

Abstract
Pulmonary fibrosis is a common and severe fibrotic lung disease with high morbidity and mortality. Recent studies have reported a large number of unwanted myofibroblasts appearing in pulmonary fibrosis, and shown that the sustained activation of myofibroblasts is essential for unremitting interstitial fibrogenesis. However, the origin of these myofibroblasts remains poorly understood. Here, we create new mouse models of pulmonary fibrosis and identify a previously unknown population of endothelial cell (EC)-like myofibroblasts in normal lung tissue. We show that these EC-like myofibroblasts significantly contribute myofibroblasts to pulmonary fibrosis, which is confirmed by single-cell RNA sequencing of human pulmonary fibrosis. Using the transcriptional profiles, we identified a small molecule that redirects the differentiation of EC-like myofibroblasts and reduces pulmonary fibrosis in our mouse models. Our study reveals the mechanistic underpinnings of the differentiation of EC-like myofibroblasts in pulmonary fibrosis and may provide new strategies for therapeutic interventions.
Summary:
New mouse models of pulmonary fibrosis are created to identify a previously unknown cell population that contributes myofibroblasts to pulmonary fibrosis. A small molecule is identified to redirect myofibroblasts and reduces pulmonary fibrosis.
Introduction
Pulmonary fibrosis is a severe fibrotic lung disease, affecting over three million people worldwide [1–6]. In the progression of pulmonary fibrosis, extensive fibrogenesis occurs in the interstitium of lung tissue where it replaces normal structural components, damages alveolar units and disrupts gas exchanges [1–8]. The survival time of patients with pulmonary fibrosis is quite poor due to the dramatic reduction of pulmonary function [4, 9–11]. Although the mechanism has not yet been determined, the deposition of abnormal extracellular fibrotic matrix is considered to be a key component in the progression of pulmonary fibrosis [12–20]. Recent studies have shown that several types of cells contribute to the stimulus of extracellular matrix deposition, such as alveolar type 2 epithelial cells and alveolar macrophages [21–24]. The studies reveal that these cells secrete a number of growth factors and cytokines that activate myofibroblasts, which produce aberrant compositions of extracellular fibrotic matrix leading to pulmonary fibrosis [3].
Myofibroblasts were initially discovered in the granulation tissue during tissue repair [25, 26]. Advanced studies have observed that myofibroblasts contain the features of both fibroblasts and smooth muscle cells, and play a critical role in quickly responding and producing the collagens needed to repair a wound site [27, 28]. Beyond normal tissue repair, overloaded myofibroblasts are found in almost all fibrotic organs, including the liver [29], kidneys [30], heart [31], and lungs [1–6], where myofibroblasts persistently produce and accumulate unwanted fibrotic components to form scar tissue [16]. In pulmonary fibrosis, excessively activated myofibroblasts have been shown to generate deleterious extracellular matrix that are dominated by fibrillar collagens, fibronectin and other fibrotic proteomes [1–6, 32, 33]. Although studies suggest that several cell lineages may be involved in the differentiation of myofibroblasts, the origin of myofibroblasts remains unclear.
Matrix Gla Protein (MGP) is a bone morphogenic protein (BMP) antagonist and highly expressed in pulmonary cells [34–38]. MGP is essential for endothelial-epithelial interactions to direct pulmonary specification. Conventional deletion of the Mgp gene in mice causes an imbalance between the pulmonary vasculature and the airways, leading to vascular malformations and underdeveloped lungs [36, 38]. Mutations in the human Mgp gene cause Keutel syndrome, which is characterized by peripheral pulmonary stenoses and other developmental defects [39]. With warfarin treatment that renders MGP non-functional by interfering with γ-carboxylation, pulmonary fibrosis deteriorates sharply [40–42]. MGP has also been found to support normal endothelial differentiation in progenitor cells and prevent ECs from unwanted differentiation [43–45].
In this study, we reveal that cell-specific deletion of Mgp gene causes severe pulmonary fibrosis. We identify previously unknown endogenous EC-like myofibroblasts, which can differentiate into myofibroblasts in normal lungs, and we show that EC-like myofibroblasts significantly contribute myofibroblasts to pulmonary fibrosis. We find that MGP binds to a unique member of the BMP family, BMP-1, to inhibit its activity in the differentiation of EC-like myofibroblasts. We also identify a small molecule that redirects the differentiation of EC-like myofibroblasts and reduces pulmonary fibrosis.
Material and methods
Animals
VE-cadherincre (B6.FVB-Tg(Cdh5-cre)7Mlia/J), SM22acre (B6.129S6-Taglntm2(cre)Yec/J) and RosatdTomato (B6;129S6-Gt(ROSA)26Sortm9(CAG-tdTomato)Hze/J) mice on C57BL/6J background were purchased from the Jackson Laboratory. VE-cadherincre/ERT2 mice were obtained as gifts from Dr. Ralf Adams. Genotypes were confirmed by PCR 12, and experiments were performed with generation F4-F6. Littermates were used as wild type controls. All mice were fed a standard chow diet (Diet 8604, Harlan Teklad Laboratory). Bleomycin (Cat# 1076308) and UK383367 (Cat# PZ0156) were obtained from MilliporeSigma. The studies were reviewed and approved by the Institutional Review Board and conducted in accordance with the animal care guidelines set by the University of California, Los Angeles (UCLA). The investigation conformed to the National Research Council, Guide for the Care and Use of Laboratory Animals, Eighth Edition (Washington, DC: The National Academies Press, 2011).
Tissue culture
The tdTomato-VE−cadherin+, tdTomatoVE+cadherin+ and tdTomatoVE+cadherin− cells were isolated by FACS and cultured as previously described [46]. Berbamine (Sigma-Aldrich, 547190), TGFβ1 (R&D system, 7666-MB) and BMP-1 (R&D system, RDC2450) treatments were performed as described in the main text. Lentiviral vectors containing CMV-ALK5, CMV-FoxA2, MGP or FoxA2 siRNA were all purchased from GeneCopeia™ and applied to the cells as per the manufacturer’s protocols.
Vector construction
The Flag-hMGP vector was constructed as previously described [46]. Briefly, to construct the N-terminally FLAG-tagged human MGP (hMGP) vector, a fragment containing the coding region for hMGP was amplified by PCR. The FLAG tag was placed in the N-terminus of the secreted, mature protein by subcloning a synthesized FLAG-coding DNA fragment between the coding regions of the signal peptide and the mature protein. The FLAG-containing hMGP DNA fragment was amplified by PCR and subcloned into the NheI and XhoI sites of pcDNA3.1(+) (Invitrogen) using restriction sites in the primers.
Pulmonary function test
Mouse pulmonary function tests were performed as previously described [35]. Mice were weighed and placed into a single chamber with a volume of 0.8 L where they were allowed to move freely and acclimate for at least 15 minutes. To provide a baseline reading, a room air-reading was taken as follows: compressed air was supplied to the chamber at a flow rate of 1 L/min for 45 minutes. At this point, the chamber was completely sealed, with air flow momentarily stopped. The changes in pressure caused by breathing were recorded and amplified by the software. Subsequently, the mice were allowed to rest for at least 5 minutes or until the breathing returned to baseline before conducting the hypercapnia phase. In the hypercapnia phase, a gas mixture containing 7% CO2, 21% O2 and balanced N2 was supplied to the chamber at a flow of 1 L/min. After 5 minutes, the chamber was sealed and ventilatory patterns recorded. During the breathing room-air and hypercapnia phase, the average tidal volume (TV) and respiratory rate (RR) were measured for a period of at least 10 consecutive breaths. To avoid an excessive build-up in CO2 concentration within the chamber due to re-breathing, a Bias Flow Regulator (Buxco Electronics, Inc.) was used. The Bias Flow Regulator provided a quiet, constant, and smooth flow through the animal chamber that prevented CO2 build-up. The following parameters were recorded: respiratory rate (RT), tidal volume (TV), peak inspiration flow (PIF), peak expiratory flow (PEF), and minute ventilation. The results were calculated and corrected for body weight. Average values were calculated one per minute for each serial 10 minutes. The machine was sanitized with alcohol between uses.
RNA analysis
Real-time PCR analysis was performed as previously described [35]. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as a control gene [35]. Primers and probes for mouse MGP, Col3a1, Fibronectin 1, VE-cadherin, Flk1, ALK5, FoxA2 and von Willebrand factor (vWF) were obtained from Applied Biosystems as part of Taqman® Gene Expression Assays.
Enzyme-linked immunosorbent assay (ELISA)
ELISA was performed as previously described [35]. ELISA kits were obtained from Abcam (ab119557 and ab222942). The assays were performed in accordance with the manufacturer’s protocols.
Single cell RNA-sequencing (scRNA-seq)
10x Library Preparation, Sequencing, and Alignment: scRNA-seq library was generated with the Chromium Single Cell 3′ v3 assay (10x Genomics). The library was sequenced using Illumina NextSeq 550 platform with a depth of 378 million reads. Raw reads were aligned to the mouse genome (mm10). The cellranger (v3.0.0) mkfastq function was used to generate FASTQ files and cells and gene counts were called using the cellranger count function.
Cell Clustering and Cell Type Annotation: The R package Seurat version 4.0.1 was used for cell clustering and differential gene expression analysis. Cells were first filtered to have more than 500 detected genes and less than 10% of mitochondrial genes. Natural-log transformation was then applied to the gene counts and data were scaled to regress out total number of RNA counts and the percentage of mitochondrial reads. The FindVariableFeatures function was used to select variable genes with default parameters and principal component analysis (PCA) was performed using the RunPCA function on the variable genes. The first 25 PCs were used for cell clustering with a resolution of 0.5. Uniform manifold approximation and projection (UMAP) dimensional reduction was applied using the RunUMAP function. The FindAllMarkers function was used to identify marker genes for each cluster. Cell identities were defined by known marker genes. The FindMarkers function was performed for differential gene expression analysis between two cell types using Wilcoxon Rank Sum test with at least log-fold difference of 0.25 between the two groups of cells.
Pseudotime Trajectory Construction: R package Monocle (v2.18.0) was used for pseudotime trajectory construction. Genes with average expression greater than 0.5 were retained for trajectory analysis in a subset of data. Variable genes were adopted from variable features identified by the Seurat FindVariableFeatures function, then were used as ordering genes. The trajectory was constructed by the reduceDimension function with default parameters. Differential expression in pseudotime was carried out by the differentialGeneTest function using likelihood ratio tests.
CMAP and compound identification
From each cell type in scRNA-seq analysis, we selected the top 100 marker genes. Connectivity Map (CMAP) from the Broad Institute is a database with a collection of gene expression profiles that were obtained from nine human cell lines treated with various small compounds. To identify a small molecule from 2429 compounds in the LINCS database (L1000), we used the next-generation CMap (CLUE) platform to directly explore the connectivity of gene signatures among perturbagens with Touchstone tool. The drug similarity was ranked according to the CMap connectivity score (from −100 to 100). Connectivity scores above 95 or below −95 were considered as strong scores to predict small candidate molecule corresponding to the query of selected gene expression signatures between different cell types.
Fluorescence-activated cell sorting (FACS)
FACS analysis was performed as previously described [35]. The cells were stained with fluorescein isothiocyanate (FITC)-, phycoerythrin (PE)-, or Alexa Fluor 488 (AF-488)-conjugated antibodies against CD34 and VE-cadherin (all from BD Biosciences, 553731 and 562243). Nonspecific fluorochrome- and isotype-matched IgGs (BD Pharmingen) served as controls.
Immunoblotting and immunofluorescence
Immunoblotting was performed as previously described [44]. Equal amounts of tissue lysates were used for immunoblotting. Blots were incubated with specific antibodies to BMP-1 and LTBP-1 (Abcam, ab205394 and ab78294), Flag (Sigma-Aldrich, F3165), ALK5 (R&D system, MAB5871), Col3a1 and Fibronectin 1 (Novus Biologicals, NB600 and NBP1–91258). β-Actin (Sigma-Aldrich, A2228) was used as a loading control. Immunofluorescence was performed as previously described in detail [44]. We used specific antibodies to CD34 (BD Bioscience, 553731 or Abcam, ab54208), Nkx2.1 (Abcam, ab76013) and VE-cadherin (BD biosciences, 562243). The nuclei were stained with 4’,6-diamidino-2-phenylindole (DAPI, Sigma-Aldrich, D9564).
Masson’s trichrome (MT) staining
Sections were deparaffinized, rehydrated and stained with Trichrome Stain Kit (Abcam, ab150686) as per the manufacturer’s protocol. After being washed in distilled water, the sections were dehydrated and cleared in xylene, then mounted with resinous mounting medium.
Statistical analysis
The analyses were performed using GraphPad Instat®, version 3.0 (GraphPad Software). Data was analyzed by either unpaired 2-tailed Student’s t test or one-way ANOVA with Tukey’s multiple-comparisons test for statistical significance.
Results
Specific MGP deletion causes pulmonary fibrosis
We have shown that a conventional gene deletion of Mgp disrupted pulmonary cell differentiation [35, 38]. To perform cell-specific deletions of Mgp, we generated Mgpflox/flox mice, in which the genomic region of exon 2 to 4 of Mgp gene was floxed by two loxP sites (Supplemental Figure S1). We bred Mgpflox/flox mice with VE-cadherincre or Smooth muscle 22αcre (SM22αcre) mice. At 10 weeks of age, Masson’s trichrome staining showed striking pulmonary fibrosis in both VE-cadherincreMgpflox/flox mice and SM22αcreMgpflox/flox mice, where quantification of collagen in lung tissues confirmed more collagen accumulated (Figure 1a–b). At one year of age, both mice had severe pulmonary fibrosis (Figure 1a–b). We examined the pulmonary function of the mice at one year of age using unrestricted whole body barometric plethysmography. The results showed that the respiratory rates of VE-cadherincreMgpflox/flox mice and SM22αcreMgpflox/flox mice were significantly higher than that of Mgpflox/flox control mice during room air breathing and hypercapnia phase (Figure 1c). VE-cadherincreMgpflox/flox and SM22αcreMgpflox/flox mice also displayed a significant decrease in tidal volume and lower peaks of expiratory and inspiratory flow (Figure 1c). Bleomycin-injected wild type mice and Mgpflox/flox mice were used as controls [47]. The results suggested that progressive pulmonary fibrosis occurred in both VE-cadherincreMgpflox/flox mice and SM22αcreMgpflox/flox mice.
