

Abstract
Patient-derived xenograft models (PDX) are an effective preclinical in vivo platform for testing the efficacy of novel drugs and drug combinations for cancer therapeutics. Here we describe a repository of 79 genomically and clinically annotated lung cancer PDXs available from The Jackson Laboratory that have been extensively characterized for histopathological features, mutational profiles, gene expression, and copy number aberrations. Most of the PDXs are models of non-small cell lung cancer (NSCLC), including 37 lung adenocarcinoma (LUAD) and 33 lung squamous cell carcinoma (LUSC) models. Other lung cancer models in the repository include four small cell carcinomas, two large cell neuroendocrine carcinomas, two adenosquamous carcinomas, and one pleomorphic carcinoma. Models with both de novo and acquired resistance to targeted therapies with tyrosine kinase inhibitors are available in the collection. The genomic profiles of the LUAD and LUSC PDX models are consistent with those observed in patient tumors from The Cancer Genome Atlas (TCGA) and to previously characterized gene expression-based molecular subtypes. Clinically relevant mutations identified in the original patient tumors were confirmed in engrafted PDX tumors. Treatment studies performed in a subset of the models recapitulated the responses expected based on the observed genomic profiles. These models therefore serve as a valuable preclinical platform for translational cancer research.
Keywords: Patient-derived xenograft, NSCLC, mouse models, NSG
Introduction
Lung cancer is the leading cause of cancer deaths worldwide (1). Non-small cell lung cancer (NSCLC) differs from most other cancer types both quantitatively and qualitatively for its high level of mutational burden and genomic complexity. Further, the two major histologic subtypes of NSCLC: lung adenocarcinoma (LUAD) and lung squamous cell carcinoma (LUSC), have distinctive genomic alteration signatures, pathway disruption, and immune host response (2,3). Transcriptional subtypes for both LUAD and LUSC have been reported that are associated with differences in patient prognosis, response to treatment, and survival (4,5).
Genomic characterization of tumors has been instrumental in precision medicine strategies for NSCLC through the identification of “druggable” oncogene drivers which, in turn, has expanded treatment options and a growing number of targeted therapy approaches (6). A prominent example of molecularly-guided therapy in NSCLC relates to the finding of activating mutations in the epidermal growth factor receptor (EGFR) gene, resulting in constitutive, ligand-independent receptor activity and a high degree of sensitivity to EGFR-targeted tyrosine kinase inhibitors (TKIs) (7,8). Targeted therapies have also been effective in treating other molecular subtypes, including ALK-EML4 and ROS1 fusions (9). Although advances in targeted therapies for NSCLC have transformed treatment options, not all patients respond to treatment and the development of acquired resistance is almost universal. Resistance mechanisms in some treatment settings for oncogene-driven NSCLC are well established, such as development of the T790M “gatekeeper” mutation after therapy with first and second generation EGFR TKIs (10). However, resistance mechanisms are much more complex in most other therapeutic settings, generally characterized as either secondary mutations or bypass mechanisms. Testing novel treatment strategies and new therapeutic agents to overcome acquired resistance remains a high priority for translational cancer research.
Human tumors or circulating tumor cells (CTCs) engrafted into transplant-compliant recipient mouse hosts to produce Patient-Derived Xenografts (PDXs) and Circulating tumor cell-derived explant models (CDXs) retain critical biological properties of a patient’s tumor, including tumor heterogeneity and genomic complexity (11,12). PDXs have demonstrated utility as preclinical models for testing therapeutic strategies for many cancers, including lung cancer. Previous studies have demonstrated that lung cancer PDX models recapitulate faithfully many aspects of the original patient tumor for histology, karyotype, and genomics (13,14), as well as expected sensitivity and resistance patterns to targeted therapies, including clinical responses observed in patients. These models have provided insights into therapies based on other molecular markers (15,16). Collections of PDX models have allowed further studies on understanding the contributing factors affecting engraftment rates, new treatment combinations for lung cancer models which developed resistance, and discovery of new biomarkers for lung cancer treatment (12,17,18).
In collaboration with the University of California Davis Comprehensive Cancer Center and Northern Light Eastern Maine Medical Center, we generated and characterized (19) a repository of 79 lung cancer PDX models to use as a platform for research for basic research on mechanisms of treatment response and to facilitate translational pre-clinical and co-clinical trial research. The PDX models were generated using the NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mouse strain as the host and includes models of high clinical relevance, including EGFR- and KRAS-mutated LUADs and PI3K-mutant LUSCs. Clinical demographic information, histology, immunohistochemistry images, summarized genomic data, and treatment response data for these PDX models are freely available from The Jackson Laboratory (JAX) PDX web portal hosted by the Mouse Models of Human Cancer database (MMHCdb, http://tumor.informatics.jax.org/mtbwi/pdxSearch.do) (20) and from PDX Finder, a global catalog of thousands of PDX models (21).
Materials and Methods
Establishing xenografts
An overview of the PDX model generation process is shown in Fig. 1. All animal procedures were performed at The Jackson Laboratory Sacramento facility under IACUC protocol 12027. Tumor samples from biopsies, pleural effusions, or surgical resections were obtained from lung cancer patients and implanted subcutaneously by trocar in the right flank of up to five, 6–8 week-old female Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG; JAX Stock 005557) mice without intervening in vitro culturing of the tumor cells. Written informed consent was obtained for patients by the donating institution to allow creation and unrestricted use of the models and associated data. Most tumors were implanted within 24 hours of surgery and the maximum post-surgery time allowed for implantation was 48 hours. Solid tumors were divided into 3–5 mm3 fragments in RPMI medium before implantation. Pleural effusion samples were centrifuged, and the supernatant was removed with a pipet. Pellets were then re-suspended in Dulbecco’s Phosphate Buffered Saline (DPBS), and 200 μl were implanted subcutaneously into the host mouse as a 1:1 bolus of pleural effusion cells in RPMI media and growth factor free Matrigel. Matrigel was not used for subsequent passages.
Figure 1.
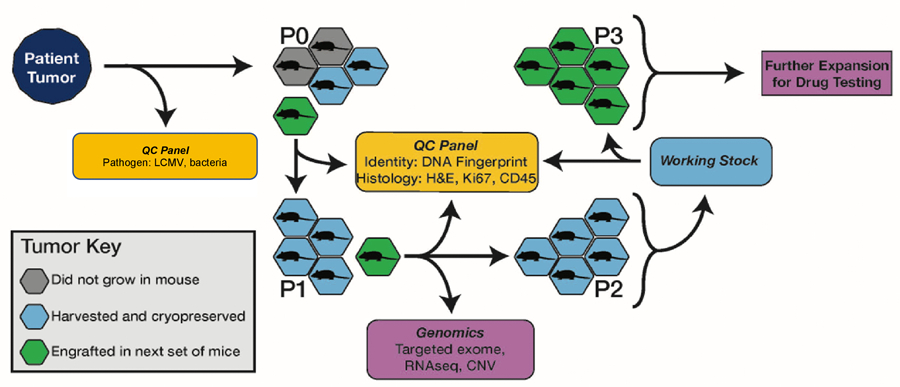
Schematic overview of the process for PDX model generation and characterization for the JAX PDX Resource.
When an implanted tumor reached 2000 mm3, it was harvested and subdivided into 3–5 mm3 fragments which were implanted into five, 6–8 week old female NSG mice for expansion to P1. For quality control assessment (see below), a 50 mm3 fragment was collected in 10% neutral buffered formalin and a formalin-fixed, paraffin-embedded block was generated. The remaining fragments were cryopreserved in 10% DMSO. When P1 tumors reached ~2000mm3, they were harvested and subdivided into 3–5 mm3 fragments which were subsequently embedded in FFPE for quality control, snap-frozen for genomics, placed into RNALater (Ambion) for RNA-Seq, and viably cryopreserved in a solution of 10% DMSO in Dulbecco’s Modified Eagle Medium (DMEM) with 1% penicillin-streptomycin.
To establish cohorts of tumor-bearing mice for drug treatment studies, 3–5 mm3 tumor fragments or 40 µl of minced tumor were subcutaneously implanted in the right rear flank of NSG mice by trocar or a 14-gauge disposable needle. Low passage tumor fragments (P3-P6) were used to establish cohorts of tumor-bearing animals for dosing studies. Tumor volumes were monitored with ULTRA-Cal IV digital calipers (Fowler, Newton, MA). Individual tumor-bearing mice were randomized into treatment cohorts of 8–12 mice each on an accrual (asynchronous growth) basis once individual tumors reached an initial volume of 70–300 mm3. For some studies, tumors were removed and divided, with half of the material preserved in neutral-buffered formalin and half flash-frozen.
PDX model quality control
The quality control procedures employed for PDX models included testing the patient tumor for LCMV (lymphocytic choriomeningitis virus) and bacterial contamination. Engrafted tumors at P0 and P1 were DNA fingerprinted using a Short Tandem Repeat (STR) assay (22) and then compared to the profile of the patient sample to ensure correct tissue provenance. Immunohistochemistry (IHC) for human CD45 antibodies (IR75161–2, Agilent Technologies) was performed on FFPE blocks of engrafted tumors to identify cases of lymphomagenesis, which have been reported previously in PDXs (23). IHC for human Ki67 (IR62661–2, Agilent Technologies) was used to ensure the propagated tumors were comprised of human cells. H&E sections of engrafted tumors were evaluated by a board-certified pathologist (RGE) to verify the concordance of the morphological features of the engrafted tumor to the patient tumor. Engrafted tumors were assessed by sequencing or digital droplet PCR to determine if diagnostic/therapeutic molecular markers identified in patient samples were present in the engrafted tumors (Supplementary Table S1).
