

Abstract
Over the past decade, multiple trials, including the precision medicine trial NCI-MATCH (National Cancer Institute-Molecular Analysis for Therapy Choice, EAY131, NCT02465060) have sought to determine if treating cancer based on specific genomic alterations is effective, irrespective of the cancer histology. Although many therapies are now approved for the treatment of cancers harboring specific genomic alterations, most patients do not respond to therapies targeting a single alteration. Further, when antitumor responses do occur, they are often not durable due to the development of drug resistance. Therefore, there is a great need to identify rational combination therapies that may be more effective. To address this need, the National Cancer Institute (NCI) and National Clinical Trials Network have developed NCI-ComboMATCH, the successor to NCI-MATCH. Like the original trial, NCI-ComboMATCH is a signal-seeking study. The goal of ComboMATCH is to overcome drug resistance to single-agent therapy and/or utilize novel synergies to increase efficacy by developing genomically-directed combination therapies, supported by strong preclinical in vivo evidence. While NCI-MATCH was mainly comprised of multiple single-arm studies, NCI-ComboMATCH tests combination therapy, evaluating both combination of targeted agents as well as combinations of targeted therapy with chemotherapy. While NCI-MATCH was histology agnostic with selected tumor exclusions, ComboMATCH has histology-specific and histology-agnostic arms. While NCI-MATCH consisted of single arm studies, ComboMATCH utilizes single-arm as well as randomized designs. NCI-MATCH had a separate, parallel Pediatric MATCH trial, whereas ComboMATCH will include children within the same trial. We present rationale, scientific principles, study design and logistics supporting the ComboMATCH study.
Keywords: Precision oncology, Personalized cancer therapy, genomic testing, Next generation sequencing, basket trial
Introduction
Advances in both molecular diagnostics and targeted drug development have ushered in an era of precision oncology. In this therapeutic paradigm, comprehensive genomic profiling (CGP) identifies actionable genomic alterations, including mutations, copy number changes, and fusions/rearrangements, to suggest rational, patient-specific anticancer treatments. The National Cancer Institute (NCI) Molecular Analysis for Therapy Choice (NCI-MATCH) trial,1 that consisted of 39 single-arm, phase 2 protocols, was launched in August 2015 to use tumor genotyping to ‘match’ patients with biomarker-selected therapies and to evaluate the antitumor activity of these therapies in both common and rare cancers.
The NCI-MATCH trial identified the very small fractions of patients with any given cancer type, who had prespecified oncogenic driver mutations or loss-of-function tumor suppressor gene mutations and assigned them to therapy with a corresponding targeted agent. Each NCI-MATCH protocol therefore determined the activity of the same therapy within molecularly defined cancer subsets, regardless of tumor histology. All but three of the treatment arms (pertuzumab/trastuzumab, BRAF/MEK, and PD-1/LAG-3) were single agent therapies as there were few validated combinations at that time.
For the first 6391 patients screened in NCI-MATCH, 5954 fresh tumor biopsy specimens or recent archival tumor tissue were obtained for analysis in one of the four central laboratories. Molecular alterations that would have qualified a patient for one of the arms (assuming arm was open, and patient met baseline eligibility criteria) were present in 37.6% of patients. After clinical and molecular exclusions, 17.8% of patients were assigned to a matched therapy.1 The trial’s success in rapidly accruing patients (6000 patients in 15 months total) demonstrated the great interest in precision oncology trials among patients and oncologists.
A similar study for pediatric cancer, the National Cancer Institute-Children’s Oncology Group Pediatric Molecular Analysis for Therapy Choice (NCI-COG Pediatric MATCH) trial, was launched in July 2017 and consisted of 13 single-arm, phase 2 subprotocols.2 The Pediatric MATCH was met with similar enthusiasm, and a had higher sub-arm enrollment rate among those with prior CGP (20.4%) suggested preselection of study participants.
The subsequent rare variant initiative in NCI-MATCH further demonstrated that the adoption of commercial and academic laboratory genomic testing as a standard-of-care had reached a point at which patients could successfully be accrued using “local” CGP by a designated network of sequencing laboratories that had been carefully selected for their ability to accurately identify actionable variants. Over the past decade, multiple industry-sponsored genomically-selected trials and other programmatic initiatives, such as the American Society of Clinical Oncology Targeted Agent and Profiling Utilization Registry (TAPUR), have successfully accrued patients through local testing.3–8 With the increasing use of CGP in cancer care, it is now feasible to launch genomically-driven basket and umbrella trials looking for rare alterations as well as disease-specific trials, even in rare cancers.
However, the initial wave of precision oncology trials revealed that precision oncology is a greater challenge than many had initially imagined for several reasons: 1) most tumors do not have a single genomic ‘driver’; 2) although many genes have been proposed as cancer drivers, only a few can be targeted by single-agent therapies that can achieve tumor regression; 3) objective responses with single-agent targeted therapies are often not durable due to emergence of resistance; and 4) driver mutations may have differing responses to targeted therapies based on differing adaptive responses in different tumors; and 5) responses may be limited due to concurrent or emergent genomic alterations in different cancers. In short, the one tumor–one gene–one drug approach oversimplified cancer biology and did not take into account tumor heterogeneity, clonal evolution or other mechanisms of resistance that tumors employ to overcome single-target blockade. Indeed, therapeutic success, as defined in the NCI-MATCH trial, was demonstrated in only 6 of the initial 27 completed arms (22%); 10 of these have been published in peer-reviewed journals, with another 10 reported in abstract form.1,9–43 Thus, the clinical outcomes of most of the single-agent NCI-MATCH arms, as well as a growing body of evidence regarding tumor mechanisms of adaptation and resistance, provided the impetus to pursue combinations of targeted therapies instead of continuing to focus on single-agent therapies in this new iteration of precision oncology trials.
Just as the concept of a platform for trials of combination therapies addresses the complexity of tumor response to therapy, so also does it increase the complexity of developing such trials; multiple considerations emerged. Since the number of potential combinations is nearly unlimited and a theoretical rationale could be developed for many of them, a strategy was required to select the most promising combinations to be tested in the trial. Since the contribution of individual drugs to the activity of a combination therapy may not be known, many arms would require randomization. Since most drug combinations required safety testing prior to efficacy determination, criteria for safety testing had to be developed, and mechanisms for doing so provided when necessary. Since drugs would be sought from different pharmaceutical partners, new mechanisms for developing agreements between NCI and pharmaceutical collaborators would be required. Since local CGP testing would be used for screening, support for pretreatment biopsies and central retrospective comprehensive testing to facilitate correlative science objectives, would need to be developed.
Facing all of these challenges, NCI developed NCI-ComboMATCH, a successor to the NCI-MATCH precision medicine trial, in collaboration with five cooperative groups in the NCI National Clinical Trials Network (NCTN): ECOG-ACRIN Cancer Research Grou; SWOG Cancer Research Network, ; NRG Oncology; the Alliance for Clinical Trials in Oncology; and the Children’s Oncology Group (COG). Whereas NCI-MATCH was comprised of parallel single-arm signal-seeking trials, NCI-ComboMATCH is a blend of signal-seeking single-arm trials and randomized trials dissecting the contributions of individual agents in combination treatments.
