

Abstract
Vps13 is a large, conserved protein that transports lipids between membranes. Its localization at multiple organelle membranes and membrane contact sites suggests its important physiological roles. In addition, the high correlation of mutant VPS13 with certain diseases, especially those involving neurodegeneration, makes this protein of considerable biomedical interest. Taking advantage of the fact that yeasts only have one Vps13 protein, the roles of yeast Vps13 have been well studied. However, whether and how Vps13 functions in macroautophagy/autophagy, a process of degradation of cytoplasmic cargoes, have been elusive questions. In this paper, we investigated the role of Vps13 in both non-selective and selective autophagy and found that this protein participates in non-selective autophagy, reticulophagy and pexophagy, but not mitophagy, and that Vps13 plays a role in the late stage of autophagy.
Keywords: Membrane, mitophagy, protein trafficking, stress, vacuole
Introduction
Vps13 is a large, evolutionarily conserved protein that exists in all eukaryotes. Only one Vps13 protein exists in yeast, but in mammalian cells there are four isoforms, VPS13A to VPS13D, with VPS13A having the highest similarity with the yeast protein [1]. VPS13 proteins are of intense biomedical interest because mutations in each VPS13 protein are associated with human diseases, such as VPS13A in chorea-acanthocytosis [2, 3], VPS13B in Cohen syndrome [4], VPS13C in Parkinson disease [5] and VPS13D in spastic ataxia [6].
Both the yeast and mammalian Vps13/VPS13 proteins are present at multiple sites. Yeast Vps13 has been found in various membrane contact sites, including vacuole-mitochondrial patches/vCLAMPs and nucleus-vacuole junctions/NVJs, and on individual organelle membranes, including endosomes and prospore membranes [7]. Vps13 is recruited through its interaction with adaptors on the organelle membrane, such as Mcp1 on mitochondria, Spo71 on the prospore membrane and Ypt35 on the vacuole and endosome membrane [8]. The common feature of these adaptors is the proline-X-proline (PxP) motif which binds to a site on Vps13 that has six repeated regions named Vps13 adaptor binding (VAB) domain [8]. Even though the functions of C-terminal domains, including APT1, ATG2_C and PH domains, are not well indicated in yeast Vps13, they are proposed to contribute to membrane tethering together with the adaptor proteins possibly through lipid binding [7]. In mammalian cells, the C terminal region is better studied, with the DH-like/DHL-PH domain at the C terminus found to be responsible for VPS13A binding to mitochondria and VPS13A and VPS13C to lipid droplets [9]. At the same time, FFAT motifs in mammalian VPS13 proteins interact with a VAP (VAMP associated protein) on the endoplasmic reticulum (ER), together generating their localization at membrane contact sites [9].
Besides the localization at membrane contact sites, the crystal structure of the first 335 residues of Chaetomium thermophilum Vps13 shows a scoop-like structure with hydrophobic residues facing the concave side, which is suited for solubilizing lipids [9]. A more recent structural analysis of residue 1-1390 of C. thermophilum Vps13 reveals that the groove is extended beyond the N-terminal chorein domain and formed as a basket handle-like structure [10]. Additionally, the purified N terminus of Vps13 (residues 1-1350) from Saccharomyces cerevisiae binds multiple types of lipids at the same time and transports lipids in vitro [9]. All these features together allow Vps13 to be proposed as a channel for the bulk transport of lipids between membranes [10, 11].
Even though the roles of Vps13 in lipid transport, membrane contact sites and prospore expansion have been well studied, its role in autophagy is less clear [7]. Autophagy is a process in which parts of the cytoplasm are sent to the vacuole for degradation and recycling [12]. Autophagy can be non-selective, sequestering random or phase-separated portions of the cytoplasm, and selective, which involves the targeted degradation of protein aggregates and organelles [13]. The morphological hallmark of autophagy is the formation of a double-membrane structure named an autophagosome, which is the terminal product of the dynamic sequestering compartment, the phagophore [14]. Even though it is not clear how this double-membrane structure is generated, it is widely accepted that phagophore expansion requires lipid addition. Because Vps13 plays a critical role in lipid transport between membranes, it is reasonable to propose that it has a role in autophagy. One previous study shows that the deletion of tipC, the homolog of VPS13 in Dictyostelium discoideum, leads to decreased numbers of GFP-Atg8 and GPF-Atg18 puncta during starvation. Additionally, knocking down VPS13A in mammalian cells results in more GFP-LC3 and GFP-WIPI1 puncta compared to wild-type cells under nutrient-rich conditions but the puncta numbers do not increase when the cells are shifted to starvation conditions [15]. These discoveries suggest a potential role of Vps13 in autophagy. However, whether yeast Vps13 functions similarly and at which stage of autophagy the protein acts are unclear. In addition, Vps13 is demonstrated to function in reticulophagy, selective ER degradation through autophagy, where Vps13 facilitates the packaging of ER into phagophores [16]. With regard to mitophagy, the selective autophagic degradation of mitochondria, Park et al reported that Vps13 is important for mitochondria integrity and the loss of Vps13 leads to increased mitophagy [17], and Chen et al reported a similar result, even though the increase was not as large [16]. VPS13 apparently functions differently in mammalian cell mitophagy as deletion of VPS13A impairs mitophagy [18]. Similarly, two independent studies indicated that VPS13D is required for mitochondrial clearance and the deletion of VPS13D leads to increased ER-mitochondria contact sites and a large and round mitochondrial morphology [19, 20]. To have a better idea of the function of Vps13 in yeast cells with regard to autophagy, we investigated the role of Vps13 in both non-selective and selective autophagy and determined the step in which it may act. Because we obtained different conclusions compared with certain previous studies and noticed different phenotypes between colonies having the same apparent genotype, we suggest an explanation for some of the different conclusions about the role of this protein.
Results
Vps13 is essential for efficient autophagy
To test whether Vps13 is important for autophagy, we used three well-established assays to measure non-selective autophagy activity in wild-type and vps13Δ cells. First, we performed the green fluorescent protein (GFP)-Atg8 processing assay. The principle of this assay is that the N-terminal GFP-tagged Atg8 (GFP-Atg8) on the inner membrane of autophagosomes will be delivered to the vacuole after the autophagosome and vacuole fuse. Atg8 is degraded by hydrolases, while GFP is more resistant to degradation; therefore, free GFP is generated and the conversion of GFP-Atg8 to free GFP can be a readout of autophagy activity [21]. After 2 h of starvation, we observed free GFP in both wild-type and vps13Δ cells, with less free GFP seen in vps13Δ cells (Fig. 1A and B), indicating deficient autophagy activity in the mutant cells. We then used the Pho8Δ60 assay to measure autophagy activity. Pho8 is a vacuolar phosphatase that is delivered to the vacuole through the secretory pathway, where it gets cleaved and activated. The truncated form of Pho8, Pho8Δ60, which lacks the first 60 amino acids including the transmembrane domain, can only be transported to the vacuole through non-selective autophagy and, therefore, the phosphatase activity of Pho8Δ60 reflects non-selective autophagy activity [21]. After 4-h starvation, significantly decreased Pho8Δ60 activity was seen in the cells lacking VPS13 compared to the wild type (Fig. 1C), again suggesting a deficiency in autophagy.
Figure 1.