Genomic characterization of engrafted tumors
The DNA and RNA of engrafted tumors were characterized at the P0 and/or P1 passage (Fig. 1) using a targeted gene panel (JAX Cancer Treatment Profile (CTP)) sequencing (24), Affymetrix Human SNP array 6.0, and expression microarrays or RNA-Seq. Detailed protocols for nucleic acid extraction, library preparation, and data analysis are described elsewhere (19). The JAX Clinical Knowledge Base (CKB) was used to annotate clinical relevance of variants and gene expression (25).
Analysis of genomic data from engrafted tumors
Mutation, copy number (CN) and gene expression analyses were performed as described in Woo et al. (19). Tumor mutation burden (TMB) was calculated using variants that met all quality criteria (coverage, strand bias, mapping quality, and read rank position) and were not present on a curated list of false-positive variants (loci prone to sequencing and analysis errors and/or associated with highly polymorphic genes: MUC4, MUC5B, MUC16, MUC17, and HLA-A). Only somatic mutations based on germline filtering criteria that were predicted with high or moderate functional impact (i.e., non-synonymous changes, frame shifts, stop losses/gains, and splice-site acceptor/donor changes) were retained. TMB was estimated by dividing the number of variants that met the quality criteria by the length (in Mb) of the CTP gene panel. High TMB was defined as 22 mutations/Mb (calculated based on the TMB distribution of all PDX models analyzed as follows: third quartile + 1.5 x inter-quartile range). The MSIsensor2 (26) algorithm was used to determine microsatellite instability (MSI) status of engrafted tumors. Tumors with MSI percentage > 20% were classified as MSI-High and those < 20% as MSI-Stable.
To summarize the mutations prevalent in the PDX models, oncoplots (maftools v2.2.10) (27) for both subtypes were created using all the somatic and clinically relevant point mutations and indels that occurred at a frequency >30%. For PDXs with more than one sequenced sample data from all samples were combined to derive a unique list of mutations per model. Gene mutation frequencies of LUAD and LUSC in the TCGA PanCancer Atlas were obtained from cBioPortal (28) for comparison with the PDX data. Genes were classified as oncogenes or tumor suppressor genes (TSGs) based on OncoKB annotations (Download date: 2020/9/17) (29).
Copy number data were visualized using GenVisR (30) on a per-sample basis and an overall gain and loss frequency basis within the subtypes. For frequency calculation, one sample was selected to represent each model, and log2(CN ratio) = ±0.5 was used as a cut-off to call CN gain and loss.
To summarize the expression data, the percentile rank z-score values from stranded RNA-Seq, non-stranded RNA-Seq, and microarray platforms were combined and a correlation heatmap was plotted using the Pretty Heatmaps package in R (https://cran.r-project.org/web/packages/pheatmap/index.html).
Gene expression-based subtypes of LUAD and LUSC PDXs
To determine if previously identified transcriptional subtypes for LUAD (TRU: terminal respiratory unit, PIF: proximal inflammatory, PPR: proximal proliferative) and LUSC (CLA: classical, PRI: primitive, BAS: basal, and SEC: secretory) were represented in our repository of lung cancer PDX models, we used Nearest Template Prediction (NTP) implemented in the R package CMScaller. We selected genes from publicly available RNA-seq data to enrich for classification of the subtypes, following similar methods reported in Eide et al. (31). TCGA raw gene expression data (non-stranded RNA-Seq) were downloaded from the Broad GDAC Firehose repository for LUAD (7) and LUSC (32). Molecular subtype annotations for LUAD and LUSC samples from TCGA were downloaded using the TCGAquery_subtype function in the R package TCGAbiolinks (33). Gene expression data were harmonized between TCGA and PDX samples and filtered for lowly expressed genes (mean normalized expression ≤ 1). We estimated differential expression between subtypes in TCGA samples using the R package DESeq2 v. 1.28.1 (34). Genes were classified as differentially expressed for each subtype using a threshold of an FDR-adjusted P-value of ≤ 0.01 and an absolute log2 fold-change value > 1. All differentially expressed genes that showed discrimination between at least one subtype and the others (i.e., not commonly identified as differentially expressed among all subtypes) were included to generate custom templates for NTP. We also selected genes that showed a large range of expression values among LUAD and LUSC cell lines in the Cancer Cell Line Encyclopedia (35) and highly expressed in at least a subset of the cell lines. Finally, we selected genes to exclude from the training set by performing a differential expression analysis between matched lung cancer patients (7 samples) and PDX samples (41 samples) obtained from the NCI Patient-Derived Model Repository (PDMR, NCI-Frederick, Frederick National Laboratory for Cancer Research, https://pdmr.cancer.gov/), using DESeq2. The genes with an absolute log2 fold change ≥ 1 were excluded from the model training set as they may be lost upon engraftment.
The final list of genes used to train the NTP classifiers included 3,525 genes for LUAD and 3,544 genes for LUSC. Raw expression data from TCGA for these genes were used to generate templates for NTP. TCGA samples were split randomly into 80% training and 20% validation sets stratified by labeled subtype. Custom templates were then prepared for LUAD and LUSC training sets using subDEG and ntpMakeTemplates in the R package CMScaller. Subtype prediction performance was estimated using the validation sets with the function ntp in the CMScaller, specifying 1000 permutations. Performance of subtype predictions from NTP was calculated using 20% of the labeled TCGA data held back for validation. For each subtype, the performance of the NTP classification was measured by precision, recall, and F1-score. The overall accuracy of the predictions was calculated as the unweighted average of the proportion of true positives to samples of each subtype. Finally, we used the custom templates to generate subtype predictions for the unlabeled PDX models with non-stranded RNA-Seq data, and we considered high confidence subtype classifications with FDR-adjusted P-values < 0.05.
PDX treatment studies
Tumor-bearing mice at low passage (P3-P6) were assigned to cohorts (8–10 mice per treatment group) and treated with single and combination agent therapies depending on the lung cancer subtype and presence of targetable molecular markers. Vehicle treated mice were used as controls. Treatment was initiated when tumors reached approximately 70–300 mm3. Tumors were monitored until the end of the dosing study (typically 28 days) or when the tumors reached 2000 mm3. To monitor for toxicity effects from treatment, animal body weight was monitored three times weekly throughout the study, and percent body weight loss was calculated for each mouse. Animals with >20% body weight loss were euthanized and recorded as treatment-related deaths. Tumor volumes were calculated from digital caliper raw data by using the formula: Volume (mm3) = (l × w2) / 2.
Treatment responses were calculated based on the percentage of tumor volume change (ΔVol) at the final study day (i.e., seven days after the last treatment) compared with the baseline tumor volume at Day 0 or Day 1. Responses were classified using a modified Response Evaluation Criteria In Solid Tumors (RECIST) method adapted from Gao et al. (36). Classification method details are available from the PDX data portal via MMHCdb. Graphical summaries of treatment responses for individual models were generated using custom software (https://github.com/TheJacksonLaboratory/PDX-SOC) are also available from MMHCdb. Graphical summaries of treatment responses across all models were generated using the R package Xeva (version 1.6.0) (37).
Western blots
Immunoblotting was performed on treated tumors using methods described previously (38).
Data availability
Information and data for the PDX models described in the manuscript are freely available from the Mouse Models of Human Cancer database (MMHCdb, http://tumor.informatics.jax.org/mtbwi/pdxSearch.do) and from PDX Finder (https://pdxfinder.org)
Results
Enrollment and patient characteristics
The clinical and demographic data for the patients from whom the lung PDX models were generated are summarized in Table 1. The median age for patients was 63 (range 42–85). Slightly more female (n=44) versus male patients (n=35) are represented in the patient population from which the models were derived. Most patients (80%) reported their race as White. The self-reported smoking status of the patient cohort was as follows: former (44%), current (20%), and never (8%). Most of the patients (30 of 33) diagnosed with LUSC were treatment naïve at the time their tumor tissue was acquired to generate a PDX model. For LUAD, half of the patients (19 of 37) were treatment-naïve at the time of PDX model generation.
Table 1.