The NCI facilitated ComboMATCH in two important ways. First, as with the original MATCH study, ComboMATCH could only be possible if it was conducted under NCI IND sponsorship. Ordinarily, new NCI IND agents must be approved by NCI leadership through the NExT program. To streamline the incorporation of agents into ComboMATCH, NCI allowed the collaborative NCI-extramural ComboMATCH governance to approve agents for ComboMATCH and therefore NCI sponsorship without going through additional leadership review and approval. Second, the NCI provided support for pre-treatment biopsies, specimen storage, and genomic analyses of those biopsies; this is a level of support that is not usually provided for NCTN studies.
ComboMATCH also served as a vehicle to establish consensus among NCTN groups and with NCI regarding evaluation of the strength of preclinical data and in the design of signal- seeking studies of drug combinations. Representatives of the five lead trial organizations and NCI worked together in joint governance committees to evaluate drug combination proposals and clinical trial designs for those combinations. In working together on a common project on common ground, the clinician scientists and statisticians involved in ComboMATCH gradually achieved consensus on issues that undoubtedly will lead to more consistency in study designs, including NCI-funded studies beyond ComboMATCH.
In this paper, we review the rationale for, and design of, ComboMATCH, including the rationale, hypothesis, objectives and design of the ComboMATCH trial, the process by which potential combination therapy arms were selected, as well as statistical and biomarker considerations. We will also review its approach to patient matching, the qualified laboratory network, and the cooperative group logistics.
ComboMATCH: Hypothesis and Objectives
The ComboMATCH trial aims to establish whether patients with selected tumor mutations or amplifications are likely to demonstrate clinical activity (e.g., objective response, progression-free survival) from treatment with combinations of agents targeting that specific pathway. The central hypothesis of each individual study is that the evidence-based addition of a genomically-targeted agent to another anticancer therapy, whether that therapy is a single targeted agent or a standard chemotherapy agent or regimen, will produce greater clinical activity than will the standard treatment without the added targeted agent. The contribution of the additional targeted agent may be directly measured in a randomized cohort or in a single-arm cohort when there are adequate data on activity of the component agents individually to assess with confidence the activity of the added targeted agent. An important additional hypothesis to be tested in this trial is that preclinical data from in vivo models of drug combinations in the setting of known tumor genomic variants can predict clinical activity in defined patient groups. The central hypothesis of the overall ComboMATCH project is that a precision medicine initiative organized around principles of reliance on pre-clinical in vivo evidence will efficiently generate clinical studies that are likely to reach their clinical activity endpoints.
Studying drug combinations presents the challenge of identifying the best combination to test clinically for each molecular target. In NCI-MATCH, the choice of drug assigned for each target was limited by the number of drugs in each class, and their availability. However, in choosing a drug combination for a molecular target, the number of potential combinations could be in the dozens, or even in the hundreds. The selection of treatment arms for ComboMATCH, therefore, requires robust in vivo evidence, either preclinical or clinical, of the activity of a drug combination. For preclinical evidence, xenograft studies must show a significant combinatorial effect of both agents in comparison to each single agent in a molecularly relevant model. For clinical evidence, usually both agents in the combination need to show individual clinical activity in the same molecular context.
The ComboMATCH trial has a patient registration protocol managed by ECOG-ACRIN and several treatment protocols, that may contain one or more distinct substudies (Figure 1). The registration protocol contains rules for assigning patients to treatments and other guidelines. Pediatric and adult candidates who have undergone genomic profiling will be screened to assign those whose tumors harbor specific molecular abnormalities to a relevant substudy treatment arm. Some treatment arms are tumor agnostic, assigning treatments regardless of tumor type, while others may be limited to specific tumor types.
Figure 1.
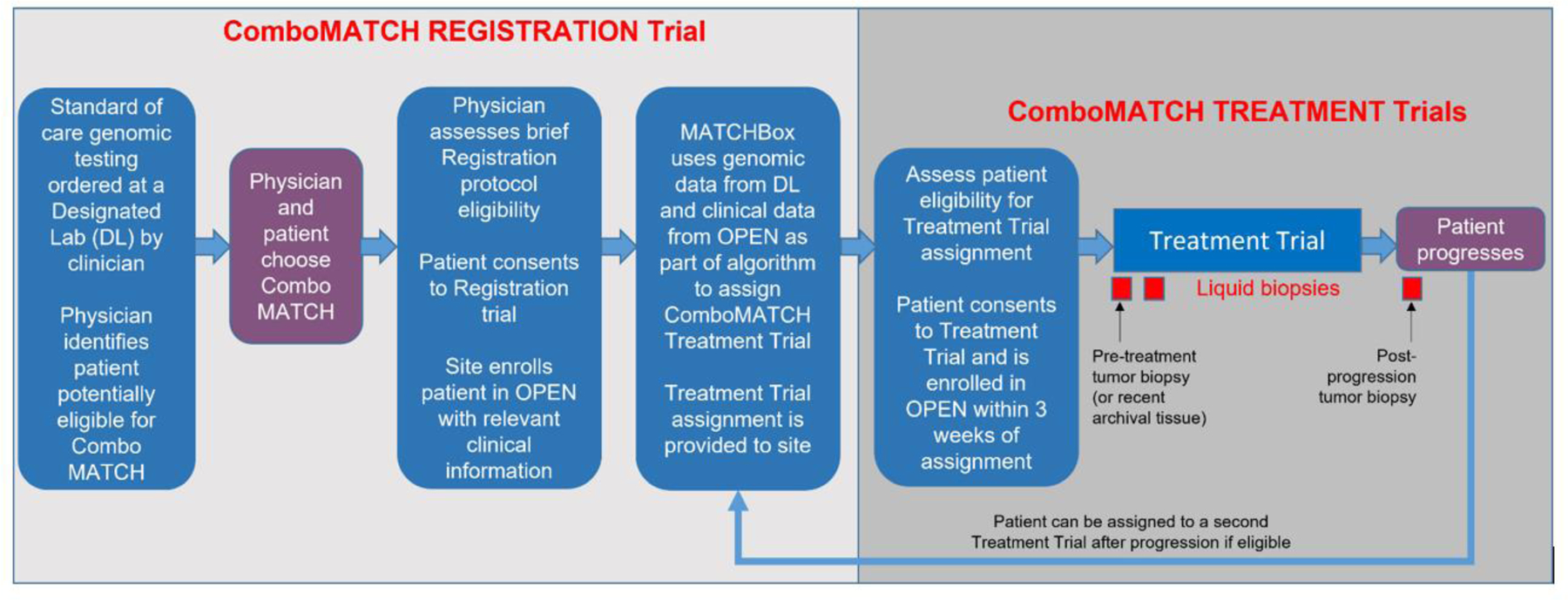
ComboMATCH Study Schema. Patients may enter ComboMATCH registration system and treatment trials through one of two routes. A patient’s oncologist may be aware of genomic eligibility as a result of prior genomic testing, or may be informed of genomic eligibility via a referral from a designated laboratory. The physician will then discuss the potentially available ComboMATCH treatment trial(s) with the patient. The first step towards enrollment on a ComboMATCH treatment trial is enrollment on the ComboMATCH registration trial and submission of eligibility information. These data will be analyzed in the ComboMATCH Precision Medicine Analysis and Coordination Center (PMACC) by a rules-based algorithm called the MATCHBox. The physician will be notified by the PMACC if the patient is potentially eligible for a ComboMATCH treatment trial. The physician will then determine whether the patient meets the specific ComboMATCH treatment trial eligibility requirements. If so, and if physician and patient agree, the patient may then enroll on the ComboMATCH treatment trial. Patients will be treated on the assigned arm until disease progression. Adult patients will undergo tumor biopsies prior to treatment initiation and at progression as well as longitudinal plasma collections.