Figure 1. Pulmonary fibrosis in VE-cadherincreMgpflox/flox and Sm22αcreMgpflox/flox mice.
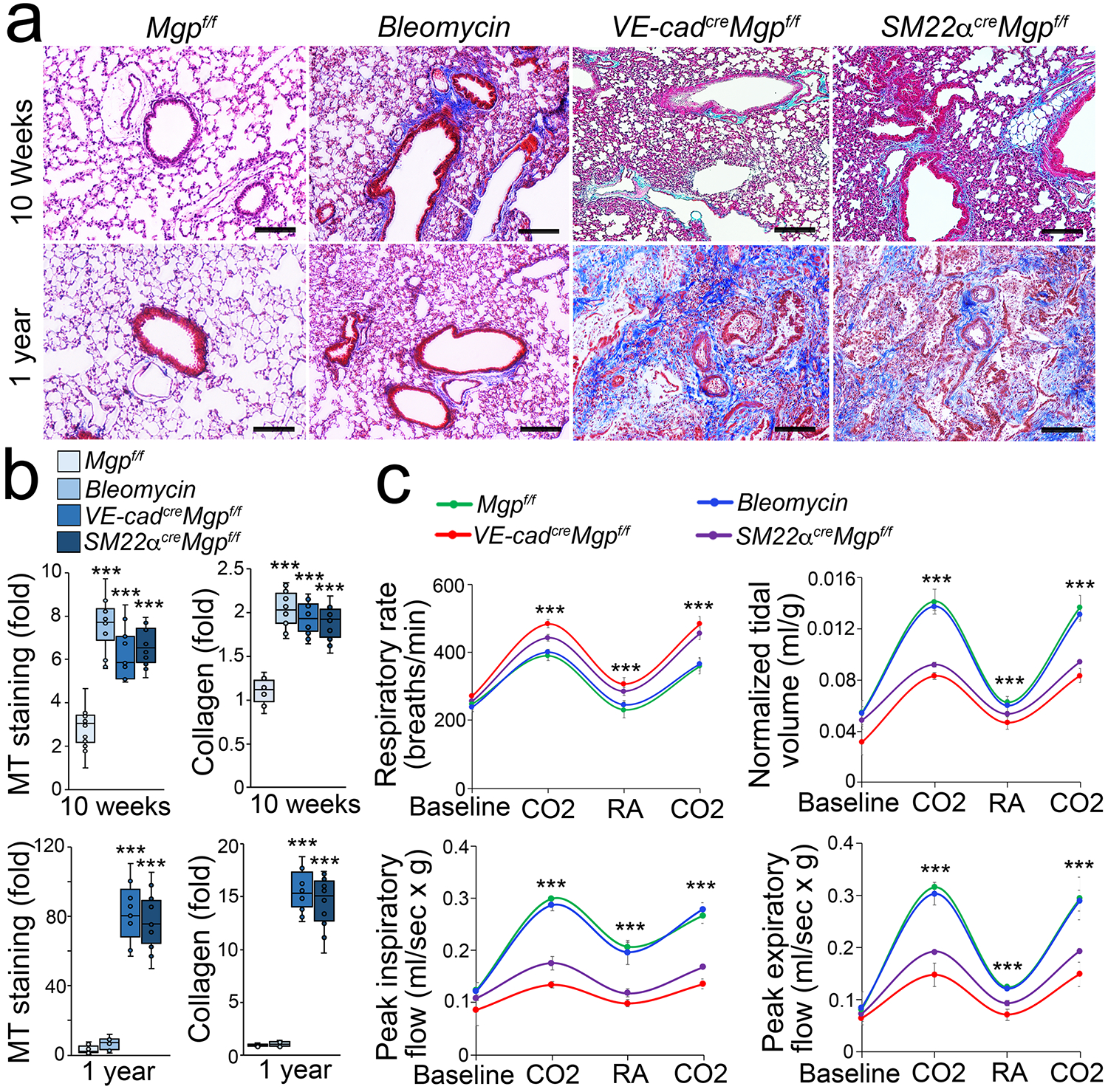
a, Masson’s trichrome (MT) staining of pulmonary tissues (n=10). f/f, flox/flox. Bleomycin, wild type mice injected with bleomycin. VE-cad, VE-cadherin. Scale bar, 100 μm.
b, Quantification of MT staining and collagen accumulation in pulmonary tissues (n=9).
c, Mouse pulmonary tests (n=6). CO2, hypercapnia phase with 7% CO2, 21% O2, and balanced N2. RA, room air.
b and c were analyzed for statistical significance by ANOVA with post hoc Tukey’s test. The bounds of the boxes are upper and lower quartiles with data points. The line in the box is the median. Error bars are maximal and minimal values (b). Error bars are mean ± standard deviation (SD)(c). ***, P<0.0001.
Endogenous EC-like myofibroblasts in normal lungs
Interestingly, when we examined the expression of endothelial markers, immunostaining and FACS both showed a robustly increased CD34+ cell population in the lungs of VE-cadherincreMgpflox/flox mice and SM22αcreMgpflox/flox mice (Figure 2a). We verified this result using different anti-CD34 antibodies (Supplemental Figure S2). We also showed that the increase of CD34+ cell population in the lungs of VE-cadherincreMgpflox/flox mice and SM22αcreMgpflox/flox mice persisted at one year of age (Supplemental Figure S2). Immunostaining also showed an increase of the CD34+ cell population in human pulmonary fibrosis (Figure 2b). We used Nkx2.1 staining as control and showed no overlap between CD34+ cells and pulmonary epithelial cells.
Figure 2. Enlarged CD34+ cell population in pulmonary fibrosis.
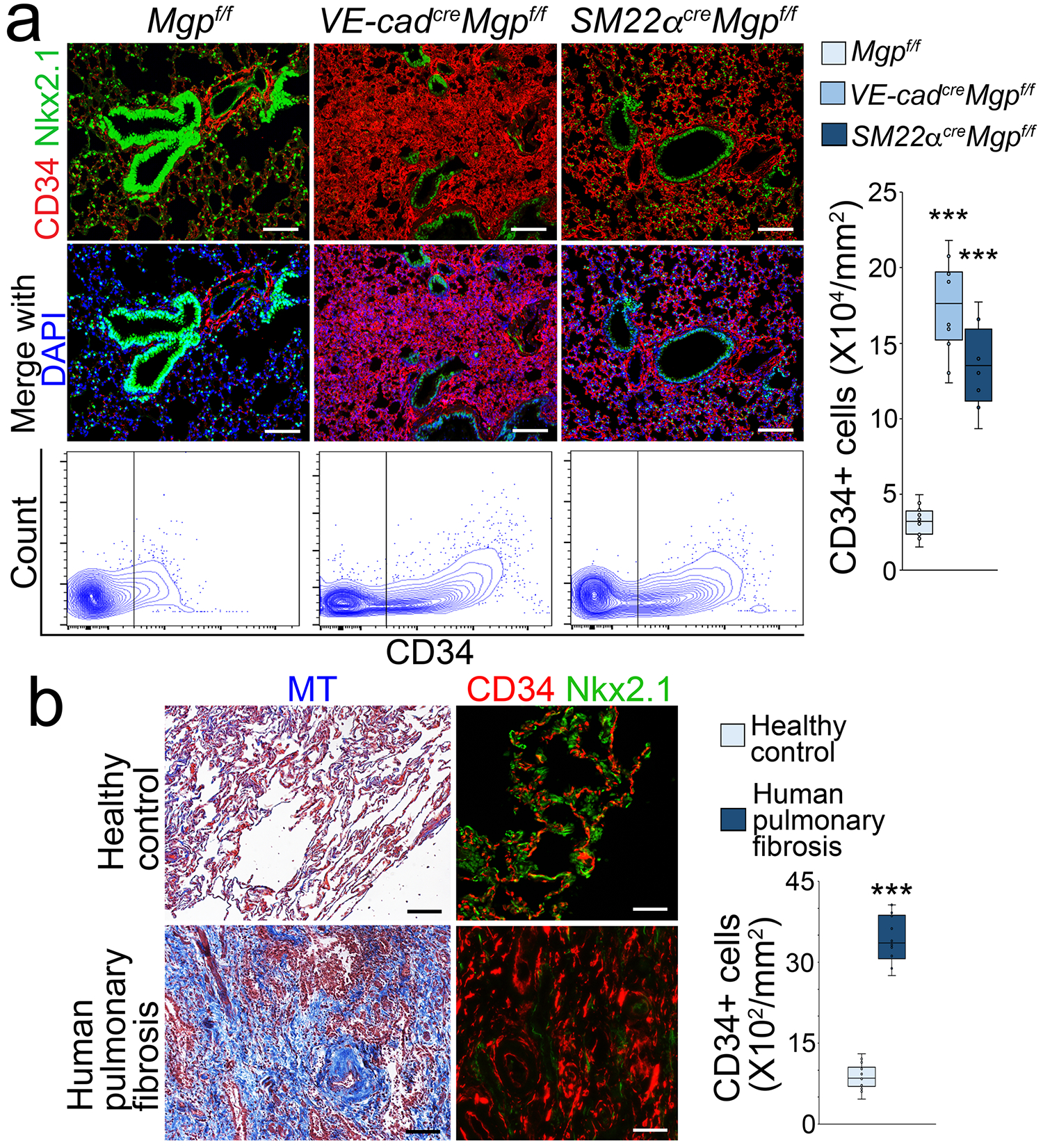
a, Immunostaining with quantification of CD34+ cells and FACS in pulmonary tissues of VE-cadherincreMgpflox/flox and Sm22αcreMgpflox/flox mice (n=8). Mgpflox/flox was used as control. Scale bar, 100 μm.
b, MT staining and immunostaining of human tissues with pulmonary fibrosis (left), and quantification of CD34+ cells in human pulmonary fibrosis (right) (n=6). Control, healthy lungs. Scale bar, 100 μm. The data was analyzed for statistical significance by unpaired 2-tailed Student’s t test. The bounds of the boxes are upper and lower quartiles with data points. The line in the box is the median. Error bars are maximal and minimal values. ***, P<0.0001.
We isolated the lungs of wild type mice and used FACS to select pulmonary CD34+ population, in which immune cells were excluded. We examined the transcriptional profile of CD34+CD45− cells at single-cell resolution. The single-cell RNA sequencing (scRNA-seq) uncovered specific cell clusters in the CD34+CD45− population (Figure 3a). Differential gene expression divided these clusters into three major types of cells: 1) ECs that only expressed endothelial markers; 2) EC-like myofibroblasts that expressed endothelial and myofibroblast markers, and MGP, SM22α, Snai1, Snai2, Zeb1, Zeb2; 3) myofibroblasts that expressed myofibroblast markers and Twist1 and 2 (Figure 3a). Cell differentiation trajectory projected a clear differential path from ECs to EC-like myofibroblasts ending with myofibroblasts (Figure 3b). Interestingly, none of the clusters expressed the typical smooth muscle cell marker Smooth Muscle Myosin Heavy Chain (SMMHC), ruling out the involvement of smooth muscle cells (Figure 3a). We examined wild-type mouse lungs and healthy human lungs. Immunostaining showed the co-localization of VE-cadherin and PDGFRα around the vessels and small airways in the mouse lungs (Figure 3c). The co-localization was also identified in the structures of alveolar units in healthy human lungs (Figure 3c). The results suggested that the endogenous EC-like myofibroblasts in normal lungs contribute to myofibroblasts.
Figure 3. Endogenous population of EC-like myofibroblasts in wild type pulmonary tissues.
