

Abstract
Intracellular oncoproteins can be inhibited with targeted therapy, but responses are not durable. Immune therapies can be curative, but most oncogene-driven tumors are unresponsive to these agents. Fragments of intracellular oncoproteins can act as neoantigens presented by the major histocompatibility complex (MHC) but recognizing minimal differences between oncoproteins and their normal counterparts is challenging. We have established a platform technology that exploits hapten-peptide conjugates generated by covalent inhibitors to create distinct neoantigens that selectively mark cancer cells. Using the FDA-approved covalent inhibitors sotorasib and osimertinib, we developed “HapImmune™” antibodies that bind to drug-peptide conjugate/MHC complexes but not to the free drugs. A HapImmune™-based bispecific T cell engager selectively and potently kills sotorasib-resistant lung cancer cells upon sotorasib treatment. Notably, it is effective against KRASG12C mutant cells with different HLA supertypes, HLA-A*02 and A*03/11, suggesting loosening of MHC restriction. Our strategy creates targetable neoantigens by design, unifying targeted and immune therapies.
Introduction
The past twenty years have witnessed a revolution in cancer therapeutics along two major fronts. First, “targeted therapies” (e.g., small molecule signal transduction inhibitors, antibodies against receptor tyrosine kinases) have been developed against specific mutant oncogenes or components of their downstream signal transduction cascades (1,2). Even KRAS, long viewed as “undruggable”, has now been targeted in tumors bearing the specific mutant allele KRASG12C (3–5). Targeted therapies can cause remarkable regressions, but unfortunately, some mutant cells are able to resist the initial drug onslaught via “adaptive resistance” (6–10) or as drug-tolerant persisters (11–16). Such cells can serve as reservoirs for the eventual development of stable resistance, which leads to disease recurrence and ultimately, patient demise. In parallel, “immune therapies” emerged (e.g., immune checkpoint blockade, adoptive T-cell transfer, CAR-T cells, CAR-NK cells). These modalities, unlike targeted therapies, can sometimes induce durable remissions (and likely cures), but most patients, including those with oncogene-driven tumors, fail to respond (17–25). Therefore, achieving durable responses and ultimately cures for metastatic cancers driven by intracellular oncogenes remains a major unmet medical need.
Conceivably, targeted therapies fail because they fail to evoke a sustained anti-tumor immune response. Thus, a key question is how we can effectively combine the benefits of targeted therapies as debulking agents with the durability of immune therapies. In principle, aberrant intracellular oncoproteins could be recognized by the immune system. Specifically, mutant peptides derived from oncoproteins and presented on class I major histocompatibility complex (class I MHC, hereafter, MHC) molecules might be recognized by cytotoxic T cells with cognate T cell receptors (TCRs). That tumors are present, presumably due to immune escape, indicates that such T cells must be few in number, exhausted, senescent, or otherwise dysfunctional. Targeting mutant peptide/MHC complexes (hereafter pmutant/MHC; e.g., KRAS mutants) with TCRs or antibodies is conceptually feasible and has been demonstrated in some cases (26,27). Recognizing the typically minimal differences between the mutant and wild-type peptides in the context of the p/MHC complex makes this approach quite challenging (28).
To address these challenges, we developed a technology platform, “HapImmune™”, that capitalizes on covalent targeted therapies to create drug-peptide conjugates as cancer neoantigens (Fig. 1A). The bulky chemical moiety of the conjugated inhibitor substantially alters the surface topography and chemistry with respect to unconjugated peptides. Thus, inhibitor-p/MHC should be a distinctly different and unique antigen, which could be more readily recognized by antibodies (or TCRs), leading to high selectivity. We utilized antibody engineering technologies to develop human antibodies that recognize such neoantigens on MHC and are minimally inhibited by the free inhibitor or inhibitor-p in the absence of MHC, a prerequisite for co-administration with the inhibitor. Such antibodies could kill tumor cells by engaging immune cells (e.g., T lymphocytes, NK cells, tumoricidal macrophages) or delivering toxic cargos (24,29,30). Importantly, the small molecule drug need not act as an inhibitor of cancer cell growth or even as an inhibitor of the target protein, so long as it forms a stable covalent bond with the target protein and the inhibitor-p/MHC is presented on the surface of cancer cells. We present proof-of-concept data by developing highly specific human antibodies that specifically recognize complexes of inhibitor-peptide conjugates and their matched MHCs generated by two FDA-approved covalent drugs, sotorasib, which targets KRAS(G12C), and osimertinib, which targets activated EGFR. We also present initial data showing similar reagents can be generated for a third FDA-approved agent, ibrutinib, conjugated to a fragment of its target BTK. Our concept enables the development and targeting of any drug-peptide conjugate capable of presentation on MHC and could substantially enhance the effectiveness of both targeted therapy and biologics against cancer.
Figure 1.
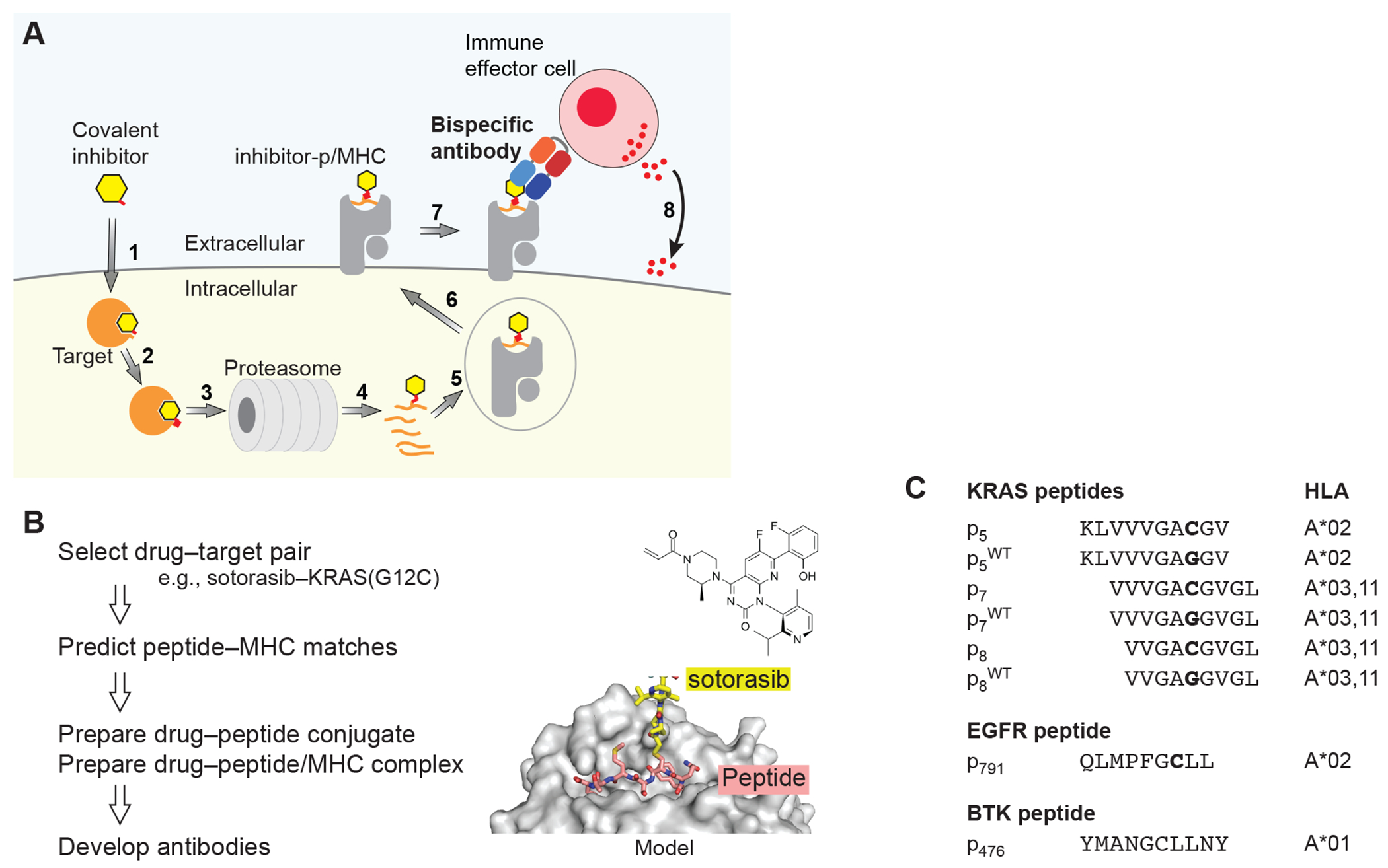
The HapImmune™ concept. (A) A covalent inhibitor enters the cell (step 1) and binds and forms a covalent bond with its target (2). As a part of natural protein turnover, the target-drug conjugate is degraded, and peptides with the conjugated drug are produced (3, 4). A drug-peptide conjugate is incorporated into a compatible MHC molecule (5). The drug-peptide/MHC complex translocates to the cell surface (6). A HapImmune™ antibody binds the complex (7) and recruits an immune effector cell, which initiates cell killing (8). Alternatively, the HapImmune™ antibody can serve as the recognition element for antibody conjugates or cellular therapies. (B) Overview of antibody development strategy. The molecular model was based on PDB ID 3RL1 (67). (C) Peptides used in this study and their predicted HLA matches.
Results
Antigen design for inhibitor-peptide conjugates originating from sotorasib and KRAS(G12C)
We chose KRAS(G12C) as an initial target for testing the HapImmune™ concept. RAS mutations at codon 12 are among the most common oncogenic drivers, and these and other RAS mutant proteins had long been viewed as challenging, if not “undruggable” targets. Recent breakthroughs led to the development of multiple covalent inhibitors for KRAS(G12C), hereafter termed G12Cis. Sotorasib (AMG510) is the first FDA-approved G12Ci, and it evokes therapeutic responses and extends progression-free survival in a significant fraction of non-small cell lung cancer patients whose tumors express the target oncoprotein (31). Unfortunately, as is the case for other targeted therapies, resistance to G12Ci develops quickly, and cures remain elusive (32–34).
We previously developed biologics (synthetic antibodies and monobodies), that directly target KRAS(G12C) and its covalent complex with ARS1620 (35,36). Although these reagents are effective tools for mechanistic studies, their inability to enter cells made them ineffective as potential therapeutics. Nevertheless, the relatively high abundance (~1 μM) of KRAS(G12C) in cells (37), the effective target engagement by G12Ci, and the emerging mechanisms of sotorasib resistance all suggested that the sotorasib-KRAS(G12C) peptide conjugates might be amenable to the HapImmune™ approach.