Each of the five lead protocol organizations in the NCTN (ECOG-ACRIN, the Alliance for Clinical Trials in Oncology, NRG Oncology, SWOG Cancer Research Network, and COG) will manage a cassette of treatment protocols. Each subprotocol will specify the drug combination, genomic targets, trial design, and statistical endpoints to be used. As in NCI-MATCH, multiple investigators will lead each treatment arm in ComboMATCH. There will be opportunities for both early career and senior investigators to work alongside translational researchers.
The ComboMATCH study aims to determine if combination therapies shown to have enhanced activity compared to single agent therapy in in vivo preclinical studies, will predict clinical activity as defined by the individual treatment trials. If successful, we expect this paradigm to be more routinely implemented for early development of oncology drug combination regimens for molecularly targeted therapies in the future and may assist in identifying beneficial therapies for patients with rare malignancies and/or rare molecular abnormalities in their tumors.
Selection of Study Arms
Solicitation of Study Arms
The NCI-ComboMATCH trial was introduced to the broader scientific community on June 2, 2019, at the annual meeting of the American Society of Clinical Oncology, and meetings were held with interested investigators and with potential pharmaceutical collaborators. After these meetings, the Agents and Genes Working Group (AGWG) began accepting concepts for NCI-ComboMATCH subprotocols.
Study Arm Selection and Approval
The NCI ComboMATCH trial uses the AGWG to solicit and review subprotocol concept proposals. The AGWG includes four members from each NCTN lead protocol organization, additional members with expertise in developmental therapeutics and precision oncology, as well as the NCI Cancer Therapy Evaluation Program (CTEP) and other NCI investigators (Supplementary Table 1). Biomarker-linked drug combinations approved by AGWG are next reviewed by the Statistical Design Development Working Group (SDDWG) for statistical considerations and by the Molecular Biomarkers and Specimen Management (MBSM) Committee for correlative science. The SDDWG-approved study designs are then presented to the ComboMATCH Steering Committee for approval and then for CTEP review. In parallel with the ComboMATCH committee approval process, all concepts are also discussed with relevant pharmaceutical collaborators and with relevant CTEP drug monitors and CTEP disease leads. This work will culminate with the activation of ComboMATCH with ten initial treatment protocols, with at least one from each of the four adult NCTN groups and one from COG. The combinations and patient populations included in the first wave of the ComboMATCH trial are shown in Figure 2 and listed in Table 1. Additional concepts have already been approved, some with safety studies under way.
Figure 2.
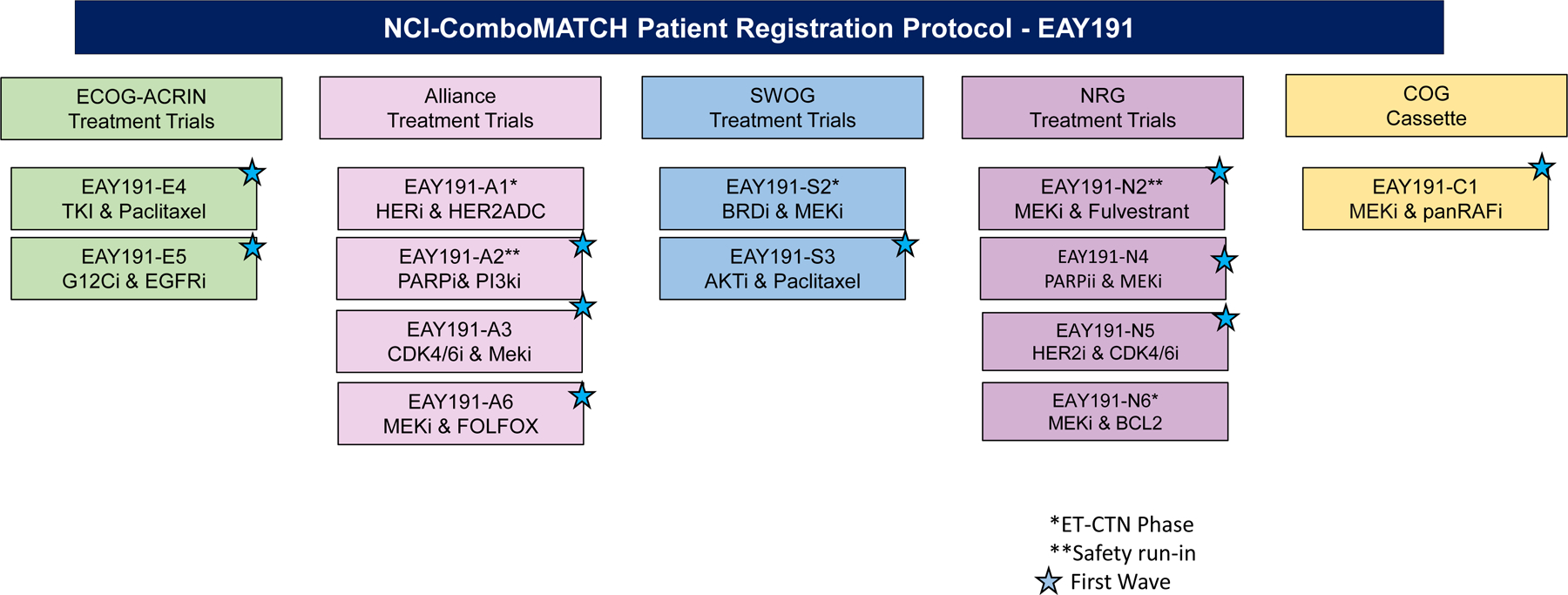
Initial ComboMATCH Subprotocols.
Table 1.
