

Abstract
Purpose:
Immune checkpoint therapy (ICT) is currently ineffective in a majority of patients. Tumor drug exposure measurements can provide vital insights into mechanisms involved in the resistance of solid tumors to those therapeutics, however, tools to quantify in situ drug exposure are few. We have investigated the potential of programmed death ligand- 1 (PD-L1) pharmacodynamics, quantified using positron emission tomography (PET), to inform on the tumor exposure of anti-PD-L1 therapeutics.
Experimental Design:
To non-invasively quantify PD-L1 levels, we first developed a novel peptide-based gallium-68 labeled binder, [68Ga]Ga-DK223, and evaluated its in vivo distribution, pharmacokinetics, and PD-L1 specificity in preclinical models of triple-negative breast cancer (TNBC) and urothelial carcinoma (UC) with variable PD-L1 expression. We then quantified baseline and accessible PD-L1 levels in tumors as a non-invasive pharmacodynamic measure to assess tumor exposure to two anti-PD-L1 antibodies (avelumab and durvalumab).
Results:
DK223 exhibited a KD of 1.01±0.83 nM for PD-L1 and inhibited the PD-1:PD-L1 interaction in a dose-dependent manner. [68Ga]Ga-DK223 provides high-contrast PET images within 60 min of administration and detects PD-L1 in an expression-dependent manner in xenograft models. PD-L1 pharmacodynamics measured using [68Ga]Ga-DK223-PET revealed that avelumab and durvalumab had similar exposure early during therapy, but only durvalumab exhibited sustained exposure at the tumor.
Conclusions:
[68Ga]Ga-DK223 detected variable PD-L1 levels and exhibited salient features required for clinical translation. [68Ga]Ga-DK223-PET could be useful for quantifying total PD-L1 levels at baseline and accessible PD-L1 levels during therapy to understand drug exposure at the tumor, thus supporting its use for guiding and optimizing ICT.
STATEMENT OF TRANSLATIONAL RELEVANCE
Monoclonal antibodies (mAbs) against immune checkpoint proteins have transformed cancer treatment, but only a subset of cancer patients benefit from such immunotherapy. Although much research has focused on the genetic and molecular basis of resistance to immune checkpoint therapeutics, failure to achieve sufficient drug exposure at the tumor site also contributes to acquired resistance. In situ drug exposure is difficult to measure, and recognizing the need for real-time quantitation of drug exposure at the tumor, we developed a gallium-68 labeled peptide radiotracer, [68Ga]Ga-DK223, which provides high-contrast images of programmed death ligand-1 (PD-L1) expression by positron emission tomography (PET). Using [68Ga]Ga-DK223 PET, we demonstrated that the pharmacodynamics of PD-L1 can be used to noninvasively evaluate the exposure of anti-PD-L1 mAbs at the tumor. Our pharmacodynamics-based approach has broader implications for patient selection, guiding therapy, and accelerating the development of new checkpoint inhibitor mAbs.
INTRODUCTION
The past two decades have seen an improved understanding of the roles of the host immune system in fighting cancer1. Monoclonal antibodies (mAb) targeting immune checkpoints have taken center stage in efforts to leverage that understanding for improving cancer therapy, and increasing numbers of agents are in development. Population pharmacokinetic (PK) studies are currently used to optimize the dosing regimens of those therapeutics2. However, the exposure and activity of mAbs in tumors are poorly understood. Although PK data guide dosing regimens, they provide virtually no information about drug exposure at the disease site. Given the poor penetration of mAbs within solid tumors and the heterogeneity of target expression within and across patients, in situ drug exposure measurements could enable personalized dosing and improved efficacy3–5. Measures of drug exposure that are agnostic to the disease type and location, independent of the biophysical properties of mAbs, enable repeat sampling, and encompass all of the factors associated with heterogeneity of tumors may address an unmet clinical need6,7.
Immune checkpoint protein programmed death-ligand 1 (PD-L1, CD274) is one important target for therapeutic efforts in immuno-oncology, with three mAbs now regulatory approved (atezolizumab, avelumab, and durvalumab). PD-L1 expression is currently used as a biomarker to guide immune checkpoint therapy in some cancers8–11, but PD-L1 detection by immunohistochemistry (IHC) involves sampling a small fraction of the tumor and fails to capture total PD-L1 levels or heterogeneity in PD-L1 expression within and across patients12,13. Moreover, PD-L1 expression is dynamic and changes during the course of immunotherapy, suggesting that it could be beneficial to capture total PD-L1 levels and dynamics with use of noninvasive technologies, such as positron emission tomography (PET) 14,15. For example, clinical evaluation of [89Zr]atezolizumab showed that radiotracer uptake is heterogeneous within and across patients and that [89Zr]atezolizumab uptake in lesions is a better predictor of response to anti-PD-L1 (aPD-L1) therapy than genomics-derived biomarkers16.
To complement these efforts, we and others have reported small-protein- and peptide-based imaging agents that provide high-contrast images much sooner than mAb-derived radiotracers17,18. One such peptide-based radiotracer, [68Ga]WL12, showed PD-L1 dependent uptake in preclinical studies and in patients with NSCLC within 120 min of injection19. In addition, our peptide-based radiotracers bind PD-L1 in a manner similar to that of endogenous receptor programmed death protein 1 (PD-1) and aPD-L1 therapeutics20. This unique binding mode enabled us to quantify the target engagement of aPD-L1 therapeutics by measuring accessible PD-L1 levels (target levels not occupied by mAbs) using PET. All three approved PD-L1 mAbs fit a two-compartment linear clearance PK model, but their temporal kinetics in the tumor remain to be characterized21.
Here, we report the development of a new gallium-68 labeled peptide-based radiotracer for PD-L1 that shows substantially improved pharmacokinetic properties compared to previously reported agents. We evaluated the pharmacokinetics, biodistribution, and PD-L1 specificity of [68Ga]Ga-DK223 in vitro and in vivo using multiple cell lines and xenografts with variable PD-L1 levels derived from triple-negative breast cancer (TNBC) and urothelial carcinoma (UC). Moreover, we evaluated whether accessible PD-L1 levels within the tumor microenvironment, measured using [68Ga]Ga-DK223 PET, inform the temporal kinetics of drug exposure of two PD-L1 mAbs (avelumab and durvalumab) in the tumor, where it is most relevant1.
MATERIALS AND METHODS
Chemicals.
DK221 ((Cyclo-(-Ac-Tyr-NMeAla-Asn-Pro-His-Glu-Hyp-Trp-Ser-Trp(Carboxymethyl)-NMeNle-NMeNle-Lys)-Gly-NH2)) was custom synthesized by CPC Scientific (Sunnyvale, CA) with >95% purity. 1,4,7,10-Tetraazacyclododecane-1,4,7,10-tetraacetic acid mono-N-hydroxysuccinimide ester (DOTA-NHS ester) and 2,2′,2”-(10-(2,6-dioxotetrahydro-2H-pyran-3-yl)-1,4,7,10-tetraazacyclododecane-1,4,7-triyl)triacetic acid (DOTA-GA anhydride) were purchased from Macrocyclics (# B-280) and CheMatech (#C109), respectively. All other chemicals were purchased from Sigma-Aldrich or Fisher Scientific.
Synthesis of DK223:
In a reaction vial, to a stirred solution of DK221 (5.0 mg, 2.55 μmol) in Dimethylformamide (1.0 mL), was added Diisopropylethylamine (0.66 mg, 5.1 μmol) followed by DOTA-NHS Ester (3.0 mg, 3.8 μmol). The reaction mixture was stirred for 2 h at room temperature. The reaction mixture was concentrated using a rotavapor before purification on a reverse-phase high-performance liquid chromatography (RP-HPLC) system using a semi-preparative C-18 Luna column (5 mm, 10 × 250 mm Phenomenex, Torrance, CA). The HPLC method used gradient elution starting with 20% acetonitrile (0.1% TFA) and reaching 60% in 25 min at a flow rate of 5 mL/min co-buffered with water (0.1% TFA). The desired DK223 was collected at 8.3 min, solvent evaporated under reduced pressure, residue reconstituted in 20% acetonitrile, and lyophilized to powder form with 68% yield. Purified DK223 was characterized by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS). Calculated [M+H]+:2343.61; observed:2343.62.
Synthesis of DK397:
In a reaction vial, to a stirred solution of DK223 (1.0 mg, 0.42 μmol) in 1M NaOAc buffer pH 4.0 was added 10 μL of 0.1M GaCl3 solution. The reaction mixture was then heated at 95°C for 10 min. After cooling reaction mixture was loaded onto activated C18 sep-pack, washed with 5 mL DI water, and eluted with 90% acetonitrile. The solvent was evaporated under reduced pressure, residue reconstituted in 20% acetonitrile and lyophilized to form an off-white powder with 90% yield. The final product was characterized using MALDI-TOF. Calculated [M+H]+:2410.31; observed:2410.95. The DK397 complex was then used to optimize RP-HPLC conditions as a standard for radiolabeling and for PD-L1 and PD-1 competition binding assays.