Although no data explicitly demonstrate that sotorasib-peptide conjugates are presented on MHCs, much evidence suggested that this was likely. First, MHC-presentation of RAS peptides that include residues 12 has been reported (27,38). Cys12 in these p/MHC complexes should be located outside the anchor positions of the presented peptides that are crucial for MHC binding, suggesting that drug conjugation would minimally affect peptide presentation (Fig. 1B). Second, NetMHCpan (39) predicts that peptides containing Trp at the 12th position, mimicking the bulky side chain of sotorasib-conjugated Cys12, can be presented on HLA-A*03, -A*11, and -A*02, with the highest score for the 9-residue peptide corresponding to residues 8-16 (hereafter termed p8 where the subscripted number indicates the position of the N-terminal residue of the peptide within the full-length, parental protein) on HLA-A*03 (Fig. 1B, 1C). For brevity, we will use abbreviations to refer to an inhibitor-peptide conjugate in complex with an MHC molecule: for example, soto-p8/A03 refers to the sotorasib-p8 conjugate in complex with HLA-A*03.
We conjugated sotorasib to these peptides and produced their MHC complexes using a standard refolding procedure (40). Size exclusion chromatography showed the formation of stable MHC complexes (Fig. S1). As controls, we also prepared the corresponding complexes harboring the cognate wild-type peptides. Hereafter, peptides with the wild-type sequence are denoted as pWT (see Fig. 1C for nomenclature of peptides used in this study).
Development of antibodies selective for sotorasib-KRAS(G12C) conjugates in complex with MHC
We set out to develop antibodies that selectively recognize inhibitor-peptide conjugates in the context of MHCs. Using the soto-p8/A03 complex as a target and the p8WT/A03 complex as an off-target control for negative selection, we performed selections on a human synthetic antibody phage-display library and identified a clone, R001, that preferentially bound to soto-p8/A03 (Fig. 2A, Supplementary Fig. S2A). To facilitate characterization and improvement of its properties, we transferred the phage-displayed Fab clone into a yeast display vector in the single-chain Fv (scFv) format. Consistent with its preliminary characterization as a phage-displayed Fab, R001 specifically bound to soto-p8/A03 with an apparent dissociation constant (KD,APP) of 2.7 nM and showed no binding to p8WT/A03 or p7WT/A03) (Supplementary Fig. S2A). This antibody was highly selective to soto-p8/A03. We detected significant but weak binding to soto-p7/A03, the p/MHC complex with a longer, 10mer peptide (KD,APP >100 nM) but no binding to soto-p7/A11 and soto-p8/A11 complexes (Supplementary Fig S2A). R001 also was highly selective to sotorasib, showing no cross-reactivity to two other G12Ci-p8/A03 complexes, ARS1620-p8/03 or MRTX849 (adagrasib)-p8/03 (Supplementary Fig. S2B).
Figure 2.
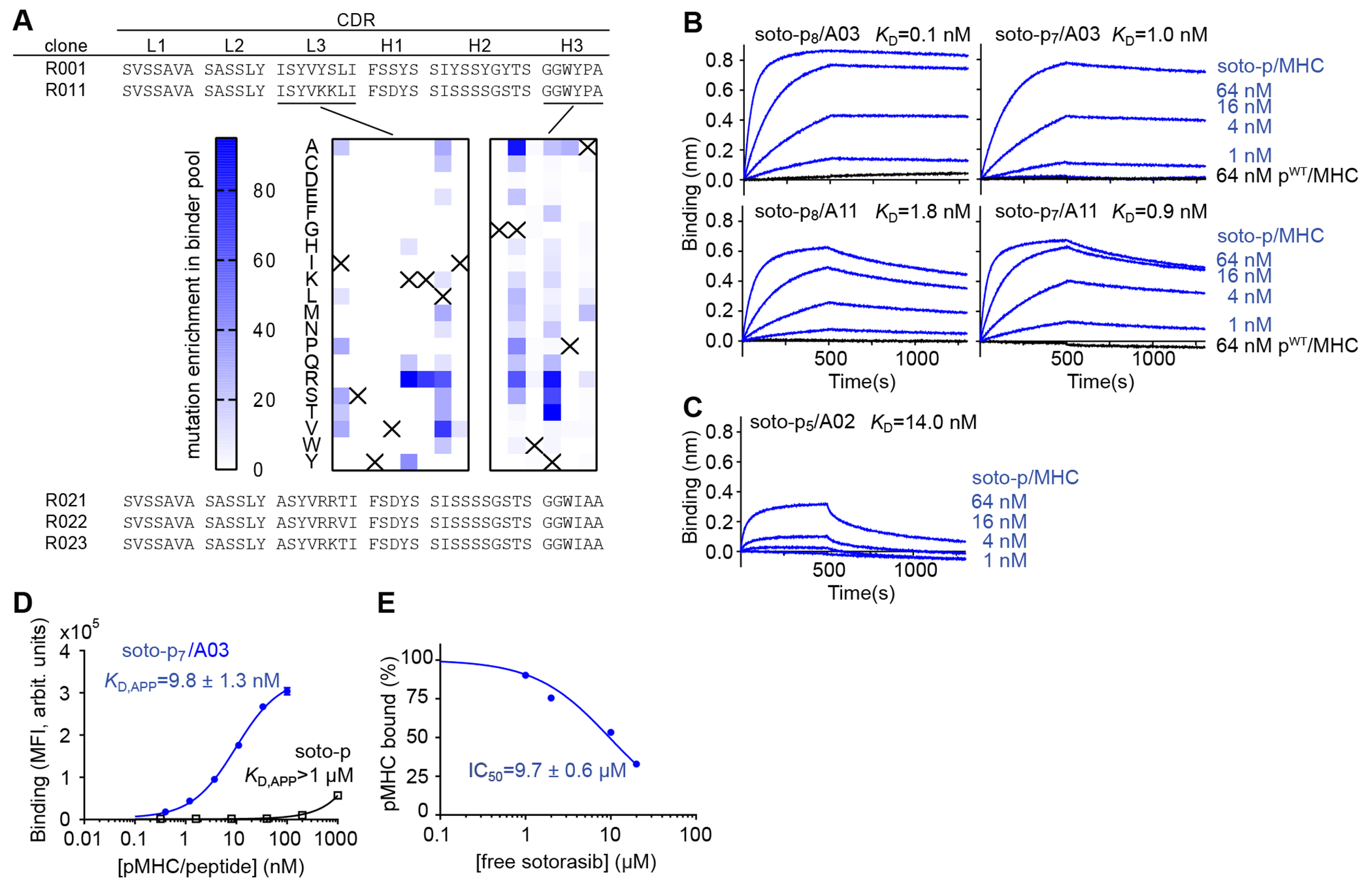
Development and binding properties of the R023 antibody. (A) CDR sequences of R023 and its precursors and related clones. The middle images show the results of deep mutational scanning of clone R011. The numbers indicate the total numbers of sequencing reads for each mutation, divided by the total number of reads for all mutations at the position, multiplied by 1000. The crosses show the wild-type residue. (B) BLI sensorgrams of the interaction between R023 Fab and the indicated MHC complexes. Biotinylated R023 Fab was immobilized, and binding of soluble p/MHC samples was measured. KD values from global fitting are shown. (C) BLI sensorgrams of the interaction between R023 Fab and the soto-p5/A02 complex. (D) Binding titration of scFv R023 displayed on the yeast cell surface to soto-p7/03 (blue) and the soto-p7 conjugate in the absence of an MHC (open squares). (E) Inhibition by free sotorasib of the interaction between soto-p7/A03 (10 nM) and scFv R023 displayed on the yeast cell surface. The binding signal intensity was normalized using the value without sotorasib (100%) and that in the absence of soto-p7/A03 (0%). IC50 values are reported ± standard error. In B and C, each data point shows the mean (n = 3; technical replicates) of the median fluorescence intensity. Error bars represent the standard deviation.
To improve upon the affinity of R001 and to explore whether it is possible to expand its recognition spectrum to the related sotorasib-conjugated peptides on HLA-A*11 while maintaining selectivity for soto-p/MHC complexes, we performed rounds of affinity maturation (Fig. 2A, Supplementary Fig. S3). Following mutagenesis and library sorting of CDR residues, we developed clone R011, which showed increased affinity towards soto-p7/03 and weak but detectable binding to the soto-p8 and soto-p7 conjugates presented by HLA-A*11 (Fig. 2A, Supplementary Fig. S2A). We then performed deep mutational scanning (41) of the CDR-L3 and H3 residues of clone R011 to identify permissible substitutions (Fig. 2A, Supplementary Fig. S3). This step allowed us to define the sequence landscape of antibodies towards different soto-p/MHC antigens. Based on these data, we designed a tailored library that combined permissible residues in CDR-L3 and -H3, and identified three clones, named R021, R022 and R023, that bound with low nanomolar affinity to all four targets, soto-p8/A03, soto-p7/A03, soto-p8/A11 and soto-p7/A11) (Supplementary Fig. S2A). We chose clone R023 for further characterization and produced it in the Fab format for biophysical characterization. BLI experiments using purified Fab confirmed its high affinity to all four soto-p/MHCs, with KD values ranging from 110 pM to 1.8 nM (Fig. 2B), and lack of detectable binding to pWT/MHCs (Fig. 2B, black traces). Intriguingly, BLI experiments also revealed that R023 bound, though with lower affinity, to sotorasib conjugated with a distinct peptide, p5, presented on a different HLA supertype, HLA-A*02 (Fig. 2C).
Tthese antibodies bound only minimally to the free sotorasib-conjugated peptide in the absence of MHC or to free sotorasib. Binding to the free conjugate was observed only at very high concentrations (free soto-p KD,APP > 1μM; Fig. 2D, Supplementary Fig. S2C). Furthermore, antibody binding to soto-p/MHCs was inhibited only marginally by free sotorasib (IC50 of 7–12 μM, Fig. 2E and Supplementary Fig. S2D). Remarkably, despite their ability to bind the sotorasib-conjugated peptides in a manner not restricted to a single HLA, their specificity toward the inhibitor-peptide conjugates in complex with MHCs, over free sotorasib, was maintained. Taken together, these data establish the feasibility of developing potent and selective antibodies to the complex of an inhibitor-peptide conjugate and its matched HLA that are minimally inhibited by the free inhibitor. These data also demonstrate the potential to expand the patient population who could be treated with this approach (see Discussion).
T-cell engaging bispecific antibodies selectively kill cells presenting drug-peptide conjugates as MHC complexes
Direct detection of specific p/MHC complexes on the cell surface using standard immunochemical methods such as flow cytometry is extremely challenging because of their low copy number (38). Likewise, low copy numbers are expected for sotorasib-KRAS(G12C) conjugates presented by MHCs on the cell surface. Therefore, to detect these neoantigen complexes and maximize the efficacy of target cell killing by our antibodies, we utilized a T cell-engaging bispecific antibody platform. Specifically, we constructed a single-chain diabody (scDb) (42) comprising a HapImmune™ antibody for recognizing the target cell and the UCHT1 clone as the component that engages CD3ε on T lymphocytes (43), and used cell killing as a sensitive readout of p/MHC on the cell surface.