ComboMATCH substudies being activated in the first wave of trials.
| Substudy | Disease | Disease exclusions | Biomarker Inclusion | Biomarker Exclusion | Prior Treatment Requirement | Study Design | Arm 1 | Arm 2 | |
|---|---|---|---|---|---|---|---|---|---|
| EAY191- E4 | Solid tumors | Platinum-resistant serous histology ovarian cancer | None | cKIT mutation | Taxane resistant* | Single arm | Paclitaxel+ TKI | ||
| EAY191-E5 | Cohort A | Solid tumors | Colorectal cancer and NSCLC | KRAS G12C | KRAS G12C inhibitor naive | Randomized: | KRAS G12Ci | KRAS G12Ci + EGFRi | |
| EAY191-E5 | Cohort B | Solid tumors | KRAS G12C | KRAS G12C exposed | Single arm | KRAS G12Ci + EGFRi | |||
| EAY191-A2 | Cohort 1 | Breast cancer | Germline or somatic deleterious mutations in BAP1, BARD1, BRCA1, BRCA2, BRIP1, FANCA, FANCC, FANCD2, FANCE, FANCF, FANCM, MRE11, PALB2, RAD50, RAD51B, RAD51C, RAD51D | HER2 positive | PARP inhibitor naïve Prior PI3K inhibitor allowed Patients with ER+ tumors should have received prior endocrine therapy |
Randomized | PARPi + PI3Ki | PARPi | |
| EAY191-A2 | Cohort 2 | Breast cancer | Germline or somatic deleterious mutations in BAP1, BARD1, BRCA1, BRCA2, BRIP1, FANCA, FANCC, FANCD2, FANCE, FANCF, FANCM, MRE11, PALB2, RAD50, RAD51B, RAD51C, RAD51D | HER2 positive | PARP inhibitor exposed Prior PI3K inhibitor allowed Patients with ER+ tumors should have received prior endocrine therapy |
Single arm | PARPi + PI3Ki | ||
| EAY191-A3 | Cohort 1 A3.C1 |
Low grade serous ovarian cancer | KRAS, NRAS or BRAF activating mutation | MEK and CDK4/6 inhibitor naïve | Randomized | MEKi | MEKi+CDK4/6i | ||
| EAY191-A3 | Cohort 2 A3.C2 |
Low grade serous ovarian cancer | None | Progression on a MEK inhibitor No prior CDK4/6 inhibitor |
Single arm | MEKi+CDK4/6i | |||
| EAY191-A3 | Cohort 3 A3.C3 |
Pancreatic cancer | KRAS, NRAS, HRAS activating mutation, BRAF fusion | MEK and CDK4/6 inhibitor naïve | Single arm | MEKi+CDK4/6i | |||
| EAY191-A3 | Cohort 4 A3.C4 |
LGSOC, NSCLC, CRC, pancreatic and melanoma | KRAS, NRAS, HRAS activating mutation, BRAF fusion | MEK and CDK4/6 inhibitor naïve | Single arm | MEKi+CDK4/6i | |||
| EAY191-A6 | Intrahepatic or extrahepatic cholangiocarcinoma or gall bladder cancer | RAS, RAF, MEK, ERK pathway mutation | BRAF V600E | Prior gemcitabine therapy MEK and KRAS inhibitor naive |
Randomized | FOLFOX* | FOLFOX+MEKi | ||
| EAY191-S3 | Solid tumors | Breast cancer | AKT1, AKT2, or AKT3 mutation | KRAS, NRAS, HRAS, or BRAF mutation | Prior taxane No more than 2 prior lines of chemotherapy |
Single arm | Paclitaxel+AKTi | ||
| EAY191-N2 | Cohort 1 N2.C1 |
Breast cancer | Estrogen or progesterone receptor positive and HER2 negative NF1 nonsense or frameshift mutation or NF1 deletion |
Fulvestrant naïve Prior CDK4/6 encouraged. Up to one line of chemo |
Randomized | Fulvestrant | Fulvestrant+MEKi | ||
| EAY191-N2 | Cohort 2 N2.C2 |
Breast cancer | Estrogen or progesterone receptor positive and HER2 negative NF1 nonsense or frameshift mutation or NF1 deletion |
Fulvestrant-exposed Up to one line of chemo |
Single arm | Fulvestrant + MEKi | |||
| EAY191-N4 | Ovarian cancer | Activating KRAS/NRAS, HRAS BRAF activating mutations, inactivating NF1 | MEK, PARP inhibitor naive | Randomized; | MEKi | MEK1+PARPi | |||
| EAY191-N4 | Endometrial cancer | Activating KRAS/NRAS, HRAS BRAF activating mutations, inactivating NF1 | MEK, PARP inhibitor naive | Randomized: | MEKi | MEKi+PARPi | |||
| EAY191-C1 | C1. Cohort 1 |
Low grade glioma | BRAF, HRAS, KRAS, NF1, NRAS, or RAF1 | Patients with a BRAFV600E mutation with documented progression on a BRAFV600E inhibitor. Patients with a BRAF fusion or non-V600 SNV with documented progression on a MEK inhibitor. Patients with a deletion or deleterious mutation in NF1 with documented progression on a MEK inhibitor. Patients who have stopped MEKi due to the inability to tolerate the treatment will be excluded |
Single arm | pan-RAFi+MEKi | |||
| EAY191-C1 | C1. Cohort 2 | Malignant tumors excluding LGG | Activating SNVs or fusions in BRAF or | Single arm | pan-RAFi+ MEKi | ||||
| EAY191-C1 | C1. Cohort 3 |
Malignant tumors excluding LGG | Activating mutations in HRAS, KRAS, or NRAS | Single arm | Pan-RAFi + MEKi | ||||
| EAY191-C1 | C1. Cohort 4 | Malignant tumors Excluding LGG |
Deletions or other loss of function alterations in NF1 | Single arm | Pan-RAF1+MEKi |
Patients who previously responded to prior taxane therapy must have received their last dose of taxane therapy within 6 months prior to EAY191-E4 registration and have had no other intervening treatment prior to EAY191-E4 registration.
EAY191-C1 will be available for individuals 3–40 years of age.
All ComboMATCH arms will be reviewed by the FDA for design and potential outcome. However, ComboMATCH trials are signal-seeking studies, and not conducted with registrational intent. Trials that meet their primary clinical endpoint are expected to be followed by larger, confirmatory studies.
Brief Scientific Principles for Study Arm Selection
Subprotocol concepts are solicited and reviewed based on the following scientific principles:
Proposals must be supported by both a strong scientific rationale and in vivo evidence of activity in relevant models or in patients.
Combination therapy proposals are desired. There should be evidence that both agents in the combination are required for higher efficacy. The demonstrable activity in preclinical studies should be at least prolonged stable disease and preferably tumor regression.
Proposals should have safety data available, but justification of a short run-in design or plans for a phase 1 study may be considered if adequately justified.
Combinations with strong preclinical data but lacking safety data will be considered for future arms after safety data are obtained. Such combinations are referred by the ComboMATCH AGWG to the NCI Experimental Therapeutics Clinical Trials Network (ETCTN) for phase 1 study, or may have investigator-initiated or industry-sponsored phase 1 studies completed outside of the NCI network.