Summary of clinical and demographic data for patients whose tumor material was used to generate the JAX collection of lung cancer PDX models.
| Characteristics | Adenocarcinoma (LUAD) | Squamous Cell Carcinoma (LUSC) | All other Lung Cancers | |
|---|---|---|---|---|
| N= | 37 | 33 | 9 | |
| Age | Median (range) | 58 (42–79) | 67 (50–85) | 60 (50–780 |
| Sex | Female/Male | 24/13 | 12/21 | 8/1 |
| Race/Ethnicity | White/Not Hispanic | 22 | 16 | 6 |
| White/Not Reported | 5 | 11 | 1 | |
| White/Hispanic | 1 | 0 | 0 | |
| Asian or Pacific Islander/Not Hispanic | 5 | 1 | 0 | |
| American Indian or Alaskan Native/Not Hispanic | 1 | 1 | 0 | |
| American Indian or Alaskan Native/Hispanic | 0 | 1 | 0 | |
| Not Reported/Not Hispanic | 1 | 1 | 0 | |
| Not Reported/Not Reported | 2 | 2 | 2 | |
| Tumor Type | Primary/Relapse/Metastatic | 22/1/14 | 31/1/1 | 7/0/2 |
| Stage | I (A,B) | 4 | 7 | 2 |
| II (A,B) | 5 | 9 | 0 | |
| III (A,B) | 5 | 9 | 3 | |
| IV (A,B) | 23 | 4 | 3 | |
| Not reported | 0 | 4 | 1 | |
| Smoking Status | Smoker | 7 | 6 | 3 |
| Former | 15 | 17 | 3 | |
| Never | 6 | 0 | 0 | |
| Unknown | 9 | 10 | 3 | |
| Treatment Naïve? | Y/N/Unknown | 19/16/2 | 30/2/1 | 5/3/1 |
PDX models
A summary of the 79 lung PDX models described in this report is presented in Supplementary Table S1. The collection of models is comprised mostly of LUAD (37 models) and LUSC (33 models). The collection includes two LUAD models developed from a primary and metastatic lesion from the same patient (TM00233 and TM01357). Models for other lung cancer types available from the repository include four small cell carcinomas, two large cell neuroendocrine carcinomas, two adenosquamous carcinomas, and one pleomorphic carcinoma. Only the LUAD and LUSC models were used in the analyses described in this report. On average, 38% of implantations resulted in successful engraftment, similar to other reports on lung cancer PDXs (34 – 39%) (17).
Quality Control (QC)
All patient tumor samples were negative for LCMV. All engrafted tumors demonstrated positive labeling for human Ki67 protein. Results of STR analysis for each model confirmed the engrafted tumor originated from the expected patient tumor. All models for which hematoxylin and eosin (H&E) stained slides were available for both patient and engrafted tumors data were determined to have moderate to high concordance following visual evaluation of the images by a board-certified pathologist (RGE).
Of 95 tumors engrafted, 13 (16%) were identified as lymphoid tumors based on positive staining for human CD45 antigen. These tumors likely arose from transplanted Epstein-Barr Virus (EBV)-infected human B-cells (39). The corresponding PDX models were removed from the JAX repository resulting in the final set of 79 models described here. A similar percentage of lymphomagenesis was reported in another PDX lung cancer model collection (40).
The Xenome (41) algorithm was used to determine human and mouse origins of sequence data generated from engrafted tumor samples. The average percentage of human sequences was 87% (53% - 99%) and 79% (50% - 89%) for the CTP assay and RNA-Seq, respectively (Supplementary Fig. S1). The average percentage of mouse sequences identified by Xenome were ~12% (0.5% - 47%), and the percentage of sequence reads that were classified as “both” or “ambiguous” were <0.2% and ~0.6% for both platforms, respectively. The “neither human nor mouse” category averaged 0.04% for CTP and 7.8% for RNA-Seq. The observed differences in the percentages of mouse and human sequences are likely due to platform differences (sequence capture method for CTP compared to direct sequencing for RNA-Seq). The difference between the platforms was significant for human reads (Welch Two Sample t-test; p-value = 2.19×10−8) but not for reads classified as mouse.
Genomic characterization: Somatic mutation
Genes on the JAX CTP panel that were mutated in at least 30% of the LUAD and LUSC PDX models are summarized in Fig. 2A and B (see Supplementary Table S2 for complete gene list). As has been observed previously in many human cancers, TP53 is the most commonly mutated gene in both LUAD and LUSC (7,32).
Figure 2.
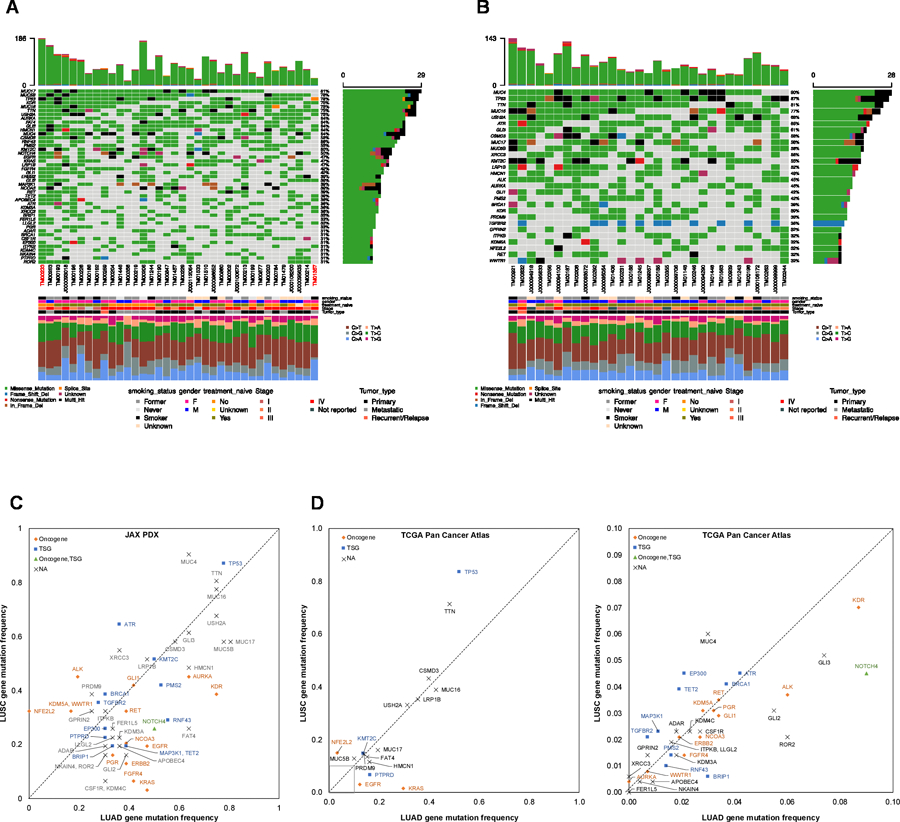
Oncoplot of the most frequently mutated genes in (A) LUAD and (B) LUSC PDX models. The oncoplot shows the PDX models in a horizontal orientation, annotated with smoking status, gender, treatment naïve status, stage of cancer and tumor type. Genes with mutation frequency > 30% is shown on the vertical axis. The barplot at the top has the frequency of mutations for each PDX model, while the right barplot has the frequency of mutations in each gene. Colors in the oncoplot columns indicate different mutation types (see legend for details). The bottom panel shows the classification of the SNPs into transitions and transversions. Model IDs in red originate from the same patient. (C) Comparison of gene mutation frequency in LUAD and LUSC PDX models (frequency >30%). (D) Comparison of gene mutation frequency in LUAD and LUSC TCGA samples (left: frequency >10%, right: frequency < 10%). Oncogene and Tumor Suppressor Gene (TSG) annotations from OncoKB.
An evaluation of mutation frequencies between LUAD and LUSC in the JAX PDX repository revealed that mutation frequencies in some genes are characteristic of the NSCLC subtype (Fig. 2C). Because of the relatively small number of PDX models in this analysis, these trends cannot be considered definitive. However, several of the patterns in the JAX collection are also observed in the TCGA PanCancer Atlas from cBioPortal (28) (Fig. 2D). Genes that are more frequently mutated in LUSC in both JAX PDX and TCGA datasets include NFE2L2, TP53, and MUC4. Genes that show higher mutation frequencies in LUAD in both datasets include KRAS, EGFR, NOTCH4, HMCN1, and MUC17. Other genes identified as being characteristic of LUAD and LUSC in one but not both data sets. For example, mutations in AURKA and FER1L5 were characteristic of LUAD tumor in the JAX data set but not TCGA; mutations in TET2 were characteristic of LUAD in the JAX data but is reported as being more frequently mutated in LUSC in the TCGA data; ALK mutations were higher in LUSC tumor in the JAX data but occur more frequently in LUAD tumors in the TCGA data set. Several factors could explain these differences. First, the JAX PDX samples were sequenced at very high coverage (mean coverage = 941x) compared to the whole-exome sequencing of TCGA samples (~100x) (42). Second, the types of samples used to generate the two resources differ. The tumor types used to generate the JAX resource were often selected by collaborating clinical oncologists based on known clinical (e.g., stage, prior treatment, metastasis, relapse) and/or genomic features. Finally, a greater proportion of late-stage tumors (Stage II or later) are found in the JAX PDX models (LUAD: 89%, LUSC: 63%) compared to TCGA PanCancer Atlas (LUAD: 41%, LUSC: 51%) (Supplementary Fig. S2).
For 22 of the LUAD PDX models, genomic testing data for the patient tumor was provided. All the engrafted tumors retained the clinically relevant mutations of the donor patient’s tumor (Supplementary Table S1). For model TM01244, the expected EGFR T790M mutation was not observed in the sequence data from the CTP targeted gene panel, but the presence of the mutation was confirmed by droplet digital PCR (ddPCR). The failure of the targeted gene sequencing to identify the mutation in this case could be due to the random sampling of subclones from a heterogeneous patient tumor in the sample used to establish the PDX model (11,43).