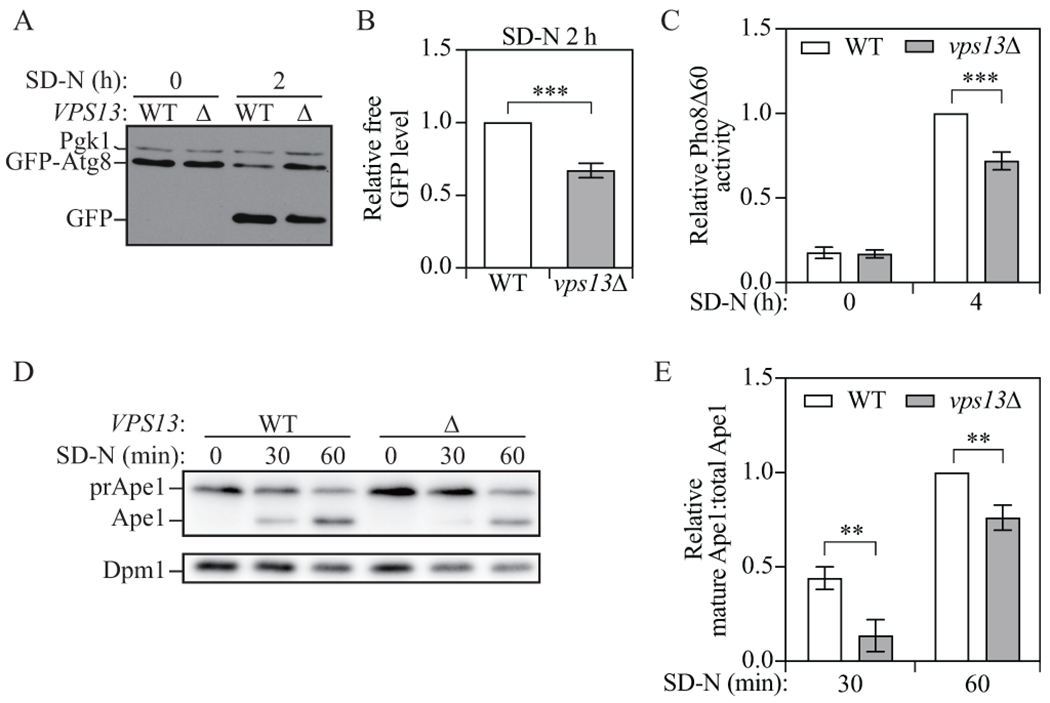
Vps13 is important for an efficient autophagy. (A) Protein extracts were collected from wild-type (YLY085) and vps13Δ (YLY086) cell cultures in YPD grown to mid-log phase (SD-N 0 h) and 2 h after nitrogen starvation (SD-N 2 h). Western blot was performed with anti-YFP and anti-Pgk1 antibodies or antisera. (B) Quantitative analysis of the relative free GFP level. Free GFP levels were normalized to Pgk1 protein level then the relative free GFP levels were normalized to wild-type cells which were set to 1.0. (C) Protein samples were collected from the cell culture of wild-type (WLY176) and vps13Δ (WXY206) cells grown to mid-log phase in YPD (SD-N 0 h) and 4 h after nitrogen starvation (SD-N 4 h). Autophagy activity was measured with the Pho8Δ60 assay. Pho8Δ60 activities were normalized to wild-type cells after 4-h nitrogen starvation. (D) Protein extracts were collected from wild-type (YLY113, vac8Δ ) and vps13Δ (YLY114, vac8Δ) cell cultures grown in YPD to OD600 = 0.4-0.5 (SD-N 0 h) and 30 and 60 min after nitrogen starvation. Western blots were probed with anti-Ape1 and anti-Dpm1 antibodies. (E) Quantitative analysis of the relative mature Ape1 to total Ape1 (Ape1 + prApe1) ratio. The ratio of wild-type cells after 60-min starvation was set to 1 and other samples were normalized accordingly. In quantitative analysis, the error bar represents the standard deviation (SD) of three independent experiments. Two-tailed t test was used for statistical significance. **p < 0.01, ***p < 0.001.
To further confirm the conclusion from the two assays mentioned above, another assay, which relies on the processing of precursor Ape1 (aminopeptidase I; prApe1), was performed. Ape1 is a vacuolar hydrolase that is initially synthesized as a precursor with an N-terminal propeptide. The propeptide is proteolytically cleaved from prApe1 upon vacuolar delivery through the cytoplasm-to-vacuole targeting/Cvt pathway or non-selective autophagy, resulting in a change in the protein size. The processing of prApe1 under nutrient-rich conditions can be prohibited by deleting VAC8; upon autophagy induction, prApe1 is efficiently delivered to the vacuole in a vac8Δ strain through an autophagic process that requires a specific receptor (Atg19) and a scaffold protein (Atg11) to reach a high efficiency [21]. As expected, there was no prApe1 processing under the growing condition (SD-N, 0 min), but mature Ape1 was generated after the cells were shifted to starvation medium. In vps13Δ cells, less mature Ape1 was seen after starvation for both 30 and 60 min compared to the wild type (Fig. 1D and E), which is consistent with what we observed in the other two assays. Together, these results indicate that Vps13 is important for efficient autophagic processes in yeast.
The deficient autophagy activity in vps13Δ cells could be rescued by expressing a plasmid bearing VPS13 (Fig. S1A and B), further suggesting the autophagy deficiency in the mutant cells resulted from the lack of Vps13 rather than an unknown arbitrary mutation. As mentioned above, VPS13 is a very large gene. To diminish the potential influence of deleting a big region on the genome, we also generated another strain, lacking most of VPS13 but retaining the first 300 and last 402 base pairs (vps13Δ2). These mutant cells also showed decreased autophagy activity (Fig. S1C and D), further suggesting that Vps13 is crucial for autophagy.
Vps13 functions in a late step of autophagy
Because we found that Vps13 is critical for efficient autophagy, we decided to investigate the step of autophagy in which Vps13 functions. Vps13 has a similar structure to Atg2, and many functional domains exist in both proteins including the chorein domain at the N terminus, and the APT1 and ATG2_C domains close to the C terminus [7]. More importantly, a chimeric protein with the first 235 residues of Vps13 and the rest composed of Atg2 (residues 236-1592) functions similarly as the wild-type Atg2 protein in autophagy [22], suggesting Vps13 has a similar function to Atg2. Because Vps13 is a lipid transport protein and Atg2 is responsible for membrane tethering and lipid transport to the phagophore for its expansion [22, 23], we hypothesized that the deletion of VPS13 may cause a delay in lipid transport to the phagophore and a deficiency in a late stage of autophagy. To test this hypothesis, we performed an experiment to determine whether the GFP-Atg8 puncta formed in the vps13Δ strain during starvation are incomplete structures (e.g., phagophores) or essentially complete autophagosomes that are not capable of fusing with the vacuole. Accordingly, wild-type or vps13Δ cells were starved in SD-N for 1 h to induce autophagy before shifting back to nutrient-rich medium to stop the formation of new phagophores and allow the completion of those that had already formed. In the wild-type cells, 5 min after shifting back to nutrient-rich medium, the cytoplasmic signal for most puncta that had accumulated after starvation disappeared (Fig. 2A–C). In contrast, 30% of the GFP-Atg8 puncta remained even after 15 min in nutrient-rich medium in vps13Δ cells compared to approximately 15% seen in the WT. These results suggest that in cells without Vps13, more GPF-Atg8 puncta are abnormal autophagosomes that are defective in fusion with the vacuole.
Figure 2.
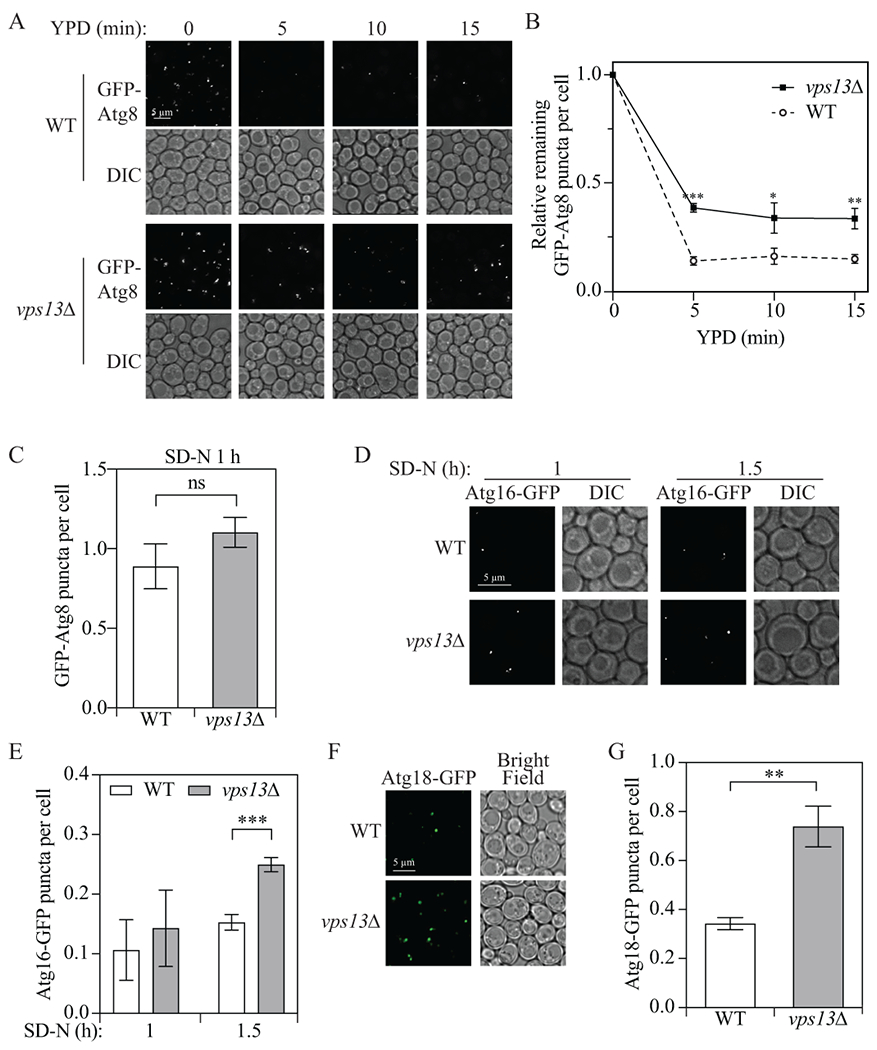
Vps13 functions in a late stage of autophagy. (A and B) Wild-type (YLY085) and vps13Δ (YLY086) strains were grown in YPD to mid-log phase and shifted to nitrogen-starvation medium. After starvation for 1 h (YPD 0 min), cells were shifted back to YPD and samples were collected after 5, 10 and 15 min and imaged. Total puncta number to total cell number ratio was quantified at every time point and the ratio was normalized to the samples at 0 min, which was set to 1.0. (C) Quantification of the total GFP-Atg8 puncta to total cell number ratio after 1-h starvation as described in (A). (D and E) Wild-type (UNY174, Atg16-GFP) and vps13Δ (YLY144, Atg16-GFP) cell samples were collected at 1 and 1.5 h after nitrogen starvation (SD-N) and imaged. Total puncta to total cell number ratio was quantified at every time point. (F and G) Wild-type (YLY394, Atg18-GFP) and vps13Δ (YLY395, Atg18-GFP) cell samples were collected 1.5 h after nitrogen starvation and imaged. The total puncta to total cell number ratio was quantified. In the quantitative analyses, the error bar represents the SD of three independent experiments. Two-tailed t test was used for statistical significance. *p < 0.05, **p < 0.01, ***p < 0.001, ns, not significant. DIC: differential interference contrast.