ComboMATCH substudies may have single-arm, or randomized designs.
Combinations involving immunotherapy agents are not allowed for two reasons. First, no consensus has emerged regarding the utilization of preclinical in vivo models for therapeutic testing of immunotherapy agents, so it was not possible to establish a preclinical evidence threshold that could be broadly applied for evaluating proposed combinations. Second, a separate NCI initiative (iMATCH) is in development to provide relevant diagnostic support for prospective immune-oncology signal-seeking studies.
Assessing Preclinical Data
Combinations chosen for ComboMATCH are expected to have preclinical data demonstrating that the combination had efficacy greater than that of either single agent in at least two clinically relevant in vivo models (cell line–derived xenografts or patient-derived xenografts [PDXs]) at clinically relevant doses. Although preclinical data for the agents proposed in the clinical trial are preferred, data generated for agents from the same class are allowed. The data for several accepted treatment arms were generated by PDXNet, a Cancer Moonshot–funded initiative designed to advance precision cancer medicine through rational testing of novel agent combinations in patient-derived models. Similarly, it is expected that the NCI-supported Pediatric Preclinical in Vivo Testing (PIVOT) program will generate preliminary data for future pediatric concepts.
Preclinical combination treatment experiments are expected to include cohorts treated with each single agent to demonstrate whether combination treatment enhances antitumor activity not only compared to untreated or vehicle-treated controls, but also compared to both of the single agents. Prolonged duration of treatment or follow-up is encouraged to determine the durability of treatment activity and safety. Combinations are expected to have more growth-inhibitory activity compared to either monotherapy alone and to achieve durable stabilization of tumor growth or, preferably, tumor regression.
Safety Considerations in Combination Therapy
In considering combination therapies, combinations for which safety data were already available were prioritized for the first wave of trials. Safety data for each combination were reviewed, along with any existing clinical activity data from monotherapy trials and combination therapy trials involving the proposed agents. For each proposed combination, planning included careful review of the safety of each agent and the potential for overlapping toxic effects of the two agents. Notably, some of the proposed preclinical combinations did not move forward because of emerging safety signals observed in early-phase trials of those combinations.
Statistical design of ComboMATCH studies
Design of the ComboMATCH signal-finding platform trial required tailoring subprotocols to the available evidence for each drug combination of interest, often by focusing on whether known single-agent activity data for specific genotypes or tumor types were available, and on the strength of those data. For each combination, the intended patient population was partitioned into sub-study cohorts (usually 2–4 per protocol) on the basis of histology, molecular characteristics, and prior treatment experience, so that a relevant activity signal can be isolated by an appropriate randomized or single-arm phase 2 design according to the principles outlined below.
Randomized designs
A key question for any study of a combination therapy is whether the combination offers superior antitumor activity to that of either of the individual agents alone. This question necessitated incorporation of randomized designs for some ComboMATCH cohorts, which were not required in the single-agent arms of the original NCI-MATCH study. In most scenarios encountered in ComboMATCH, there is evidence of clinical activity of one of the agents individually, and the hypothesis is that the combination will substantially enhance activity. In this situation, a design that randomizes patients previously untreated with the active agent in the combination to active single-agent and combination arms is felt to be appropriate and informative about added benefit of the combination.
An implicit advantage of randomized designs in the advanced cancer setting is that they allow use of time-to-event endpoints like progression-free survival (PFS). Time-to-event endpoints are generally not appropriate for single-arm studies because they cannot isolate the activity signal from the varying natural course of most cancers. PFS-based designs are more statistically efficient (i.e., they require a smaller sample size) than are designs using binary endpoints, such as tumor response. Moreover, PFS captures more clinically meaningful information, including not only whether a tumor responds and/or progresses, but also the durability of the response or the rate of progression. In settings where death usually proceeds quickly after progression, overall survival (OS) could theoretically be used as it avoids challenges in determining time of progression, but PFS is often preferred in signal seeking studies because it typically results in nontrivially smaller designs owing to more rapid event accumulation than OS and ability to target a larger hazard ratio (HR). Most randomized designs within ComboMATCH allocate about 30 to 40 patients per arm to yield at least 80% power for an one-sided 0.10 level log-rank test to detect an PFS HR of about 0.5, corresponding roughly to median PFS of 6 months on single-agent versus 12 months on combination treatment. Crossover to combination treatment may be allowed for patients who experience disease progression on a monotherapy arm. Randomization is stratified (sometimes with accrual caps) by tumor histology and/or clinical or molecular characteristics when these factors are thought to be substantially prognostic but their treatment effects (i.e., HR) are similar. All randomized designs include an interim stopping rule for futility, typically at half of total expected events44
Single-arm designs
Single-arm designs with a response endpoint are considered appropriate when eligible patients have already had disease progression while on the agents involved in the combination or when background data strongly suggest that monotherapy activity is minimal at best. Although prior progression on both single agents in the combination when each given as monotherapy would maximize interpretability, accrual of such a cohort in certain disease settings may not be considered feasible. Therefore, a combination of evidence suggesting minimal or no monotherapy activity and/or a requirement for prior progression can be used to justify single-arm designs in ComboMATCH. The possibility for a response upon re-challenge after progression with the single agent or another in same class must also be considered. Some uncertainty may remain about the extent to which single-agent activity might explain responses to combinations. Accordingly, targeted response rates considered “promising” for ComboMATCH are set higher in cohorts where there is greater uncertainty about monotherapy activity. All single-arm designs in ComboMATCH use an objective response endpoint, defined as complete response or partial response based on RECIST v1.1 criteria.45
Single-arm designs in ComboMATCH employ two-stage strategies (e.g., Simon two-stage) to permit early stopping if the observed response rate in the first stage is low. Null response rates are set in the 10% to 30% range, with lower null values corresponding to stronger evidence that single-agent response rates are minimal. Targeted promising response rates are typically set 15% to 20% higher (absolute) than null values. The resulting single-arm designs typically require a maximum of 20 to 30 patients, with the possibility of early stopping for futility, usually after the first 10 to 15 patients.
Designated (Qualified) Laboratory Network and Biomarker Considerations
Molecular eligibility for ComboMATCH trials will be determined by using archival specimens analyzed by laboratories in the Designated Laboratory Network (DLN) developed for the NCI-MATCH trial. This network of academic and commercial Clinical Laboratory Improvement Amendments–accredited laboratories was validated previously to perform high-quality next-generation sequencing (NGS). The Precision Medicine Analysis and Coordination Committee (PMACC) will use information provided by patients’ treating physicians as well as data from the DLN for initial study and cohort assignment.