We used Raji cells harboring HLA-A*03 (Fig. 3A, Supplementary Fig. S4A) and pulsed with the soto-p7 and -p8 conjugates to ask whether the R023 scDb could have cytotoxic effects on cells displaying soto-p/A03 complexes. Raji cells express the transporter associated with antigen processing (TAP), which is required for assembly of p/MHC complexes and their consequent transport to the cell surface (44). Consequently, MHC molecules on the Raji cell surface are already bound with endogenous peptides, and only a small fraction of HLA-A*03 on the surface of these cells can be loaded with exogenously added peptide-drug conjugates. When cocultured with T cells, the R023 scDb showed potent cytotoxic effects on cells pulsed with soto-p7 or soto-p8 (EC50 = 2.8 pM and 5.2 pM, respectively), but not with the p7WT or p8WT peptides, indicating selective killing (Fig. 3B). As predicted by our binding studies (Fig. 2B, Supplementary Fig. S2A), the cell killing efficacy of the R023 scDb was substantially higher than that of the original R001 clone in the scDb format, particularly for cells pulsed with soto-p7. Importantly, the R023 scDb showed no cytotoxic effect on sotorasib-treated, unpulsed Raji cells, which do not express KRAS(G12C) (Fig. 3C), indicating that the killing depends on the presence of KRAS(G12C) peptides. Notably, the R023 scDb also killed OCI-AML3 cells (expressing HLA-A*02) pulsed with soto-p5 but not cells pulsed with p5WT, although the efficacy was lower than Raji cells pulsed with soto-p7 or soto-p8 (Fig. 3D). This result is consistent with the weaker affinity of R023 for the soto-p5/A02 complex than for soto-p7/A03 (Fig. 2B, 2C). In concert, these data show that the R023 scDb can induce potent, highly selective killing of cells presenting sotorasib-KRAS(G12C) peptide conjugates bound to MHC complexes on the cell surface. They also provide further evidence that the range of actionable MHCs can be extended via the HapImmune™ approach.
Figure 3.
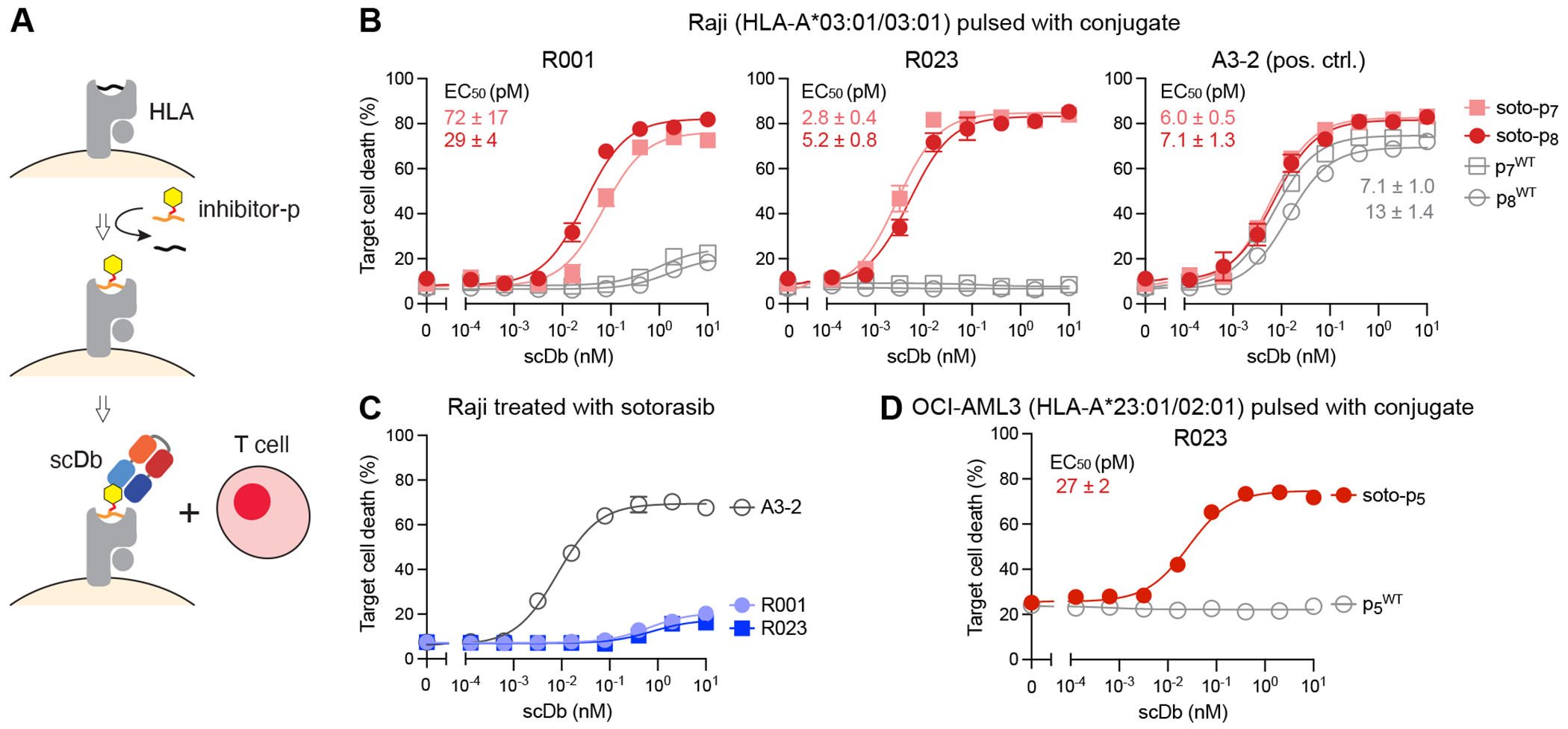
Cytotoxic effects of R023 scDb on cells pulsed with a sotorasib-KRAS(G12C) conjugate. (A) Schematic representation of the assay. Cells are pulsed with a conjugate or a negative control peptide, and then cocultured with T cells in the presence of scDb. (B) Cytotoxic effects of scDbs on Raji cells pulsed with soto-p7, soto-p8, p7WT or p8WT. (C) Cytotoxic effects of the R001 and R023 scDbs on sotorasib-treated Raji cells, which do not possess KRAS(G12C). (D) Cytotoxic effects of R023 on OCI-AML3 cells pulsed with soto-p5 and p5WT. Data are from triplicate measurements, and calculated EC50 values are shown. A3-2 is a positive-control antibody that binds to HLA-A3 irrespective of the bound peptide. Data shown are representative of ≥2 equivalent measurements.
Sotorasib-treated tumor cells can be killed selectively by HapImmune™ antibodies
We next asked whether the R023 scDb can target sotorasib-treated KRAS(G12C)-harboring tumor cells that are resistant to the inhibitor. The NCI-H358 cell line commonly used for studies of sotorasib and other G12Cis is highly sensitive to these agents and therefore is not suitable for evaluating our approach. Instead, we identified a sotorasib-insensitive cell line, NCI-H2122 (hereafter H2122) that expresses KRAS(G12C) and HLA-A*03 (Supplementary Fig. S4B). H2122 is resistant to sotorasib at up to ~10 μM in two-dimensional culture (Fig 4A), a therapeutically relevant concentration range, even though sotorasib at concentrations as low as 0.1 μM fully engages KRAS(G12C) in these cells (Fig. 4B). By contrast, nearly all of the H358 cells were killed with 0.1 μM sotorasib (Fig. 4A). Therefore, we chose to assess HapImmune™ scDb-induced killing in H2122 cells exposed to 0.1–1.0 μM sotorasib. The sotorasib concentration in plasma remains higher than 0.1 μM after a single administration of the standard 960 mg dose for nearly the entire 24 h dosing interval (5). To specifically measure target cell death in the presence of T cells (some of which also die), we generated a variant of H2122 cells, H2122-Nluc, with intracellular expression of NanoLuc. Luciferase released into the media by dying cells can then be quantified, providing an accurate assessment of cancer cell death (45). In this manner, we avoid artifacts caused by cell death during detachment, which would be required for flow cytometry-based analysis.
Figure 4.
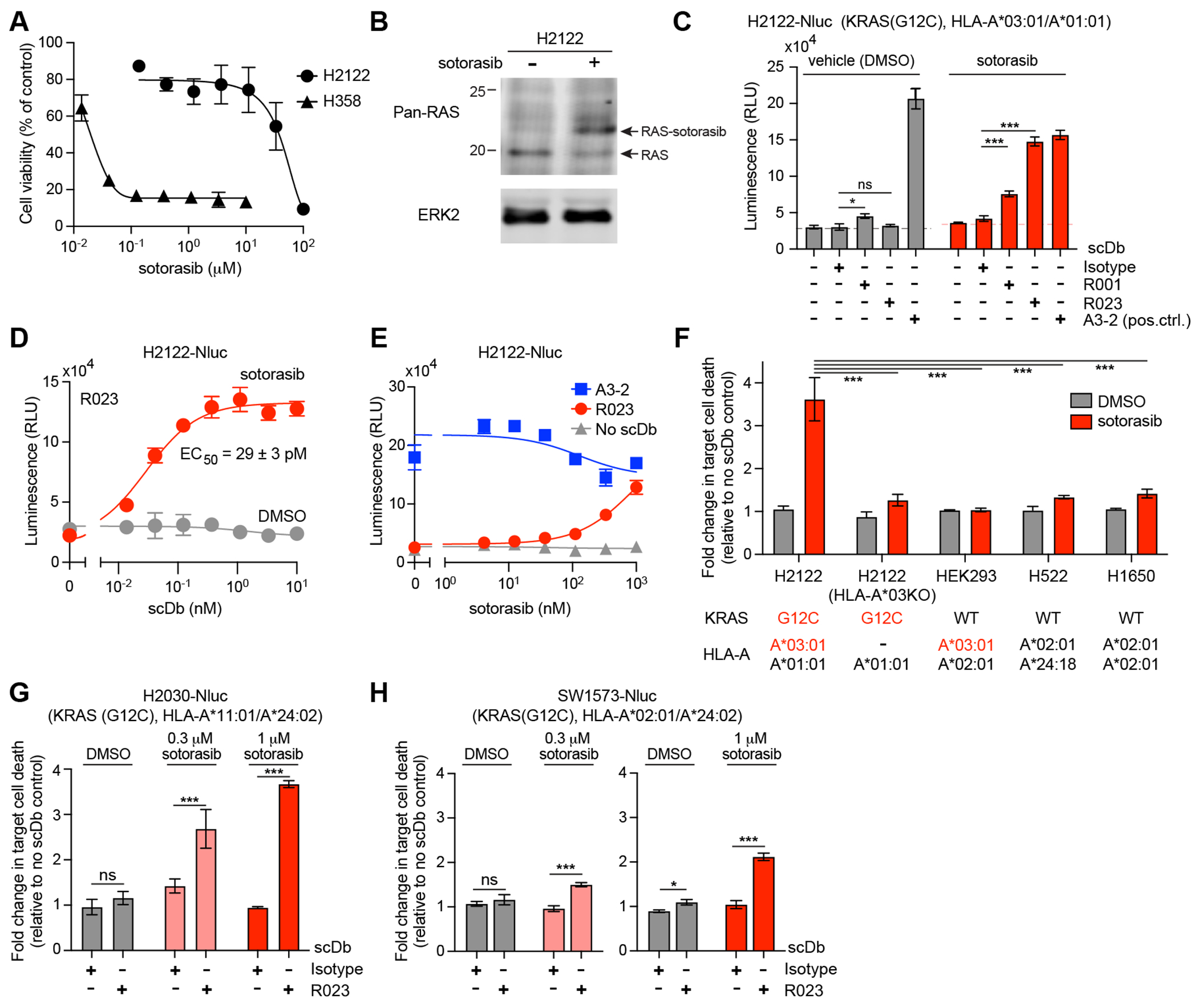
Cytotoxic effect of R023 scDb on sotorasib-treated tumor cells. (A) Dose response curves of the viability of H358 and H2122 cells following exposure to sotorasib for 72 hr. (B) Analysis of sotorasib conjugation to KRAS(G12C) in H2122 cells by Western blot. H2122 cells were incubated with 100 nM sotorasib for 24hr. The arrow indicates KRAS(G12C) conjugated to sotorasib. Note that the anti-pan-RAS antibody detects KRAS, HRAS, and NRAS, so complete shift of the original band is not expected. (C) Cytotoxic effects of the indicated scDbs on H2122-Nluc cells treated with 1 μM sotorasib. The scDb concentration was 10 nM except for the A3-2 scDb (1 nM). (D) Cell killing titration curve of the R023 scDb on H2122-Nluc cells treated with 1 μM sotorasib. (E) Dependence of cell killing on sotorasib concentration with the indicated scDbs at 1 nM. (F) HLA dependence of cell killing by R023 scDb. The normalized luminescence intensity (see Supplementary Fig. S5A for the procedure) is shown for cell lines treated with 0.3 μM sotorasib and cocultured with T cells in the presence of 1 nM scDbs and 0.3 μM sotorasib. KRAS mutation state and HLA alleles for the cell lines are shown. (G,H) Cytotoxic effects of the R023 scDb (1 nM) on H2030-Nluc (G) and SW1573-Nluc (H) cells treated with sotorasib. Data shown are from technical quadruplicate measurements, representative of ≥2 equivalent measurements. Data represent mean ± s.d., one-way ANOVA with Tukey’s multiple comparison test; *P < 0.05, **P < 0.01, ***P <0.001. See Supplementary Fig. S5 for raw data for panels F–H.