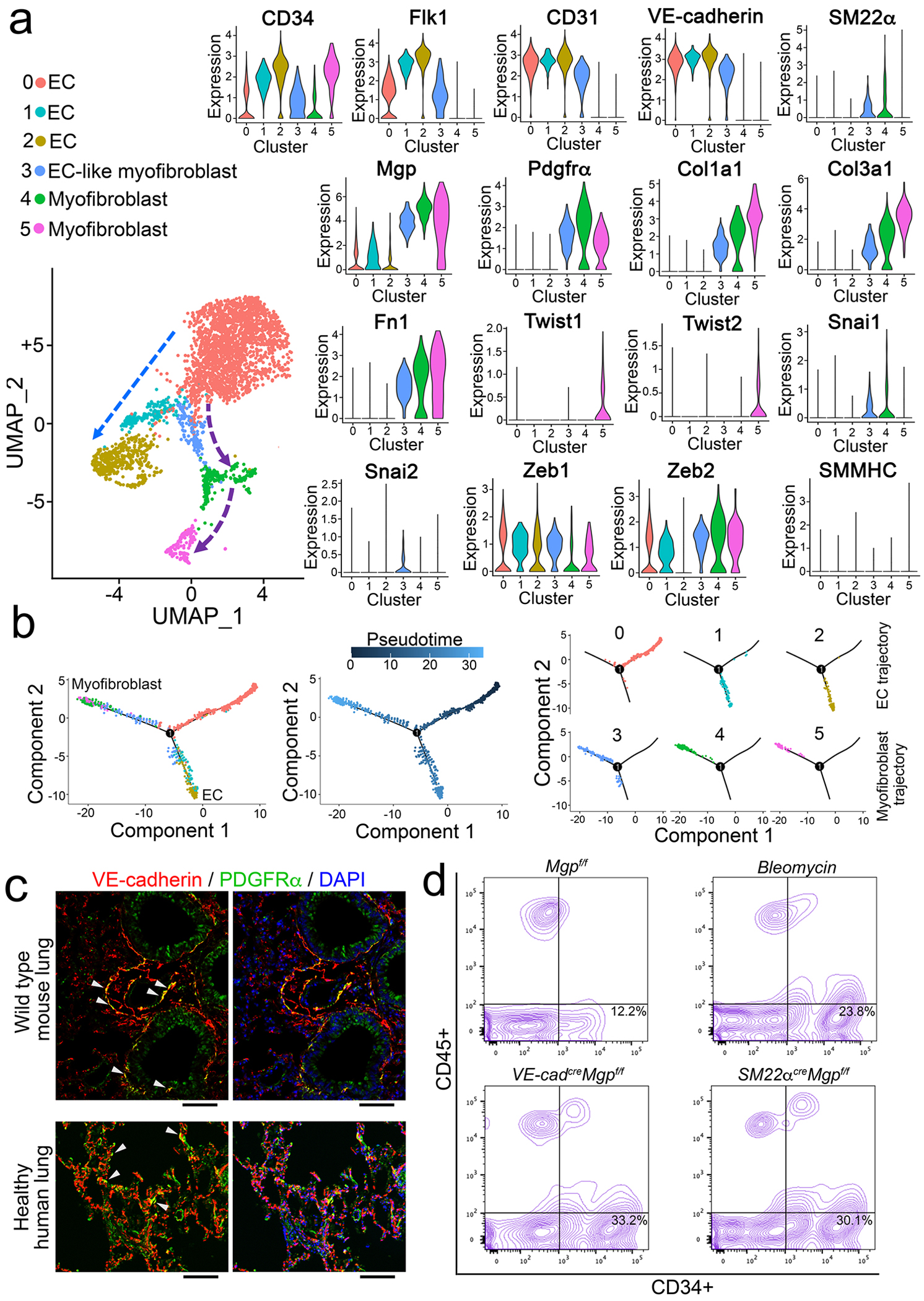
a, UMAP for the cell populations subclustered from the whole population of CD34+CD45− pulmonary cells and violin plots of the gene expression of the lineage markers in the cell clusters. Fn1, fibronectin 1.
b, Pseudotemporal trajectories of the cell clusters.
c, Immunostaining of the lungs of wild type mice and healthy human (n=3). Scale bar, 100 μm.
d, FACS of whole lung cells isolated from bleomycin-injected mice, VE-cadherincreMgpflox/flox mice and Sm22αcreMgpflox/flox mice (n=6).
We examined CD34+CD45− cells in the lungs of VE-cadherincreMgpflox/flox, SM22αcreMgpflox/flox and bleomycin-injected wild type mice. FACS showed an increased number of CD34+CD45− cells in all three models (Figure 3d), suggesting a similar induction of CD34+CD45− cells in mouse models of pulmonary fibrosis.
EC-like myofibroblasts contribute to human pulmonary fibrosis
To determine the role of EC-like myofibroblasts in pulmonary fibrosis, we re-analyzed publicly available scRNA-seq data of 10 healthy human lungs and 20 patients with pulmonary fibrosis [15, 32]. The analysis identified the cluster of EC-like myofibroblasts that co-expressed endothelial and myofibroblast markers, MGP, and SM22α in healthy lungs and pulmonary fibrosis (Figure 4a–b). Cell differential trajectory showed that EC-like myofibroblasts were potentially derived from ECs and differentiated into myofibroblasts (Figure 4a–c). Strikingly, differential expression revealed lower expression of the endothelial markers VE-cadherin and von Willebrand factor (vWF) but higher expression of the myofibroblast markers α-smooth muscle actin, Col3a1 and fibronectin 1 (Fn1) in EC-like myofibroblasts of pulmonary fibrosis samples compared to those of healthy control samples (Figure 4b). A decrease of EC differentiation together with an increase of EC-like myofibroblasts and myofibroblasts was also detected in pulmonary fibrosis (Figure 4d). The results suggested that EC-like myofibroblasts contributed myofibroblasts to human pulmonary fibrosis.
Figure 4. EC-like myofibroblasts are present in healthy human lungs and contribute to human pulmonary fibrosis.
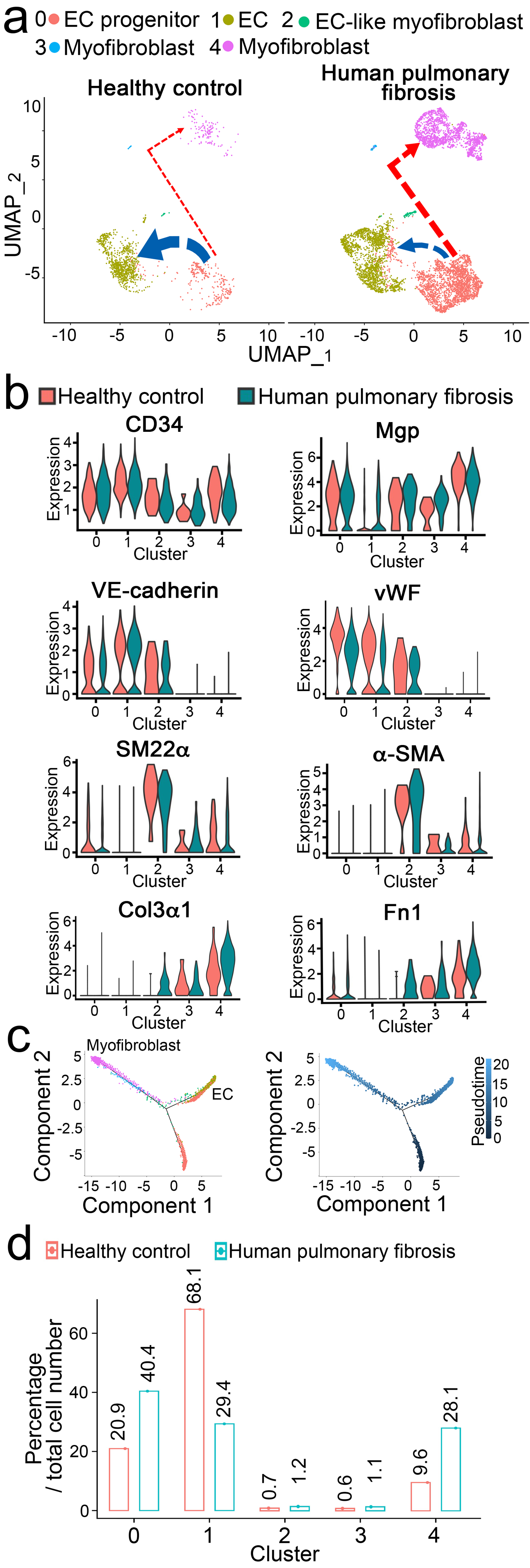
a, UMAP for cell populations subclustered from the whole population of CD34+CD45− pulmonary cells of healthy human lungs and human pulmonary fibrosis.
b, Violin plots of the gene expression of the lineage markers. Fn1, fibronectin 1.
c, Pseudotemporal trajectories of the cell clusters.
d, Alterations in cell compositions of different populations in healthy human lungs and human pulmonary fibrosis.
Cell lineage tracings reveal EC-like myofibroblasts contributing to pulmonary fibrosis
To further determine if endogenous ECs and EC-like myofibroblasts contributed to pulmonary fibrosis, we preformed cell lineage tracings in the mouse model of bleomycin-induced pulmonary fibrosis. We used VE-cadherincre/ERT2RosatdTomato mice where founder cells expressing VE-cadherin could be traced by detecting tdTomato after tamoxifen injections. We also used SM22αcreRosatdTomato mice, in which we could trace the tdTomato-labeled cells that reflected the SM22α expression. At 6 weeks of age, VE-cadherincre/ERT2RosatdTomato mice received tamoxifen injections (75 μg/g, daily) for 5 consecutive days to induce tdTomato expression. At 8 weeks of age, tamoxifen-treated VE-cadherincre/ERT2RosatdTomato mice and SM22αcreRosatdTomato mice received bleomycin injections as previously described [47] to induce pulmonary fibrosis. Saline-treated mice were used as controls. At 11 weeks of age, we examined the lung tissues. Masson’s trichrome staining showed pulmonary fibrosis in both tamoxifen-treated VE-cadherincre/ERT2RosatdTomato mice and SM22αcreRosatdTomato mice after bleomycin injections (Figure 5a–b), confirming the induction of pulmonary fibrosis. In tamoxifen-treated VE-cadherincre/ERT2RosatdTomato mice, immunostaining showed more tdTomato-positive cells expressing higher levels of PDGFRα and Fn1 in the bleomycin-treated group than the controls, suggesting ECs and EC-like myofibroblasts that expressed VE-cadherin contributed to bleomycin-induced pulmonary fibrosis (Figure 5a). In SM22αcreRosatdTomato mice, more tomato-positive cells were identified to express VE-cadherin and a higher level of Fn1 in the bleomycin-treated group than the controls, suggesting again that EC-like myofibroblasts contributed to the bleomycin-induced pulmonary fibrosis (Figure 5b).
Figure 5. Cell lineage tracings reveal EC-like myofibroblasts contributing to pulmonary fibrosis.
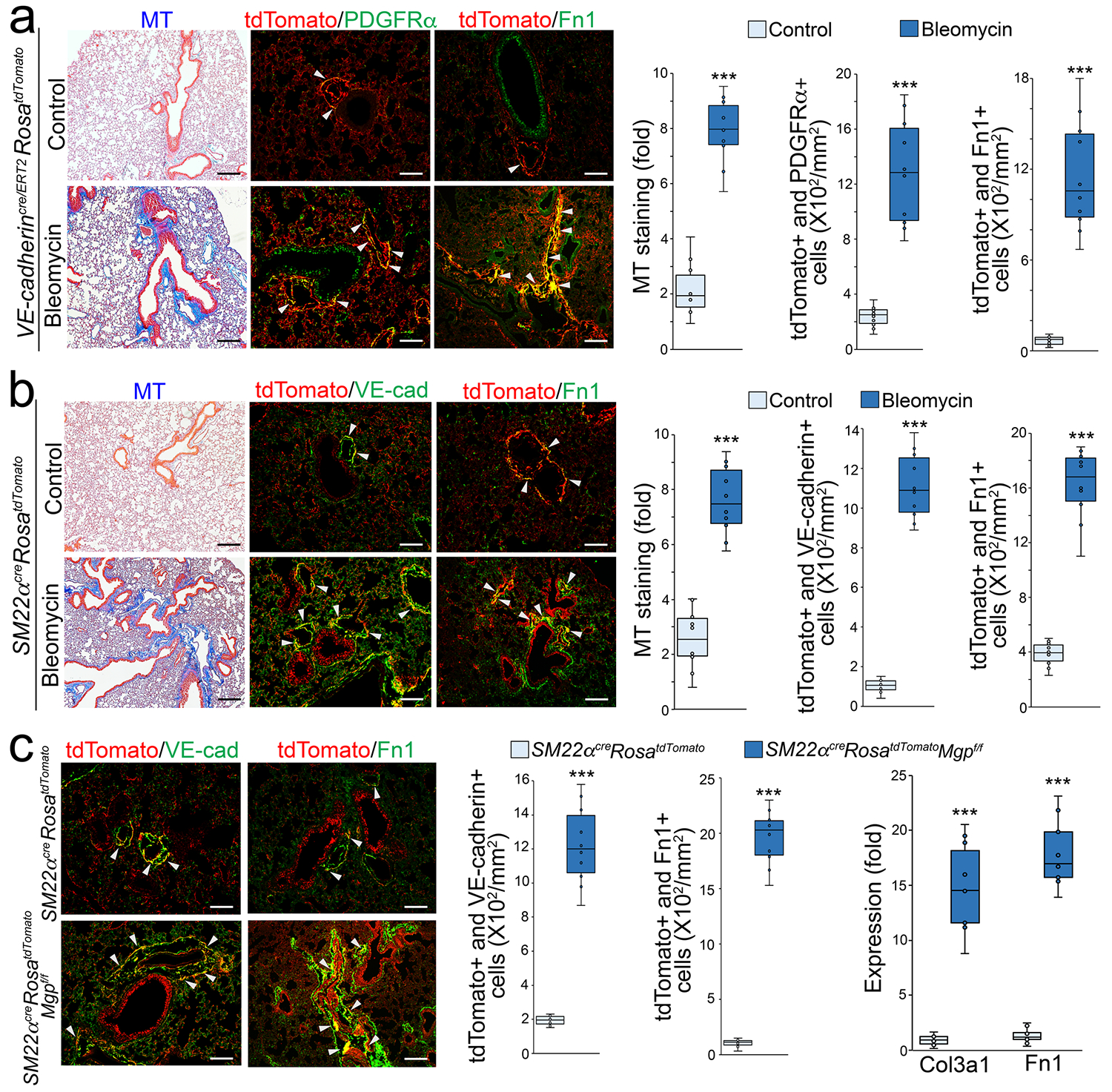
a, Masson’s trichrome (MT) staining and immunostaining with cell quantification of the lungs of tamoxifen-injected VE-cadherincre/ERT2RosatdTomato mice after administration of bleomycin (n=10).
b, Masson’s trichrome (MT) staining and immunostaining with cell quantification of the lungs of Sm22acreRosatdTomato mice after administration of bleomycin (n=10).
c, Immunostaining with cell quantification of the lungs of Sm22acreRosatdTomato and Sm22acreRosatdTomatoMgpFlox/Flox mice. The expression of Col3a1 and Fn1 in tdTomato and VE-cadherin double positive cells isolated from lungs of Sm22acreRosatdTomato and Sm22acreRosatdTomatoMgpFlox/Flox mice (n=10).
