

Abstract
Peripheral neuropathy, and specifically distal peripheral neuropathy (DPN), is one of the most frequent and troublesome complications of diabetes mellitus. It is the major reason for morbidity and mortality among diabetic patients. It is also frequently associated with debilitating pain. Unfortunately, our knowledge of the natural history and pathogenesis of this disease remains limited. For a long time hyperglycemia was viewed as a major, if not the sole factor, responsible for all symptomatic presentations of DPN. Multiple clinical observations and animal studies supported this view. The control of blood glucose as an obligatory step of therapy to delay or reverse DPN is no longer an arguable issue. However, while supporting evidence for the glycemic hypothesis has accumulated, multiple controversies accumulated as well. It is obvious now that DPN cannot be fully understood without considering factors besides hyperglycemia. Some symptoms of DPN may develop with little, if any, correlation with the glycemic status of a patient. It is also clear that identification of these putative non-glycemic mechanisms of DPN is of utmost importance for our understanding of failures with existing treatments and for the development of new approaches for diagnosis and therapy of DPN. In this work we will review the strengths and weaknesses of the glycemic hypothesis, focusing on clinical and animal data and on the pathogenesis of early stages and triggers of DPN other than hyperglycemia.
Keywords: Diabetes, Pre-diabetes, Neuropathy, Impaired glucose tolerance, Hyperglycemia, Insulinopenia, Insulin-resistance
INTRODUCTION
Diabetes mellitus is a complex of metabolic disorders associated with insufficiency of insulin secretion, insulin action or both, and is manifested by hyperglycemia[1–3]. Diabetes is diagnosed when fasting blood glucose exceeds 6.9 mmol/L, or casual or 2-h glucose in a glucose tolerance test exceeds 11 mmol/L[4]. Control of blood glucose in vertebrate organisms is accomplished essentially by the action of two pancreatic hormones, i.e., insulin and glucagon, with the participation of epinephrine, ACTH, growth hormone and glucocorticoids occurring under special circumstances, such as stress. Insulin is released by islet beta cells in response to an increase of blood glucose (usually after a meal). It suppresses glucose production and stimulates the uptake and storage of glucose in skeletal muscle and liver (Figure 1). It also suppresses lipogenesis in the fat tissue and stimulates amino-acid synthesis in skeletal muscle. During a fasting state, when blood glucose is low or during stress requiring mobilization of energy, insulin secretion is suppressed and glucagon is released into the circulation by pancreatic α cells, opposing the action of insulin to increase the release of stored energy resources for use by the organism (Figure 1)[5,6].
Figure 1.
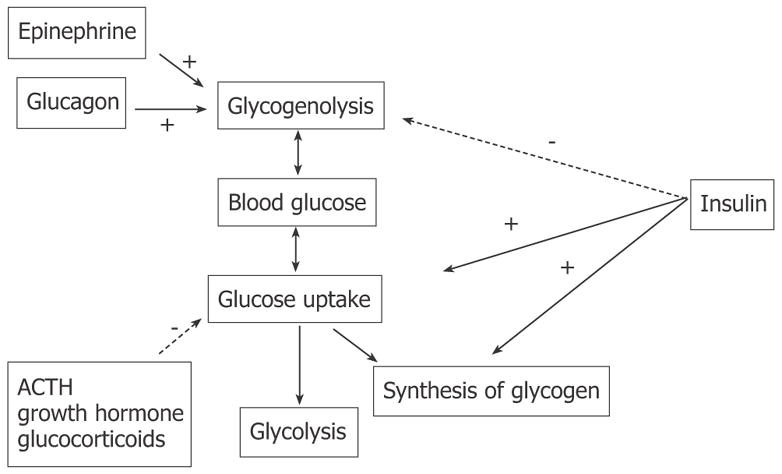
Hormonal regulation of systemic blood glucose.
The metabolic effects of insulin are mediated by activation of its cognate receptors that are expressed in target tissues (skeletal muscle, liver, fat) in large excess compared to the amount needed for normal regulation of glucose metabolism (spare receptors[7–10]). This lays a background for and signifies the paramount importance of the glucose control mechanisms. Indeed, in type 1 diabetes, which is usually associated with idiopathic autoimmune attack and destruction of islet beta cells, more than 90% of islet cells need to be destroyed or less than 10% of insulin production should remain for overt hyperglycemia to manifest[11]. Similarly, in type 2 diabetes, associated in its early stages with decreased sensitivity of insulin-responsive tissues to the action of the hormone (insulin resistance) and compensatory hyperinsulinemia, no clinical diabetes develops before muscle and fat tissue sensitivity to insulin is decreased below 50%–35% of normal[9]. In rats, and similarly in humans, plasma insulin may vary over the range of 1 to 4 ng/mL with fasting blood glucose exceeding the diabetic threshold of 6.9 mmol/L (solid line in Figure 2)[12–17] only in rare cases. In most cases, insulin must decrease to less than 0.5 ng/mL level (15% of control, 3.5 ng/mL level) in order to manifest overt hyperglycemia and diabetes.
Figure 2.
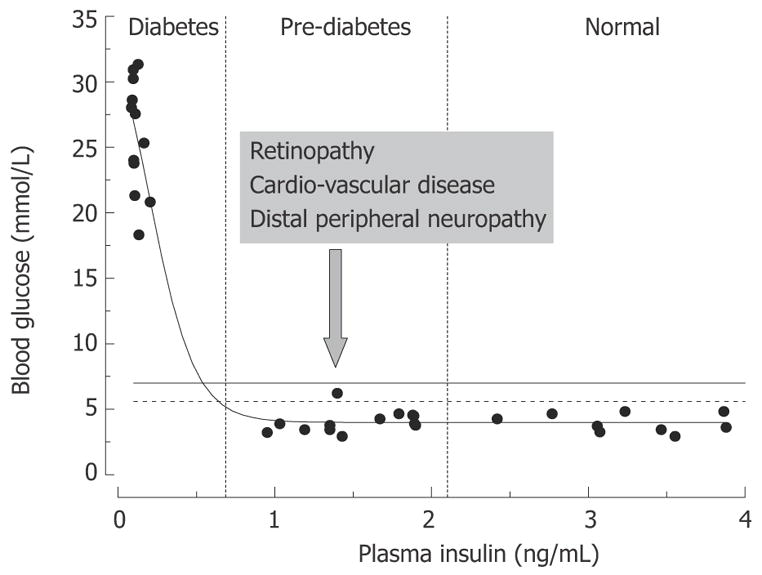
Relationships between rat plasma insulin and fasting blood glucose concentrations. Data are from normal and streptozotocin-injected adult Sprague-Dawley rats (the authors’ unpublished observations). STZ-rats having moderate pancreatic impairment and moderately decreased plasma insulin (vertical dashed lines) do not develop overt hyperglycemia.
Such a large safety factor for glucose control is of critical biological importance; however, clinically the impairment of insulin signaling in this disease process starts long before it manifests in overt hyper glycemia. With the discovery of insulin and improvement in techniques for blood glucose measurement, diabetes is not a life threatening disease by itself[18]. Therefore the long pre-clinical progression of diabetes could be a relatively minor issue; however, diabetes is associated with a variety of life threatening complications, among which distal peripheral neuropathy (DPN), cardio-vascular disease (CVD), retinopathy and renal disease are most frequent[2]. The problem is signs of these complications are frequently present prior to overt hyperglycemia and diabetes (Figure 2). Realization of this fact led to the definition of pre-diabetes as a state with moderate impairment of blood glucose control and high risk of development of overt diabetes, retinopathy and CVD[3,19]. Pre-diabetes is diagnosed when fasting blood glucose exceeds 5.6 mmol/L and/or 2-h glucose in a glucose tolerance test (GTT) exceeds 6.9 mM (impaired fasting glucose (IFG) and impaired glucose tolerance (IGT) dashed and solid horizontal lines in Figure 2, respectively)[4].
Establishing the correct lower limit for diagnosis of pre-diabetes is important because it determines whether clinical tests for complications should be performed and recommendations in life style and diet modifications should be presented to patients. In a recent study of young adult men, it was shown that fasting glucose exceeding 4.82 mmol/L constitutes an independent risk factor for developing type 2 diabetes in otherwise healthy subjects[20]. In another study of adult healthy men without diabetes or pre-diabetes, progressive loss of β-cell function and decrease in the first-phase of insulin secretion were detected at fasting glucose between 5.0 and 5.4 mmol/L[21]. Another clinically important issue is to understand the pathogenesis of diabetic complications, starting with pre-diabetic patients. This knowledge is required for early detection, prognosis and treatment of diabetic complications. Here we will discuss data and hypotheses for the triggers and progression of one such complication, which is distal peripheral neuropathy.
DISTAL PERIPHERAL NEUROPATHY
The prevalence of peripheral neuropathy in diabetic subjects approaches 70% and about 50% of these are cases of DPN[22–24]. The disease usually progresses to involve cardiac autonomic nerves, and as a result it is a major factor in mortality of diabetic subjects. DPN is also the major reason for loss of protective limb mechanical sensations, traumatic ulceration injures and therefore amputations[23,24]. Finally, about 11% of DPN cases are associated with chronic pain symptoms that severely diminish the quality of life and are frequently associated with depression[22,23]. The etiology of DPN is unknown and prediction of progression and treatments of the symptoms of DPN are limited[22,25–27].
Perhaps the largest problem associated with DPN, complicating its classification and treatment, is the variety of clinical presentations of this disease (Figure 3)[28–30]. Aside from a generally bilateral manifestation, distal to proximal advance and prevalence of sensory over motor impairment signs and symptoms, any two randomly selected cases of DPN may have nothing in common at the time of diagnosis and, to the extent it is known, their history and the pattern of their future progression may be very different. There are two broad categories of sensory symptoms of DPN, positive and negative[26,29]. Positive symptoms include pain, paresthesias and aberrant, exaggerated sensitivity to normally painless or moderately painful stimuli (allodynia and hyperalgesia). Negative symptoms consist of loss of sensory perception in one or several modalities. Motor symptoms, if present, manifest as muscle weakness, and thus they are also negative. Finally, a classical electrophysiological, and also negative, sign of DPN is a decrease in nerve conduction velocity (NCV) and amplitude of compound action potentials (APs) in peripheral sensory and motor nerves[23,24,26,27,31]. Furthermore, three categories of DPN, acute painful remitting neuropathy, chronic painful neuropathy and painless neuropathy with ulcer can be outlined as separate clinical entities[24,32,33]. The relationships among these entities, however, if any exist, are not known. Within each of these groups, signs of demyelination of large peripheral fibers (decrease in NCV) may or may not co-exist with signs and symptoms of large fiber axonopathy (decrease in amplitude of compound APs, loss of vibration sensation and/or loss of stretch reflexes). Loss of warm and cold perception, impairment of unmyelinated and small myelinated fibers, may or may not co-exist with signs of large fiber abnormalities[24,31,32,34–36]. Furthermore, the pain normally conducted by small unmyelinated peripheral axons may or may not be present at the same time with any of the above mentioned symptoms[22–24,26,36,37]. Finally, the modalities of pain and the degree of involvement of autonomic nervous system impairment constitute another large set of variables[22,24,33,38–40].
Figure 3.

Signs and symptoms of distal peripheral neuropathy. Categories of symptoms most frequently manifested in humans with diabetes are given in bold.
From experiments in animals, and by analogy with other neuropathies, it can be suggested that the pathogenesis of negative symptoms and signs of DPN is likely to be associated with demyelination and axonal atrophy and degeneration[41,42]. Failure of re-innervation will make these symptoms essentially irreversible[42–44]. Mechanisms of neuropathic pain, paresthesias and hyperalgesia are less understood[27,40]. However, it is generally accepted that abnormally intense spontaneous input from primary afferent fibers to the spinal cord is a primary trigger of these symptoms[24,27,45,46]. At least three usually overlapping conditions, resulting in such abnormal activity of peripheral axons, are well established. First is impairment of endoneurial circulation and following it ischemia[47,48]. Second is impairment of axon-glia relationships and segmental or paranodal demyelination[49,50]. The third condition is an axonal injury and following it Wallerian degeneration and neuroma[51,52]. In addition, increased excitability of regenerating, and therefore not yet properly myelinated, nerve fibers may add to the generation of aberrant peripheral discharge and pain[53,54]. At least at advanced stages, evidence for axonal, glial and vascular injuries are detectable in most cases of DPN[41,55].
Further insight into the pathogenesis of DPN is provided by its diffuse, bilateral presentation and distal to proximal progression. The former suggests that systemic rather then local conditions underlie the clinical pathology, while the latter could indicate two possibilities (Figure 4). First, DPN may be a manifestation of dying back degeneration, with the primary insult consisting of the impairment of synthesis or efferent axonal transport of proteins, therefore affecting the function of the longest axons in the body that are most dependent on these mechanisms[42]. Failure of protein synthesis, including synthesis of some important neurotrophic molecules, in diabetes could result in impaired nerve regeneration and dying back axonopathy[43,44,55,56]. Alternatively, accumulation of the effects of multiple injuries randomly located along the axon, for example demyelinating injures, may result in a clinical picture that is practically indistinguishable from dying back neuropathy[41,42,57]. The longest axons in the body will most likely be hit by a critical number of such local insults and their function will fail first. Micro-vascular disease followed by local impairment of blood supply to the nerve fascicules may be a basis for random demyelinating insults progressing later to axonal degeneration[58–60]. It is also conceivable that both mechanisms are operating at the same time. Both demyelination and axonal degeneration were detected in human DPN by electrophysiological tests and biopsy studies[24,41,61]. Peripheral axons are the longest in the human body, and this might be an important factor explaining PNS involvement superseding the CNS complications. Another potentially important reason for the high vulnerability of PNS to diabetic injury is the relatively weak anti-oxidative defenses of peripheral neurons (see Section 3).
Figure 4.
