

Abstract
Background:
Thrombospondin-1 (TSP1), a well-known angiogenesis inhibitor, mediates differential effects via interacting with cell surface receptors including CD36 and CD47. However, the role of TSP1 in regulating lymphangiogenesis is not clear. Our previous study suggested the importance of cell-specific CD47 blockade in limiting atherosclerosis. Further, our experiments revealed CD47 as a dominant TSP1 receptor in lymphatic endothelial cells (LEC). As the lymphatic vasculature is functionally linked to atherosclerosis, we aimed to investigate the effects of LEC TSP1-CD47 signaling inhibition on lymphangiogenesis and atherosclerosis.
Methods:
Murine atherosclerotic and non-atherosclerotic arteries were utilized to investigate TSP1 expression using Western blotting and immunostaining. LEC-specific knockout mice were used to determine the in vivo role of LEC Cd47 in lymphangiogenesis and atherosclerosis. Various in vitro cell-based assays, in vivo Matrigel plug implantation, molecular biological techniques, and immunohistological approaches were employed to evaluate the underlying signaling mechanisms.
Results:
Elevated TSP1 expression was observed in mouse atherosclerotic aortic tissue compared with non-atherosclerotic control tissue. TSP1 at pathological concentrations suppressed both in vitro and in vivo lymphangiogenesis. Mechanistically, TSP1 inhibited VEGF-C-induced AKT and eNOS activation in LEC, and attenuated nitric oxide production. Further, CD47-silencing in LEC prevented the effects of TSP1 on lymphangiogenic AKT-eNOS signaling and lymphangiogenesis. Atheroprone AAV8-PCSK9-injected LEC-specific CD47 knockout mice (CD47ΔLEC) had reduced atherosclerosis in both aorta and aortic root compared with control mice (CD47ΔWT). However, no differences in metabolic parameters including body weight, plasma total cholesterol levels, and fasting blood glucose were observed. Additional immunostaining experiments performed on aortic root cross-sections indicated higher lymphatic vessel density in CD47ΔLEC mice in comparison to controls.
Conclusions:
These findings demonstrate that TSP1 inhibits lymphangiogenesis via activation of CD47 in LEC, and loss of LEC Cd47 attenuates atherosclerotic lesion formation. Collectively, these results identify LEC CD47 as a potential therapeutic target in atherosclerosis.
Keywords: atherosclerosis, thrombospondin-1, CD47, lymphatic endothelial cells, lymphangiogenesis
Graphical Abstract
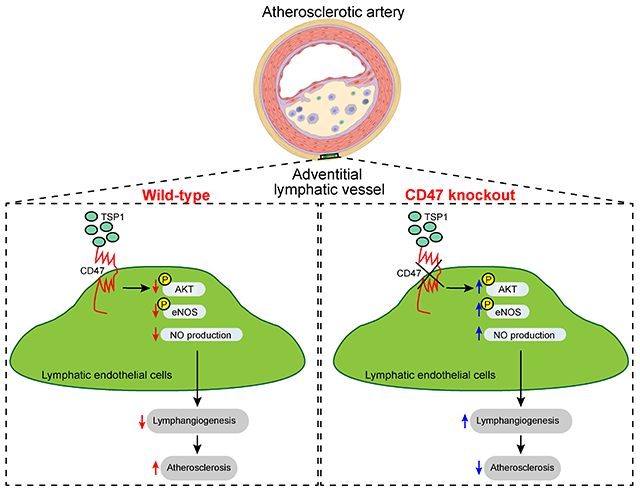
1. Introduction
Atherosclerosis, a chronic vascular inflammatory disease, is characterized by the accumulation of lipid-loaded cells in the subendothelial space of medium- and large-sized arteries and develops at certain focal points. Being an underlying cause of myocardial infarction, stable and unstable angina, and stroke, it accounts for the majority of deaths worldwide 1. Earlier studies have demonstrated that adventitial lymphatic vasculature represents the primary route of cholesterol removal from atherosclerotic vessels and improved lymphatic function leads to atherosclerosis regression 2,3. Further, inhibition of arterial lymphatic drainage via surgical and genetic approaches induces atherosclerotic lesion formation 2,4,5. Moreover, early treatment with lymphangiogenic factor VEGF-C (152S) has been shown to improve lymphatic function and attenuate high-fat diet-induced atherosclerosis in Ldlr knockout mice 6. Therefore, arterial lipid accumulation is an outcome of the disturbed equilibrium between the influx of plasma lipoproteins and the efflux of lipids from the vessel wall via reverse cholesterol transport (RCT) mediated by lymphatic vessels (LV). Despite this information, the molecular factors and downstream signaling mechanisms that govern arterial lymphatic vessel formation (lymphangiogenesis) are understudied.
