

Abstract
With the combination of KRAS G12C and EGFR inhibitors, KRAS is becoming a druggable target in colorectal cancer. However, secondary resistance limits its efficacy. Using cell lines, patient-derived xenografts, and patient samples, we detected a heterogeneous pattern of putative resistance alterations expected primarily to prevent inhibition of ERK signaling by drugs at progression. Serial analysis of patient blood samples on treatment demonstrates that most of these alterations are detected at a low frequency except for KRAS G12C amplification, a recurrent resistance mechanism that rises in step with clinical progression. Upon drug withdrawal, resistant cells with KRAS G12C amplification undergo oncogene-induced senescence, and progressing patients experience a rapid fall in levels of this alteration in circulating DNA. In this new state, drug resumption is ineffective as mTOR signaling is elevated. However, our work exposes a potential therapeutic vulnerability, whereby therapies that target the senescence response may overcome acquired resistance.
Introduction
KRAS is the most mutated oncogene in human cancer(1). It acts as a signaling switch that, when bound to GTP, orchestrates a program of cell proliferation and survival. Until recently, efforts to target KRAS have been unsuccessful due to its small binding pocket, high affinity for GTP, and redundant mechanisms of posttranslational processing. The development of allele specific KRAS G12C inhibitors that trap KRAS in the inactive, GDP-bound state(2,3) led to a paradigm change, with clinical responses in 30-50% of non-small cell lung cancer (NSCLC) patients harboring KRAS G12C mutations(4,5).
These agents are not as effective in colorectal cancers (CRC) with KRAS G12C mutation. We have previously shown that the activity of these drugs in KRAS G12C CRC is limited because activation of epidermal growth factor receptor (EGFR) reactivates ERK signaling and consequently combinatorial KRAS G12C and EGFR inhibition more effectively targets KRAS G12C CRC(6). Early trial data provide clinical support for this observation: the response rate for sotorasib was 7-10% in CRC(7) and, in the first report of sotorasib plus the EGFR antibody panitumumab, the response rate was 27%(8). For adagrasib monotherapy, it was ~20% and, for adagrasib with the EGFR antibody cetuximab, it increased to ~40%(9). Based on these data, combination treatments based on KRAS G12C inhibitors and EGFR antibodies are being evaluated in registrational, phase 3 trials.
Nonetheless, patients treated with these agents eventually acquire resistance and the response to single agent or combination treatment is brief. Several studies have characterized resistance to KRAS G12C monotherapy(10–12). Remarkably, these alterations are highly heterogeneous, including KRAS, BRAF, or MEK mutations, as well as gene amplifications and fusions, and circulating tumor DNA (ctDNA) analysis typically identifies multiple resistance alterations in the same patient. Here we sought to determine the landscape of genetic mechanisms of resistance to EGFR-KRAS G12C inhibition in gastrointestinal cancer and to identify novel approaches to potentially overcome resistance.
Results
Mechanisms of resistance to combined KRAS G12C and EGFR inhibition
To identify mechanisms of resistance to the combination of KRAS G12C and EGFR inhibitors, we grew the CRC cell lines C106 and RW7213, both of which are sensitive to this treatment(6) in drugs until the emergence of secondary resistance (Fig. 1A). Treatment with sotorasib 3μM and cetuximab 50μg/mL led to massive cell death of both cell lines with few viable cells. Cells were therefore subjected to increasing doses of sotorasib (from 0.1μM to 3μM) with cetuximab 50μg/mL to generate resistance (Supplementary Fig. S1A). Resistant sublines grew well in drugs after a period of 4 months for C106 cells and 2 months for RW7213 cells (Supplementary Figs. S1A–S1C).
Figure 1. Mechanisms of resistance to combined KRAS G12C and EGFR inhibition in colorectal cancer.
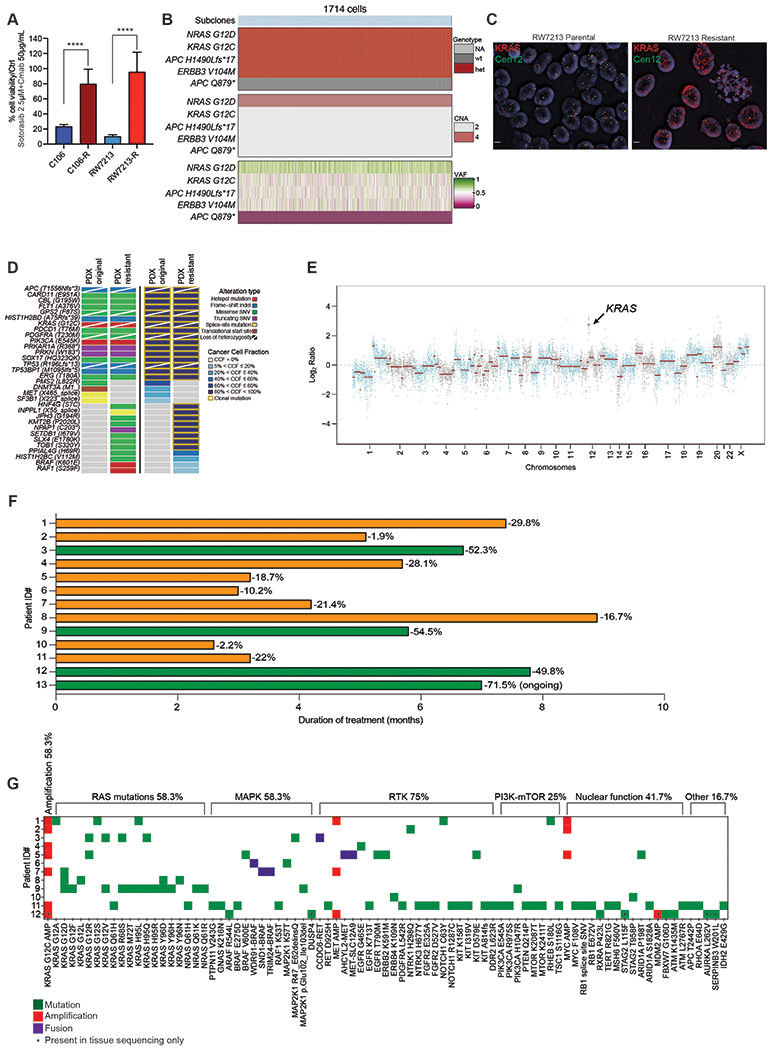
A. Graph showing cell viability of parental and resistant C106 and RW7213 cells. Statistical analyses and P-values represent Mann Whitney Test (T-Test), ****= P-value ≤0.0001. B. Heatmap of KRAS G12C and NRAS G12D alleles detected by single cell sequencing of C106 resistant subline. VAF: variant allelic frequency; GQ: genotyping quality score from GATK; DP: sequencing depth. C. FISH staining for KRAS gene in RW7213 parental and resistant subline. Manual review of parental RW7213 cells indicated no amplification (mean KRAS (red)/Cen12 (green) ratio of 1.1; 50 cells counted) in approximately 90% of the hybridized area and approximately 10% hybridized area with increased KRAS copies (mean red/green ratio of 3.5; 50 cells counted). Mean red/green ratio in the resistant subline, based on manual counting of 20 cells, was 6.4 with >20 KRAS (red) signals in all cells. Scale bars 5μm. D. Nonsynonymous somatic mutations identified by MSK-IMPACT in the CLR-113 original and resistant PDX. Mutation types (left) and cancer cell fraction (CCF) of mutations identified (right) are color coded according to the legend. E. Copy-number alterations (CNAs) of the CLR-113 original and resistant PDX (top). Copy-number log2 ratios are shown on the y-axis according to the chromosomes on the x-axis. The arrow shows KRAS amplification. F. Plot showing duration of response to KRAS G12C inhibitor (adagrasib/sotorasib) plus EGFR inhibitor (cetuximab/panitumumab) by patient ID number. Best response by RECIST is noted at the end of each bar, and partial responses are shaded green and stable disease shaded orange. G. Oncoprint of emergent alterations detected in circulating tumor DNA (ctDNA) of CRC patients at time of radiographic or clinical progression through combined KRAS G12C and EGFR inhibition. Patient 12 had both ctDNA and tumor tissue analyzed at progression, and emergent alterations identified only in tissue are marked with an asterisk.