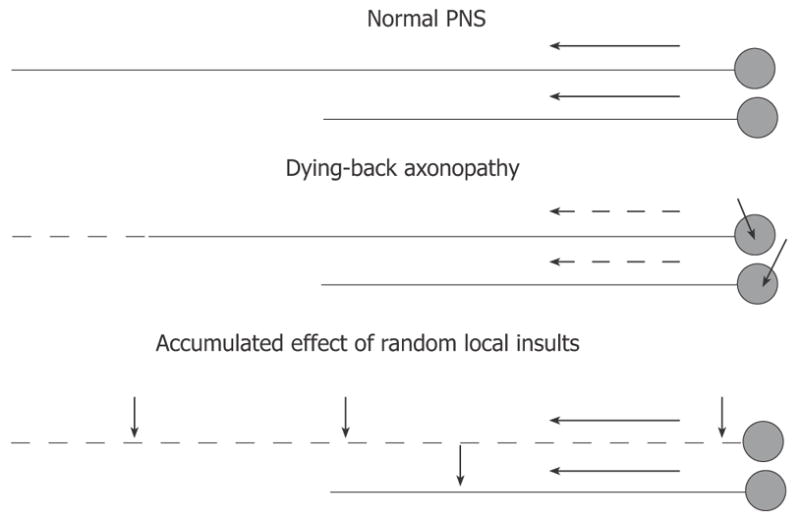
Hypotheses on distal-to-proximal progression of DPN. In normal PNS (top), neurons synthesize proteins in the cell body and transport them down the axon at the rate determined by axonal structural and functional needs. Impairment of synthesis or axonal transport of proteins will result in dying-back neuropathy, in which neurons with longest processes are affected first (middle). Alternatively, the neuropathy may result from effect of random local insults to the axon, with probability of accumulation of a critical number of such insults being higher for neurons having longer axons (bottom). Short arrows indicate non-specified axonal or neuronal injuries. Long solid or dashed arrows indicate normal or compromised axonal transport (respectively).
While the discussion above appears to encompass all the major features of DPN, multiple questions related to the pathogenesis of DPN remain unresolved. Thus whether or not dying back axonopathy or multiple local injuries or both mechanisms lead to the disease, it is not clear why, in most cases of neuropathy, sensory symptoms prevail over signs of impairment to motor axons innervating the same distal areas of the human body. Furthermore, neither of these mechanisms explains a variety of clinical presentations of DPN nor answers the question of why some diabetic patients live without any symptoms of DPN for years[62]. Finally, major questions that remain to be answered include identifying pathogenic triggers of DPN, pathogenesis of individual symptoms, and relationships among different symptoms and signs of the disease[22,24].
To illustrate the importance of the latter question, two hypothetical scenarios of DPN are shown in Figure 5. In the first scenario (Figure 5, top), there is a single pathogenic process triggering and maintaining the disease. The course of the disease and its clinical manifestations at the time of diagnosis and neurological evaluation (ovals in the Figure 5) will be determined by the duration of DPN and individual differences in the genetic backgrounds of patients. From the point of view of the symptoms revealed by a neurological exam, the second scenario (Figure 5, bottom) is identical to the first one; the critically important difference, however, is that different sets of symptoms in this scenario are triggered and driven by entirely independent pathogenic mechanisms (symptoms “a”, “c”, and “d” vs symptom “b”). Some of the branches of the pathogenic process (“b” in the first scenario) may enter an irreversible stage. Therefore, the early detection of DPN is an obligatory condition for the successful treatment of this disease. The cartoons demonstrate also that identification of all participating triggering mechanisms and symptoms associated with them is another critical step for the efficient treatment of DPN.
Figure 5.
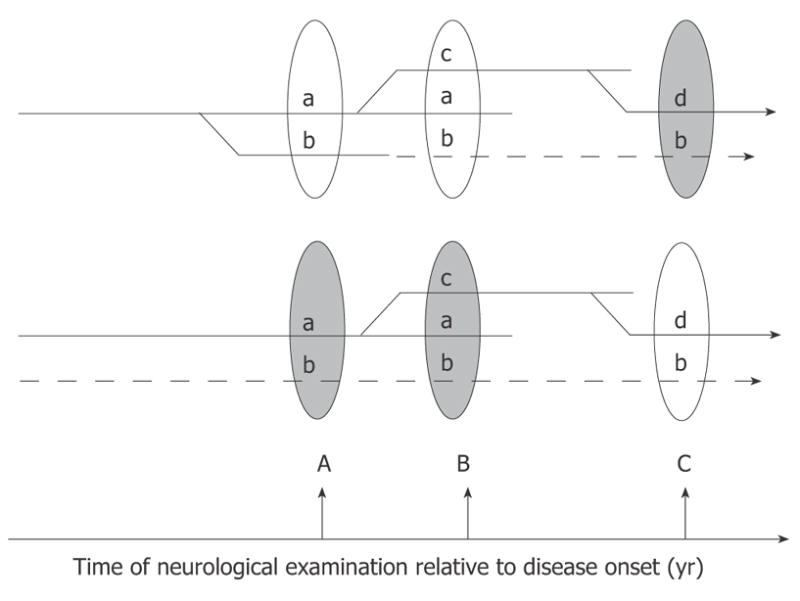
Hypothetical branching (top) and multi-trigger (bottom) pathogenesis of DPN. In the first scenario (top) all manifestations of the disease result from the unique branching pathogenic process, and symptom “b” discovered at the time of neurological exam B is not corrected by treatment because it has already progressed to an irreversible stage (dashed lines). Earlier diagnosis and institution of treatment (at time A) may critically change the outcome of therapy in this scenario. Alternatively (lower scenario), several independent factors may trigger and maintain the progression of DPN. In this case the therapy may fail to treat symptoms not because they are irreversible, but because the correct cause of the pathology is not identified and is not treated (dashed lines).
CHRONIC HYPERGLYCEMIA AND PATHOGENESIS OF DPN
DPN follows both type 1 and type 2 diabetes, and systemic hyperglycemia is the most obvious symptom that these types of the disease have in common[24,63,64], suggesting hyperglycemia as a universal trigger for DPN. Indeed, insulin treatment or treatment with insulin-sensitizing drugs to control hyperglycemia reverses some symptoms of DPN and delays its progression in general[65,66]. The Diabetes Control and Complications Trial (DCCT) data show that strict control of hyperglycemia in type 1 diabetes patients without clinical neuropathy decreased development of DPN in 60% of cases over 5 years of follow up study[67,68]. Well within the framework of the glycemic hypothesis, the failure to prevent all cases of DPN could be explained by the fact that glucose control can never be perfect. Type 1 and 2 patients are, on average, euglycemic for only about 62% of the day, while during the remaining 30% and 8% of the day they have various degrees of hyperglycemia and hypoglycemia, respectively[69]. Therefore, to avoid hypoglycemic crisis, the acceptable target value for blood glucose in controlled subjects is usually set to values above normal (6.7 to 10 mmol/L in DCCT and 6 mmol/L in U.K. Prospective Diabetes Study; of type 2 diabetic patients[65,68]). Another explanation for incomplete efficacy of glucose control is that, after longstanding diabetes, some neuropathic mechanisms may enter either an irreversible stage or a stage of progression that is already independent of the original trigger. With the limitations imposed by generally late diagnosis of diabetes and DPN[24,70,71], slowing of NCV, paraesthesia and painful symptoms appear to be the earliest (closest to the initiating pathogenic insult) manifestations of DPN. Therefore, it appears to be in good agreement with the glycemic hypothesis that in patients with newly diagnosed diabetes, NCV slowing and paresthesias (hyperglycemic neuropathy) can be frequently completely recovered with the establishment of euglycemia[31,42].
Further support for the glycemic hypothesis comes from research in animals, specifically rat models of type 1 diabetes (STZ-induced or spontaneous in BB-rats) and spontaneous type 2 diabetes in Zucker fatty rats[66,72] that appear to be the best studied animal models with regard to neuropathy. The short life span of rodents severely limits evaluation of chronic human diseases in these animals. Another limitation is that in behavioral tests, evoked pain manifested by limb withdrawal can be tested, but neither spontaneous pain nor changes in non-nociceptive sensory thresholds can be reliably measured in animals. Nonetheless, in general agreement with both clinical data on the earliest signs and symptoms of human DPN and the glycemic hypothesis, slowing of sensory[73–76] and motor[74,75,77–83] NCV and manifestations of evoked pain (hyperalgesia[75,84–87] and allodynia[85]) were shown to develop within the first month of onset of hyperglycemia in diabetic rats. With a longer time allowed (six to twelve months of diabetes) signs of axonopathy, demyelination and nerve degeneration can also be detected in diabetic animals[56,74,80,82,88–90]. Finally, early in the course of diabetes in rat models impairment of endoneurial blood flow and micro- and macrovascular reactivity are reported by many investigators[75,91–94]. Skin and arterial blood flow is abnormal early in diabetic patients[95–98], but no reduction in sural nerve blood flow was detected in humans with diabetes and mild DPN[99]. Thus, it is not clear whether impaired endoneurial blood flow represents a rat-specific component of DPN or if it is missed in humans because of its transient character and usually late detection of DPN in diabetic patients. In agreement with the glycemic hypothesis, all abnormalities found in rat models are reversible with normalization of blood glucose in insulin replacement experiments, and some of them (decreased pain pressure threshold) can be induced in normal rats by chronic in vivo perfusion of a DRG, or a segment of sciatic nerve with hyperglycemic solution[100,101].
Finally, support for the glycemic hypothesis is provided also by studies of cellular pathology associated with experimental diabetes. These studies show that practically all signs and symptoms of DPN observed in animal models may be linked to hyperglycemia-induced metabolic impairment of nerve, glial and endothelial cells in PNS. A detailed description of these studies is beyond the scope of the present work and can be found in a number of recent reviews[94,102–109]. The purpose of Figure 6 is only to provide a brief outline of cell metabolic abnormalities associated with diabetes and emphasize the findings directly relevant to the following discussion of triggers of early DPN.
Figure 6.
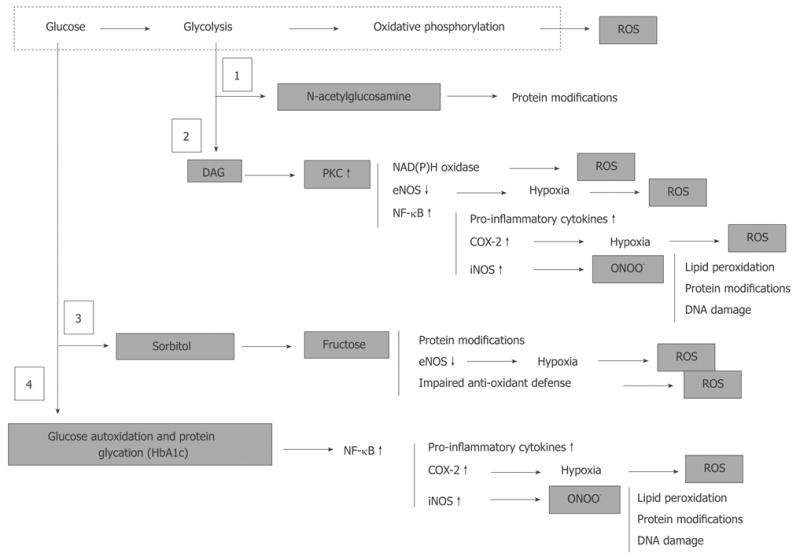
Hyperglycemia, derangement of cell metabolism and oxidative/nitrosative stress. Hyperglycemia associates with accumulation of fructose-6-phosphate and hexosamine pathway (1) to N-acetylglucosamine, accumulation of dihydroxyacetone phosphate and associated activation of PKC (2), activation of polyol sugar pathway (3), and glucose autoxidation and non-enzymatic protein glycation (4). These metabolic events are either regular physiologically important components cell metabolism (1, 2 and 3) or are normally under strict control of intrinsic intracellular defense mechanisms (4). However, under conditions of chronic hyperglycemia activation of these pathways leads to a global derangement of the cell and tissue homeostasis, which culminates in an uncontrolled cascade of abnormal protein modifications, oxidative/nitrosative stress and pro-inflammatory conditions.
Although the scheme in Figure 6 is simplified and omits many essential steps for the cascade of events [the poly (ADP-ribose) polymerase (PARP)[108]; activation and consequences of lipid peroxidation[110] are not shown], it clearly demonstrates the complexity of this cascade. Some events appear to be more specific to one type of cell than to another, and there is as yet no agreement on which events (for example activation of polyol pathway[111] or abnormally intensified oxidative phosphorylation[106]) plays a leading role in cellular impairment. Nonetheless, most of the data available indicate that all the various pathways activated by hyperglycemia converge in generation of excess reactive oxygen species (ROS). This process eventually overwhelms the intrinsic anti-oxidant mechanisms of the cell and ends in oxidative/nitrosative stress and pro-inflammatory conditions in the tissues[112–115]. The efficacy of treatment with anti-oxidants in correction of DPN in animals and humans supports this view[116–119].
In diabetic animals, oxidative stress injury develops in parallel in all major cellular elements of PNS. Injury to glial cells is responsible for the demyelination component of DPN, which may explain the decrease in NCV and painful manifestations of the disease. Oxidative stress in neurons might be responsible for axonopathy, impaired regenerative capacity of axons and negative symptoms of DPN. Glial cell injury will affect the nerve neurotrophism adding to the progression of neuronal defects. Finally, oxidative stress and impairment of nitric oxide (NO) production in the endothelium of epi- and endoneurial blood vessels results in impairment of endoneurial circulation and endoneurial hypoxia, exaggerating and speeding up the direct effect of hyperglycemic conditions on glial cells and neurons. Oxidative stress is pro-inflammatory, which affects the production of cytokines by glial cells, and provokes the recruitment of immune response cells into the affected tissue. This might be another important component of the pathogenesis and progression of DPN. Thus, combining these data and observations lays the foundation for a view of the natural history of DPN similar to that depicted in Figure 7 according to an expanded view of the glycemic hypothesis.
Figure 7.
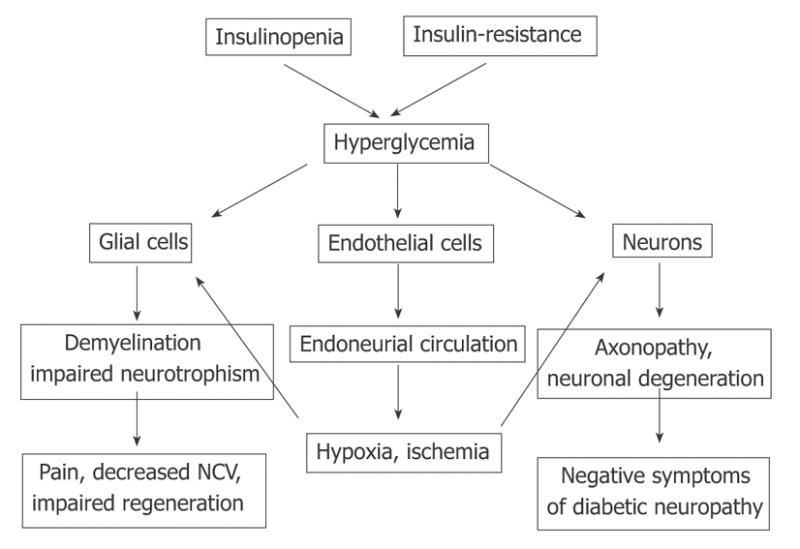
Pathogenesis of DPN with hyperglycemia as a major trigger of PNS injury.