Synthesis of [68Ga]Ga-DK223 and [68Ga]Ga-DK385.
The 68Ge/68Ga generator was eluted manually using 6 mL of 0.1M HCl (ultrapure trace-metal-free) by fractional elution (1 mL/fraction). To a reaction vial (5 mL) containing 200 μL of 1 M sodium acetate buffer (NaOAc; pH 5.5) and 20-40 μg of DK223, 7-19 mCi 68GaCl3 was added (1 mL fraction eluate). Ascorbic acid (50 μg) was added as a stabilizer to quench free radicals. The reaction mixture was then incubated for 10 min at 100 °C in a temperature-controlled heating block. The reaction vial was cooled initially at room temperature for 1 min followed by 2-3 min in cold water and quenched with 20% acetonitrile in water. The crude reaction mixture was purified on an RP-HPLC system using a semi-preparative C-18 Luna column (5 mm, 10 × 250 mm Phenomenex, Torrance, CA, USA). The HPLC gradient elution started with 25% acetonitrile (0.1% formic acid) and reached 60% in 15 min at a flow rate of 5 mL/min, co-buffered with water (0.1% formic acid). The radiolabeled product was collected at 6.63 min, evaporated under stream of N2 at 60°C and formulated in 5% EtOH in saline. [68Ga]Ga-DK223 was obtained with a non-decay radiochemical yield of 20±5% (n=37) and >95% radiochemical purity. The formulated tracer was used for both in vitro and in vivo studies. [68Ga]Ga-DK385 was similarly synthesized using fractional elution method with a non-decay radiochemical yield of 26±7% (n=7) and >95% radiochemical purity. For stability studies, see supplementary methods.
Surface plasmon resonance (SPR) analysis.
SPR analysis was carried out using the Biacore Molecular Interaction Shared Resource at Georgetown University. All experiments were performed using a Biacore T200 instrument equipped with a CM5 chip at 25°C. Human PD-L1 Fc-tagged (52 kDa, 1.93 μM stock concentration) was used as a ligand for capture onto the CM5 chip. DK223 (2342.6 Da, 10 mM stock concentration), DK385 (2414.7 Da, 10 mM stock concentration), and DK397 (2409.31 Da, 10 mM stock concentration) were used as analytes to flow over the ligand-capture surface. Flow cell (FC) 1 was used as the reference for FC2. Protein A/G was diluted (1:25 dilution, 1 μM diluted concentration) in 10 mM sodium acetate buffer at pH 4.5, and immobilized on FC1 and FC2 to a level of ~4400 RU using standard amine coupling chemistry. PBS-P (20 mM phosphate buffer pH 7.4, 137 mM NaCl, 2.7 mM KCl, 0.05% v/v surfactant P20) was used as the immobilization running buffer. Human PD-L1 was diluted (1:10 dilution) in PBS-P captured on FC2 to ~1200 RU. PBS-P was used as the capture buffer. Based on these captured response values, theoretical Rmax values were calculated assuming a 1:1 interaction mechanism. The flow rate of all the analyte solutions was maintained at 50 μL/min. The contact and dissociation times used were 120 s and 360 s, respectively. Glycine pH 2.0 was injected for 15 seconds for surface regeneration. This regeneration condition removes all the captured ligands from FC2. Therefore, fresh ligands are captured at the beginning of each injection cycle. The injected analyte concentrations ranged from 20 nM to 0.625 nM (two-fold dilutions). All analytes were injected in duplicates. For analysis, sensorgrams from the overnight kinetics were evaluated using 1:1 kinetics model fitting.
PD-L1 and PD-1 binding inhibition assay.
A competitive inhibition assay for PD-L1 binding to PD-1 was optimized from a previously described fluorescence resonance energy transfer (FRET)-based assay in discussion with Cisbio22 using PD-1-Ig and PD-L1-His-tag that were detected by anti-human IgG-Eu3+ cryptate and anti-6HIS-XL665 monoclonal antibodies using a Perkin Elmer Victor3 1420 multi-label counter (PerkinElmer, Waltham, MA).
Cell culture reagents and antibodies.
All cell culture reagents were purchased from Invitrogen (Carlsbad, CA, USA). The aPD-L1 mAbs avelumab and durvalumab were purchased from the Johns Hopkins School of Medicine Pharmacy.
Cell culture.
All cell lines were purchased from ATCC and cultured in the recommended medium in an incubator at 37°C in an atmosphere containing 5% CO2. MDAMB231, SCaBER, and BFTC909 cells were maintained in DMEM. SUM149 was a gift from Dr. Stephen Ethier and maintained in Ham’s F-12K medium. A549, A549iPD-L1, CHO, CHOhPD-L1 was cultured as described previously20. All cells were supplemented with 10% Fetal Bovine serum and 1% P/S antibiotic. All cell lines were routinely tested for mycoplasma. Freshly thawed cells were cultured for less than 3 months or authenticated at JHU Genomics Core.
Detection of PD-L1 expression by flow cytometry.
Adherent cells were detached using enzyme-free cell dissociation buffer (Thermo Fisher Scientific, Waltham, MA). PD-L1 surface expression was evaluated by direct staining of 1x106 cells in 100 μL FACS buffer (PBS with 0.1% FBS and 2 mM ethylenediaminetetraacetic acid) with anti-PD-L1 antibody (clone: MIH1 BD # 558065) for 30 min at 4°C. The cells were then washed and analyzed for mean fluorescence intensity (MFI) by flow cytometry.
In vitro binding assays with [68Ga]Ga-DK223.
In vitro binding of [68Ga]Ga-DK223 to MDAMB231, BFTC909, SUM149, and SCaBER cells was determined by incubating 1×106 cells with approximately 1 μCi [68Ga]Ga-DK223 for 45 min at 4°C. After incubation, cells were washed three times with ice cold PBS containing 0.1% tween 20 and counted on an automated gamma counter. Percentage of incubated activity (%IA) was calculated based on signal decay correction and normalization to external [68Ga]Ga-DK223 standards measured in quadruplicate. All cell radioactivity uptake studies were performed in quadruplicate for each cell line and repeated thrice.
Receptor density measurements.
Phycoerythrin (PE) Fluorescence Quantitation Kit (BD Biosciences #340495) containing four levels of PE/bead were used. Beads were reconstituted, with 0.5 mL of PBS containing sodium azide and 0.5% bovine serum albumin, just before use. Cells were stained for 30 min at 4°C with PE labeled anti-PD-L1 antibody (clone: MIH1 BD # 558065) and run along with the beads to estimate receptor density using flow cytometry. Calibration curve of geometric mean vs PE/bead from different bead populations was generated as per the manufacturer’s protocol. This calibration curve was used to derive receptors/cell for each cell type from their respective geometric means. Isotype controls were used to eliminate any non-specific staining. PD-L1 receptor density measured above was then correlated with [68Ga]Ga-DK223 %IA uptake measured in in vitro binding assays.
In vivo studies.
All mouse studies were conducted using protocols approved by the Johns Hopkins University Animal Care and Use Committee (ACUC). Xenografts were established in five-to-six-week-old, male (UC) or female (TNBC), non-obese, diabetic, severe-combined immunodeficient gamma (NSG) mice obtained from Johns Hopkins University Immune Compromised Animal Core.
Xenograft models.
Mice were subcutaneously injected with cancer cells in 100 μL saline (top right flank, unless otherwise noted) in all tumor models. The following cell lines were used: A549 (5 M), A549iPD-L1 (5 M), CHO (10 M), CHOhPD-L1 (10 M), MDAMB231 (1 M), SUM149 (2 M), BFTC909 (1 M), and SCaBER (5 M). Mice with tumor volumes of 100-200 mm3 were used for treatment, PET imaging, and ex vivo biodistribution experiments.
PET-CT imaging of mouse xenografts.
Mice with 100-200 mm3 tumor volume were injected with 200 μCi (7.4 MBq) of [68Ga]Ga-DK223 in 200 μL of 5% ethanol in saline intravenously and anesthetized with 2.5% isoflurane. PET images were acquired 60 min after radiotracer injection for 10 min in one or two 2 bed positions (whole body) using an ARGUS small-animal PET/CT scanner (Sedecal, Madrid, Spain). PET data were reconstructed using the two-dimensional ordered subset expectation maximization algorithm (2D-OSEM) and corrected for radioactive decay and dead time. The percentage of incubated dose per cubic centimeter (%ID/cc) values were calculated based on the calibration factor obtained from a known radioactive quantity. Image fusion, visualization, and 3D rendering were accomplished using Amira 6.1® (FEI, Hillsboro, OR, USA).
Ex vivo biodistribution.
