

Abstract
The search for susceptibility loci in hereditary prostate cancer (HPC) is challenging due to locus and disease heterogeneity. One approach to reduce disease heterogeneity is to stratify families based on the occurrence of multiple cancer types. This method may increase power for detecting susceptibility loci, including those with pleiotropic effects. We have completed a genome-wide SNP linkage analysis of 96 HPC families, each of which has one or more first-degree relatives with colon cancer (CCa), and further analyzed the subset of families with two or more CCa cases (n=27). When only a prostate cancer (PCa) phenotype was considered affected, we observed suggestive evidence for linkage (LOD≥1.86) at 15q14, 18q21 and 19q13 in all families, and at 1p32 and 15q11-q14 in families with two or more CCa cases. When both the PCa and CCa phenotypes were considered affected, suggestive evidence for linkage was observed at 11q25, 15q14 and 18q21 in all families, and at 1q31, 11q14, and 15q11-14 in families with two or more CCa cases. The strongest linkage signal was identified at 15q14 when both the PCa and CCa phenotypes were considered affected in families with two or more CCa cases (recessive HLOD=3.88). These results provide further support for the presence of HPC susceptibility loci on chromosomes 11q14, 15q11-q14 and 19q13 and highlight loci at 1q31, 11q, 15q14 and 18q21 as having possible pleiotropic effects. This study demonstrates the benefit of utilizing a comprehensive family cancer history to create more genetically homogenous subsets of HPC families for linkage analyses.
Keywords: Prostate cancer, colon cancer, linkage analysis, pleiotropic effects, hereditary
Introduction
Family-based genome-wide linkage studies have highlighted over two dozen putative loci for hereditary prostate cancer (HPC) with significant or suggestive signals; however due to the joint contribution of locus and disease heterogeneity, few have been confirmed and most have not yielded clear evidence of specific genes or causative variants (reviewed in 1,2). To overcome some of these challenges and create more homogenous subsets of pedigrees, linkage studies have focused on refining the phenotypic definition of prostate cancer (PCa)3,4.
One approach to overcome heterogeneity in a complex disease such as HPC is to stratify families by the presence of other primary cancers. This method is supported by evidence that some inherited susceptibility genes have a pleiotropic effect5. While a cancer syndrome that includes PCa has not been identified, there is evidence that relatives of PCa probands are at an increased risk for other primary cancers such as colon6. Analysis of a subset of HPC families where there is an occurrence of an additional cancer type offers the advantage of reducing locus heterogeneity, and thereby improving power for finding susceptibility loci.
Stratifying families based on the presence of multiple cancers has been used successfully to map loci and then characterize the BRCA1 and BRCA2 genes for breast cancer7–10. While specific loci have not yet been identified, PCa linkage studies of families with multiple cancers have also suggested regions that may contain HPC susceptibility genes. The putative 1p36 HPC susceptibility region was initially mapped using families with first-degree relatives diagnosed with primary brain cancer11, with subsequent evidence for this locus reported in other studies12,13. Linkage analyses of HPC families stratified by the presence of kidney cancer14 or pancreas cancer15 in first-degree relatives of PCa probands have also suggested susceptibility loci, two of which, 7p21 and 16q23, have also been identified in other HPC linkage analyses4,16,17. Evidence for a shared genetic etiology of PCa and colon cancer (CCa) has emerged from studies showing an excess risk of CCa in families ascertained for PCa, and vice versa6,18,19. In addition, several susceptibility regions, including 3p14, 8q24 and 15q13-q14, have been suggested in both PCa16,20–24 and CCa20,25–27 studies, providing support for a potential shared genetic disposition for developing these two cancers.
Based on the evidence presented above, we have selected 96 HPC families with at least one first-degree relative of a PCa proband with CCa and performed genome-wide parametric and nonparametric linkage analyses.
Materials and Methods
Hereditary prostate cancer families with colon cancer members
HPC families included in these analyses are participating in the Prostate Cancer Genetic Research Study (PROGRESS)24,28. Participants have completed a baseline and two follow-up surveys that queried personal and family cancer history. From these questions, we identified families that had one or more members with CCa. An individual was considered to have a diagnosis of CCa if: 1) it was self-reported; 2) there was at least one first-degree relative report; and/or 3) there were at least two second-degree relative reports. A random sample of 27 CCa cases was chosen for reporting validation. For the eleven individuals for whom a medical record or death certificate was obtained, ten CCa (91%) reports were confirmed. In addition to the above 27 cases, death certificates were available for another 23 CCa cases, 12 of whom also had PCa. Of these, 100% of the 23 CCa reports were confirmed (five self-reports and 18 relative-reports). Families were only considered eligible for this study if the individual(s) with CCa had a first-degree relative with PCa. An individual with CCa was also considered affected if they had a first-degree relative with CCa who in turn, had a first-degree relative with PCa. All other CCa cases were considered unknown. Using the above criteria, 136 CCa cases were identified in 99 PROGRESS families. Fifty-seven of the CCa cases (41.9%) also had PCa. Ninety-eight CCa cases had genotyping data, of which 43 were inferred. Families were of Caucasian (N=95), Hispanic (N=1), or other (N=3) ancestry. The analyses reported here included only the 96 Caucasian/Hispanic HPC families with one or more CCa cases as summarized in Table 1.
Table 1.