Both resistant sublines expressed higher RAS-GTP levels than parental cells and drug treatment led to incomplete inhibition of RAS-GTP (Supplementary Fig. S1D and S1E). Drug treatment of C106 resistant cells failed to suppress activity of downstream effectors in the RAS/ERK pathway, whereas RW7213 resistant cells experienced a reduction in pathway activity but continued to have high levels of phospho-MEK and phospho-ERK due to elevated baseline pathway activation. Targeted sequencing of the resistant sublines using MSK-IMPACT(13) identified acquired clonal NRAS G12D mutation and subclonal APC Q879* nonsense mutation in C106 resistant cells and amplification of KRAS G12C in RW7213 resistant cells, which have a homozygous (through loss of heterozygosity) and clonal KRAS G12C mutation (Fig. 1B; Supplementary Figs. S2A–S2E and S3A and S3B). Single cell sequencing of the C106 resistant subline indicated that the NRAS G12D mutation occurred in the same cells bearing the KRAS G12C alteration (Fig. 1B), with an acquired gain of the mutant NRAS allele occurring in all cells (Fig. 1B). In addition, the subclonal APC Q879* nonsense mutation was found to be a late event in tumor evolution (Fig. 1B; Supplementary Figs. S2A–S2E). Copy number analysis of the resistant RW7213 cells revealed the presence of more than 20 copies of KRAS, further validated by fluorescence in situ hybridization (Fig. 1C; Supplementary Fig. S3A and S3B). In parallel, a KRAS G12C mutant CRC patient-derived xenograft (PDX) model (CLR113) that was initially sensitive to sotorasib and cetuximab combination treatment(6) developed acquired resistance after about 10 months that was associated with amplification of KRAS G12C (variant allelic frequency [VAF] 1.00, cancer cell fraction (CCF) 100%), BRAF K601E (VAF 0.03, CCF 13%), and RAF1 S259F (VAF 0.03, CCF 10%) acquired alterations (Fig. 1D and 1E; Supplementary Fig.e S4). These data indicate that multiple resistance mechanisms can contribute to the survival of KRAS G12C mutant cells and that KRAS G12C amplification may be a recurrent alteration at resistance.
To evaluate resistance mechanisms to KRAS G12C inhibitor (KRAS G12Ci) and anti-EGFR antibody (EGFRi) in patients, we collected circulating free DNA (cfDNA) from twelve CRC patients treated with combination treatment (adagrasib plus cetuximab [n=8] or sotorasib plus panitumumab [n=4]). who initially experienced tumor regression and then developed either radiographic (RECIST) or clinical progression (Fig. 1F). Patient characteristics and response information are summarized in Supplementary Table S1 and baseline tumor tissue sequencing results are shown in Supplementary Figure S5. Emergent alterations identified at resistance (Fig. 1G; Supplementary Table S2) included KRAS G12C amplification, KRAS mutations (G12A/D/F/LR/S/V, H95L/Q/R, and Y96D/H/N), NRAS mutations (Q61K/R), downstream ERK pathway alterations (BRAF mutations/fusions, MEK1 mutations), RTK (receptor tyrosine kinase) activation (MET amplification/fusion, RET fusion, EGFR mutations), and MYC amplification. Similar to what was previously reported for resistance to KRAS G12C inhibitor monotherapy and in accordance with our preclinical models treated with sotorasib-cetuximab combination, multiple resistance-associated alterations were identified in individual patients, with the majority predicted to prevent inhibition of ERK signaling by drug(12).
Resistance dynamics in ctDNA of colorectal cancer patients on KRAS G12C-EGFR inhibition
Similar to resistance alterations to KRAS G12C inhibitor monotherapy(10,11), all acquired alterations were identified at low VAF, at one-tenth or one-hundredth of the frequency of alterations identified at baseline. To better understand clonal dynamics of resistance to combination treatment in CRC, ctDNA was serially collected during treatment and sequenced about every six weeks in four patients (Figs. 2A–2C; Supplementary Table S3). Longitudinal analysis confirmed the emergence of multiple resistance alterations. Resistant alterations often emerged many weeks before the development of clinical resistance and remained at a low frequency, largely <1% VAF, while the baseline alterations and tumor marker (CEA) rose higher during treatment. In multiple patients, once resistance alterations were first detected, each successive time point identified new resistance alterations with only modest changes in the VAF of the pre-existing resistant alterations (Figs. 2A–2C; Supplementary Table S3). We did not observe a clonal sweep with emergence of a dominant resistance alteration in any patient. In several patients, the VAF of the putative resistance alterations actually decreased and became undetectable despite continued treatment. These included alterations expected to cause resistance to a KRAS G12C inhibitor, such as the KRAS G13D, NRAS Q61K, and BRAF V600E mutations detected in patient 1 (Fig. 2A) and the BRAF V600E mutation and BRAF fusion in patient 3 (Fig. 2C). These data together suggest that resistant subclones do not grow effectively and are unable to grow out to dominate the population. Indeed, among the many low frequency resistance alterations detected in patients, the only putative resistance genetic event that increased steadily in step with tumor marker response was amplification of the KRAS G12C variant. Clinical resistance to KRAS G12C and EGFR inhibition is thus characterized by the accumulation and loss of many low frequency resistance alterations, while KRAS G12C amplification drives a higher portion of the resistance phenotype.
Figure 2. Longitudinal analysis of ctDNA in colorectal cancer patients on KRAS G12C-EGFR inhibition.
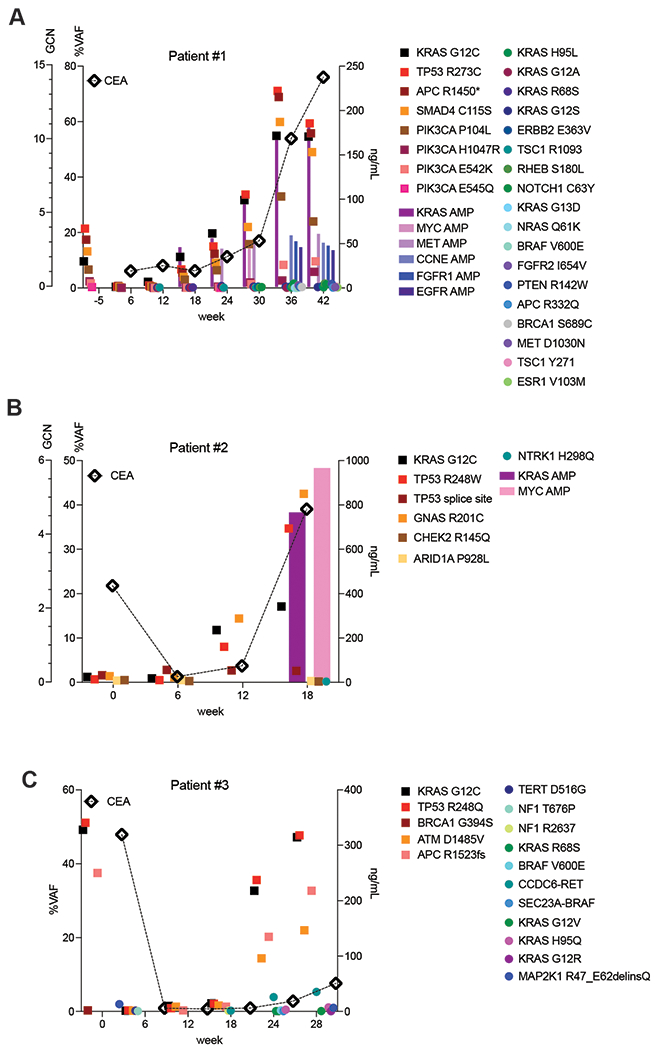
A,B,C. CRC patients treated with combined KRAS G12C and EGFR inhibitors: circles indicate emergent alterations on treatment; bars indicate emergent copy number changes; tumor biomarker (CEA) indicated with a diamond. In all graphs, KRAS G12C is marked with solid black square and TP53 alterations marked with red square to track these truncal alterations.
Effect of drug withdrawal on KRAS G12C amplified resistant cells
Intrigued by the correlation of KRAS G12C amplification with clinical resistance, we used the RW7213-resistant cells (RW7213-R) harboring high-grade KRAS G12C amplification to investigate the characteristics of this resistance mechanism. The resistant cells grow as colonies in medium containing cetuximab and sotorasib, maintaining the same morphology of the parental RW7213 cell line. We then grew these cells in the absence of drugs to mimic the effect of stopping treatment at clinical progression. We found that KRAS amplification was maintained in the resistant RW7213 cells with short-term drug withdrawal and analysis of RAS downstream effectors signaling in RW7213 parental, resistant, and resistant cells off drugs showed instead further increase of MAPK (mitogen activated protein kinase) and Pi3K-mTOR (phosphatidylinositol 3-kinase-mammalian target of rapamycin) pathway activation upon drug withdrawal in the resistant cells, compared to the resistant cells on drugs and parental RW7213 cells (Fig. 3A).