Perhaps the most attractive aspect of such a view of the pathogenesis of DPN is that while it suggests a unifying trigger and mechanism (hyperglycemia and oxidative stress) for all symptoms of DPN, it nevertheless remains flexible enough to leave room for individual variability in the rates of progression and spectrum of manifestations of the disease. Indeed, the actual effects of uniform pro-oxidative and pro-inflammatory conditions may differ sharply depending on differences of individual cells and tissues in their intrinsic anti-oxidant defenses and individual organisms in their immune defenses. The injuries to a single myelinating or non-myelinating Schwann cell, endothelial cell or neuron will unlikely have even subclinical significance. The death of several myelinating cells will result in a decrease of NCV in a given axon and injury to several endothelial cells may cause the closure of a given capillary. Yet there are many axons and there is regeneration of damaged axons, and there are many capillaries and regeneration of damaged and collateral capillaries. It is only after cellular defenses against the oxidative stress are overwhelmed in many cells that the sub-clinical signs of the disease may be expected to appear, and it is anticipated to take an even longer time before clinical manifestations will themselves appear. In agreement with this, en mass CNS cells appear to have a much higher capacity for anti-oxidative mechanisms than do PNS cells and this might be one of the reasons why diabetic neuropathy affects the CNS much later than it affects peripheral nervous functions[90,113,120].
DIFFICULTIES OF THE GLYCEMIC HYPOTHESIS
While apparently logical and consistent with many clinical observations and the results of animal studies, the glycemic hypothesis cannot completely explain all the data. Thus, experiments in rodents consistently demonstrate that the polyol pathway (conversion of glucose to sorbitol by aldose reductase and then to fructose by sorbitol dehydrogenase; pathway 3 in Figure 6) is an important source of reactive oxygen species, and inhibition of aldose reductase prevents or reverses many signs of DPN seen in diabetic animals[121]. The same treatment in humans, however, has shown questionable efficacy so far[27,53,122,123]. Other problems include the failure of a pre-clinical slowing of NCV[24,124] or increase in glycosylated hemoglobin[22] (HA1c, integral measure of persistent hyperglycemia; pathway 4 in Figure 6) to predict development or severity of symptoms of DPN. Also, the finding of an inverse correlation between hyperglycemia and pain severity in diabetic patients with remitting painful neuropathy at presentation[33] is inconsistent with the glycemic hypothesis. These and other similar inconsistencies certainly could be attributed to the complexity of the disease, differences in studied patient populations, or inadequate design of drug trials in terms of timing, duration or dose[121]. Similarly, the lack of absolute efficacy in glucose control in preventing DPN in humans with diabetes[68] could also be explained within the framework of the glycemic hypothesis considering the difficulty of maintaining blood glucose concentrations within the relatively narrow range of normal values, which suggests that the actual threshold for neuropathic effects of hyperglycemia is lower than was previously thought. Therefore, recent findings of increased incidence of DPN in patients with pre-diabetes, many of whom have an impaired glucose tolerance (IGT) but not fasting hyperglycemia[22,25,70,71,125–127], appear to present the most serious challenge for the glycemic hypothesis.
The possibility that PNS injury may be triggered by exaggerated and prolonged postprandial hyperglycemic episodes, without necessarily requiring chronic hyperglycemia, should be considered to reconcile the glycemic hypothesis with observations of DPN in glucose intolerant patients[70,128–130]. Indeed, indices of large fiber function (ankle and knee reflexes and vibratory perception thresholds) were shown to decay with impairment of glucose tolerance in humans without diabetes and neuropathy[131]. Furthermore, pain is a frequent symptom of pre-diabetic DPN. An acute glucose infusion, which could be considered as an analog to a postprandial glucose surge, decreased thresholds to electrical stimulation in healthy adult volunteers[132] and decreased pain pressure thresholds in type 1 diabetic patients without clinical neuropathy[133]. In the latter study, however, no association between acute hyperglycemia and heat pain, warmth/cooling or vibration perception thresholds, was found[133]. Inconsistent with the idea that postprandial glucose changes have a neuropathic effect, no correlation was detected between short-term fluctuations in blood glucose and pain scores or heat pain thresholds in the study of type 1 and type 2 diabetic subjects with painful neuropathy[134]. Furthermore, no correlation between the number of glycemic excursions and the number of painful episodes was found in the study of type 1 patients with painless neuropathy[135].
Difficulties with the glycemic hypothesis are not unique to the human clinic. Thus in the STZ-rat model of diabetes, NCV could be corrected by a low level of insulin therapy below that required to correct hyperglycemia[89,136]. Pain pressure and von Frey filament thresholds studied in the same model demonstrate no correlation with the degree of hyperglycemia[84,101,137]. Furthermore, aldose reductase inhibitors (blockers of the polyol sugar pathway; Figure 6), given at doses sufficient to correct nerve sorbitol and fructose and heat pain thresholds, do not correct von Frey filament threshold in STZ-hyperglycemic rats[138,139]. As another example, pain pressure thresholds in type 2 diabetic Zucker rats could be corrected with insulin-like-growth factor II (IGF-II) that has no effect on blood glucose[86]. All these examples are taken from experiments in overtly diabetic and hyperglycemic animals. Therefore, formally the possibility remains that hyperglycemia was the triggering event for the observed abnormalities, but it is not required for the progression and maintenance of these pathologies. Whether DPN develops in pre-diabetic rats as it does in humans has not yet been studied. Since previous work has focused on diabetes, little attention has been devoted to the development of pre-diabetic animal models and studies of DPN in these models. Nonetheless, neuropathic decreases of mechanical and thermal nociceptive thresholds[140] and slowing of motor NCV[141] were observed in studies in Zucker-fatty rats. Since these are insulin-resistant but normoglycemic animals (type 2 pre-diabetes) the impaired glucose tolerance could be responsible for DPN in these animals. Recently, we described decreased pain pressure threshold in rats that were injected with STZ but remained normoglycemic[84]. These rats also had normal glucose tolerance, maintained normal levels of HbA1c, and normal concentrations of sorbitol in the nerve, suggesting that not only fasting but casual glucose also was maintained within physiological limits (Figure 8).
Figure 8.
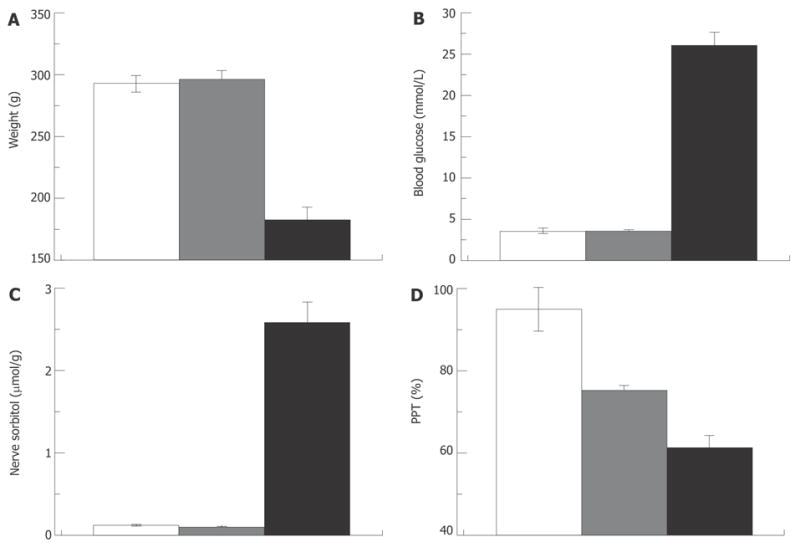
Pain pressure thresholds are not correlated with hyperglycemia. Weight (A), blood glucose (B), nerve sorbitol (C) and pain pressure thresholds (D) in control, STZ-normoglycemic and hyperglycemic rats (white, grey and black columns respectively; 2 wk after injection of 65 mg/kg STZ).
Thus there is solid evidence that hyperglycemia is an important factor of DPN. However, there are also both clinical and animal studies indicating that in addition to chronic and/or postprandial hyperglycemia, other pathogenic mechanisms must exist that trigger and maintain at least some of the symptoms of DPN. Identification of these factors is of critical importance for our understanding of both the natural history of pre-diabetic DPN and the pathogenesis of diabetic DPN in general[27,88].
INSULIN SIGNALING AND DPN
In the search for triggers of DPN other than hyperglycemia, it is important to note that successful reversion or postponing of DPN in clinical glucose control trials does not necessarily prove the glucose hypothesis[142]. Glucose metabolism is regulated by insulin via type B receptors (IR-B) abundantly expressed by liver, skeletal muscle and fat cells. Insulin receptors, however, are also expressed in central and peripheral nervous systems[143,144]. Furthermore, in PNS the highest densities of IRs are located on endothelial cells, paranodal loops of Schwann cells and medium and small size primary sensory neurons[143,144]. All these locations are strategically critical points of PNS function considering what is known about the pathogenic mechanisms of DPN. While the nervous system has mostly type A receptors (IR-A), the insulin affinities of these and IR-B receptors are similar (K0.5 is 3 to 6 ng/mL[145]) and well within the range of circulating concentrations of insulin (1 to 6 ng/mL[12–17]). Thus, it is very possible that correction of insulin levels in treatments for Type 1 diabetes or insulin-sensitizing therapy in cases of type 2 diabetes not only corrects glucose metabolism, but also has independent effects on the function of PNS. Serious consideration of this hypothesis is warranted, first because it provides an explanation for at least some failures of glycemic controls to reverse DPN, and second because it may explain the development of DPN in pre-diabetic patients.
Insulinopenia (Type 1 diabetes and pre-diabetes)
As described in the Introduction, overt type 1 diabetes is preceded by a state of partial pancreatic damage and moderate insulinopenia in which insulin production is still satisfactory for blood glucose control[11]. The type 1 pre-diabetic state is probably short and little is known about neuropathy in these patients. The results of a recently completed follow-up to DCCT study of type 1 diabetic patients, however, has shown that regardless of the level of glycemic control, neuropathy is less prevalent in the group of patients that maintained intensive vs. conventional insulin therapy[146]. Further support for the possibility of a direct role of insulinopenia in DPN comes from experiments in the STZ-rat model of type 1 diabetes. It was demonstrated that local insulin application to the nerve prevents motor NCV slowing in STZ-hyperglycemic rats[147]. Similarly, systemic[136] or intrathecal[89] application of insulin can correct sensory and motor NCV in STZ-treated rats without having an effect on hyperglycemia. Finally, in our experiments in rats that were injected with STZ and developed moderate insulinopenia but not fasting hyperglycemia (Figure 8), pressure pain thresholds were decreased in proportion to the degree of insulinopenia, and low dose insulin-replacement therapy corrected this defect without changes in the systemic blood glucose level (Figure 9). Taken together these data suggest that at least some signs of neuropathy (slowing of NCV, pressure-evoked pain in rats) may indeed be triggered by insulinopenia with no relevance to the blood glucose level. Another notable aspect of these experiments is that correction of nerve conduction[89,136,147] and pain pressure thresholds (Figure 9) with insulin treatment could be achieved without changes in systemic blood glucose levels, leading to an important implication that the thresholds of “metabolic” and “neuropathic” effects of insulinopenia may differ.
Figure 9.
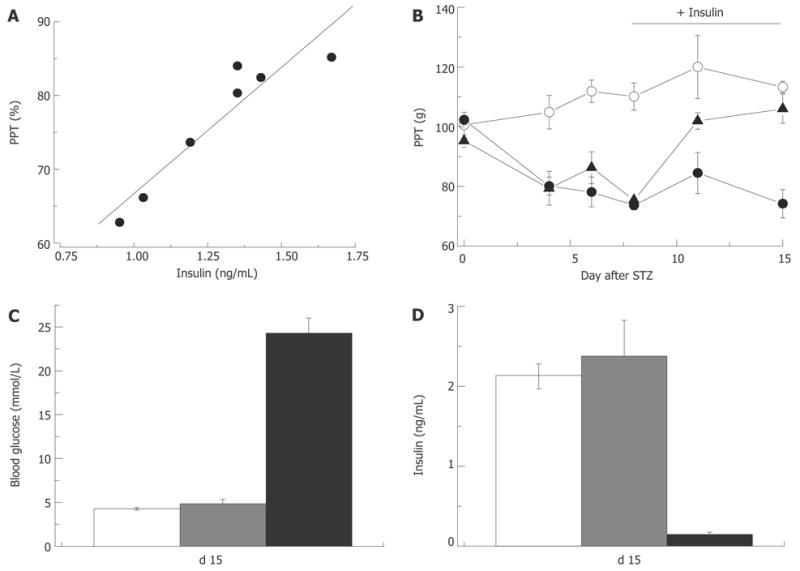
Insulin-dependence of pain pressure threshold in STZ-normoglycemic rats. Two weeks after injection of STZ pain pressure threshold of STZ-NG rats is decreased in proportion to the plasma insulin level (R = 0.97; A). Insulin replacement initiated one week after STZ injection (horizontal bar in panel B) does not change PPT of control or hyperglycemic rats, but corrects it in STZ-normoglycemic animals (empty and filled circles, and filled triangles, respectively). Insulin replacement, does not affect blood glucose (C), but normalizes plasma insulin level (D) in STZ-NG rats. In C and D white, grey and black columns represent control, STZ-NG and STZ-HG rats, respectively.
To date, detailed information on the relation between insulin and nerve conduction is not available. However, comparison of better studied “dose-response” relationships between insulin, blood glucose and pain pressure thresholds (Figure 10A) allows speculation that in the rat, control of glucose metabolism may tolerate at least five times lower insulin levels than does nerve function. Given that this difference was confirmed in both animals and humans, the outcome of these studies is of a great importance. This finding may explain the development of neuropathy in pre-diabetes and also suggests that neuropathy may start at stages preceding pre-diabetes, and some therapeutic interventions to correct insulin levels or insulin resistance (see next section) are warranted in pre-diabetic patients.
Figure 10.