After treatment assignment, pretreatment tumor biopsies will be mandatory for adults (age≥18) on all ComboMATCH substudies, and biopsies at the time of progression will be optional. In nearly all substudies, biopsy samples will be freshly retrieved and then fixed in neutral-buffered formalin and embedded in paraffin for use in subsequent assays. Tumor biopsy specimens that are obtained within 12 months of study entry may substitute for fresh ones if there has been no intervening therapy. To assess determinants of response and of intrinsic or acquired resistance to the various drug combinations, whole exome sequencing (WES) and whole-transcriptome sequencing (RNA-Seq) will be performed as integrated biomarkers. WES will also permit retrospective confirmation of variants identified by NGS performed by one of the laboratories in the DLN for patient eligibility and enrollment. For WES, hybrid capture technology will be used on the Illumina NovaSeq 6000 sequencing platform to compare tumor and germline genomes. This technology can perform (1) calling of single-nucleotide variants and indels; (2) estimation of copy number variations; (3) determination of loss of heterozygosity; (4) assessment of microsatellite instability and tumor mutational burden; and (5) assessment of mutational signatures. RNA-Seq will use transcript-based probe capture technology on a similar platform to facilitate (1) gene expression analyses; (2) calling of RNA variants; and (3) calling of gene fusions, including intragenic fusion events.
For trials involving PARP inhibitors or other inhibitors of DNA repair, WES will also be used to determine a pretreatment homologous recombination deficiency score, calculated using a sum of scores for loss of heterozygosity, telomeric allelic imbalance, and large-scale state transitions. It is recognized that homologous recombination deficiency may cause these characteristic genomic scar signatures, which may persist even if the homologous recombination pathway is restored as acquired resistance to previous treatments emerges. For this reason, in selected sub-studies, biopsy samples will also be assessed for the presence of RAD51 foci, which will allow genomic and functional information to be correlated in the characterization of the tumor immediately before exposure to study drug combinations.
ComboMATCH will also incorporate circulating tumor DNA (ctDNA) sequencing across sub-studies, typically at 3 timepoints: (1) baseline, (2) after 1 cycle, and (3) at time of progression. The methodology involves hybrid capture technology with unique molecular indices and error correction algorithms. Illumina TruSight Oncology 500 ctDNA (TSO500) library preparation will be used to interrogate 523 genes for single-nucleotide variants, indels, and copy number variants and to estimate tumor mutational burden and microsatellite instability. Pretreatment ctDNA analysis will be compared to tumor profiles generated by WES and targeted NGS and may capture tumor heterogeneity that could be missed on analysis of a single core biopsy sample. Changes (or lack thereof) in ctDNA alterations after one cycle of combination treatment will be correlated with ultimate clinical outcomes. Among patients who have a response and/or clinical benefit, ctDNA analysis at time of progression may provide insight into the mechanisms of acquired resistance.
All central assays (WES, RNA-Seq, ctDNA analysis) will be performed by NCI’s Molecular Diagnostic Network (MDNet), a group of laboratories supported by NCI for several precision medicine trials. In addition, principal investigators may incorporate additional trial-specific biomarkers that have been approved by the Molecular Biomarker and Specimen Management Committee. For example, some of the trials using a CDK4/6 inhibitor will incorporate assays for serum thymidine kinase 1 (TK1) activity, which can provide a pharmacodynamic measure of CDK4/6 inhibitor exposure.46 The kinetics and degree of decline on treatment can then be linked to clinical outcomes. Similar to NCI-MATCH, deidentified sequencing data and associated clinical data will be deposited in the NCI Cancer Research Data Commons, upon publication of primary publications.
ComboMATCH Trial Matching
As with NCI-MATCH, treatments will be assigned with a validated NCI-designed computational platform (MATCHBox). In most cases, ComboMATCH treatment assignments will be driven by actionable alterations reported by the Designated Laboratory Network. Treatment assignments issued by MATCHBox will consider priorities of histology, gene/variants, and mutational type, as well as variant allele frequency, levels of evidence, and accrual balance. While NCI-MATCH leveraged a locked assignment algorithm, ComboMATCH will allow for a more adaptive algorithmic approach.
ComboMATCH will take advantage of capabilities developed to import data from the Designated Laboratory Network under NCI-MATCH. These capabilities support the automated downloading and harmonized annotation of molecular sequencing data. Additionally, to facilitate more precise treatment assignments, NCI is collecting histology and prior therapy data capture at registration.
An initial list of actionable variants has been selected prior to activation of ComboMATCH and flexibility to update these variants regularly will be permitted without protocol amendment. ComboMATCH has also inherited the novel aMOI process of NCI-MATCH. This process allows for the real-time submission and review of potentially actionable alterations as a patient is registered. New for ComboMATCH will be a physician’s choice option at registration. This option will allow a one-time selection at registration that will take effect if the algorithm provides more than one option, including the indicated preference, or if there is clinical justification to support modification of the algorithm. Reassignment of patients with disease progression, or another reason for therapy discontinuation, to a second ComboMATCH subprotocol will be permitted.
Conclusion
ComboMATCH is being launched as a large precision medicine initiative designed collaboratively with NCI/CTEP and representatives from all NCTN lead protocol organizations. The design of the sub-studies is being developed with wide engagement across academia, including basic and translational researchers and clinical trialists. ComboMATCH leverages the CTEP Investigational New Drug program to sponsor innovative trials of combinations of agents from different pharmaceutical partners. Coming together to build this next-generation precision medicine trial are NCI’s ETCTN, which is generating phase 1 safety data that will enable a number of the second wave of ComboMATCH studies; NCI’s Cancer Moonshot-funded PDXNet, which produced the preclinical data that led to several of the ComboMATCH arms; the DLN that was originally built to support NCI-MATCH and now will support this effort; and of course, the NCI-funded NCTN. In many ways, the timing is right for ComboMATCH. While NCI-MATCH was launched when CGP was just being explored for the care of patients with cancer, CGP is now part of routine care for patients with advanced/metastatic disease who may benefit from genomic biomarker–matched therapy.47 ComboMATCH is also more patient-centric than NCI-MATCH as patients do not need to undergo genomic testing through Combo-MATCH. Instead, patients who undergo routine CGP in any of the laboratories in the Designated Laboratory Network can access a genomically-matched combination therapy at a ComboMATCH site near them through the large NCTN.
This is the era of team science and biomarker-directed therapy. It is only through continued collaborations among health care providers, clinical and translational researchers, statisticians, patient advocates, and the NCI as well as Industry partners that such an important initiative can come to fruition. It is the great promise to improve patient outcomes that makes these efforts absolutely worthwhile.
Supplementary Material
Supplementary Table 1: The ComboMATCH study team.
Translational Relevance.
With a few notable exceptions, genomically-matched, single-agent anticancer therapies rarely achieve durable clinical benefit. Therefore, the National Cancer Institute (NCI) and National Clinical Trials Network have developed ComboMATCH, a coordinated set of clinical trials that each test a rational, targeted drug combination therapy selected on the basis of strong preclinical evidence of efficacy. ComboMATCH will succeed the MATCH trial as the NCI platform for advancing cancer precision medicine. We here present the rationale, scientific principles, study design and logistics supporting the ComboMATCH study, emphasizing how ComboMATCH has served as a framework for achieving consensus among all participants on issues of preclinical and clinical evidence, and clinical trial design. As the number of available ComboMATCH trials grows over time, we expect that ComboMATCH will provide promising treatment options for patients while serving as a vehicle for translating robust preclinical evidence of therapeutic efficacy of novel drug combinations into the clinic.