Figure 3. Drug withdrawal drives senescent phenotype in resistant CRC cell line with acquired KRAS G12C amplification.
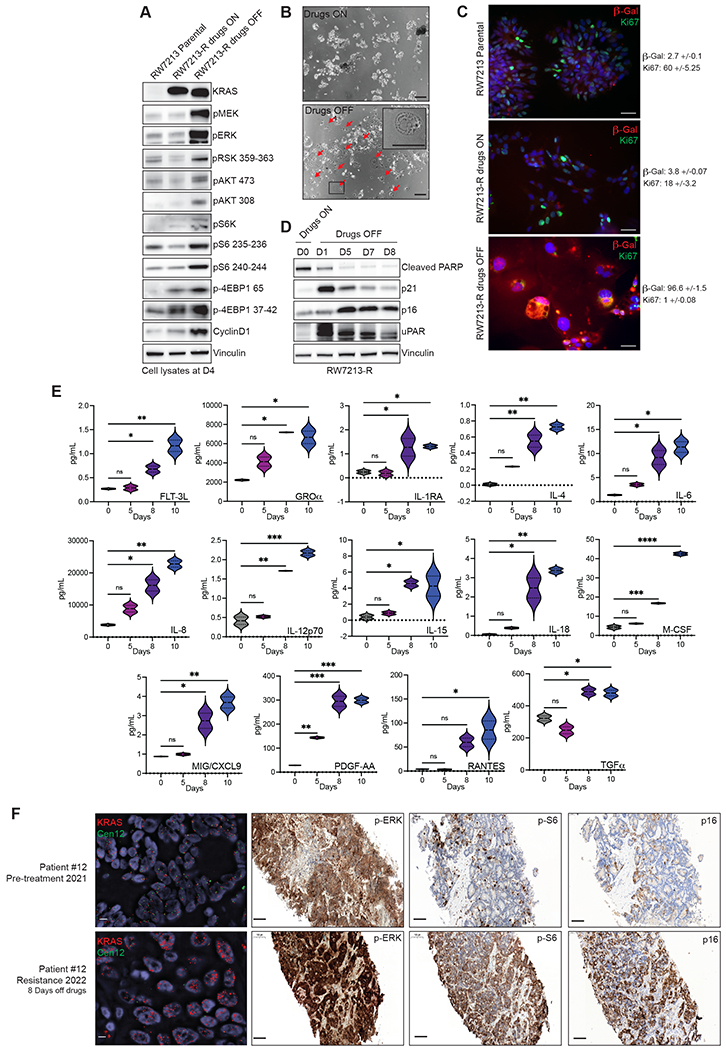
A. Western blot analyses of the effects on MAPK and mTOR pathway regulation in RW7213 parental cells and in RW7213-R with and without cetuximab-sotorasib combination; vinculin is included as loading control. B. Microscopy images of RW7213-R with and without Cetuximab/Sotorasib combo: 10x magnification, scale bars 100μm. In the right panel, the black square represents the area magnified in the upper-right corner inset. C. Ki67 and β-Gal staining by immunofluorescence (time point 4 days). 10x magnification, scale bars 100μm. Quantification represents percentage of β-Gal and Ki67 positive cells per total number of cells, +/− symbol indicates variation between pictures. 10 independent pictures have been quantified per condition. D. Western blot analyses of p16, p21, caspase-3, cleaved PARP and uPAR expression upon drug withdrawal, vinculin is included as loading control. E. SASP cytokine array time course experiment. Data shown represent duplicates. Statistical analyses and P-values represent 2way ANOVA with Dunnett’s multiple comparison test. ns (not significant) = P-value >0.05, *=P-value ≤0.05, **=P-value ≤0.01, ***=P-value ≤0.001, ****= P-value ≤0.0001. F. FISH staining for KRAS (scale bar 5μm) and immunohistochemistry for phospho-ERK, phospho-S6 (S235), and p16 in tissue samples collected from patient 12, consisting of pre-treatment liver metastasis biopsy (pre-treatment 2021) and progression liver metastasis biopsy collected 8 days after stopping KRAS G12C and EGFR inhibitors (resistance 2022). Mean KRAS (red)/Cen12 (green) ratio, based on manual counting of 50 cells from each time point, was 1.8 for the pre-treatment specimen and 13.2 for the resistance specimen. Phospho-ERK staining was 2+ involving >90% of cells pre-treatment and 3+ involving >90% of cells at progression; phospho-S6 staining was absent pre-treatment and 2+ involving 70% of cells at progression; and p16 staining was 2+ involving 5% of cells pre-treatment and 2+ involving 65% of cells at progression. Magnification of all immunohistochemistry slides is 20x scale bars 100μm.
Together with these effects on signaling, we also observed that 24 to 48 hours after drug withdrawal, RW7213 KRAS G12C amplified cells acquired a large and flat morphology, and this phenotype was maintained over time (Fig. 3B). This feature is reminiscent of cellular senescence, a program that can be triggered by excessive oncogenic signaling(14). RW7213-R cells in which drug was withdrawn showed an increase in beta-galactosidase activity and decrease in cellular proliferation, as measured by Ki-67 staining, both consistent with a senescent phenotype (Fig. 3C). Senescent cells acquire a new metabolic state, are protected from apoptosis, and activate a secretory program known as the senescence associated secretory phenotype (SASP)(15–17). In line with a senescent phenotype, RW7213-R cells taken off the drug combination downregulated apoptosis markers, increased expression of cyclin-dependent kinase inhibitors and uPAR, a marker of senescence(18), and accumulated cytokines indicative of SASP (Fig. 3D and 3E). By contrast, C106-R cells harboring KRAS G12C and NRAS G12D mutations did not exhibit the senescence phenotype or markers (Supplementary Fig. S6A and S6B), suggesting that this effect is specific for KRAS G12C amplification. These data suggest that high levels of KRAS signaling, which are needed to drive resistance to the drugs, trigger oncogene-induced senescence upon drug withdrawal.
To check for similar changes in the clinical setting, we evaluated activation of the MAPK and mTOR pathways in tissue samples from a patient who developed KRAS G12C amplification at resistance (Fig. 1E). Several CRC patients had identifiable KRAS G12C amplification at resistance, and patient 12 underwent biopsy of a liver metastasis immediately before starting KRAS G12C and EGFR inhibitors (pre-treatment 2021) and again at progression where tissue was collected eight days after the stop of KRAS G12C and EGFR inhibitor drug therapy and before start of any new therapy (resistance 2022) (Fig. 3F). Sequencing and fluorescence in situ hybridization showed acquired high level KRAS amplification at progression (Fig. 3F; Supplementary Fig. S6C). Phosphorylated ERK levels were high pre-treatment (2+ staining involving >90% of cells) and further increased at progression (3+ staining involving >90% of cells). Phosphorylated ribosomal protein S6 (S235) levels were low prior to treatment (absent staining) and elevated (2+ staining involving 70% of cells) in the progression sample collected after eight days of drug withdrawal (Fig. 3F). These data demonstrate in a patient progressing with KRAS G12C amplification, that, after drug stop, tumor tissue has elevated MAPK and mTOR pathway signaling. We further checked changes in p16 levels, which commonly associates with senescence(19), in these clinical samples as a marker for senescence and found rare staining pre-treatment (2+ staining, involving 5% of cells) and increased p16 expression at progression (2+ staining, involving 65% of cells) (Fig. 3F). These data provide support that, in patients, tumors with acquired KRAS amplification may also undergo senescence changes upon drug withdrawal.
Exploiting the new steady state after drug withdrawal to overcome resistance
Given the dramatic effect of drug withdrawal, we wondered if KRAS G12C amplification produces a selective disadvantage upon drug withdrawal in the clinical setting and monitored circulating tumor DNA in two of the CRC patients with acquired KRAS G12C amplification at resistance. We followed the ctDNA of patients 1 and 5, who both harbored KRAS G12C mutant CRC that had developed multiple resistance alterations, including KRAS G12C amplification (Fig. 1E and 2A). Comparison of ctDNA from before and about 4 weeks after drug withdrawal in each of these patients showed a 2-fold reduction of the signal from KRAS amplification. By contrast, the relative frequency of the other pre-existing alterations and emergent mutations remained mostly unchanged (Fig. 4A and 4B; Supplementary Table S3). Together these data suggest that KRAS G12C amplification is a mechanism of secondary resistance that shows fitness only in the presence of the selective pressure mediated by drug treatment.
Figure 4. Effect of treatment withdrawal in resistant colorectal cancers with amplified KRAS G12C.