Characteristics of the PROGRESS prostate cancer (PCa) pedigrees with one or more colon cancer (CCa) cases
| Affected PCa Phenotype | Affected CCa Phenotype | Affected PCa + CCa Phenotypes | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Families | # Peds |
Total # unaffected relatives genotyped |
Total # affected subjects available1 |
Average # affected per family1 |
Average age at diagnosis (± SD) |
Total # affected subjects available1 |
Average # affected per family1 |
Average age at diagnosis (± SD)2 |
Total # affected subjects available1 |
Average # affected per family1 |
Average age at diagnosis (± SD) |
| Families with 1+ CCa | 96 | 478 | 422 | 3.7 (2–11) | 66.6 (4.6) | 97 | 0.6 (0–3) | 67.6 (12.7) | 473 | 3.9 (2–11) | 66.6 (4.7) |
| Diagnosis Age | |||||||||||
| < 65 years | 31 | 119 | 132 | 3.8 (2–11) | 61.3 (2.8) | 29 | 0.5 (0–2) | 63.6 (14.6) | 148 | 3.9 (2–11) | 61.7 (3.0) |
| ≥ 65 years | 65 | 359 | 290 | 3.7 (2–7) | 69.2 (2.8) | 68 | 0.6 (0–3) | 69.5 (11.4) | 325 | 3.9 (2–9) | 69.0 (3.3) |
| # of Affected | |||||||||||
| < 5 | 40 | 145 | 126 | 2.9 (2–4) | 66.5 (4.9) | 42 | 0.7 (0–3) | 67.3 (13.1) | 147 | 3 (2–5) | 66.4 (5.2) |
| ≥ 5 | 56 | 333 | 296 | 4.3 (2–11) | 66.8 (4.5) | 55 | 0.5 (0–2) | 67.8 (12.6) | 326 | 4.5 (2–11) | 66.8 (4.4) |
| Aggressiveness | |||||||||||
| < 3 More Aggr PC | 67 | 285 | 252 | 3.2 (2–7) | 66.2 (4.8) | 73 | 0.6 (0–3) | 65.9 (12.7) | 288 | 3.4 (2–7) | 66.1 (4.8) |
| 3 + More Aggr PC | 29 | 193 | 170 | 4.8 (2–11) | 67.7 (4.1) | 24 | 0.4 (0–2) | 71.1 (12.4) | 185 | 5.0 (2–11) | 67.9 (4.5) |
| Families with 2+ CCa | 27 | 168 | 121 | 3.7 (2–7) | 66.9 (5.4) | 45 | 1.1 (0–3) | 66.6 (8.2) | 141 | 4 (2–9) | 67.0 (5.2) |
| Diagnosis Age | |||||||||||
| < 65 years | 8 | 30 | 31 | 3.8 (3–5) | 60.1 (2.6) | 9 | 0.8 (0–2) | 64.7 (10.4) | 35 | 3.9 (3–5) | 60.8 (4.0) |
| ≥ 65 years | 19 | 138 | 90 | 3.6 (2–7) | 69.7 (3.1) | 36 | 1.3 (0–3) | 67.4 (7.3) | 106 | 4.1 (2–9) | 69.6 (3.0) |
| # of Affected | |||||||||||
| < 5 | 11 | 43 | 34 | 2.9 (2–4) | 66.1 (5.9) | 19 | 1.3 (0–3) | 66.8 (9.9) | 42 | 3.2 (2–5) | 66.2 (5.6) |
| ≥ 5 | 16 | 125 | 87 | 4.2 (2–7) | 67.4 (5.1) | 26 | 1.0 (0–2) | 66.4 (7.1) | 99 | 4.6 (2–9) | 67.5 (5.0) |
| Aggressiveness | |||||||||||
| < 3 More Aggr PC | 21 | 110 | 80 | 3.2 (2–5) | 66.7 (5.6) | 36 | 1.2 (0–3) | 64.7 (7.8) | 94 | 3.5 (2–5) | 66.5 (5.4) |
| 3 + More Aggr PC | 6 | 58 | 41 | 5.3 (3–7) | 67.6 (4.8) | 9 | 0.8 (0–2) | 73.6 (5.3) | 47 | 5.7 (3–9) | 68.6 (4.3) |
Affected subjects who are genotyped or inferred;
An age at diagnosis was not available for all CCa cases.
Genotyping and quality control
Genotyping and quality control analyses were performed as a part of a larger study described previously24. Briefly, samples were genotyped using the Illumina Linkage Panel IVb (5,867 SNPs) and analyses were performed on a total of 4,743 tag-SNPs with a median minor allele frequency (MAF) of 0.40 (range: 0.05 – 0.50), median call rate of 99% (range: 97 – 100%), median intermarker distance over all chromosomes of 0.60 cM (range: 0.001 – 5.97 cM) and median overall information content of 86% (range: 63 – 90%).
Linkage analysis
Both parametric and nonparametric allele-sharing linkage analyses were performed using Merlin software29. The parametric LOD scores were computed using an assumed PCa susceptibility allele frequency of 0.003 and 0.15 for autosomal dominant and recessive models, respectively, with a fixed phenocopy rate of 15%. Penetrances of 0.001 for non-carriers and 1.0 for carriers of a putative risk allele were assumed. Parametric LOD scores allowed for linkage heterogeneity (HLOD) by estimating the fraction of linked pedigrees. Nonparametric LOD scores were calculated using the Kong and Cox exponential allele sharing model score (KCLOD)30.
Parametric LOD scores were based on an “affecteds-only” analysis. For this approach, the genotypes of all pedigree members were used, but only affecteds were coded as such: unaffected members were coded as unknown phenotype. This strategy eliminates modeling the penetrance for unaffected subjects, who might not be thoroughly screened for disease. Furthermore, the absolute values of the assumed penetrances are not critical, only the ratio of penetrances (e.g., as a genotype relative risk). Including the genotypes of subjects with unaffected or unknown phenotypes, however, does increase the linkage information content by increasing the identity-by-descent information among the affected subjects.
Our analyses considered PCa and CCa in two different ways: 1) only individuals with PCa were coded as affected (subjects with CCa were coded as unknown); and, 2) individuals with either PCa or CCa were coded as affected. This allowed us to evaluate the contribution of CCa to the linkage results. The parametric linkage models were the same for both analyses (in terms of assumed allele frequencies and penetrances).
Although Merlin software can analyze SNPs that are in linkage disequilibrium (LD) by treating them as multi-allelic markers and employing the expectation-maximization algorithm31, this option requires an excessive amount of memory and analytical time for large pedigrees. We thus eliminated much of the LD by selecting 4,743 tag-SNPs using LDselect32 with a low r2 threshold (r2 ≤ 0.10); tags for each bin were chosen considering Illumina QC measures, MAF, and SNP call rate. Marker allele frequencies were estimated across the pool of all subjects, ignoring genetic relationships.
Pedigrees were stratified as follows: 1) families with one or more colon cancer cases (96 families); and 2) families with two or more colon cancer cases (27 families). One HPC-colon cancer pedigree had to be split into two sub-pedigrees to “fit” the data into the memory limits of Merlin software.
Results
The findings presented here are based on 96 HPC-colon cancer pedigrees in which 900 individuals were genotyped (n=835) or genotype information was reconstructed from relatives (n=65). These individuals included 422 PCa cases, 67 male CCa cases (46 of whom also had a diagnosis of PCa) and 30 female CCa cases (Table 1). Twenty-seven of the 96 pedigrees had at least two affected subjects with CCa.