Thrombospondin-1 (TSP1) is a secreted matricellular glycoprotein, which mediates differential physio- or pathological effects via interacting with integrins and cell surface receptors including CD36 and CD47 7. It regulates various biological processes, including cell proliferation, migration, angiogenesis, and inflammation 8,9. TSP1 expression is low in healthy blood vessels, however, elevated TSP1 levels have been associated with cardiovascular disease including injury-induced neointima formation and atherosclerosis 10–12. Human TSP1 gene polymorphism and its protein expression are linked with the increased risk of myocardial infarction and atherogenesis, respectively 12,13. Additional studies utilizing global genetic deletion of Tsp1 in mice have reported contradictory roles of TSP1 in atherosclerosis development 14,15. However, the mechanisms by which TSP1 contributes to atherosclerotic plaque formation and its cell-specific role in its pathogenesis are understudied. TSP1 after binding to CD47 present in vascular endothelial cells suppresses angiogenesis via blocking required nitric oxide (NO) signaling 16. NO signaling in lymphatic endothelial cells (LEC) is also pivotal for lymphangiogenesis and the maintenance of lymphatic vessels 17. Kojima et al. have shown that the administration of CD47-blocking antibodies in murine models of atherosclerosis inhibits plaque formation via promoting efferocytosis and reduces vascular inflammation in human cancer patients 18,19. We recently reported that global deletion of Cd47 in mice protects from atherosclerosis, however, myeloid cell-specific Cd47 loss augments lesion formation suggesting the importance of cell-specific CD47 blockade in atherosclerosis 20. Nonetheless, the importance of TSP1-induced LEC CD47 activation in regulating arterial lymphangiogenesis and atherosclerosis remains unexplored.
In the current study, we examined the role of LEC TSP1-CD47 signaling axis in governing both in vitro and in vivo lymphangiogenesis and development of atherosclerosis. Our data demonstrate that: 1) TSP1-mediated LEC CD47 activation inhibits lymphangiogenesis and 2) blockade of LEC CD47 signaling suppresses atherosclerosis. Altogether, the findings of the present study identify LEC CD47 as a potential therapeutic target to promote arterial lymphangiogenesis and reduce atherosclerosis.
2. Material and methods
The data that support the findings of this study are available from the corresponding author upon reasonable request. Please see the extended Methods and Major Resources Table in the Supplemental Material.
2.1. Animals
All animal experimental procedures were approved by the Institutional Animal Care and Use Committee of the University of Tennessee Health Science Center (UTHSC), Memphis, TN, and/or Augusta University. The procedures were conducted following the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Eight- to ten-week-old male wild-type C57BL/6J (stock # 000664), Apoe−/− (stock # 002052), and Lyve-1 Cre+ (stock # 012601) mice were procured from the Jackson Laboratory (Bar Harbor, USA). Cd47-floxed (Cd47f/f) mice were cryorecovered from frozen germplasm maintained in the KOMP Repository (Cd47tm1a(KOMP)Mbp, Mouse Biology Program, University of California, Davis) 20. LEC-specific Cd47 knockout mice (Cd47f/f Lyve-1 Cre+/−, referred as Cd47ΔLEC) were generated by crossing Cd47f/f mice with Lyve-1 Cre+ mice. Littermate Cd47f/f Lyve-1 Cre−/− and Lyve-1 Cre+/− were used as controls (Cd47WT). All mice were genotyped by PCR amplification of tail genomic DNA using primers listed in Table S1 and LEC-specific deletion was also confirmed using double immunostaining experiments.
