

Abstract
A hallmark of cholesteatoma is hyperproliferation of keratinocytes with abundant production of keratins in the middle ear under chronic inflammatory conditions. We demonstrated in this study that Id1, an inhibitor of DNA-binding protein, is involved in the hyperproliferation of keratinocytes, positively via the nuclear factor-kappa B (NF-κB)/cyclin D1 pathway which is linked to cell cycle progression and negatively via the p16Ink4a which is linked to cell cycle inhibition. Id1 significantly increased the transcription of NF-κB which, in turn, upregulated the expression of cyclin D1 and keratin 10 in keratinocytes. Specific NF-κB inhibitors (pyrrolidine dithiocarbamate, PDTC), or dominant-negative inhibitor (I kappa B alpha mutant, IκBαM) abrogated the Id1-induced cell proliferation and keratin 10 production whereas p65, a subunit of the NF-κB heterodimer and an enhancer of the NF-κB activity strengthened the Id1-induced cell proliferation and keratin 10 production. It was concluded that Id1 contributed to hyperproliferation of keratinocytes via enhancement of cell cycle progression and removal of cell cycle inhibition and simultaneously increased keratin production.
Keywords: keratinocyte, hyperproliferation, Id1, cyclin D1
Introduction
Chronic otitis media (COM) is linked to cholesteatoma, a condition of aggressive proliferation of keratinocytes and abundant production of keratins in the middle ear. Morphologically, cholesteatoma forms an epithelial cyst which grows aggressively and erodes the ossicular chain and bony wall of the middle ear. It represents a threat to the neighboring tissues or organs. However, little is known about the driving force for abnormal growth of the epithelial cyst and abundant production of keratins in cholesteatoma. Recently, we found that the expression of Id1 was upregulated in the middle ear mucosa of rats following bacterial infection (1) and in the middle ear mucosa of humans with COM and/or cholesteatoma (2). It suggests that Id1 plays a role in the pathogenesis of middle ear cholesteatoma.
The Id gene family encodes four related proteins, from Id1 to Id4, which are involved in the control of cell-cycle progression in organisms ranging from flies to man (3, 4). Id proteins are involved in cell growth and proliferation (3, 5, 6) by antagonizing the action of basic helix-loop-helix (bHLH) transcription factors which are essential for cell differentiation. In the literature, enforced expression of Id1 in primary human keratinocytes leads to lifespan extension and cell immortalization (7). Transfection of the rat middle ear with Id1 in vivo resulted in proliferative responses in the mucosal layer (8). These led us to examine the role of Id1 in the proliferation of keratinocytes and production of keratins.
In this study, we hypothesized that Id1 induced the proliferation of keratinocytes through up-regulation of NF-κB/cyclin D1, a signaling pathway leading to Go- to S-phase progression, and down-regulation of p16Ink4a, an inhibitor for suppression of the cyclin-dependent kinase (cdk) activity. In addition, we hypothesized that Id1 induced the production of keratin 10, one of the major keratin products of keratinocytes via NF-κB. To test these hypotheses, cellular and molecular biologic experiments were performed (i) on human middle ear cholesteatoma specimens for evaluation of the importance of Id1-induced signaling in the pathogenesis of cholesteatoma and (ii) on cultured human keratinocytes for verification of the signaling pathway.
Methods
Materials
Fourteen cholesteatoma tissue specimens were procured from Ichinomiya City hospital at Nagoya, Japan, Cincinnati Children's Hospital Medical Center (CCHMC), and University of Minnesota Clinics and Hospitals (UMHC). Eight normal (no history of otitis media) middle ear mucosal and normal head and neck skin specimens procured from UMHC served as controls. The diagnosis of these specimens was made clinically and verified pathologically. All tissues were procured, handled, and maintained according to the protocols approved by each Institutional Review Board (9).
The Rhek-1A cell line, representative epidermal keratinocytes (10), was maintained in Eagle's minimal essential medium (MEM, Invitrogen) supplemented with 10% fetal bovine serum (FBS), penicillin/streptomycin (50 μg/mL) and 2 mM L-glutamine (herein referred to as full growth medium, FGM). During transient transfection of cells, Opti-MEM supplemented with 6 μg/mL of Polybrene® was used (Invitrogen, herein referred to as transfection medium). Rhek-1A was chosen because it is derived from human skin keratinocytes as is cholesteatoma.