To determine whether the delay in fusion between autophagosomes and the vacuole resulted from a deficiency in phagophore closure, we carried out a protease protection experiment by monitoring sensitivity of the prApe1 propeptide. In a mutant defective for autophagosome formation, the prApe1 complex is accessible to exogenously added protease; in this case, the proteolytically sensitive is removed, generating the mature form of the enzyme. In contrast, in a strain that has normal autophagosome completion, the prApe1 is protected from proteinase K degradation within the autophagosome and can only be degraded when the autophagosome membrane is disrupted by the addition of detergent (Fig. S2A). In wild-type cells, or in the ypt7Δ strain where autophagosomes cannot fuse with the vacuole [24], prApe1 was protected from proteinase K degradation, indicating that these cells had completed autophagosome formation (Fig. S2B). In contrast, in the control atg9Δ cells where autophagosomes cannot normally form [25], prApe1 was degraded even without the addition of the detergent. In vps13Δ cells, prApe1 was protected from proteinase K degradation, suggesting that the autophagosomes in these cells had sealed.
Before or immediately after the completion of the autophagosome, early-acting Atg proteins dissociate from the vesicle [26]. Because we found a higher percentage of GFP-Atg8 puncta that could not fuse with the vacuole in vps13Δ cells but autophagosome formation was apparently completed, we next sought to determine whether early Atg protein dissociation was affected. Thus, we carried out a disassembly assay, where we checked the localization of several Atg proteins that act in the early stages of autophagy. C-terminal GFP-tagged Atg16, which forms a complex with Atg5 and Atg12 and is recruited to the phagophore to facilitate Atg8 conjugation to phosphatidylethanolamine [27], should be detectable as discrete puncta instead of a diffuse weak cytosolic signal when the protein is present on the phagophore membrane. The quantification revealed that after a 1-h starvation, vps13Δ cells had slightly more puncta than the wild-type cells; however, after 1.5-h starvation, only 15% of the wild-type cells contained Atg16-GFP puncta whereas 25% of the vps13Δ cells had punctate structures (Fig. 2D and 2E). We also monitored Atg18, which is recruited to the phagophore through its binding to phosphatidylinositol-3-phosphate and regulates phagophore assembly site recruitment of Atg2 [28]. After 1.5-h starvation, approximately 35% of the wild-type cells contained Atg18-GFP puncta whereas 75% of the vps13Δ cells had punctate structures (Fig. 2F and 2G). Finally, we examined puncta for Atg14, a component of phosphatidylinositol 3-kinase complex I, but no significant difference was found between wild-type and vps13Δ cells after 1.5-h starvation (Fig. S2C and 2D). Because phagophore formation was not affected as shown by the similar number of GFP-Atg8 puncta per cell after 1-h starvation (Fig. 2A and 2C) and VPS13 deletion led to a deficiency in the fusion between autophagosomes and the vacuole, we suggest that Vps13 is important for the proper disassembly of some early-acting Atg proteins from the autophagosome, which may result in a subsequent defect in autophagosome-vacuole fusion.
Vps13 is important for reticulophagy and pexophagy, but not mitophagy
As mentioned above, Vps13 localizes to multiple membrane contact sites [7]. Previous studies have revealed that membrane contact sites are essential for the full activation of several types of selective autophagy, including ER-mitochondria encounter structure (ERMES) in mitophagy and pexophagy [29, 30] and nucleus-vacuole junctions in the cytoplasm-to-vacuole targeting pathway [31]. Accordingly, we asked whether Vps13 functions in selective autophagy. To monitor selective autophagy in yeast, we took advantage of assays that are similar to GFP-Atg8 processing, where GFP is fused to a protein that targets to a specific organelle, which will be sent to the vacuole when the appropriate type of selective autophagy is induced. In the vacuole, the organelle marker protein will be proteolytically degraded, but the GFP moiety will accumulate. Therefore, the free GFP level can again be used to measure the activity of selective autophagy [21]. We first focused on reticulophagy, and to measure the corresponding activity we chose the ER membrane protein Sec63 and tagged GFP at its C terminus (Sec63-GFP); because the autophagic degradation of ER occurs slowly relative to other types of selective autophagy, we needed to starve the cells for a longer time to detect the free GFP band. Sec63-GFP was not cleaved under growing conditions in wild-type cells, but after 8 h of starvation, free GFP was detected (Fig. 3A). We also examined processing of Sec63-GFP in two control strains, atg40Δ [32] and atg1Δ, which are defective for reticulophagy and autophagy, respectively. Both strains showed a block in Sec63-GFP processing at the 8-h timepoint, indicating that the generation of free GFP was not the result of non-specific cleavage of the chimera. The vps13Δ strain showed a lower level of Sec63-GFP processing compared to the wild type, indicating deficient reticulophagy in the mutant cells, consistent with a published study [16].
Figure 3.
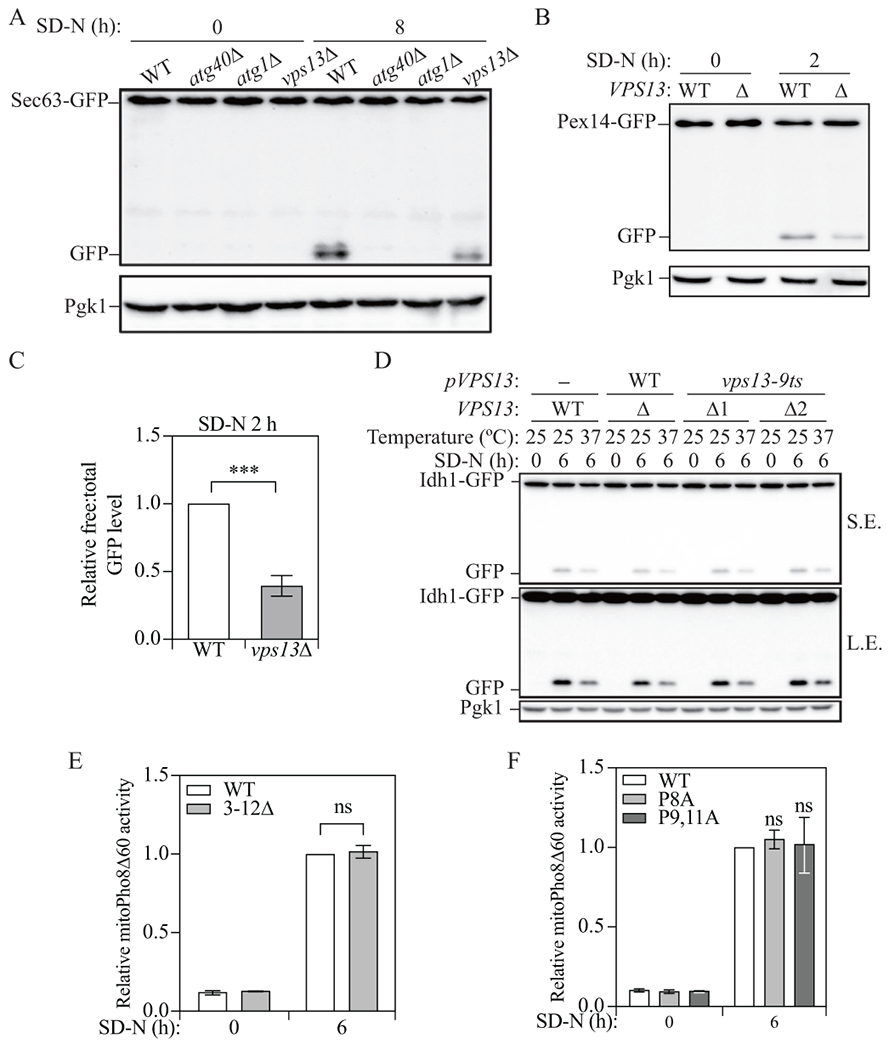
Vps13 is important for reticulophagy and pexophagy, but not for mitophagy. (A) Protein samples were collected from wild-type (WXY233, Sec63-GFP), atg40Δ (DGY147, Sec63-GFP), atg1Δ (WXY283, Sec63-GFP), and vps13Δ (WXY234, Sec63-GFP) cells that were grown in YPD to mid-log phase (SD-N 0 h) and 8 h after nitrogen starvation. Western blots were probed with anti-YFP and anti-Pgk1 antibodies or antisera. (B) Wild-type (WXY201, Pex14-GFP) and vps13Δ (WXY203, Pex14-GFP) strains were cultured as indicated in Materials and Methods to induce pexophagy. The protein samples were collected from the culture in YTO (SD-N 0 h) and 2 h after nitrogen starvation (SD-N). Western blots were probed with anti-YFP and anti-Pgk1 antibodies or antisera. (C) Quantitative analysis of the relative free GFP to total GFP (Pex14-GFP + free GFP) level. The ratio of wild-type cells after 2 h starvation was set to 1 and other samples were normalized accordingly. (D) Wild-type cells with an empty pRS313 vector (YLY205) and vps13Δ cells with a pRS313 vector bearing wild-type Vps13 (YLY209) or the temperature-sensitive mutant Vps13 (YLY211, two colonies were included) were cultured as indicated in Materials and Methods to induce mitophagy. Protein samples were collected from the culture in SML-His (SD-N 0 h) at 25°C and 6 h after nitrogen starvation (SD-N) at either 25°C or 37°C. Western blots were probed with anti-YFP and anti-Pgk1 antibodies or antisera. (E and F) Cells expressing wild-type Mcp1 (YLY110, Mcp1-PA) and cells with truncated Mcp1 (YLY111) or single mutant (YLY115) and double mutant Mcp1 (YLY116) were cultured as indicated in Materials and Methods to induce mitophagy. Protein samples were collected from the culture in YPL (SD-N 0 h) and 6 h after nitrogen starvation. Mitophagy activity was monitored with the mitoPho8Δ60 assay. The mitoPho8Δ60 activities were normalized to wild-type cells after 6 h starvation. The error bar represents the SD of three independent experiments. Two-tailed t test was used for statistical significance. ***p < 0.001, ns, not significant.