We cultured H2122-Nluc cells in the presence of sotorasib for a week to allow adequate time for the processes of sotorasib engagement with KRAS(G12C), degradation of the sotorasib-KRAS(G12C) conjugate, and loading of the conjugates on HLA to reach a steady state (Fig. 1A). We chose this incubation period in order to account for the slow turnover of KRAS (tt/2 ≥ 24h), which might limit the presentation of sotorasib-peptide conjugates by MHCs. Remarkably, coculture of sotorasib pre-treated H2122-Nluc cells with T cells in the presence of sotorasib and the R023 scDb resulted in efficient cell killing (Fig. 4C, Supplementary Fig. S5A). Although we predict that the copy number of soto-p/A03 on the cell surface is low, cell killing by the sotorasib/scDb combination was comparable to that evoked by a positive control scDb made with an antibody targeting all cell surface-expressed HLA-A*03, irrespective of its bound peptides, clone A3-2 (Fig. 4C, Supplementary Fig. S6). The EC50 of the R023 scDb on sotorasib-treated H2122-Nluc was 29 pM (Fig. 4D), whereas it showed no killing of vehicle-treated H2122. Cell killing was dependent on sotorasib concentration, as expected (Fig. 4E). These results support the notion that selective targeting of inhibitor-p/MHC complexes could lead to a new immunotherapeutic approach.
We performed a series of rigorous control experiments to validate the proposed mechanism of tumor cell killing. Deletion of the HLA-A3 allele in H2122-Nluc by means of CRISPR/Cas9 technology (Supplementary Fig. S4B) rendered these cells resistant to killing by the sotorasib/R023 scDb combination (Fig. 4F, Supplementary Fig. S5B). Likewise, the R023 scDb had no cytotoxic effects on cells harboring KRAS(WT) either with matched or mismatched HLAs (Fig. 4F, Supplementary Fig. S5B). Hence, killing of H2122 cells by the R023 scDb depends on the presence of the covalent targeted therapy drug, its target, and an appropriately matched HLA, providing strong support for the HapImmune™ concept (Fig. 1A).
Intriguingly, the R023 scDb also killed sotorasib-treated H2030-Nluc (expressing HLA-A11), and SW1573-Nluc (expressing HLA-A02), as anticipated from the binding profile of R023 Fab to purified soto-p/MHC complexes (Fig. 2B–C, 4G–H, Supplementary Fig. S5C–D). These results demonstrate the potential of the HapImmune™ approach to enable a single antibody to effectively target cancer cells with distinct HLA supertypes.
Development of HapImmune™ antibodies selective to other drug-target conjugates in complex with an MHC
To test the general applicability of our HapImmune™ approach, we also developed antibodies selective to the osimertinib-EGFR conjugate presented on an MHC. Osimertinib covalently binds to endogenous C797 of activated EGFRs, such as EGFR(T790M) (46). NetMHCpan predicted a fragment corresponding to residues 791–799 (hereafter p791) to be a strong ligand for HLA-A*02 (Fig. 1C). Using essentially the same methods, we first identified an antibody that specifically recognizes the osim-p791/A02 complex over the p791WT/A02 complex. By employing iterative approaches similar to those described above, including deep mutational scanning to define a sequence landscape of potential binders (Fig. 5A), we improved its affinity and specificity and developed clone E021, which showed strong binding to the osim-p791/A02 complex (KD,app ~ 17 nM) and no binding to the p791/A02 complex (Fig. 5B). Binding was minimally inhibited by free osimertinib, with an IC50 value of ~100 μM (Fig. 5C).
Figure 5.
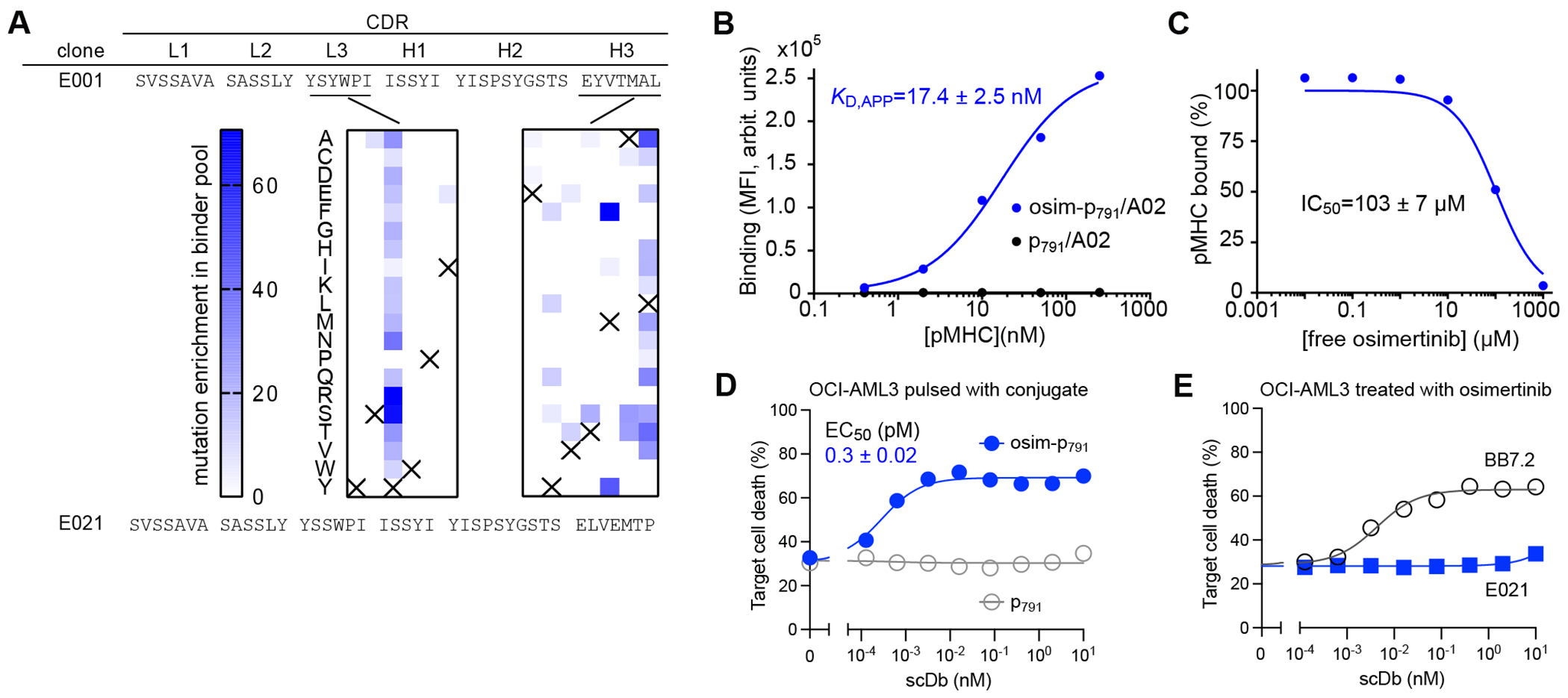
Binding and cell killing analyses of HapImmune™ antibody E021 against osimertinib-EGFR peptide conjugate in complex with HLA-A*02. (A) CDR sequences of clones E001 and E021. The middle panel shows the enrichment profiles of amino acid substitutions deduced from deep mutational scanning of CDR-L3 and -H3 positions. Data are presented as in Fig. 2A. (B) Binding analysis of E021 using yeast display. (C) Effect of free osimertinib on binding of E021 to osim-p791/A02. Binding signals were normalized to that in the absence of osimertinib. The IC50 value is the mean ± s.d. (n=3, technical replicates). (D) Cytotoxic effect of the E021 scDb on OCI-AML3 cells pulsed with osim-P791 or P791. Note that the E021 scDb showed potent cytotoxic effect on cells pulsed with the osimertinib-EGFR conjugate but not with the control peptide. (E) Cytotoxic effects of E021 scDb on osimertinib-treated OCI-AML3 cells, negative control cells that do not possess activating EGFR mutants. Data are from triplicate measurements, and calculated EC50 values are shown (mean ± s.d.; n=3, technical replicates). BB7.2 is a positive control that binds to HLA-A2 irrespective of the bound peptide. Data shown are representative of ≥2 equivalent measurements.
We then produced E021 in scDb format. The resultant E021 scDb potently killed OCI-AML3 cells pulsed with the osim-p791 conjugate with EC50 of 0.3 pM (Fig. 5D), but not cells pulsed with the p791WT peptide (Fig. 5D) or un-pulsed cells treated with osimertinib (Fig. 5E). Finally, we tested our approach with yet another inhibitor-target pair, ibrutinib and BTK (47). Taking an equivalent approach, we identified initial antibody hits that bound to the ibru-p476/HLA-A*01 complex but not to p476/HLA-A*01 without inhibitor conjugation (Supplementary Fig. S7). These data provide strong evidence for the general applicability of the HapImmune™ approach.