The data were analyzed statistical significance by unpaired 2-tailed Student’s t test. The bounds of the boxes are upper and lower quartiles with data points. The line in the box is median. Error bars are maximal and minimal values. Scale bar, 100 μm. ***, P<0.0001.
We bred and examined SM22αcreRosatdTomatoMgpflox/flox mice. At 11 weeks of age, we found that more tomato-positive cells expressed VE-cadherin and a higher level of Fn1 than that in SM22αcreRosatdTomato mice (Figure 5c). We isolated the tdTomato and VE-cadherin double positive cells from the lungs. Real-time PCR showed higher expression of Col3a1 and Fn1 in the cells isolated from SM22αcreRosatdTomatoMgpflox/flox mice than from SM22αcreRosatdTomato mice (Figure 5c). Together, multiple cell lineage tracings in the mouse models revealed that EC-like myofibroblasts contributed to pulmonary fibrosis.
MGP controls EC-like myofibroblast differentiation by regulating BMP-1 activity and TGFβ1 maturation
To explore the differentiation of EC-like myofibroblasts in lungs, we isolated tdTomato and VE-cadherin double positive cells (tdTomato+VE-cadherin+), tdTomato negative and VE-cadherin positive cells (tdTomato-VE-cadherin+), and tdTomato positive and VE-cadherin negative cells (tdTomato+VE-cadherin−) from the lungs of SM22αcreRosatdTomato mice. We characterized the gene expression of these cells. Real-time PCR showed the expression of MGP, VE-cadherin, SM22α, Col3a1 and Fn1 in tdTomato+VE-cadherin+ cells. TdTomato-VE-cadherin+ cells expressed VE-cadherin with low level of MGP but not SM22α, Col3a1 and Fn1. TdTomato+VE-cadherin− expressed MGP, SM22α, Col3a1 and Fn1 and high level of SMMHC (Figure 6a–b). The results suggested that tdTomato+VE-cadherin+ cells were the endogenous EC-like myofibroblasts, tdTomato-VE-cadherin+ cells were the pulmonary ECs and tdTomato+VE-cadherin− cells could be a mixture of myofibroblasts and smooth muscle cells. We then depleted MGP in these cells using specific siRNA (Figure 6c), treated the cells with TGFβ1 (1 ng/ml), and examined the myofibroblast markers Col3a1 and Fn1 and endothelial markers VE-cadherin and vWF. Real-time PCR showed the induction of myofibroblast markers with the reduction of endothelial markers in MGP-depleted or TGFβ1-treated tdTomato+VE-cadherin+ cells. The combination of MGP-depletion and excess TGFβ1 significantly magnified the changes in the expression of these markers (Figure 6c). The results suggested that lack of MGP or excess TGFβ1 promoted the differentiation of EC-like myofibroblasts toward myofibroblasts. Real-time PCR also showed the induction of myofibroblast markers in MGP-depleted or TGFβ1-treated tdTomato+VE-cadherin− cells and the combination of MGP-depletion and excess TGFβ1 enhanced the induction of these markers (Figure 6c), suggesting that lack of MGP or excess TGFβ1 further drove the myofibroblasts towards fibrogenesis. Real-time PCR showed no significant alterations in myofibroblast markers in tdTomato-VE-cadherin+ cells. Interestingly, the data showed that TGFβ1 reduced endothelial markers in this cells and MGP-depletion further reduced the expression of endothelial markers (Figure 6c), suggesting that MGP-depletion and excess TGFβ1 caused ECs to lose their identity but gain potential towards EC-like myofibroblasts.
Figure 6. TGFβ1 drives EC-like myofibroblasts towards myofibroblast differentiation.
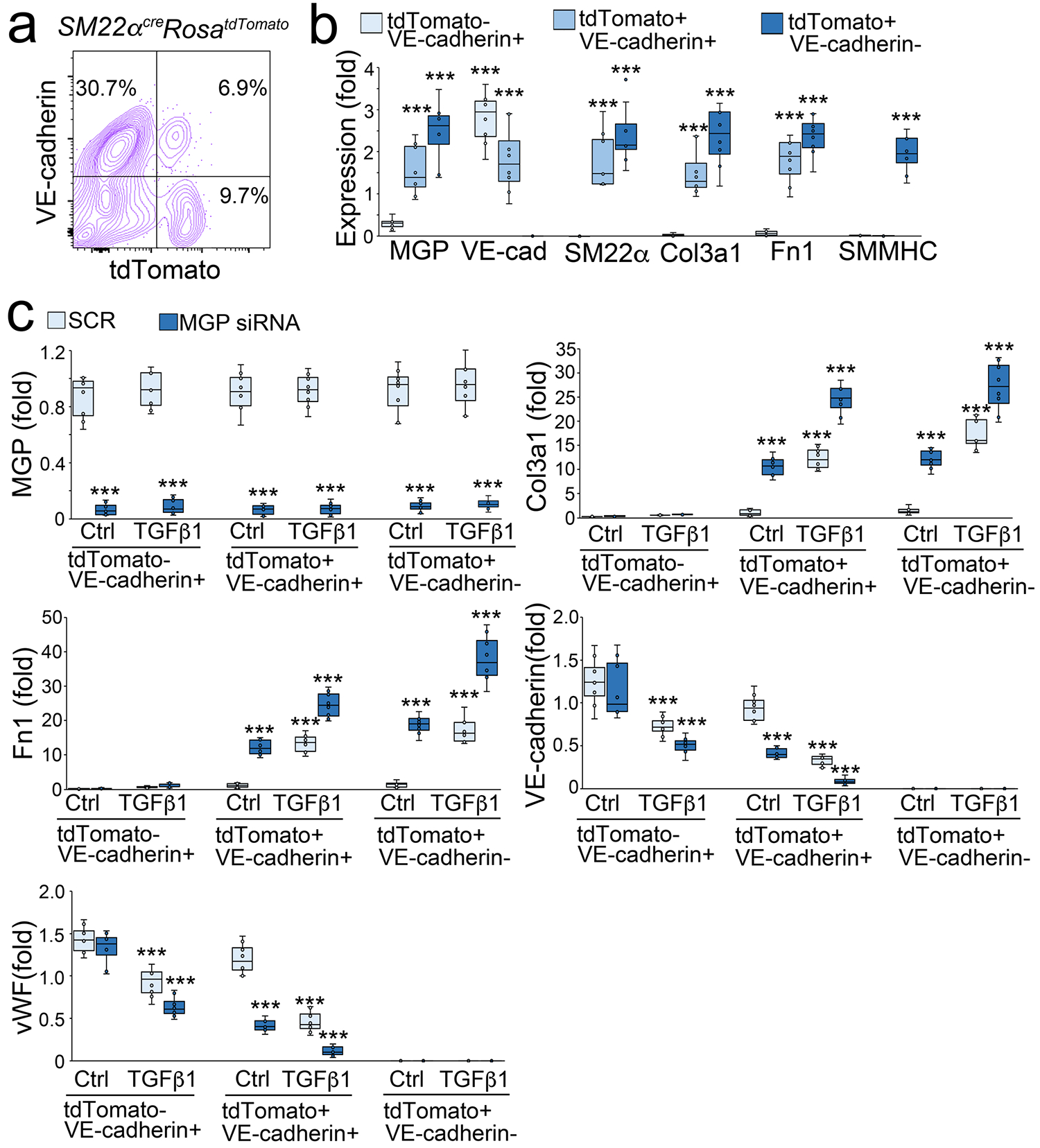
a, FACS of whole lung cells of Sm22acreRosatdTomato mice.
b, Gene expression of tdTomato negative and VE-cadherin positive (tdTomato-VE-cadherin+), tdTomato positive and VE-cadherin positive (tdTomato+VE-cadherin+), and tdTomato positive and VE-cadherin negative (tdTomato+VE-cadherin−) cells isolated from lungs of Sm22acreRosatdTomato mice (n=6).
c, Gene expression of tdTomato-VE-cadherin+, tdTomato+VE-cadherin+, and tdTomato+VE-cadherin− cells after MGP deletion with siRNA and treated with or without TGFβ1 (1 ng/ml) (n=6).
Data were analyzed for statistical significance by ANOVA with post hoc Tukey’s test. The bounds of the boxes are upper and lower quartiles with data points. The line in the box is median. Error bars are maximal and minimal values. ***, P<0.0001.
BMP-1 is known to play a role in the maturation of TGFβ1 [48]. Therefore, we isolated tdTomato+VE-cadherin+ cells from the lungs of SM22αcreRosatdTomato mice and treated the cells with BMP-1 in combination with over-expression of FLAG-tagged MGP. Co-immunoprecipitation followed by immunoblotting uncovered an interaction between BMP-1 and MGP (Figure 7a). Chemical crosslinking of BMP-1 and FLAG-tagged MGP resulted in the formation of a complex detected by immunoblotting with both anti-FLAG and anti-BMP-1 antibodies (Figure 7b), strongly supporting an interaction between BMP-1 and MGP. We used BMP-1 to treat tdTomato+VE-cadherin+ cells with or without MGP deletion. The results showed that excess BMP-1 induced Col3a1 and Fn1. With MGP depletion, BMP-1 treatment further induced Col3a1 and Fn1 (Figure 7c), suggesting that MGP interfered with the activity of BMP-1.
Figure 7. MGP inhibits BMP-1, which regulates maturation of TGFβ1 and in turn controls the differentiation of EC-like myofibroblasts.
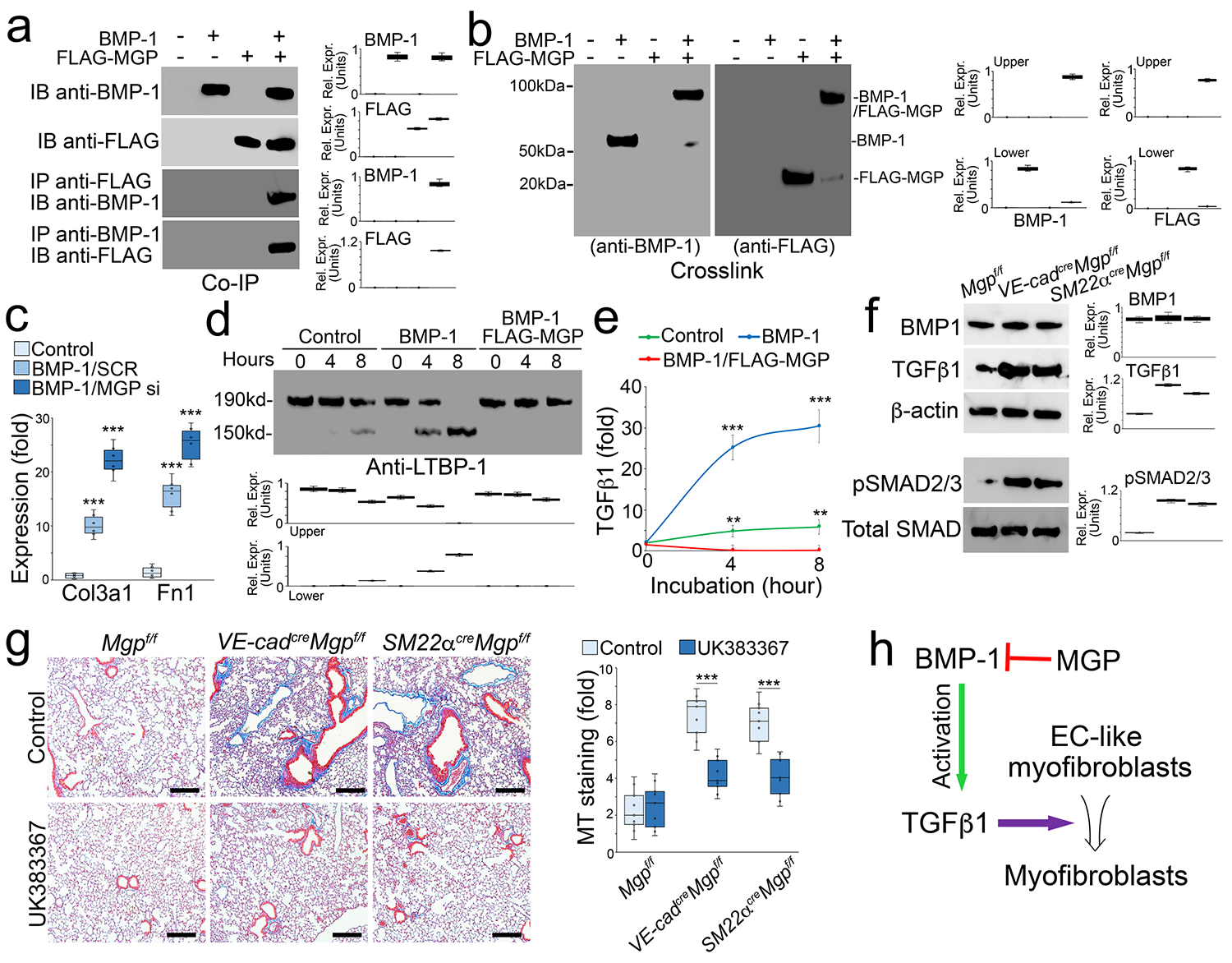
a, Binding of BMP-1 to MGP shown by immunoprecipitation. BMP-1 (200 ng) and conditioned medium containing N-FLAG-MGP (100 μl; approximately 200 ng) were combined as indicated in the top panel, and the presence of the respective protein was confirmed by immunoblotting (IB) (top 2 blots). Interactions between the proteins were analyzed by immunoprecipitation (IP) followed by immunoblotting with antibodies as indicated and quantified by densitometry.
b, Binding of BMP-1 to MGP shown by chemical crosslinking. BMP-1 (200 ng) and conditioned medium containing N-FLAG-MGP (100 μl; approximately 200 ng) were combined as indicated in lane 2 and 3. Interactions were analyzed using chemical crosslinking followed by immunoblotting with antibodies as indicated and quantified by densitometry.
c, Expression of Col3a1 and Fn1 in tdTomato+VE-cadherin+ cells isolated from the lungs of SM22acreRosatdTomato mice and treated with BMP-1 (200 ng/ml) with transfection of MGP siRNA or SCR (n=6).
d, Time-course immunoblotting with densitometry of LTBP-1 in tdTomatoVE-cadherin+ cell lysates after treatment with BMP-1 (200 ng/ml) with or without conditioned medium containing N-FLAG-MGP (n=6).
e, Levels of TGFβ1 in culture media of tdTomato+VE-cadherin+ cell after treatment with BMP-1 with or without conditioned medium containing N-FLAG-MGP (n=3).
f, Immunoblotting with densitometry of lung tissues of VE-cadherincreMgpflox/flox and Sm22acreMgpflox/flox and control mice (n=3).
g, Masson’s trichrome (MT) staining with quantification of the lungs of VE-cadherincreMgpflox/flox and Sm22acreMgpflox/flox mice and control mice after treatment with UK388367 (n=8). Scale bar, 100 μm.
h, A schematic diagram.
c, e and g were analyzed for statistical significance by ANOVA with post hoc Tukey’s test. The bounds of the boxes are upper and lower quartiles with data points. Error bars are maximal and minimal values (c and g). Error bars are mean ± standard deviation (SD)(e). ***, P<0.0001.