Although patients with LUSC have limited targeted treatment options compared to those diagnosed with LUAD, recent findings of recurrent genomic alterations that are characteristic of this histologic subtype, including activating alterations in PIK3CA, KRAS, and MET, provide future therapeutic avenues for research (44). Within the JAX PDX collection, clinically relevant alterations of these genes were found in 26 of the LUSC models (Supplementary Table S1).
Patient tumor genomics was tested before establishing the PDX model, in which the KRAS G12C mutation was also observed in the engrafted tumor (TM00231). Patient tumors of TM01243 and TM01448 were assayed on the same platforms, and 2 out of 4 of the alterations can be detected in the PDXs.
Considering both LUAD and LUSC models, 97% (n=68/70) of the engrafted tumors harbored clinically relevant alterations based on annotations from the JAX CKB database (25). As TMB and MSI are used as biomarkers for immunotherapy response (45–49), we observed trends similar to other lung cancer data sets, where lung tumors are rarely MSI-high, but do have high TMB scores (Supplementary Fig. S3, Supplementary Table S1). Within the JAX PDX collection, none of the lung cancer PDX models are MSI-high (MSI score > 20); while ten of the models are classified as high tumor mutation burden (TMB score > 22). The MSI and TMB scores are similar across multiple samples and passages of the same model, indicating that these genomic features are maintained throughout passaging and expansion.
Genomic characterization: Copy Number Alteration (CNA)
Recurrent gains and losses of chromosomal regions have been documented in NSCLC previously, including gains in MYC, EGFR, CCND1 and losses in LRP1B and CDKN2A (7,32). In LUSC, gains in 3q and losses in 3p and 5q occur more frequently than in LUAD (50). An overview of the gains and losses observed in engrafted tumors from LUAD and LUSC PDX models is provided in Figure 3A and 3B, respectively. Gain and loss frequencies of individual genes are provided in Supplementary Table S3. The frequently amplified and deleted chromosomal regions are consistent with previous studies.
Figure 3.
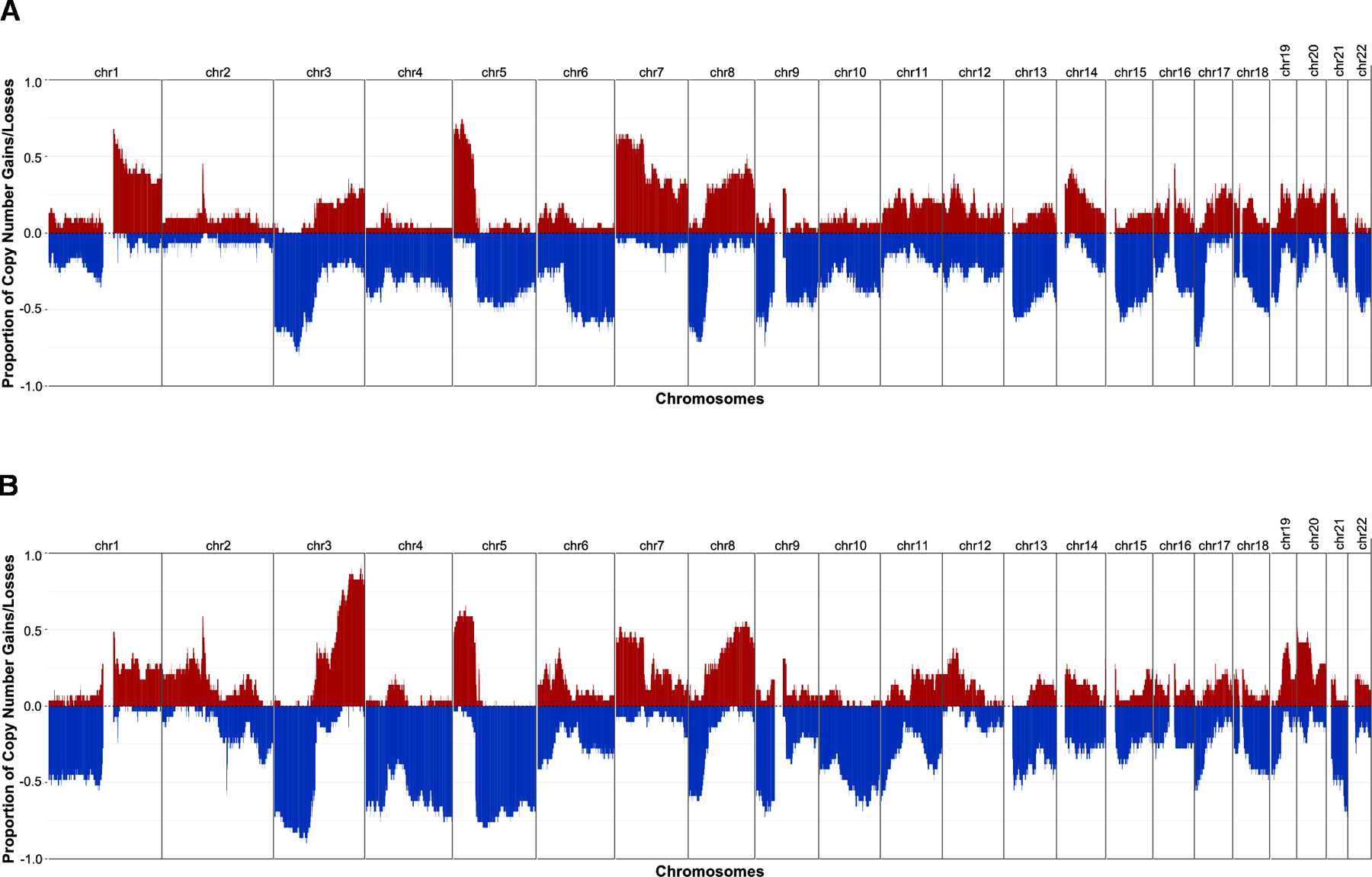
Frequency of copy number gain and loss for (A) LUAD and (B) LUSC PDX models. CN Gain: log2(CN/ploidy) > 0.5; CN Loss: log2(CN/ploidy) < −0.5. One sample per model was used to calculate the frequency.
Copy number profiles of individual PDX samples for LUAD, LUSC, and other lung cancer subtypes are shown in Supplementary Fig. S4A, S4B and S4C,respectively. Amplifications reported for patient tumors were also observed in the corresponding PDX model. For example, MET, EGFR, and MYC amplifications reported in a patient tumor were recapitulated in the corresponding PDX model (TM00784). Different engrafted tumor samples derived from the same PDX model had high concordance in copy number (11).
Genomic characterization: Transcriptional profiling
Unsupervised hierarchical clustering of gene expression data for engrafted tumors revealed that samples clustered primarily by the platform (RNA-Seq or microarray) and then by the diagnosis (Fig. 4A). Tumor samples derived from the same PDX model were highly correlated regardless of platform (Supplementary Fig. S5), indicating that the expression profile is retained during engraftment, expansion, and passaging.
Figure 4.
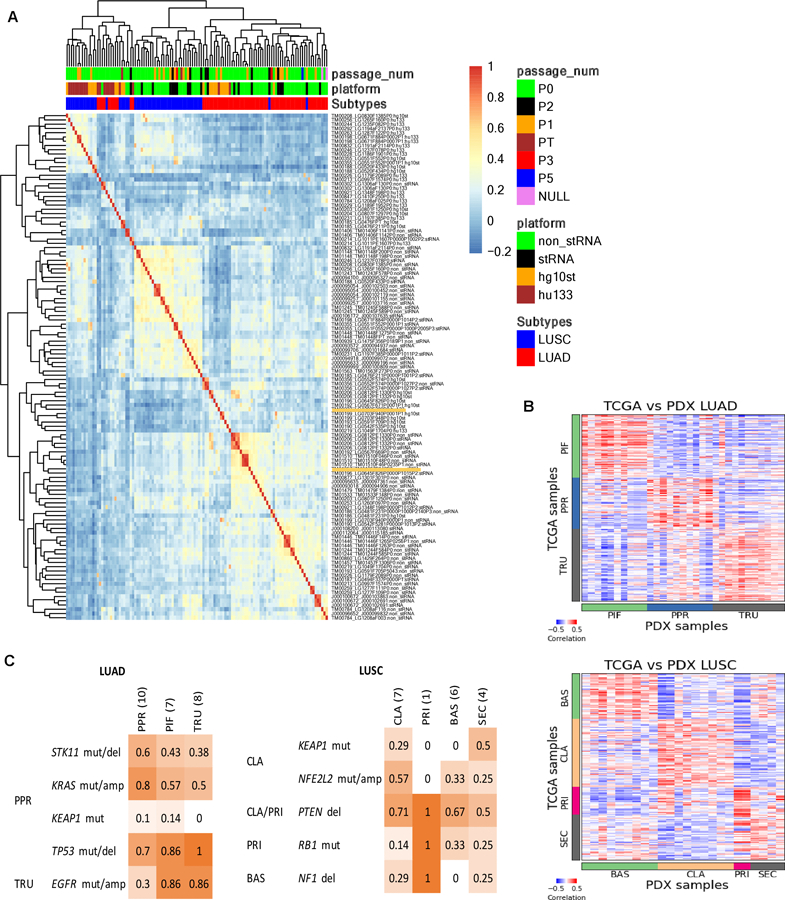
Gene expression in lung cancer PDX models. (A) Hierarchical clustering of gene expression percentile rank z-score for all lung cancer PDX samples and platforms. The heatmap is based on correlation values of expression percentile rank z-score expression for LUSC and LUAD PDX samples from sequencing and array platforms. The top horizontal color bars indicate the library preparation methods and platforms, subtype designation and passage number. Sample labels are indicated by model ID, sample ID and library preparation/platform. (B) Expression (quantile-normalized raw RSEM counts) correlation of nearest template prediction genes between TCGA (LUAD: n=230, LUSC: n=178) and PDX (LUAD: n=36, LUSC: n=24) samples for LUAD and LUSC. The color bars indicate the subtype labels for TCGA and subtype predictions for PDX. (C) Proportion of PDX models with mutations reported to be enriched in LUAD and LUSC subtypes as indicated on the left. LUAD subtypes: proximal-inflammatory (PIF), proximal-proliferative (PPR), terminal respiratory unit (TRU). LUSC subtypes: basal (BAS), classical (CLA), primitive (PRI) and secretory (SEC).