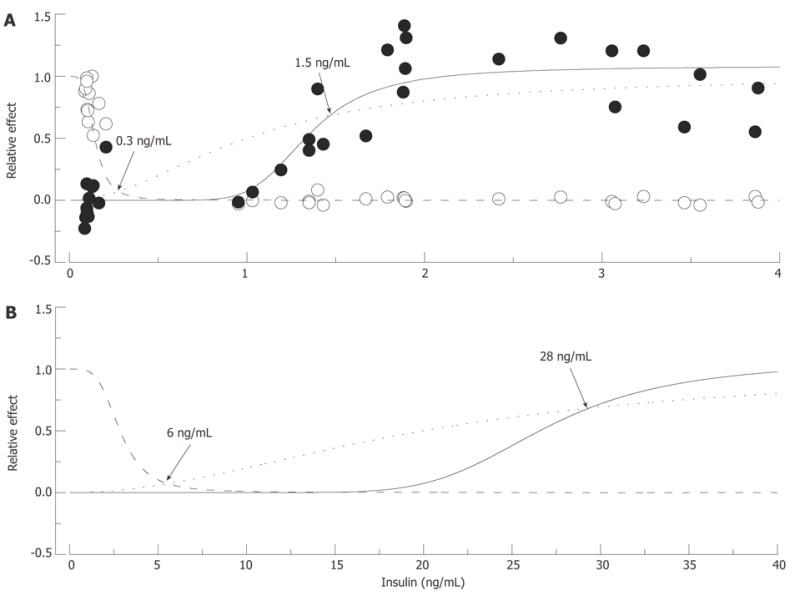
Putative “insulin-glucose metabolism” and “insulin-nerve function” relationships in normal and insulin-resistant rats. In A: Normal rats: Empty circles represent fasting glucose and filled circles pain pressure thresholds measured in control, STZ-NG and STZ-HG rats, pooled together and normalized to show relative changes of these parameter between control and diabetic animals. The data were fitted by Hill curves calculated based on the assumption that insulin binds to the receptor with an affinity of 1 ng/mL (dotted curve) and 10% and 65% occupancy of these receptors is required for maximum metabolic (intersection with dashed curve) and nerve (intersection with solid curve) effect of the hormone, respectively. In B: Insulin-Resistant Rats: Insulin resistant state was simulated by increasing K0.5 of insulin binding to the receptor to 20 ng/mL. Points of intersections of the dotted curve with the dashed and solid curves recalculated with the new K0.5 parameter show (arrows and labels) that maintaining glucose metabolism now requires about 6 ng/mL of plasma insulin, and nerve function requires at least 28 ng/mL of the hormone.
Differences in threshold concentrations of a ligand are usually determined by the differences in receptor properties. However, insulin affinities of IR-B and IR-A isoforms of the insulin receptor expressed in cells of organs responsible for glucose metabolism and in nerve tissue are too close to account for apparent differences in the concentrations of hormone required for maintaining normal blood glucose concentrations and normal pain pressure thresholds[145]. On the other hand, strong correlative relationships between insulin and pain pressure thresholds (Figure 9A) suggest a nearly direct link between insulin regulation and pressure pain mechanisms. The hypothesis of spare receptors[7,8,10] is the easiest way to explain this discrepancy. Thus as shown in Figure 10A, both metabolic and neuropathic effects of insulin may be described adequately within the concept of insulin binding with the same affinity (K0.5 = 1 ng/mL) in nerve and in glucose controlling organs, if 65% of the receptor occupancy is needed to control nerve function, and only 10% of occupancy is required for glucose metabolism. Hill equations were used in this simulation; however, since the purpose was merely to illustrate the potential possibility of the given scenario, parameters of the equations were adjusted by a trial and error approach and no attempt was made to optimize the parameters. Some alternatives to this scenario will be discussed in section 6.1 of this review.
Insulin resistance
Prevalence of type 2 to type 1 diabetes is about 9 to 1 and most of the cases of human pre-diabetes are type 2 pre-diabetes or metabolic syndrome cases[24,125]. Over the long-term, insulin production is impaired in type 2 diabetes further increasing the incidence of DPN in this population by mechanisms described above. In a ten-year study of the natural history of type 2 diabetic patients, it was found that decreased serum insulin and increased blood glucose concentrations are independent predictors of DPN[148]. However, in early type 2 diabetes and pre-diabetes there is a compensatory hyperinsulinemia. Because of this hyperinsulinemia type 2 pre-diabetes usually spans a much longer period of time than does type 1 pre-diabetes. Thus, after 5 years only 20% to 35% of patients with impaired fasting glucose or impaired glucose tolerance develop overt hyperglycemia (see[70]). Despite this compensatory hyperinsulinemia, however, many type 2 pre-diabetic patients do develop DPN[125,126]. This latter observation suggests that in terms of neuropathic outcome, insulin resistance and insulinopenia may be equivalent states. It also suggests that increased production of insulin may fail to compensate for decreased sensitivity of PNS to regulation by insulin.
Not much experimental data exists to verify the validity of either of these suggestions. In studies in normal human volunteers, warmth detection threshold correlated with insulin but not fasting or 2-h GTT glucose, leading the authors to suggest that insulin resistance may determine some sensory functions of PNS[149]. This is also supported by observations of decreased NCV[141] and pressure pain thresholds[86] and the authors’ unpublished observations in the Zucker fatty rat model of type 2 pre-diabetes. However, whether it is possible that hyperinsulinemia is effective in regulating glucose metabolism but fails to compensate for nerve insulin resistance is at this time absolutely not known. Furthermore, it is unknown if this mechanism plays a role in DPN associated with type 2 diabetes. In theory such a possibility does exist, and Figure 10B illustrates the scenario that we believe provides a useful working hypothesis for future experiments. Using “dose-response” relations as a starting point, shown in Figure 10A, the insulin resistant state that leads to type 2 diabetes may be modeled as a decrease in affinity of IR from 1 to 20 ng/mL, which leads to proportional rightward shifts of insulin-glucose metabolism and insulin-nerve function relationships. If the requirements of 10% and 65% occupancy of IR remain unchanged, maintenance of normal glucose metabolism under these new conditions will need about 6 ng/mL of circulating insulin and nearly 30 ng/mL insulin concentration will be the minimum needed to maintain normal PNS function. The calculated 6 ng/mL insulin concentration is in the range of insulin concentrations measured in Zucker fatty rats (5.1 to 11.7 ng/mL; model of compensated insulin resistance[150–152]). These latter numbers are, however, significantly lower than predicted by the model insulin concentration needed to maintain nerve function. Therefore, it may be speculated that the “set-point” or natural goal of compensatory hyperinsulinemia is merely to correct glucose metabolism, which is vitally important for the organism, with no concern about the less significant problem of nerve function.
Cellular mechanisms
Thus, while the connection remains speculative, the data above suggest that impairment of insulin signaling in PNS (because of decreased insulin production, insulin resistance or both) may be an important factor in the pathogenesis of DPN. Further studies are needed to confirm this hypothesis and further studies are also needed to understand the cellular mechanisms of insulin action in PNS.
Glucose is a major fuel for neurons of peripheral and central nervous systems. However, unlike that in major target tissues of insulin regulation, uptake of glucose in nervous tissue is an insulin-independent process. Therefore, the simple explanation of the neural effects of insulin to regulate the energy supply does not appear to be applicable. There should be some other role of IR in the nervous system. In the CNS, these receptors are involved in the insulin control of feeding behavior, reproductive and cognitive functions and neuromodulation[27,153]. Insulin also clearly has neurotrophic functions. It stimulates neurite outgrowth, is involved in peripheral nerve regeneration and is required for survival of sympathetic neurons (see[27]). These effects are likely very important for regeneration, which is suppressed in long-term DPN. For short term diabetes, the possibility of insulin regulation of axon-glia relationships, vascular permeability, and function of nociceptive primary afferent neurons[143,144] may be of importance. The possibility of a selective acute effect of insulin on endoneurial blood flow (decrease) was also demonstrated by experiments in normal rats[154].
Note also that the negative cellular effects of hyperglycemia and insufficiency of insulin signaling seem to converge at some point. Pain pressure thresholds are decreased in rat models of local hyperglycemia with a time course and to a degree that is very similar to those in STZ-normoglycemic rats[101]. Since many studies support oxidative/nitrosative stress as a central event of hyperglycemic impairment, it is reasonable to suggest that oxidative/nitrosative stress can interfere with insulin regulation in some or all of the hyperglycemia-induced steps of the pathogenic cascade depicted in Figure 6. Insulin directly regulates inner mitochondrial membrane potentials and may affect oxidative phosphorylation[136]. Insulin also suppresses expression of NADPH oxidase[155] and controls expression of Nf-κB and associated inflammatory reactions[155,156]. In fact, the anti-inflammatory effects of insulin have been known since the discovery of the benefits of insulin therapy in systemic inflammatory responses to trauma or bacterial infection[157]. Insulin signaling also was shown to be linked to the regulation of Na, K-ATPase[158,159] and endothelial NO production[158,160]. These data suggest that insulinopenia does have the potential to produce the same or very similar neuropathic effects as were attributed previously solely to hyperglycemia.
OTHER FACTORS OF SIGNIFICANCE
While the above outlined insulin-signaling hypothesis of DPN appears compelling, it will certainly be corrected and modified in many of its segments to conform to the results of future studies. It can also be stated here, without reservation, that no complete picture of the pathogenesis of DPN will be created unless the roles of insulin-like growth factors and C-peptide are considered in addition to hyperglycemia and insulin signaling in PNS[142].
Insulin-like growth factors (IGFs)
IGFs are produced in the kidney, spinal cord, skeletal muscle and peripheral glia. IGFs possess multiple neurotrophic functions, including control of neuronal survival, neurite outgrowth and regeneration, and expression of genes encoding axonal cytoskeletal proteins (tubulins and neurofilaments)[142,161–166]. Interestingly, IGF-1 appears to be involved in the regulation of resistance to oxidative stress[167]. IGFs primarily act via specific receptors, but since IGFs are present in the circulation in a 100-fold excess compared to insulin, they may also bind to and activate IR, mimicking some but not all affects of insulin[168,145,165]. It might be important in this regard that, in peripheral nerve, IGF-I receptors are co-localized with IR in sensory neurons[169,170] and Schwann cells[171] and a large fraction of them are likely hybrids composed of protein subunits of both IR-A and IGF-I receptors[143,169]. These hybrid IR/IGF receptors have substantially higher affinity to IGF-I than to insulin[172,173]. However, even when present in physiological concentrations, insulin may still bind to and activate some portion of hybrid as well as homomeric IGF-I receptors[161,174]. In addition, IGF-I may act by suppressing growth hormone and improving insulin sensitivity and insulin production in type 1 and type 2 diabetic subjects[174–178]. Insulin on the other hand, may modify kidney production of IGFs, and via regulation of IGF binding proteins, it may control the activity of circulating IGFs[161,175,176]. Whether, any of these multiple mechanisms participate in the apparent dissociation of the metabolic and neuropathic effects of insulinopenia remains to be determined.
Abnormal expression and levels of circulating IGFs and/or changes in expression of receptors for IGF were measured in diabetic human subjects[174], in STZ-rats (see[179–181]), and in the type 2 diabetes Zucker diabetic fatty (ZDF) rat model[182]. Furthermore, in obese Zucker rats, both insulin- and IGF-I resistances were shown to develop and mediate impaired glucose tolerance in this model of pre-diabetes[183]. Thus potentially, via impairment of protein synthesis, insufficiency of IGFs may add to the pathogenesis of regenerative capacity, neurodegeneration and irreversible stages of DPN[142]. This suggestion is supported by observations of recovery of NCV and reversion of atrophy of myelinated sensory axons in the sural nerve of STZ rats treated with intrathecal IGF-I[89]. In addition, the defect in IGFs or IGF-receptor expression could also add to the pathogenesis of early symptoms of DPN either directly or through modulation of insulin production or nerve sensitivity to insulin. Indeed, down-regulation of IGF-I receptors, which is observed in nerves of STZ-diabetic rats, occurs comparatively early, within 1 week after the onset of hyperglycemia[181]. Furthermore, continuous subcutaneous infusion of IGF-II was shown to recover pain pressure thresholds to a normal level after 6 wk of diabetes in the ZDF rat model[86]. The latter observation is interesting because, in STZ-diabetic rats, a similar magnitude and early decrease in pain pressure thresholds seems to result from insulinopenia (see previous section). Affinity of IGF-II binding to brain type insulin receptors or to IGF-I-R is two-three times lower than that of respective natural ligands[145]. Therefore, the effect of IGF-II on mechanical hyperalgesia may still be explained within the framework of our insulin signaling hypothesis of early neuropathy. However, the possibility of a more complex regulation of pain pressure thresholds cannot be excluded. It is important also that in all the examples above the effects of treatments with IGFs occurred with no measurable changes in the glycemic status of studied animals; once again suggesting that the pathogenesis of DPN is multifactorial.
C-peptide
C-peptide is a segment of the proinsulin molecule sliced off to form insulin. Acting through both its own receptors and modulating activity of insulin receptors, C-peptide produces multiple insulin and IGF-like effects[27,184,185]. C-peptide also enhances autophosphorylation of IR and effects of insulin, and treatment with C-peptide reverses decreased expression of IGF-I, NGF and neurotrophin-3 receptors in type 1 spontaneously diabetic rats[27,184,186]. Considering these effects and the fact that C-peptide is secreted in equimolar concentrations with insulin, it can be concluded that C-peptide insufficiency may have an important role in the pathogenesis of type 1 diabetes[184,75,185]. In agreement with this implication, in patients with recently diagnosed type 1 diabetes, C-peptide treatment was shown to correct sensory NCV and vibration perception[187]. C-peptide treatment also corrects skin microcirculation in diabetic patients[188] and endoneurial blood flow and NCV slowing[75], thermal hyperalgesia, atrophy and degeneration of C-fibers[186] in BB/Wor type 1 diabetes rat model.
As expected, no differences in plasma C-peptide levels were found in pre-diabetic patients and patients with type 2 diabetes of short duration (less than 5 years from diagnosis), even though the number and severity of the signs and symptoms of DPN differed substantially between these groups[128]. What is less clear in the same study is a lack of detectable deficiency of C-peptide in advanced type 2 diabetes patients (more than 5 years) who could have been expected to start developing insulinopenia. In general, however, multiple questions remain to be resolved in relation to C-peptide and its role and mechanisms of action in DPN. Similar to conditions of chronic or postprandial hyperglycemia or impaired insulin- or IGF-signaling, C-peptide deficiency appears to affect activity of Na,K-ATPase, NO production and neurotrophism, but the C-peptide mediated regulation does not appear to depend on oxidative stress, which is apparently important in the pathogenesis of these conditions[75,189]. It is also not clear whether any of the symptoms may be directly attributed to C-peptide insufficiency in DPN. Endoneurial blood flow, NCV and heat nociception appear to be the foremost candidates[187,188], but antioxidant treatments and insulin replacement correct these abnormalities in STZ rats at least as efficiently as does C-peptide replacement in the BB/Wor rat model of type 1 diabetes.