Discussion
We have shown that a covalent inhibitor conjugated to a peptide can be presented on HLA molecules, and “TCR-like” antibodies that bind selectively to such an inhibitor-p/MHC complex can be developed. These antibodies, when formatted as bispecific T cell engagers, can efficiently and selectively kill inhibitor-resistant cancer cells. Although we did not attempt to directly detect the drug-p/MHC complexes using immunopeptidomics, the efficient, MHC-dependent killing of sotorasib-treated H2122 cells by the R023 scDb provides strong evidence for its presence on the cell surface at sufficient density. Importantly, because HapImmune™ antibodies bind the drug/peptide/MHC complex, not the hapten itself, binding and T cell-dependent killing occurs even in the presence of a large excess of the free drug. These properties suggest that covalent drugs and HapImmune™ antibodies could be deployed concurrently. Furthermore, we were able to generate antibodies with these properties against drug-peptide conjugates generated with structurally distinct covalent inhibitors in complex with different MHCs, confirming this approach as a novel therapeutic strategy that unifies targeted and immune therapies.
We were successful in developing HapImmune™ antibodies against all three initial targets comprising chemically diverse drugs conjugated to distinct peptides that are presented on different HLA supertypes. Deep mutational scanning identified a total of 73 single-point mutants of clone R011 targeting soto-p/MHC (Fig. 2A), suggesting that many more antibodies can be developed for this antigen. In parallel to developing HapImmune™ antibodies, we attempted to generate antibodies selective to other nonconjugated neoantigen peptides corresponding to other KRAS mutations (p8 peptides harboring G12D, G12V or G13D) presented on HLA-A*03. Despite using the same antigen designs with the same synthetic antibody library and the same overall library sorting strategy, we failed to identify clones selective to any of these neoantigens over the p8WT/A03 complex. This stark contrast suggests that the conjugation with bulky drugs creates neoantigens that are more readily targetable by antibodies and potentially TCRs than conventional, unmodified neoantigen peptides.
Like other approaches to target MHC-presented antigens, HLA restriction limits the HapImmune™ approach to the subset of patients harboring appropriately matched HLA alleles. However, unlike previous attempts to target MHC complexes bearing unmodified peptides (27), it appears that HapImmune™ antibodies can recognize drug-peptide conjugates presented by multiple HLA supertypes, as exemplified by the R023 clone (Fig. 2, 4). First, R023 recognizes sotorasib conjugated to two different peptides. Second, it recognizes both of these peptides presented by homologous but distinct HLA molecules, HLA-A*03 and -A*11. Third, and most significantly, it also recognizes sotorasib conjugated to a substantially different peptide presented on a different HLA supertype, A*02. This degree of cross-HLA targeting is greater than that exhibited by recently reported peptide-centric CARs, which recognize a peptide presented by two divergent HLAs (48). These unique properties of HapImmune™ antibodies mean that a therapeutic like R023 could, in principle, be deployed in 40–50% of the US patient population with tumors bearing KRAS(G12C) (49). Remarkably, R023 achieves this level of cross-reactivity while maintaining strong discrimination between sotorasib-p/MHC and free sotorasib (Fig. 2). These results demonstrate another aspect of the impact of inhibitor conjugation in shifting the focus of p/MHC recognition toward the inhibitor in addition to making neoantigens more targetable. Our ongoing antibody-engineering, structural, and immunopeptidomics efforts promise to elucidate the molecular basis for this intriguing mode of inhibitor-p/MHC recognition and test the feasibility of developing antibodies that potently target sotorasib, adagrasib, and other covalent drug-peptide conjugates displayed on even broader repertoire of HLA molecules.
Our ability to develop HapImmune™ antibodies against multiple inhibitor-p/MHC complexes suggests that the same principles might be applied even more generally to target inhibitor-p/MHC complexes formed by existing (and future) covalent inhibitors that target intracellular proteins. If so, then comprehensive analysis of the immunopeptidome of inhibitor-treated cells could substantially accelerate the HapImmune™ discovery process. As we demonstrated with sotorasib and H2122 cells, a covalent inhibitor need not inhibit the target cell to be accessible to HapImmune™ antibody-mediated killing, so long as the inhibitor forms the intended covalent complex and the inhibitor-p/MHC is presented on the cell surface at a sufficient density for antibody recognition. Notably, several recent studies of patient samples showed that sotorasib-resistant tumors almost always retain sotorasib-KRASG12C engagement (33). Consequently, HapImmune™ antibodies might be useful in the setting of primary or acquired resistance. Even if tumors initially respond well, drug tolerant persisters or small reservoirs of intrinsically resistant cells could be eradicated by frontline sotorasib/HapImmune™ antibody administration.
In the Codebreak-200 trial (50), a randomized trial for second line disease comparing sotorasib with the standard of care-docetaxel, progression-free survival was improved by one month, but there was no impact on overall survival. In addition, 10% of patients had grade 3/4 liver toxicity, forcing six patients to be removed from the study, while two patients experienced drug-induced liver injury. However, this toxicity was mitigated by dose reduction, which may have impacted overall survival. These limitations of sotorasib may be mitigated by lowering its dose combined with a HapImmune™ therapeutic.
We also note that the HapImmune™ concept is not restricted to hapten-peptides generated by covalent inhibitors. Rather, any protein that is selectively and specifically accessible by a drug-like reactive agent and can be processed to a hapten-peptide and presented on MHC can be targeted. For example, the immunogenicity of cancer-specific proteins such as cancer testis antigens (51) might be augmentable by haptenization. Moreover, reactive amino acids other than cysteine (e.g., lysine, aspartate) are potentially targetable. In this context, emerging mass spectrometric databases of reactive proteins (52,53) could identify new targets and lead small molecules.
We used diabodies as an initial platform to evaluate T cell-dependent killing, but the “recognition end” of HapImmune™ antibodies can be assembled into multiple formats, including other types of T cell engagers (bi-specific, tri-specific), NK cell engagers, antibody-drug conjugates, radio-conjugates, cytokine conjugates, and even CAR-T/NK cells. The optimal “effector arm” could be tumor- or tumor site-dependent. In principle, it might also be possible to engineer TCRs or TCR derivatives (54) specific for drug-peptide/MHC complexes. Identifying the optimal format for killing tumors in mice is the major focus of current work in our laboratories. Interestingly, preclinical studies suggest that sotorasib efficacy is greater in the presence of competent immune system and can be further enhanced by immune checkpoint inhibition (4). It will be of interest to determine whether sotorasib treatment induces T cells bearing HapImmune™-like TCRs.
While this manuscript was in preparation, Zhang et al. published a related study targeting another covalent G12Ci, ARS1620, presented on HLAs (55). They developed antibodies using an inhibitor-peptide conjugate in the absence of MHC complex as the antigen, whereas we used inhibitor-p/MHC complexes. Consequently, the main antibody analyzed by Zhang et al., P1A4, does not discriminate between inhibitor-p and inhibitor-p/MHC, and it cannot be used to target cell surface antigen in the presence of the free inhibitor at a therapeutically relevant concentration. By contrast, as shown in Fig. 4, our scDb effectively killed H2122 cells in the presence of 1 μM sotorasib. We also demonstrate that our approach can be generalized to other covalent drug-peptide complexes. These differences suggest that our strategy may be more useful for lead antibody development and have greater potential for therapeutic application.
Materials and Methods
Antigen preparation
Peptides were synthesized and purified by Genemed Synthesis (San Antonio, TX). Sotorasib, osimertinib, ARS1620, and MRTX849 were purchased from Selleckchem (Radnor, PA). Covalent conjugation reactions of peptides to sotorasib, osimertinib, ARS1620, or MRTX849 (referred to as soto-, osim-, ARS-, and MRTX-peptides, respectively) were performed in solution as follows: Soto-peptides were prepared by adding 50 μl of 4 mM peptide dissolved in H2O to 50 μl of 8 mM sotorasib in 20% dimethylformamide (DMF) in H2O, followed by the addition of 5 μl 2 M Tris-HCl buffer, pH8, and subsequently incubating the mixture at 25°C overnight in the dark. All reaction mixtures listed below were incubated in the same manner. Osim-peptides were prepared by mixing 50 μl of 4 mM peptide dissolved in H2O and 50 μl of 8 mM osimertinib in 50% acetonitrile in H2O, followed by the addition of 5 μl 2 M Tris-HCl buffer, pH8 and incubation. ARS-peptides were prepared by mixing 50 μl of 4 mM peptide dissolved in H2O and 50 μl of 8 mM ARS 1620 in 50% acetonitrile in H2O, followed by the addition of 5 μl 2 M Tris-HCl buffer, pH8, and incubation. MRTX-peptides were prepared by mixing 50 μl of 4 mM peptide dissolved in H2O and 50 μl of 8 mM MRTX 849 in 100% DMF, followed by the addition of 5 μl 2 M Tris-HCl buffer, pH8, and subsequent incubation. The efficiency of all reactions was verified by reversed-phase chromatography with a C18 Eclipse column (Agilent) using an acetonitrile gradient in 0.1% trifluoroacetic acid.
Recombinant MHC heavy chains, HLA-A*02, A*03, and A*11, and beta-2-microglobulin (β2m) were expressed using pET-based vectors containing synthetic genes, generally following published procedures (40). All HLA constructs contained a C-terminal His6 tag and Avi-tag (Avidity). HLA proteins were produced as inclusion bodies in E. coli BL21(DE3) co-expressing BirA with 50 μM biotin in the culture medium, resulting in their biosynthetic biotinylation. β2m containing an N-terminal His6 tag and a TEV protease cleavage site and was expressed as inclusion bodies in E. coli BL21(DE3). HLA proteins were solubilized in urea, purified using a Ni-affinity chromatography on Ni-Sepharose column (Cytiva), and stored in 100 mM Tris-HCl buffer pH 8, containing 8 M urea. β2m was refolded on an Ni-Sepharose column using 50 mM Tris-HCl buffer pH 8 containing 25 mM NaCl and eluted in 50 mM Tris-HCl buffer pH 8 containing 25 mM NaCl and 0.5 M imidazole. After removing the N-terminal tag with TEV protease, the sample was purified further using a Superdex S75 column (Cytiva) in 10 mM sodium phosphate and 1.8 mM potassium phosphate buffer, pH 7.4, containing 138 mM NaCl (PBS). Biotinylation of HLA samples was confirmed using gel shift assays (56). Peptide-HLA-β2m complexes were assembled by refolding as follows: Briefly, a refolding mixture consisting of 30 μM peptide and 3 μM β2m was prepared in PBS. Next, concentrated HLA in 0.1 M Tris-HCl buffer, pH 8, containing 8 M urea and 0.5 M NaCl was quickly injected into the refolding mixture at a final concentration of 3 μM resulting in the final HLA:β2m:peptide ratio of 1:1:10. After incubation at 4°C overnight, the solution was centrifuged at 20,000xg, and the supernatant was concentrated with an Amicon centrifugal filter unit with a 10kDa cutoff (Millipore) and further purified using a Superdex75 10/300 GL column (Cytiva) equilibrated in PBS. Sample purity was typically >95% as verified using SDS-PAGE. Purified p/MHC complexes were concentrated to >2 μM and stored at −80°C until use.