Figure 1 presents the LOD scores for the HPC-colon cancer families analyzed for the PCa only phenotype. LOD scores of at least 1.86 were considered suggestive for linkage33 and these are summarized in Table 2. When considering families with one or more CCa cases, suggestive evidence for linkage was seen on chromosomes 15 and 19 under a recessive model (15q14 HLOD=1.90 at 33 Mb and 19q13.33 HLOD=1.96 at 56 Mb) and on 18q21.2 with a dominant HLOD of 1.87 at 49 Mb. Considering the 27 families with two or more CCa cases, the most striking linkage signal was detected on chromosome 15q14, with a recessive HLOD of 3.47 at 33 Mb. Further suggestive evidence for linkage on chromosome 15 was present at 15q11-q14 (KCLOD=2.85 on 15q11.2 at 22 Mb and dominant HLOD=2.22 on 15q13.3-q14 at 31 Mb). There was also suggestive evidence for linkage on chromosome 1p32.3, with a recessive HLOD of 2.45 at 54 Mb.
Figure 1.
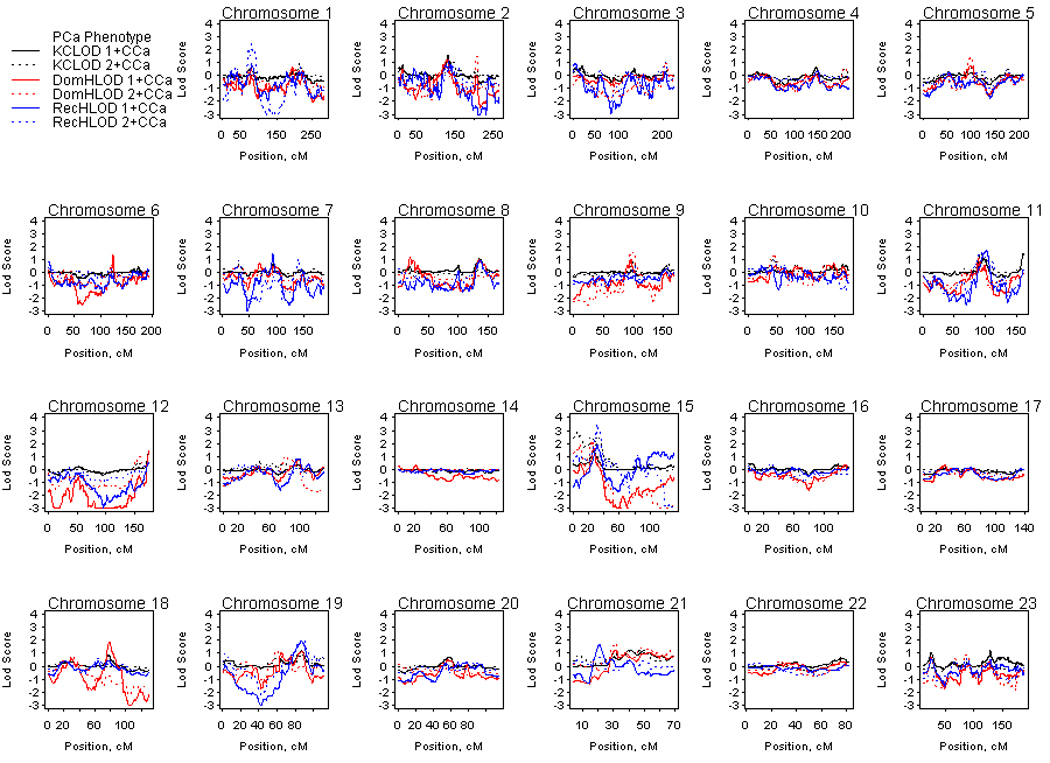
Genome-wide linkage results for the prostate cancer phenotype in hereditary prostate cancer – colon cancer families. Solid lines denote analyses considering all 96 families and dashed lines denote analyses considering the 27 hereditary prostate cancer families with two or more colon cancer cases. DomHLOD (red) and RecHLOD (blue) are the dominant and recessive heterogeneity LOD scores under an affecteds only model. KCLOD (black) is the Kong and Cox exponential allele sharing model LOD score.
Table 2.
Suggestive LOD scores (LOD ≥1.86) for prostate cancer and for prostate and colon cancer (CCa) phenotypes
| Phenotype | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Prostate Cancer | Prostate and Colon Cancer | ||||||||
| Chromosome | cM | Left Flanking SNP (bp) |
Right Flanking SNP (bp) |
K&C1 LOD |
Dominant HLOD2 |
Recessive HLOD2 |
K&C1 LOD |
Dominant HLOD2 |
Recessive HLOD2 |
| Families with 1+ CCa (n=96) | |||||||||
| 11q25 | 162.07 | rs1824832 (134313007) | rs4430531 (134447835) | 1.46 | −0.06 | 0.22 | 1.94 | −0.04 | 0.43 |
| 15q13-14 | 25.7 | rs343913 (31133168) | rs2033610 (31542203) | 1.46 | 1.76 | 0.75 | 0.99 | 0.40 | 1.02 |
| 15q14 | 31.04 | rs732165 (33141489) | rs1989223 (33914397) | 0.97 | 0.43 | 1.90 | 0.91 | 0.33 | 2.13 |
| 18q21 | 78.54 | rs869224 (49231796) | rs11455315 (49942953) | 0.86 | 1.87 | 0.40 | 1.16 | 2.67 | 0.44 |
| 79.39 | rs1145315 (49942953) | rs1942569 (50997722) | 0.7 | 1.84 | 0.39 | 1.08 | 2.82 | 0.59 | |
| 19q13 | 87.21 | rs897783 (56722974) | rs1993104 (56932060) | 1.21 | 1.10 | 1.96 | 1.14 | 0.55 | 0.79 |
| Families with 2+ CCa (n=27) | |||||||||
| 1p32 | 80.95 | rs731828 (54140841) | rs1537323 (54255111) | 0.53 | 0.53 | 2.45 | 0.75 | −0.23 | −0.47 |
| 1q31 | 202.08 | rs1052238 (196901248) | rs18924333 (197228191) | 0.06 | 0.34 | −1.51 | 0.12 | 0.06 | 2.19 |
| 11q14 | 93.01 | rs1459937 (81203361) | rs1459952 (81260386) | 1.4 | 0.71 | 1.63 | 1.67 | 1.05 | 2.35 |
| 15q11 | 4.93 | rs6576326 (22336292) | rs975963 (22568288) | 2.85 | 1.56 | 1.06 | 2.14 | 0.74 | −0.13 |
| 15q13-14 | 25.7 | rs343913 (31133168) | rs2033610 (31542203) | 2.38 | 2.22 | 1.36 | 1.86 | 0.52 | 1.65 |
| 15q14 | 31.04 | rs732165 (33141489) | rs1989223 (33914397) | 2.45 | 0.77 | 3.47 | 2.29 | 0.41 | 3.88 |
K&C=Kong & Cox exponential allele sharing model;
Model = Affecteds-only model (see text).