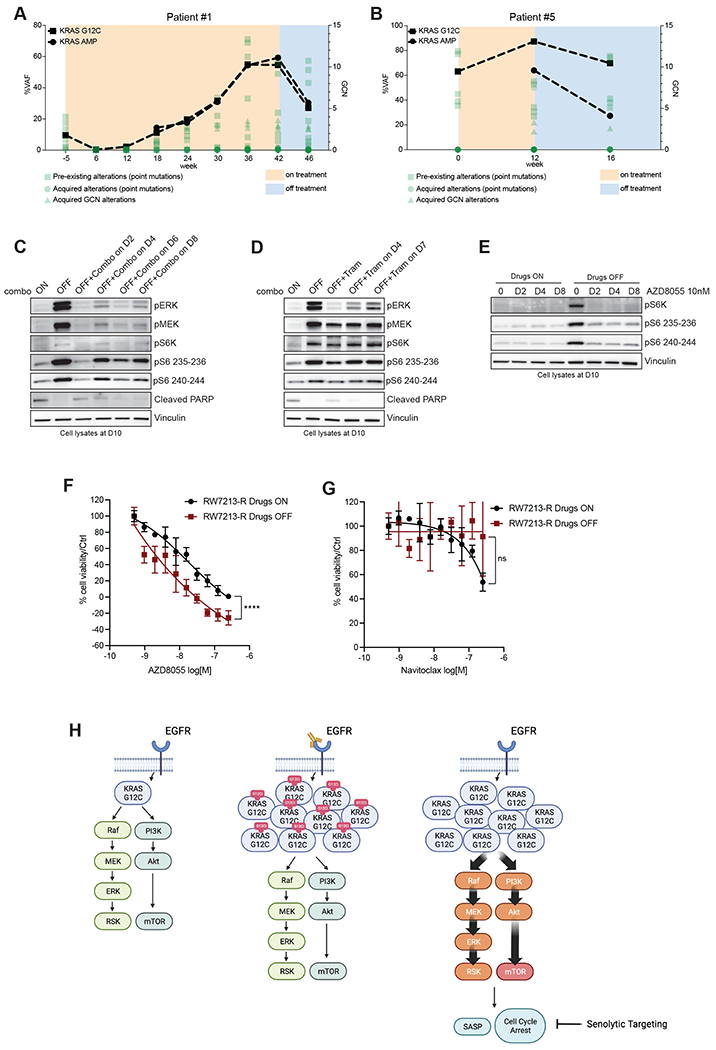
A.Longitudinal analysis of ctDNA in a CRC patient who held KRAS and EGFR inhibition for approximately four weeks after progression. B. Longitudinal analysis of ctDNA in a CRC patient who held KRAS and EGFR inhibition for approximately four weeks after progression. KRAS G12C ctDNA variant allelic frequencies are marked with squares, and KRAS plasma copy numbers are marked with circles. All the other variants are reported in green. C. Western blot analyses of p-ERK, p-MEK, p-S6K, p-S6 and cleaved PARP expression upon drug withdrawal and re-challenge with cetuximab 50μg/mL-sotorasib 3μM combination; vinculin is included as loading control. D. Western blot analyses of p-ERK, p-MEK, p-S6K, p-S6 and cleaved PARP expression upon drug withdrawal and re-challenge with 10nM trametinib; vinculin is included as loading control. E. Western blot analyses of p-S6K and p-S6 upon drug withdrawal or in drug-containing medium after treatment with 10nM AZD8055; vinculin is included as loading control. F. Short term proliferation assay RW7213-R cells in medium containing cetuximab-sotorasib (black) and in senescent conditions (dark red). Cells were seeded in absence or presence of drugs for 4 days and then treated for 96 hours with increasing concentration of AZD8055 and then ATP content was measured using CellTiterGlo. Data represents the average and standard deviation of 3 biological replicates. G. Short term proliferation assay RW7213-R cells in medium containing cetuximab-sotorasib (black) and in senescent conditions (dark red). Cells were seeded in absence or presence of drugs for 4 days and then treated for 96 hours with increasing concentration of Navitoclax and then ATP content was measured using CellTiterGlo. Data represent the average and standard deviation of 3 biological replicates. H. Proposed model: KRAS G12C mutant signaling is maintained at similar level in parental cells and in resistant cells in presence of concomitant EGFR and KRAS G12C blockade. Upon drugs removal, KRAS G12C amplified signaling drives oncogene-induced senescence characterized by elevated mTOR activity creating a new steady state that may be targeted by senolytic treatments.
Hence, we used the RW7213 resistant cells to investigate the effects of MAPK signaling suppression after a period of drug removal. After stopping cetuximab-sotorasib combination treatments, we re-challenged the cells with either the same combination (Fig. 4C) or with the MEK inhibitor trametinib (Fig. 4D) as function of time. While drug treatment was able to significantly decrease MAPK signaling, the cells maintained higher levels of p-S6K and p-S6 that were not suppressed by drug treatment, suggesting a new steady state after drug withdrawal with higher mTOR pathway signaling. mTOR signaling activation has been associated with senescence, as it regulates several senescence-associated phenotypes(15). Phospho-ERK was inhibited best after the shortest time off drug (2 days) and this time point was associated with a lower induction of p-S6 levels. However, re-challenge at longer time points, could not suppress mTOR signaling or restore apoptotic potential (cleaved PARP).
These data suggest that periods of drug withdrawal and retreatment will be unable to re-establish drug sensitivity and cell death. However, we hypothesized that the senescent state and associated activation of mTOR-dependent signaling in these cells may provide a therapeutic vulnerability. Indeed, the mTOR inhibitor AZD8055, which has been previously proposed as a senolytic agent(20), was able to inhibit S6K and S6 phosphorylation selectively in RW7213-R cells where drug was removed (Fig. 4E). Proliferation assays show that RW7213 cells off drug are more sensitive to AZD8055 than cells maintained with continuous drug exposure (Fig. 4F). To support the specificity of mTOR inhibition, we used another senolytic drug, the bcl2 inhibitor navitoclax(21) and this compound failed to block cell proliferation in the RW7213-R cells after drug withdrawal or with continuous drug exposure (Fig. 4G).
Altogether, these data suggest that there may be enhanced activity and selectivity of mTOR blockade in resistant KRAS G12C cancers with KRAS G12C amplification following drug withdrawal and nominates further exploration of a one-two punch approach of drug withdrawal and senolytic therapy as a potential strategy to overcome acquired resistance to EGFR-KRAS G12C combination in those tumors that acquired KRAS G12C amplification (Fig. 4H).
Discussion
Here we report the genetic mechanisms of secondary resistance to concomitant EGFR and KRAS G12C blockade in KRAS G12C mutant CRC. In agreement with previous studies, our patients show sub-clonal heterogeneity and acquired mutations at low variant allele frequencies. This may be due in part to the evaluation of ctDNA as this method exposes tumor heterogeneity more than single biopsy specimens(22). Sequencing data from our study and others’ (10–12) may suggest that a small fraction of cells may be sufficient to drive clinical resistance. This concept is in line with recent studies of metastatic behavior where, for example, single cell RNA sequencing of small cell lung cancers identified a rare population of stem-like cells that appears to drive metastatic outcomes in this cancer(23). However, our study highlights that these low frequency alterations appear and disappear during treatment, and this may support an induction of mutagenesis due to drug(24) and also that these lesions can be characterized by low fitness and may not effectively drive resistance.
In contrast to the low frequency resistance mechanisms, we identify KRAS G12C amplification as a recurrent resistance mechanism that tracks with tumor markers and response and engages oncogene-induce senescence when drug is removed. Previous reports showed that BRAF amplification upon RAF inhibitor resistance can be modulated by intermittent treatment(25) and we show that amplified KRAS G12C recedes in the absence of drug in patients’ plasma. Here, we describe the transition to a senescence state in a cell line and patient’s tumor and hypothesize that the drop of KRAS G12C in ctDNA is due to the protective effect of senescence from apoptosis and consequently the release of cell-free DNA. We also show in the cell line that the senescent phenotype which has high mTOR pathway activation prevents a strategy of intermittent therapy from overcoming resistance and that in clinical samples, from a patient with acquired KRAS amplification, mTOR pathway signaling is induced after drug release and correlates with p16 induction as a marker of senescence. These results provide a possible mechanistic explanation for the worse outcomes seen with intermittent RAF inhibition for melanoma treatment in the clinic(26).