Full-length Id1 cDNA was cloned into a protein-expressing vector (pEGFG, Clontech) using standard protocols as previously described (11). The functionality of this Id1 construct was recently verified (12). I kappa B alpha mutant (IκBαM, a kind gift of Dr. Inder Verma at Salk Institute, La Jolla, CA), in which the serine at position 36 had been changed to alanine, thus preventing phosphorylation and subsequent proteosomal degradation in response to stimuli (13, 14), is a dominant-negative inhibitor of NF-κB activity. Pyrrolidine dithiocarbamate (PDTC), a proteasome inhibitor purchased from Calbiochem, was used as a potent inhibitor of NF-κB (15) and proven as an effective inhibitor of the NF-κB activity in keratinocytes (16). The cyclin D1 reporters were generous gifts of Dr. Richard Pestell (Department of Developmental and Molecular Biology, Albert Einstein College of Medicine, Bronx, NY). They contain the wild-type and mutant cyclin D1 reporter constructs, as previously described (17). Briefly, the wild-type cyclin D1 reporter construct is consisted of a fragment of the cyclin D1 promoter sequence (from −1745 to 0) flanking the cyclin D1 gene. The mutant cyclin D1 (CDmt) reporter construct has a fragment of the cyclin D1 promoter sequence (from −66 to 0) with the NF-κB binding site truncated. The NF-κB reporter, provided by Dr. Keith Brown at the National Institute of Allergy and Infectious Diseases, contains three repeats of κB sites for the immunoglobulin κ-light chain (18). The β-galactosidase (β-gal) reporter was purchased from Stratagene and used as an internal control for NF-κB and cyclin D1 reporters. The Id1 signaling pathway and relationship among IκBαM, NF-κB (p65 and p50 heterodimer), PDTC, cyclin D1, and their effectors are schematically presented in Fig. 1.
Figure 1. Schematic representation of the Id1-induced cellular proliferation and keratin 10 production pathways in keratinocytes.

Id1 activates the translocation of NF-κB subunit p65 into the nucleus and increase the transcription of cyclin D1 (CD1). CD1, together with cyclin-dependent kinases (cdk 4/6), phosphorylate the retinoblastoma (Rb) which releases E2F from the Rb-E2F complex. E2F, a transcription factor that drives the Go to S phase transition of cells, promotes cell cycle progression. Ki67 is a proliferating cell antigen from G1 to M phase. With the expression of Ki67, a cell proliferates. In addition, the phosphorylation of NF-κB subunit p65 and transcription of p65 by Id1 increases the production of keratins. Id1-induced downregulatin of P16Ink4a, an inhibitor of cdks, also promotes cell cycle progression. IκBαM inhibits the NF-κB activity by forming a firm complex with p65 and p50 whereas PDTC inhibits the NF-κB activity by suppressing formation and degradation of IκB (an inhibitor of NF-κB). NF-κB, consisting of a heterodimer: p65 and p50; T-bar, indicating inhibition; arrow, symbolizing upregulation.
Immunohistochemistry
Cholesteatoma and control specimens were fixed in 10% formalin, embedded in paraffin and cut to a thickness of 5 μ. Sections were deparaffinized, incubated with primary antibodies to Id1 (rabbit anti-human Id1, Santa Cruz, CA), keratin 10 (rabbit anti-human K10, NeoMarker, CA, mouse anti-human pan-cytokeratin, Abcam), “activated” NF-κB (anti-NF-κB subunit p65, Chemicon), cyclin D1 (BD Sciences), and Ki67 (Abcam) for 90 minutes followed by secondary antibodies conjugated to fluorescein isothiocyanate (19) or tetramethylrhodamine isothiocyanate (TRITC), using protocols previously described (20). Tissue sections incubated with non-specific antibodies (mouse or rabbit IgG isotope controls from Zymed) or antibody absorbed with specific antigen (Id1 blocking peptide, Santa Cruz Biotechnology Inc, 5-fold excess, 4° C overnight or room temperature for 2 h) served as controls.
3H-thymidine incorporation and Trypan Blue exclusion
Cells were cultured in 24-well plates until 40% confluence, transfected with Id1 and empty vector, respectively, at a concentration of 1.4 μg/ml for 16 h in transfection medium, recovered in FGM for 24 h after transfection, incubated with 3H-Thymidine (1 μCi/well) for 5, 10, and 24 h, respectively, and harvested for measurement of radioactivity in a scintillation counter, as previously described (21). Simultaneously, cells in duplicated 24-well plates were harvested for cell counts by Trypan Blue exclusion. Radioactivity in a well was divided by its cell numbers in a duplicated well yielding radioactivity on average per cell. Results are presented as CPM/104 cells for 3H-thymidine incorporation. Cell growth rate (0 vs. 5, 10, and 24 h, respectively) was calculated using total cell numbers at 0 h against 5, 10, and 24 h, respectively.