Since BMP-1 has been shown to cleave latent TGFβ1 binding protein-1 (LTBP-1) for TGFβ1 maturation [48], we used BMP-1 to treat tdTomato+VE-cadherin+ cells with or without the over-expression of MGP and examined LTBP-1 in the cell matrix. Immunoblotting showed that excess BMP-1 efficiently cleaved LTBP-1, and that over-expression of MGP inhibited the BMP-1 cleavage of LTBP-1 (Figure 7d). We also determined the levels of TGFβ1 in the media. ELISA showed that BMP-1 increased the TGFβ1 level and that over-expression of MGP decreased the TGFβ1 level even with BMP-1 treatment (Figure 7e). We examined the expression of BMP-1 and TGFβ1 in the lung tissues of VE-cadherincreMgpflox/flox and SM22αcreMgpflox/flox mice. Immunoblotting showed higher levels of TGFβ1 in VE-cadherincreMgpflox/flox and SM22αcreMgpflox/flox mice than in Mgpflox/flox control mice (Figure 7f). Increased phosphorylation of SMAD2/3 confirmed the induction of TGFβ1 signaling (Figure 7f). No change in BMP-1 expression was detected, supporting that the interaction between MGP and BMP-1 inhibited the BMP-1 activity. To determine if inhibition of BMP-1 affected the pulmonary fibrosis in the new mouse models, we treated VE-cadherincreMgpflox/flox and SM22αcreMgpflox/flox mice with the BMP-1 inhibitor UK383367 (5 μg/g, daily) for two weeks. Masson’s trichrome staining showed a reduction of pulmonary fibrosis in both VE-cadherincreMgpflox/flox and SM22αcreMgpflox/flox mice after the treatment with UK383367 (Figure 7g). Together, the results suggested that MGP interacted with BMP-1, inhibited the production of mature TGFβ1 and in turn regulated the differentiation of EC-like myofibroblasts towards myofibroblasts (Figure 7h).
Berbamine prevents the shift of EC-like myofibroblasts towards myofibroblasts and ameliorates pulmonary fibrosis in mouse models
In our studies, the scRNA-seq revealed a clear trajectory of cell differentiation from ECs towards EC-like myofibroblasts and myofibroblasts. By analyzing the differential gene expression between myofibroblasts and ECs, we identified 537 genes with increased expression and 668 genes with decreased expression using adjusted p value less than 0.01 cutoff (Figure 8a). These alterations in gene expression detailed the genetic signature of the shift from ECs to myofibroblasts. To identify a compound that prevented this cell shift, we input the differential expression profile into the Connectivity Map (CMap) platform. The CMap platform is generated to connect genetic variants with small molecule treatments [49, 50]. Using a connectivity score from designed measurements of expression profiles, a query of CMap can be made to search compounds that cause similar genetic perturbations [51–56]. With this advantage, The CMap platform provides a possible approach to identify compounds that modify the transcriptional landscape towards a desired differentiation direction. To find a compound to reverse the genetic alteration from ECs to myofibroblasts, we input the top 100 differential expression profiles with an opposite direction of alteration to generate a novel query, which allowed the platform to find the compound with the capability to reverse the genetic alteration. CMap identified a small molecule, berbamine, as a potential candidate.
Figure 8. Berbamine ameliorates pulmonary fibrosis in mouse models.
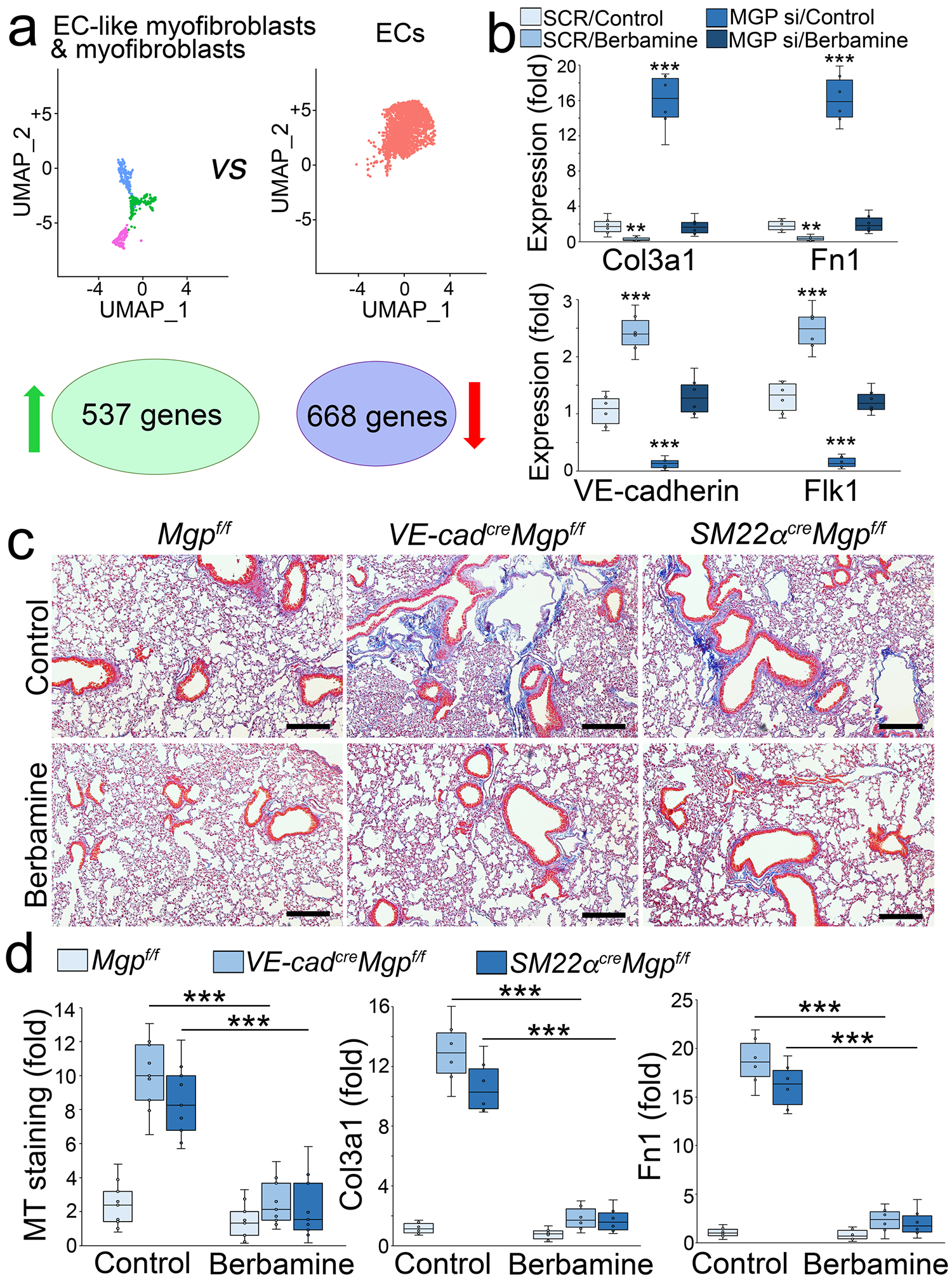
a, UMAP and differential gene expression between EC-like myofibroblasts and ECs.
b, Gene expression in tdTomato+VE-cadherin+ cells isolated from the lungs of SM22acreRosatdTomato mice and treated with berbamine (20 μM) with transfection of MGP siRNA or SCR (n=6). Fn1, fibronectin 1.
c, MT staining of pulmonary tissues from VE-cadherincreMgpflox/flox and Sm22acreMgpflox/flox mice after berbamine treatment (100 ng/g, daily) (n=12).
d, Quantification of MT staining and expression of Col3a1 and Fn1 in pulmonary tissues from VE-cadherincreMgpflox/flox and Sm22acreMgpflox/flox mice after berbamine treatment (n=9 for quantification of MT and n=6 for gene expression).
b and d were analyzed for statistical significance by ANOVA with post hoc Tukey’s test. The bounds of the boxes are upper and lower quartiles with data points. The line in the box is median. Error bars are maximal and minimal values. ***, P<0.0001.
To determine if berbamine affected the differentiation of EC-like myofibroblasts, we isolated tdTomato+VE-cadherin+ cells from the lungs of SM22acreRosatdTomato mice. We treated the cells with 20 μM berbamine for 24 hours and found a reduction of myofibroblast markers and an increase of endothelial markers (Figure 8b). We also depleted MGP in the cells and treated them with berbamine. Real-time PCR showed that berbamine prevented the induction of myofibroblast markers and restored the expression of endothelial markers in MGP-depleted tdTomato+VE-cadherin+ cells (Figure 8b). The results suggested that berbamine redirected EC-like myofibroblasts back to EC differentiation.
We treated VE-cadherincreMgpflox/flox mice and SM22αcreMgpflox/flox mice at 10 weeks of age with berbamine (100 ng/g, daily) for 4 weeks. At 14 weeks of age, we scarified the mice and analyzed the lungs. Masson’s trichrome staining showed that berbamine significantly decreased the pulmonary fibrosis in both mice (Figure 8c–d). Real-time PCR of lung tissue also showed that the expression of Col3a1 and Fn1 was reduced in the berbamine-treated group compared to saline-treated controls (Figure 8d). The results suggested that berbamine reduced pulmonary fibrosis.
To determine if berbamine affects TGFβ signaling, we isolated tdTomato+VE-cadherin+ cells from the lungs of SM22acreRosatdTomato mice and treated the cells with berbamine (20 μM). Real-time PCR and immunoblotting both showed that berbamine reduced the expression of the TGFβ1 receptor activin receptor-like kinase 5 (ALK5) (Figure 9a). We used lentiviral vectors containing cytomegalovirus (CMV) promoter-driven ALK5 to infect the cells and treated the cells with berbamine. Real-time PCR showed that excess ALK5 restored the induction of Col3a1 and Fn1 in MGP-depleted tdTomato+VE-cadherin+ cells (Figure 9b), suggesting that berbamine-reduced ALK5 prevented the differentiation of EC-like myofibroblasts to myofibroblasts. Interestingly, excess ALK5 did not affect berbamine-induced endothelial markers (Figure 9b), suggesting that berbamine targets an additional factor to drive EC-like myofibroblasts towards EC differentiation.
Figure 9. Berbamine induces FoxA2 to regulate TGFβ signaling and control EC-like myofibroblast differentiation.
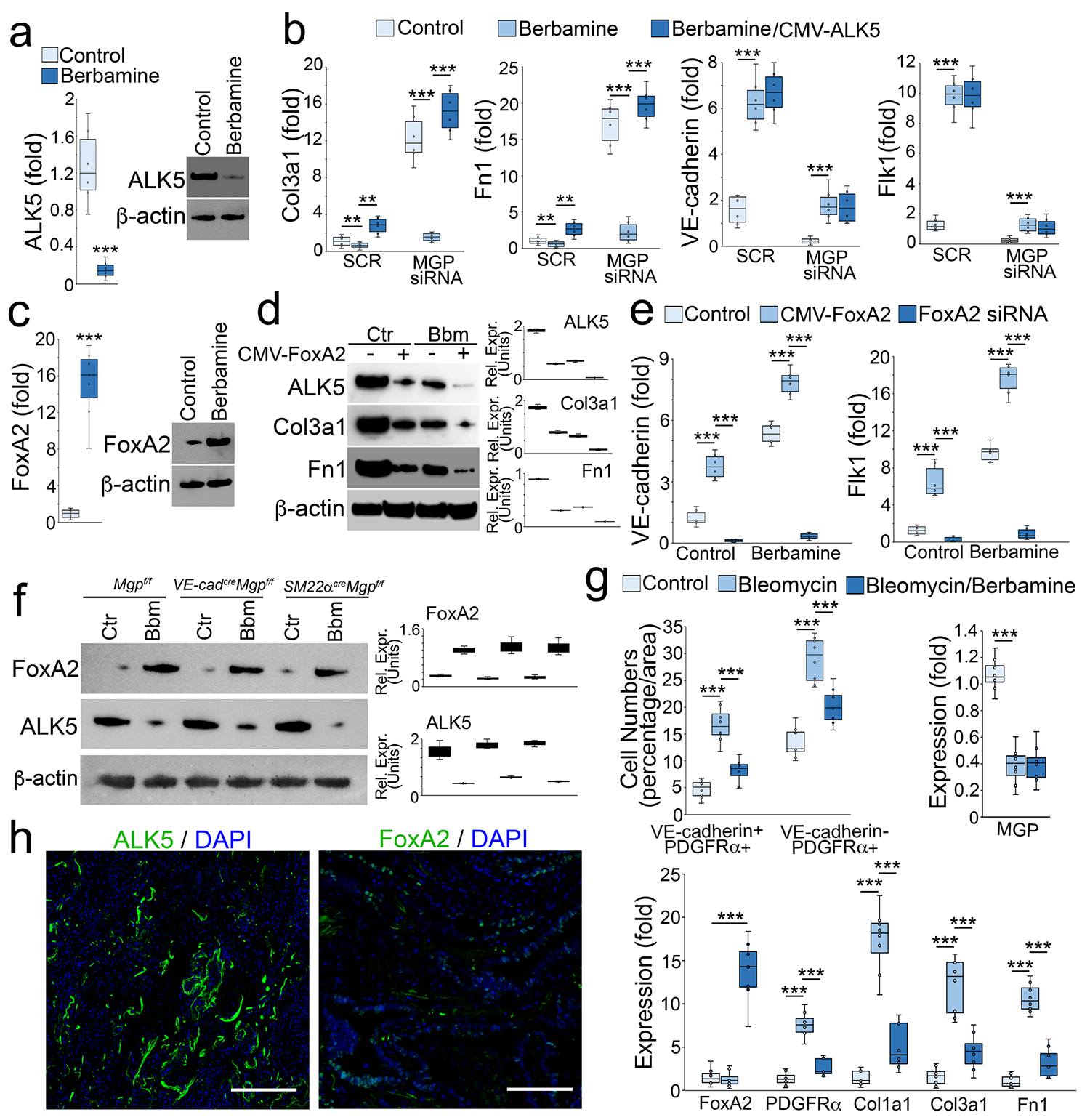
a, Expression of ALK5 in tdTomato+VE-cadherin+ cells isolated from the lungs of SM22acreRosatdTomato mice and treated with berbamine (20 μM).