2.2. Atherosclerotic lesion analysis
Male mice were used in the present study because sex hormones in females are known to affect atherosclerotic lesion formation and plasma cholesterol levels 21,22. Male mice were injected with a recombinant adeno-associated viral vector expressing a gain-of-function mutant form of human proprotein convertase subtilisin/kexin type 9 (pAAV-hPCSK9-D374Y, Vigene Biosciences, Rockville, referred as AAV8-PCSK9, 1×1011 VG, i.p.) once 23 and fed a Western diet (Envigo, Indianapolis, IN, TD.88137) for 16 weeks to induce liver-specific low-density lipoprotein receptor (Ldlr) degradation, hypercholesterolemia, and atherosclerosis. Whole-body fat, lean and fluid mass (Bruker Minispec Live Mice Analyzer, LF90), and fasting blood glucose (ReliOn Prime Blood Glucose Monitoring System) were determined. Mice were fasted overnight (16 h) prior to euthanasia to determine fasting blood glucose and plasma total cholesterol levels. Mice were anesthetized by isoflurane inhalation (3%) and blood was collected in a heparinized syringe via cardiac puncture for further analysis. In situ images of the aortic arch and aorta were captured using an Olympus SZX16 stereomicroscope. Mouse aorta and heart were harvested and fixed in 4% paraformaldehyde (PFA) for 24 h. Heart tissue was embedded in the optimum cutting temperature compound (OCT) (Fisher Healthcare, Houston, TX, 23-730-571) and stored at −80 °C until use. Amplex Red cholesterol assay (Molecular Probes, Eugene, OR, A12216) was used to quantify plasma total cholesterol levels. Plasma triglyceride levels were examined using reagents and standards obtained from Wako Chemicals, Richmond, VA.
For en face analysis, whole PFA-fixed aortas were stained with 2% Oil Red O (ORO) solution (Sigma-Aldrich, MO, USA) for 30 minutes at room temperature, washed with 60% isopropanol (30 sec) and H2O (5 min). Then, aortas were opened longitudinally and imaged with an Olympus SZX16 stereomicroscope, and ORO-positive areas were quantified. Further, the upper halves of fixed hearts were dissected and embedded in the OCT compound. Serial aortic root frozen sections (9 μm) were stained with 2% ORO solution, washed with 60% isopropanol and counterstained with hematoxylin (Fisher Healthcare, Pittsburgh, PA, 22-220-100) 20. Images were acquired using an Olympus BX43 phase-contrast microscope to determine the lipid deposition in the aortic sinus. Image analysis was performed utilizing the Image-Pro Plus software (Media Cybernetics, Bethesda, MD).
2.3. Cell culture and gene silencing
Primary human dermal LEC isolated from adult skin and human aortic endothelial cells (HAoEC) were purchased from PromoCell GmbH (Heidelberg, Germany) and cultured in endothelial cell growth medium MV 2 (PromoCell) containing 5% heat-inactivated fetal bovine serum (FBS), 100 IU/mL of penicillin, 100 μg/mL streptomycin, and growth factors bullet kit. Cells were maintained in a humidified incubator at 37°C and used till passage 7. Primary mouse lung LEC were isolated after digestion of lungs with collagenase type 2 (1 mg/mL) for 1 h at 37°C as we described previously 24. LYVE-1-positive cells were separated and cultured.
A smart pool of siRNA for human CD47 (M-019505-01-0005) and a non-targeting control siRNA (D-001210-01-05) were purchased from Horizon Discovery (Cambridge, United Kingdom). Cells were transfected with siRNAs using the TransIT-TKO transfection reagent (Mirus Bio LLC, Madison, WI, USA) according to the manufacturer’s instructions. Cells were used for further experiments 48 h post-transfection and gene silencing was confirmed using qRT-PCR. Protein knockdown was validated by Western blot experiments.