Luciferase assays
Cells were cultured in 6-well plates until 60% confluence, transfected with empty vector, Id1, p65, and Id1+p65, co-transfected with NF-κB/β-galactosidase reporters and cyclin D1/β-galactosidase reporters, respectively, at 1.4 μg/mL for 7–16 h, recovered in FGM for 24 h, and harvested for luciferase assays, as previously described (22). The activities of target luciferase reporters vs. β-galactosidase are presented as relative luciferase activity (RLA).
Western blot
Cells were cultured in T-75 flasks until 60% confluence, transfected with empty vector, Id1, p65, IκBαM, Id1+p65, Id1+ IκBαM, and Id1+PDTC, respectively, in transfection medium for 16 h, recovered in FGM for 24 h, and harvested for cytosol and nuclear protein isolation. Forty μg of cytosol or nuclear protein were electrophoresed and blotted on a nitrocellulose membrane. Specific antigens (cyclin D1, Keratin 10, p16Ink4a, and GAPDH) on the membrane were detected by antibodies (anti-cyclin D1, BD Biosciences product; anti-K10; anti-p16Ink4a, BioMarkers product; anti-GAPDH, Novus Biological product), respectively using ECL kit (Amersham Biosciences) according to the manufacturer's instructions.
Statistical analysis
The Student's t-test for unequal variances was used for evaluation of two-group studies whereas analysis of variance (ANOVA) was used for evaluation of multiple-group studies. P values less than 0.05 were considered significant.
Results
Id1, NF-κB, cyclin D1, Ki67, and keratin 10, are highly expressed in the human middle ear cholesteatoma specimens
By immunohistochemistry, Id1, NF-kB, cyclin D1, Ki67, and keratin 10 were detected in the epithelial layer of cholesteatoma specimens (11 out of 14). Three out of 14 middle ear specimens contained onion peel-like substances but no epithelial layer. Normal middle ear mucosa showed negative staining for Id1 and activated NF-κB in the epithelial layer (Fig. 2a & b) whereas normal head and neck skin showed a baseline expression of Id1 and NF-kB in the basal cell layer (limited to few basal cells, data not shown). Unlike normal head and neck skin specimens, Id1 and NF-κB were extensively expressed in the cholesteatoma specimens. Id1 and NF-κB, NF-κB and cyclin D1, cyclin D1 and Ki67 antigens were co-expressed in the basal cells (Fig. 2d-h, nuclei) of cholesteatoma whereas NF-κB and keratin 10 were co-expressed in the suprabasal cell layer (Fig. 2i, nuclei and cytosol) of cholesteatomatous epithelium.
Figure 2. Id1, NF-κB, cyclin D1, and Ki67 are highly expressed in the cholesteatoma tissues.

Normal middle ear epithelium was negative for Id1 (a) and activated NF-κB (b) whereas cholesteatoma produced abundant pan-cytokeratins (pan-ck) in the basal cell (37), suprabasal (SB), and stratum cornium (SC) layers (c, between green arrowheads). Id1 (d) and activated NF-κB (e) were co-expressed in the basal cell (37) layer (f) of cholesteatoma tissues. Similarly, activated NF-κB and cyclin D1 (g) as well as cyclin D1 and Ki67 (h) were co-expressed in the basal layer; while keratin 10 and NF-κB (i) were co-expressed in the suprabasal layer of cholesteatoma epithelium. Bar=10 μ, applying to the same row; Ctrl, control middle ear tissue (a and b); and cholesteatoma tissue (c–i).
Id1 induces the proliferation of keratinocytes via NF-κB in vitro
To study the role of Id1 in cholesteatomatous epithelium, Id1 was transfected into Rhek-1A cells and cell proliferation was studied by using 3H-tymidine incorporation and cell growth rate methods. Id1 significantly increased DNA synthesis (Fig. 3A) and cell growth rate (Fig. 3B), associated with an increase of NF-κB luciferase activity (Fig. 3C) and translocation of NF-κB from the cytosol to the nuclei (Fig. 3D) compared with controls.