Figure 2 presents the LOD scores for HPC-colon cancer families analyzed for PCa and CCa phenotypes. Suggestive LOD scores of at least 1.86 are presented in Table 2. Considering all families with one or more CCa cases, suggestive evidence for linkage was observed at chromosome 11q25 (KCLOD=1.94 at 134 Mb), 15q14 (recessive HLOD=2.13 at 33 Mb) and 18q21.2 (dominant HLOD=2.82 at ~ 50 Mb). In the subset of families with two or more CCa cases, suggestive evidence for linkage under a recessive model was observed on chromosome 1q31.3 (HLOD=2.19 at ~197 Mb) and 11q14.1 (HLOD=2.35 at 81 Mb). Again, the most striking evidence for linkage was observed on chromosome 15q14 with a recessive HLOD of 3.88 at 33 Mb, however, the KCLOD decreased slightly at this locus compared to that observed for the PCa only phenotype (KCLOD of 2.29 and 2.45 at 33 Mb, respectively). An additional suggestive linkage peak was observed at 15q11.2 with a KCLOD of 2.14 at 22 Mb, although evidence for linkage again decreased slightly when the CCa phenotype was considered as affected.
Figure 2.
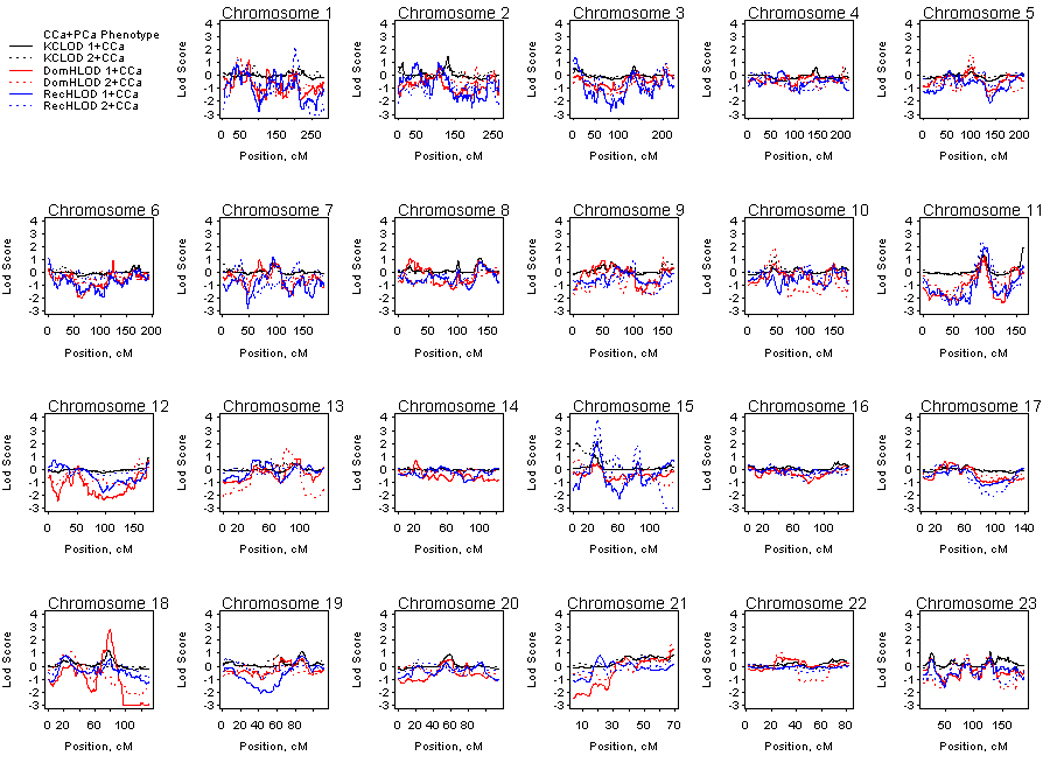
Genome-wide linkage results for the prostate and colon cancer phenotypes in hereditary prostate cancer – colon cancer families. Solid lines denote analyses considering all 96 families and dashed lines denote analyses considering the 27 hereditary prostate cancer families with two or more colon cancer cases. DomHLOD (red) and RecHLOD (blue) are the dominant and recessive heterogeneity LOD scores under an affecteds only model. KCLOD (black) is the Kong and Cox exponential allele sharing model LOD score.
Since CCa, like PCa, is a relatively common cancer with both hereditary and sporadic forms, we investigated whether the stratification of families by number of CCa cases impacted our strongest linkage signal at 15q11-14. We reanalyzed the data according to the number of CCa cases: one case vs. two or more cases per family. Each stratum was analyzed for the PCa only phenotype and then the PCa and CCa phenotypes. Figure 3 demonstrates that under both a nonparametric and recessive model, HPC families with only one CCa case provided no evidence for linkage to 15q11-14, whilst families with two or more CCa cases provided all of the evidence for linkage to this region.
Figure 3.