Acknowledgements
We thank Amy Ninetto from the MD Anderson Research Medical Library for her editorial assistance and Susanna Brisendine for administrative assistance.
Financial Support:
This work was supported in part by the The MD Anderson Cancer Center Support Grant (P30 CA016672) and the UT MD Anderson Cancer Center Network Lead Academic Participating Site (LAP) UG1 grant (UG1 CA233329). REO is funded in part by the NIH/NCI Cancer Center Support Grant P30 CA008748.
References
- 1.Flaherty KT, Gray RJ, Chen AP, et al. Molecular Landscape and Actionable Alterations in a Genomically Guided Cancer Clinical Trial: National Cancer Institute Molecular Analysis for Therapy Choice (NCI-MATCH). Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2020; 38(33): 3883–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Parsons DW, Janeway KA, Patton DR, et al. Actionable Tumor Alterations and Treatment Protocol Enrollment of Pediatric and Young Adult Patients With Refractory Cancers in the National Cancer Institute-Children’s Oncology Group Pediatric MATCH Trial. J Clin Oncol 2022; 40(20): 2224–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mangat PK, Halabi S, Bruinooge SS, et al. Rationale and Design of the Targeted Agent and Profiling Utilization Registry (TAPUR) Study. JCO Precis Oncol 2018; 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hyman DM, Piha-Paul SA, Won H, et al. HER kinase inhibition in patients with HER2- and HER3-mutant cancers. Nature 2018; 554(7691): 189–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hainsworth JD, Meric-Bernstam F, Swanton C, et al. Targeted Therapy for Advanced Solid Tumors on the Basis of Molecular Profiles: Results From MyPathway, an Open-Label, Phase IIa Multiple Basket Study. J Clin Oncol 2018; 36(6): 536–42. [DOI] [PubMed] [Google Scholar]
- 6.Meric-Bernstam F, Bahleda R, Hierro C, et al. Futibatinib, an Irreversible FGFR1–4 Inhibitor, in Patients with Advanced Solid Tumors Harboring FGF/FGFR Aberrations: A Phase I Dose-Expansion Study. Cancer Discov 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Subbiah V, Puzanov I, Blay JY, et al. Pan-Cancer Efficacy of Vemurafenib in BRAF (V600)-Mutant Non-Melanoma Cancers. Cancer Discov 2020; 10(5): 657–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hong DS, DuBois SG, Kummar S, et al. Larotrectinib in patients with TRK fusion-positive solid tumours: a pooled analysis of three phase 1/2 clinical trials. Lancet Oncol 2020; 21(4): 531–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bedard PL, Li S, Wisinski KB, et al. Abstract CT139: NCI Molecular Analysis for Therapy Choice (NCI-MATCH EAY131) arm B: Phase II study of afatinib in patients (pts) with HER2 (ERBB2) activating mutations. Cancer Research 2019; 79(13_Supplement): CT139–CT. [Google Scholar]
- 10.Mansfield AS, Wei Z, Mehra R, et al. Crizotinib in patients with tumors harboring ALK or ROS1 rearrangements in the NCI-MATCH trial. npj Precision Oncology 2022; 6(1): 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Salama AKS, Li S, Macrae ER, et al. Dabrafenib and trametinib in patients with tumors with BRAF V600E/K mutations: Results from the molecular analysis for therapy choice (MATCH) Arm H. Journal of Clinical Oncology 2019; 37(15_suppl): 3002-. [Google Scholar]
- 12.Salama AKS, Li S, Macrae ER, et al. Dabrafenib and Trametinib in Patients With Tumors With BRAF(V600E) Mutations: Results of the NCI-MATCH Trial Subprotocol H. J Clin Oncol 2020; 38(33): 3895–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Krop IE, Jegede O, Grilley-Olson JE, et al. Results from molecular analysis for therapy choice (MATCH) arm I: Taselisib for PIK3CA-mutated tumors. Journal of Clinical Oncology 2018; 36(15_suppl): 101-.29220288 [Google Scholar]
- 14.Krop IE, Jegede OA, Grilley-Olson JE, et al. Phase II Study of Taselisib in PIK3CA-Mutated Solid Tumors Other Than Breast and Squamous Lung Cancer: Results From the NCI-MATCH ECOG-ACRIN Trial (EAY131) Subprotocol I. JCO Precision Oncology 2022; (6): e2100424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Connolly RM, Wang V, Hyman DM, et al. Activity of trastuzumab and pertuzumab (HP) in patients with non-breast/gastroesophgeal HER2-amplified tumours: Results of the NCI-MATCH trial (EAY131) subprotocol J. Annals of Oncology 2020; 31. [Google Scholar]
- 16.Janku F, Jegede O, Puhalla S, et al. NCI-MATCH Arms N & P: phase II study of PI3K beta inhibitor GSK2636771 in patients (pts) with cancers (ca) with PTEN mutation/deletion (mut/del) or PTEN protein loss. Annals of Oncology 2018; 29. [Google Scholar]
- 17.Jhaveri KL, Makker V, Wang XV, et al. Ado-trastuzumab emtansine (T-DM1) in patients (pts) with HER2 amplified (amp) tumors excluding breast and gastric/gastro-esophageal junction (GEJ) adenocarcinomas: Results from the National Cancer Institute (NCI) Molecular Analysis for Therapy Choice (MATCH) trial. Journal of Clinical Oncology 2018; 36(15_suppl): 100-. [Google Scholar]
- 18.Jhaveri KL, Wang XV, Makker V, et al. Ado-trastuzumab emtansine (T-DM1) in patients with HER2-amplified tumors excluding breast and gastric/gastroesophageal junction (GEJ) adenocarcinomas: results from the NCI-MATCH trial (EAY131) subprotocol Q. Ann Oncol 2019; 30(11): 1821–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Johnson DB, Zhao F, Noel M, et al. Trametinib Activity in Patients with Solid Tumors and Lymphomas Harboring BRAF Non-V600 Mutations or Fusions: Results from NCI-MATCH (EAY131). Clin Cancer Res 2020; 26(8): 1812–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chae YK, Hong F, Vaklavas C, et al. Phase II Study of AZD4547 in Patients With Tumors Harboring Aberrations in the FGFR Pathway: Results From the NCI-MATCH Trial (EAY131) Subprotocol W. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2020; 38(21): 2407–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chae YK, Vaklavas C, Cheng HH, et al. Molecular analysis for therapy choice (MATCH) arm W: Phase II study of AZD4547 in patients with tumors with aberrations in the FGFR pathway. Journal of Clinical Oncology 2018; 36(15_suppl): 2503-. [Google Scholar]