Figure 3. Id1 induces the proliferation of keratinocytes via NF-κB in vitro.
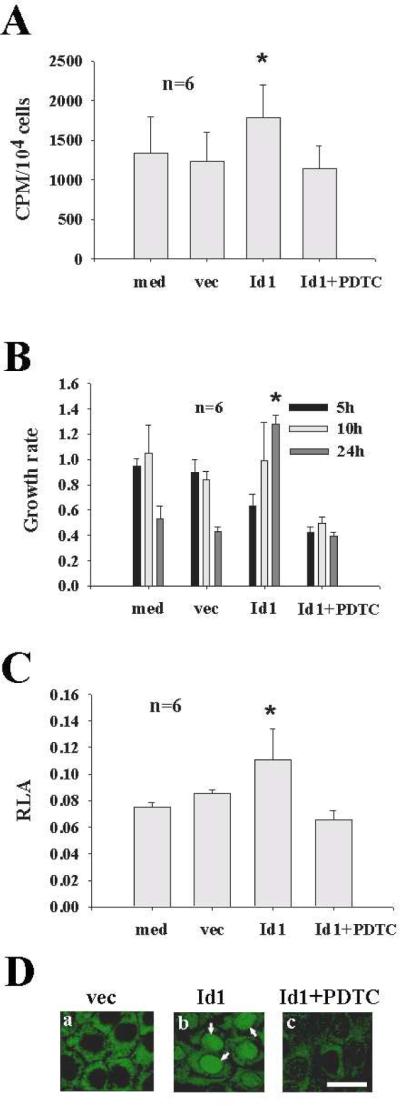
A: Id1 significantly increased DNA synthesis (CPM, counts per minute) compared with vec whereas PDTC cancelled out the effect of Id on DNA synthesis of keratinocytes. B: Id1 increased the cell growth rate in a time-dependent manner. At 24 hour, Id1 significantly increased the growth rate compared with vec. PDTC significantly inhibited the effect of Id1 on the growth rate. Note that empty vector-transfected cells had a high growth rate at 5 h but declined thereafter whereas Id1-transfected cells had a low growth rate at 5 h but increased thereafter. C: Id1 significantly increased the promoter activity of NF-κB (RLA, relative luciferase activity) compared with vec and PDTC cancelled out the effect of Id1 on the promoter activity of NF-κB. D: Id1 increased the NF-κB translocation into the nuclei of Rhek-1A keratinocytes after 1 hour transfection of Id1 compared with vec (arrows in panel b pointing to the nuclei of Rhek-1A keratinocytes which are positive for activated NF-κB after stain with anti-activated NF-κB). Bar=10 μ, applying to the same row in D. med, medium alone as blank control; vec, empty vector as transfection control; *p<0.05
Id1 upregulates cyclin D1 via NF-κB in keratinocytes
The immunohistochemistry data demonstrated the co-expression of NF-κB and cyclin D1 in the basal cells of cholesteatoma tissues. To study the relationship between NF-κB and cyclin D1, Rhek-1A cells were transfected with Id1, p65, and Id1+p65, respectively, co-transfected with wildtype or mutant cyclin D1/β-galactosidase reporters, and harvested for luciferase assays and Western blot. It was demonstrated that Id1 significantly increased the luciferase activity of wildtype cyclin D1 reporter (Fig. 4A, Id1 vs. vec). Mutation of the NF-κB binding site at the cyclin D1 promoter fully abrogated the Id1-induced cyclin D1 promoter activity (Fig. 4B, Id1 vs. Id1+CDmt). This suggests that Id1 drives the transcription of cyclin D1 via NF-κB. In addition, p65 significantly increased the Id1-induced promoter activity of cyclin D1 (Id+p65 vs. Id1), adding support to the notion that Id1 regulates cyclin D1 via NF-κB.
Figure 4. Id1 upregulates cyclin D1 via NF-κB in keratinocytes in vitro.

A: Id1 significantly increased the luciferase activity of cyclin D1 in Rhek-1A cells (Id1 vs. vec); p65 further increased Id1-induced action (Id1+p65 vs. Id1); p65 itself significantly increased the luciferase activity of cyclin D1 (p65 vs. vec); and Id1+p65 significantly increased the luciferase activity of cyclin D1 compared with Id1 or p65 alone (Id1+p65 vs. Id1 or p65). B: Mutation of the NF-κB binding site at the promoter of cyclin D1 (CDmt) fully abrogated the Id1-indued cyclin D1 promoter activity (Id1 vs. Id1+CDmt); and CDmt itself did not increase or decrease the luciferase activity (CDmt vs. vec). *p<0.05 when compared with vec in A or Id1 in B.