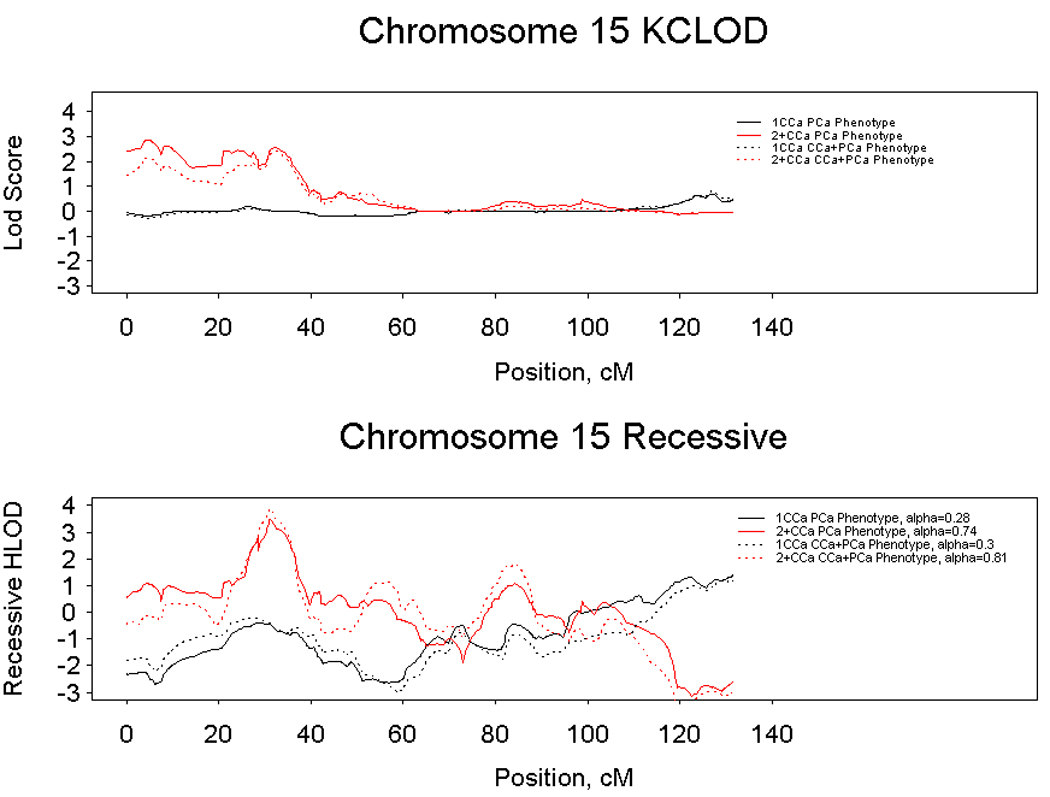
Linkage results for chromosome 15 under a non-parametric (top panel) and recessive (lower panel) model considering a prostate cancer phenotype (solid lines) and both prostate and colon cancer phenotypes (dashed lines). Analyses of hereditary prostate cancer families with one colon case are denoted with a black line and analyses of hereditary prostate cancer families with two or more colon cancer cases are denoted with a red line.
Discussion
In order to create a comparatively more homogeneous subset of HPC pedigrees, we analyzed data from 96 HPC families with at least one CCa case among first-degree relatives of a PCa proband. Families were initially analyzed using a PCa only phenotype. To search for loci with possible pleiotropic effects, analyses were repeated considering both PCa and CCa phenotypes as affected. Several suggestive linkage signals were identified, with the most striking evidence observed at 15q14 in families with two or more CCa cases and both PCa and CCa phenotypes considered affected (recessive HLOD=3.88).
Several of the linkage peaks observed in this study have been noted in previous PCa linkage studies, including 11q1434,35, 19q1336,37 and, of particular interest, 15q11-14. The International Consortium for Prostate Cancer Genetics (ICPCG) presented suggestive evidence for linkage to 15q11 (recessive LOD=2.10) in a microsatellite analysis of 1,233 HPC families, including 254 PROGRESS families38. A study of 230 multiplex PCa sibships from the U.S.16 found a Z score of 2.77 at 15q13.3 and when investigating 139 families from Germany23, a Z score of 2.23 was observed at 15q13.1. In 2004, a study of 44 Japanese PCa families highlighted a linkage peak at 15q14 (Zlr=1.75)12 and most recently, 15q13-q14 was highlighted in a dense SNP genome-wide scan of 289 Caucasian HPC families (HLOD=1.99), which included the 96 pedigrees presented here24. Interestingly, the strongest evidence for linkage at this region was in a subset analysis of 128 families with younger ages at diagnosis (KCLOD=2.82)24, eight of which overlap with the 27 HPC-colon cancer families that provided the strongest evidence for linkage in the current analyses (recessive HLOD=3.87). While stratifying by age at diagnosis appeared to create some homogeneity in the original analyses of the PROGRESS dataset24, the method of selecting families based on the presence of another primary cancer, in this case CCa, clearly created greater homogeneity among the families, especially in families with two or more CCa cases. Interestingly, families contributing evidence for linkage to one locus rarely provided evidence for linkage to any of the other loci, further suggesting that this study achieved both inter- and intra-familial heterogeneity.
Based on several lines of evidence, our a priori hypothesis was that a shared genetic etiology exists for PCa and CCa. First, an excess risk of CCa has been reported in families ascertained for PCa6,18 and conversely, an excess of PCa (RR=1.3, 95% CI 1.1–1.4) has been observed in families ascertained for CCa19. Second, a study by Ozden and colleagues (2003) found a 10.7% 10-year cumulative incidence of PCa in men with a history of CCa compared to 3.8% in men without CCa39. Third, mutations in two of the mismatch repair genes (MLH1, MSH3) that are associated with predisposition to hereditary nonpolyposis colon cancer (HNPCC), have been associated with an increased relative risk of PCa40,41. Of particular interest to the work reported here are prior studies that have highlighted several common susceptibility regions for both PCa and CCa, including 3p14.221,26, 15q13-q1416,23–25 and 18q2124,42. Finally, a PCa linkage study highlighted the 8q24 locus22, which has since been shown to harbor a SNP, rs6983267, that is associated with increased relative risks of both PCa and CCa20,45.
The fact that several of the regions observed in this study have been previously observed in PCa or CCa studies, or both, is noteworthy. The 15q11-q14 region was first suggested as a CCa susceptibility locus based on linkage analyses of Ashkenazi Jewish pedigrees27. More recently, a linkage scan of colon neoplasia families also highlighted 15q14-q2225. Jaeger and colleagues (2008) have restricted the location of the CCa susceptibility locus to a region spanning approximately 1 Mb (30.7 – 31.4 Mb), which overlaps with the larger region of linkage presented here (22.3 – 33.9 Mb)46. Then in a large association study of CCa in cases from the U.K, a SNP from this same region was shown to increase risk in cases with a family history or early onset of disease (rs4779584; p=4.44×10−14)46. This is particularly interesting as 15q13-14 has been highlighted previously in PROGRESS families with a younger mean age at diagnosis24.
The 15q11-14 peak observed in this study encompasses an interesting candidate gene, FMN1, which is highly expressed in the prostate. The formin (FMN) family of proteins govern cytoskeletal organization in cells and is known to play an essential role in cell division and polarity47,48. While a link between FMN1 and cancer has not yet been described, the peak signals for the abovementioned U.S.16 and German 23 PCa linkage analyses also fall within the transcript region of the FMN1 gene. Intriguingly, the colorectal susceptibility locus identified in the study by Jaeger and colleagues (2008), also spans the FMN1 gene (30.7 – 31.4 Mb)46.