Oncogene overexpression in vitro is challenging as cells select against this hyper-activation over time, and we were unable to generate KRAS G12C overexpressing cell lines. This suggests that KRAS G12C amplified cells need the adaptation underlying long-term drug exposure and acquired resistance to therapy and raises questions about what are the mechanisms that regulate this adaptation and how can we target them.
Our data, however, nominate a new potential approach to overcome acquired resistance, by exploiting vulnerabilities due to the senescence program during periods of drug withdrawal to target resistant cells more broadly. In this work, we took advantage of the new dependency on mTOR signaling that develops when cells enter the senescent state because of excessive oncogenic RAS signaling. Activation of mTOR dependent signaling is required for maintenance of cellular senescence (15,27), and drug withdrawal can be combined with senolytic approaches(28) to facilitate tumor clearance, as shown by us and others, by inhibition of mTOR signaling(20). Additional mechanistic insights into mTOR activation and longer-term drug-off kinetics will be important to examine further in future studies. Other potential senolytic approaches can be based on exploiting SASP chemokines that can recruit immune cells suggesting the potential to target resistant cancer cells with immune checkpoint inhibitors(29–32). Moreover, senescence-driven expression of uPAR could also become a target for CART cell therapy(33). Further studies will provide important insights on how to effectively target KRAS G12C mutant cancers that developed secondary resistance to KRAS G12C inhibitors as single agents and in combination.
Methods
Cell lines and Compounds
RW7213 cell line was cultured in RPMI (Lonza). C106 cell line was cultured in Iscove’s modified medium (Lonza). Each media was supplemented with 10% FBS, 2mM L-glutamine, 100U/ml penicillin and 100mg/mL streptomycin. Resistant derivatives were grown in media containing cetuximab (50μg/mL) and sotorasib (3μM). C106 cells were purchased from ECACC, RW7213 were provided by Dr. Diego Arando.
All the cell lines were determined to be mycoplasma free using the Venor® GeM Classic kit (Minerva biolabs, last test July 2022) and tested by Short Tandem Repeats profiling at 10 different loci.
Sotorasib and trametinib were purchased from SelleckChem. Cetuximab was purchased from the Pharmacy at Memorial Sloan Kettering Cancer Center.
Cell viability assay
For dose-response proliferation assays, 4000 cells were seeded in 96-well culture plates in complete medium. After 24 hours, the indicated concentrations of sotorasib and cetuximab were added to cell. After 72 hours, cell viability was determined by measuring ATP content using Cell Titer-Glo® Luminescent Cell Viability assay (Promega). DMSO-only treated cells were used as control. Assays were performed with 3 replicates and were each repeated three times. For senolytic proliferation assays with RW7213-R, 2000 cells were seeded in 96-well culture plates in medium containing cetuximab 50μg/mL and sotorasib 3μM combo or medium only to induce senescence. After 96 hours, serial dilutions of AZD8055 or navitoclax were added to cells and cell viability was determined at baseline by measuring ATP content using Cell Titer-Glo® Luminescent Cell Viability assay (Promega). After 96 hours cell viability was determined again by measuring ATP content using Cell Titer-Glo® Luminescent Cell Viability assay (Promega). DMSO-only treated cells were used as control and all the values were normalized on baseline measurements. In all the experiments, plates were incubated at 37°C in 5% CO2.
Antibodies and Western blotting
After seeding and drug treatments, cells were washed with cold PBS and lysed in RIPA buffer (Pierce #89901) plus phosphatase and protease inhibitors (Thermo Scientific #1861277, #1861278). Lysates were cleared by centrifugation at 14000rpm at 4°C and quantified using BCA method (Pierce #2322).
Samples were prepared using LDS+Reducing agent Novex buffers (Invitrogen #NP0008, #NP0009). 10 to 20μg of lysates were loaded and run on NuPageTM 4-12% Bis-Tris gels (ThermoFisher #NP0321BOX) followed by transfer to nitrocellulose membranes (Biorad #1620233). Membranes were incubated over night with the indicated antibodies, washed and incubated again for 45 minutes with anti-rabbit or anti-mouse secondary antibodies. Detection was performed using Immobilion Western (Millipore #WBKLS0500).
Primary antibodies were obtained from Cell Signaling Technology and were used at a concentration of 1:1000: anti-p16 (#80772), anti-p21 12D1 (#2947), anti-Actin (#4970), anti-phospho-MEK1/2 S217/221 (#9154), anti-phospho-p44/42 MAPK T202/204 (#9101), anti-total ERK1/2 (#9102),anti-p-AKT 473 (#4060), anti p-S6K(#9204), anti p-S6 235-236(#4858), anti pS6 240-244(#5364), anti uPAR (#12713), anti p-RSK (#9944), anti p-4EBP1 65 (#9451), anti p-4EBP1 37/42 (#2855) and anti-Vinculin (#13901S). Anti-CyclinD1 antibody was purchased from Thermo-Scientific (PA516777) and used at 1:1000 dilution. Anti-KRAS and anti-NRAS from Santa Cruz Biotechnology and used at 1:500; anti-p338-CRAF from Millipore and used at 1:1000.
RAS-GTP pull-down assay
RAS-GTP pull –down assay was performed according to manufacturer’s protocol (Thermo Scientific #16117). Briefly, 500μg of lysates were loaded into columns together with agarose beads and RAS-RBD bait and incubated for 1 hour at 4°C. After the incubation, beads were washed three times and resuspended in LDS+Reducing agent Novex buffers (Invitrogen #NP0008, #NP0009). A fraction of lysates was used to measure total RAS amount. Pull-Down and total lysates were subjected to western blotting procedure as described above. The kit provided primary antibody against pan-RAS.
SASP Cytokine array
Conditioned media was collected from cells that were cultured in presence or absence of the drug combination. Aliquots of the media were analyzed with a multiplex immunoassay designed for human samples, “Human Cytokine Array / Chemokine Array 48-Plex HD48” (Eve technologies). Cytokine concentration was normalized by cell count.
In vivo studies
The CLR113 PDX was derived from liver metastasis. Tumor tissue was transplanted orthotopically into NSG mice to establish the PDX (IRB protocols 06-107, 14-091). Once a tumor became visible in the first mouse, it was transplanted and expanded to other animals. Tumor tissue was implanted subcutaneously in the flank of 4–6-week-old NSG female mice and treatment of the mice began when tumor reached approximately 100mm3 in size. Mice were randomized (n = 5 mice per group) to receive drug treatments or vehicle as control.
Sotorasib (100mg/kg) and trametinib (3mg/kg) were given daily by gavage. Cetuximab was administered 50mg/kg twice a week, by intra-peritoneal injections.
Studies were performed in compliance with institutional guidelines under an IACUC approved protocol. The animals were immediately euthanized as soon as the tumors reached the IACUC set limitations.
Patients
All patients were treated on KRAS inhibitor clinical trials approved by MSKCC Institutional Review Board/Privacy Board (protocols 19-408 [NCT03785249], 20-183 [NCT04185883]). Collection of patient samples were conducted under appropriate Institutional Review Board/Privacy Board protocols and waivers (protocols 06-107, 12-245, 14-019). Participating patients signed written informed consent for these clinical trials and biospecimen protocols. This study was conducted in accordance with ethical guidelines in the Declaration of Helsinki.
Fluorescence In Situ Hybridization (FISH)
FISH analysis was performed on adherent cells. KRAS FISH analysis was performed using a 2-color KRAS/Cen12 probe mix (developed at MSKCC). The probe mix consisted of bacterial artificial chromosomes (BAC) clones containing the full length KRAS gene (clones RP11-29515 and RP11-707F18; labelled with red dUTP) and a centromeric repeat plasmid for chromosome 12 served as the control (clone pa12H8; labelled with green dUTP). Probe labelling, hybridization, washing, and fluorescence detection were performed according to standard procedures. Slides were scanned using a Zeiss Axioplan 2i epifluorescence microscope equipped with a megapixel CCD camera (CV-M4+CL, JAI) controlled by Isis 5.5.9 imaging software (MetaSystems Group Inc, Waltham, MA). The entire section was scanned through 63X or 100X to assess signal pattern and select representative regions for imaging. Amplification was defined as >10 copies of each locus.
Combined β-galactosidase and immunofluorescence staining
For immunofluorescent staining, 20000 cell/well were seeded in Nunc™ Lab-Tek™ II Chamber Slide™ System (Thermo Fisher, 154526). β-gal staining was performed using ImaGeneRed C12RG (I-2906) according to manufacturer instruction with a final concentration of 33mM C12RG compound for 2 hours. To stop b-galactosidase activity, PETG was added to the medium. Cells were fixed with 4% PFA for 10 minutes at RT, permeabilized using 0.02% Triton/ PBS and incubated with Ki67 antibody (Abcam, ab16667) overnight in 0.02% Triton/ 5% BSA/ PBS. Secondary antibody was added the day after and Dapi was used to stain the nucleus. Slides were mounted using ProLong Gold Antifade Mountant (Thermo Fisher, P10144). Pictures were quantified using Fiji Software and 10 independent pictures have been quantified per condition.