Id1 inhibits the expression of p16Ink4a but induces the expression of keratin 10 via an NF-κB dependent mechanism in keratinocytes
p16Ink4a is an inhibitor of cyclin-dependent kinases, preventing cells entering into cell cycles (23). To study whether Id1 inhibits the expression of p16Ink4a in keratinocytes, Id1 and empty vector were transfected into Rhek-1A cells and the expression of p16Ink4a and keratin 10 was evaluated by Western blot. The data demonstrated that Id1 inhibited the expression of p16Ink4a (Fig. 5A, middle row, Id1 vs. vec) but increased the expression of keratin 10 (Fig. 5, top row, Id1 vs. vec). The same applied to p65 and Id1+p65. IκBαM, a dominant-negative inhibitor of NF-κB, inhibited the expression of keratin 10 but rescued the expression of p16Ink4a. However, Id1 antagonized the action of IκBαM on the expression of keratin 10 and the action of IκBαM on the expression of p16Ink4a. To study whether Id1-induced keratin 10 expression is also dependent upon NF-κB, Id1+p65 as well as Id1+ IκBαM were co-transfected into cells. The results demonstrated that Id1+p65 strengthened the expression of keratin 10 compared with p65 and Id1 alone whereas IκBαM weakened the expression of keratin 10 induced by Id1 compared with Id1.
Figure 5. Id1 positively regulates the expression of keratin 10 via NF-κB but negatively regulates the expression of p16Ink4a in keratinocytes in vitro.

Western blot demonstrated that Id1 increased the expression of keratin 10 (lane 1, top) compared with its control (lane 2, top). p65 further increased the Id1-induced expression of keratin 10 (lane 3 vs. lane 1, top) but IκBαM abrogated the Id1-induced expression of keratin 10 (lane 5 vs. lane 1, top). p65 itself also increased the expression of keratin 10 (lane 4) compared with empty vector (lane 2, top). Id1, p65, and Id1+p65 inhibited the expression of p16Ink4a (lanes 1, 3, and 4, middle) compared with empty vector (lane 2, middle) in Rhek-1A cells. In contrary, IκBαM increased the expression of p16Ink4a (lane 6, middle) but Id1 overcame the action of IκBαM on the expression of p16Ink4a (lane 5, middle). med, blank control (without any transfection). GAPDH, glyceraldehyde 3-phosphate dehydrogenase (loading control).
Discussion
Id1 was originally recognized as a negative regulator of helix-loop-helix DNA binding proteins (5). Later on, it was shown to be involved in neurogenesis and angiogenesis of tumor xenogrants (24). We demonstrated for the first time that Id1 was involved in mucosal infectious diseases (1) and tied to proliferative middle ear diseases in this study.
As expected, Id1 is linked to the aggressive growth of acquired cholesteatoma through the Id1→NF-κB→cyclin D1→Ki67 signaling pathway (Fig. 1). First, Id1 activates the activity of NF-κB. NF-κB, in turn, activates the transcription of cyclin D1. Cyclin D1 then increases the progression of cell cycles from Go to S phase. In addition, Id1 inhibits the expression of p16Ink4a, liberating cells from cell cycle inhibition. The fact of Id1 acting through the NF-κB/cyclin D1/Ki67 signaling pathway provides an explanation as to why middle ear infection is linked to cholesteatoma. It is not clear at the moment whether physical damage and trauma to the tympanic membrane trigger the expression of Id family.
Numerous mechanisms are proposed to underlie acquired cholesteatoma. All of them invoke inflammation of keratinocytes in some way. We demonstrated in this study that activation of NF-κB is actively linked to cellular proliferation in keratinocytes which frequently originate from the external auditory canal skin. The molecular mechanism involves upregulation of cyclin D1 and downregulation p16Ink4a in keratinocytes. The former is a cell cycle progression protein and the latter is cell cycle progression inhibitor. Through this mechanism, Id1 opens a window for keratinocytes to actively grow and proliferate. Our in vitro studies demonstrate that PDTC, an inhibitor of NF-κB, blocks the Id1-induced proliferation of keratinocytes, indicating that Id1-induced cell proliferation is dependent upon NF-κB.