CONCLUSION
DPN is a frequent and troublesome complication of diabetes mellitus. Diabetes manifests in a case-specific variety of signs and symptoms, and associates with complex biochemical, functional and structural abnormalities of the peripheral nervous system. While the obvious hyper-glycemia present in diabetes can explain the development of these abnormalities, data suggest that other factors may also contribute. We have discussed the evidence for insulinopenia in type 1 diabetes or insulin resistance in type 2 diabetes as causal factors in the development of DPN. We have suggested that these two cases actually represent a single cause of impaired insulin signaling. Considering the role of insulin signaling in DPN more completely explains the changes in nerve function in pre-diabetic or early diabetic patients and animal models. This does not preclude hyperglycemia also as a factor in DPN, but allows a more complete picture of the disease process (Figure 11). In addition to insulin signaling, some evidence also exists for a role of IGF and C-peptide in mediating DPN. The pathogenesis of DPN is obviously multifactorial, and despite long-standing efforts, remains poorly understood. This work has also suggested that insulin signaling has effects that occur with different levels of receptor occupancy. Thus insulin action on nerve function requires a higher level of receptor occupancy than does insulin action on glycemic control. This could be explained by different levels of receptor coupling to second messenger signaling pathways in nerve versus liver or muscle. Future studies should elucidate these mechanisms so that a clearer picture of DPN can be obtained, especially in pre-diabetes where early detection could improve therapeutic outcomes.
Figure 11.
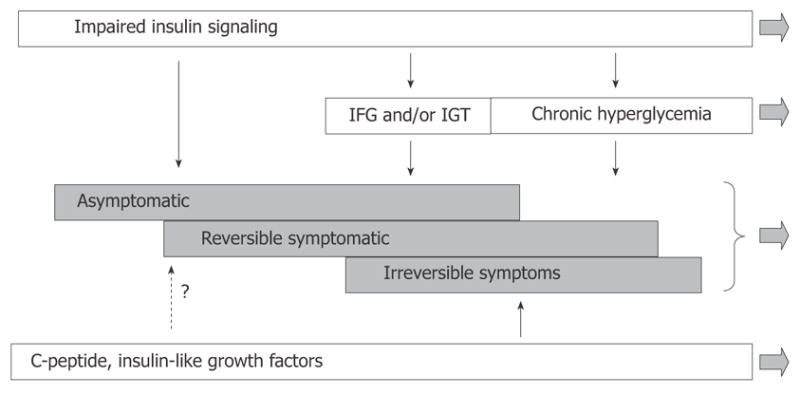
Modified scheme of pathogenesis of diffuse diabetic neuropathy with inclusion of insulin signaling. According to this view, the disease process starts before impairment of glucose metabolism becomes apparent. Derangement of insulin signaling in PNS triggers and maintains the neuropathy at this stage. Postprandial and chronic hyperglycemia is not the least important factor of DPN, but they become involved at relatively advanced stages of DPN. C-peptide and IGFs insufficiencies represent another important set of pathogenic mechanisms; however, the role of these mechanisms in early DPN remains undetermined.
Footnotes
S- Editor Liu Y L- Editor Lutze M E- Editor Bi L
Supported by NIH National Institute of Diabetes and Digestive and Kidney Diseases, No. DK067248
Contributor Information
Maxim Dobretsov, Departments of Anesthesiology, Neurobiology and Developmental Sciences, University of Arkansas for Medical Sciences, 4301 West Markham Street, Little Rock, AR 72205, United States.
Dmitry Romanovsky, Department of Anesthesiology, University of Arkansas for Medical Sciences, 4301 West Markham Street, Little Rock, AR 72205, United States.
Joseph R Stimers, Department of Pharmacology and Toxicology, University of Arkansas for Medical Sciences, 4301 West Markham Street, Little Rock, AR 72205, United States.
References
- 1.American Diabetes Association. Diagnosis and classification of diabetes mellitus. Diabetes Care. 2006;29 (Suppl 1):S43–S48. [PubMed] [Google Scholar]
- 2.Diabetes in America. Bethesda MR: National Diabetes Data Group, NIH; 1995. pp. 1–733. [Google Scholar]
- 3.American Diabetes Association. Report of the Expert Committee on the Diagnosis and Classification of Diabetes Mellitus. Diabetes Care. 1997;20:1183–1197. doi: 10.2337/diacare.20.7.1183. [DOI] [PubMed] [Google Scholar]
- 4.American Diabetes Association. Standards of medical care in diabetes--2006. Diabetes Care. 2006;29 (Suppl 1):S4–S42. [PubMed] [Google Scholar]
- 5.Stryer L. Biochemistry. New York: W.H. Freeman and Company; 1988. pp. 1–1089. [Google Scholar]
- 6.Zierler K. Whole body glucose metabolism. Am J Physiol. 1999;276:E409–E426. doi: 10.1152/ajpendo.1999.276.3.E409. [DOI] [PubMed] [Google Scholar]
- 7.Kono T, Barham FW. The relationship between the insulin-binding capacity of fat cells and the cellular response to insulin. Studies with intact and trypsin-treated fat cells. J Biol Chem. 1971;246:6210–6216. [PubMed] [Google Scholar]
- 8.Gliemann J, Gammeltoft S, Vinten J. Time course of insulin-receptor binding and insulin-induced lipogenesis in isolated rat fat cells. J Biol Chem. 1975;250:3368–3374. [PubMed] [Google Scholar]
- 9.Kahn BB, Rossetti L. Type 2 diabetes--who is conducting the orchestra? Nat Genet. 1998;20:223–225. doi: 10.1038/3018. [DOI] [PubMed] [Google Scholar]
- 10.Olefsky JM. Insensitivity of large rat adipocytes to the antilipolytic effects of insulin. J Lipid Res. 1977;18:459–464. [PubMed] [Google Scholar]
- 11.Eisenbarth GS. Type I diabetes mellitus. A chronic autoimmune disease. N Engl J Med. 1986;314:1360–1368. doi: 10.1056/NEJM198605223142106. [DOI] [PubMed] [Google Scholar]
- 12.Kobayashi T, Kamata K. Effect of insulin treatment on smooth muscle contractility and endothelium-dependent relaxation in rat aortae from established STZ-induced diabetes. Br J Pharmacol. 1999;127:835–842. doi: 10.1038/sj.bjp.0702554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burcelin R, Eddouks M, Kande J, Assan R, Girard J. Evidence that GLUT-2 mRNA and protein concentrations are decreased by hyperinsulinaemia and increased by hyperglycaemia in liver of diabetic rats. Biochem J. 1992;288 (Pt 2):675–679. doi: 10.1042/bj2880675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rossetti L, Giaccari A. Relative contribution of glycogen synthesis and glycolysis to insulin-mediated glucose uptake. A dose-response euglycemic clamp study in normal and diabetic rats. J Clin Invest. 1990;85:1785–1792. doi: 10.1172/JCI114636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rossetti L, Shulman GI, Zawalich W, DeFronzo RA. Effect of chronic hyperglycemia on in vivo insulin secretion in partially pancreatectomized rats. J Clin Invest. 1987;80:1037–1044. doi: 10.1172/JCI113157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Russell JW, Golovoy D, Vincent AM, Mahendru P, Olzmann JA, Mentzer A, Feldman EL. High glucose-induced oxidative stress and mitochondrial dysfunction in neurons. FASEB J. 2002;16:1738–1748. doi: 10.1096/fj.01-1027com. [DOI] [PubMed] [Google Scholar]
- 17.Lin CY, Higginbotham DA, Judd RL, White BD. Central leptin increases insulin sensitivity in streptozotocin-induced diabetic rats. Am J Physiol Endocrinol Metab. 2002;282:E1084–E1091. doi: 10.1152/ajpendo.00489.2001. [DOI] [PubMed] [Google Scholar]
- 18.Patlak M. New weapons to combat an ancient disease: treating diabetes. FASEB J. 2002;16:1853. [PubMed] [Google Scholar]
- 19.Genuth S, Alberti KG, Bennett P, Buse J, Defronzo R, Kahn R, Kitzmiller J, Knowler WC, Lebovitz H, Lernmark A, Nathan D, Palmer J, Rizza R, Saudek C, Shaw J, Steffes M, Stern M, Tuomilehto J, Zimmet P. Follow-up report on the diagnosis of diabetes mellitus. Diabetes Care. 2003;26:3160–3167. doi: 10.2337/diacare.26.11.3160. [DOI] [PubMed] [Google Scholar]
- 20.Tirosh A, Shai I, Tekes-Manova D, Israeli E, Pereg D, Shochat T, Kochba I, Rudich A. Normal fasting plasma glucose levels and type 2 diabetes in young men. N Engl J Med. 2005;353:1454–1462. doi: 10.1056/NEJMoa050080. [DOI] [PubMed] [Google Scholar]
- 21.Godsland IF, Jeffs JA, Johnston DG. Loss of beta cell function as fasting glucose increases in the non-diabetic range. Diabetologia. 2004;47:1157–1166. doi: 10.1007/s00125-004-1454-z. [DOI] [PubMed] [Google Scholar]
- 22.Argoff CE, Cole BE, Fishbain DA, Irving GA. Diabetic peripheral neuropathic pain: clinical and quality-of-life issues. Mayo Clin Proc. 2006;81:S3–11. doi: 10.1016/s0025-6196(11)61474-2. [DOI] [PubMed] [Google Scholar]
- 23.Vinik AI, Newlon P, Milicevic Z, McNitt P, Stansberry KB. Diabetic neuropathies: an overview of clinical aspects. In: LeRoith D, Taylor SI, Olefsky JM, editors. Diabetes Mellitus. Philadelphia, New-York: Lippincott-Raven Publishers; 1996. pp. 737–751. [Google Scholar]
- 24.Vinik AI, Mehrabyan A. Diabetic neuropathies. Med Clin North Am. 2004;88:947–999. doi: 10.1016/j.mcna.2004.04.009. xi. [DOI] [PubMed] [Google Scholar]
- 25.Polydefkis M, Griffin JW, McArthur J. New insights into diabetic polyneuropathy. JAMA. 2003;290:1371–1376. doi: 10.1001/jama.290.10.1371. [DOI] [PubMed] [Google Scholar]
- 26.Horowitz SH. Diabetic neuropathy. Clin Orthop Relat Res. 1993;296:78–85. [PubMed] [Google Scholar]
- 27.Sugimoto K, Murakawa Y, Sima AA. Diabetic neuropathy--a continuing enigma. Diabetes Metab Res Rev. 2000;16:408–433. doi: 10.1002/1520-7560(200011/12)16:6<408::aid-dmrr158>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 28.Bastyr EJ, 3rd, Price KL, Bril V. Development and validity testing of the neuropathy total symptom score-6: questionnaire for the study of sensory symptoms of diabetic peripheral neuropathy. Clin Ther. 2005;27:1278–1294. doi: 10.1016/j.clinthera.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 29.Dyck PJ. Detection, characterization, and staging of polyneuropathy: assessed in diabetics. Muscle Nerve. 1988;11:21–32. doi: 10.1002/mus.880110106. [DOI] [PubMed] [Google Scholar]
- 30.Boulton AJ, Vinik AI, Arezzo JC, Bril V, Feldman EL, Freeman R, Malik RA, Maser RE, Sosenko JM, Ziegler D. Diabetic neuropathies: a statement by the American Diabetes Association. Diabetes Care. 2005;28:956–962. doi: 10.2337/diacare.28.4.956. [DOI] [PubMed] [Google Scholar]
- 31.Thomas PK. Classification, differential diagnosis, and staging of diabetic peripheral neuropathy. Diabetes. 1997;46 (Suppl 2):S54–S57. doi: 10.2337/diab.46.2.s54. [DOI] [PubMed] [Google Scholar]
- 32.Young RJ, Zhou YQ, Rodriguez E, Prescott RJ, Ewing DJ, Clarke BF. Variable relationship between peripheral somatic and autonomic neuropathy in patients with different syndromes of diabetic polyneuropathy. Diabetes. 1986;35:192–197. doi: 10.2337/diab.35.2.192. [DOI] [PubMed] [Google Scholar]
- 33.Young RJ, Ewing DJ, Clarke BF. Chronic and remitting painful diabetic polyneuropathy. Correlations with clinical features and subsequent changes in neurophysiology. Diabetes Care. 1988;11:34–40. doi: 10.2337/diacare.11.1.34. [DOI] [PubMed] [Google Scholar]
- 34.Maser RE, Nielsen VK, Bass EB, Manjoo Q, Dorman JS, Kelsey SF, Becker DJ, Orchard TJ. Measuring diabetic neuropathy. Assessment and comparison of clinical examination and quantitative sensory testing. Diabetes Care. 1989;12:270–275. doi: 10.2337/diacare.12.4.270. [DOI] [PubMed] [Google Scholar]
- 35.Navarro X, Kennedy WR, Fries TJ. Small nerve fiber dysfunction in diabetic neuropathy. Muscle Nerve. 1989;12:498–507. doi: 10.1002/mus.880120611. [DOI] [PubMed] [Google Scholar]
- 36.Ellenberg M. Diabetic neuropathy: clinical aspects. Metabolism. 1976;25:1627–1655. doi: 10.1016/0026-0495(76)90115-3. [DOI] [PubMed] [Google Scholar]
- 37.Boulton AJ. Management of diabetic peripheral neuropathy. Clinical Diabetes. 2005;23:9–15. [Google Scholar]
- 38.Belgrade MJ, Cole BE, McCarberg BH, McLean MJ. Diabetic peripheral neuropathic pain: case studies. Mayo Clin Proc. 2006;81:S26–S32. doi: 10.1016/s0025-6196(11)61476-6. [DOI] [PubMed] [Google Scholar]
- 39.Pfeifer MA, Ross DR, Schrage JP, Gelber DA, Schumer MP, Crain GM, Markwell SJ, Jung S. A highly successful and novel model for treatment of chronic painful diabetic peripheral neuropathy. Diabetes Care. 1993;16:1103–1115. doi: 10.2337/diacare.16.8.1103. [DOI] [PubMed] [Google Scholar]
- 40.Benbow SJ, MacFarlane IA. Painful diabetic neuropathy. Baillieres Best Pract Res Clin Endocrinol Metab. 1999;13:295–308. doi: 10.1053/beem.1999.0021. [DOI] [PubMed] [Google Scholar]
- 41.Dyck PJ, Giannini C. Pathologic alterations in the diabetic neuropathies of humans: a review. J Neuropathol and Exp Neurol. 1996;55:1181–1193. doi: 10.1097/00005072-199612000-00001. [DOI] [PubMed] [Google Scholar]
- 42.Thomas PK. Diabetic neuropathy: mechanisms and future treatment options. J Neurol Neurosurg Psychiatry. 1999;67:277–279. doi: 10.1136/jnnp.67.3.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zochodne DW. Neurotrophins and other growth factors in diabetic neuropathy. Semin Neurol. 1996;16:153–161. doi: 10.1055/s-2008-1040971. [DOI] [PubMed] [Google Scholar]