DNA sequencing
Circulating free DNA (cfDNA) Analysis
cfDNA analysis was performed using the commercially available, targeted next-generation sequencing assays Guardant360 (Guardant Health) (patients 1-5, 11) and ctDx FIRST (Resolution Bioscience) (patients 6-10) and the MSK-ACCESS assay (patient 12). Guardant360 CDx is a CLIA-accredited, New York State Department of Health-approved cfDNA assay with analytic and clinical validation previously reported(34,35). During this study, the assay included assessment of 74-83 genes (depending on panel version ordered) with coverage of single nucleotide variants (SNVs) and select insertions/deletions, amplifications, and fusions. Resolution Bioscience ctDx FIRST assay includes assessment of 113 genes and detects SNV/Indel Hotspots, SNV/Indel Full CDS, amplifications, deletions, gene rearrangements, and gene fusions. The ctDx FIRST assay uses a custom bioinformatics pipeline to call variants associated with genomic targets. The MSK-ACCESS assay is a custom, ultra-deep assay that includes key exons and domains of 129 genes and introns of 10 genes harboring recurrent breakpoints. It uses duplex unique molecular identifiers (UMIs) and dual index barcodes to minimize background sequencing errors and sample-to-sample contaminations, and alterations are called against matched normal DNA. The longitudinal ctDNA analysis in this study was performed with the Guardant360 CDx assay.
Bulk Tissue Sequencing
Genomic DNA was extracted from cell lines, frozen xenograft tumors, or formalin fixed paraffin embedded (FFPE) patient tissues obtained from biopsies or resections and sequenced with the MSK-IMPACT next-generation sequencing assay(13). Copy number alterations (CNAs) and loss of heterozygosity were defined using FACETS(36). The cancer cell fraction (CCFs) of somatic mutations identified in the cell lines and frozen xenograft tumors were computed using ABSOLUTE (v1.0.6)(37), and a mutation was classified as clonal if its probability of being clonal was >50% or if the lower bound of the 95% confidence interval of its CCF >90%, as previously described(38). For the construction of phylogenetic trees based on CNAs, major and minor copy number computed by FACETS(36) were modeled using transducer-based pairwise comparison functions using MEDICC(39) assuming a diploid state with no CNAs to root the phylogenies.
Single cell DNA sequencing
The C106-R cell line was subjected to single cell sequencing. The cell line was washed with PBS and quantified by combining 5uL of cell suspension with an equal amount of Trypan Blue, loaded on chamber slides, and counted with the Countess automated cell counter (Invitrogen). A total of 250,000 cells were used for the barcoding run. In brief, cells were encapsulated with lysis buffer (100 mM Tris at pH 8.0, 0.5% IGEPAL, proteinase K 1.0 mg/ml) in a Tapestri platform (Mission Bio, San Francisco, CA, USA) and further lysed on the thermal cycler with the following conditions: 60 minutes at 50°C and 10 minutes at 80°C. The DNA from the encapsulated cell lysate was then primed and barcoded using a custom panel (Mission Bio, San Francisco, CA, USA), which targets hotspot variants of 54 oncogenes and tumour suppressor genes, for a total of 317 amplicons. After exposure to UV light, droplet PCR reactions were thermocycled with the following conditions: 6 minutes at 98°C, 10 cycles of 30 seconds at 95°C, 10 seconds at 72°C, 9 minutes at 61°C, and 20 seconds at 72°C; 10 cycles of 30 seconds at 95°C, 10 seconds at 72°C, 9 minutes at 48°C, and 20 seconds at 72°C; and a final step of 2 minutes at 72°C. PCR products were digested at 37°C for 60 minutes and posteriorly purified using 0.63x of SPRI beads (Beckman Coulter). Sample indices and illumina adapter sequences were then added via a 9 cycle PCR reaction, and a second 0.63x SPRI purification was then performed on the completed PCR reactions. Libraries were analyzed on a DNA 1000 assay chip with a Bioanalyzer (Agilent Technologies) and sequenced on a NextSeq 550 instrument (Illumina, Inc., San Diego, CA, USA; 150 bp paired-end reads). Sequence data were analyzed using the proprietary software provided by Mission Bio(40). In brief, sequence reads were trimmed for adapter sequences using Cutadapt(41) and mapped to the hg19 human genome using the Burrows-Wheeler Aligner (BWA)(42) after extracting barcode information. Following mapping, on target sequences were selected using standard bioinformatics tool (SAMtools)(43) and barcode sequences were error-corrected based on a white list of known sequences(40). The number of cells was determined from barcodes based on number of reads assigned to each barcode and amplicon read completeness. HaplotypeCaller (GATK v4.2.1.0)(44,45) was used to genotype the mutations present in all single-cells with a joint-calling approach. The mutations identified in each cell were further intersected with MuTect2 (GATK v4.2.1.0)(44,46) to obtain high confidence mutations. Genotyping calls were further examined and corrected according to variant allele frequency. Potential doublets or multiplets characterized by the existence of 2 or more cells that are captured within a droplet and linked to a single barcode, were identified using DoubletD(47) and further excluded from the analysis. For genotype clustering analysis of the five known variants (KRAS G12C, NRAS G12D, APC H1490Lfs*17, ERBB3 V104M and APC Q879*), cells were included when these five variants met the criteria of read depth (≥10) and genotyping quality (≥60)(48). In addition, subclones with a higher allele dropout (ADO) rate compared to the overall ADO rate of all cells were further excluded(49). For clonal architectures, fishplot was created using the fishplot package(50).
To estimate allele specific copy number alterations, we used a pool of single, non-neoplastic, diploid cells identified in endometrioid endometrial tumors (51). Read counts of amplicons for C106-R and non-neoplastic cells were obtained from MissionBio’s pipeline and further used for allele specific copy number estimation. Amplicon read counts for cells with no coverage were imputed according to neighbouring cells using MAGIC(52). Imputed read counts were normalized to total library size for each cell. Amplicon copy number ratio was calculated by dividing C106-R read counts with the median of non-neoplastic counts. Because C106-R cells were considered to be diploid in the matched bulk sequencing data, amplicon copy number ratio was further transformed into non-integer copy number by multiplying by 2. Finally, gene integer copy number was obtained by taking the median value of amplicon copy number for each gene, and by taking the nearest integer value.
Immunohistochemistry (IHC)
Samples were loaded into Leica Bond RX and pretreated with EDTA-based epitope retrieval ER2 solution (Leica, AR9640) for 20 minutes at 95°C. The rabbit monoclonal antibodies against pMAPK (Cell Signaling Technologies, Cat# 4060, 0.5ug/ml) or pS6R (Cell Signaling Technologies, Cat# 4858, 0.17ug/ml) were applied for 60 minutes and detected with Polymer Refine Detection Kit (Leica, DS9800). Antibody Leica Bond Polymer anti-rabbit HRP was used, followed Refine Detection Kit Mixed DAB Refine for 10 mins, and Refine Detection Kit Hematoxylin counterstaining for 20 mins. After staining, sample slides were washed in water, dehydrated using ethanol gradient (70%, 90%, 100%), washed three times in HistoClear II (National Diagnostics, HS-202), and mounted in Permount (Fisher Scientific, SP15). For p16 IHC, samples were loaded into Leica Bond RX and pre-treated with EDTA-based epitope retrieval ER2 solution (Leica, AR9640) for 20 minutes at 95°C. The mouse monoclonal antibody against p16 (Santa Cruz, sc-56330, 0.2ug/ml) was applied for 60 minutes. Next Rabbit anti-mouse linker antibody (Leica Bond, Post Primary 1/5 dilution) and Leica Bond Polymer anti-rabbit HRP (Leica, DS9800) was used, followed by Refine Detection Kit Mixed DAB Refine for 10 mins, and Refine Detection Kit Hematoxylin counterstaining for 20 mins. After staining, sample slides were washed in water, dehydrated using ethanol gradient (70%, 90%, 100%), washed three times in HistoClear II (National Diagnostics, HS-202), and mounted in Permount (Fisher Scientific, SP15).
Supplementary Material
Statement of significance:
Clinical resistance to KRAS G12C-EGFR inhibition primarily prevents suppression of ERK signaling. Most resistance mechanisms are subclonal, while KRAS G12C amplification rises over time to drive a higher portion of resistance. This recurrent resistance mechanism leads to oncogene-induced senescence upon drug withdrawal and creates a potential vulnerability to senolytic approaches.