Three other regions highlighted in this study, 11q14, 11q25 and 18q21 have also been observed in PCa and/or CCa studies. The 11q14 region has been noted not only in PCa linkage studies34,35, but also in a genome-wide linkage analysis of 18 Swedish CCa families49. While 11q25 has been highlighted in previous PCa linkage studies23,24, recent evidence suggests that 11q25 may contain a locus that is involved in multiple malignancies. Cui and colleagues (2008) have shown that the 11q25 tumor-suppressor gene, OPCML, is down-regulated by promoter methylation in several tumor cell lines including prostate, colon and breast50. Chromosome 18q21 has also been suggested as both a PCa and CCa susceptibility region through LOH studies43,44, a previous linkage analysis of PROGRESS families with a comparatively more clinically aggressive PCa phenotype and an older mean age at diagnosis24, and, finally, a GWAS of familial CCa cases and controls (rs4939827; p=1.0×10−12)51.
There were a number of linkage peaks noted in this study that were either stronger or only present when analyzing a specific phenotype. For example, peaks at 19q13 and 1p32 were stronger or only present, respectively, when analyzing the PCa phenotype, while peaks at 11q14 and 1q31 were stronger or only present, respectively, when analyzing both the PCa and CCa phenotypes. While none of these regions reached genome-wide statistical significance, these results suggest that specific phenotype analyses may reveal susceptibility loci that have either disease specific effects (1p32 and 19q13) or potential pleiotropic effects (1q31 and 11q14).
One limitation of this study is that not all CCa cases were confirmed by medical records or death certificates. Such documentation was obtained for only 34 (25.6%) of the CCa cases, but these records confirmed 97% of the CCa diagnoses in this subset. Previous studies have found from 17 – 100% of self-reported CCa52,53 and 48 – 93% of first-degree relative-reported CCa were confirmed by medical records54,55. Therefore, there may be individuals with CCa that were missed or, more rarely, individuals with an incorrect assignment of CCa. The first scenario should not contribute to false findings of linkage, since all our analyses relied on phenotype data only from affected individuals.
To our knowledge, in only rare instances does PCa metastasize to the colon, or vice versa, however, two additional issues that need to be considered when studying these two cancers are the potential effects of screening or early detection and treatment. Once a diagnosis of either PCa or CCa is made, the patient may be exposed to greater medical attention, increasing the likelihood of screening for additional cancer types. The diagnosis of the first primary cancer could also influence the patient’s lifestyle choices and positive changes (e.g., cessation of smoking, weight loss, increase in exercise), which may alter risk of a subsequent cancer. Treatment for the first primary cancer may also affect the risk for subsequent cancers. A recent study found that PCa patients treated with external beam radiotherapy had an increased risk of CCa in the period of 5–9 years after treatment56, however, studies previous to this have found discordant results regarding the occurrence of CCa and PCa and the subsequent risk of the other cancer (reviewed in 57). Of the 57 men in this study with prior diagnoses of both primary PCa and primary CCa, we have information regarding the temporal relationship between these diagnoses for 46 men. Of these, two men (4%) were diagnosed with both cancers within the same year and five (11%) men initially diagnosed with PCa and treated with primary radiation therapy were subsequently diagnosed with CCa 5–9 years post-radiation treatment.
In summary, to create a more homogeneous subset of HPC families, a genome-wide linkage analysis of 96 families with at least one first-degree relative of a PCa proband with CCa was performed. This study has provided further support for the presence of HPC susceptibility regions on chromosomes 11q14, 15q11-q14, 18q21, 19q13, with the most striking evidence for a region on 15q14. Thus, these results support both previously suggested HPC regions, as well as novel candidate HPC regions with potential pleiotropic effects. In fact, four well recognized CCa susceptibility regions, 11q14, 11q25, 15q13-q14 and 18q21, were suggested as candidate PCa susceptibility regions in this study. Future work will focus on refining the interval of linkage at 15q11-q14 in HPC-colon families, with the aim of identifying the underlying susceptibility locus (loci). While the search for highly penetrant, rare PCa susceptibility loci has proven difficult, the combination of approaches utilized here is likely to elucidate unique regions containing genes that are responsible for the heritable form of this common and complex disease.
Acknowledgements
We thank all the men and women who are participating in the PROGRESS study for their time, effort and cooperation. We also thank the study staff for help with ongoing data collection and processing. This work was supported by grants RO1 CA080122 and P50-CA097186 from the National Cancer Institute, with additional support from the Fred Hutchinson Cancer Research Center. Genotyping services were provided by the Center for Inherited Disease Research at Johns Hopkins University (contract N01-HG-65403 from the National Institutes of Health). We acknowledge the Prostate Cancer Foundation and the Intramural Program of the National Human Genome Research Institute.
Footnotes
The authors declare that they have no conflicts of interest.
References
- 1.Ostrander EA, Markianos K, Stanford JL. Finding prostate cancer susceptibility genes. Annu Rev Genomics Hum Genet. 2004;5:151–175. doi: 10.1146/annurev.genom.5.061903.180044. [DOI] [PubMed] [Google Scholar]
- 2.Schaid DJ. The complex genetic epidemiology of prostate cancer. Hum Mol Genet. 2004;13(Spec No 1):R103–R121. doi: 10.1093/hmg/ddh072. [DOI] [PubMed] [Google Scholar]
- 3.Chang BL, Isaacs SD, Wiley KE, et al. Genome-wide screen for prostate cancer susceptibility genes in men with clinically significant disease. Prostate. 2005;64:356–361. doi: 10.1002/pros.20249. [DOI] [PubMed] [Google Scholar]