- 44.Liuzzi FJ, Bufton SM, Vinik AI. Streptozotocin-induced diabetes mellitus causes changes in primary sensory neuronal cytoskeletal mRNA levels that mimic those caused by axotomy. Exp Neurol. 1998;154:381–388. doi: 10.1006/exnr.1998.6938. [DOI] [PubMed] [Google Scholar]
- 45.Koltzenburg M, Scadding J. Neuropathic pain. Curr Opin Neurol. 2001;14:641–647. doi: 10.1097/00019052-200110000-00014. [DOI] [PubMed] [Google Scholar]
- 46.Torebjork HE, Lundberg LE, LaMotte RH. Central changes in processing of mechanoreceptive input in capsaicin-induced secondary hyperalgesia in humans. J Physiol. 1992;448:765–780. doi: 10.1113/jphysiol.1992.sp019069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ochoa JL, Torebjork HE. Paraesthesiae from ectopic impulse generation in human sensory nerves. Brain. 1980;103:835–853. doi: 10.1093/brain/103.4.835. [DOI] [PubMed] [Google Scholar]
- 48.Mogyoros I, Bostock H, Burke D. Mechanisms of paresthesias arising from healthy axons. Muscle Nerve. 2000;23:310–320. doi: 10.1002/(sici)1097-4598(200003)23:3<310::aid-mus2>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 49.Wallace VC, Cottrell DF, Brophy PJ, Fleetwood-Walker SM. Focal lysolecithin-induced demyelination of peripheral afferents results in neuropathic pain behavior that is attenuated by cannabinoids. J Neurosci. 2003;23:3221–3233. doi: 10.1523/JNEUROSCI.23-08-03221.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sima AA, Nathaniel V, Bril V, McEwen TA, Greene DA. Histopathological heterogeneity of neuropathy in insulin - dependent and non-insulin-dependent diabetes, and demonstration of axo- glial dysjunction in human diabetic neuropathy. J Clin Invest. 1988;81:349–364. doi: 10.1172/JCI113327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dyck PJ, Lambert EH, O’Brien PC. Pain in peripheral neuropathy related to rate and kind of fiber degeneration. Neurology. 1976;26:466–471. doi: 10.1212/wnl.26.5.466. [DOI] [PubMed] [Google Scholar]
- 52.Bonica JJ. In: The Managment of Pain. Loeser JD, Chapman CR, Fordyce WE, editors. Philadelphia, London: Lea & Febiger; 1990. pp. 1–958. [Google Scholar]
- 53.Hotta N, Toyota T, Matsuoka K, Shigeta Y, Kikkawa R, Kaneko T, Takahashi A, Sugimura K, Koike Y, Ishii J, Sakamoto N. Clinical efficacy of fidarestat, a novel aldose reductase inhibitor, for diabetic peripheral neuropathy: a 52-week multi-center placebo- controlled double-blind parallel group study. Diabetes Care. 2001;24:1776–1782. doi: 10.2337/diacare.24.10.1776. [DOI] [PubMed] [Google Scholar]
- 54.Brown MJ, Martin JR, Asbury AK. Painful diabetic neuropathy. A morphometric study. Arch Neurol. 1976;33:164–171. doi: 10.1001/archneur.1976.00500030020004. [DOI] [PubMed] [Google Scholar]
- 55.Britland ST, Young RJ, Sharma AK, Clarke BF. Association of painful and painless diabetic polyneuropathy with different patterns of nerve fiber degeneration and regeneration. Diabetes. 1990;39:898–908. doi: 10.2337/diab.39.8.898. [DOI] [PubMed] [Google Scholar]
- 56.Sima AA. Peripheral neuropathy in the spontaneously diabetic BB-Wistar-rat. An ultrastructural study. Acta Neuropathol (Berl) 1980;51:223–227. doi: 10.1007/BF00687389. [DOI] [PubMed] [Google Scholar]
- 57.Waxman SG. Pathophysiology of nerve conduction: relation to diabetic neuropathy. Ann Intern Med. 1980;92:297–301. doi: 10.7326/0003-4819-92-2-297. [DOI] [PubMed] [Google Scholar]
- 58.Johnson PC, Doll SC, Cromey DW. Pathogenesis of diabetic neuropathy. Ann Neurol. 1986;19:450–457. doi: 10.1002/ana.410190505. [DOI] [PubMed] [Google Scholar]
- 59.Dyck PJ, Karnes JL, O’Brien P, Okazaki H, Lais A, Engelstad J. The spatial distribution of fiber loss in diabetic polyneuropathy suggests ischemia. Ann Neurol. 1986;19:440–449. doi: 10.1002/ana.410190504. [DOI] [PubMed] [Google Scholar]
- 60.Dyck PJ, Lais A, Karnes JL, O’Brien P, Rizza R. Fiber loss is primary and multifocal in sural nerves in diabetic polyneuropathy. Ann Neurol. 1986;19:425–439. doi: 10.1002/ana.410190503. [DOI] [PubMed] [Google Scholar]
- 61.Dyck PJ, Gutrecht JA, Bastron JA, Karnes WE, Dale AJ. Histologic and teased-fiber measurements of sural nerve in disorders of lower motor and primary sensory neurons. Mayo Clin Proc. 1968;43:81–123. [PubMed] [Google Scholar]
- 62.Malik RA. The pathology of human diabetic neuropathy. Diabetes. 1997;46 (Suppl 2):S50–S53. doi: 10.2337/diab.46.2.s50. [DOI] [PubMed] [Google Scholar]
- 63.Nathan DM. The pathophysiology of diabetic complications: how much does the glucose hypothesis explain? Ann Intern Med. 1996;124:86–89. doi: 10.7326/0003-4819-124-1_part_2-199601011-00002. [DOI] [PubMed] [Google Scholar]
- 64.Vinik AI. Advances in diabetes for the millennium: new treatments for diabetic neuropathies. MedGenMed. 2004;6:13. [PMC free article] [PubMed] [Google Scholar]
- 65.Skyler JS. Effect of glycemic control on diabetes complications and on the prevention of diabetes. Clinical Diabetes. 2004;22:162–166. [Google Scholar]
- 66.Sima AA, editor. Chronic Complications in Diabetes. Animal Models and Chronic Complications. Amsterdam: Harwood Academic Publishers; 2000. pp. 1–277. [Google Scholar]
- 67.Lasker RD. The diabetes control and complications trial. Implications for policy and practice. N Engl J Med. 1993;329:1035–1036. doi: 10.1056/NEJM199309303291410. [DOI] [PubMed] [Google Scholar]
- 68.DCCT. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. The Diabetes Control and Complications Trial Research Group. N Engl J Med. 1993;329:977–986. doi: 10.1056/NEJM199309303291401. [DOI] [PubMed] [Google Scholar]
- 69.Bode BW, Schwartz S, Stubbs HA, Block JE. Glycemic characteristics in continuously monitored patients with type 1 and type 2 diabetes: normative values. Diabetes Care. 2005;28:2361–2366. doi: 10.2337/diacare.28.10.2361. [DOI] [PubMed] [Google Scholar]
- 70.Singleton JR, Smith AG, Bromberg MB. Increased prevalence of impaired glucose tolerance in patients with painful sensory neuropathy. Diabetes Care. 2001;24:1448–1453. doi: 10.2337/diacare.24.8.1448. [DOI] [PubMed] [Google Scholar]
- 71.Novella SP, Inzucchi SE, Goldstein JM. The frequency of undiagnosed diabetes and impaired glucose tolerance in patients with idiopathic sensory neuropathy. Muscle Nerve. 2001;24:1229–1231. doi: 10.1002/mus.1137. [DOI] [PubMed] [Google Scholar]
- 72.Sima AA, Shafrir E, editors. Animal Models of Diabetes. A Primer. Amsterdam: Harwood Academic Publishers; 2001. pp. 1–364. [Google Scholar]
- 73.Moore SA, Peterson RG, Felten DL, O’Connor BL. A quantitative comparison of motor and sensory conduction velocities in short- and long-term streptozotocin- and alloxan-diabetic rats. J Neurol Sci. 1980;48:133–152. doi: 10.1016/0022-510x(80)90156-2. [DOI] [PubMed] [Google Scholar]
- 74.Weis J, Dimpfel W, Schroder JM. Nerve conduction changes and fine structural alterations of extra- and intrafusal muscle and nerve fibers in streptozotocin diabetic rats. Muscle Nerve. 1995;18:175–184. doi: 10.1002/mus.880180205. [DOI] [PubMed] [Google Scholar]
- 75.Stevens MJ, Zhang W, Li F, Sima AA. C-peptide corrects endoneurial blood flow but not oxidative stress in type 1 BB/Wor rats. Am J Physiol Endocrinol Metab. 2004;287:E497–E505. doi: 10.1152/ajpendo.00048.2004. [DOI] [PubMed] [Google Scholar]
- 76.Zochodne DW, Ho LT, Allison JA. Dorsal root ganglia micro-environment of female BB Wistar diabetic rats with mild neuropathy. J Neurol Sci. 1994;127:36–42. doi: 10.1016/0022-510x(94)90132-5. [DOI] [PubMed] [Google Scholar]
- 77.Ferreira LD, Huey PU, Pulford BE, Ishii DN, Eckel RH. Sciatic nerve lipoprotein lipase is reduced in streptozotocin-induced diabetes and corrected by insulin. Endocrinology. 2002;143:1213–1217. doi: 10.1210/endo.143.4.8723. [DOI] [PubMed] [Google Scholar]
- 78.Coppey LJ, Davidson EP, Dunlap JA, Lund DD, Yorek MA. Slowing of motor nerve conduction velocity in streptozotocin-induced diabetic rats is preceded by impaired vasodilation in arterioles that overlie the sciatic nerve. Int J Exp Diabetes Res. 2000;1:131–143. doi: 10.1155/EDR.2000.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Qiang X, Satoh J, Sagara M, Fukuzawa M, Masuda T, Sakata Y, Muto G, Muto Y, Takahashi K, Toyota T. Inhibitory effect of troglitazone on diabetic neuropathy in streptozotocin-induced diabetic rats. Diabetologia. 1998;41:1321–1326. doi: 10.1007/s001250051072. [DOI] [PubMed] [Google Scholar]
- 80.Sima AA, Brismar T. Reversible diabetic nerve dysfunction: structural correlates to electrophysiological abnormalities. Ann Neurol. 1985;18:21–29. doi: 10.1002/ana.410180105. [DOI] [PubMed] [Google Scholar]
- 81.Sima AA, Lattimer SA, Yagihashi S, Greene DA. Axo-glial dysjunction. J Clin Invest. 1986;77:474–484. doi: 10.1172/JCI112326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Greene DA, Chakrabarti S, Lattimer SA, Sima AA. Role of sorbitol accumulation and myo-inositol depletion in paranodal swelling of large myelinated nerve fibers in the insulin-deficient spontaneously diabetic bio-breeding rat. Reversal by insulin replacement, an aldose reductase inhibitor, and myo-inositol. J Clin Invest. 1987;79:1479–1485. doi: 10.1172/JCI112977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Shimoshige Y, Ikuma K, Yamamoto T, Takakura S, Kawamura I, Seki J, Mutoh S, Goto T. The effects of zenarestat, an al-dose reductase inhibitor, on peripheral neuropathy in Zucker diabetic fatty rats. Metabolism. 2000;49:1395–1399. doi: 10.1053/meta.2000.17723. [DOI] [PubMed] [Google Scholar]
- 84.Romanovsky D, Hastings SL, Stimers JR, Dobretsov M. Relevance of hyperglycemia to early mechanical hyperalgesia in streptozotocin-induced diabetes. J Peripher Nerv Syst. 2004;9:62–69. doi: 10.1111/j.1085-9489.2004.009204.x. [DOI] [PubMed] [Google Scholar]
- 85.Chen SR, Pan HL. Hypersensitivity of spinothalamic tract neurons associated with diabetic neuropathic pain in rats. J Neurophysiol. 2002;87:2726–2733. doi: 10.1152/jn.2002.87.6.2726. [DOI] [PubMed] [Google Scholar]
- 86.Zhuang HX, Wuarin L, Fei ZJ, Ishii DN. Insulin-like growth factor (IGF) gene expression is reduced in neural tissues and liver from rats with non-insulin-dependent diabetes mellitus, and IGF treatment ameliorates diabetic neuropathy. J Pharmacol Exp Ther. 1997;283:366–374. [PubMed] [Google Scholar]
- 87.Piercy V, Banner SE, Bhattacharyya A, Parsons AA, Sanger GJ, Smith SA, Bingham S. Thermal, but not mechanical, nociceptive behavior is altered in the Zucker Diabetic Fatty rat and is independent of glycemic status. J Diabetes Complications. 1999;13:163–169. doi: 10.1016/s1056-8727(99)00034-3. [DOI] [PubMed] [Google Scholar]
- 88.Schmidt RE, Dorsey DA, Beaudet LN, Parvin CA, Zhang W, Sima AA. Experimental rat models of types 1 and 2 diabetes differ in sympathetic neuroaxonal dystrophy. J Neuropathol Exp Neurol. 2004;63:450–460. doi: 10.1093/jnen/63.5.450. [DOI] [PubMed] [Google Scholar]
- 89.Brussee V, Cunningham FA, Zochodne DW. Direct insulin signaling of neurons reverses diabetic neuropathy. Diabetes. 2004;53:1824–1830. doi: 10.2337/diabetes.53.7.1824. [DOI] [PubMed] [Google Scholar]
- 90.Schmeichel AM, Schmelzer JD, Low PA. Oxidative injury and apoptosis of dorsal root ganglion neurons in chronic experimental diabetic neuropathy. Diabetes. 2003;52:165–171. doi: 10.2337/diabetes.52.1.165. [DOI] [PubMed] [Google Scholar]
- 91.Cameron NE, Cotter MA, Low PA. Nerve blood flow in early experimental diabetes in rats: relation to conduction deficits. Am J Physiol. 1991;261:E1–E8. doi: 10.1152/ajpendo.1991.261.1.E1. [DOI] [PubMed] [Google Scholar]
- 92.Nagamatsu M, Nickander KK, Schmelzer JD, Raya A, Wittrock DA, Tritschler H, Low PA. Lipoic acid improves nerve blood flow, reduces oxidative stress, and improves distal nerve conduction in experimental diabetic neuropathy. Diabetes Care. 1995;18:1160–1167. doi: 10.2337/diacare.18.8.1160. [DOI] [PubMed] [Google Scholar]
- 93.Cameron NE, Cotter MA, Jack AM, Basso MD, Hohman TC. Protein kinase C effects on nerve function, perfusion, Na(+), K(+)-ATPase activity and glutathione content in diabetic rats. Diabetologia. 1999;42:1120–1130. doi: 10.1007/s001250051280. [DOI] [PubMed] [Google Scholar]
- 94.Way KJ, Katai N, King GL. Protein kinase C and the development of diabetic vascular complications. Diabet Med. 2001;18:945–959. doi: 10.1046/j.0742-3071.2001.00638.x. [DOI] [PubMed] [Google Scholar]