- 22.Kalinsky K, Hong F, McCourt CK, et al. Effect of Capivasertib in Patients With an AKT1 E17K-Mutated Tumor: NCI-MATCH Subprotocol EAY131-Y Nonrandomized Trial. JAMA Oncol 2021; 7(2): 271–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cleary JM, Wang V, Heist RS, et al. Differential Outcomes in Codon 12/13 and Codon 61 NRAS-Mutated Cancers in the Phase II NCI-MATCH Trial of Binimetinib in Patients with NRAS-Mutated Tumors. Clin Cancer Res 2021; 27(11): 2996–3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Azad NS, Gray RJ, Overman MJ, et al. Nivolumab Is Effective in Mismatch Repair-Deficient Noncolorectal Cancers: Results From Arm Z1D-A Subprotocol of the NCI-MATCH (EAY131) Study. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2020; 38(3): 214–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Damodaran S, Zhao F, Deming DA, et al. Phase II study of copanlisib in patients with tumors with PIK3CA mutations (PTEN loss allowed): NCI MATCH EAY131-Z1F. Journal of Clinical Oncology 2020; 38(15_suppl): 3506-. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Damodaran S, Zhao F, Deming DA, et al. Phase II Study of Copanlisib in Patients With Tumors With PIK3CA Mutations: Results From the NCI-MATCH ECOG-ACRIN Trial (EAY131) Subprotocol Z1F. J Clin Oncol 2022: Jco2101648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mita AC, Wei Z, Mayer IA, et al. Abstract LBA003: Erdafitinib in patients with tumors harboring FGFR gene mutations or fusions: Results from the NCI-MATCH ECOG-ACRIN Trial (EAY131) Sub-protocol K2. Molecular Cancer Therapeutics 2021; 20(12_Supplement): LBA003–LBA. [Google Scholar]
- 28.Jackman DM, Jegede O, Zauderer MG, et al. A phase 2 study of defactinib (VS-6063) in patients with NF2 altered tumors: Results from NCI-match (EAY131) subprotocol U. Journal of Clinical Oncology 2021; 39(15_suppl): 3087-.34228512 [Google Scholar]
- 29.Cella D, Wagner L. Re-Personalizing Precision Medicine: Is there a role for patient-reported outcomes? Journal of Community and Supportive Oncology 2015; 13(8): 3.25839059 [Google Scholar]
- 30.Chen A, Flaherty K, O’Dwyer PJ, et al. Tumor Genomic Profiling Practices and Perceptions: A Survey of Physicians Participating in the NCI-MATCH Trial. JCO precision oncology 2020; 4: PO.20.00217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen AP, O’dwyer PJ, Harris L, et al. Abstract PL03–01: NCI-MATCH: A new paradigm in the era of genomic oncology 2018. [Google Scholar]
- 32.Conley BA, Gray R, Chen A, et al. Abstract CT101: NCI-molecular analysis for therapy choice (NCI-MATCH) clinical trial: interim analysis. Cancer Research 2016; 76(14_Supplement): CT101–CT. [Google Scholar]
- 33.Conley BA, Doroshow JH. Molecular analysis for therapy choice: NCI MATCH. Semin Oncol 2014; 41(3): 297–9. [DOI] [PubMed] [Google Scholar]
- 34.Conley BA, Chen AP, O’Dwyer PJ, et al. NCI-MATCH (Molecular Analysis for Therapy Choice) – a national signal finding trial. Journal of Clinical Oncology 2016; 34(15_suppl): TPS2606–TPS. [Google Scholar]
- 35.Conley BA, Hamilton SR, Li S, et al. Abstract A053: Prevalence of mismatch repair deficiency (dMMR) in the NCI Molecular Analysis for Therapy Choice (NCI-MATCH or EAY131) population. Molecular Cancer Therapeutics 2018; 17(1_Supplement): A053–A. [Google Scholar]
- 36.Coyne GOS, Takebe N, Chen AP. Defining precision: The precision medicine initiative trials NCI-MPACT and NCI-MATCH. Current Problems in Cancer 2017; 41(3): 182–93. [DOI] [PubMed] [Google Scholar]
- 37.Do K, O’Sullivan Coyne G, Chen AP. An overview of the NCI precision medicine trials-NCI MATCH and MPACT. Chin Clin Oncol 2015; 4(3): 31. [DOI] [PubMed] [Google Scholar]
- 38.Flaherty KT, Gray R, Chen A, et al. The Molecular Analysis for Therapy Choice (NCI-MATCH) Trial: Lessons for Genomic Trial Design. J Natl Cancer Inst 2020; 112(10): 1021–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Harris LN, Gray RJ, Conley BA, et al. Abstract A079: National Cancer Institute Molecular Analysis for Therapy Choice (NCI-MATCH): A successful precision medicine signal-seeking trial in patients (pts) with rare variants and refractory malignancies. Molecular Cancer Therapeutics 2019; 18(12_Supplement): A079–A. [Google Scholar]
- 40.Khoury JD, Wang W-L, Prieto VG, et al. Validation of Immunohistochemical Assays for Integral Biomarkers in the NCI-MATCH EAY131 Clinical Trial. Clin Cancer Res 2018; 24(3): 521–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lih CJ, Takebe N. Considerations of developing an NGS assay for clinical applications in precision oncology: The NCI-MATCH NGS assay experience. Curr Probl Cancer 2017; 41(3): 201–11. [DOI] [PubMed] [Google Scholar]
- 42.Lih C-J, Harrington RD, Sims DJ, et al. Analytical Validation of the Next-Generation Sequencing Assay for a Nationwide Signal-Finding Clinical Trial: Molecular Analysis for Therapy Choice Clinical Trial. J Mol Diagn 2017; 19(2): 313–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tricoli JV, Zane L, Harrington R, et al. Design and development of the molecular analysis for Therapy Choice (NCI-MATCH) Designated Laboratory Network. Journal of Clinical Oncology 2019; 37(15_suppl): 3016-. [Google Scholar]
- 44.Wieand S, Schroeder G, O’Fallon JR. Stopping when the experimental regimen does not appear to help. Statistics in Medicine 1994; 13(13–14): 1453–8. [DOI] [PubMed] [Google Scholar]
- 45.Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 2009; 45(2): 228–47. [DOI] [PubMed] [Google Scholar]
- 46.Knudsen ES, Shapiro GI, Keyomarsi K. Selective CDK4/6 Inhibitors: Biologic Outcomes, Determinants of Sensitivity, Mechanisms of Resistance, Combinatorial Approaches, and Pharmacodynamic Biomarkers. American Society of Clinical Oncology Educational Book 2020; (40): 115–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chakravarty D, Johnson A, Sklar J, et al. Somatic Genomic Testing in Patients with Metastatic or Advanced Cancer: ASCO Provisional Clinical Opinion. Journal of Clinical Oncology 2021. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1: The ComboMATCH study team.