- 4.Stanford JL, McDonnell SK, Friedrichsen DM, et al. Prostate cancer and genetic susceptibility: a genome scan incorporating disease aggressiveness. Prostate. 2006;66:317–325. doi: 10.1002/pros.20349. [DOI] [PubMed] [Google Scholar]
- 5.Fearon ER. Human cancer syndromes: clues to the origin and nature of cancer. Science. 1997;278:1043–1050. doi: 10.1126/science.278.5340.1043. [DOI] [PubMed] [Google Scholar]
- 6.Damber L, Gronberg H, Damber JE. Familial prostate cancer and possible associated malignancies: nation-wide register cohort study in Sweden. Int J Cancer. 1998;78:293–297. doi: 10.1002/(SICI)1097-0215(19981029)78:3<293::AID-IJC5>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 7.Hall JM, Lee MK, Newman B, et al. Linkage of early-onset familial breast cancer to chromosome 17q21. Science. 1990;250:1684–1689. doi: 10.1126/science.2270482. [DOI] [PubMed] [Google Scholar]
- 8.Miki Y, Swensen J, Shattuck-Eidens D, et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. 1994;266:66–71. doi: 10.1126/science.7545954. [DOI] [PubMed] [Google Scholar]
- 9.Thorlacius S, Tryggvadottir L, Olafsdottir GH, et al. Linkage to BRCA2 region in hereditary male breast cancer. Lancet. 1995;346:544–545. doi: 10.1016/s0140-6736(95)91383-1. [DOI] [PubMed] [Google Scholar]
- 10.Wooster R, Bignell G, Lancaster J, et al. Identification of the breast cancer susceptibility gene BRCA2. Nature. 1995;378:789–792. doi: 10.1038/378789a0. [DOI] [PubMed] [Google Scholar]
- 11.Gibbs M, Stanford JL, McIndoe RA, et al. Evidence for a rare prostate cancer-susceptibility locus at chromosome 1p36. Am J Hum Genet. 1999;64:776–787. doi: 10.1086/302287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Matsui H, Suzuki K, Ohtake N, et al. Genomewide linkage analysis of familial prostate cancer in the Japanese population. J Hum Genet. 2004;49:9–15. doi: 10.1007/s10038-003-0099-y. [DOI] [PubMed] [Google Scholar]
- 13.Badzioch M, Eeles R, Leblanc G, et al. Suggestive evidence for a site specific prostate cancer gene on chromosome 1p36. The CRC/BPG UK Familial Prostate Cancer Study Coordinators and Collaborators. The EU Biomed Collaborators. J Med Genet. 2000;37:947–949. doi: 10.1136/jmg.37.12.947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Johanneson B, Deutsch K, McIntosh L, et al. Suggestive genetic linkage to chromosome 11p11.2-q12.2 in hereditary prostate cancer families with primary kidney cancer. Prostate. 2007;67:732–742. doi: 10.1002/pros.20528. [DOI] [PubMed] [Google Scholar]
- 15.Pierce BL, Friedrichsen-Karyadi DM, McIntosh L, et al. Genomic scan of 12 hereditary prostate cancer families having an occurrence of pancreas cancer. Prostate. 2007;67:410–415. doi: 10.1002/pros.20527. [DOI] [PubMed] [Google Scholar]
- 16.Suarez BK, Lin J, Burmester JK, et al. A genome screen of multiplex sibships with prostate cancer. Am J Hum Genet. 2000;66:933–944. doi: 10.1086/302818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Witte JS, Suarez BK, Thiel B, et al. Genome-wide scan of brothers: replication and fine mapping of prostate cancer susceptibility and aggressiveness loci. Prostate. 2003;57:298–308. doi: 10.1002/pros.10304. [DOI] [PubMed] [Google Scholar]
- 18.Albright LA, Schwab A, Camp NJ, Farnham JS, Thomas A. Population-based risk assessment for other cancers in relatives of hereditary prostate cancer (HPC) cases. Prostate. 2005;64:347–355. doi: 10.1002/pros.20248. [DOI] [PubMed] [Google Scholar]
- 19.Enblad P, Adami HO, Glimelius B, Krusemo U, Pahlman L. The risk of subsequent primary malignant diseases after cancers of the colon and rectum. A nationwide cohort study. Cancer. 1990;65:2091–2100. doi: 10.1002/1097-0142(19900501)65:9<2091::aid-cncr2820650934>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 20.Haiman CA, Le Marchand L, Yamamato J, et al. A common genetic risk factor for colorectal and prostate cancer. Nat Genet. 2007;39:954–956. doi: 10.1038/ng2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Larson GP, Ding Y, Cheng LS, et al. Genetic linkage of prostate cancer risk to the chromosome 3 region bearing FHIT. Cancer Res. 2005;65:805–814. [PubMed] [Google Scholar]
- 22.Amundadottir LT, Sulem P, Gudmundsson J, et al. A common variant associated with prostate cancer in European and African populations. Nat Genet. 2006;38:652–658. doi: 10.1038/ng1808. [DOI] [PubMed] [Google Scholar]
- 23.Maier C, Herkommer K, Hoegel J, Vogel W, Paiss T. A genomewide linkage analysis for prostate cancer susceptibility genes in families from Germany. Eur J Hum Genet. 2005;13:352–360. doi: 10.1038/sj.ejhg.5201333. [DOI] [PubMed] [Google Scholar]
- 24.Stanford JL, FitzGerald LM, McDonnell SK, et al. Dense genome-wide SNP linkage scan in 301 hereditary prostate cancer families identifies multiple regions with suggestive evidence for linkage. Hum Mol Genet. 2009;18:1839–1848. doi: 10.1093/hmg/ddp100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Daley D, Lewis S, Platzer P, et al. Identification of susceptibility genes for cancer in a genome-wide scan: results from the colon neoplasia sibling study. Am J Hum Genet. 2008;82:723–736. doi: 10.1016/j.ajhg.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ohta M, Inoue H, Cotticelli MG, et al. The FHIT gene, spanning the chromosome 3p14.2 fragile site and renal carcinoma-associated t(3;8) breakpoint, is abnormal in digestive tract cancers. Cell. 1996;84:587–597. doi: 10.1016/s0092-8674(00)81034-x. [DOI] [PubMed] [Google Scholar]
- 27.Tomlinson I, Rahman N, Frayling I, et al. Inherited susceptibility to colorectal adenomas and carcinomas: evidence for a new predisposition gene on 15q14-q22. Gastroenterology. 1999;116:789–795. doi: 10.1016/s0016-5085(99)70061-2. [DOI] [PubMed] [Google Scholar]
- 28.Janer M, Friedrichsen DM, Stanford JL, et al. Genomic scan of 254 hereditary prostate cancer families. Prostate. 2003;57:309–319. doi: 10.1002/pros.10305. [DOI] [PubMed] [Google Scholar]