Activity of the cyclin D/retinoblastoma (Rb) pathway leads to proliferation of cells (25). Cyclin D1, together with cyclin-dependent kinases 4/6 (cdk 4/6), overcomes the function of Rb protein that promotes cell cycle progression. It is generally accepted that normal Rb function must be removed, one way or another, for a cell to divide. Down-regulation of p16Ink4a is one of the mechanisms for removing Rb function. Cyclin D1 is a well-established positive regulator of early cell cycle progression from G0/G1-to-S phase transition (26) through phosphorylation of Rb and dissociation of E2F (a check point protein for S phase entry). The up-regulation of cyclin D1 and removal of p16Ink4a inhibition in Rhek-1A cells by Id1 potentiates the growth and proliferation of keratinocytes and in part explains the behavior of keratinocytes in cholesteatoma— aggressive growth (27–29) which destroys the ossicular chain and temporal bone and causes serious complications such as deafness and intracranial lesions. The Id1-induced cyclin D1 upregulation and p16Ink4a downregualtion may represent a disease mechanism for cholesteatoma epithelial growth under chronic inflammatory conditions.
Activation of NF-κB not only increases the proliferation of keratinocytes but also upregulates the production of keratins. Keratin 10 is a product of mature keratinocytes (30–32) and highly expressed in the external auditory canal skin. The abundant expression of keratin 10 suggests that keratinocytes in the middle ear cholesteatoma are originated from the external auditory canal epidermis possibly via a migration process under chronic inflammatory conditions (33). Biologically, cell migration is coupled with cellular proliferation. Our data indicate that Id1 regulates NF-κB and NF-κB, in turn, upregulates the expression of keratin 10, which represents a pathological mechanism for accumulation of the onion peel-like substances in cholesteatoma. p65 strengthens this process whereas IκBαM attenuates this process.
It is noted that NF-κB at the suprabasal layer and above may not be related to cell growth and proliferation because cells at the suprabasal layer and above do not grow and proliferate but commit to keratin production. Referenced studies point out that NF-κB at the suprabasal layer and above may be related to survival and protection of cells from apoptosis (34–36). Why Id1-induced NF-κB in the basal layer leads to cell proliferation while NF-κB alone beyond the basal layer results in cell survival and resistance to apoptosis is an interesting question. It is also not clear in this study how Id1 inhibits the expression of p16Ink4a. It is warranted to address these questions in the future due to their importance in the pathogenesis of cholesteatoma and the physiology of the epidermis.
Acknowledgements
This study is in part supported by the NIH grants (R01008165 and R03CA107989) and the Minnesota Medical Foundation. We would like to express our thanks to Jack D. Wang, Eileen P. Schlentz, and Beverly Wuertz for their editorial assistance in the preparation of this manuscript.
References
- 1.Lin J, Tsuboi Y, Pan W, Giebink SG, Adams GL, Kim Y. Analysis by cDNA microarrays of altered gene expression in middle ears of rats following penumococcal infection. Int J Pedatr Otorhinolaryngol. 2002;65:203–11. doi: 10.1016/s0165-5876(02)00130-1. [DOI] [PubMed] [Google Scholar]
- 2.Zhang QA, Hamajima Y, Zhang Q, Lin J. Identification of Id1 in acquired middle ear cholesteatoma. Arch Otolaryngol. 2008;134:306–310. doi: 10.1001/archotol.134.3.306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Norton JD, Atherton GT. Coupling of cell growth control and apoptosis functions of Id proteins. Mol Cell Biol. 1998;18:2371–81. doi: 10.1128/mcb.18.4.2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Norton JD, Deed RW, Craggs G, Sablitzky F. Id helix-loop-helix proteins in cell growth and differentiation. Trends Cell Biol. 1998;8:58–65. [PubMed] [Google Scholar]