To validate the imaging studies, ex vivo biodistribution studies were conducted in mice with at least 100-200 mm3. The mice received 80 μCi (2.96 MBq) [68Ga]Ga-DK223 for dosimetry studies and 50 μCi (1.85 MBq) for all other biodistribution studies. Total mass of non-radioactive peptide varied from 0.03 to 0.15 μg/kg. All injections were in 200 μL of 5% ethanol in saline intravenously, and biodistribution studies were conducted at specified time points for radiotracer pharmacokinetics studies and at 60 min after [68Ga]Ga-DK223 injection for all other studies (n=4-5). Selected tissues (the heart, lung, liver, stomach, spleen, pancreas, kidney, small intestine, testicles, and muscle), tumors, and blood were collected, weighed, and counted, and their incubated dose per gram (%ID/g) values were calculated based on signal decay correction and normalization to external [68Ga]Ga standards measured in triplicate.
Immunohistochemistry.
Tumor sections were evaluated for PD-L1 expression by immunohistochemistry (IHC) as reported previously11. Briefly, 4 μm tumor sections were treated with 3% H2O2 for 10 minutes, blocked with 5% goat serum for 1 h, and then incubated with a primary anti-human PD-L1 antibody (#13684, Cell Signaling) at 1:500 dilution at 4°C overnight. Subsequently, using Dako CSAII Biotin-free Tyramide Signal Amplification System kit, slides were incubated with secondary antibody, amplification reagent, and with anti-fluorescein-HRP. Final staining was carried out by adding DAB chromogen.
In vivo stability of [68Ga]Ga-DK223.
[68Ga]Ga-DK223 (Rt = 7.43 min) was generated with more than 99% radiochemical purity. Stability studies were conducted by injecting 250±17 μCi of [68Ga]Ga-DK223 intravenously (n=3). At 60 min, 6 μL of urine was collected having 24±8 μCi of activity which was diluted 3x with cold ethanol followed by syringe filtration. The filtered urine sample further diluted 10x with water and injected into HPLC. The radio HPLC chromatogram of urine sample showed intact tracer (Rt = 7.34 min) and no radioactive side peaks.
Effect of non-radioactive DK223 on [68Ga]Ga-DK223 uptake in tumors.
[68Ga]Ga-DK223 was co-administered with non-radioactive DK223 at doses of 0, 1.2, 4, 12, and 1200 μg/kg, and uptake was assessed in BFTC909 UC xenografts using PET imaging.
iPD-L1 treatment in A549 xenografts.
NSG male mice harboring subcutaneous A549 and A549hPD-L1 tumors were treated daily with a 200 μL of 10 mg/mL doxycycline solution by oral gavage (n=3/group). PET-CT imaging was performed 48 h after treatment.
aPD-L1 treatment in BFTC909 xenografts.
NSG male mice harboring subcutaneous BFTC909 tumors were treated with a 10 mg/kg dose of avelumab, durvalumab, or saline injected intravenously (n=6/group). PET-CT imaging was performed every day after treatment for up to 5 days. For biodistribution studies, mice were treated on days 1, 2, 3, 4, and 5 and sacrificed on day 6, resulting in 120, 96, 72, 48, and 24 h of treatment, respectively.
Statistical analysis.
All statistical analyses were performed using Prism 8 (GraphPad Software, La Jolla, CA, USA). Unpaired Student’s t-test and one- or two-way ANOVA were utilized for column, multiple column, and grouped analyses, respectively. Statistical significance was set at P<0.05. Correlation was performed using simple linear regression without keeping the term constant at zero.
Data Availability:
All study data are included in the article and/or supplementary information. The additional data are available upon request from the corresponding author.
RESULTS
Synthesis and characterization of DK223 as PD-L1 binding moiety
DK223 is derived from DK221, a 14 amino acid cyclic peptide with a free lysine amine, that has demonstrated specificity for human PD-L123,24. To evaluate the effect of chelators on binding affinity and in vivo pharmacokinetics, the free lysine amine of DK221 was conjugated to DOTA or DOTAGA chelators to effectively bind gallium-68 to produce [68Ga]Ga-DK223 or [68Ga]Ga-DK385, respectively, as well as the non-radioactive analog DK397 (Figure 1A and S1A–L). DK223 and DK385 bound to PD-L1 with an affinity of 1.01±0.83 nM and 5.7±0.91 nM, respectively, as per surface plasmon resonance (SPR) analysis (Figure 1B). DK223 and DK385 also inhibited the PD-1:PD-L1 interaction in a dose-dependent manner, with an IC50 of 27±4.9 nM (Figure 1C). The radiolabeled products [68Ga]Ga-DK223 and [68Ga]Ga-DK385 were obtained with non-decay corrected radiochemical yields of 20±5% (n=37) and 26±7% (n=7), respectively, with >95% radiochemical purity. We observed a moderate specific activity (molar activity) of 10.36 GBq/μmol (280 mCi/μmol) with [68Ga]Ga-DK223. The formulated [68Ga]Ga-DK223 dose was stable for 4h (Figure S1M).
Figure 1. Synthesis and in vitro characterization of DK221 derivatives.
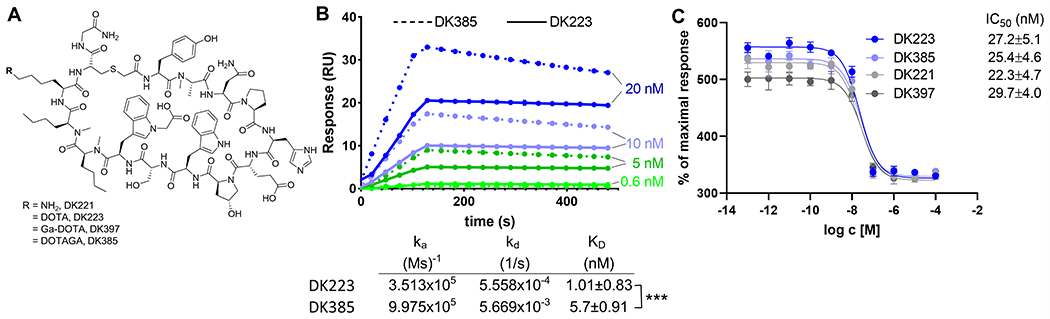
A, Structures of parent peptide DK221 and its analogs. B, Surface plasmon resonance (SPR) analysis of DK223 (n=3) and DK385 (n=3) binding to PD-L1 C, Fluorescence resonance energy transfer (FRET)-based assay showing sub-nanomolar affinities for DK221 (n=3), DK223 (n=3), DK385 (n=3) and DK397 (n=3) for inhibiting PD1:PD-L1 interaction. ***P<0.001 by student’s t test
Pharmacokinetics of [68Ga]Ga-DK223
Biodistribution studies conducted with [68Ga]Ga-DK385 showed less tumor uptake (8.50±1.67 %ID/g) and tumor-to-muscle (19.86±2.15) and tumor-to-blood (2.32±0.38) ratios at 60 min compared to [68Ga]Ga-DK223 (Figure S2A). Thus, we pursued detailed evaluation of [68Ga]Ga-DK223.
[68Ga]Ga-DK223 is specific to hPD-L1 and lacks reactivity towards mPD-L1 (Figure S2B–D). Therefore, to evaluate the pharmacokinetics, biodistribution, and in vivo specificity of [68Ga]Ga-DK223 for binding PD-L1, we used immunodeficient NSG mice with MDAMB231 human TNBC xenografts expressing PD-L1 (Figure 2A). PET images of MDAMB231 tumor-bearing mice injected with [68Ga]Ga-DK223 revealed high radiotracer accumulation in tumors as early as 5 minutes after injection. A higher tumor contrast was observed at 60 min owing to background radiotracer clearance. Administration of 2 mg/kg non-radioactive DK223 reduced tracer uptake in MDAMB231 xenografts demonstrating the specificity of [68Ga]Ga-DK223 for PD-L1. In comparison, low PD-L1 expressing SUM149 tumors showed low levels of uptake, similar to levels seen in non-PD-L1 expressing tissues such as muscle. Kidney and bladder tissues showed the highest radiotracer accumulation among normal tissues, consistent with the renal clearance mechanism for low-molecular-weight peptides. To confirm the PET results, ex vivo biodistribution studies were performed (Table 1). [68Ga]Ga-DK223 uptake reached a maximum of 15.21±1.43% %ID/g in MDAMB231 tumors at 60 min and demonstrated high tumor retention even at 180 min (Figure 2B). Blood pool activity showed consistent washout, with nearly 90% of activity cleared by 60 min. Similar radioactivity washouts were observed in all other non-specific tissues. Consequently, the tumor-to-blood and tumor-to-muscle ratios were the highest between 60 and 180 min (Figure 2C). The in vivo stability of [68Ga]Ga-DK223 was also measured in urine collected at 60 min, which showed no metabolic cleavage (Figure S2E). Therefore, we used the 60-minute time point for all further experiments, as it provided good contrast and fits within the standard PET clinical workflow.