Consistent with previous reports we observed a positive correlation between the copy number of amplified and deleted genes and gene expression level across 63 lung PDX tumor samples assayed for both copy number and expression profiles (Pearson correlation coefficient = 0.54, p < 10−15) for a subset of frequently amplified and deleted genes in both LUAD and LUSC PDX models (Supplementary Fig. S6A) (51). Supplementary Fig. S6B shows the concordance between gene expression and the copy number for these genes individually. We observed that most tumors with copy number alterations in oncogenes exhibit both copy number and expression gains. Conversely, tumor samples with copy number alterations in tumor suppressor genes exhibit both copy number and expression losses. Based on these examples, it is evident that the amplification status elevates the gene expression levels of these frequently amplified oncogenes. Similarly, the deletion status decreases the gene expression levels of these frequently deleted TSGs.
The patient tumor associated with model TM01244 was noted as having elevated MET expression, but this property was only recapitulated as low-level over-expression (percentile rank z-score = 0.44, 0.51) in the engrafted tumors. Given that the EGFR T790M mutation present in the patient tumor was detected in the PDX tumor only after ddPCR, it is plausible that the patient tumor fragment used to establish the model was not representative of the clonal population used for genomic testing of the patient or that the tumor cells that were positive for the markers were out competed by other cancer cells in the mouse host.
Transcriptional subtyping
We adapted a PDX molecular subtyping tool developed for colorectal cancer PDX models, CMScaller (31), to classify the expression subtypes for the LUAD and LUSC models (4,5,52). Using the training set from TCGA yielded 793 and 1224 template genes to classify LUAD and LUSC subtypes respectively (Supplementary Table S7 and S8), resulting in high accuracies of 93% and 92% for the TCGA validation set (Supplementary Table S4). For the JAX PDX models, 31 out of 36 LUAD samples and 24 out of 24 LUSC samples were classified in expression subtype categories with high confidence (FDR-adjusted P-values < 0.05) (Supplementary Tables S5 and S6). Among the LUAD samples, 32% were classified as PIF, 32% as PPR and 35% as TRU. Among the LUSC samples, 38% were classified as BAS, 38% as CLA, 8% as PRI, and 17% as SEC. For models with multiple samples, all were predicted as the same subtype within each model, except for LUSC model TM01448 in which PT and P0 were classified as BAS and SEC, respectively. We hypothesize that spatial tumor heterogeneity in the tumor sample that was used to establish the PDX model is the source of these classification differences (53). The patient and P0 tumor samples of this model share the same clinically relevant mutations except for a PTEN nonsense mutation that was detected only in the PT sample.
To further confirm the reliability of the classifications, we compared the expression of the template genes between TCGA samples with known subtype labels and the predicted subtypes of the samples of the PDX models. We observed high correlation within the template genes of each respective LUAD or LUSC subtype (Fig. 4B). This confirms that the expression level of the template genes is replicated in the lung cancer PT/PDX samples. The subtypes were also enriched in other genomic alteration profiles (5,7,32,52). Despite the limited number of samples, we observed higher proportion in some of the reported subtype-enriched alterations within the respective predicted PDX subtypes (Fig. 4C). In particular, the LUAD PPR subtype was reported to be enriched in STK11 and KRAS alterations in other PT datasets, and the PDX models classified as PPR subtype showed higher frequencies of these alterations compared other subtypes. The same observation can also be made for the NFE2L2 alteration enriched in LUSC CLA subtype. As such, the PDX models displayed subtype-specific expression and/or alteration profiles similar to those reported in patient tumor subtyping studies.
Treatment responses in PDX models
Many of the tumors submitted for PDX generation were selected based on the presence of clinically relevant mutations per National Comprehensive Cancer Network (NCCN) guidelines (Supplementary Table S1) (10,25,54). Cohorts of tumor-bearing mice of a subset of the lung cancer PDX models were enrolled in dosing studies to evaluate responses to drug treatment. A summary of treatment responses is illustrated for LUAD models (Supplementary Fig. 7A), LUSC models (Supplementary Fig. 7B), and other lung models in the collection (Supplementary Fig. 7C).
Targeted treatment of EGFR mutant PDXs.
Nine PDX models in the JAX collection harbor activating mutations in EGFR (L858R, exon 19 deletion, exon 20 insertion) and were tested for response to tyrosine kinase inhibition. Six of the models (TM00199, TM00204, TM00219, TM00253, and TM00784) were derived from patients at the time of progression on either single-agent or combinations of erlotinib; two of the models (J000100672 and TM00193) were derived from treatment-naïve patients. Both TM00204 and TM00219 harbor the EGFR T790M mutation, and TM00784 harbors MET amplification. These markers are associated with acquired resistance to treatment with TKIs (10,54). J000100672 harbors the exon 20 insertion associated with de novo resistance to TKI inhibitors (55). TM00253 harbors the mutation EGFR V834L, which is associated with decreased response to erlotinib (56). Cohorts of tumor-bearing mice of these models, except for TM00193, were treated with single-agent erlotinib. TM00199 displayed partial response (PR), TM00253 displayed stable disease (SD), and the other four models with TKI treatment resistance mutations displayed progressive disease (PD) (Fig. 5A, Supplementary Fig. S7A and S8). The lack of complete response (CR) recapitulated the treatment response observed in the patients and the response expected from the EGFR mutation status of these models.
Figure 5.
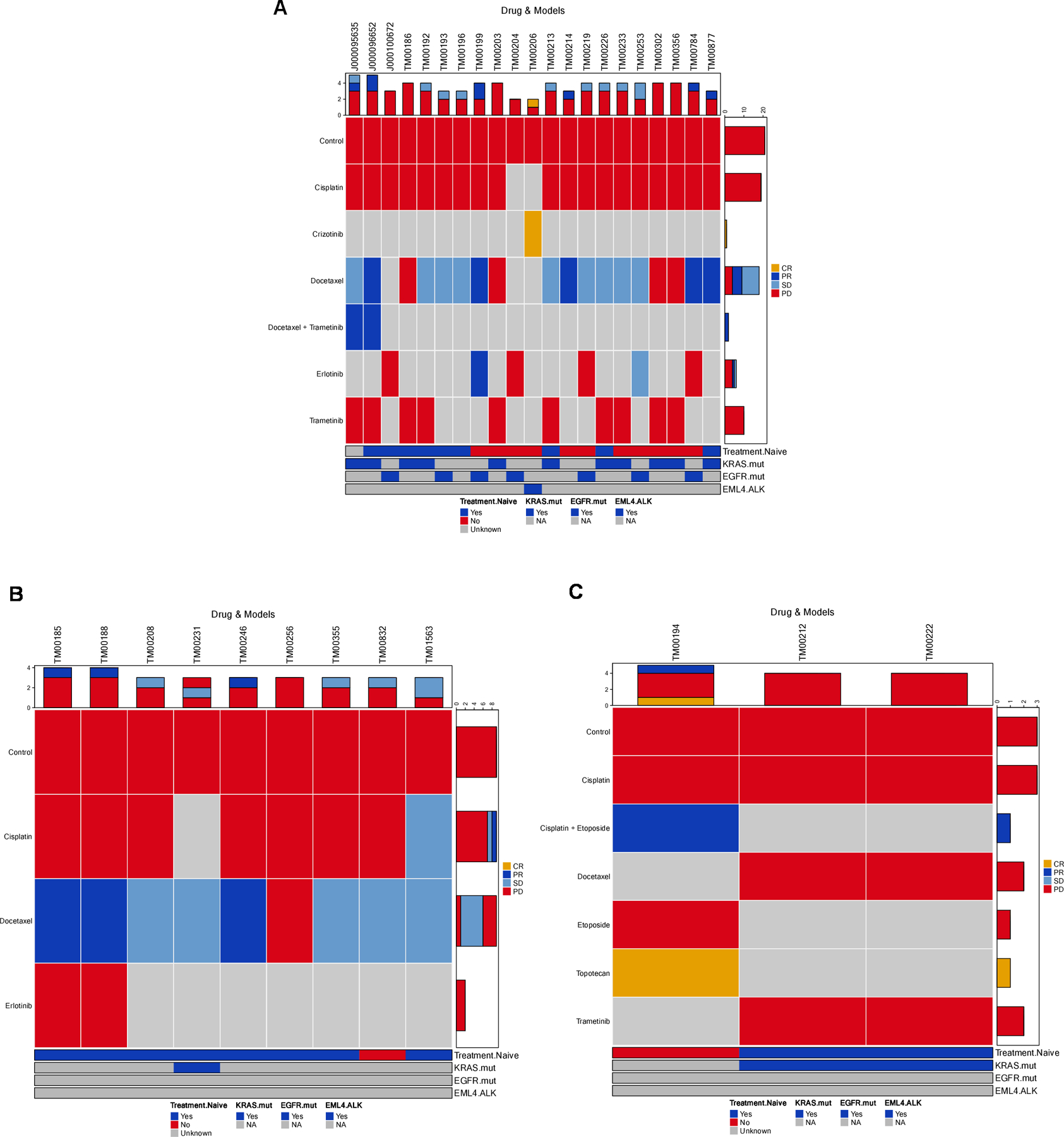
Summary of treatment responses in lung PDX models. (A) LUAD, (B) LUSC, and (C) all other lung cancer types. Treatment responses were calculated as percent change in tumor volume for each animal as V: ((end_volume – start_volume)/start_volume) * 100) at day 21. Within each group the minimum (Vm) and average volume (Va) were calculated with response classifications calculated as follows: Complete response (CR): Vm < −95%, Va < −40%; Partial response (PR): Vm < −50%, Va < −20%; Stable disease (SD): Vm < 35%, Va < 30%; Progressive disease (PD): Anything else. The number of treatments in each RECIST category is shown at the top, and number of models in each RECIST category is shown on the right side of each plot. The color bar at the bottom indicates the treatment naïve and mutation status (KRAS, EGFR and EML4-ALK fusion) of each model. Plots were generated with the R package Xeva (version 1.6.0).