Acknowledgements
Supported by National Institutes of Health R01 CA233736 (R.Y., N.R.), U54 OD020355 (E. DS.), and Cancer Center Core Grant P30 CA008748. R.M. is a Cancer Research Institute Irvington Fellow supported by the Cancer Research Institute (CRI Award 3441). Y.R.M-G. is supported by the Kristina M. Day Young Investigator Award from Conquer Cancer, the ASCO Foundation, and received training through an NIH K30 institutional grant (CTSA UL1TR00457). This research is the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Disclosures of potential conflict of interest
R.Y. has served as an advisor for Natera, Array BioPharma/Pfizer, Mirati Therapeutics, and Zai Lab and has received research support to her institution from Array BioPharma/Pfizer, Boehringer Ingelheim, Daiichi Sankyo and Mirati Therapeutics. E.T and S.C. are employees of Boundless Bio. M.F.B. has received consulting fees from PetDx and Eli Lilly and research funding from GRAIL. H.D., K.A., J.C.G. are employees of Mirati Therapeutics. G.R. has been an uncompensated consultant to Daiichi, Pfizer, Merck, Verastem, Novartis, and Mirati and has institutional research support from Mirati, Takeda, Merck, Roche, Pfizer, and Novartis. Y.R.M-G. has received honoraria from Virology Education and has received travel, accommodation, and expenses from AstraZeneca. She acknowledges associated research funding to the institution from Loxo Oncology at Eli Lilly, Elucida Oncology, Taiho Oncology, Hengrui USA, Ltd/ Jiangsu Hengrui Pharmaceuticals, Luzsana Biotechnology, and Endeavor Biomedicines. She acknowledges royalties from Rutgers University Press and Wolters Kluwer. B.T.L. has served as a consultant/advisory board member for Roche, Biosceptre International, Thermo Fisher Scientific, Mersana Therapeutics, Hengrui Therapeutics, Guardant Health and has received research funds to his institution from Genentech, Daiichi Sankyo, Hengrui Therapeutics, Illumina, BioMed Valley Discoveries, AstraZeneca, GRAIL, and Amgen. J.S.R-F. is a member of the scientific advisory board (with paid honoraria) of Volition Rx, Paige.AI, Invicro, Roche, Genentech, and Ventana, and a consultant with paid fees of Goldman Sachs Merchant Banking. D.B.S. has consulted with and received honoraria from Pfizer, Loxo Oncology, Lilly Oncology, Vivideon Therapeutics, Q.E.D. Therapeutics and Illumina. N.R. is on the scientific advisory board (SAB) of Chugai, BeiGene, Fortress Biotech, Daiichi-Sankyo, AstraZeneca, F-Prime, Zai Lab, Arvinas, and Array BioPharma; and he is a past SAB member of Millennium-Takeda, Kadmon, Kura Oncology, and Araxes. N.R. is also a consultant to Novartis Biomed, Boehringer Ingelheim, Tarveda, Foresite Capital, Array BioPharma, and Revolution Medicines; and in recent years has also consulted with Eli Lilly, Merrimack, Kura Oncology, Araxes, and Kadmon. N.R. owns equity in BeiGene, Zai Lab, Fortress Biotech, Kura Oncology, Araxes, Kadmon, and Effector. S.M. has served as a consultant for Boehringer Ingelheim and has received research support to her institution from Boehringer Ingelheim and Daiichi Sankyo. All other authors declare no potential conflicts.
Data Availability
The human sequence raw data generated in this study are protected and not publicly available due to patient privacy requirements but are available upon reasonable request from the corresponding author subject to institutional approvals. Cell lines and patient tumor somatic mutations, identified by MSK-IMPACT sequencing, are available in the cBioPortal (https://www.cbioportal.org/study/summary?id=coadread_mskresistance_2022). All relevant cell-free DNA sequencing data are included within the article and supplementary data files.
References
- 1.Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med 2017;23(6):703–13 doi 10.1038/nm.4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013;503(7477):548–51 doi 10.1038/nature12796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lito P, Solomon M, Li LS, Hansen R, Rosen N. Allele-specific inhibitors inactivate mutant KRAS G12C by a trapping mechanism. Science 2016;351(6273):604–8 doi 10.1126/science.aad6204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Skoulidis F, Li BT, Dy GK, Price TJ, Falchook GS, Wolf J, et al. Sotorasib for Lung Cancers with KRAS p.G12C Mutation. N Engl J Med 2021;384(25):2371–81 doi 10.1056/NEJMoa2103695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ou SI, Janne PA, Leal TA, Rybkin II, Sabari JK, Barve MA, et al. First-in-Human Phase I/IB Dose-Finding Study of Adagrasib (MRTX849) in Patients With Advanced KRAS(G12C) Solid Tumors (KRYSTAL-1). J Clin Oncol 2022;40(23):2530–8 doi 10.1200/JCO.21.02752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Amodio V, Yaeger R, Arcella P, Cancelliere C, Lamba S, Lorenzato A, et al. EGFR Blockade Reverts Resistance to KRAS. Cancer Discov 2020;10(8):1129–39 doi 10.1158/2159-8290.CD-20-0187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fakih MG, Kopetz S, Kuboki Y, Kim TW, Munster PN, Krauss JC, et al. Sotorasib for previously treated colorectal cancers with KRAS(G12C) mutation (CodeBreaK100): a prespecified analysis of a single-arm, phase 2 trial. Lancet Oncol 2022;23(1):115–24 doi 10.1016/S1470-2045(21)00605-7. [DOI] [PubMed] [Google Scholar]
- 8.Fakih GSF M, Hong DS, Yaeger RD, Chan E, Mather O, Cardona P, Dai T, Strickler J. 434P CodeBreaK 101 subprotocol H: Phase Ib study evaluating combination of sotorasib (Soto), a KRASG12C inhibitor, and panitumumab (PMab), an EGFR inhibitor, in advanced KRAS p.G12C-mutated colorectal cancer (CRC). Annals of Oncology 2021;Volume 32:S551 doi 10.1016/j.annonc.2021.08.955. [DOI] [Google Scholar]
- 9.Weiss J, Yaeger RD, Johnson ML, Spira A, Klempner SJ, Barve MA, et al. LBA6 KRYSTAL-1: Adagrasib (MRTX849) as monotherapy or combined with cetuximab (Cetux) in patients (Pts) with colorectal cancer (CRC) harboring a KRASG12C mutation. Annals of Oncology 2021;Volume 32:S1294 doi 10.1016/j.annonc.2021.08.2093. [DOI] [Google Scholar]
- 10.Tanaka N, Lin JJ, Li C, Ryan MB, Zhang J, Kiedrowski LA, et al. Clinical Acquired Resistance to KRAS. Cancer Discov 2021;11(8):1913–22 doi 10.1158/2159-8290.CD-21-0365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Awad MM, Liu S, Rybkin II, Arbour KC, Dilly J, Zhu VW, et al. Acquired Resistance to KRAS G12C Inhibition in Cancer. N Engl J Med 2021;384(25):2382–93 doi 10.1056/NEJMoa2105281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhao Y, Murciano-Goroff YR, Xue JY, Ang A, Lucas J, Mai TT, et al. Diverse alterations associated with resistance to KRAS(G12C) inhibition. Nature 2021;599(7886):679–83 doi 10.1038/s41586-021-04065-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A, et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A Hybridization Capture-Based Next-Generation Sequencing Clinical Assay for Solid Tumor Molecular Oncology. J Mol Diagn 2015;17(3):251–64 doi 10.1016/j.jmoldx.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997;88(5):593–602 doi 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- 15.Saoudaoui S, Bernard M, Cardin GB, Malaquin N, Christopoulos A, Rodier F. mTOR as a senescence manipulation target: A forked road. Adv Cancer Res 2021;150:335–63 doi 10.1016/bs.acr.2021.02.002. [DOI] [PubMed] [Google Scholar]
- 16.Sharpless NE, Sherr CJ. Forging a signature of in vivo senescence. Nat Rev Cancer 2015;15(7):397–408 doi 10.1038/nrc3960. [DOI] [PubMed] [Google Scholar]
- 17.Collado M, Serrano M. The power and the promise of oncogene-induced senescence markers. Nat Rev Cancer 2006;6(6):472–6 doi 10.1038/nrc1884. [DOI] [PubMed] [Google Scholar]