b, Gene expression in tdTomato+VE-cadherin+ cells transfected with MGP siRNA in combination with berbamine treatment with or without overexpression of ALK5 (n=5). CMV, cytomegalovirus promoter. Fn1, fibronectin 1. SCR, scrambled siRNA.
c, Expression of FoxA2 in tdTomato+VE-cadherin+ cells isolated from the lungs of SM22acreRosatdTomato mice and treated with berbamine (20 μM).
d, Immunoblotting with densitometry using tdTomato+VE-cadherin+ cell lysates treated with berbamine (Bbm) with or without overexpression of FoxA2 (CMV-FoxA2) (n=3).
e, Expression of endothelial markers in tdTomato+VE-cadherin+ cells treated with berbamine in combination with FoxA2 overexpression or FoxA2 knockdown (FoxA2 siRNA) (n=6).
f, Immunoblotting with densitometry of the lungs of VE-cadherincreMgpflox/flox mice, Sm22acreMgpflox/flox mice and control mice after treatment of berbamine (Bbm) (n=6).
g, Quantification of VE-cadherin positive and PDGFRα positive (VE-cadherin+PDGFRα+) cells and VE-cadherin negative and PDGFRα positive (VE-cadherin-PDGFRα+) cells after immunostaining of the lungs of bleomycin-injected wild-type mice, where gene expression was examined by real-time PCR (n=8).
h, Immunostaining of ALK5 and FoxA2 of human pulmonary fibrosis (n=3).
a and c were analyzed for statistical significance by unpaired 2-tailed Student’s t test. b, e and g were analyzed for statistical significance by ANOVA with post hoc Tukey’s test. The bounds of the boxes are upper and lower quartiles with data points. The line in the box is median. Error bars are maximal and minimal values. ***, P<0.0001.
Berbamine has previously been identified as an inhibitor of calmodulin 1, a calcium channel blocker [57, 58]. We depleted calmodulin 1 in tdTomato+VE-cadherin+ cells and found no change in the expression of endothelial or myofibroblast markers (data not shown), suggesting that inhibition of calmodulin 1 was not involved in the redirection of myofibroblasts. Therefore, we screened the expression of transcription factors that were involved in endothelial differentiation. We found that berbamine significantly induced FoxA2 expression (Figure 9c) and overexpression of FoxA2 alone reduced the expression of ALK5 and the myofibroblast markers (Figure 9d). The combination of berbamine and FoxA2 overexpression further reduced ALK5 and the myofibroblast markers (Figure 9d).
We overexpressed or depleted FoxA2 in tdTomato+VE-cadherin+ cells and treated the cells with berbamine. The results showed that the overexpression of FoxA2 alone induced the endothelial markers (Figure 9e). FoxA2 overexpression with berbamine treatment enhanced the induction of endothelial markers whereas FoxA2 depletion abolished the induction of endothelial markers in tdTomatoVE-cadherin+ cells (Figure 9e). We examined the expression of ALK5 and FoxA2 in the lungs of VE-cadherincreMgpflox/flox mice and SM22αcreMgpflox/flox mice after berbamine treatment. Immunoblotting showed that berbamine increased FoxA2 expression and reduced ALK5 expression in the lungs of these mice (Figure 9f). We then treated wild type mice with berbamine (100 ng/g, daily) for 4 weeks after bleomycin administration. Berbamine significantly decreased the numbers of both PDGFRα+ cells and PDGFRα+VE-cadherin+ cells in the lungs of bleomycin-treated mice. Real-time PCR showed a reduction of PDGFRα, Col1a1, Col3a1 and Fn1 (Figure 9g). The results also showed that berbamine induced FoxA2, but had no effect on bleomycin-reduced MGP expression (Figure 9g). To determine if ALK5 and FoxA2 were expressed in human pulmonary fibrosis, we performed immunostaining that showed strong expression of ALK5 and less of FoxA2 (Figure 9h). Together, these results suggested that berbamine induced FoxA2 to suppress ALK5 expression, in turn redirecting EC-like myofibroblasts towards EC differentiation. The results also suggested berbamine as a new treatment strategy for pulmonary fibrosis.
Discussion
Pulmonary fibrosis is a severe fibrotic lung disease with high morbidity and mortality worldwide [1–6]. Although the precise mechanism has not been determined, pulmonary fibrosis is characterized by progressive interstitial fibrogenesis that causes distortion of the alveolar architecture leading to loss of pulmonary function [1–6, 59, 60]. Recent studies have shown ill-fated myofibroblasts emerging in pulmonary fibrosis. These unwanted myofibroblasts are strongly activated during interstitial fibrogenesis, where they produce and deposit excessive amounts of extracellular fibrotic matrix in the tissue [12–20]. In this study, we found that cell-specific deletions of Mgp cause pulmonary fibrosis. We showed that MGP regulates the differentiation of a previously unknown population of EC-like myofibroblasts that contributed to the differentiation of myofibroblasts in pulmonary fibrosis. We also identified a small molecule which could redirect the differentiation of the EC-like myofibroblasts and reduce pulmonary fibrosis.
Pulmonary ECs are essential in lung tissue. Endothelial defects result in poor induction of pulmonary lineages from progenitor cells and impaired epithelial repair after lung injury [61]. Pulmonary ECs both support pulmonary epithelial cells and guide their differentiation and maturation [35, 62]. Previous studies have reported co-expression of endothelial and mesenchymal markers in pulmonary disease, suggesting that ECs transition into other lineages under disease conditions [63, 64]. In this study, we uncovered a novel differentiation trajectory in normal lung tissue, where pulmonary ECs may differentiate into EC-like myofibroblasts and ultimately myofibroblasts. We showed that the dysregulation of this differentiation trajectory dramatically increased the production of myofibroblasts that when activated, contributed to pulmonary fibrosis. We found a robust increase of CD34+ cells in the lungs of VE-cadherincreMgpflox/flox mice and SM22αcreMgpflox/flox mice. We showed CD34 to be expressed in all the cells in the differentiation trajectory from ECs to myofibroblasts. There was also an interesting trend of more CD34+ cells in VE-cadherincreMgpflox/flox mice than that in SM22αcreMgpflox/flox mice (Figure 2). The differentiation trajectory projected that ECs were the upstream of EC-like myofibroblasts. Deletion of MGP in ECs could drive more ECs to EC-like myofibroblasts and myofibroblasts than MGP deletion in EC-like myofibroblasts. That would cause more CD34+ cells to accumulate in VE-cadherincreMgpflox/flox mice where MGP was deleted in ECs and EC-like myofibroblasts than in SM22αcreMgpflox/flox mice where MGP was deleted in EC-like myofibroblasts and myofibroblasts.
MGP, a BMP inhibitor, is essential for regulating vascular BMP activity [34, 46, 65–67]. Loss of MGP causes arteriovenous malformation in cerebrum, lungs, kidneys, and retina in mice, which resemble the mouse model for hereditary hemorrhagic telangiectasia type 2 [38, 66–68]. Mutations in the human Mgp gene cause Keutel syndrome, a rare disease, that involves cardiovascular defects and peripheral pulmonary stenoses [39, 69, 70]. However, pulmonary fibrosis has not been reported in these rare patients [39, 69, 70]. In addition, severe side effects of warfarin treatment in pulmonary fibrosis patients suggest interference by MGP in pulmonary fibrosis [40–42]. Warfarin inhibits the vitamin K–dependent γ-carboxylation, which is essential for the function of MGP [34]. Warfarin treatment prevents the modification of Glu to Gla residues in MGP, resulting in impaired BMP binding [34]. Warfarin treatment was reported to rapidly worsen the progression of pulmonary fibrosis and a ban of warfarin in pulmonary fibrosis patents has been suggested [40–42]. In this study, we find that the Mgp deletion in VE-cadherin or SM22α positive cells causes pulmonary fibrosis in mice, suggesting an important role of MGP in the fibrotic process. These mice also provide new animal models for the study of pulmonary fibrosis.
BMP-1 was initially classified as a BMP based on bone induction [71–73]. However, BMP-1 encodes a metalloprotease with few similarities to other BMPs, and is not included in the TGFβ superfamily of growth factors [74]. BMP-1 executes its activity by modifying the protein precursors to mature proteins [74–76]. For example, BMP-1 cleaves the BMP antagonist Chordin to regulate BMP activity [77]. BMP-1 also processes extracellular matrix proteins such as collagens, biglycan, and osteoglycin [76, 78]. Interestingly, fibroblasts strongly increase the deposition of collagens onto the insoluble extracellular matrix when incubated with BMP-1 [79]. In addition, BMP-1 cleaves LTBP-1 to facilitate the maturation of TGFβ1, a master regulator of myofibroblast differentiation in pulmonary fibrosis [48]. Our study suggests that MGP binds to BMP-1 and reduces the production of mature TGFβ1, thereby regulating the differentiation of EC-like myofibroblasts to myofibroblasts. We showed that inhibition of BMP-1 decreased the fibrosis in the lungs of VE-cadherincreMgpflox/flox mice and SM22αcreMgpflox/flox mice. The deletion of BMP-1 was unable to reduce bleomycin-induced pulmonary fibrosis [80], which indicated the mechanism of fibrosis in VE-cadherincreMgpflox/flox mice and SM22αcreMgpflox/flox mice may differ from bleomycin-induced fibrosis. The precise mechanism of bleomycin-induced fibrosis is yet to be revealed. The previous studies uncovered that bleomycin injured lung cells and triggered the influx of inflammatory and immune cells, which may lead to the fibrogenesis [81]. To our knowledge, no studies have shown whether or not BMP-1 is involved in the inflammatory response towards fibrogenesis. The fibrosis in VE-cadherincreMgpflox/flox mice and SM22αcreMgpflox/flox mice occurred during development and we did not observe accumulation of inflammatory cells in the lungs of these mice. During lung development, BMP-1 acts as an important factor for TGFβ1 production, which is essential to balance the differentiation of ECs and myofibroblasts [48, 82]. We argue that lack of MGP unleashes BMP-1 activity, enhances the production of mature TGFβ1, pushes ECs and EC-like myofibroblasts towards myofibroblast differentiation, subsequently causing fibrosis.
Our results suggested an interaction between MGP and BMP-1. MGP has been previously shown to bind to BMP-2, 4 and 7 through Proline-64 and surrounding Gla residues [34]. MGP has also been shown to interact with several other protein, such as Fn1, vitronectin, through its C-terminus [83, 84] and elastin with its N-terminus [85]. Further studies would be necessary to identify what region of MGP interacts with BMP-1. The balance between BMP-1 activity and other BMPs regulated by MGP would also be interesting to explore in fibrosis. Since BMP-1 is a metalloprotease, the effect of BMP-1 on fibrosis would be different from that of other BMPs. Some BMPs are reported to reduce fibrogenesis through suppression of TGFβ signaling [86], but our results suggest that enhanced BMP-1 activity increases TGFβ1 signaling to cause fibrosis. Differences in affinity between MGP and various BMPs might also be important to explain variations in BMPs’ activity after loss of MGP. The complexity of extracellular cell matrix may also play an important role.
Berbamine is a small molecule extracted from the plant named Berberis [87]. Berbamine was initially identified as a calcium channel blocker with anti-arrhythmic effects and ischemic protective activity through the inhibition of calmodulin 1 [57, 58]. Recent studies also found that berbamine inhibits the nuclear factor-kappa B (NF-κB) signaling pathway for anti-myeloma effects and reduces the activity of signal transducer and activator of transcription 3 (STAT3) in hepatocellular carcinoma [88, 89]. Our results showed that berbamine induced the transcription factor FoxA2, which not only prevented EC-like myofibroblasts from differentiating into myofibroblast but also shifted EC-like myofibroblasts towards ECs. FoxA2 is essential for the development of the cardiovascular system [90]. FoxA2 is highly expressed at an early stage of EC differentiation and the progenitors of FoxA2+ endoderm contribute ECs [91, 92]. Therefore, it is not surprising that FoxA2 induced by berbamine redirected EC-like myofibroblasts back to EC differentiation, decreasing myofibroblasts and fibrosis.