Two models (TM00199 and TM00219) were tested using second-generation agents to evaluate treatment options following the development of resistance to first-generation TKIs (56). The models were treated with a combination of afatinib and cetuximab along with single-agent erlotinib, afatinib, cetuximab, and vehicle controls. As expected, TM00199 did not exhibit significant tumor growth inhibition upon treatment with erlotinib. Single-agent afatinib-treated animals subsequently progressed after a succession of treatment, whereas animals treated with cetuximab (with or without afatinib) exhibited complete response. The treatment effects on EGFR expression and phosphorylation were examined at six and 24-hour timepoints (Figure 6). After a single treatment, the EGFR TKIs erlotinib and afatinib resulted in notable downregulation of EGFR phosphorylation within six hours, rebounding to control levels after 24 hours (Figure 6A and Supplementary Figure S11). In contrast, cetuximab showed evidence of moderate downregulation by six hours and 24 hours accompanied by diminished total protein expression. The combination of afatinib plus cetuximab resulted in ablated phosphorylation at six hours in two of three models, maintained at the 24 timepoints, associated with reduced protein expression.
Figure 6.
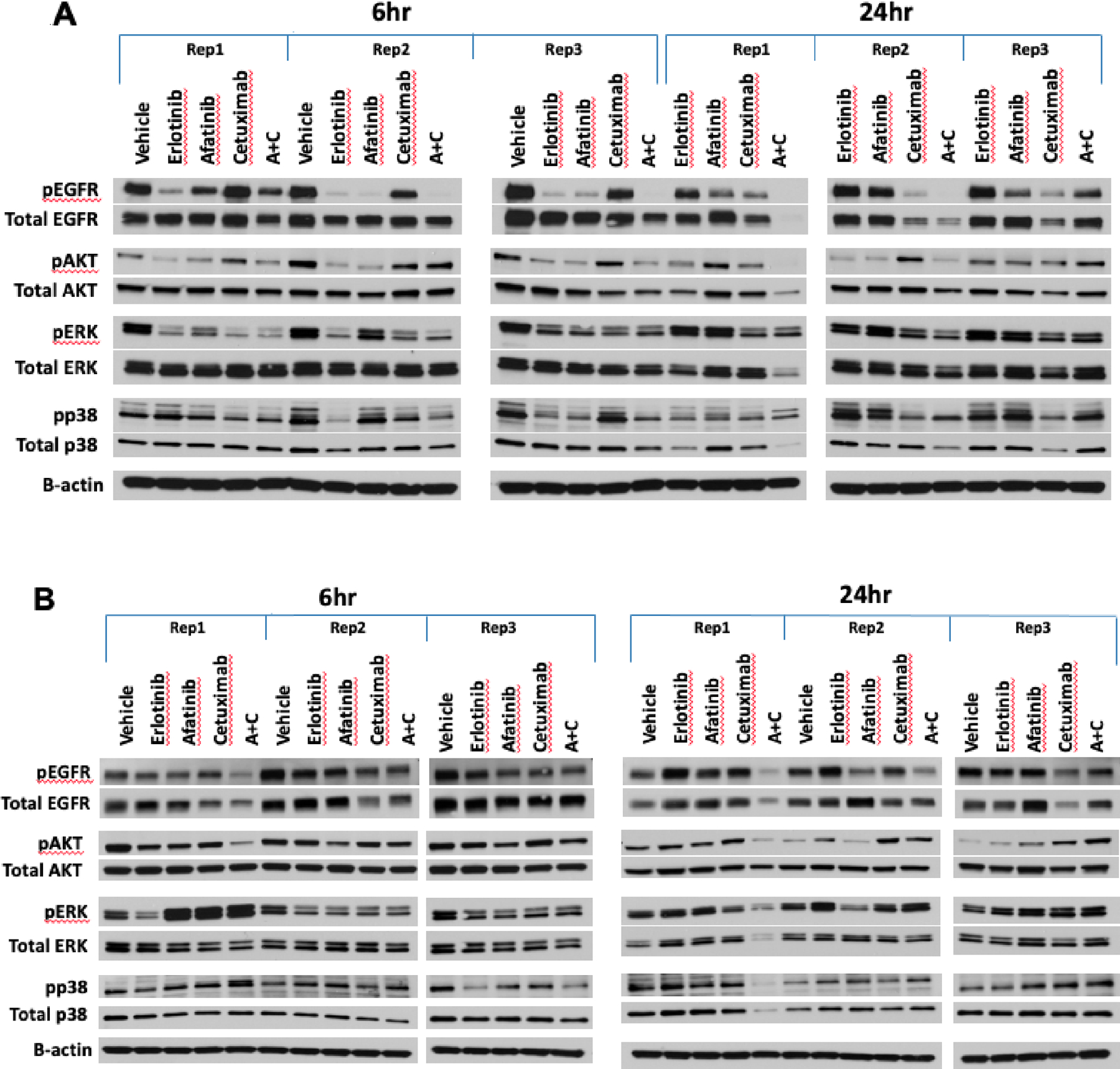
Treatment-induced changes in phosphorylation of EGFR, AKT, ERK and p38 at 6 and 24 hours in lung PDX models treated with erlotinib, afatinib, cetuximab, and afatinib+cetuximab. Studies were conducted in triplicate for each time point, independently in different animals. AKT, ERK1 and p38 phosphorylation state were examined to evaluate changes in downstream signal transduction, with the understanding that the timing of effects will be subject to variation across models. (A) In model TM00199, a single treatment of erlotinib and afatinib induced downregulation of EGFR phosphorylation within six hours, rebounding to control levels after 24 hours. Treatment with cetuximab demonstrated moderate downregulation by six hours in two of three models and substantial downregulation at 24 hours in three of three models, accompanied by diminished total protein expression. The combination of afatinib plus cetuximab resulted in ablated phosphorylation at six hours in two of three models, maintained at the 24 timepoints, associated with reduced protein expression. (B) In model TM00219, none of the EGFR-targeted agents could entirely suppress EGFR phosphorylation at 6 or 24 hours.
The TM00219 model was derived from a patient at the time of erlotinib progression, associated with the emergence of the T790M EGFR resistance mutation which was observed in both the patient post-erlotinib treatment biopsy and the engrafted tumor. This model showed no benefit from erlotinib or cetuximab, with marginal activity from afatinib. In this unresponsive model, none of the EGFR-targeted agents could entirely suppress EGFR phosphorylation at six or 24 hours (Figure 6B and Supplementary Figure S11).
Targeted treatment of an EML4-ALK fusion PDX model.
LUAD model TM00206 harbors the EML4-ALK fusion and, as expected, had a robust response to treatment with the ALK TKI, crizotinib (Fig. 5A, Supplementary Fig. S7A and S9) (57). The response at the cohort level was categorized as a complete response (CR). However, 2 of the 9 mice in the treatment cohort were classified as partial response (PR). Acquired crizotinib resistance has been reported in ALK-rearranged NSCLCs (58) and the clinical records for the patient reveal that the individual’s cancer progressed while on treatment. The variability in the response in the corresponding individual mice may be due to the presence of resistant subclones. Although the treated PDXs were not tested for known resistance variants, the genomic data from two early passage (P0) tumors for this model revealed the presence of reported resistance mutations at a subclonal level. The ALK L1196M mutation was detected at an allele frequency of 22% and a low-level KIT amplification (log(CN/ploidy) = 0.43) was detected in one of the P0 tumors (LG0812PE1330P0), while low-level amplification in ALK and EML4, possibly the fusion, was detected in another P0 sample (LG0812PE1332P0).