- 95.Hamdy O, Ledbury S, Mullooly C, Jarema C, Porter S, Ovalle K, Moussa A, Caselli A, Caballero AE, Economides PA, Veves A, Horton ES. Lifestyle modification improves endothelial function in obese subjects with the insulin resistance syndrome. Diabetes Care. 2003;26:2119–2125. doi: 10.2337/diacare.26.7.2119. [DOI] [PubMed] [Google Scholar]
- 96.Huvers FC, De Leeuw PW, Houben AJ, De Haan CH, Hamulyak K, Schouten H, Wolffenbuttel BH, Schaper NC. Endothelium-dependent vasodilatation, plasma markers of endothelial function, and adrenergic vasoconstrictor responses in type 1 diabetes under near-normoglycemic conditions. Diabetes. 1999;48:1300–1307. doi: 10.2337/diabetes.48.6.1300. [DOI] [PubMed] [Google Scholar]
- 97.Khan F, Elhadd TA, Greene SA, Belch JJ. Impaired skin microvascular function in children, adolescents, and young adults with type 1 diabetes. Diabetes Care. 2000;23:215–220. doi: 10.2337/diacare.23.2.215. [DOI] [PubMed] [Google Scholar]
- 98.Caballero AE, Arora S, Saouaf R, Lim SC, Smakowski P, Park JY, King GL, LoGerfo FW, Horton ES, Veves A. Microvascular and macrovascular reactivity is reduced in subjects at risk for type 2 diabetes. Diabetes. 1999;48:1856–1862. doi: 10.2337/diabetes.48.9.1856. [DOI] [PubMed] [Google Scholar]
- 99.Theriault M, Dort J, Sutherland G, Zochodne DW. Local human sural nerve blood flow in diabetic and other polyneuropathies. Brain. 1997;120 (Pt 7):1131–1138. doi: 10.1093/brain/120.7.1131. [DOI] [PubMed] [Google Scholar]
- 100.Dobretsov M, Hastings SL, Stimers JR, Zhang J-M. Mechanical hyperalgesia in rats with chronic perfusion of lumbar dorsal root ganglion with hyperglycemic solution. J Neurosci Methods. 2001;110:9–15. doi: 10.1016/s0165-0270(01)00410-1. [DOI] [PubMed] [Google Scholar]
- 101.Dobretsov M, Hastings SL, Romanovsky D, Stimers JR, Zhang JM. Mechanical hyperalgesia in rat models of systemic and local hyperglycemia. Brain Res. 2003;960:174–183. doi: 10.1016/s0006-8993(02)03828-3. [DOI] [PubMed] [Google Scholar]
- 102.King RH. The role of glycation in the pathogenesis of diabetic polyneuropathy. Mol Pathol. 2001;54:400–408. [PMC free article] [PubMed] [Google Scholar]
- 103.Srivastava SK, Ramana KV, Bhatnagar A. Role of aldose reductase and oxidative damage in diabetes and the consequent potential for therapeutic options. Endocr Rev. 2005;26:380–392. doi: 10.1210/er.2004-0028. [DOI] [PubMed] [Google Scholar]
- 104.Yabe-Nishimura C. Aldose reductase in glucose toxicity: a potential target for the prevention of diabetic complications. Pharmacol Rev. 1998;50:21–33. [PubMed] [Google Scholar]
- 105.Reusch JE. Diabetes, microvascular complications, and cardiovascular complications: what is it about glucose? J Clin Invest. 2003;112:986–988. doi: 10.1172/JCI19902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Brownlee M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes. 2005;54:1615–1625. doi: 10.2337/diabetes.54.6.1615. [DOI] [PubMed] [Google Scholar]
- 107.Pieper GM. Review of alterations in endothelial nitric oxide production in diabetes: protective role of arginine on endothelial dysfunction. Hypertension. 1998;31:1047–1060. doi: 10.1161/01.hyp.31.5.1047. [DOI] [PubMed] [Google Scholar]
- 108.Pacher P, Szabo C. Role of poly(ADP-ribose) polymerase-1 activation in the pathogenesis of diabetic complications: endothelial dysfunction, as a common underlying theme. Antioxid Redox Signal. 2005;7:1568–1580. doi: 10.1089/ars.2005.7.1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wang Y, Schmeichel AM, Iida H, Schmelzer JD, Low PA. Ischemia-reperfusion injury causes oxidative stress and apoptosis of Schwann cell in acute and chronic experimental diabetic neuropathy. Antioxid Redox Signal. 2005;7:1513–1520. doi: 10.1089/ars.2005.7.1513. [DOI] [PubMed] [Google Scholar]
- 110.Chait A, Brunzell JD. Diabetes, lipids, and atherosclerosis. In: LeRoith D, Taylor SI, Olefsky JM, editors. Diabetes Mellitus. Philadelphia, New York: Lippincott-Raven Publishers; 1996. pp. 772–777. [Google Scholar]
- 111.Obrosova IG. Increased sorbitol pathway activity generates oxidative stress in tissue sites for diabetic complications. Antioxid Redox Signal. 2005;7:1543–1552. doi: 10.1089/ars.2005.7.1543. [DOI] [PubMed] [Google Scholar]
- 112.Kellogg AP, Pop-Busui R. Peripheral nerve dysfunction in experimental diabetes is mediated by cyclooxygenase-2 and oxidative stress. Antioxid Redox Signal. 2005;7:1521–1529. doi: 10.1089/ars.2005.7.1521. [DOI] [PubMed] [Google Scholar]
- 113.Greene DA, Stevens MJ, Obrosova I, Feldman EL. Glucose-induced oxidative stress and programmed cell death in diabetic neuropathy. Eur J Pharmacol. 1999;375:217–223. doi: 10.1016/s0014-2999(99)00356-8. [DOI] [PubMed] [Google Scholar]
- 114.Feldman EL. Oxidative stress and diabetic neuropathy: a new understanding of an old problem. J Clin Invest. 2003;111:431–433. doi: 10.1172/JCI17862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ceriello A, Motz E. Is oxidative stress the pathogenic mechanism underlying insulin resistance, diabetes, and cardiovascular disease? The common soil hypothesis revisited. Arterioscler Thromb Vasc Biol. 2004;24:816–823. doi: 10.1161/01.ATV.0000122852.22604.78. [DOI] [PubMed] [Google Scholar]
- 116.Kishi Y, Schmelzer JD, Yao JK, Zollman PJ, Nickander KK, Tritschler HJ, Low PA. Alpha-lipoic acid: effect on glucose uptake, sorbitol pathway, and energy metabolism in experimental diabetic neuropathy. Diabetes. 1999;48:2045–2051. doi: 10.2337/diabetes.48.10.2045. [DOI] [PubMed] [Google Scholar]
- 117.Ametov AS, Barinov A, Dyck PJ, Hermann R, Kozlova N, Litchy WJ, Low PA, Nehrdich D, Novosadova M, O’Brien PC, Reljanovic M, Samigullin R, Schuette K, Strokov I, Tritschler HJ, Wessel K, Yakhno N, Ziegler D. The sensory symptoms of diabetic polyneuropathy are improved with alpha-lipoic acid: the SYDNEY trial. Diabetes Care. 2003;26:770–776. doi: 10.2337/diacare.26.3.770. [DOI] [PubMed] [Google Scholar]
- 118.Maritim AC, Sanders RA, Watkins JB., 3rd Diabetes, oxidative stress, and antioxidants: a review. J Biochem Mol Toxicol. 2003;17:24–38. doi: 10.1002/jbt.10058. [DOI] [PubMed] [Google Scholar]
- 119.Johansen JS, Harris AK, Rychly DJ, Ergul A. Oxidative stress and the use of antioxidants in diabetes: linking basic science to clinical practice. Cardiovasc Diabetol. 2005;4:5–25. doi: 10.1186/1475-2840-4-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Petersen M, LaMotte RH. Relationships between capsaicin sensitivity of mammalian sensory neurons, cell size and type of voltage gated Ca-currents. Brain Res. 1991;561:20–26. doi: 10.1016/0006-8993(91)90744-g. [DOI] [PubMed] [Google Scholar]
- 121.Obrosova IG, Van Huysen C, Fathallah L, Cao XC, Greene DA, Stevens MJ. An aldose reductase inhibitor reverses early diabetes-induced changes in peripheral nerve function, metabolism, and antioxidative defense. FASEB J. 2002;16:123–125. doi: 10.1096/fj.01-0603fje. [DOI] [PubMed] [Google Scholar]
- 122.Young RJ, Ewing DJ, Clarke BF. A controlled trial of sorbinil, an aldose reductase inhibitor, in chronic painful diabetic neuropathy. Diabetes. 1983;32:938–942. doi: 10.2337/diab.32.10.938. [DOI] [PubMed] [Google Scholar]
- 123.Martyn CN, Reid W, Young RJ, Ewing DJ, Clarke BF. Six-month treatment with sorbinil in asymptomatic diabetic neuropathy. Failure to improve abnormal nerve function. Diabetes. 1987;36:987–990. doi: 10.2337/diab.36.9.987. [DOI] [PubMed] [Google Scholar]
- 124.Boulton AJ, Malik RA, Arezzo JC, Sosenko JM. Diabetic somatic neuropathies. Diabetes Care. 2004;27:1458–1483. doi: 10.2337/diacare.27.6.1458. [DOI] [PubMed] [Google Scholar]
- 125.Singleton JR, Smith AG, Russell J, Feldman EL. Polyneuropathy with Impaired Glucose Tolerance: Implications for Diagnosis and Therapy. Curr Treat Options Neurol. 2005;7:33–42. doi: 10.1007/s11940-005-0004-4. [DOI] [PubMed] [Google Scholar]
- 126.Russell JW, Feldman EL. Impaired glucose tolerance--does it cause neuropathy? Muscle Nerve. 2001;24:1109–1112. doi: 10.1002/mus.1122. [DOI] [PubMed] [Google Scholar]
- 127.Singleton JR, Smith AG, Bromberg MB. Painful sensory polyneuropathy associated with impaired glucose tolerance. Muscle Nerve. 2001;24:1225–1228. doi: 10.1002/mus.1136. [DOI] [PubMed] [Google Scholar]
- 128.Pittenger GL, Mehrabyan A, Simmons K, Rice A, Dublin C, Barlow P, Vinik AI. Small fiber neuropathy is associated with the metabolic syndrome. Metabolic Syndrome and Related Disorders. 2005;3:113–121. doi: 10.1089/met.2005.3.113. [DOI] [PubMed] [Google Scholar]
- 129.Sumner CJ, Sheth S, Griffin JW, Cornblath DR, Polydefkis M. The spectrum of neuropathy in diabetes and impaired glucose tolerance. Neurology. 2003;60:108–111. doi: 10.1212/wnl.60.1.108. [DOI] [PubMed] [Google Scholar]
- 130.Monnier L, Mas E, Ginet C, Michel F, Villon L, Cristol JP, Colette C. Activation of oxidative stress by acute glucose fluctuations compared with sustained chronic hyperglycemia in patients with type 2 diabetes. JAMA. 2006;295:1681–1687. doi: 10.1001/jama.295.14.1681. [DOI] [PubMed] [Google Scholar]
- 131.de Neeling JN, Beks PJ, Bertelsmann FW, Heine RJ, Bouter LM. Peripheral somatic nerve function in relation to glucose tolerance in an elderly Caucasian population: the Hoorn study. Diabet Med. 1996;13:960–966. doi: 10.1002/(SICI)1096-9136(199611)13:11<960::AID-DIA268>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 132.Morley GK, Mooradian AD, Levine AS, Morley JE. Mechanism of pain in diabetic peripheral neuropathy. Effect of glucose on pain perception in humans. Am J Med. 1984;77:79–82. doi: 10.1016/0002-9343(84)90439-x. [DOI] [PubMed] [Google Scholar]
- 133.Thye-Ronn P, Sindrup SH, Arendt-Nielsen L, Brennum J, Hother-Nielsen O, Beck-Nielsen H. Effect of short-term hyperglycemia per se on nociceptive and non-nociceptive thresholds. Pain. 1994;56:43–49. doi: 10.1016/0304-3959(94)90148-1. [DOI] [PubMed] [Google Scholar]
- 134.Chan AW, MacFarlane IA, Bowsher D. Short term fluctuations in blood glucose concentrations do not alter pain perception in diabetic-patients with and without painful peripheral neuropathy. Diabetes Res. 1990;14:15–19. [PubMed] [Google Scholar]
- 135.Oyibo SO, Prasad YD, Jackson NJ, Jude EB, Boulton AJ. The relationship between blood glucose excursions and painful diabetic peripheral neuropathy: a pilot study. Diabet Med. 2002;19:870–873. doi: 10.1046/j.1464-5491.2002.00801.x. [DOI] [PubMed] [Google Scholar]
- 136.Huang TJ, Price SA, Chilton L, Calcutt NA, Tomlinson DR, Verkhratsky A, Fernyhough P. Insulin prevents depolarization of the mitochondrial inner membrane in sensory neurons of type 1 diabetic rats in the presence of sustained hyperglycemia. Diabetes. 2003;52:2129–2136. doi: 10.2337/diabetes.52.8.2129. [DOI] [PubMed] [Google Scholar]
- 137.Maneuf YP, Blake R, Andrews NA, McKnight AT. Reduction by gabapentin of K+-evoked release of [3H]-glutamate from the caudal trigeminal nucleus of the streptozotocin-treated rat. Br J Pharmacol. 2004;141:574–579. doi: 10.1038/sj.bjp.0705579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Calcutt NA, Jorge MC, Yaksh TL, Chaplan SR. Tactile allodynia and formalin hyperalgesia in streptozotocin-diabetic rats: effects of insulin, aldose reductase inhibition and lidocaine. Pain. 1996;68:293–299. doi: 10.1016/s0304-3959(96)03201-0. [DOI] [PubMed] [Google Scholar]
- 139.Calcutt NA, Freshwater JD, Mizisin AP. Prevention of sensory disorders in diabetic Sprague-Dawley rats by aldose reductase inhibition or treatment with ciliary neurotrophic factor. Diabetologia. 2004;47:718–724. doi: 10.1007/s00125-004-1354-2. [DOI] [PubMed] [Google Scholar]
- 140.Roane DS, Porter JR. Nociception and opioid-induced analgesia in lean (Fa/-) and obese (fa/fa) Zucker rats. Physiol Behav. 1986;38:215–218. doi: 10.1016/0031-9384(86)90156-3. [DOI] [PubMed] [Google Scholar]
- 141.Oltman CL, Coppey LJ, Gellett JS, Davidson EP, Lund DD, Yorek MA. Progression of vascular and neural dysfunction in sciatic nerves of Zucker diabetic fatty and Zucker rats. Am J Physiol Endocrinol Metab. 2005;289:E113–E122. doi: 10.1152/ajpendo.00594.2004. [DOI] [PubMed] [Google Scholar]
- 142.Ishii DN. Implication of insulin-like growth factors in the pathogenesis of diabetic neuropathy. Brain Res Brain Res Rev. 1995;20:47–67. doi: 10.1016/0165-0173(94)00005-a. [DOI] [PubMed] [Google Scholar]
