : Phase I dose escalation study of the PI3kinase pathway inhibitor BKM120 and the oral poly (ADP ribose) polymerase (PARP) inhibitor olaparib for the treatment of high-grade serous ovarian and breast cancer. Ann Oncol 28:512–518, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eikesdal HP, Yndestad S, Elzawahry A, et al. : Olaparib monotherapy as primary treatment in unselected triple negative breast cancer. Ann Oncol 32:240–249, 2021 [DOI] [PubMed] [Google Scholar]
- 29.Fisher S, Barry A, Abreu J, et al. : A scalable, fully automated process for construction of sequence-ready human exome targeted capture libraries. Genome Biology 12:R1, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yates LR, Gerstung M, Knappskog S, et al. : Subclonal diversification of primary breast cancer revealed by multiregion sequencing. Nat Med 21:751–759, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.NCI Genomic Data Commons [Internet][cited 2021 May 2] Available from: https://gdc.cancer.gov/
- 32.Ellrott K, Bailey MH, Saksena G, et al. : Scalable Open Science Approach for Mutation Calling of Tumor Exomes Using Multiple Genomic Pipelines. Cell Syst 6:271–281.e7, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Van der Auwera GA, Carneiro MO, Hartl C, et al. : From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics 43:11.10.1–11.10.33, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Favero F, Joshi T, Marquard AM, et al. : Sequenza: allele-specific copy number and mutation profiles from tumor sequencing data. Ann Oncol 26:64–70, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mermel CH, Schumacher SE, Hill B, et al. : GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol 12:R41, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sztupinszki Z, Diossy M, Krzystanek M, et al. : Migrating the SNP array-based homologous recombination deficiency measures to next generation sequencing data of breast cancer. NPJ Breast Cancer 4:16, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Knijnenburg TA, Wang L, Zimmermann MT, et al. : Genomic and Molecular Landscape of DNA Damage Repair Deficiency across The Cancer Genome Atlas. Cell Rep 23:239–254.e6, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Carter SL, Cibulskis K, Helman E, et al. : Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol 30:413–421, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Islam SMA, Wu Y, Díaz-Gay M, et al. : Uncovering novel mutational signatures by de novo extraction with SigProfilerExtractor. bioRxiv 2020.12.13.422570, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Campbell PJ, Getz G, Korbel JO, et al. : Pan-cancer analysis of whole genomes. Nature 578:82–93, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.D’Andrea AD: Mechanisms of PARP inhibitor sensitivity and resistance. DNA Repair (Amst) 71:172–176, 2018 [DOI] [PubMed] [Google Scholar]
- 42.Li H, Liu Z-Y, Wu N, et al. : PARP inhibitor resistance: the underlying mechanisms and clinical implications. Mol Cancer 19:107, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ning J-F, Stanciu M, Humphrey MR, et al. : Myc targeted CDK18 promotes ATR and homologous recombination to mediate PARP inhibitor resistance in glioblastoma. Nat Commun 10:2910, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jaspers JE, Kersbergen A, Boon U, et al. : Loss of 53BP1 Causes PARP Inhibitor Resistance in Brca1-Mutated Mouse Mammary Tumors. Cancer Discov 3:68–81, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors support the dissemination of research data that has been generated, and increased cooperation between investigators. The data that support the findings of this study are available at doi:10.17632/zg5xvm3f3w.1 Raw data for this study were generated at the Dana-Farber Cancer Center (Boston, MA) and Memorial Sloan Kettering Cancer Center (New York, NY). Derived data supporting the findings of this study are available at the database of Genotypes and Phenotypes (dbGaP) via study accession: phs003019.v1.p1. Further de-identified individual participant data will be provided according to institutional procedures. Requests addressed to G.M. Wulf must include a description of the nature of the proposed research and extent of data requirements. Data recipients are required to enter a formal data sharing agreement that describes the conditions for release and requirements for data transfer, storage, archiving, publication and intellectual property. Requests are reviewed by the study team in terms of scientific merit and ethical considerations, including patient consent.
Haukeland University Hospital and the University of Bergen support the dissemination of research data and cooperation between investigators nationally and internationally. Based on current Norwegian laws and regulations, and the biobank approval for the PETREMAC trial given by the Regional Ethics Committee Western Norway before the study commenced, genomic data are not to be made openly available. After publication and upon formal request, raw sequencing data, including de-identified individual participant data and a data dictionary defining each field in the data set, may be shared according to institutional guidelines, pending project-specific approvals from the Regional Ethics Committee in Norway. Requests are via a standard pro forma describing the nature of the proposed research and extent of data requirements. Data recipients are required to enter a formal data sharing agreement that describes the conditions for release and requirements for data transfer, storage, archiving, publication and intellectual property. Requests are reviewed by the PETREMAC study team in terms of scientific merit and ethical considerations, including patient consent. An evaluation as described above will typically take three months. Requests may be directed to H.P. Eikesdal.