Treatment of KRAS-mutant PDXs.
Twelve models in the collection harbor loss-of-function KRAS mutations that result in downstream pathway activation (Supplementary Table 1) and were treated with a MEK1/2 inhibitor (trametinib). Trametinib acts through the inhibition the MAPK pathway downstream of KRAS. While most models exhibited no growth inhibition from trametinib and no cohort-level responses surpassed the threshold of progressive disease, the per animal response data for five of the models (TM00226, J000095635, TM00186, TM00203, TM00233) had at least 2 animals in the cohort with a response classification of stable disease (Supplementary Fig. S7A), suggesting moderate activity of the drug in some animals. This result is consistent with clinical trials that demonstrated single-agent trametinib showed no improved efficacy compared to docetaxel in patients with advanced KRAS-mutant NSCLC (Fig. 5B, Supplementary Fig. S7A) (59). Partial response was observed for the combination of docetaxel and trametinib for model J000095635 (KRAS G12D) compared to single-agents (stable and progressive disease, respectively). For model J000096652 (KRAS G12C), the combination docetaxel and trametinib showed no additional benefit over single-agent docetaxel. Both treatment arms were classified as partial response (PR) (Fig. 5A, Supplementary Fig. S7A and S10).
Discussion
The treatment of NSCLC has rapidly evolved, with chemotherapy being replaced by either targeted therapies or immunotherapy in many cases. For treatment advances to continue, preclinical models which accurately reflect the complexity and heterogeneity of human cancers, as well as being predictive of drug sensitivity and resistance patterns observed in patients, are mandatory. Given the complexities of drug-tumor interactions together with inter- and intra-patient tumor heterogeneity, PDX models stand apart in the preclinical arena as experimentally tractable and reproducible models that recapitulate the clinically relevant genomic properties and treatment responses of the patient tumors from which they are derived.
The repository of lung cancer PDX models described here was generated, characterized, and annotated in collaboration with clinical investigators. For models with corresponding patient tumor genomic data, the implanted tumors in the lung PDXs maintained the histological characteristics and genomic properties of patient tumors from which they were derived. Treatment responses for targeted agents in the models were consistent with expectations based on the presence of specific molecular targets and also recapitulated the treatment responses of patients treated with the same agents. The PDX models available from the JAX repository are a validated resource for preclinical investigations into the efficacy of new cancer treatments and for basic research into the mechanisms of acquired resistance to target-directed therapies and for developing strategies to overcome treatment resistance.
Supplementary Material
Significance:
Patient-derived xenografts of lung cancer retain key features observed in the originating patient tumors and show expected responses to treatment with standard-of-care agents, providing experimentally tractable and reproducible models for preclinical investigations.
Acknowledgements
The authors express their deep appreciation to the patients who generously consented to have their tumor tissues used to generate the patient-derived cancer models described in this manuscript and to make these models broadly available for basic and pre-clinical cancer research. The generation and characterization of models in the JAX lung cancer PDX repository was funded by The Jackson Laboratory Director’s Innovation Fund, the Maine Cancer Foundation, the Kleeberg Foundation, the Hope Foundation, and the Integrated Translational Science Center (ITSC) (5U01 CA180944). Genome data analysis was supported, in part, by the JAX Cancer Center Computational Sciences Shared Service (P30 CA034196). The PDX Web portal was developed with support from R01 CA089713. Additional support provided by the UCD Comprehensive Cancer Center (P30CA093373), the UCaMP Minority PDX Center (U54CA233306), the Susan and Jerry Knapp and the Addario Foundation, and Stand Up to Cancer (SU2C 201502309). The UCD Pathology Biorepository assisted with collection and histological interpretation of specimens. RNA-seq data for patient and PDX tumors used as part of the expression-based subtype analysis were obtained from the NCI Patient-Derived Models Repository (PDMR). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
P30 CA034196 (ETL), P30 CA093373 (PNL), SU2C 201502309 (DRD), 5U01 CA180944 (ETL), R01 CA089713 (CJB). The Jackson Laboratory Director’s Innovation Fund (CJB and SDA), the Maine Cancer Foundation (CJB), the Kleeberg Foundation (CJB), and the Hope Foundation (ETL).
Footnotes
The authors declare no potential conflicts of interest.
References
- 1.Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA: A Cancer Journal for Clinicians 2021;71:209–49 [DOI] [PubMed] [Google Scholar]
- 2.Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, Tian Ng AW, Wu Y, et al. The repertoire of mutational signatures in human cancer. Nature 2020;578:94–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013;499:214–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hayes DN, Monti S, Parmigiani G, Gilks CB, Naoki K, Bhattacharjee A, et al. Gene Expression Profiling Reveals Reproducible Human Lung Adenocarcinoma Subtypes in Multiple Independent Patient Cohorts. Journal of Clinical Oncology 2006;24:5079–90 [DOI] [PubMed] [Google Scholar]
- 5.Wilkerson MD, Yin X, Hoadley KA, Liu Y, Hayward MC, Cabanski CR, et al. Lung Squamous Cell Carcinoma mRNA Expression Subtypes Are Reproducible, Clinically Important, and Correspond to Normal Cell Types. Clinical Cancer Research 2010;16:4864–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chan BA, Hughes BGM. Targeted therapy for non-small cell lung cancer: current standards and the promise of the future. Translational Lung Cancer Research 2014;4:36–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Collisson EA, Campbell JD, Brooks AN, Berger AH, Lee W, Chmielecki J, et al. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014;511:543–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee CS, Milone M, Seetharamu N. Osimertinib in EGFR-Mutated Lung Cancer: A Review of the Existing and Emerging Clinical Data. Onco Targets Ther 2021;14:4579–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kwak EL, Bang Y-J, Camidge DR, Shaw AT, Solomon B, Maki RG, et al. Anaplastic Lymphoma Kinase Inhibition in Non–Small-Cell Lung Cancer. New England Journal of Medicine 2010;363:1693–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yun C-H, Mengwasser KE, Toms AV, Woo MS, Greulich H, Wong K-K, et al. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proceedings of the National Academy of Sciences 2008;105:2070–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Woo XY, Giordano J, Srivastava A, Zhao Z-M, Lloyd MW, de Bruijn R, et al. Conservation of copy number profiles during engraftment and passaging of patient-derived cancer xenografts. Nature Genetics 2021;53:86–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Simpson KL, Stoney R, Frese KK, Simms N, Rowe W, Pearce SP, et al. A biobank of small cell lung cancer CDX models elucidates inter- and intratumoral phenotypic heterogeneity. Nat Cancer 2020;1:437–51 [DOI] [PubMed] [Google Scholar]
- 13.Ilie M, Nunes M, Blot L, Hofman V, Long-Mira E, Butori C, et al. Setting up a wide panel of patient-derived tumor xenografts of non–small cell lung cancer by improving the preanalytical steps. Cancer Medicine 2015;4:201–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Drapkin BJ, George J, Christensen CL, Mino-Kenudson M, Dries R, Sundaresan T, et al. Genomic and Functional Fidelity of Small Cell Lung Cancer Patient-Derived Xenografts. Cancer Discovery 2018;8:600–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang X-c, Zhang J, Li M, Huang X-s, Yang X-n, Zhong W-z, et al. Establishment of patient-derived non-small cell lung cancer xenograft models with genetic aberrations within EGFR, KRAS and FGFR1: useful tools for preclinical studies of targeted therapies. Journal of Translational Medicine 2013;11:168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stewart EL, Mascaux C, Pham N-A, Sakashita S, Sykes J, Kim L, et al. Clinical Utility of Patient-Derived Xenografts to Determine Biomarkers of Prognosis and Map Resistance Pathways in EGFR-Mutant Lung Adenocarcinoma. Journal of Clinical Oncology 2015;33:2472–80 [DOI] [PubMed] [Google Scholar]