- 29.Abecasis GR, Cherny SS, Cookson WO, Cardon LR. Merlin--rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- 30.Kong A, Cox NJ. Allele-sharing models: LOD scores and accurate linkage tests. Am J Hum Genet. 1997;61:1179–1188. doi: 10.1086/301592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Abecasis GR, Wigginton JE. Handling marker-marker linkage disequilibrium: pedigree analysis with clustered markers. Am J Hum Genet. 2005;77:754–767. doi: 10.1086/497345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carlson CS, Eberle MA, Rieder MJ, Yi Q, Kruglyak L, Nickerson DA. Selecting a maximally informative set of single-nucleotide polymorphisms for association analyses using linkage disequilibrium. Am J Hum Genet. 2004;74:106–120. doi: 10.1086/381000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lander E, Kruglyak L. Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nat Genet. 1995;11:241–247. doi: 10.1038/ng1195-241. [DOI] [PubMed] [Google Scholar]
- 34.Schaid DJ, McDonnell SK, Zarfas KE, et al. Pooled genome linkage scan of aggressive prostate cancer: results from the International Consortium for Prostate Cancer Genetics. Hum Genet. 2006;120:471–485. doi: 10.1007/s00439-006-0219-9. [DOI] [PubMed] [Google Scholar]
- 35.Schleutker J, Baffoe-Bonnie AB, Gillanders E, et al. Genome-wide scan for linkage in finnish hereditary prostate cancer (HPC) families identifies novel susceptibility loci at 11q14 and 3p25-26. Prostate. 2003;57:280–289. doi: 10.1002/pros.10302. [DOI] [PubMed] [Google Scholar]
- 36.Eeles RA, Kote-Jarai Z, Giles GG, et al. Multiple newly identified loci associated with prostate cancer susceptibility. Nat Genet. 2008;40:316–321. doi: 10.1038/ng.90. [DOI] [PubMed] [Google Scholar]
- 37.Slager SL, Schaid DJ, Cunningham JM, et al. Confirmation of linkage of prostate cancer aggressiveness with chromosome 19q. Am J Hum Genet. 2003;72:759–762. doi: 10.1086/368230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu J, Dimitrov L, Chang BL, et al. A combined genomewide linkage scan of 1,233 families for prostate cancer-susceptibility genes conducted by the international consortium for prostate cancer genetics. Am J Hum Genet. 2005;77:219–229. doi: 10.1086/432377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ozden N, Saruc M, Smith LM, Sanjeevi A, Roy HK. Increased cumulative incidence of prostate malignancies in colorectal cancer patients. Int J Gastrointest Cancer. 2003;34:49–54. doi: 10.1385/IJGC:34:1:49. [DOI] [PubMed] [Google Scholar]
- 40.Burmester JK, Suarez BK, Lin JH, et al. Analysis of candidate genes for prostate cancer. Hum Hered. 2004;57:172–178. doi: 10.1159/000081443. [DOI] [PubMed] [Google Scholar]
- 41.Hirata H, Hinoda Y, Kawamoto K, et al. Mismatch repair gene MSH3 polymorphism is associated with the risk of sporadic prostate cancer. J Urol. 2008;179:2020–2024. doi: 10.1016/j.juro.2008.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Curtin K, Lin WY, George R, et al. Meta association of colorectal cancer confirms risk alleles at 8q24 and 18q21. Cancer Epidemiol Biomarkers Prev. 2009;18:616–621. doi: 10.1158/1055-9965.EPI-08-0690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Frattini M, Balestra D, Suardi S, et al. Different genetic features associated with colon and rectal carcinogenesis. Clin Cancer Res. 2004;10:4015–4021. doi: 10.1158/1078-0432.CCR-04-0031. [DOI] [PubMed] [Google Scholar]
- 44.Ueda T, Komiya A, Emi M, et al. Allelic losses on 18q21 are associated with progression and metastasis in human prostate cancer. Genes Chromosomes Cancer. 1997;20:140–147. doi: 10.1002/(sici)1098-2264(199710)20:2<140::aid-gcc4>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 45.Tomlinson I, Webb E, Carvajal-Carmona L, et al. A genome-wide association scan of tag SNPs identifies a susceptibility variant for colorectal cancer at 8q24.21. Nat Genet. 2007;39:984–988. doi: 10.1038/ng2085. [DOI] [PubMed] [Google Scholar]
- 46.Jaeger E, Webb E, Howarth K, et al. Common genetic variants at the CRAC1 (HMPS) locus on chromosome 15q13.3 influence colorectal cancer risk. Nat Genet. 2008;40:26–28. doi: 10.1038/ng.2007.41. [DOI] [PubMed] [Google Scholar]
- 47.Wallar BJ, Alberts AS. The formins: active scaffolds that remodel the cytoskeleton. Trends Cell Biol. 2003;13:435–446. doi: 10.1016/s0962-8924(03)00153-3. [DOI] [PubMed] [Google Scholar]
- 48.Zhou F, Leder P, Martin SS. Formin-1 protein associates with microtubules through a peptide domain encoded by exon-2. Exp Cell Res. 2006;312:1119–1126. doi: 10.1016/j.yexcr.2005.12.035. [DOI] [PubMed] [Google Scholar]
- 49.Djureinovic T, Skoglund J, Vandrovcova J, et al. A genome wide linkage analysis in Swedish families with hereditary non-familial adenomatous polyposis/non-hereditary non-polyposis colorectal cancer. Gut. 2006;55:362–366. doi: 10.1136/gut.2005.075333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cui Y, Ying Y, van Hasselt A, et al. OPCML is a broad tumor suppressor for multiple carcinomas and lymphomas with frequently epigenetic inactivation. PLoS One. 2008;3:e2990. doi: 10.1371/journal.pone.0002990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Broderick P, Carvajal-Carmona L, Pittman AM, et al. A genome-wide association study shows that common alleles of SMAD7 influence colorectal cancer risk. Nat Genet. 2007;39:1315–1317. doi: 10.1038/ng.2007.18. [DOI] [PubMed] [Google Scholar]
- 52.Navarro C, Chirlaque MD, Tormo MJ, et al. Validity of self reported diagnoses of cancer in a major Spanish prospective cohort study. J Epidemiol Community Health. 2006;60:593–599. doi: 10.1136/jech.2005.039131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sijmons RH, Boonstra AE, Reefhuis J, et al. Accuracy of family history of cancer: clinical genetic implications. Eur J Hum Genet. 2000;8:181–186. doi: 10.1038/sj.ejhg.5200441. [DOI] [PubMed] [Google Scholar]
- 54.Chang ET, Smedby KE, Hjalgrim H, Glimelius B, Adami HO. Reliability of self-reported family history of cancer in a large case-control study of lymphoma. J Natl Cancer Inst. 2006;98:61–68. doi: 10.1093/jnci/djj005. [DOI] [PubMed] [Google Scholar]
- 55.Theis B, Boyd N, Lockwood G, Tritchler D. Accuracy of family cancer history in breast cancer patients. Eur J Cancer Prev. 1994;3:321–327. doi: 10.1097/00008469-199407000-00004. [DOI] [PubMed] [Google Scholar]
- 56.Rapiti E, Fioretta G, Verkooijen HM, et al. Increased risk of colon cancer after external radiation therapy for prostate cancer. Int J Cancer. 2008;123:1141–1145. doi: 10.1002/ijc.23601. [DOI] [PubMed] [Google Scholar]
- 57.Bostrom PJ, Soloway MS. Secondary cancer after radiotherapy for prostate cancer: should we be more aware of the risk? Eur Urol. 2007;52:973–982. doi: 10.1016/j.eururo.2007.07.002. [DOI] [PubMed] [Google Scholar]