- 143.Sugimoto K, Murakawa Y, Zhang W, Xu G, Sima AA. Insulin receptor in rat peripheral nerve: its localization and alternatively spliced isoforms. Diabetes Metab Res Rev. 2000;16:354–363. doi: 10.1002/1520-7560(200009/10)16:5<354::aid-dmrr149>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 144.Sugimoto K, Murakawa Y, Sima AA. Expression and localization of insulin receptor in rat dorsal root ganglion and spinal cord. J Peripher Nerv Syst. 2002;7:44–53. doi: 10.1046/j.1529-8027.2002.02005.x. [DOI] [PubMed] [Google Scholar]
- 145.Frasca F, Pandini G, Scalia P, Sciacca L, Mineo R, Costantino A, Goldfine ID, Belfiore A, Vigneri R. Insulin receptor isoform A, a newly recognized, high-affinity insulin-like growth factor II receptor in fetal and cancer cells. Mol Cell Biol. 1999;19:3278–3288. doi: 10.1128/mcb.19.5.3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Martin CL, Albers J, Herman WH, Cleary P, Waberski B, Greene DA, Stevens MJ, Feldman EL. Neuropathy among the diabetes control and complications trial cohort 8 years after trial completion. Diabetes Care. 2006;29:340–344. doi: 10.2337/diacare.29.02.06.dc05-1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Singhal A, Cheng C, Sun H, Zochodne DW. Near nerve local insulin prevents conduction slowing in experimental diabetes. Brain Res. 1997;763:209–214. doi: 10.1016/s0006-8993(97)00412-5. [DOI] [PubMed] [Google Scholar]
- 148.Partanen J, Niskanen L, Lehtinen J, Mervaala E, Siitonen O, Uusitupa M. Natural history of peripheral neuropathy in patients with non-insulin-dependent diabetes mellitus. N Engl J Med. 1995;333:89–94. doi: 10.1056/NEJM199507133330203. [DOI] [PubMed] [Google Scholar]
- 149.Delaney CA, Mouser JV, Westerman RA. Insulin sensitivity and sensory nerve function in non-diabetic human subjects. Neurosci Lett. 1994;180:277–280. doi: 10.1016/0304-3940(94)90538-x. [DOI] [PubMed] [Google Scholar]
- 150.Jacob S, Streeper RS, Fogt DL, Hokama JY, Tritschler HJ, Dietze GJ, Henriksen EJ. The antioxidant alpha-lipoic acid enhances insulin-stimulated glucose metabolism in insulin-resistant rat skeletal muscle. Diabetes. 1996;45:1024–1029. doi: 10.2337/diab.45.8.1024. [DOI] [PubMed] [Google Scholar]
- 151.Broca C, Breil V, Cruciani-Guglielmacci C, Manteghetti M, Rouault C, Derouet M, Rizkalla S, Pau B, Petit P, Ribes G, Ktorza A, Gross R, Reach G, Taouis M. Insulinotropic agent ID-1101 (4-hydroxyisoleucine) activates insulin signaling in rat. Am J Physiol Endocrinol Metab. 2004;287:E463–E471. doi: 10.1152/ajpendo.00163.2003. [DOI] [PubMed] [Google Scholar]
- 152.Wong V, Stavar L, Szeto L, Uffelman K, Wang CH, Fantus IG, Lewis GF. Atorvastatin induces insulin sensitization in Zucker lean and fatty rats. Atherosclerosis. 2006;184:348–355. doi: 10.1016/j.atherosclerosis.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 153.Freychet P. Insulin receptors and insulin actions in the nervous system. Diabetes Metab Res Rev. 2000;16:390–392. doi: 10.1002/1520-7560(200011/12)16:6<390::aid-dmrr161>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 154.Kihara M, Zollman PJ, Smithson IL, Lagerlund TD, Low PA. Hypoxic effect of exogenous insulin on normal and diabetic peripheral nerve. Am J Physiol. 1994;266:E980–E985. doi: 10.1152/ajpendo.1994.266.6.E980. [DOI] [PubMed] [Google Scholar]
- 155.Dandona P, Mohanty P, Chaudhuri A, Garg R, Aljada A. Insulin infusion in acute illness. J Clin Invest. 2005;115:2069–2072. doi: 10.1172/JCI26045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Nedrebo T, Karlsen TV, Salvesen GS, Reed RK. A novel function of insulin in rat dermis. J Physiol. 2004;559:583–591. doi: 10.1113/jphysiol.2004.067751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Jeschke MG, Einspanier R, Klein D, Jauch KW. Insulin attenuates the systemic inflammatory response to thermal trauma. Mol Med. 2002;8:443–450. [PMC free article] [PubMed] [Google Scholar]
- 158.Davel AP, Rossoni LV, Vassallo DV. Effects of ouabain on the pressor response to phenylephrine and on the sodium pump activity in diabetic rats. Eur J Pharmacol. 2000;406:419–427. doi: 10.1016/s0014-2999(00)00679-8. [DOI] [PubMed] [Google Scholar]
- 159.Sweeney G, Klip A. Regulation of the Na+/K+-ATPase by insulin: why and how? Mol Cell Biochem. 1998;182:121–133. [PubMed] [Google Scholar]
- 160.Steinberg HO, Brechtel G, Johnson A, Fineberg N, Baron AD. Insulin-mediated skeletal muscle vasodilation is nitric oxide dependent. A novel action of insulin to increase nitric oxide release. J Clin Invest. 1994;94:1172–1179. doi: 10.1172/JCI117433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Ranke MB. Insulin-like growth factor-I treatment of growth disorders, diabetes mellitus and insulin resistance. Trends Endocrinol Metab. 2005;16:190–197. doi: 10.1016/j.tem.2005.03.011. [DOI] [PubMed] [Google Scholar]
- 162.Recio-Pinto E, Rechler MM, Ishii DN. Effects of insulin, insulin-like growth factor-II, and nerve growth factor on neurite formation and survival in cultured sympathetic and sensory neurons. J Neurosci. 1986;6:1211–1219. doi: 10.1523/JNEUROSCI.06-05-01211.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Mill JF, Chao MV, Ishii DN. Insulin, insulin-like growth factor II, and nerve growth factor effects on tubulin mRNA levels and neurite formation. Proc Natl Acad Sci U S A. 1985;82:7126–7130. doi: 10.1073/pnas.82.20.7126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Near SL, Whalen LR, Miller JA, Ishii DN. Insulin-like growth factor II stimulates motor nerve regeneration. Proc Natl Acad Sci U S A. 1992;89:11716–11720. doi: 10.1073/pnas.89.24.11716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Dupont J, Khan J, Qu BH, Metzler P, Helman L, LeRoith D. Insulin and IGF-1 induce different patterns of gene expression in mouse fibroblast NIH-3T3 cells: identification by cDNA microarray analysis. Endocrinology. 2001;142:4969–4975. doi: 10.1210/endo.142.11.8476. [DOI] [PubMed] [Google Scholar]
- 166.Meier C, Parmantier E, Brennan A, Mirsky R, Jessen KR. Developing Schwann cells acquire the ability to survive without axons by establishing an autocrine circuit involving insulin-like growth factor, neurotrophin-3, and platelet-derived growth factor-BB. J Neurosci. 1999;19:3847–3859. doi: 10.1523/JNEUROSCI.19-10-03847.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Holzenberger M, Dupont J, Ducos B, Leneuve P, Geloen A, Even PC, Cervera P, Le Bouc Y. IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature. 2003;421:182–187. doi: 10.1038/nature01298. [DOI] [PubMed] [Google Scholar]
- 168.Rajkumar K, Krsek M, Dheen ST, Murphy LJ. Impaired glucose homeostasis in insulin-like growth factor binding protein-1 transgenic mice. J Clin Invest. 1996;98:1818–1825. doi: 10.1172/JCI118982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Karagiannis SN, King RH, Thomas PK. Colocalisation of insulin and IGF-1 receptors in cultured rat sensory and sympathetic ganglion cells. J Anat. 1997;191 (Pt 3):431–440. doi: 10.1046/j.1469-7580.1997.19130431.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Craner MJ, Klein JP, Black JA, Waxman SG. Preferential expression of IGF-I in small DRG neurons and down-regulation following injury. Neuroreport. 2002;13:1649–1652. doi: 10.1097/00001756-200209160-00016. [DOI] [PubMed] [Google Scholar]
- 171.Cheng HL, Randolph A, Yee D, Delafontaine P, Tennekoon G, Feldman EL. Characterization of insulin-like growth factor-I and its receptor and binding proteins in transected nerves and cultured Schwann cells. J Neurochem. 1996;66:525–536. doi: 10.1046/j.1471-4159.1996.66020525.x. [DOI] [PubMed] [Google Scholar]
- 172.Jonas HA, Harrison LC. The human placenta contains two distinct binding and immunoreactive species of insulin-like growth factor-I receptors. J Biol Chem. 1985;260:2288–2294. [PubMed] [Google Scholar]
- 173.Moxham CP, Duronio V, Jacobs S. Insulin-like growth factor I receptor beta-subunit heterogeneity. Evidence for hybrid tetramers composed of insulin-like growth factor I and insulin receptor heterodimers. J Biol Chem. 1989;264:13238–13244. [PubMed] [Google Scholar]
- 174.Bondy CA, Underwood LE, Clemmons DR, Guler HP, Bach MA, Skarulis M. Clinical uses of insulin-like growth factor I. Ann Intern Med. 1994;120:593–601. doi: 10.7326/0003-4819-120-7-199404010-00011. [DOI] [PubMed] [Google Scholar]
- 175.Saukkonen T, Amin R, Williams RM, Fox C, Yuen KC, White MA, Umpleby AM, Acerini CL, Dunger DB. Dose-dependent effects of recombinant human insulin-like growth factor (IGF)-I/IGF binding protein-3 complex on overnight growth hormone secretion and insulin sensitivity in type 1 diabetes. J Clin Endocrinol Metab. 2004;89:4634–4641. doi: 10.1210/jc.2004-0243. [DOI] [PubMed] [Google Scholar]
- 176.Clemmons DR, Moses AC, McKay MJ, Sommer A, Rosen DM, Ruckle J. The combination of insulin-like growth factor I and insulin-like growth factor-binding protein-3 reduces insulin requirements in insulin-dependent type 1 diabetes: evidence for in vivo biological activity. J Clin Endocrinol Metab. 2000;85:1518–1524. doi: 10.1210/jcem.85.4.6559. [DOI] [PubMed] [Google Scholar]
- 177.Thrailkill KM, Quattrin T, Baker L, Kuntze JE, Compton PG, Martha PM., Jr Cotherapy with recombinant human insulin-like growth factor I and insulin improves glycemic control in type 1 diabetes. RhIGF-I in IDDM Study Group. Diabetes Care. 1999;22:585–592. doi: 10.2337/diacare.22.4.585. [DOI] [PubMed] [Google Scholar]
- 178.Cusi K, DeFronzo R. Recombinant human insulin-like growth factor I treatment for 1 week improves metabolic control in type 2 diabetes by ameliorating hepatic and muscle insulin resistance. J Clin Endocrinol Metab. 2000;85:3077–3084. doi: 10.1210/jcem.85.9.6827. [DOI] [PubMed] [Google Scholar]
- 179.Schmidt RE, Dorsey DA, Beaudet LN, Peterson RG. Analysis of the Zucker Diabetic Fatty (ZDF) type 2 diabetic rat model suggests a neurotrophic role for insulin/IGF-I in diabetic autonomic neuropathy. Am J Pathol. 2003;163:21–28. doi: 10.1016/S0002-9440(10)63626-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Busiguina S, Fernandez AM, Barrios V, Clark R, Tolbert DL, Berciano J, Torres-Aleman I. Neurodegeneration is associated to changes in serum insulin-like growth factors. Neurobiol Dis. 2000;7:657–665. doi: 10.1006/nbdi.2000.0311. [DOI] [PubMed] [Google Scholar]
- 181.Chen HS, Shan YX, Yang TL, Lin HD, Chen JW, Lin SJ, Wang PH. Insulin deficiency downregulated heat shock protein 60 and IGF-1 receptor signaling in diabetic myocardium. Diabetes. 2005;54:175–181. doi: 10.2337/diabetes.54.1.175. [DOI] [PubMed] [Google Scholar]
- 182.Zhuang HX, Wuarin L, Fei ZJ, Ishii DN. Insulin-like growth factor (IGF) gene expression is reduced in neural tissues and liver from rats with non-insulin-dependent diabetes mellitus, and IGF treatment ameliorates diabetic neuropathy. J Pharmacol Exp Ther. 1997;283:366–374. [PubMed] [Google Scholar]
- 183.Jacob RJ, Sherwin RS, Greenawalt K, Shulman GI. Simultaneous insulinlike growth factor I and insulin resistance in obese Zucker rats. Diabetes. 1992;41:691–697. doi: 10.2337/diab.41.6.691. [DOI] [PubMed] [Google Scholar]
- 184.Sima AA. Diabetic neuropathy in type 1 and type 2 diabetes and the effects of C-peptide. J Neurol Sci. 2004;220:133–136. doi: 10.1016/j.jns.2004.03.014. [DOI] [PubMed] [Google Scholar]
- 185.Sima AA, Zhang W, Grunberger G. Type 1 diabetic neuropathy and C-peptide. Exp Diabesity Res. 2004;5:65–77. doi: 10.1080/15438600490424541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Kamiya H, Zhang W, Sima AA. C-peptide prevents nociceptive sensory neuropathy in type 1 diabetes. Ann Neurol. 2004;56:827–835. doi: 10.1002/ana.20295. [DOI] [PubMed] [Google Scholar]
- 187.Ekberg K, Brismar T, Johansson BL, Jonsson B, Lindstrom P, Wahren J. Amelioration of sensory nerve dysfunction by C-Peptide in patients with type 1 diabetes. Diabetes. 2003;52:536–541. doi: 10.2337/diabetes.52.2.536. [DOI] [PubMed] [Google Scholar]
- 188.Forst T, Kunt T, Pohlmann T, Goitom K, Engelbach M, Beyer J, Pfutzner A. Biological activity of C-peptide on the skin microcirculation in patients with insulin-dependent diabetes mellitus. J Clin Invest. 1998;101:2036–2041. doi: 10.1172/JCI2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Rola R, Szulczyk P. Quantitative differences between kinetic properties of Na(+) currents in postganglionic sympathetic neurones projecting to muscular and cutaneous effectors. Brain Res. 2000;857:327–336. doi: 10.1016/s0006-8993(99)02318-5. [DOI] [PubMed] [Google Scholar]