Figure 2. Pharmacokinetics of [68Ga]Ga-DK223.
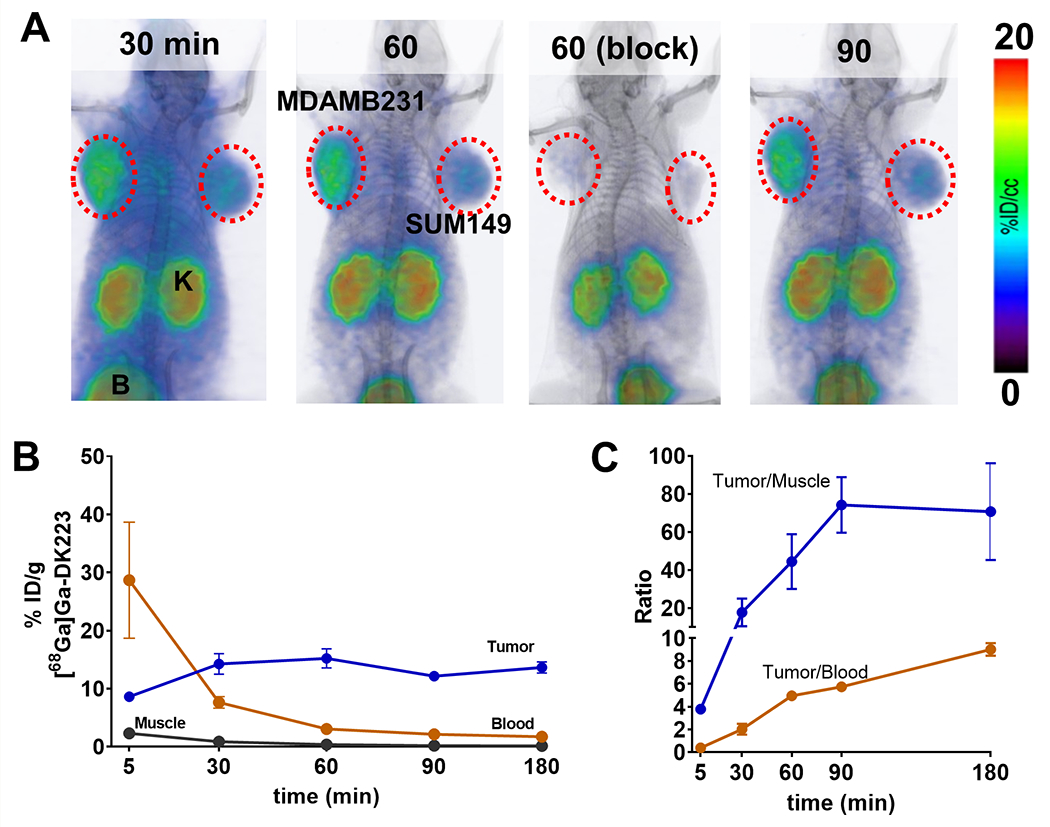
A, PET-CT images in NSG mice bearing MDAMB231 (left flank) and SUM149 (right flank) tumors at different time-points show high tumor-to-background ratio at 60 min after injecting [68Ga]Ga-DK223. Radioactivity uptake reduced in MDAMB231 tumors after blocking with unlabeled 50 μg DK223 confirming PD-L1 specificity. Mice were injected with ~ 7.4 MBq (~200 μCi) [68Ga]Ga-DK223 (K: Kidney; B: bladder) (n=3). B, Pharmacokinetics of [68Ga]Ga-DK223 in tumor, blood and muscle, and C, tumor-to-tissue ratios. For B and C, mice were injected with ~ 740 kBq (~50 μCi) [68Ga]Ga-DK223 and sacrificed at different time-points after injection (n=4-5).
Table 1. Pharmacokinetics of [68Ga]Ga-DK223 in mice with MDAMB231 xenografts and predicted residence times in human organs with respective absorbed doses.
Ex vivo biodistribution studies in NSG mice bearing MDAMB231 tumors. Mice were injected with ~ 740 kBq (~50 μCi) [68Ga]Ga-DK223 and sacrificed after corresponding time-point (n=4-5). Blocking was performed by treating mice with non-radioactive 50 μg DK223 confirming PD-L1 specificity. Residence time and equivalent human organ dose was calculated using OLINDA/XEM software.
| Tissues | %ID/g [68Ga]Ga-DK223 time (min) | Residence time (h) | Organ Dose (rem/mCi) | |||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| 5 | 30 | 60 | 60 (block) | 90 | 180 | |||
|
| ||||||||
| Blood | 28.67 ± 8.65 | 7.63 ± 0.87 | 3.05 ± 0.11 | 1.09 ± 0.18 | 2.14 ± 0.08 | 1.72 ± 0.04 | 0.17533 | n/a |
| Muscle | 2.29 ± 0.09 | 0.85 ± 0.09 | 0.36 ± 0.04 | 0.85 ± 0.07 | 0.17 ± 0.01 | 0.08 ± 0.02 | 0.06168 | 0.0083 |
| Tumor | 8.62 ± 0.29 | 14.26 ± 1.54 | 15.21 ± 1.43 | 0.04 ± 0.01 | 12.15 ± 0.32 | 11.59 ± 0.79 | n/a | n/a |
| Lung | 14.83 ± 0.4 | 11.03 ± 2.29 | 4.7 ± 0.42 | 0.57 ± 0.1 | 5.3 ± 1.68 | 2.98 ± 0.71 | 0.03245 | 0.0705 |
| Thymus | 9.96 ± 0.82 | 7.02 ± 1.06 | 3.53 ± 0.27 | 2.76 ± 0.16 | 1.26 ± 0.47 | 1.52 ± 0.75 | 0.00047 | 0.0434 |
| Heart | 6.39 ± 0.35 | 2.49 ± 0.3 | 0.95 ± 0.07 | 2.42 ± 0.4 | 0.73 ± 0.03 | 0.46 ± 0.03 | 0.0151 | 0.0558 |
| Liver | 5.63 ± 0.29 | 2.85 ± 0.41 | 1.31 ± 0.08 | 0.99 ± 0.14 | 0.91 ± 0.11 | 0.82 ± 0.09 | 0.01633 | 0.0252 |
| Stomach (with contents) | 2.2 ± 0.16 | 1.39 ± 0.31 | 0.65 ± 0.09 | 0.35 ± 0.07 | 0.46 ± 0.09 | 0.37 ± 0.04 | 0.00072 | 0.0061 |
| Pancreas | 3.04 ± 0.11 | 2.01 ± 0.54 | 0.45 ± 0.05 | 0.41 ± 0.02 | 0.39 ± 0.01 | 0.24 ± 0.03 | 0.00052 | 0.015 |
| Spleen | 3.69 ± 0.13 | 2.45 ± 0.51 | 1.05 ± 0.07 | 0.91 ± 0.07 | 0.71 ± 0.06 | 0.58 ± 0.03 | 0.00129 | 0.0196 |
| Small Intestine (with contents) | 3.41 ± 0.37 | 1.64 ± 0.32 | 0.65 ± 0.2 | 0.44 ± 0.04 | 0.96 ± 0.09 | 0.74 ± 0.17 | 0.00453 | 0.013 |
| Large Intestine (with contents) | 2.01 ± 0.16 | 0.79 ± 0.1 | 0.52 ± 0.09 | 0.29 ± 0.06 | 0.42 ± 0.1 | 1.74 ± 0.37 | 0.00099 | 0.0084 |
| Adrenals | 4.18 ± 0.26 | 2.22 ± 0.19 | 4.74 ± 3.64 | 1.32 ± 0.09 | 0.18 ± 0.03 | 0.16 ± 0.11 | 0.00014 | 0.0181 |
| Kidney | 28.59 ± 2.4 | 24.4 ± 1.5 | 22.99 ± 2.01 | 26.34 ± 0.19 | 16.23 ± 0.8 | 19.14 ± 1.32 | 0.0337 | 0.21 |
| Ovaries | 6.1 ± 0.55 | 2.69 ± 0.34 | 0.93 ± 0.08 | 2.92 ± 0.79 | 0.47 ± 0.09 | 0.27 ± 0.18 | 0.00011 | 0.017 |
| Uterus | 7.48 ± 1.37 | 3.82 ± 0.57 | 1.52 ± 0.26 | 1.03 ± 0.06 | 1.13 ± 0.09 | 0.47 ± 0.03 | 0.00114 | 0.0253 |
| Bladder | 5.74 ± 0.71 | 4.09 ± 0.49 | 1.42 ± 0.12 | 2.69 ± 1.82 | 0.78 ± 0.07 | 1.28 ± 0.32 | 0.00187 | 0.0081 |
| Femur | 10.58 ± 5.79 | 1.74 ± 0.14 | 0.89 ± 0.06 | 0.34 ± 0.03 | 0.59 ± 0.04 | 0.64 ± 0.07 | 0.03956 | 0.0333 |
| Brain | 0.65 ± 0.08 | 0.22 ± 0.05 | 0.08 ± 0.01 | 0.44 ± 0.09 | 0.05 ± 0.01 | 0.04 ± 0.01 | 0.00118 | 0.0029 |
Human Radiation Dosimetry Estimates
Pharmacokinetic data evaluated from MDAMB231 tumor-bearing mice were decay-corrected and used to calculate the residence times of [68Ga]Ga-DK223 in human organs (Table 1 and Figure S2F). We then used these residence times as inputs for OLINDA/EXM software to obtain absorbed organ doses for gallium-68 in the adult reference human female phantom. The predicted absorbed doses in the human organs are listed in Table 1 and Figure S2G. The organs receiving the maximum dose were the kidneys (0.21 rem/mCi), followed by the lungs (0.07 rem/mCi), heart wall (0.06 rem/mCi), and thymus (0.04 rem/mCi). Based on these findings, a 20 mCi dose can be administered with an effective dose equivalent estimate of less than 5 rem to acquire PET images in human subjects.