We next tested pexophagy, the selective autophagic degradation of peroxisomes, using the GFP-tagged peroxisome membrane protein Pex14 (Pex14-GFP). Before autophagy induction, cells grown in nutrient-rich medium containing glucose (YPD) were transferred to medium with glycerol and a minimal level of glucose (SGd), followed by 20-h growth in medium with oleic acid as the sole carbon source (YTO) to promote the proliferation of peroxisomes, which otherwise, are of very low abundance in S. cerevisiae. After starvation for 2 h, we observed much less Pex14-GFP cleavage in the vps13Δ cells, indicating a partial defect in pexophagy (Fig. 3B and 3C).
To test mitophagy activity in the vps13Δ strain, cells were first grown in YPD and were then cultured in medium containing the nonfermentable carbon source lactate (YPL) to promote mitochondria accumulation. After the cells are shifted to starvation medium (SD-N) containing glucose as the carbon source, the elevated population of mitochondria becomes unnecessary and mitophagy is activated to degrade the surplus organelles. To measure mitophagy activity, we tagged the mitochondrial matrix protein Idh1 with GFP at its C terminus (Idh1-GFP). We detected free GFP in the wild-type cells after 6 h of starvation but no free GFP in vps13Δ cells (Fig. S3A), which suggested a major deficiency or even a complete block of mitophagy in these cells. However, we noticed that these vps13Δ cells had a substantially longer doubling time in YPD medium compared to the wild-type cells and displayed nearly no growth in YPL medium. This phenotype is previously observed in cells with deletions for the genes encoding ERMES members and it is usually accompanied with defects in mitochondrial structure [33, 34]. Along these lines, in these vps13Δ cells, a lower Idh1 (Idh1-GFP) protein level after growing in YPL could also been seen (Fig. S3A). These phenotypes suggested that VPS13 deletion possibly leads to a functional deficiency in mitochondria; the organelle may not have proper biogenesis and therefore, may not accumulate when growing in YPL. To better understand the nature of the mitochondria in the mutant cells, we examined the Idh1-GFP and MitoTracker Red signal by fluorescence microscopy. In the wild-type cells, both Idh1-GFP and MitoTracker Red showed punctate and reticular structures (Fig. S3B); however, in vps13Δ cells, there were fewer Idh1-GFP puncta, and the MitoTracker Red signals were weaker and more diffuse, indicating fewer and less healthy mitochondria in these cells. We suggest that the mitophagy deficiency in these cells is not simply due to the deletion of VPS13 because expressing a plasmid bearing wild-type VPS13 could not rescue the slow growth or the mitophagy deficiency (Fig. S3C) and the expression of the plasmid bearing VPS13 was confirmed by quantitative PCR (data not shown).
During the construction of the vps13Δ mitophagy strains we noticed different growth rates among the colonies; these colonies were independent from each other, with neither colony of either phenotype obviously arising from the other. We first examined vps13Δ cells that grew normally and tested their mitophagy activity using two additional assays. In one of the assays, the mitochondrial outer membrane protein Om45 was tagged with GFP at its C terminus (Om45-GFP), and we observed similar free GFP levels between the wild-type and vps13Δ cells after a 6-h starvation (Fig. S3D). In the other assay, Pho8Δ60 was fused to a mitochondrial inner membrane protein, Cox4, targeting the chimera to the mitochondria [35]; this construct is sent to the vacuole as part of the mitochondrial cargo when mitophagy is induced, eventually allowing proteolytic activation of the Pho8Δ zymogen. Thereby, mitoPho8Δ60-dependent phosphatase activity reflects mitophagy activity. As expected, there was a clear increase in mitoPho8Δ60 activity after 6 h in SD-N conditions in the wild-type cells (Fig. S3E). Consistent with the Om45-GFP assay, we did not see an obvious difference in the mitoPho8Δ60 activity between wild-type and vps13Δ cells that came from colonies with a normal growth rate, suggesting that mitophagy in these cells was normal. In contrast, vps13Δ cells from slow-growing colonies displayed a consistent defect in mitophagy (data not shown). Considering these results together, we propose that because VPS13 is a very large gene and Vps13 has multiple localizations, some possibly not yet characterized, the deletion of this gene may result in unexpected consequences for the cells including a functional deficiency in mitochondria, which cannot be properly maintained. With fewer mitochondria and mitochondrial proteins in the cells, the readout of mitophagy, such as the free GFP level and mitoPho8Δ60 activity, may appear to be lower.
These different phenotypes seen in vps13Δ cells may explain the discrepancy we noticed between colonies concerning the role of Vps13 in mitophagy. However, this does not answer the question of whether Vps13 has a role in this process. To avoid the potential effect of VPS13 deletion on mitochondrial proliferation, we tested mitophagy activity in cells expressing a temperature-sensitive mutant VPS13 (vps13-9ts) [36], allowing us to conditionally inhibit the function of this protein. The temperature sensitivity of the mutant protein was first confirmed through the use of a colony blot Prc1/carboxypeptidase Y (CPY) assay, in which Vps13 loss of function leads to the secretion of Prc1/CPY, which can be retained on a nitrocellulose membrane and detected by immunoblot (Fig. S3F; Δ1 and Δ2 are two independent colonies expressing the conditional mutation) [37]. Secreted Prc1/CPY was not detected in the wild-type strain but was clearly seen in vps10Δ cells lacking the Prc1/CPY receptor, which was included as a positive control [38]. The vps13-9ts cells displayed a secretion phenotype similar to that of the vps10Δ strain indicating that the mutant Vps13 protein was inactivated under these conditions.
Next, we carried out a mitophagy assay and compared cells that had been cultured at a permissive temperature (25°C) and then starved under the permissive or non-permissive temperature (37°C) for 6 h. We observed that there was less free GFP at 37°C than at 25°C after starvation in vps13-9ts mutants; however, the reduced Idh1-GFP cleavage at 37°C was also seen in the cells expressing wild-type Vps13 or in the wild-type cells with an empty plasmid (Fig. 3D), suggesting that the decreased mitophagy activity was more likely due to the temperature rather than the loss of function of Vps13. It is important to note the possibility that the temperature-sensitive mutation only results in a partial loss of Vps13 function at the non-permissive temperature; however, these data when considered together with our findings that vps13Δ cells have a normal accumulation of mitochondria suggest that Vps13 does not play a significant role in mitophagy.
Vps13 and Mcp1 interaction is not important for mitophagy
Mcp1 is a mitochondrial outer membrane protein and interacts with Vps13 through its PxP motif at the N terminus [8]. Overexpressing Mcp1 can suppress the growth defects in ERMES mutant cells, which depends on the recruitment of Vps13 to mitochondria [39]. Because ERMES not only has an important tethering activity, but also has lipid exchange functions [29, 40], we decided to investigate whether Mcp1 and its interaction with Vps13 are critical for mitophagy. When creating mcp1Δ strains, we again observed an inconsistency in the colonies; some of the mcp1Δ cells grew slowly while some grew at the normal rate, and mcp1Δ cells with a slow growth rate showed an almost complete block in mitophagy (Fig. S3A and mcp1Δ-2 in Fig. S3D), whereas cells with a normal growth rate displayed mitophagy activities comparable to the wild-type cells (mcp1Δ-1 in Fig. S3D and Fig. S3E). The lower Idh1 and Om45 levels in the cells growing slowly again suggested a deficiency in mitochondrial biogenesis after growth in the nonfermentable medium (Fig. S3A and S3D). Consistent with the low expression, we noticed substantially fewer Idh1 puncta and weaker MitoTracker Red signals in the slow-growing mcp1Δ cells, indicating fewer and less healthy mitochondria (Fig. S3B). The morphology of mitochondria in these cells was also not the same as that in wild-type cells, with Om45 showing a diffuse signal rather than the punctate structures in the wild-type cells (mcp1Δ-2 in Fig. S3G). These observations suggest that the mitophagy blockage we saw in these cells was more likely due to the smaller population of mitochondria, which led to mitophagy signals (free GFP) that were too low to be detected.