Supplementary Material
ACKNOWLEDGMENTS:
Funding for this work was provided in part by NIH grants NS79353 (Y.Y.), HL139675 (Y.Y.), HL162643 (Y.Y.), HL81397 (K.I.B.) and HL154548 (K.I.B.)
Footnotes
COMPETING FINANCIAL INTERESTS:
The authors have declared that no conflict of interest exists.
Reference
- 1.Hutchinson J, Fogarty A, Hubbard R, McKeever T. Global incidence and mortality of idiopathic pulmonary fibrosis: a systematic review. Eur Respir J 2015: 46(3): 795–806. [DOI] [PubMed] [Google Scholar]
- 2.Lederer DJ, Martinez FJ. Idiopathic Pulmonary Fibrosis. N Engl J Med 2018: 379(8): 797–798. [DOI] [PubMed] [Google Scholar]
- 3.Martinez FJ, Collard HR, Pardo A, Raghu G, Richeldi L, Selman M, Swigris JJ, Taniguchi H, Wells AU. Idiopathic pulmonary fibrosis. Nat Rev Dis Primers 2017: 3: 17074. [DOI] [PubMed] [Google Scholar]
- 4.Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, Colby TV, Cordier JF, Flaherty KR, Lasky JA, Lynch DA, Ryu JH, Swigris JJ, Wells AU, Ancochea J, Bouros D, Carvalho C, Costabel U, Ebina M, Hansell DM, Johkoh T, Kim DS, King TE Jr., Kondoh Y, Myers J, Muller NL, Nicholson AG, Richeldi L, Selman M, Dudden RF, Griss BS, Protzko SL, Schunemann HJ, Fibrosis AEJACoIP. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 2011: 183(6): 788–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Raghu G, Weycker D, Edelsberg J, Bradford WZ, Oster G. Incidence and prevalence of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2006: 174(7): 810–816. [DOI] [PubMed] [Google Scholar]
- 6.Richeldi L, Collard HR, Jones MG. Idiopathic pulmonary fibrosis. Lancet 2017: 389(10082): 1941–1952. [DOI] [PubMed] [Google Scholar]
- 7.Barratt SL, Creamer A, Hayton C, Chaudhuri N. Idiopathic Pulmonary Fibrosis (IPF): An Overview. J Clin Med 2018: 7(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hamanaka RB, Mutlu GM. Metabolic requirements of pulmonary fibrosis: role of fibroblast metabolism. The FEBS journal 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim HJ, Perlman D, Tomic R. Natural history of idiopathic pulmonary fibrosis. Respiratory medicine 2015: 109(6): 661–670. [DOI] [PubMed] [Google Scholar]
- 10.Hewson T, McKeever TM, Gibson JE, Navaratnam V, Hubbard RB, Hutchinson JP. Timing of onset of symptoms in people with idiopathic pulmonary fibrosis. Thorax 2017. [DOI] [PubMed] [Google Scholar]
- 11.Ley B, Ryerson CJ, Vittinghoff E, Ryu JH, Tomassetti S, Lee JS, Poletti V, Buccioli M, Elicker BM, Jones KD, King TE Jr., Collard HR. A multidimensional index and staging system for idiopathic pulmonary fibrosis. Ann Intern Med 2012: 156(10): 684–691. [DOI] [PubMed] [Google Scholar]
- 12.King TE Jr., Pardo A, Selman M. Idiopathic pulmonary fibrosis. Lancet 2011: 378(9807): 1949–1961. [DOI] [PubMed] [Google Scholar]
- 13.Tsukui T, Sun KH, Wetter JB, Wilson-Kanamori JR, Hazelwood LA, Henderson NC, Adams TS, Schupp JC, Poli SD, Rosas IO, Kaminski N, Matthay MA, Wolters PJ, Sheppard D. Collagen-producing lung cell atlas identifies multiple subsets with distinct localization and relevance to fibrosis. Nature communications 2020: 11(1): 1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Valenzi E, Bulik M, Tabib T, Morse C, Sembrat J, Trejo Bittar H, Rojas M, Lafyatis R. Single-cell analysis reveals fibroblast heterogeneity and myofibroblasts in systemic sclerosis-associated interstitial lung disease. Annals of the rheumatic diseases 2019: 78(10): 1379–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Peyser R, MacDonnell S, Gao Y, Cheng L, Kim Y, Kaplan T, Ruan Q, Wei Y, Ni M, Adler C, Zhang W, Devalaraja-Narashimha K, Grindley J, Halasz G, Morton L. Defining the Activated Fibroblast Population in Lung Fibrosis Using Single-Cell Sequencing. American journal of respiratory cell and molecular biology 2019: 61(1): 74–85. [DOI] [PubMed] [Google Scholar]
- 16.Pakshir P, Noskovicova N, Lodyga M, Son DO, Schuster R, Goodwin A, Karvonen H, Hinz B. The myofibroblast at a glance. J Cell Sci 2020: 133(13). [DOI] [PubMed] [Google Scholar]
- 17.Phan SH. Genesis of the myofibroblast in lung injury and fibrosis. Proc Am Thorac Soc 2012: 9(3): 148–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Phan SH. The myofibroblast in pulmonary fibrosis. Chest 2002: 122(6 Suppl): 286S–289S. [DOI] [PubMed] [Google Scholar]
- 19.Xie T, Wang Y, Deng N, Huang G, Taghavifar F, Geng Y, Liu N, Kulur V, Yao C, Chen P, Liu Z, Stripp B, Tang J, Liang J, Noble PW, Jiang D. Single-Cell Deconvolution of Fibroblast Heterogeneity in Mouse Pulmonary Fibrosis. Cell reports 2018: 22(13): 3625–3640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Habiel DM, Hogaboam CM. Heterogeneity of Fibroblasts and Myofibroblasts in Pulmonary Fibrosis. Curr Pathobiol Rep 2017: 5(2): 101–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Uhal BD, Joshi I, Hughes WF, Ramos C, Pardo A, Selman M. Alveolar epithelial cell death adjacent to underlying myofibroblasts in advanced fibrotic human lung. Am J Physiol 1998: 275(6): L1192–1199. [DOI] [PubMed] [Google Scholar]
- 22.Jablonski RP, Kim SJ, Cheresh P, Williams DB, Morales-Nebreda L, Cheng Y, Yeldandi A, Bhorade S, Pardo A, Selman M, Ridge K, Gius D, Budinger GRS, Kamp DW. SIRT3 deficiency promotes lung fibrosis by augmenting alveolar epithelial cell mitochondrial DNA damage and apoptosis. FASEB J 2017: 31(6): 2520–2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Misharin AV, Morales-Nebreda L, Reyfman PA, Cuda CM, Walter JM, McQuattie-Pimentel AC, Chen CI, Anekalla KR, Joshi N, Williams KJN, Abdala-Valencia H, Yacoub TJ, Chi M, Chiu S, Gonzalez-Gonzalez FJ, Gates K, Lam AP, Nicholson TT, Homan PJ, Soberanes S, Dominguez S, Morgan VK, Saber R, Shaffer A, Hinchcliff M, Marshall SA, Bharat A, Berdnikovs S, Bhorade SM, Bartom ET, Morimoto RI, Balch WE, Sznajder JI, Chandel NS, Mutlu GM, Jain M, Gottardi CJ, Singer BD, Ridge KM, Bagheri N, Shilatifard A, Budinger GRS, Perlman H. Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. J Exp Med 2017: 214(8): 2387–2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Naikawadi RP, Disayabutr S, Mallavia B, Donne ML, Green G, La JL, Rock JR, Looney MR, Wolters PJ. Telomere dysfunction in alveolar epithelial cells causes lung remodeling and fibrosis. JCI Insight 2016: 1(14): e86704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gabbiani G, Hirschel BJ, Ryan GB, Statkov PR, Majno G. Granulation tissue as a contractile organ. A study of structure and function. J Exp Med 1972: 135(4): 719–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gabbiani G, Ryan GB, Majne G. Presence of modified fibroblasts in granulation tissue and their possible role in wound contraction. Experientia 1971: 27(5): 549–550. [DOI] [PubMed] [Google Scholar]
- 27.Majno G, Gabbiani G, Hirschel BJ, Ryan GB, Statkov PR. Contraction of granulation tissue in vitro: similarity to smooth muscle. Science 1971: 173(3996): 548–550. [DOI] [PubMed] [Google Scholar]
- 28.Darby I, Skalli O, Gabbiani G. Alpha-smooth muscle actin is transiently expressed by myofibroblasts during experimental wound healing. Lab Invest 1990: 63(1): 21–29. [PubMed] [Google Scholar]
- 29.Karin D, Koyama Y, Brenner D, Kisseleva T. The characteristics of activated portal fibroblasts/myofibroblasts in liver fibrosis. Differentiation 2016: 92(3): 84–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Di Carlo SE, Peduto L. The perivascular origin of pathological fibroblasts. J Clin Invest 2018: 128(1): 54–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tallquist MD, Molkentin JD. Redefining the identity of cardiac fibroblasts. Nature reviews Cardiology 2017: 14(8): 484–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Adams TS, Schupp JC, Poli S, Ayaub EA, Neumark N, Ahangari F, Chu SG, Raby BA, DeIuliis G, Januszyk M, Duan Q, Arnett HA, Siddiqui A, Washko GR, Homer R, Yan X, Rosas IO, Kaminski N. Single-cell RNA-seq reveals ectopic and aberrant lung-resident cell populations in idiopathic pulmonary fibrosis. Sci Adv 2020: 6(28): eaba1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reyfman PA, Walter JM, Joshi N, Anekalla KR, McQuattie-Pimentel AC, Chiu S, Fernandez R, Akbarpour M, Chen CI, Ren Z, Verma R, Abdala-Valencia H, Nam K, Chi M, Han S, Gonzalez-Gonzalez FJ, Soberanes S, Watanabe S, Williams KJN, Flozak AS, Nicholson TT, Morgan VK, Winter DR, Hinchcliff M, Hrusch CL, Guzy RD, Bonham CA, Sperling AI, Bag R, Hamanaka RB, Mutlu GM, Yeldandi AV, Marshall SA, Shilatifard A, Amaral LAN, Perlman H, Sznajder JI, Argento AC, Gillespie CT, Dematte J, Jain M, Singer BD, Ridge KM, Lam AP, Bharat A, Bhorade SM, Gottardi CJ, Budinger GRS, Misharin AV. Single-Cell Transcriptomic Analysis of Human Lung Provides Insights into the Pathobiology of Pulmonary Fibrosis. Am J Respir Crit Care Med 2019: 199(12): 1517–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yao Y, Shahbazian A, Bostrom KI. Proline and gamma-carboxylated glutamate residues in matrix Gla protein are critical for binding of bone morphogenetic protein-4. Circ Res 2008: 102(9): 1065–1074. [DOI] [PubMed] [Google Scholar]
- 35.Yao J, Guihard PJ, Wu X, Blazquez-Medela AM, Spencer MJ, Jumabay M, Tontonoz P, Fogelman AM, Bostrom KI, Yao Y. Vascular endothelium plays a key role in directing pulmonary epithelial cell differentiation. J Cell Biol 2017: 216(10): 3369–3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yao Y, Nowak S, Yochelis A, Garfinkel A, Bostrom KI. Matrix GLA protein, an inhibitory morphogen in pulmonary vascular development. J Biol Chem 2007: 282(41): 30131–30142. [DOI] [PubMed] [Google Scholar]
- 37.Gilbert KA, Rannels SR. Matrix GLA protein modulates branching morphogenesis in fetal rat lung. Am J Physiol Lung Cell Mol Physiol 2004: 286(6): L1179–1187. [DOI] [PubMed] [Google Scholar]
- 38.Yao Y, Jumabay M, Wang A, Bostrom KI. Matrix Gla protein deficiency causes arteriovenous malformations in mice. J Clin Invest 2011: 121(8): 2993–3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Meier M, Weng LP, Alexandrakis E, Ruschoff J, Goeckenjan G. Tracheobronchial stenosis in Keutel syndrome. Eur Respir J 2001: 17(3): 566–569. [DOI] [PubMed] [Google Scholar]
- 40.Alagha K, Secq V, Pahus L, Sofalvi T, Palot A, Bourdin A, Chanez P. We should prohibit warfarin in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2015: 191(8): 958–960. [DOI] [PubMed] [Google Scholar]
- 41.Cottin V, Crestani B, Valeyre D, Wallaert B, Cadranel J, Dalphin JC, Delaval P, Israel-Biet D, Kessler R, Reynaud-Gaubert M, Cordier JF, Aguilaniu B, Bouquillon B, Carre P, Danel C, Faivre JB, Ferreti G, Just N, Kouzan S, Lebargy F, Marchand Adam S, Philippe B, Prevot G, Stach B, Thivolet-Bejui F, French National R, Competence Centers for Rare D, Societe de Pneumologies de Langue F. [French practical guidelines for the diagnosis and management of idiopathic pulmonary fibrosis. From the National Reference and the Competence centers for rare diseases and the Societe de Pneumologie de Langue Francaise]. Rev Mal Respir 2013: 30(10): 879–902. [DOI] [PubMed] [Google Scholar]
- 42.Noth I, Anstrom KJ, Calvert SB, de Andrade J, Flaherty KR, Glazer C, Kaner RJ, Olman MA, Idiopathic Pulmonary Fibrosis Clinical Research N. A placebo-controlled randomized trial of warfarin in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2012: 186(1): 88–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yao J, Guihard PJ, Blazquez-Medela AM, Guo Y, Liu T, Bostrom KI, Yao Y. Matrix Gla protein regulates differentiation of endothelial cells derived from mouse embryonic stem cells. Angiogenesis 2016: 19(1): 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yao Y, Jumabay M, Ly A, Radparvar M, Cubberly MR, Bostrom KI. A role for the endothelium in vascular calcification. Circ Res 2013: 113(5): 495–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yao J, Guihard PJ, Blazquez-Medela AM, Guo Y, Moon JH, Jumabay M, Bostrom KI, Yao Y. Serine Protease Activation Essential for Endothelial-Mesenchymal Transition in Vascular Calcification. Circ Res 2015: 117(9): 758–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yao Y, Zebboudj AF, Shao E, Perez M, Bostrom K. Regulation of bone morphogenetic protein-4 by matrix GLA protein in vascular endothelial cells involves activin-like kinase receptor 1. J Biol Chem 2006: 281(45): 33921–33930. [DOI] [PubMed] [Google Scholar]