- 5.Benezra R, Davis RL, Lockshon D, Turner DL, Weintraub H. The protein Id: a negative regulator of helix-loop-helix DNA binding proteins. Cell. 1990;61:49–59. doi: 10.1016/0092-8674(90)90214-y. [DOI] [PubMed] [Google Scholar]
- 6.Norton JD. ID helix-loop-helix proteins in cell growth, differentiation and tumorigenesis. J Cell Sci. 2000;113:3897–905. doi: 10.1242/jcs.113.22.3897. [DOI] [PubMed] [Google Scholar]
- 7.Alani RM, Hasskarl J, Grace M, Hernandez MC, Israel MA, Munger K. Immortalization of primary human keratinocytes by the helix-loop-helix protein, Id-1. Proc Natl Acad Sci U S A. 1999;96:9637–41. doi: 10.1073/pnas.96.17.9637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hamajima Y, Toyama K, Zhao Z, Kim Y, Ondrey FG, Lin J. The Eighth International Symposium on Recent Advances in Otitis Media. BC Decker; Fort Lauderdale, Florida, USA: 2003. Id1 induces the proliferation of middle ear epithelial cells in rats. [Google Scholar]
- 9.Diehn M, Cho RW, Lobo NA, Kalisky T, Dorie MJ, Kulp AN, Qian D, Lam JS, Ailles LE, Wong M, Joshua B, Kaplan MJ, Wapnir I, Dirbas FM, Somlo G, Garberoglio C, Paz B, Shen J, Lau SK, Quake SR, Brown JM, Weissman IL, Clarke MF. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. 2009;458:780–3. doi: 10.1038/nature07733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gamou S, Shimizu N. Change in metabolic turnover is an alternate mechanism increasing cell surface epidermal growth factor receptor levels in tumor cells. J Biol Chem. 1987;262:6708–13. [PubMed] [Google Scholar]
- 11.Ozeki M, Hamajima Y, Feng L, Ondrey FG, Zheng M, Schlentz EP, Lin J. Id1 induces the proliferation of cochlear sensorineural epithelial cells via the NF-kB/cyclin D1 pathway in vitro. J Neurosci Res. 2007;85:515–524. doi: 10.1002/jnr.21133. [DOI] [PubMed] [Google Scholar]
- 12.Lin J, Guan Z, Wang C, Feng L, Zheng Y, Granados E, Bearth E, Peng J, Gaffney P, Ondrey F. Id1 regulates the survival of HNSCC via the NF-kB/survivin and PI3K/Akt signaling pathways. 2009 accepted. [Google Scholar]
- 13.Van Antwerp DJ, Martin SJ, Kafri T, Green DR, Verma IM. Suppression of TNF-alpha-induced apoptosis by NF-kappaB. Science. 1996;274:787–9. doi: 10.1126/science.274.5288.787. [DOI] [PubMed] [Google Scholar]
- 14.Gilmore TD, Koedood M, Piffat KA, White DW. Rel/NF-kappaB/IkappaB proteins and cancer. Oncogene. 1996;13:1367–78. [PubMed] [Google Scholar]
- 15.Schreck R, Meier B, Mannel DN, Droge W, Baeuerle PA. Dithiocarbamates as potent inhibitors of nuclear factor kappa B activation in intact cells. J Exp Med. 1992;175:1181–94. doi: 10.1084/jem.175.5.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chun KS, Cha HH, Shin JW, Na HK, Park KK, Chung WY, Surh YJ. Nitric oxide induces expression of cyclooxygenase-2 in mouse skin through activation of NF-kappaB. Carcinogenesis. 2004;25:445–54. doi: 10.1093/carcin/bgh021. [DOI] [PubMed] [Google Scholar]
- 17.Albanese C, Johnson J, Watanabe G, Eklund N, Vu D, Arnold A, Pestell RG. Transforming p21ras mutants and c-Ets-2 activate the cyclin D1 promoter through distinguishable regions. J Biol Chem. 1995;270:23589–97. doi: 10.1074/jbc.270.40.23589. [DOI] [PubMed] [Google Scholar]
- 18.Fujita T, Nolan GP, Liou HC, Scott ML, Baltimore D. The candidate proto-oncogene bcl-3 encodes a transcriptional coactivator that activates NF-kappa B p50 homodimers. Genes Dev. 1993;7:1354–63. doi: 10.1101/gad.7.7b.1354. [DOI] [PubMed] [Google Scholar]
- 19.Atchley WR, Fitch WM. A natural classification of the basic helix-loop-helix class of transcription factors. Proc Natl Acad Sci U S A. 1997;94:5172–6. doi: 10.1073/pnas.94.10.5172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lin J, Tsprun V, Kawano H, Paparella MM, Zhang Z, Andway R, Ho SB. Characterization of mucins in human middle ear and eustachian tube. Am J Physiol Lung Cell Mol Physiol. 2001;280:L1157–67. doi: 10.1152/ajplung.2001.280.6.L1157. [DOI] [PubMed] [Google Scholar]