To provide further information on the potential role of Mcp1 and Vps13 in mitophagy we decided to determine whether the interaction between these two proteins is important for this process. To disrupt the interaction between Mcp1 and Vps13, we truncated the first 3-12 amino acids, which includes the interacting PxP motif, or mutated the two prolines, P9 and P11 in the PxP motif to alanines. Both the truncation and mutation have been shown to weaken the Mcp1-Vps13 interaction [8]. In the cells with the putative binding motif truncated or mutated, we did not find mitoPho8Δ60 activities significantly different from wild-type cells (Fig. 3E, 3F, S3H and S3I), indicating mitophagy was not affected. We noted that there is another proline close to the PxP motif, at position 8. When we mutated all three prolines to alanines simultaneously (Mcp1P8,9,11A), we noticed a significant decrease in Mcp1 protein level (Fig. S3I), slower growth and deficient mitophagy activity (Fig. S3J). These data suggest that the triple mutation led to the instability and degradation of Mcp1, which prohibited normal mitochondrial proliferation and, thereby, the mitophagy deficiency in the triple mutant cells was less likely to have resulted from the loss of interaction with Vps13 per se. Therefore, we suggest that the interaction between Mcp1 and Vps13 is not important for efficient mitophagy.
Discussion
Studies from the past decades have revealed multiple membrane contact sites and they appear to be important for various aspects of cellular physiology [41]. Along these lines, membrane contact sites play critical roles in autophagy [42]. For instance, in mammalian cells, ER-mitochondria contact sites are important for autophagosome formation [43], and ERMES in yeast is critical for mitophagy and pexophagy [29, 30]. Vps13 is a very large protein that locates at membrane contact sites and is responsible for the lipid transport between membranes, so it is likely that Vps13 also functions in autophagy. Additionally, mutations in human VPS13 genes are highly correlated with human diseases and autophagy plays important roles in some of these diseases [44], which further pushed us to investigate whether Vps13 functions in autophagy. Here, we show that Vps13 is important for efficient non-selective autophagy (Fig. 1) and the deletion of VPS13 leads to the generation of more phagophores or autophagosomes that cannot fuse with the vacuole as evidenced by the relative accumulation of autophagosomes following a “pulse-chase” shift from SD-N to rich medium (Fig. 2A, B) and delayed disassociation of some early Atg proteins (Fig. 2D–G). Vps13 also functions in some types of selective autophagy, such as reticulophagy and pexophagy, but not in mitophagy (Fig. 3). Because the role of Vps13 in pexophagy has not been reported previously, it would be interesting to further explore the mechanism of Vps13 in this process. Pex6, Pex7 and Pex25 are three peroxisome proteins containing the putative Vps13-binding PxP motif [8]; these proteins may interact with Vps13 to obtain an efficient level of pexophagy activity.
Previous studies have attempted to determine the role of Vps13 in autophagy, and two previous studies show an increased level of mitophagy in vps13Δ cells [16, 17]. During our examination of mitophagy, we noticed discrepancies between colonies even though they theoretically had the same vps13Δ genotype. Because we could not rescue the phenotype by expressing a VPS13-bearing plasmid (Fig. S3C), we suggest that slowly growing colonies are not the result of only VPS13 deletion, even though the deletion of VPS13 has been suggested to result in a functional deficiency in some organelles, such as mitochondria in both yeast and mammalian cells and lipid droplets in mammalian cells [17, 18] together with a recent study identifying Vps13 as being responsible for peroxisome biogenesis in pex11Δ cells [45]. Instead, we propose that the damage of other genes when deleting VPS13 is more likely to be the explanation considering the very large size of this gene. Because the concomitant deficiency in mitochondria accumulation would affect the analysis of mitophagy assays, we bypassed the problems associated with the deletion of VPS13 by using a temperature-sensitive mutant and found that the loss of Vps13 function does not affect mitophagy activity, in contrast to what has been reported previously. Whole genome sequencing can be done to determine the cause of the slow growth phenotype, identifying which gene or genes are altered in these cells. These genes could be further investigated regarding their roles in mitochondria synthesis and integrity.
Even though in our study we found evidence supporting the conclusion that Vps13 functions in a late stage of autophagy, the precise mechanism needs further investigation. Because Vps13 is critical for lipid transport between membranes, it is possible that this protein directly participates in lipid transfer to the phagophore. However, Vps13 is also located at multiple membrane contact sites. Therefore, the deletion of the VPS13 gene may result in a change in lipid metabolism in one or more organelles, which will indirectly affect autophagy. This latter possibility can also explain why Vps13 functions in the general process of autophagy, but mitophagy is not affected by the absence of this protein; Vps13 could affect lipid transport and metabolism differently under the different growing conditions used for various selective autophagy assays. Another possible explanation for the participation of Vps13 in only some types of autophagy is that even though Vps13 plays a role in transporting lipids to the phagophore for its expansion, it is a minor contributor, and some other protein or protein complexes are also carrying out a similar function; in fact, a substantial amount of the membrane flow during starvation appears to be directed to autophagy. Thus, a complex such as ERMES, which is critical for mitophagy via its function in transporting lipids to the phagophore [29], or some other membrane junction proteins may be able to compensate for the loss of Vps13. Hence, the deletion of VPS13 or disrupting the interaction between Vps13 and Mcp1 do not lead to a clear change in mitophagy activity in the wild-type background. It would be interesting to see whether overexpressing Vps13 can rescue the mitophagy deficiency in ERMES mutant cells. In addition, some dominant mutations are found in VPS13 and expressing the mutant gene can rescue the growth defects of ERMES mutants [17, 46]. Thus, it is also worth trying to determine whether these mutants can rescue the mitophagy phenotype in these cells.
To further examine whether the lipid transport function of Vps13 is critical for autophagy, we can take advantage of several mutations within the Vps13 channel, which disrupt lipid transfer [10], and analyze their influence on autophagy activity. Additionally, recently published papers found that some corresponding disease-related mutations in VPS13 lead to the loss of its function in vacuolar sorting or in binding to adaptor proteins [47, 48]. These mutations may be interesting for further examination to explore their effect on autophagy, which will shed light on how Vps13 functions in this process, as well as on the relationship of these diseases to autophagy.
Material and Methods
Yeast strains, media and culture conditions
All yeast strains used are listed in Table S1. Gene deletions were generated according to standard methods [49]. Yeast cells were cultured at 30°C in rich (YPD; 1% yeast extract, 2% peptone, and 2% glucose) or synthetic minimal medium (SMD; 0.67% yeast nitrogen base, 2% glucose, and auxotrophic amino acids and vitamins as needed) as indicated. For mitochondria proliferation, cells were grown in lactate medium (YPL; 1% yeast extract, 2% peptone, and 2% lactic acid) or synthetic minimal medium with lactic acid (SML; 0.67% yeast nitrogen base, 2% lactic acid, and auxotrophic amino acids and vitamins as needed) for 18 h. For peroxisomal proliferation, cells were shifted to glycerol medium (SGd; 0.67% yeast nitrogen base, 0.1% glucose, and 3% glycerol) for 16 h after growth in YPD to OD600 of approximately 0.5. Then, yeast extract and peptone were added into the cultures in the SGd medium, and cells were cultured for 4 h. Then the cells were shifted to oleic acid medium (YTO; 0.67% yeast nitrogen base, 0.1% oleic acid, 0.1% Tween 40 and auxotrophic amino acids as needed) for 20 h. To induce autophagy, cells in mid-log phase (OD600 = 0.4-0.5 for the prApe1 processing assay and OD600 = 0.8-1.0 for other autophagy assays) were shifted to nitrogen starvation medium with glucose (SD-N; 0.17% yeast nitrogen base without ammonium sulfate or amino acids, and 2% glucose) for the indicated times. For the strains with temperature-sensitive proteins, cells were cultured at a permissive temperature (25°C) before shifting to a restrictive temperature for the indicated time.
Plasmids
The pRS405-CUP1p-GFP-ATG8 plasmid was constructed by replacing the endogenous ATG8 promoter with the CUP1 promoter and contains the open reading frame of GFP-Atg8 [50]. The plasmid was linearized and integrated into the corresponding strains. pRS313 plasmids expressing wild-type VPS13 with the endogenous promoter and terminator used in the rescue experiment or temperature-sensitive Vps13 (vps13-9ts) are gifts from Dr. Roberta Fuller, University of Michigan. The construction of the temperature-sensitive VPS13 plasmid is described elsewhere [36].