Antibody development
Sorting of a synthetic human Fab library was performed as described previously (57) with small modifications. Briefly, a phage library was incubated with drug-p/HLA complexes at the concentrations of 100 nM (first and second rounds), 50 nM (third round), and 20 nM (fourth round). In the second and later rounds, phage solutions were first reacted with respective pWT-/MHC complexes immobilized on the Streptavidin MagneSphere particles (Promega), to eliminate cross-reactive clones. Sorted phage clones were assessed by multiplex bead binding assay (MBBA) (58).
The A3-2 antibody, selective to HLA-A*03 (and 02), and the A11-1 antibody, selective to HLA-A*11, were developed in an equivalent manner, except that negative selection was performed using p/MHC samples of the other supertype to enrich for supertype-selective clones, e.g., p/A11 was used for enriching clones selective for p/A03. Their binding profiles were characterized using the phage MBBA assay (Supplementary Fig. S6).
Affinity maturation of R001 was performed using yeast display following published general procedures (59). The Fab genes were reformatted into the scFv format and cloned the yeast display vector, pGalAga (60). We first constructed libraries in which two adjacent residues in CDR-H3 and -L3 of R001 were mutated to all amino acid combinations except for Cys, Met, Phe, Asn and Gln, using oligo pools (Twist Bioscience). Target concentrations for sorting were determined based on the apparent KD; 10x, 2x, <1x and <1x KD,APP for the first, second, third and fourth rounds, respectively; and 100 nM of the non-conjugated p/MHC complexes were used for negative sorting throughout. An IntelliCyt iQue Screener PLUS flow cytometer (Sartorius) and a S3e fluorescence-activated cell sorter (Bio-Rad) were used for analysis and cell sorting. The second library was constructed by introducing amino acid diversity in the CDR-H1 and -H2 positions following a published design (61) in the enriched pools from the first library. The second library was sorted three times using target concentrations described above, which yielded clone R011.
Deep mutational scanning (DMS) was performed by constructing a third library, in which we diversified each of the CDR-H3 and -L3 residues of clone R011, one amino acid at a time, using the NNK codon, where N is a mixture of A, T, G, and C, and K is a mixture of G and T. The library was sorted using soto-p8/A03 and soto-p7/A03 at target concentrations of 50 nM, 10 nM, and 3 nM for the first, second, and third rounds, respectively. Plasmids containing scFv genes were purified from the enriched pool of yeast cells, using Zymoprep Yeast Plasmid Miniprep II (Zymo Research Corporation), and the scFv genes were amplified and sequenced on a MiSeq sequencer (Illumina). Sequencing data were analyzed using a set of in-house developed UNIX and Python scripts to deduce the number of reads for each mutation. Finally, a fourth library was constructed using oligo pools (Twist Bioscience), introducing CDR L3 and H3 mutations based on the DMS results. After four rounds of sorting using soto-p8/03, soto-p7/03, soto-p8/11 and soto-p7/11 as targets, single clones were analyzed.
Affinity maturation of Fab E001 for osim-p791/A02 by the use of deep mutational scanning was carried out in the equivalent manner, using p791/A02 for negative sorting.
Fab Expression
Fabs were expressed and purified as reported previously (57). Briefly, genes encoding antibody clones were cloned into a vector that expresses a Fab with Avi-tag at the C-terminus of the heavy chain. Fabs were produced in E. coli strain 55244 (ATCC) and purified using a HiTrap Protein G affinity column (Cytiva). Purity >90% was confirmed using SDS-PAGE.
Expression and purification of scDbs
A synthetic gene encoding an anti-human CD3ε monoclonal antibody (clone UCHT1) in single-chain Fv format was synthesized (Integrated DNA Technologies). To construct expression vectors encoding scDbs, genes encoding the variable domains of heavy and light chains of the HapImmune™ antibodies and UCHT1 with a His-tag at the C-terminus were cloned into the mammalian expression vector pBCAG. Expi293F cells (Thermo Fisher) were transiently transfected with expression vectors using the ExpiFectamine 293 Transfection Kit (Thermo Fisher), according to the manufacturer’s protocol. Transfected cells were incubated at 37°C with 8% CO2 for 7 days, and scDbs were purified from supernatants using a HisTrap excel column (Cytiva) followed by size exclusion chromatography using a Superdex 200 10/300 column (Cytiva). The purity of the scDb proteins was analyzed by SDS-PAGE.
Biolayer interferometry
Binding kinetics of Fabs were assessed by using an Octet RED96e instrument (Sartorius). Briefly, biotinylated Fabs at 50nM in TBS were loaded on streptavidin SA biosensors to a final immobilization level ranging from 0.5 to 0.8 nm, and binding kinetics were measured against p/MHCs at 1, 4, 6, 16 and 64 nM in TBS buffer, pH 7.4, containing 1% BSA, 5 μM biotin, and 0.005% Tween-20, with an association period of 500s and a dissociation period of 800s. Data were analyzed using global fitting of a 1:1 binding model with Octet Data Analysis software, version 12.0.2.59.
Mammalian cell culture
Raji, NCI-H2030, and HEK293 cells were purchased from ATCC. NCI-H2122 and OCI-AML3 cells were obtained from Drs. Thales Papagiannakopoulos and Christopher Park, respectively (NYU Grossman School of Medicine). NCI-H358, NCI-H522, NCI-H1650, and SW1573 cells were obtained as described previously (62). HLA types of these cells were obtained from the TCLP database (63). The cell lines were tested negative for Mycoplasma monthly using a PCR-based mycoplasma testing kit (LiLIF). They have not been authenticated since the first acquisition. Raji, NCI-H358, NCI-H2122, NCI-H522, NCI-H1650, SW1573, and NCI-H2030 cells were maintained in Roswell Park Memorial Institute (RPMI)-1640 media (Thermo Fisher) supplemented with 10% fetal bovine serum (FBS, Gemini Bio) and penicillin/streptomycin (Thermo Fisher) at 37°C with 5% CO2. HEK293 cells were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM, Thermo Fisher) supplemented with 10% FBS (Gemini Bio) and penicillin/streptomycin (Thermo Fisher) at 37°C with 5% CO2. OCI-AML3 cells were maintained with Iscove’s Modified Dulbecco’s Medium (IMDM) with 20% FBS (Gemini Bio) and penicillin/streptomycin (Thermo Fisher) at 37°C with 5% CO2. Expi293F cells (Thermo Fisher) were maintained in Expi293 Expression Medium (Thermo Fisher) at 37°C with 8% CO2. All cell lines were used within 15 passages after thawing frozen stocks.
Peripheral blood mononuclear cells were purchased from STEMCELL Technologies. T cells were expanded by using CTS OpTimizer T-cell Expansion SFM (Thermo Fisher) and following the manufacturer’s protocol. T cells were cultured in CTS OpTimizer T-cell Expansion SFM supplemented with L-glutamine (Thermo Fisher) and penicillin/streptomycin, or in RPMI with 10% FBS and penicillin/streptomycin, in the presence of human IL-7 and human IL-15 (PeproTech), each at 10 ng/ml.
Cell line generation
A lentiviral vector containing Nanoluc engineered from Oplophorus gracilorostris (Nluc) (45) was kindly provided by Dr. Preet Chaudhary (USC Keck School of Medicine). Nluc lentivirus was produced as described (35). To generate stable cell lines, 1 ml of viral supernatant with 10 μg/ml polybrene was added to 1x106 cells in a 6-well plate and cultured for 8 hours. Media were exchanged and two days later, cells were selected in 20 μg/ml Blasticidin (InvivoGen). After selection, cell lines expressing Nanoluc were maintained in the media described above in the presence of 10 μg/ml Blasticidin (InvivoGen).
To generate H2122 lacking HLAA3, H2122-Nluc cells (1x106) were transfected with 2 μg of the Cas9/sgRNA vector PX458 (Addgene; plasmid 48138) using Lipofectamine 3000 (Thermo Fisher) in a 6-well plates. The following oligonucleotides were used for cloning sgRNAs into pX458: HLAA3 forward, 5’-CACCGCATCCTGGATACTCACGACG-3’; HLAA3 reverse, 5’-aaacCGTCGTGAGTATCCAGGATGC-3’. Two days after transfection, GFP+ cells were purified by FACS using a FACSAria IIu SORP cell sorter (BD Bioscience), and single cells were seeded into a 96-well plate. Clones were screened for HLA-A*03 expression by flow cytometric analysis (see below). These N-Luc cell lines were tested monthly for Mycoplasma infection.
Analysis of HLA-A*03 expression
Raji, H2122, and H2122(HLA-A*03KO) cells were stained with PE-conjugated anti-human HLA-A*03 (clone GAP-A3, BD Bioscience) or PE-conjugated mouse IgG2a isotype (clone MOPC-173, BioLegend) at the manufacturer’s recommended concentration. After staining, cells were washed with PBS supplemented with 1% BSA, and analyzed by using a ZE5 Cell Analyzer (Bio-Rad) or IntelliCyt iQue Screener PLUS (Sartorius).
Cell viability and killing assays
To assess their sotorasib sensitivity, H2122 and H358 cells (5x103 cells per well) were seeded in 96-well plates. The next day, media were replaced with fresh media containing serial dilutions of sotorasib. After 72 hr of incubation, cell viability was assessed by the PrestoBlue assay (Thermo Fisher), following the manufacturer’s protocol. Fluorescence was detected by using a FlexStation 3 multi-mode microplate reader (Molecular Devices).
The cytotoxic effects of scDbs on Raji and OCI-AML3 cells were measured by following a published protocol (64). Briefly, cells were stained with carboxyfluorescein succinimidyl ester (CSFE, Thermo Fisher) and then incubated with 10 μM sotorasib-KRAS(G12C) conjugates or 1 μM osimertinib-EGFR conjugate in the presence of 10 μg/ml human β2m for 4hr. Cells were incubated with control peptides in the same manner. For testing non-specific cell killing of drug-treated cells, Raji or OCI-AML3 cells were incubated with 1 μM sotorasib or 1 μM osimertinib in the presence of 10 μg/ml human β2m for 4 hr. Cells were then harvested using centrifugation and washed in media. Cells pulsed with peptides or treated with drugs were co-cultured with human T cells (E:T = 5:1) in the presence of scDbs for 18-21hr. We used E:T ratios of 5:1 and 10:1, because these ratios are commonly used for bispecific T-cell engagers (65,66). After incubation, cells were harvested and washed with PBS, then stained with Fixable Viability Dye eFluor660 (Thermo Fisher). After washing, cells were analyzed on an IntelliCyt iQue Screener (Sartorius).