Evaluation of [68Ga]Ga-DK223 in UC and TNBC cancer xenografts
Next, we validated the ability of [68Ga]Ga-DK223 to detect variable levels of PD-L1 in vivo, using xenografts of two TNBC cell lines (MDAMB231 and SUM149) and two UC cell lines (BFTC909 and SCaBER). PD-L1 levels in these cell lines measured using flow cytometry (Figures 3A and S3A) were in the following order: BFTC909 > MDAMB231 > SCaBER > SUM149. Cells incubated with [68Ga]Ga-DK223 showed highest levels of uptake in BFTC909 (28.9±0.55 %IA), followed by MDAMB231 (21.4±0.57), and less than 2 %IA in SCaBER and SUM149 cells, in agreement with flow cytometry data (Figure 3B). Cells incubated with 1 μM non-radioactive DK223 showed significantly reduced [68Ga]Ga-DK223 uptake (P<0.0001), confirming the uptake specificity of the radiotracer. To establish relevance of PD-L1 expression to radioactivity uptake, we measured cell surface PD-L1 density (Figure 3C) and found a strong correlation between PD-L1 density and [68Ga]Ga-DK223 uptake (R2=0.9370). Taken together, these cell-based assays provide evidence for the potential of [68Ga]Ga-DK223 to detect variable PD-L1 levels.
Figure 3. Evaluation of [68Ga]Ga-DK223 in UC and TNBC xenografts.
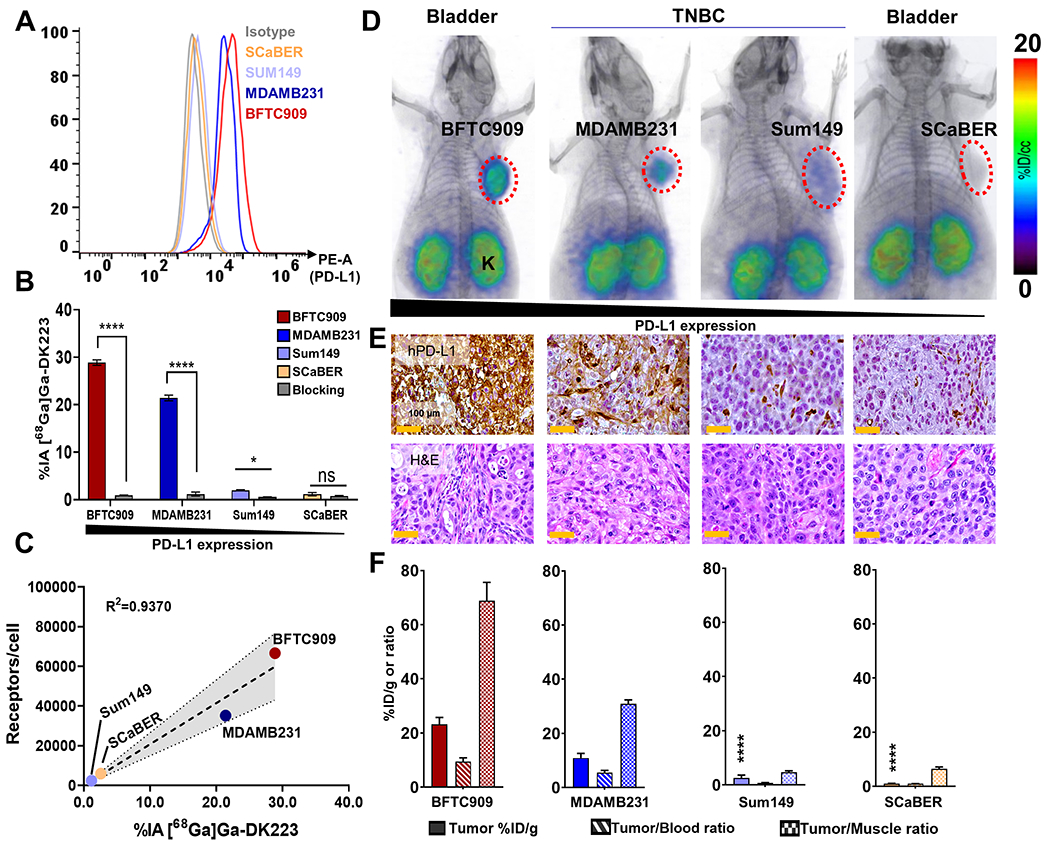
A, Flow cytometry analysis of surface expression of PD-L1 in different cancer cell lines. B, In vitro uptake of [68Ga]Ga-DK223 in different cell lines. [68Ga]Ga-DK223 uptake is PD-L1 expression dependent, and co-incubation with 1 μM of unlabeled peptide reduced uptake confirming PD-L1 specificity. C, In vitro [68Ga]Ga-DK223 uptake correlates with PD-L1 receptor density. D, PET-CT images of NSG mice bearing UC and TNBC tumors at 60 min showing PD-L1 specific tumor uptake. Mice were injected with ~ 7.4 MBq (~200 μCi) (n=2-3) (K: Kidney; B: bladder). E, IHC staining for PD-L1 of the corresponding tumors. F, Tumor and tumor-to-normal tissue uptake derived using [68Ga]Ga-DK223 ex vivo biodistribution studies in NSG mice bearing respective cancer xenografts (n=3-5). Mice were injected with ~ 740 kBq (~20 μCi) and sacrificed after 60 min. **** P<0.0001 by unpaired Student’s t test when compared with either MDAMB231 or BFTC909 tumors. Simple linear regression and Pearson coefficient were used in C.
To validate those in vitro results, we performed PET imaging of tumor xenografts derived from the same four cell lines (Figure 3D). We observed the highest uptake of [68Ga]Ga-DK223 in BFTC909 tumors, followed by MDAMB231, which was also confirmed by region of interest (ROI) analyses of the tumors (Figures S3B–C). Tumors of low PD-L1-expressing SCaBER and SUM149 cell lines showed correspondingly low radiotracer uptake. IHC staining of the same xenografts for PD-L1 corroborated the PET data (Figure 3E). Next, ex vivo biodistribution studies were performed to validate the imaging findings. We found that the tumor uptake was in agreement with the in vitro and PET results, with the highest uptake observed with BFTC909 (21±2.1 %ID/g), followed by MDAMB231 (11±1.6 %ID/g). In contrast, SCaBER and SUM149 tumors showed less than 3 %ID/g of [68Ga]Ga-DK223, consistent with their low PD-L1 status. Tumor-to-muscle and tumor-to-blood ratios showed similar trends with respect to PD-L1 status (Figure 3F), and background tissues showed no significant differences between tumor models (Figures S3D–E). Further analysis showed an absence of a positive correlation between tumor mass and %ID/g, indicating that uptake of [68Ga]Ga-DK223 in BFTC909 and MDAMB231 xenografts was not size-dependent but rather PD-L1 specific (Figures S3F–G). We also evaluated the effect of precursor mass (i.e., effective specific activity) on the uptake of [68Ga]Ga-DK223 in BFTC909 xenografts. We observed that 12 μg/kg of non-radioactive DK223, a human equivalent dose of 1.15 μg/kg - had a minimal effect on [68Ga]Ga-DK223 uptake in tumors (Figure S3H).
In addition to testing human tumor xenografts of natural expressing PD-L1, we also tested [68Ga]Ga-DK223 in genetically engineered cell lines and tumors with constitutive PD-L1 expression, specifically CHOhPD-L1, and corresponding negative controls. Both in vitro cell uptake and PET imaging studies showed hPD-L1 uptake in CHOhPD-L1 and not in CHO cells and tumors (Figures S4A–C). Moreover, to evaluate the potential of [68Ga]Ga-DK223 to detect inducible PD-L1 expression that is seen during immune checkpoint therapy, we employed a genetically engineered cell line A549iPD-L1 that induces hPD-L1 upon doxycycline treatment.