Antibodies and antisera
Anti-YFP antibody, which detects GFP, was from Clontech (632381). Anti-PA antibody was from Jackson Immuno Research (323-005-024). Anti-Dpm1 antibody was from Molecular Probes/Fisher (A6429). Anti-Pgk1 antiserum is a generous gift from Dr. Jeremy Thorner (University of California, Berkeley). Prc1/CPY antibody was from Invitrogen (A-6428). Antibodies to Ape1 were described previously [51]. Goat anti-rabbit IgG secondary antibody (Fisher, ICN55676) and rabbit anti-mouse IgG secondary antibody (Jackson, 315-035-003) were from commercial sources.
Fluorescence microscopy
The images of GFP-Atg8, Atg16-GFP and Atg14-GFP puncta were collected on a Deltavision Elite deconvolution microscope (GEHealthcare/Applied Precision) with a 100× objective and CCD camera (CoolSnap HQ; Photometrics). For puncta quantification, stacks of 15 image planes were collected with a spacing of 0.3 μm to cover the entire cells.
For mitochondria imaging, the cells were stained with 50 nM MitoTracker Red CMXRos (Invitrogen, M7512) for 30 min. Images of mitochondria and Atg18-GFP puncta were collected on a Leica DMi8 microscope with a 100× objective and Leica Thunder imager. For Atg18-GFP, stacks of 51 image planes were collected with a spacing of 0.2 μm to cover the entire cell. For mitochondria imaging, single z-sections of the representative image were shown.
Prc1/CPY secretion assay
After overnight growth to mid-log phase in SMD-His or YPD media at 25°C, yeast cells were collected and diluted to 1.0 OD600/ml and five-fold serial dilutions were made in water. Two μl of each dilution were spotted on a YPD plate and a nitrocellulose membrane was placed on the colonies. The colonies were incubated with the membrane at either 37°C overnight or 25°C for another day due to the slower growth under the lower temperature. The membranes were rinsed with distilled water to remove the visible colonies and then placed in TBST (20 mM Tris, 150 mM NaCl, 0.1% Tween 20) and washed for 15 min. The membranes were then probed for secreted Prc1/CPY using anti-Prc1/CPY antibody overnight at 4°C then with rabbit anti-mouse IgG secondary antibody 1 h at room temperature. The signal was detected with a ChemiDoc Imaging System (Bio-Rad).
Protease-protection assay
Cells were cultured in 50 ml YPD overnight until mid-log phase and then transferred to nitrogen starvation medium (SD-N) for 3 h. Cells (50 OD600 units) were harvested by centrifugation at 3000 rpm for 5 min. After washing the cells with H2O, the cells were resuspended in 25 mL freshly made DTT buffer (10 mM Tris, pH 9.4, 10 mM DTT) and cultured on a platform shaker at room temperature for 30 min while shaking at 150 rpm. The cells were collected and washed with H2O twice and were then resuspended in 5 ml SP buffer (1 M sorbitol, 20 mM PIPES, pH 6.8). After adding 0.24 mg zymolyase 100T (previously dissolved in the SP buffer), the cells were incubated at 30°C while shaking at 180 rpm for 30 min to obtain spheroplasts. The spheroplasts were loaded on top of 5 ml of 1.8 M sorbitol in a 15-ml tube, centrifuged for 10 min at 2000 rpm and the supernatant discarded. The spheroplasts were then gently resuspended in 5 ml PS200 buffer (20 mM PIPES, pH 6.8, 200 mM sorbitol, 5 mM MgCl2). Next, the spheroplasts were homogenized by forcing them through a 3.0-μm Nucleopore Track-Etch membrane (Whatman, 110612). Homogenates were collected in a pre-cooled 15-ml tube then centrifuged at 2000 rpm for 10 min twice to remove the cellular debris. The supernatant was divided into four 1-ml aliquots then centrifuged at 5000 g for 5 min at 4°C. The pellets were resuspended in 300 μl PS200 containing the following: (1) PS200 alone, (2) 0.2% Triton X-100, (3) 40 μg/ml proteinase K, (4) 0.2% Triton X-100 and 40 μg/ml proteinase K and incubated on ice for 30 min. To stop the reaction, 33 μL TCA was added to the samples. The samples were thoroughly mixed by vortex and incubated on ice for 30 min. After centrifuging the samples at 13,000 rpm for 10 min, the pellets were washed with acetone twice, air dried, resuspended in 50 μl MURB buffer (50 mM sodium phophate buffer, pH 7.0, 25 mM MES, 1% SDS [w:v], 3 M urea, 1 mM NaN3, 1% β-mercaptoethanol, 0.01% bromophenol blue) and heated for 15 min at 55°C. Three μl of the sample were loaded for SDS-PAGE, and western blotting using anti-Ape1 antibody was performed to detect processed and unprocessed prApe1.
Other methods
Western blot, GFP-Atg8 processing and prApe1 processing assays were described previously [21].
Quantification and statistical analyses
Western blots were quantified by ImageJ or Bio-Rad ImageLab software. Two-tailed t test was used to determine statistical significance.
Supplementary Material
Figure S1. Vps13 is important for efficient autophagy. Protein samples were collected from (A and B) wild-type (YLY085) and vps13Δ cells (YLY086) with the indicated pRS313 plasmids or (C and D) from wild-type cells (YLY085) and cells with most of VPS13 deleted (WXY222) after growth in YPD to mid-log phase (SD-N 0 h) and (A and C) 2 h or (B and D) 4 h after nitrogen starvation. Western blots were probed and Pho8Δ60 was quantified as indicated in Figure 1. **p<0.01, ***p<0.001, ns, not significant. Related to Fig. 1.
Figure S2. Autophagosome completion is not affected by VPS13 deletion. (A) Schematic representation of the protease-protection assay. (B) Wild-type (YLY085), vps13Δ (YLY086), ypt7Δ (YLY162) and atg9Δ (YLY164) cells were cultured in YPD to mid-log phase and then transferred to nitrogen starvation medium (SD-N) for 3 h. A protease-protein assay was conducted as described in the Materials and Methods. (C and D) Wild-type (UNY176, Atg14-GFP) and vps13Δ (YLY145, Atg14-GFP) cell samples were collected 1.5 h after nitrogen starvation (SD-N) and imaged. The total puncta to total cell number ratio was quantified and normalized to wild-type cells. In the quantitative analysis, the error bar represents the SD of three independent experiments. DIC: differential interference contrast. Related to Fig. 2.
Figure S3. Neither Vps13 nor the interaction between Vps13 and Mcp1 is important for mitophagy. (A) Wild-type (YLY068, Idh1-GFP), vps13Δ (YLY123, Idh1-GFP) and mcp1Δ (YLY124, Idh1-GFP) cells were cultured as indicated in Materials and Methods to induce mitophagy. Protein samples were collected from the culture in YPL (SD-N 0 h) and 6 h after nitrogen starvation. Western blots were probed as indicated in Figure 3C. (B) The same cells as in (A) were cultured in YPD to mid-log phase, shifted to YPL for 18 h and stained with 50 nM MitoTracker Red (MTR) for 30 min before imaging. Single Z-sections of representative images were shown. (C) Wild-type (YLY068) or vps13Δ cells (YLY123) with an empty pRS313 vector or pRS313 vector bearing wild-type Vps13 were cultured as indicated in the Materials and Methods to induce mitophagy. Protein samples were collected from the culture in SML-His (SD-N 0 h) and 6 h after nitrogen starvation (SD-N). Western blots were probed with anti-YFP and anti-Pgk1 antibodies or antisera. (D) Wild-type (TKYM23, Om45-GFP), vps13Δ (YLY083, Om45-GFP) and mcp1Δ (YLY084, Om45-GFP; two different colonies were included) cells were cultured as indicated in Materials and Methods to induce mitophagy. Two mcp1Δ colonies have the same apparent genotype, but mcp1Δ-2 cells grew slower, and the mcp1Δ-1 cells grew at the normal rate. Protein samples were collected from the culture in YPL (SD-N 0 h) and 6 h after nitrogen starvation. Western blots were probed as indicated in (C). (E) Wild-type (KDM1009), vps13Δ (YLY087), mcp1Δ (YLY088) and atg1Δ (KDM1024, as control) strains were cultured as described in Materials and Methods to induce mitophagy. Protein samples were collected from the culture in YPL (SD-N 0 h) and 6 h after nitrogen starvation. mitoPho8Δ60 was used to measured mitophagy activity. mitoPho8Δ60 activity was quantified as indicated in Figure 3. (F) The same cells as in Fig. 3D and vps10Δ (YLY279, as a control) cells were cultured in SMD-His or YPD at 25°C to mid-log phase and a Prc1/CPY secretion assay was performed as described in Materials and Methods. (G) The same cells in (D) were cultured in YPD to mid-log phase and shifted to YPL for 18 h before imaging. Single Z-sections of representative images are shown. (H) Wild-type (YLY110 Mcp1-PA), MCP1 single mutant (YLY115, two colonies were included) and double mutant (YLY116, two colonies were included) cells or (I) MCP1 truncated (YLY111) and triple mutant cells (YLY112; P3A) cells were cultured as indicated in Materials and Methods to induce mitophagy. Protein samples were collected from the culture in YPL (SD-N 0 h) and 6 h after nitrogen starvation. Western blots were probed with anti-PA and anti-Dpm1 antibodies. (J) Wild-type (YLY110, Mcp1-PA), MCP1 triple mutant cells (YLY112) and atg1Δ (KDM1024, as control) cells were cultured as described in Materials and Methods to induce mitophagy. Protein samples were collected from the culture in YPL (SD-N 0 h) and 6 h after nitrogen starvation. mitoPho8Δ60 was used to measured mitophagy activity. mitoPho8Δ60 activity was quantified as indicated in Figure 3. Related to Fig. 3.