- 47.Harrison JH Jr., Lazo JS. High dose continuous infusion of bleomycin in mice: a new model for drug-induced pulmonary fibrosis. J Pharmacol Exp Ther 1987: 243(3): 1185–1194. [PubMed] [Google Scholar]
- 48.Ge G, Greenspan DS. BMP1 controls TGFbeta1 activation via cleavage of latent TGFbeta-binding protein. J Cell Biol 2006: 175(1): 111–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Subramanian A, Narayan R, Corsello SM, Peck DD, Natoli TE, Lu X, Gould J, Davis JF, Tubelli AA, Asiedu JK, Lahr DL, Hirschman JE, Liu Z, Donahue M, Julian B, Khan M, Wadden D, Smith IC, Lam D, Liberzon A, Toder C, Bagul M, Orzechowski M, Enache OM, Piccioni F, Johnson SA, Lyons NJ, Berger AH, Shamji AF, Brooks AN, Vrcic A, Flynn C, Rosains J, Takeda DY, Hu R, Davison D, Lamb J, Ardlie K, Hogstrom L, Greenside P, Gray NS, Clemons PA, Silver S, Wu X, Zhao WN, Read-Button W, Wu X, Haggarty SJ, Ronco LV, Boehm JS, Schreiber SL, Doench JG, Bittker JA, Root DE, Wong B, Golub TR. A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles. Cell 2017: 171(6): 1437–1452 e1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Duan Q, Flynn C, Niepel M, Hafner M, Muhlich JL, Fernandez NF, Rouillard AD, Tan CM, Chen EY, Golub TR, Sorger PK, Subramanian A, Ma’ayan A. LINCS Canvas Browser: interactive web app to query, browse and interrogate LINCS L1000 gene expression signatures. Nucleic acids research 2014: 42(Web Server issue): W449–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Qiu Y, Lu T, Lim H, Xie L. A Bayesian approach to accurate and robust signature detection on LINCS L1000 data. Bioinformatics 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu TP, Hsieh YY, Chou CJ, Yang PM. Systematic polypharmacology and drug repurposing via an integrated L1000-based Connectivity Map database mining. R Soc Open Sci 2018: 5(11): 181321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Brum AM, van de Peppel J, van der Leije CS, Schreuders-Koedam M, Eijken M, van der Eerden BC, van Leeuwen JP. Connectivity Map-based discovery of parbendazole reveals targetable human osteogenic pathway. Proc Natl Acad Sci U S A 2015: 112(41): 12711–12716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dyle MC, Ebert SM, Cook DP, Kunkel SD, Fox DK, Bongers KS, Bullard SA, Dierdorff JM, Adams CM. Systems-based discovery of tomatidine as a natural small molecule inhibitor of skeletal muscle atrophy. J Biol Chem 2014: 289(21): 14913–14924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Farooq F, Balabanian S, Liu X, Holcik M, MacKenzie A. p38 Mitogen-activated protein kinase stabilizes SMN mRNA through RNA binding protein HuR. Hum Mol Genet 2009: 18(21): 4035–4045. [DOI] [PubMed] [Google Scholar]
- 56.Liu J, Lee J, Salazar Hernandez MA, Mazitschek R, Ozcan U. Treatment of obesity with celastrol. Cell 2015: 161(5): 999–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xu CQ, Dong DL, Du ZM, Chen QW, Gong DM, Yang BF. [Comparison of the anti-arrhythmic effects of matrine and berbamine with amiodarone and RP58866]. Yao Xue Xue Bao 2004: 39(9): 691–694. [PubMed] [Google Scholar]
- 58.Guo ZB, Cao HY, Xu Z, Li Q. [Electrophysiological effects of berbamine on ischemic ventricular tachyarrhythmia]. Zhongguo Yao Li Xue Bao 1991: 12(1): 44–47. [PubMed] [Google Scholar]
- 59.Wolters PJ, Collard HR, Jones KD. Pathogenesis of idiopathic pulmonary fibrosis. Annu Rev Pathol 2014: 9: 157–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Selman M, Pardo A. Revealing the pathogenic and aging-related mechanisms of the enigmatic idiopathic pulmonary fibrosis. an integral model. Am J Respir Crit Care Med 2014: 189(10): 1161–1172. [DOI] [PubMed] [Google Scholar]
- 61.Lee J-H, Bhang DH, Beede A, Huang TL, Stripp BR, Bloch KD, Wagers AJ, Tseng Y-H, Ryeom S, Kim CF. Lung stem cell differentiation in mice directed by endothelial cells via a BMP4-NFATc1-thrombospondin-1 axis. Cell 2014: 156(3): 440–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ding BS, Nolan DJ, Guo P, Babazadeh AO, Cao Z, Rosenwaks Z, Crystal RG, Simons M, Sato TN, Worgall S, Shido K, Rabbany SY, Rafii S. Endothelial-derived angiocrine signals induce and sustain regenerative lung alveolarization. Cell 2011: 147(3): 539–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.El Agha E, Kramann R, Schneider RK, Li X, Seeger W, Humphreys BD, Bellusci S. Mesenchymal Stem Cells in Fibrotic Disease. Cell Stem Cell 2017: 21(2): 166–177. [DOI] [PubMed] [Google Scholar]
- 64.Lemos DR, Duffield JS. Tissue-resident mesenchymal stromal cells: Implications for tissue-specific antifibrotic therapies. Science translational medicine 2018: 10(426). [DOI] [PubMed] [Google Scholar]
- 65.Yao Y, Zebboudj AF, Torres A, Shao E, Bostrom K. Activin-like kinase receptor 1 (ALK1) in atherosclerotic lesions and vascular mesenchymal cells. Cardiovasc Res 2007: 74(2): 279–289. [DOI] [PubMed] [Google Scholar]
- 66.Guihard PJ, Guo Y, Wu X, Zhang L, Yao J, Jumabay M, Yao Y, Garfinkel A, Bostrom KI. Shaping Waves of Bone Morphogenetic Protein Inhibition During Vascular Growth. Circ Res 2020: 127(10): 1288–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bostrom KI, Guihard P, Blazquez Medela AM, Yao J, Moon JH, Penton A, Yao Y. Matrix Gla protein limits pulmonary arteriovenous malformations in ALK1 deficiency. Eur Respir J 2015: 45(3): 849–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yao Y, Yao J, Radparvar M, Blazquez-Medela AM, Guihard PJ, Jumabay M, Bostrom KI. Reducing Jagged 1 and 2 levels prevents cerebral arteriovenous malformations in matrix Gla protein deficiency. Proc Natl Acad Sci U S A 2013: 110(47): 19071–19076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Keutel J, Jorgensen G, Gabriel P. [A new autosomal-recessive hereditary syndrome. Multiple peripheral pulmonary stenosis, brachytelephalangia, inner-ear deafness, ossification or calcification of cartilages]. Deutsche medizinische Wochenschrift 1971: 96(43): 1676–1681 passim. [DOI] [PubMed] [Google Scholar]
- 70.Cancela ML, Laize V, Conceicao N, Kempf H, Murshed M. Keutel Syndrome, a Review of 50 Years of Literature. Front Cell Dev Biol 2021: 9: 642136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wozney JM. Bone morphogenetic proteins. Prog Growth Factor Res 1989: 1(4): 267–280. [DOI] [PubMed] [Google Scholar]
- 72.Wozney JM, Rosen V, Celeste AJ, Mitsock LM, Whitters MJ, Kriz RW, Hewick RM, Wang EA. Novel regulators of bone formation: molecular clones and activities. Science 1988: 242(4885): 1528–1534. [DOI] [PubMed] [Google Scholar]
- 73.Tabas JA, Zasloff M, Wasmuth JJ, Emanuel BS, Altherr MR, McPherson JD, Wozney JM, Kaplan FS. Bone morphogenetic protein: chromosomal localization of human genes for BMP1, BMP2A, and BMP3. Genomics 1991: 9(2): 283–289. [DOI] [PubMed] [Google Scholar]
- 74.Kessler E, Takahara K, Biniaminov L, Brusel M, Greenspan DS. Bone morphogenetic protein-1: the type I procollagen C-proteinase. Science 1996: 271(5247): 360–362. [DOI] [PubMed] [Google Scholar]
- 75.Mac Sweeney A, Gil-Parrado S, Vinzenz D, Bernardi A, Hein A, Bodendorf U, Erbel P, Logel C, Gerhartz B. Structural basis for the substrate specificity of bone morphogenetic protein 1/tolloid-like metalloproteases. J Mol Biol 2008: 384(1): 228–239. [DOI] [PubMed] [Google Scholar]
- 76.Scott IC, Blitz IL, Pappano WN, Imamura Y, Clark TG, Steiglitz BM, Thomas CL, Maas SA, Takahara K, Cho KW, Greenspan DS. Mammalian BMP-1/Tolloid-related metalloproteinases, including novel family member mammalian Tolloid-like 2, have differential enzymatic activities and distributions of expression relevant to patterning and skeletogenesis. Dev Biol 1999: 213(2): 283–300. [DOI] [PubMed] [Google Scholar]
- 77.Piccolo S, Sasai Y, Lu B, De Robertis EM. Dorsoventral patterning in Xenopus: inhibition of ventral signals by direct binding of chordin to BMP-4. Cell 1996: 86(4): 589–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Uzel MI, Scott IC, Babakhanlou-Chase H, Palamakumbura AH, Pappano WN, Hong HH, Greenspan DS, Trackman PC. Multiple bone morphogenetic protein 1-related mammalian metalloproteinases process pro-lysyl oxidase at the correct physiological site and control lysyl oxidase activation in mouse embryo fibroblast cultures. J Biol Chem 2001: 276(25): 22537–22543. [DOI] [PubMed] [Google Scholar]
- 79.Rosell-Garcia T, Rodriguez-Pascual F. Enhancement of collagen deposition and crosslinking by coupling lysyl oxidase with bone morphogenetic protein-1 and its application in tissue engineering. Scientific reports 2018: 8(1): 10780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ma HY, N’Diaye EN, Caplazi P, Huang Z, Arlantico A, Jeet S, Wong A, Brightbill HD, Li Q, Wong WR, Sandoval W, Tam L, Newman R, Roose-Girma M, Ding N. BMP1 is not required for lung fibrosis in mice. Scientific reports 2022: 12(1): 5466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hay J, Shahzeidi S, Laurent G. Mechanisms of bleomycin-induced lung damage. Archives of toxicology 1991: 65(2): 81–94. [DOI] [PubMed] [Google Scholar]
- 82.Goumans MJ, Liu Z, ten Dijke P. TGF-beta signaling in vascular biology and dysfunction. Cell research 2009: 19(1): 116–127. [DOI] [PubMed] [Google Scholar]
- 83.Nishimoto SK, Nishimoto M. Matrix gla protein binds to fibronectin and enhances cell attachment and spreading on fibronectin. Int J Cell Biol 2014: 2014: 807013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dufourcq P, Louis H, Moreau C, Daret D, Boisseau MR, Lamaziere JM, Bonnet J. Vitronectin expression and interaction with receptors in smooth muscle cells from human atheromatous plaque. Arterioscler Thromb Vasc Biol 1998: 18(2): 168–176. [DOI] [PubMed] [Google Scholar]
- 85.Parashar A, Gourgas O, Lau K, Li J, Muiznieks L, Sharpe S, Davis E, Cerruti M, Murshed M. Elastin calcification in in vitro models and its prevention by MGP’s N-terminal peptide. J Struct Biol 2021: 213(1): 107637. [DOI] [PubMed] [Google Scholar]
- 86.Yang G, Zhu Z, Wang Y, Gao A, Niu P, Chen L, Tian L. Bone morphogenetic protein 7 attenuates epithelial-mesenchymal transition induced by silica. Hum Exp Toxicol 2016: 35(1): 69–77. [DOI] [PubMed] [Google Scholar]
- 87.Schiff PL Jr. Bisbenzylisoquinoline alkaloids. J Nat Prod 1991: 54(3): 645–749. [DOI] [PubMed] [Google Scholar]
- 88.Liang Y, Xu RZ, Zhang L, Zhao XY. Berbamine, a novel nuclear factor kappaB inhibitor, inhibits growth and induces apoptosis in human myeloma cells. Acta Pharmacol Sin 2009: 30(12): 1659–1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhao W, Bai B, Hong Z, Zhang X, Zhou B. Berbamine (BBM), a Natural STAT3 Inhibitor, Synergistically Enhances the Antigrowth and Proapoptotic Effects of Sorafenib on Hepatocellular Carcinoma Cells. ACS Omega 2020: 5(38): 24838–24847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bardot E, Calderon D, Santoriello F, Han S, Cheung K, Jadhav B, Burtscher I, Artap S, Jain R, Epstein J, Lickert H, Gouon-Evans V, Sharp AJ, Dubois NC. Foxa2 identifies a cardiac progenitor population with ventricular differentiation potential. Nature communications 2017: 8: 14428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Pak B, Schmitt CE, Choi W, Kim JD, Han O, Alsio J, Jung DW, Williams DR, Coppieters W, Stainier DYR, Jin SW. Analyses of Avascular Mutants Reveal Unique Transcriptomic Signature of Non-conventional Endothelial Cells. Front Cell Dev Biol 2020: 8: 589717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Madeddu P FoxA2 hunting research identifies the early trail of mesenchymal differentiation. Stem Cell Res Ther 2013: 4(2): 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.