To measure death by quantification of Nanoluc release, H2122-Nluc cells were cultured in the presence of various concentrations of sotorasib for 1 week. For cytotoxicity assays, cells were seeded in 96-well flat-bottom plates and incubated at 37 °C for 24 hours in the presence or absence (DMSO only) of sotorasib. After incubation, cells were co-cultured with human T cells (E:T = 10:1) and scDbs in the presence of sotorasib or DMSO, 2 μg/ml β2m and 10 ng/ml IL-7 and IL-15 for 24 hours at 37 °C. Supernatants from each well, containing Nanoluc released by dead cells, were transferred to a new 96-well plate, and Coelenterazine (Cayman Chemical) was added to the wells at a final concentration of 10 μM. Luminescence was measured with a Synergy Neo2 hybrid multi-mode reader (BioTek). Statistical analyses were performed using Prism 9 (GraphPad software).
Analysis of sotorasib conjugation to KRAS (G12C)
H2122 cells (5x105 cells per well) were seeded in 6-well plates. Next day, supernatants were replaced with media containing 100 nM sotorasib or DMSO. After a 24 hr incubation, whole cell lysates were generated in RIPA buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 2 mM EDTA, 1% NP-40, and 0.1% SDS), supplemented with protease inhibitors (40 μg/ml PMSF, 2 μg/ml antipain, 2 μg/ml pepstatin A, 20 μg/ml leupeptin, and 20 μg/ml aprotinin) and phosphatase inhibitors (10 mM NaF, 1 mM Na3VO4, 10 mM β-glycerophosphate, and 10 mM sodium pyrophosphate). After clarification of debris by centrifugation, samples were quantified by using the Bradford Protein Assay Kit (Thermo Fisher). Total lysate protein (20μg) was resolved by SDS-PAGE and transferred onto PVDF membranes (MilliporeSigma). Membranes were incubated with appropriate primary and secondary antibodies labeled with IRDye (680 nm) and visualized by using an Odyssey CLx Imaging System (LI-COR). Antibodies used here were: monoclonal pan-RAS antibody (1:1000) (clone Ab-3, Millipore), mouse monoclonal ERK-2 antibody (1:1000) (clone D2, Santa Cruz Biotechnology), and IRDye® 680LT Goat anti-Mouse IgG (H + L) (1:10000) (Li-Cor).
Supplementary Material
Significance.
Targeted therapies against oncoproteins often have dramatic initial efficacy but lack durability. Immunotherapies can be curative, yet most tumors fail to respond. We developed a generalizable technology platform that exploits hapten-peptides generated by covalent inhibitors as neoantigens presented on MHC to enable engineered antibodies to selectively kill drug-resistant cancer cells.
Acknowledgments
We thank E. Rajek for assistance, and the Perlmutter Cancer Center Genome Technology Center (GTC) and Cytometry and Cell Sorting Laboratory (CCSL) for instrumentation and technical assistance. This work was supported by National Institutes of Health grants R21CA246457 (T.H.), R21CA267362 (B.G.N. and S.K.), R01CA248896 (B.G.N. and Kwok-kin Wong), and P30CA016087 (Cancer Center Support Grant, B.G.N), which provided pilot funding for the early stages of this work and support for the GTC and CCSL.
Authors’ Disclosures
T. Hattori, L. Maso, A. Koide, B.G. Neel and S. Koide are listed as inventors of pending patents filed by New York University (NYU). NYU has entered into a research and option agreement with ATP Research and Development to develop these inventions and potentially to form a start-up company, with B.G. Neel and S. Koide as co-founders, to license and commercialize them. B.G. Neel is co-founder and holds equity in Northern Biologics, LTD; is co-founder and holds equity in Navire Pharma; Is co-founder, receives consulting fees, and holds equity in Lighthorse Therapeutics; is a SAB member, receives consulting fees and holds equity in Arvinas, Inc; is a SAB member and holds equity in Recursion Pharma; receives Consulting fees for GLG group; and receives research funding from Repare Therapeutics.
S. Koide is and is a co-founder and holds equity in Revalia Bio; receives research funding from Puretech Health, Argenx BVBA, and Black Diamond Therapeutics.
Footnotes
The other authors declare no potential conflicts of interests.
Data Availability
Data were generated by the authors and are available upon request from the corresponding authors. Scripts for deep mutational scanning analysis have been deposited in Github (DOI: 10.5281/zenodo.7115018).
References
- 1.Ahronian LG, Corcoran RB. Strategies for monitoring and combating resistance to combination kinase inhibitors for cancer therapy. Genome Med 2017;9(1):37 doi 10.1186/s13073-017-0431-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Konieczkowski DJ, Johannessen CM, Garraway LA. A Convergence-Based Framework for Cancer Drug Resistance. Cancer Cell 2018;33(5):801–15 doi 10.1016/j.ccell.2018.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013;503(7477):548–51 doi 10.1038/nature12796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Canon J, Rex K, Saiki AY, Mohr C, Cooke K, Bagal D , et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019;575(7781):217–23 doi 10.1038/s41586-019-1694-1. [DOI] [PubMed] [Google Scholar]
- 5.Hong DS, Fakih MG, Strickler JH, Desai J, Durm GA, Shapiro GI , et al. KRAS(G12C) Inhibition with Sotorasib in Advanced Solid Tumors. N Engl J Med 2020;383(13):1207–17 doi 10.1056/NEJMoa1917239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ryan MB, Corcoran RB. Therapeutic strategies to target RAS-mutant cancers. Nat Rev Clin Oncol 2018;15(11):709–20 doi 10.1038/s41571-018-0105-0. [DOI] [PubMed] [Google Scholar]
- 7.Hymowitz SG, Malek S. Targeting the MAPK Pathway in RAS Mutant Cancers. Cold Spring Harb Perspect Med 2018;8(11) doi 10.1101/cshperspect.a031492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Manchado E, Weissmueller S, Morris JPt, Chen CC, Wullenkord R, Lujambio A , et al. A combinatorial strategy for treating KRAS-mutant lung cancer. Nature 2016;534(7609):647–51 doi 10.1038/nature18600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sun C, Hobor S, Bertotti A, Zecchin D, Huang S, Galimi F , et al. Intrinsic resistance to MEK inhibition in KRAS mutant lung and colon cancer through transcriptional induction of ERBB3. Cell reports 2014;7(1):86–93 doi 10.1016/j.celrep.2014.02.045. [DOI] [PubMed] [Google Scholar]
- 10.Xue JY, Zhao Y, Aronowitz J, Mai TT, Vides A, Qeriqi B , et al. Rapid non-uniform adaptation to conformation-specific KRAS(G12C) inhibition. Nature 2020;577(7790):421–5 doi 10.1038/s41586-019-1884-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sharma SV, Lee DY, Li B, Quinlan MP, Takahashi F, Maheswaran S , et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 2010;141(1):69–80 doi 10.1016/j.cell.2010.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Konieczkowski DJ, Johannessen CM, Abudayyeh O, Kim JW, Cooper ZA, Piris A , et al. A melanoma cell state distinction influences sensitivity to MAPK pathway inhibitors. Cancer Discov 2014;4(7):816–27 doi 10.1158/2159-8290.CD-13-0424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shaffer SM, Dunagin MC, Torborg SR, Torre EA, Emert B, Krepler C , et al. Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature 2017;546(7658):431–5 doi 10.1038/nature22794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roesch A, Vultur A, Bogeski I, Wang H, Zimmermann KM, Speicher D , et al. Overcoming intrinsic multidrug resistance in melanoma by blocking the mitochondrial respiratory chain of slow-cycling JARID1B(high) cells. Cancer Cell 2013;23(6):811–25 doi 10.1016/j.ccr.2013.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rambow F, Rogiers A, Marin-Bejar O, Aibar S, Femel J, Dewaele M , et al. Toward Minimal Residual Disease-Directed Therapy in Melanoma. Cell 2018;174(4):843–55 e19 doi 10.1016/j.cell.2018.06.025. [DOI] [PubMed] [Google Scholar]
- 16.Ku SY, Rosario S, Wang Y, Mu P, Seshadri M, Goodrich ZW , et al. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science 2017;355(6320):78–83 doi 10.1126/science.aah4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yun S, Vincelette ND, Green MR, Wahner Hendrickson AE, Abraham I. Targeting immune checkpoints in unresectable metastatic cutaneous melanoma: a systematic review and meta-analysis of anti-CTLA-4 and anti-PD-1 agents trials. Cancer Med 2016;5(7):1481–91 doi 10.1002/cam4.732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Seidel JA, Otsuka A, Kabashima K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations. Front Oncol 2018;8:86 doi 10.3389/fonc.2018.00086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schachter J, Ribas A, Long GV, Arance A, Grob JJ, Mortier L , et al. Pembrolizumab versus ipilimumab for advanced melanoma: final overall survival results of a multicentre, randomised, open-label phase 3 study (KEYNOTE-006). Lancet 2017;390(10105):1853–62 doi 10.1016/S0140-6736(17)31601-X. [DOI] [PubMed] [Google Scholar]
- 20.Turnis ME, Andrews LP, Vignali DA. Inhibitory receptors as targets for cancer immunotherapy. Eur J Immunol 2015;45(7):1892–905 doi 10.1002/eji.201344413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mahoney KM, Rennert PD, Freeman GJ. Combination cancer immunotherapy and new immunomodulatory targets. Nat Rev Drug Discov 2015;14(8):561–84 doi 10.1038/nrd4591. [DOI] [PubMed] [Google Scholar]
- 22.Pettitt D, Arshad Z, Smith J, Stanic T, Hollander G, Brindley D. CAR-T Cells: A Systematic Review and Mixed Methods Analysis of the Clinical Trial Landscape. Mol Ther 2018;26(2):342–53 doi 10.1016/j.ymthe.2017.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chow VA, Shadman M, Gopal AK. Translating anti-CD19 CAR T-cell therapy into clinical practice for relapsed/refractory diffuse large B-cell lymphoma. Blood 2018;132(8):777–81 doi 10.1182/blood-2018-04-839217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.June CH, Sadelain M. Chimeric Antigen Receptor Therapy. N Engl J Med 2018;379(1):64–73 doi 10.1056/NEJMra1706169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sharma P, Allison JP. The future of immune checkpoint therapy. Science 2015;348(6230):56–61 doi 10.1126/science.aaa8172. [DOI] [PubMed] [Google Scholar]