Acknowledgements
This work was supported by NIH grant GM131919 to DJK.
Abbreviations:
- Ape1
aminopeptidase I
- ER
endoplasmic reticulum
- ERMES
ER-mitochondria encounter structure
- GPF
green fluorescent protein
- prApe1
precursor Ape1
- PxP
proline-X-proline
- SD-N
synthetic medium containing dextrose/glucose, and lacking nitrogen
- SGd
synthetic medium containing glycerol and a low level of dextrose/glucose
- VAB
Vps13 adaptor binding
- YPD
nutrient-rich medium containing yeast extract, peptone and dextrose/glucose
- YPL
nutrient-rich medium containing yeast extract, peptone and lactate
- YTO
nutrient-rich medium containing yeast extract, Tween 40, and oleic acid
References
- [1].Velayos-Baeza A, Vettori A, Copley RR, Dobson-Stone C, Monaco AP. Analysis of the human VPS13 gene family. Genomics 2004; 84:536–49. [DOI] [PubMed] [Google Scholar]
- [2].Rampoldi L, Dobson-Stone C, Rubio JP, Danek A, Chalmers RM, Wood NW, et al. A conserved sorting-associated protein is mutant in chorea-acanthocytosis. Nat Genet 2001; 28:119–20. [DOI] [PubMed] [Google Scholar]
- [3].Tomiyasu A, Nakamura M, Ichiba M, Ueno S, Saiki S, Morimoto M, et al. Novel pathogenic mutations and copy number variations in the VPS13A gene in patients with chorea-acanthocytosis. Am J Med Genet B Neuropsychiatr Genet 2011; 156B:620–31. [DOI] [PubMed] [Google Scholar]
- [4].Balikova I, Lehesjoki AE, de Ravel TJ, Thienpont B, Chandler KE, Clayton-Smith J, et al. Deletions in the VPS13B (COH1) gene as a cause of Cohen syndrome. Hum Mutat 2009; 30:E845–54. [DOI] [PubMed] [Google Scholar]
- [5].Lesage S, Drouet V, Majounie E, Deramecourt V, Jacoupy M, Nicolas A, et al. Loss of VPS13C Function in Autosomal-Recessive Parkinsonism Causes Mitochondrial Dysfunction and Increases PINK1/Parkin-Dependent Mitophagy. Am J Hum Genet 2016; 98:500–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Durand CM, Angelini C, Michaud V, Delleci C, Coupry I, Goizet C, et al. Whole-exome sequencing confirms implication of VPS13D as a potential cause of progressive spastic ataxia. BMC Neurol 2022; 22:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Dziurdzik SK, Conibear E. The Vps13 Family of Lipid Transporters and Its Role at Membrane Contact Sites. Int J Mol Sci 2021; 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Bean BDM, Dziurdzik SK, Kolehmainen KL, Fowler CMS, Kwong WK, Grad LI, et al. Competitive organelle-specific adaptors recruit Vps13 to membrane contact sites. J Cell Biol 2018; 217:3593–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Kumar N, Leonzino M, Hancock-Cerutti W, Horenkamp FA, Li P, Lees JA, et al. VPS13A and VPS13C are lipid transport proteins differentially localized at ER contact sites. J Cell Biol 2018; 217:3625–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Li P, Lees JA, Lusk CP, Reinisch KM. Cryo-EM reconstruction of a VPS13 fragment reveals a long groove to channel lipids between membranes. J Cell Biol 2020; 219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Adlakha J, Hong Z, Li P, Reinisch KM. Structural and biochemical insights into lipid transport by VPS13 proteins. J Cell Biol 2022; 221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Lei Y, Huang Y, Wen X, Yin Z, Zhang Z, Klionsky DJ. How Cells Deal with the Fluctuating Environment: Autophagy Regulation under Stress in Yeast and Mammalian Systems. Antioxidants (Basel) 2022; 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Gatica D, Lahiri V, Klionsky DJ. Cargo recognition and degradation by selective autophagy. Nat Cell Biol 2018; 20:233–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Feng Y, He D, Yao Z, Klionsky DJ. The machinery of macroautophagy. Cell Res 2014; 24:24–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Munoz-Braceras S, Calvo R, Escalante R. TipC and the chorea-acanthocytosis protein VPS13A regulate autophagy in Dictyostelium and human HeLa cells. Autophagy 2015; 11:918–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Chen S, Mari M, Parashar S, Liu D, Cui Y, Reggiori F, et al. Vps13 is required for the packaging of the ER into autophagosomes during ER-phagy. Proc Natl Acad Sci U S A 2020; 117:18530–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Park JS, Thorsness MK, Policastro R, McGoldrick LL, Hollingsworth NM, Thorsness PE, et al. Yeast Vps13 promotes mitochondrial function and is localized at membrane contact sites. Mol Biol Cell 2016; 27:2435–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Yeshaw WM, van der Zwaag M, Pinto F, Lahaye LL, Faber AI, Gomez-Sanchez R, et al. Human VPS13A is associated with multiple organelles and influences mitochondrial morphology and lipid droplet motility. Elife 2019; 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Anding AL, Wang C, Chang TK, Sliter DA, Powers CM, Hofmann K, et al. Vps13D Encodes a Ubiquitin-Binding Protein that Is Required for the Regulation of Mitochondrial Size and Clearance. Curr Biol 2018; 28:287–95 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Shen JL, Fortier TM, Zhao YG, Wang R, Burmeister M, Baehrecke EH. Vmp1, Vps13D, and Marf/Mfn2 function in a conserved pathway to regulate mitochondria and ER contact in development and disease. Curr Biol 2021; 31:3028–39 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Klionsky DJ, Abdel-Aziz AK, Abdelfatah S, Abdellatif M, Abdoli A, Abel S, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition)(1). Autophagy 2021; 17:1–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Osawa T, Kotani T, Kawaoka T, Hirata E, Suzuki K, Nakatogawa H, et al. Atg2 mediates direct lipid transfer between membranes for autophagosome formation. Nat Struct Mol Biol 2019; 26:281–8. [DOI] [PubMed] [Google Scholar]
- [23].Chowdhury S, Otomo C, Leitner A, Ohashi K, Aebersold R, Lander GC, et al. Insights into autophagosome biogenesis from structural and biochemical analyses of the ATG2A-WIPI4 complex. Proc Natl Acad Sci U S A 2018; 115:E9792–E801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Kuchitsu Y, Fukuda M. Revisiting Rab7 Functions in Mammalian Autophagy: Rab7 Knockout Studies. Cells 2018; 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Matoba K, Kotani T, Tsutsumi A, Tsuji T, Mori T, Noshiro D, et al. Atg9 is a lipid scramblase that mediates autophagosomal membrane expansion. Nat Struct Mol Biol 2020; 27:1185–93. [DOI] [PubMed] [Google Scholar]
- [26].Yang Z, Klionsky DJ. An overview of the molecular mechanism of autophagy. Curr Top Microbiol Immunol 2009; 335:1–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Romanov J, Walczak M, Ibiricu I, Schuchner S, Ogris E, Kraft C, et al. Mechanism and functions of membrane binding by the Atg5-Atg12/Atg16 complex during autophagosome formation. EMBO J 2012; 31:4304–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Obara K, Sekito T, Niimi K, Ohsumi Y. The Atg18-Atg2 complex is recruited to autophagic membranes via phosphatidylinositol 3-phosphate and exerts an essential function. J Biol Chem 2008; 283:23972–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Bockler S, Westermann B. Mitochondrial ER contacts are crucial for mitophagy in yeast. Dev Cell 2014; 28:450–8. [DOI] [PubMed] [Google Scholar]
- [30].Liu X, Wen X, Klionsky DJ. ER-mitochondria contacts are required for pexophagy in Saccharomyces cerevisiae. Contact (Thousand Oaks) 2018; 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Wang YX, Catlett NL, Weisman LS. Vac8p, a vacuolar protein with armadillo repeats, functions in both vacuole inheritance and protein targeting from the cytoplasm to vacuole. J Cell Biol 1998; 140:1063–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Mochida K, Oikawa Y, Kimura Y, Kirisako H, Hirano H, Ohsumi Y, et al. Receptor-mediated selective autophagy degrades the endoplasmic reticulum and the nucleus. Nature 2015; 522:359–62. [DOI] [PubMed] [Google Scholar]