- 18.Ruscetti M, Leibold J, Bott MJ, Fennell M, Kulick A, Salgado NR, et al. NK cell-mediated cytotoxicity contributes to tumor control by a cytostatic drug combination. Science 2018;362(6421):1416–22 doi 10.1126/science.aas9090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin AW, Barradas M, Stone JC, van Aelst L, Serrano M, Lowe SW. Premature senescence involving p53 and p16 is activated in response to constitutive MEK/MAPK mitogenic signaling. Genes Dev 1998;12(19):3008–19 doi 10.1101/gad.12.19.3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang C, Vegna S, Jin H, Benedict B, Lieftink C, Ramirez C, et al. Inducing and exploiting vulnerabilities for the treatment of liver cancer. Nature 2019;574(7777):268–72 doi 10.1038/s41586-019-1607-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhu Y, Tchkonia T, Fuhrmann-Stroissnigg H, Dai HM, Ling YY, Stout MB, et al. Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors. Aging Cell 2016;15(3):428–35 doi 10.1111/acel.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Strickler JH, Loree JM, Ahronian LG, Parikh AR, Niedzwiecki D, Pereira AAL, et al. Genomic Landscape of Cell-Free DNA in Patients with Colorectal Cancer. Cancer Discov 2018;8(2):164–73 doi 10.1158/2159-8290.CD-17-1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chan JM, Quintanal-Villalonga A, Gao VR, Xie Y, Allaj V, Chaudhary O, et al. Signatures of plasticity, metastasis, and immunosuppression in an atlas of human small cell lung cancer. Cancer Cell 2021;39(11):1479–96 e18 doi 10.1016/j.ccell.2021.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Russo M, Crisafulli G, Sogari A, Reilly NM, Arena S, Lamba S, et al. Adaptive mutability of colorectal cancers in response to targeted therapies. Science 2019;366(6472):1473–80 doi 10.1126/science.aav4474. [DOI] [PubMed] [Google Scholar]
- 25.Das Thakur M, Salangsang F, Landman AS, Sellers WR, Pryer NK, Levesque MP, et al. Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature 2013;494(7436):251–5 doi 10.1038/nature11814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gonzalez-Cao M, Mayo de Las Casas C, Oramas J, Berciano-Guerrero MA, de la Cruz L, Cerezuela P, et al. Intermittent BRAF inhibition in advanced BRAF mutated melanoma results of a phase II randomized trial. Nat Commun 2021;12(1):7008 doi 10.1038/s41467-021-26572-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cayo A, Segovia R, Venturini W, Moore-Carrasco R, Valenzuela C, Brown N. mTOR Activity and Autophagy in Senescent Cells, a Complex Partnership. Int J Mol Sci 2021;22(15) doi 10.3390/ijms22158149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kirkland JL, Tchkonia T. Senolytic drugs: from discovery to translation. J Intern Med 2020;288(5):518–36 doi 10.1111/joim.13141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gasek NS, Kuchel GA, Kirkland JL, Xu M. Strategies for Targeting Senescent Cells in Human Disease. Nat Aging 2021;1(10):870–9 doi 10.1038/s43587-021-00121-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007;445(7128):656–60 doi 10.1038/nature05529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kang TW, Yevsa T, Woller N, Hoenicke L, Wuestefeld T, Dauch D, et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 2011;479(7374):547–51 doi 10.1038/nature10599. [DOI] [PubMed] [Google Scholar]
- 32.Kale A, Sharma A, Stolzing A, Desprez PY, Campisi J. Role of immune cells in the removal of deleterious senescent cells. Immun Ageing 2020;17:16 doi 10.1186/s12979-020-00187-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Amor C, Feucht J, Leibold J, Ho YJ, Zhu C, Alonso-Curbelo D, et al. Senolytic CAR T cells reverse senescence-associated pathologies. Nature 2020;583(7814):127–32 doi 10.1038/s41586-020-2403-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zill OA, Banks KC, Fairclough SR, Mortimer SA, Vowles JV, Mokhtari R, et al. The Landscape of Actionable Genomic Alterations in Cell-Free Circulating Tumor DNA from 21,807 Advanced Cancer Patients. Clin Cancer Res 2018;24(15):3528–38 doi 10.1158/1078-0432.CCR-17-3837. [DOI] [PubMed] [Google Scholar]
- 35.Odegaard JI, Vincent JJ, Mortimer S, Vowles JV, Ulrich BC, Banks KC, et al. Validation of a Plasma-Based Comprehensive Cancer Genotyping Assay Utilizing Orthogonal Tissue- and Plasma-Based Methodologies. Clin Cancer Res 2018;24(15):3539–49 doi 10.1158/1078-0432.CCR-17-3831. [DOI] [PubMed] [Google Scholar]
- 36.Shen R, Seshan VE. FACETS: allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res 2016;44(16):e131 doi 10.1093/nar/gkw520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carter SL, Cibulskis K, Helman E, McKenna A, Shen H, Zack T, et al. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol 2012;30(5):413–21 doi 10.1038/nbt.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Da Cruz Paula A, da Silva EM, Segura SE, Pareja F, Bi R, Selenica P, et al. Genomic profiling of primary and recurrent adult granulosa cell tumors of the ovary. Mod Pathol 2020;33(8):1606–17 doi 10.1038/s41379-020-0514-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schwarz RF, Trinh A, Sipos B, Brenton JD, Goldman N, Markowetz F. Phylogenetic quantification of intra-tumour heterogeneity. PLoS Comput Biol 2014;10(4):e1003535 doi 10.1371/journal.pcbi.1003535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pellegrino M, Sciambi A, Treusch S, Durruthy-Durruthy R, Gokhale K, Jacob J, et al. High-throughput single-cell DNA sequencing of acute myeloid leukemia tumors with droplet microfluidics. Genome Res 2018;28(9):1345–52 doi 10.1101/gr.232272.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Martin M Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 2011;17(1):3 doi 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 42.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009;25(14):1754–60 doi 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009;25(16):2078–9 doi 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010;20(9):1297–303 doi 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Poplin R, Ruano-Rubio V, DePristo MA, Fennell TJ, Carneiro MO, Van der Auwera GA, et al. Scaling accurate genetic variant discovery to tens of thousands of samples. bioRxiv 2018:201178 doi 10.1101/201178. [DOI] [Google Scholar]
- 46.Benjamin D, Sato T, Cibulskis K, Getz G, Stewart C, Lichtenstein L. Calling Somatic SNVs and Indels with Mutect2. bioRxiv 2019:861054 doi 10.1101/861054. [DOI] [Google Scholar]
- 47.Weber LL, Sashittal P, El-Kebir M. doubletD: detecting doublets in single-cell DNA sequencing data. Bioinformatics 2021;37(Suppl_1):i214–i21 doi 10.1093/bioinformatics/btab266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Demaree B, Delley CL, Vasudevan HN, Peretz CAC, Ruff D, Smith CC, et al. Joint profiling of DNA and proteins in single cells to dissect genotype-phenotype associations in leukemia. Nat Commun 2021;12(1):1583 doi 10.1038/s41467-021-21810-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Alberti-Servera L, Demeyer S, Govaerts I, Swings T, De Bie J, Gielen O, et al. Single-cell DNA amplicon sequencing reveals clonal heterogeneity and evolution in T-cell acute lymphoblastic leukemia. Blood 2021;137(6):801–11 doi 10.1182/blood.2020006996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Miller CA, McMichael J, Dang HX, Maher CA, Ding L, Ley TJ, et al. Visualizing tumor evolution with the fishplot package for R. BMC Genomics 2016;17(1):880 doi 10.1186/s12864-016-3195-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Paula ADC, Zhu Y, Bhaloo SI, Pareja F, Hoang T, Selenica P, et al. Abstract 1692: Single-cell DNA sequencing from frozen endometrial tumors to address clonal evolution of somatic mutations. Cancer Research 2022;82(12_Supplement):1692- doi 10.1158/1538-7445.Am2022-1692.35502545 [DOI] [Google Scholar]
- 52.van Dijk D, Sharma R, Nainys J, Yim K, Kathail P, Carr AJ, et al. Recovering Gene Interactions from Single-Cell Data Using Data Diffusion. Cell 2018;174(3):716–29 e27 doi 10.1016/j.cell.2018.05.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The human sequence raw data generated in this study are protected and not publicly available due to patient privacy requirements but are available upon reasonable request from the corresponding author subject to institutional approvals. Cell lines and patient tumor somatic mutations, identified by MSK-IMPACT sequencing, are available in the cBioPortal (https://www.cbioportal.org/study/summary?id=coadread_mskresistance_2022). All relevant cell-free DNA sequencing data are included within the article and supplementary data files.