Induced PD-L1 expression was first validated by flow cytometry using cells that were treated with 1 μg/mL doxycycline for 48 h (Figure S5A–B). PD-L1 expression was significantly higher in A549iPD-L1 cells treated with doxycycline, and we also observed increased [68Ga]Ga-DK223 uptake in a radiotracer binding assay (Figure S5C). Furthermore, we observed high [68Ga]Ga-DK223 uptake in vivo in A549iPD-L1 tumors by PET imaging and no increase in uptake in A549 tumors. This confirmed the ability of [68Ga]Ga-DK223 to detect inducible PD-L1 levels in vivo. Taken together, these results demonstrate that [68Ga]Ga-DK223 can detect the variability in PD-L1 levels in tumors in vivo.
PD-L1 mAb exposure in the tumor is quantified by measuring accessible PD-L1 levels using [68Ga]Ga-DK223 PET.
Three PD-L1 mAbs (atezolizumab, avelumab, and durvalumab) have been approved by the United States Food and Drug Administration for the treatment of metastatic UC. The overall response rate to these therapeutics is 13-25%, with higher response rates observed in patients with high PD-L1 expression25. The plasmatic exposure-efficacy relationship analysis of these therapeutics did not indicate a significant relationship, suggesting that more robust indicators are needed to guide therapy in UC21. We reasoned that differences in PD-L1 antibody exposure in the tumors could be estimated by quantifying accessible PD-L1 levels using [68Ga]Ga-DK223 PET (Figure 4A–B). To demonstrate the feasibility of this approach, we selected two PD-L1 antibodies, avelumab and durvalumab, with distinct biophysical properties. In particular, avelumab has a shorter half-life (6.1 vs 18 days) and weaker affinity (0.7 vs 0.02 nM) than durvalumab20. We first evaluated whether mAb binding to PD-L1 could be assessed using [68Ga]Ga-DK223 incubated with BFTC909 cells in the presence (60 nM) or absence of mAb (Figure 4C). In the absence of mAbs, a 27.9% uptake was observed, but less than 1% uptake was observed in presence of mAbs, indicating that [68Ga]Ga-DK223 can only bind accessible PD-L1 not bound by avelumab or durvalumab. No significant difference in [68Ga]Ga-DK223 binding was observed between mAb treatments. Extending this observation in in vivo experiments, we performed baseline [68Ga]Ga-DK223 PET imaging in BFTC909 xenografts, where we observed high radiotracer uptake in the tumors (Figure 4D). Mice were then treated with 10 mg/kg of avelumab or durvalumab, and [68Ga]Ga-DK223 PET images were acquired at 24, 48, 72, and 120 h after treatment (Figures 4E and S6). At 24 h, we observed a significantly lower uptake of [68Ga]Ga-DK223 in the tumors of mice treated with mAbs, reflecting the low accessibility of PD-L1 in vivo. At 72 h, avelumab-treated mice showed increased [68Ga]Ga-DK223 uptake in tumors compared with durvalumab. At 120 h, avelumab-treated mice showed [68Ga]Ga-DK223 uptake similar to that of saline controls. In contrast, [68Ga]Ga-DK223 uptake in durvalumab-treated mice was low due to the low accessibility of PD-L1, indicating that durvalumab remained bound to PD-L1.
Figure 4. [68Ga]Ga-DK223 PET reveals temporal differences in tumor exposure of PD-L1 antibodies in BFTC909 xenografts.
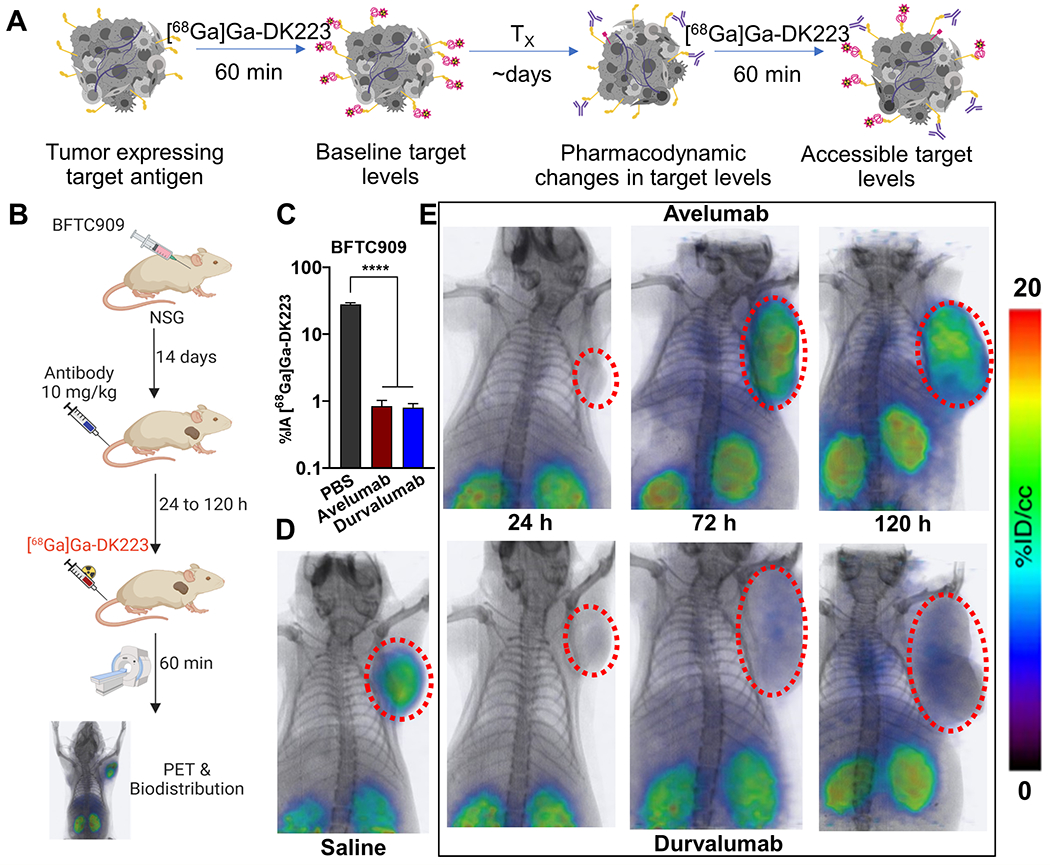
A, Schematic showing measurement of accessible target levels (PD-L1) by [68Ga]Ga-DK223 after mAb treatment (Created with BioRender.com). B, Experimental schematic C, [68Ga]Ga-DK223 uptake in BFTC909 cells with and without the presence of 60 nM of aPD-L1 mAbs. Reduced uptake is observed in the presence of mAbs, confirming the ability of [68Ga]Ga-DK223 to measure only accessible PD-L1 in presence of mAbs. D, [68Ga]Ga-DK223 PET-CT image of saline treated NSG mice bearing BFTC909 tumor at 60 min showing high tumor uptake (n=3). E, Longitudinal PET-CT study of mice bearing BFTC909 tumors and treated with 10 mg/kg of either avelumab (top) or durvalumab (bottom) for different duration of treatment (n=2-3). All images were acquired 60 min after injection of ~ 7.4 MBq (~200 μCi) [68Ga]Ga-DK223. **** P<0.0001 by unpaired Student’s t test.
To confirm pharmacodynamic effects of avelumab and durvalumab treatments, we performed biodistribution studies. Mice were treated with either avelumab or durvalumab (10 mg/kg) for 24, 48, 72, 96, or 120 h prior to radiotracer injection, and saline-treated mice were used as controls. Saline treated mice showed an average of 15 %ID/g uptake in tumors, which was significantly reduced by more than 80% (P<0.0001) after 24 h treatment with avelumab (Figure 5A). However, no significant differences in [68Ga]Ga-DK223 uptake were observed between the avelumab-treated mice and saline controls at 120 h. In contrast, durvalumab-treated mice showed a continued reduction in accessible target levels even at 120 h, corroborating the PET imaging results (Figure 5B). A similar trend was observed in tumor-to-blood and tumor-to-muscle ratios, with no significant difference in uptake in non-specific tissues between mAb treatment groups at any time point (Figures S7A–B). To measure the fraction of accessible PD-L1 bound by mAbs, we analyzed the tumor %ID/g in the treatment groups as a fraction of the saline treatment group. At 24 h, both antibodies showed ~90% reduction in accessible PD-L1 levels, indicating a significant exposure of mAbs (binding of mAbs to PD-L1) within the tumor microenvironment (Figure 5C). After 120 h of treatment, we noticed an increase in accessible PD-L1 levels in avelumab treated mice, in contrast to less than 10% accessible PD-L1 levels in durvalumab treated mice. These results demonstrate that measurements of accessible PD-L1 levels could provide insights into mAb exposure kinetics in tumors. In addition, these results show the potential of relating the dose to the exposure of mAb therapeutics in tumors over the course of treatment.