- [33].Burgess SM, Delannoy M, Jensen RE. MMM1 encodes a mitochondrial outer membrane protein essential for establishing and maintaining the structure of yeast mitochondria. J Cell Biol 1994; 126:1375–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Youngman MJ, Hobbs AE, Burgess SM, Srinivasan M, Jensen RE. Mmm2p, a mitochondrial outer membrane protein required for yeast mitochondrial shape and maintenance of mtDNA nucleoids. J Cell Biol 2004; 164:677–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Yao Z, Liu X, Klionsky DJ. MitoPho8Delta60 Assay as a Tool to Quantitatively Measure Mitophagy Activity. Methods Mol Biol 2018; 1759:85–93. [DOI] [PubMed] [Google Scholar]
- [36].Brickner J Characterization of the mechanism of localization of membrane proteins to the trans Golgi network. PhD thesis. Biochemistry. Palo Alto: Stanford University, 1998. [Google Scholar]
- [37].Bonangelino CJ, Chavez EM, Bonifacino JS. Genomic screen for vacuolar protein sorting genes in Saccharomyces cerevisiae. Mol Biol Cell 2002; 13:2486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Marcusson EG, Horazdovsky BF, Cereghino JL, Gharakhanian E, Emr SD. The sorting receptor for yeast vacuolar carboxypeptidase Y is encoded by the VPS10 gene. Cell 1994; 77:579–86. [DOI] [PubMed] [Google Scholar]
- [39].John Peter AT, Herrmann B, Antunes D, Rapaport D, Dimmer KS, Kornmann B. Vps13-Mcp1 interact at vacuole-mitochondria interfaces and bypass ER-mitochondria contact sites. J Cell Biol 2017; 216:3219–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Kornmann B, Currie E, Collins SR, Schuldiner M, Nunnari J, Weissman JS, et al. An ER-mitochondria tethering complex revealed by a synthetic biology screen. Science 2009; 325:477–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Prinz WA, Toulmay A, Balla T. The functional universe of membrane contact sites. Nat Rev Mol Cell Biol 2020; 21:7–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Kohler V, Aufschnaiter A, Buttner S. Closing the Gap: Membrane Contact Sites in the Regulation of Autophagy. Cells 2020; 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Hamasaki M, Furuta N, Matsuda A, Nezu A, Yamamoto A, Fujita N, et al. Autophagosomes form at ER-mitochondria contact sites. Nature 2013; 495:389–93. [DOI] [PubMed] [Google Scholar]
- [44].Lei Y, Klionsky DJ. The Emerging Roles of Autophagy in Human Diseases. Biomedicines 2021; 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Yuan W, Aksit A, de Boer R, Krikken AM, van der Klei IJ. Yeast Vps13 is Crucial for Peroxisome Expansion in Cells With Reduced Peroxisome-ER Contact Sites. Front Cell Dev Biol 2022; 10:842285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Lang AB, John Peter AT, Walter P, Kornmann B. ER-mitochondrial junctions can be bypassed by dominant mutations in the endosomal protein Vps13. J Cell Biol 2015; 210:883–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Dziurdzik SK, Bean BDM, Davey M, Conibear E. A VPS13D spastic ataxia mutation disrupts the conserved adaptor-binding site in yeast Vps13. Hum Mol Genet 2020; 29:635–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Park JS, Hollingsworth NM, Neiman AM. Genetic Dissection of Vps13 Regulation in Yeast Using Disease Mutations from Human Orthologs. Int J Mol Sci 2021; 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Gueldener U, Heinisch J, Koehler GJ, Voss D, Hegemann JH. A second set of loxP marker cassettes for Cre-mediated multiple gene knockouts in budding yeast. Nucleic Acids Res 2002; 30:e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Geng J, Nair U, Yasumura-Yorimitsu K, Klionsky DJ. Post-Golgi Sec proteins are required for autophagy in Saccharomyces cerevisiae. Mol Biol Cell 2010; 21:2257–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Klionsky DJ, Cueva R, Yaver DS. Aminopeptidase I of Saccharomyces cerevisiae is localized to the vacuole independent of the secretory pathway. J Cell Biol 1992; 119:287–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Vps13 is important for efficient autophagy. Protein samples were collected from (A and B) wild-type (YLY085) and vps13Δ cells (YLY086) with the indicated pRS313 plasmids or (C and D) from wild-type cells (YLY085) and cells with most of VPS13 deleted (WXY222) after growth in YPD to mid-log phase (SD-N 0 h) and (A and C) 2 h or (B and D) 4 h after nitrogen starvation. Western blots were probed and Pho8Δ60 was quantified as indicated in Figure 1. **p<0.01, ***p<0.001, ns, not significant. Related to Fig. 1.
Figure S2. Autophagosome completion is not affected by VPS13 deletion. (A) Schematic representation of the protease-protection assay. (B) Wild-type (YLY085), vps13Δ (YLY086), ypt7Δ (YLY162) and atg9Δ (YLY164) cells were cultured in YPD to mid-log phase and then transferred to nitrogen starvation medium (SD-N) for 3 h. A protease-protein assay was conducted as described in the Materials and Methods. (C and D) Wild-type (UNY176, Atg14-GFP) and vps13Δ (YLY145, Atg14-GFP) cell samples were collected 1.5 h after nitrogen starvation (SD-N) and imaged. The total puncta to total cell number ratio was quantified and normalized to wild-type cells. In the quantitative analysis, the error bar represents the SD of three independent experiments. DIC: differential interference contrast. Related to Fig. 2.
Figure S3. Neither Vps13 nor the interaction between Vps13 and Mcp1 is important for mitophagy. (A) Wild-type (YLY068, Idh1-GFP), vps13Δ (YLY123, Idh1-GFP) and mcp1Δ (YLY124, Idh1-GFP) cells were cultured as indicated in Materials and Methods to induce mitophagy. Protein samples were collected from the culture in YPL (SD-N 0 h) and 6 h after nitrogen starvation. Western blots were probed as indicated in Figure 3C. (B) The same cells as in (A) were cultured in YPD to mid-log phase, shifted to YPL for 18 h and stained with 50 nM MitoTracker Red (MTR) for 30 min before imaging. Single Z-sections of representative images were shown. (C) Wild-type (YLY068) or vps13Δ cells (YLY123) with an empty pRS313 vector or pRS313 vector bearing wild-type Vps13 were cultured as indicated in the Materials and Methods to induce mitophagy. Protein samples were collected from the culture in SML-His (SD-N 0 h) and 6 h after nitrogen starvation (SD-N). Western blots were probed with anti-YFP and anti-Pgk1 antibodies or antisera. (D) Wild-type (TKYM23, Om45-GFP), vps13Δ (YLY083, Om45-GFP) and mcp1Δ (YLY084, Om45-GFP; two different colonies were included) cells were cultured as indicated in Materials and Methods to induce mitophagy. Two mcp1Δ colonies have the same apparent genotype, but mcp1Δ-2 cells grew slower, and the mcp1Δ-1 cells grew at the normal rate. Protein samples were collected from the culture in YPL (SD-N 0 h) and 6 h after nitrogen starvation. Western blots were probed as indicated in (C). (E) Wild-type (KDM1009), vps13Δ (YLY087), mcp1Δ (YLY088) and atg1Δ (KDM1024, as control) strains were cultured as described in Materials and Methods to induce mitophagy. Protein samples were collected from the culture in YPL (SD-N 0 h) and 6 h after nitrogen starvation. mitoPho8Δ60 was used to measured mitophagy activity. mitoPho8Δ60 activity was quantified as indicated in Figure 3. (F) The same cells as in Fig. 3D and vps10Δ (YLY279, as a control) cells were cultured in SMD-His or YPD at 25°C to mid-log phase and a Prc1/CPY secretion assay was performed as described in Materials and Methods. (G) The same cells in (D) were cultured in YPD to mid-log phase and shifted to YPL for 18 h before imaging. Single Z-sections of representative images are shown. (H) Wild-type (YLY110 Mcp1-PA), MCP1 single mutant (YLY115, two colonies were included) and double mutant (YLY116, two colonies were included) cells or (I) MCP1 truncated (YLY111) and triple mutant cells (YLY112; P3A) cells were cultured as indicated in Materials and Methods to induce mitophagy. Protein samples were collected from the culture in YPL (SD-N 0 h) and 6 h after nitrogen starvation. Western blots were probed with anti-PA and anti-Dpm1 antibodies. (J) Wild-type (YLY110, Mcp1-PA), MCP1 triple mutant cells (YLY112) and atg1Δ (KDM1024, as control) cells were cultured as described in Materials and Methods to induce mitophagy. Protein samples were collected from the culture in YPL (SD-N 0 h) and 6 h after nitrogen starvation. mitoPho8Δ60 was used to measured mitophagy activity. mitoPho8Δ60 activity was quantified as indicated in Figure 3. Related to Fig. 3.