- 26.Tran E, Robbins PF, Lu YC, Prickett TD, Gartner JJ, Jia L , et al. T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N Engl J Med 2016;375(23):2255–62 doi 10.1056/NEJMoa1609279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Douglass J, Hsiue EH, Mog BJ, Hwang MS, DiNapoli SR, Pearlman AH , et al. Bispecific antibodies targeting mutant RAS neoantigens. Sci Immunol 2021;6(57) doi 10.1126/sciimmunol.abd5515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chang AY, Gejman RS, Brea EJ, Oh CY, Mathias MD, Pankov D , et al. Opportunities and challenges for TCR mimic antibodies in cancer therapy. Expert Opin Biol Ther 2016;16(8):979–87 doi 10.1080/14712598.2016.1176138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carter PJ, Lazar GA. Next generation antibody drugs: pursuit of the ‘high-hanging fruit’. Nat Rev Drug Discov 2018;17(3):197–223 doi 10.1038/nrd.2017.227. [DOI] [PubMed] [Google Scholar]
- 30.Goebeler ME, Bargou RC. T cell-engaging therapies - BiTEs and beyond. Nat Rev Clin Oncol 2020;17(7):418–34 doi 10.1038/s41571-020-0347-5. [DOI] [PubMed] [Google Scholar]
- 31.Skoulidis F, Li BT, Dy GK, Price TJ, Falchook GS, Wolf J , et al. Sotorasib for Lung Cancers with KRAS p.G12C Mutation. N Engl J Med 2021;384(25):2371–81 doi 10.1056/NEJMoa2103695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Koga T, Suda K, Fujino T, Ohara S, Hamada A, Nishino M , et al. KRAS Secondary Mutations That Confer Acquired Resistance to KRAS G12C Inhibitors, Sotorasib and Adagrasib, and Overcoming Strategies: Insights From In Vitro Experiments. J Thorac Oncol 2021;16(8):1321–32 doi 10.1016/j.jtho.2021.04.015. [DOI] [PubMed] [Google Scholar]
- 33.Awad MM, Liu S, Rybkin II, Arbour KC, Dilly J, Zhu VW , et al. Acquired Resistance to KRAS(G12C) Inhibition in Cancer. N Engl J Med 2021;384(25):2382–93 doi 10.1056/NEJMoa2105281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tanaka N, Lin JJ, Li C, Ryan MB, Zhang J, Kiedrowski LA , et al. Clinical Acquired Resistance to KRAS(G12C) Inhibition through a Novel KRAS Switch-II Pocket Mutation and Polyclonal Alterations Converging on RAS-MAPK Reactivation. Cancer Discov 2021;11(8):1913–22 doi 10.1158/2159-8290.CD-21-0365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fedele C, Li S, Teng KW, Foster CJR, Peng D, Ran H , et al. SHP2 inhibition diminishes KRASG12C cycling and promotes tumor microenvironment remodeling. J Exp Med 2021;218(1) doi 10.1084/jem.20201414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Teng KW, Tsai ST, Hattori T, Fedele C, Koide A, Yang C , et al. Selective and noncovalent targeting of RAS mutants for inhibition and degradation. Nat Commun 2021;12(1):2656 doi 10.1038/s41467-021-22969-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lim S, Khoo R, Juang YC, Gopal P, Zhang H, Yeo C , et al. Exquisitely Specific anti-KRAS Biodegraders Inform on the Cellular Prevalence of Nucleotide-Loaded States. ACS Cent Sci 2021;7(2):274–91 doi 10.1021/acscentsci.0c01337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang Q, Douglass J, Hwang MS, Hsiue EH, Mog BJ, Zhang M , et al. Direct Detection and Quantification of Neoantigens. Cancer Immunol Res 2019;7(11):1748–54 doi 10.1158/2326-6066.CIR-19-0107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jurtz V, Paul S, Andreatta M, Marcatili P, Peters B, Nielsen M. NetMHCpan-4.0: Improved Peptide-MHC Class I Interaction Predictions Integrating Eluted Ligand and Peptide Binding Affinity Data. J Immunol 2017;199(9):3360–8 doi 10.4049/jimmunol.1700893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Garboczi DN, Hung DT, Wiley DC. HLA-A2-peptide complexes: refolding and crystallization of molecules expressed in Escherichia coli and complexed with single antigenic peptides. Proc Natl Acad Sci U S A 1992;89(8):3429–33 doi 10.1073/pnas.89.8.3429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Araya CL, Fowler DM. Deep mutational scanning: assessing protein function on a massive scale. Trends Biotechnol 2011;29(9):435–42 doi 10.1016/j.tibtech.2011.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brüsselbach S, Korn T, Völkel T, Müller R, Kontermann RE. Enzyme recruitment and tumor cell killing in vitro by a secreted bispecific single-chain diabody. Tumor Targeting 1999;4:115–23. [Google Scholar]
- 43.Beverley PC, Callard RE. Distinctive functional characteristics of human “T” lymphocytes defined by E rosetting or a monoclonal anti-T cell antibody. Eur J Immunol 1981;11(4):329–34 doi 10.1002/eji.1830110412. [DOI] [PubMed] [Google Scholar]
- 44.Suh WK, Cohen-Doyle MF, Fruh K, Wang K, Peterson PA, Williams DB. Interaction of MHC class I molecules with the transporter associated with antigen processing. Science 1994;264(5163):1322–6 doi 10.1126/science.8191286. [DOI] [PubMed] [Google Scholar]
- 45.Matta H, Gopalakrishnan R, Choi S, Prakash R, Natarajan V, Prins R , et al. Development and characterization of a novel luciferase based cytotoxicity assay. Sci Rep 2018;8(1):199 doi 10.1038/s41598-017-18606-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cross DA, Ashton SE, Ghiorghiu S, Eberlein C, Nebhan CA, Spitzler PJ , et al. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov 2014;4(9):1046–61 doi 10.1158/2159-8290.CD-14-0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Honigberg LA, Smith AM, Sirisawad M, Verner E, Loury D, Chang B , et al. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc Natl Acad Sci U S A 2010;107(29):13075–80 doi 10.1073/pnas.1004594107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yarmarkovich M, Marshall QF, Warrington JM, Premaratne R, Farrel A, Groff D , et al. Cross-HLA targeting of intracellular oncoproteins with peptide-centric CARs. Nature 2021;599(7885):477–84 doi 10.1038/s41586-021-04061-6. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 49.Maiers M, Gragert L, Klitz W. High-resolution HLA alleles and haplotypes in the United States population. Hum Immunol 2007;68(9):779–88 doi 10.1016/j.humimm.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 50.Johnson ML, De Langen J, Waterhouse DM, Mazieres J, Dingemans AC, Mountzios G , et al. Sotorasib versus docetaxel for previously treated non-small cell lung cancer with KRAS G12C mutation: CodeBreaK 200 phase III study. Annal Oncol 2022;33:S808–S69 doi 10.1016/annonc/annonc1089. [DOI] [Google Scholar]
- 51.Simpson AJ, Caballero OL, Jungbluth A, Chen YT, Old LJ. Cancer/testis antigens, gametogenesis and cancer. Nat Rev Cancer 2005;5(8):615–25 doi 10.1038/nrc1669. [DOI] [PubMed] [Google Scholar]
- 52.Niphakis MJ, Cravatt BF. Enzyme inhibitor discovery by activity-based protein profiling. Annu Rev Biochem 2014;83:341–77 doi 10.1146/annurev-biochem-060713-035708. [DOI] [PubMed] [Google Scholar]
- 53.Abbasov ME, Kavanagh ME, Ichu TA, Lazear MR, Tao Y, Crowley VM , et al. A proteome-wide atlas of lysine-reactive chemistry. Nat Chem 2021;13(11):1081–92 doi 10.1038/s41557-021-00765-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lowe KL, Cole D, Kenefeck R, I OK, Lepore M, Jakobsen BK. Novel TCR-based biologics: mobilising T cells to warm ‘cold’ tumours. Cancer Treat Rev 2019;77:35–43 doi 10.1016/j.ctrv.2019.06.001. [DOI] [PubMed] [Google Scholar]
- 55.Zhang Z, Rohweder PJ, Ongpipattanakul C, Basu K, Bohn MF, Dugan EJ , et al. A covalent inhibitor of K-Ras(G12C) induces MHC class I presentation of haptenated peptide neoepitopes targetable by immunotherapy. Cancer Cell 2022;40(9):1060–9 e7 doi 10.1016/j.ccell.2022.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sorenson AE, Askin SP, Schaeffer PM. In-gel detection of biotin–protein conjugates with a green fluorescent streptavidin probe. Analytical Methods 2015;7(5):2087–92 doi 10.1039/C4AY02666G. [DOI] [Google Scholar]
- 57.Miller KR, Koide A, Leung B, Fitzsimmons J, Yoder B, Yuan H , et al. T cell receptor-like recognition of tumor in vivo by synthetic antibody fragment. PloS one 2012;7(8):e43746 doi 10.1371/journal.pone.0043746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hattori T, Koide A, Panchenko T, Romero LA, Teng KW, Corrado AD , et al. Multiplex bead binding assays using off-the-shelf components and common flow cytometers. J Immunol Methods 2020;490:112952 doi 10.1016/j.jim.2020.112952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Koide A, Koide S. Affinity maturation of single-domain antibodies by yeast surface display. Methods Mol Biol 2012;911:431–43 doi 10.1007/978-1-61779-968-6_26. [DOI] [PubMed] [Google Scholar]
- 60.Koide A, Gilbreth RN, Esaki K, Tereshko V, Koide S. High-affinity single-domain binding proteins with a binary-code interface. Proc Natl Acad Sci U S A 2007;104(16):6632–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lee CV, Liang WC, Dennis MS, Eigenbrot C, Sidhu SS, Fuh G. High-affinity human antibodies from phage-displayed synthetic Fab libraries with a single framework scaffold. J Mol Biol 2004;340(5):1073–93. [DOI] [PubMed] [Google Scholar]
- 62.Fedele C, Ran H, Diskin B, Wei W, Jen J, Geer MJ , et al. SHP2 Inhibition Prevents Adaptive Resistance to MEK Inhibitors in Multiple Cancer Models. Cancer Discov 2018;8(10):1237–49 doi 10.1158/2159-8290.CD-18-0444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Scholtalbers J, Boegel S, Bukur T, Byl M, Goerges S, Sorn P , et al. TCLP: an online cancer cell line catalogue integrating HLA type, predicted neo-epitopes, virus and gene expression. Genome Med 2015;7:118 doi 10.1186/s13073-015-0240-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yamashita M, Kitano S, Aikawa H, Kuchiba A, Hayashi M, Yamamoto N , et al. A novel method for evaluating antibody-dependent cell-mediated cytotoxicity by flowcytometry using cryopreserved human peripheral blood mononuclear cells. Sci Rep 2016;6:19772 doi 10.1038/srep19772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Aschmoneit N, Steinlein S, Kuhl L, Seifert O, Kontermann RE. A scDb-based trivalent bispecific antibody for T-cell-mediated killing of HER3-expressing cancer cells. Sci Rep 2021;11(1):13880 doi 10.1038/s41598-021-93351-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dao T, Pankov D, Scott A, Korontsvit T, Zakhaleva V, Xu Y , et al. Therapeutic bispecific T-cell engager antibody targeting the intracellular oncoprotein WT1. Nat Biotechnol 2015;33(10):1079–86 doi 10.1038/nbt.3349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang S, Liu J, Cheng H, Tan S, Qi J, Yan J , et al. Structural basis of cross-allele presentation by HLA-A*0301 and HLA-A*1101 revealed by two HIV-derived peptide complexes. Mol Immunol 2011;49(1-2):395–401 doi 10.1016/j.molimm.2011.08.015. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data were generated by the authors and are available upon request from the corresponding authors. Scripts for deep mutational scanning analysis have been deposited in Github (DOI: 10.5281/zenodo.7115018).