Figure 5. Quantification of accessible PD-L1 levels using [68Ga]Ga-DK223 in BFTC909 xenografts.
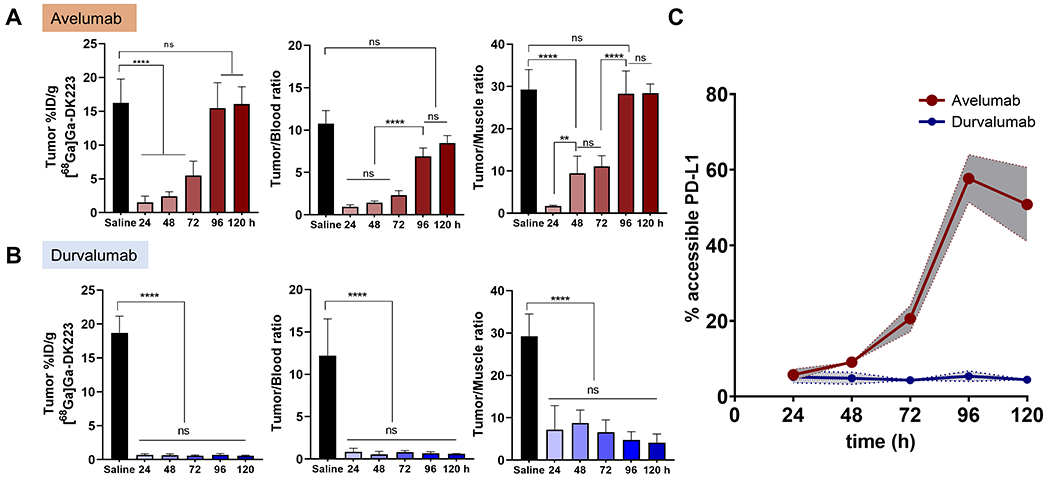
Ex vivo biodistribution studies in NSG mice bearing BFTC909 tumors treated with 10 mg/kg of either avelumab or durvalumab for 24, 48, 72, 96 or 120 h. Mice were injected with ~ 740 kBq (~20 μCi) [68Ga]Ga-DK223 and sacrificed 60 min after injection. Tumor %ID/g and tumor-to-tissue ratios over different duration of treatment of (A) Avelumab, and (B) Durvalumab (n=5-6). C, % of accessible PD-L1 levels at tumor calculated as fraction of average untreated control (n=5). Shaded region represents 95% confidence interval. ** P<0.01, *** P<0.001, ****P<0.0001 by 2-way ANOVA.
DISCUSSION
In this study, we established that a novel 68Ga-labeled radiotracer, [68Ga]Ga-DK223, binds specifically to PD-L1 with high affinity (1.01±0.83 nM) and enables noninvasive quantification of PD-L1 levels as early as 60 min post-administration, a process that takes days when using radiolabeled antibodies, such as [89Zr]atezolizumab. Although radiolabeled antibodies provide valuable information about drug distribution and accumulation in vivo, they are drug-centric and deriving target engagement information from those measurements is complex. Furthermore, measurements of radiolabeled antibodies by PET comprise a combination of target expression, antibody accumulation in the tumor, and residualization of the radionuclide, and are thus significantly influenced by factors intrinsic and extrinsic to the tumor.
Also, as antibody distribution is significantly influenced by biophysical properties, such measurements cannot be used to compare the effectiveness of different therapeutics. In contrast, [68Ga]Ga-DK223 PET measurements are target-centric, independent of antibody characteristics, and inclusive of factors intrinsic and extrinsic to the tumor.
Our experiments also demonstrated that [68Ga]Ga-DK223 can be used to quantify baseline and accessible PD-L1 levels, i.e. levels that are not occupied by mAb therapeutics during treatment. In vivo measurements using tumor xenograft models validated the ability of this radiotracer to evaluate the pharmacodynamics of two different PD-L1 therapeutics within tumors. With the growing scope of immunotherapy and more mAbs in development, this approach shows promise for establishing dose-exposure-response relationships, screening patients to predict the benefit from therapy, and monitoring the efficacy of PD-L1 treatment in a more personalized fashion. We envision that [68Ga]Ga-DK223 PET clinical application will be similar to [18F]FDG PET, once timing of the on-treatment scan is optimized, for monitoring immune checkpoint therapies.
Dose-exposure-response relationships play an important role in drug development, particularly in dose selection and optimization. The maximum tolerated dose is often used as a strategy for cytotoxic agent development, but it is difficult to apply to immune checkpoint inhibitors, as several agents do not reach MTD26. Moreover, dose-exposure-response relationships of immune checkpoint therapeutics are often confounded by a complex interplay of patent characteristics, including Eastern Cooperative Oncology Group (ECOG) status, number of metastatic sites, baseline tumor burden, cancer-related cachexia, lactate dehydrogenase (LDH) level, body weight and albumin concentration21. Exposure to these agents is often measured as acute or integrated drug concentrations in plasma and other biological fluids to model and predict occupancy at the tumor27. However, evaluation of pharmacodynamic markers focused on receptor occupancy indicates that parameters obtained from plasma samples are poor surrogates for the tumor pharmacokinetics of mAbs28. Our study demonstrates a novel and robust approach to understand the temporal kinetics of aPD-L1 mAbs in tumors using PD-L1 PET to establish dose-exposure relationships.
Patient selection for PD-1 or PD-L1 targeted therapeutics is often based on PD-L1 levels in the index lesion, as tumors with high PD-L1 levels demonstrate a better response29. mAb conjugates and small protein imaging agents have been utilized to account for the heterogeneity and dynamics of PD-L1 expression17,18. However, there is still a need for imaging agents with tractable pharmacokinetics and high tumor-to-background ratios. Here, we show that the low-molecular-weight peptide [68Ga]Ga-DK223 meets these criteria and provides high-contrast images of PD-L1 within 60 min of administration in human tumor xenografts derived from TNBC and UC. [68Ga]Ga-DK223 detects total and inducible PD-L1 levels in the tumor microenvironment, whether it is a tumor-or immune cell-based expression30. Data from the phase III KEYNOTE-355 trial in TNBC patients showed that patients with high PD-L1 expression at baseline (combined positive score [CPS]) responded better to the PD-1 inhibitor pembrolizumab31. Similarly, CPS-based stratification also guides immune checkpoint therapy (ICT) in UC32. The increasing use of CPS as a biomarker suggests that [68Ga]Ga-DK223 PET measurements could be used to quantify total PD-L1 levels in the whole body to guide therapy13.
Notably, 68Ga based imaging agents are being used increasingly in nuclear medicine, with multiple new agents recently receiving approval33,34. Gallium-68 can be obtained using a 68Ge/68Ga generator, enabling kit-based formulations that facilitate radiotracer dissemination. Recently, cyclotron-produced gallium-68 provides even larger quantities of this isotope for radiotracer synthesis.
To enable routine noninvasive pharmacodynamic measurements, a high-affinity radiotracer with in vivo target specificity is essential. In addition to meeting these needs, [68Ga]Ga-DK223 also inhibits PD-1:PD-L1 interactions with lower affinity than that of antibodies, a property that allows it to bind PD-L1 that is not bound by aPD-L1 mAbs, such as avelumab or durvalumab. Population PK models of these mAbs suggest that they have similar clearance profiles, but different half-lives35. However, the mechanism by which these biophysical properties affect exposure and target engagement at the tumor remains unknown36. Here, we have shown that [68Ga]Ga-DK223 can be used to measure baseline and accessible PD-L1 levels during treatment with these mAbs. PET imaging revealed that although both mAbs sufficiently bound accessible PD-L1 levels, as measured by [68Ga]Ga-DK223 at 24 h, there were stark differences at later time points. While these differences may appear to reflect the PK profile of the mAbs, several baseline factors, such as patient intrinsic characteristics, demographic factors, and disease-related factors, could influence exposure within the tumor but are difficult to quantify otherwise. For example, cachexia is one of the most important disease-related baseline factors that can confound the exposure-response relationships of immune checkpoint therapeutics37. Our studies were performed in immunocompromised mice due to specificity of [68Ga]Ga-DK223 towards hPD-L1. Development of new mouse and human cross-reactive analogs could provide more mechanistic insights into immunotherapy responses. Taken together, our data suggest that [68Ga]Ga-DK223 PET measurements provide a non-invasive, drug-independent approach to quantify the pharmacodynamic effects of such factors on tumor exposure to therapeutics. Based on these results, further studies in humans are warranted.
Supplementary Material
Acknowledgements.