- 17.Chen Y, Zhang R, Wang L, Correa AM, Pataer A, Xu Y, et al. Tumor characteristics associated with engraftment of patient-derived non–small cell lung cancer xenografts in immunocompromised mice. Cancer 2019;125:3738–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee P-C, Fang Y-F, Yamaguchi H, Wang W-J, Chen T-C, Hong X, et al. Targeting PKCδ as a Therapeutic Strategy against Heterogeneous Mechanisms of EGFR Inhibitor Resistance in EGFR-Mutant Lung Cancer. Cancer Cell 2018;34:954–69.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Woo XY, Srivastava A, Graber JH, Yadav V, Sarsani VK, Simons A, et al. Genomic data analysis workflows for tumors from patient-derived xenografts (PDXs): challenges and guidelines. BMC Medical Genomics 2019;12:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krupke DM, Begley DA, Sundberg JP, Richardson JE, Neuhauser SB, Bult CJ. The Mouse Tumor Biology Database: A Comprehensive Resource for Mouse Models of Human Cancer. Cancer Research 2017;77:e67–e70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Conte N, Mason JC, Halmagyi C, Neuhauser S, Mosaku A, Yordanova G, et al. PDX Finder: A portal for patient-derived tumor xenograft model discovery. Nucleic Acids Research 2018;47:D1073–D9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tsongalis GJ, Wu AH, Silver H, Ricci A, Applications of forensic identity testing in the clinical laboratory. American journal of clinical pathology 1999;112:S93–103 [PubMed] [Google Scholar]
- 23.Chen K, Ahmed S, Adeyi O, Dick JE, Ghanekar A. Human Solid Tumor Xenografts in Immunodeficient Mice Are Vulnerable to Lymphomagenesis Associated with Epstein-Barr Virus. PLOS ONE 2012;7:e39294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ananda G, Mockus S, Lundquist M, Spotlow V, Simons A, Mitchell T, et al. Development and validation of the JAX Cancer Treatment Profile™ for detection of clinically actionable mutations in solid tumors. Experimental and Molecular Pathology 2015;98:106–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Patterson SE, Liu R, Statz CM, Durkin D, Lakshminarayana A, Mockus SM. The clinical trial landscape in oncology and connectivity of somatic mutational profiles to targeted therapies. Human Genomics 2016;10:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Niu B, Ye K, Zhang Q, Lu C, Xie M, McLellan MD, et al. MSIsensor: microsatellite instability detection using paired tumor-normal sequence data. Bioinformatics 2013;30:1015–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mayakonda A, Lin DC, Assenov Y, Plass C, Koeffler HP. Maftools: efficient and comprehensive analysis of somatic variants in cancer. Genome research 2018;28:1747–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discovery 2012;2:401–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chakravarty D, Gao J, Phillips S, Kundra R, Zhang H, Wang J, et al. OncoKB: A Precision Oncology Knowledge Base. JCO Precision Oncology 2017:1–16 [DOI] [PMC free article] [PubMed]
- 30.Skidmore ZL, Wagner AH, Lesurf R, Campbell KM, Kunisaki J, Griffith OL, et al. GenVisR: Genomic Visualizations in R. Bioinformatics 2016;32:3012–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eide PW, Bruun J, Lothe RA, Sveen A. CMScaller: an R package for consensus molecular subtyping of colorectal cancer pre-clinical models. Scientific Reports 2017;7:16618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hammerman PS, Lawrence MS, Voet D, Jing R, Cibulskis K, Sivachenko A, et al. Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012;489:519–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Colaprico A, Silva TC, Olsen C, Garofano L, Cava C, Garolini D, et al. TCGAbiolinks: an R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Research 2015;44:e71-e [DOI] [PMC free article] [PubMed]
- 34.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biology 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ghandi M, Huang FW, Jané-Valbuena J, Kryukov GV, Lo CC, McDonald ER, et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 2019;569:503–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gao H, Korn JM, Ferretti S, Monahan JE, Wang Y, Singh M, et al. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nature Medicine 2015;21:1318–25 [DOI] [PubMed] [Google Scholar]
- 37.Mer AS, Ba-Alawi W, Smirnov P, Wang YX, Brew B, Ortmann J, et al. Integrative Pharmacogenomics Analysis of Patient-Derived Xenografts. Cancer Res 2019;79:4539–50 [DOI] [PubMed] [Google Scholar]
- 38.Holland WS, Chinn DC, Lara PN, Jr., Gandara DR, Mack PC. Effects of AKT inhibition on HGF-mediated erlotinib resistance in non-small cell lung cancer cell lines. J Cancer Res Clin Oncol 2015;141:615–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bondarenko G, Ugolkov A, Rohan S, Kulesza P, Dubrovskyi O, Gursel D, et al. Patient-Derived Tumor Xenografts Are Susceptible to Formation of Human Lymphocytic Tumors. Neoplasia 2015;17:735–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.John T, Yanagawa N, Kohler D, Craddock KJ, Bandarchi-Chamkhaleh B, Pintilie M, et al. Characterization of Lymphomas Developing in Immunodeficient Mice Implanted With Primary Human Non–Small Cell Lung Cancer. Journal of Thoracic Oncology 2012;7:1101–8 [DOI] [PubMed] [Google Scholar]
- 41.Conway T, Wazny J, Bromage A, Tymms M, Sooraj D, Williams ED, et al. Xenome—a tool for classifying reads from xenograft samples. Bioinformatics 2012;28:i172–i8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang VG, Kim H, Chuang JH. Whole-exome sequencing capture kit biases yield false negative mutation calls in TCGA cohorts. PLOS ONE 2018;13:e0204912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jamal-Hanjani M, Wilson GA, McGranahan N, Birkbak NJ, Watkins TBK, Veeriah S, et al. Tracking the Evolution of Non–Small-Cell Lung Cancer. New England Journal of Medicine 2017;376:2109–21 [DOI] [PubMed] [Google Scholar]
- 44.Gandara DR, Hammerman PS, Sos ML, Lara PN, Hirsch FR. Squamous Cell Lung Cancer: From Tumor Genomics to Cancer Therapeutics. Clinical Cancer Research 2015;21:2236–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bonneville R, Krook MA, Kautto EA, Miya J, Wing MR, Chen H-Z, et al. Landscape of Microsatellite Instability Across 39 Cancer Types. JCO Precision Oncology 2017:1–15 [DOI] [PMC free article] [PubMed]
- 46.Goodman AM, Kato S, Bazhenova L, Patel SP, Frampton GM, Miller V, et al. Tumor Mutational Burden as an Independent Predictor of Response to Immunotherapy in Diverse Cancers. Molecular Cancer Therapeutics 2017;16:2598–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Marabelle A, Fakih M, Lopez J, Shah M, Shapira-Frommer R, Nakagawa K, et al. Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: prospective biomarker analysis of the multicohort, open-label, phase 2 KEYNOTE-158 study. The Lancet Oncology 2020;21:1353–65 [DOI] [PubMed] [Google Scholar]
- 48.André T, Shiu K-K, Kim TW, Jensen BV, Jensen LH, Punt C, et al. Pembrolizumab in Microsatellite-Instability–High Advanced Colorectal Cancer. New England Journal of Medicine 2020;383:2207–18 [DOI] [PubMed] [Google Scholar]
- 49.Chalmers ZR, Connelly CF, Fabrizio D, Gay L, Ali SM, Ennis R, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Medicine 2017;9:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Qiu Z-W, Bi J-H, Gazdar AF, Song K. Genome-wide copy number variation pattern analysis and a classification signature for non-small cell lung cancer. Genes, Chromosomes and Cancer 2017;56:559–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shao X, Lv N, Liao J, Long J, Xue R, Ai N, et al. Copy number variation is highly correlated with differential gene expression: a pan-cancer study. BMC Medical Genetics 2019;20:175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wilkerson MD, Yin X, Walter V, Zhao N, Cabanski CR, Hayward MC, et al. Differential Pathogenesis of Lung Adenocarcinoma Subtypes Involving Sequence Mutations, Copy Number, Chromosomal Instability, and Methylation. PLOS ONE 2012;7:e36530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tavernari D, Battistello E, Dheilly E, Petruzzella AS, Mina M, Sordet-Dessimoz J, et al. Nongenetic Evolution Drives Lung Adenocarcinoma Spatial Heterogeneity and Progression. Cancer Discovery 2021;11:1490–507 [DOI] [PubMed] [Google Scholar]
- 54.Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, et al. MET Amplification Leads to Gefitinib Resistance in Lung Cancer by Activating ERBB3 Signaling. Science 2007;316:1039–43 [DOI] [PubMed] [Google Scholar]
- 55.Yasuda H, Kobayashi S, Costa DB. EGFR exon 20 insertion mutations in non-small-cell lung cancer: preclinical data and clinical implications. The Lancet Oncology 2012;13:e23–e31 [DOI] [PubMed] [Google Scholar]
- 56.Sarcar B, Gimbrone NT, Wright G, Remsing Rix LL, Gordian ER, Rix U, et al. Characterization of epidermal growth factor receptor (EGFR) P848L, an unusual EGFR variant present in lung cancer patients, in a murine Ba/F3 model. FEBS Open Bio 2019;9:1689–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shaw AT, Kim D-W, Nakagawa K, Seto T, Crinó L, Ahn M-J, et al. Crizotinib versus Chemotherapy in Advanced ALK-Positive Lung Cancer. New England Journal of Medicine 2013;368:2385–94 [DOI] [PubMed] [Google Scholar]
- 58.Katayama R, Shaw AT, Khan TM, Mino-Kenudson M, Solomon BJ, Halmos B, et al. Mechanisms of Acquired Crizotinib Resistance in ALK-Rearranged Lung Cancers. Science Translational Medicine 2012;4:120ra17–ra17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Blumenschein GR, Smit EF, Planchard D, Kim DW, Cadranel J, De Pas T, et al. A randomized phase II study of the MEK1/MEK2 inhibitor trametinib (GSK1120212) compared with docetaxel in KRAS-mutant advanced non-small-cell lung cancer (NSCLC)†. Annals of Oncology 2015;26:894–901 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Information and data for the PDX models described in the manuscript are freely available from the Mouse Models of Human Cancer database (MMHCdb, http://tumor.informatics.jax.org/mtbwi/pdxSearch.do) and from PDX Finder (https://pdxfinder.org)