- 21.Toyama K, Kim Y, Paparella MM, Lin J. Temperature-sensitive SV40 immortalized rat middle ear epithelial cells. Ann Otol Rhino Laryngol. 2004;113:967–74. doi: 10.1177/000348940411301206. [DOI] [PubMed] [Google Scholar]
- 22.Tsuchiya K, Kim Y, Ondrey FG, Lin J. Characterization of a temperature-sensitive mouse middle ear epithelial cell line. Acta Otolaryngol. 2005;125:823–9. doi: 10.1080/00016480510031533. [DOI] [PubMed] [Google Scholar]
- 23.Shapiro GI, Rollins BJ. p16INK4A as a human tumor suppressor. Biochim Biophys Acta. 1996;1242:165–9. doi: 10.1016/0304-419x(95)00011-4. [DOI] [PubMed] [Google Scholar]
- 24.Lyden D, Young AZ, Zagzag D, Yan W, Gerald W, O'Reilly R, Bader BL, Hynes RO, Zhuang Y, Manova K, Benezra R. Id1 and Id3 are required for neurogenesis, angiogenesis and vascularization of tumour xenografts. Nature. 1999;401:670–7. doi: 10.1038/44334. [DOI] [PubMed] [Google Scholar]
- 25.Weinberg RA. The retinoblastoma protein and cell cycle control. Cell. 1995;81:323–30. doi: 10.1016/0092-8674(95)90385-2. [DOI] [PubMed] [Google Scholar]
- 26.Sherr CJ. Mammalian G1 cyclins. Cell. 1993;73:1059–65. doi: 10.1016/0092-8674(93)90636-5. [DOI] [PubMed] [Google Scholar]
- 27.Michaels L. Biology of cholesteatoma. Otolaryngol Clin North Am. 1989;22:869–81. [PubMed] [Google Scholar]
- 28.Broekaert D, Coucke P, Leperque S, Ramaekers F, Van Muijen G, Boedts D, Leigh I, Lane B. Immunohistochemical analysis of the cytokeratin expression in middle ear cholesteatoma and related epithelial tissues. Ann Otol Rhinol Laryngol. 1992;101:931–8. doi: 10.1177/000348949210101109. [DOI] [PubMed] [Google Scholar]
- 29.Ergun S, Zheng X, Carlsoo B. Antigen expression of epithelial markers, collagen IV and Ki67 in middle ear cholesteatoma. An immunohistochemical Study. Acta Otolaryngol. 1994;114:295–302. doi: 10.3109/00016489409126059. [DOI] [PubMed] [Google Scholar]
- 30.Roop DR, Krieg TM, Mehrel T, Cheng CK, Yuspa SH. Transcriptional control of high molecular weight keratin gene expression in multistage mouse skin carcinogenesis. Cancer Res. 1988;48:3245–52. [PubMed] [Google Scholar]
- 31.Stoler A, Kopan R, Duvic M, Fuchs E. Use of monospecific antisera and cRNA probes to localize the major changes in keratin expression during normal and abnormal epidermal differentiation. J Cell Biol. 1988;107:427–46. doi: 10.1083/jcb.107.2.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fuchs E. Epidermal differentiation: the bare essentials. J Cell Biol. 1990;111:2807–14. doi: 10.1083/jcb.111.6.2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sade J. Epithelial invasion of intraossicular spaces. J Laryngol Otol. 1972;86:15–21. doi: 10.1017/s0022215100074880. [DOI] [PubMed] [Google Scholar]
- 34.Seitz CS, Freiberg RA, Hinata K, Khavari PA. NF-kappaB determines localization and features of cell death in epidermis. J Clin Invest. 2000;105:253–60. doi: 10.1172/JCI7630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kaufman CK, Fuchs E. It's got you covered. NF-kappaB in the epidermis. J Cell Biol. 2000;149:999–1004. doi: 10.1083/jcb.149.5.999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Qin JZ, Chaturvedi V, Denning MF, Choubey D, Diaz MO, Nickoloff BJ. Role of NF-kappaB in the apoptotic-resistant phenotype of keratinocytes. J Biol Chem. 1999;274:37957–64. doi: 10.1074/jbc.274.53.37957. [DOI] [PubMed] [Google Scholar]
- 37.Schnaper HW, Hayashida T, Hubchak SC, Poncelet AC. TGF-beta signal transduction and mesangial cell fibrogenesis. Am J Physiol Renal Physiol. 2003;284:F243–52. doi: 10.1152/ajprenal.00300.2002. [DOI] [PubMed] [Google Scholar]