This study was funded by NIH R01CA269235 (SN and SPR), NIH 1R01CA236616 (SN), the Allegheny Health Network-Johns Hopkins Cancer Research Fund (SN), and NIH P41EB024495. Core resources (histology and imaging) were supported by NIH P30CA006973. We thank Drs. Martin Pomper and Seulki Lee for their support, Precision Molecular, Inc./D&D Pharmatech for providing the Germanium-68/Gallium-68 generator and Drs. John T. Poirier and Charles Rudin for the gift of A549iPD-L1 cell line. We thank Dr. Ravindra DeSilva for assistance with installation and initial evaluation of Germanium-68/Gallium-68 generator. We thank Dr. Aykut Üren and Dr. Purushottam Tiwari at Georgetown University for performing SPR experiments.
Conflict of Interest:
D.K. and S.N. are co-inventors on a pending U.S. patent covering [68Ga]Ga-DK223, and as such are entitled to a portion of any licensing fees and royalties generated by this technology. This arrangement was reviewed and approved by Johns Hopkins University in accordance with its conflict-of-interest policies. S. N. received funding and was a consultant for Precision Molecular, Inc. S.P.R and R.F.D. are consultants for Precision Molecular Inc. S.P.R. and S.N. have equity in D&D Pharmatech, the parent company of Precision Molecular, Inc. All other authors declare no COIs pertaining to the described work.
REFERENCES
- 1.Bartelink IH et al. Tumor Drug Penetration Measurements Could Be the Neglected Piece of the Personalized Cancer Treatment Puzzle. Clin. Pharmacol. Ther 106, 148–163 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Glassman PM & Balthasar JP Physiologically-based pharmacokinetic modeling to predict the clinical pharmacokinetics of monoclonal antibodies. J. Pharmacokinet. Pharmacodyn 43, 427–446 (2016). [DOI] [PubMed] [Google Scholar]
- 3.Jain RK Physiological barriers to delivery of monoclonal antibodies and other macromolecules in tumors. Cancer Res. 50, (1990). [PubMed] [Google Scholar]
- 4.Vasan N, Baselga J & Hyman DM A view on drug resistance in cancer. Nature 575, 299–309 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Trédan O, Galmarini CM, Patel K & Tannock IF Drug resistance and the solid tumor microenvironment. J. Natl. Cancer Inst 99, 1441–1454 (2007). [DOI] [PubMed] [Google Scholar]
- 6.Weissleder R, Schwaiger MC, Gambhir SS & Hricak H Imaging approaches to optimize molecular therapies. Sci. Transl. Med 8, 1–8 (2016). [DOI] [PubMed] [Google Scholar]
- 7.de Vries EGE et al. Integrating molecular nuclear imaging in clinical research to improve anticancer therapy. Nat. Rev. Clin. Oncol 16, 241–255 (2019). [DOI] [PubMed] [Google Scholar]
- 8.Gandini S, Massi D & Mandalà M PD-L1 expression in cancer patients receiving anti PD-1/PD-L1 antibodies: A systematic review and meta-analysis. Crit. Rev. Oncol. Hematol 100, 88–98 (2016). [DOI] [PubMed] [Google Scholar]
- 9.Gong J, Chehrazi-Raffle A, Reddi S & Salgia R Development of PD-1 and PD-L1 inhibitors as a form of cancer immunotherapy: A comprehensive review of registration trials and future considerations. J. Immunother. Cancer 6, 1–18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bellmunt J, Powles T & Vogelzang NJ A review on the evolution of PD-1/PD-L1 immunotherapy for bladder cancer: The future is now. Cancer Treat. Rev 54, 58–67 (2017). [DOI] [PubMed] [Google Scholar]
- 11.Chatterjee S et al. A humanized antibody for imaging immune checkpoint ligand PD-L1 expression in tumors. Oncotarget 7, 10215–10227 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mansfield AS & Dong H Implications of Programmed Cell Death 1 Ligand 1 Heterogeneity in the Selection of Patients With Non-Small Cell Lung Cancer to Receive Immunotherapy. Clin. Pharmacol. Ther 100, 220–222 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Nimmagadda S Quantifying pd-l1 expression to monitor immune checkpoint therapy: Opportunities and challenges. Cancers (Basel). 12, 1–26 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Soares H, Lasserre R & Alcover A Orchestrating cytoskeleton and intracellular vesicle traffic to build functional immunological synapses. Immunol. Rev 256, 118–132 (2013). [DOI] [PubMed] [Google Scholar]
- 15.Onnis A, Finetti F & Baldari CT Vesicular trafficking to the immune synapse: How to assemble receptor-tailored pathways from a basic building set. Front. Immunol 7, 1–9 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bensch F et al. 89Zr-atezolizumab imaging as a non-invasive approach to assess clinical response to PD-L1 blockade in cancer. Nat. Med 24, 1852–1858 (2018). [DOI] [PubMed] [Google Scholar]
- 17.Donnelly DJ et al. Synthesis and biologic evaluation of a novel 18 F-labeled adnectin as a PET radioligand for imaging PD-L1 expression. J. Nucl. Med 59, 529–535 (2018). [DOI] [PubMed] [Google Scholar]
- 18.Lesniak WG et al. Development of [18F]FPy-WL12 as a PD-L1 Specific PET Imaging Peptide. Mol. Imaging 18, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou X et al. First-in-human evaluation of a PD-L1-binding peptide radiotracer in non-small cell lung cancer patients with PET. Journal of Nuclear Medicine (2021). doi: 10.2967/jnumed.121.262045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kumar D et al. Peptide-based PET quantifies target engagement of PD-L1 therapeutics. J. Clin. Invest 129, 616–630 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Centanni M, Moes DJAR, Trocóniz IF, Ciccolini J & van Hasselt JGC Clinical Pharmacokinetics and Pharmacodynamics of Immune Checkpoint Inhibitors. Clin. Pharmacokinet 58, 835–857 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Woodard LE et al. Bridged cyclams as imaging agents for chemokine receptor 4 (CXCR4). Nucl. Med. Biol 41, 552–561 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kumar D et al. Pharmacodynamic measures within tumors expose differential activity of PD(L)-1 antibody therapeutics. Proc. Natl. Acad. Sci. U. S. A 118, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.MATTHEW MM et al. MACROCYCLIC INHIBITORS OF THE PD-1/PD-L1 AND CD80(B7-1)/PD-L1 PROTEIN/PROTEIN INTERACTIONS. (2014). [Google Scholar]
- 25.Lu S et al. Comparison of Biomarker Modalities for Predicting Response to PD-1/PD-L1 Checkpoint Blockade: A Systematic Review and Meta-analysis. JAMA Oncol. 5, 1195–1204 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Balar AV & Weber JS PD-1 and PD-L1 antibodies in cancer: current status and future directions. Cancer Immunol. Immunother 66, 551–564 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Deng R et al. Preclinical pharmacokinetics, pharmacodynamics, tissue distribution, and tumor penetration of anti-PD-L1 monoclonal antibody, an immune checkpoint inhibitor. MAbs 8, 593–603 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brahmer JR et al. Safety and Activity of Anti–PD-L1 Antibody in Patients with Advanced Cancer. N. Engl. J. Med 366, 2455–2465 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Buttner R et al. Programmed death-ligand 1 immunohistochemistry testing: A review of analytical assays and clinical implementation in non-small-cell lung cancer. J. Clin. Oncol 35, 3867–3876 (2017). [DOI] [PubMed] [Google Scholar]
- 30.Liu Y et al. Immune cell PD-L1 colocalizes with macrophages and is associated with outcome in PD-1 pathway blockade therapy. Clin. Cancer Res 26, 970–977 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Heeke AL & Tan AR Checkpoint inhibitor therapy for metastatic triple-negative breast cancer. Cancer Metastasis Rev. 40, 537–547 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Powles T et al. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature 515, 558–562 (2014). [DOI] [PubMed] [Google Scholar]
- 33.Banerjee SR & Pomper MG Clinical applications of Gallium-68. Appl. Radiat. Isot 76, 2–13 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Willmann JK, van Bruggen N, Dinkelborg LM & Gambhir SS Molecular imaging in drug development. Nat. Rev. Drug Discov 7, 591–607 (2008). [DOI] [PubMed] [Google Scholar]
- 35.Sheng J et al. Clinical Pharmacology Considerations for the Development of Immune Checkpoint Inhibitors. J. Clin. Pharmacol 57, S26–S42 (2017). [DOI] [PubMed] [Google Scholar]
- 36.Simon GM, Niphakis MJ & Cravatt BF Determining target engagement in living systems. Nat. Chem. Biol 9, 200–205 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roch B et al. Cachexia - sarcopenia as a determinant of disease control rate and survival in non-small lung cancer patients receiving immune-checkpoint inhibitors. Lung Cancer 143, 19–26 (2020). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All study data are included in the article and/or supplementary information. The additional data are available upon request from the corresponding author.