2.4. Western blot
Cells/tissues were lysed in RIPA lysis buffer supplemented with protease and phosphatase inhibitors. Equal amounts of proteins (10-15 μg/lane) were separated using SDS-PAGE gels, transferred onto nitrocellulose membranes (Li-Cor Biosciences, Lincoln, NE), and membranes blocked with Intercept blocking buffer (Li-Cor Biosciences) were probed with the following primary antibodies at indicated concentrations: TSP1 (1:400), vinculin 1 (1:1000), GAPDH (1:2000), pAKTSer473 (1:1000), total AKT (1:1000), peNOSSer1177 (1:1000), Total eNOS (1:1000), pERK1/2Thr202/Tyr204 (1:1000), total ERK1/2 (1:1000), CD47 (1:500), VEGFR3 (1:500), Epsin 1 (1:500), p53 (1:1000) and p21 (1:1000). Total AKT (2920S), pAKTSer473 (4060S), peNOSSer1177 (9571S), Total eNOS (5880S), pERK1/2Thr202/Tyr204 (9101S), total ERK1/2 (4695S) and p21 (2947) were purchased from Cell Signaling Technology (Danvers, MA). CD47 antibody (Novus Biologicals, LLC, Centennial, CO, NBP2-31106) and (Abcam, Cambridge, MA, ab175388) were used. We obtained VEGFR3 (sc-321), epsin 1 (sc-55556), p53 (sc-126), and GAPDH (sc-365062) antibodies from Santa Cruz Biotechnology (Dallas, TX). TSP1 (18304-1-AP) antibody was acquired from Proteintech (Rosemont, IL). Antibody for vinculin 1 (V4505) was purchased from Sigma-Aldrich, Inc. (St. Louis, MO). After washing, membranes were incubated with IRDye-conjugated secondary antibodies (Li-Cor Biosciences) and scanned with the Odyssey CLx/DLx Infrared Imaging System (Li-Cor Biosciences) to detect proteins of interest. The densitometry analysis was performed utilizing the NIH ImageJ software.
2.5. Immunohistochemistry
Paraffin sections were deparaffinized, rehydrated, and underwent antigen retrieval by boiling in citrate buffer at 98°C for 10 min, while frozen sections were washed with PBS twice and permeabilized with 0.1% Triton X-100 (10 min). Then, sections were blocked with 10% goat serum (1 h) at room temperature and incubated with primary antibodies against TSP1 (Proteintech, 18304-1-AP, 1:80), SMA (Abcam, ab7817, 1:100), CD68 (Thermo Fisher Scientific, Rockford, IL, MA5-13324, 1:100), LYVE-1 (Abcam, ab14917, 1:100), CD11c (Thermo Fisher Scientific, 53-0114-80, 1:80), CD45 (Cell Signaling Technology, 70257S 1:70), CD34 (Abcam, ab81289, 1:70), iNOS (Abcam, ab3523, 1:100), Arg1 (Sigma-Aldrich, AV45673, 1:100), podoplanin (R & D Systems, AF3244, 1:40), CD31 (Abcam, ab28364, 1:70), CD3e (Thermo Fisher Scientific, 53-0031-80, 1:80) and CD47 (Novus Biologicals, NBP2-31106, 1:100 or R & D Systems, AF1866, 1:70) overnight at 4 °C. Next, sections were incubated with Alexa Fluor 488/Alexa Fluor 594-labeled secondary antibodies (Life Technologies Corporation, Eugene, OR; A11008, 1:700, A11032, 1:500, A11037, 1:500, A11001, 1:1000 and A11055, 1:400) for 1 h at room temperature, followed by mounting with DAPI containing Fluoromount-G (Thermo Fisher Scientific). Images were captured using a Zeiss 710 inverted confocal microscope.
Mouse aortic root frozen sections were stained with hematoxylin (Fisher Healthcare) and eosin (Fisher Healthcare, 22-220-104) (H & E) and Masson’s trichrome (Richard Allan Scientific LLC, Kalamazoo, MI, 22-110-648) employing standard protocols. Epredia UltraVision LP HRP polymer and DAB Detection System (Richard Allan Scientific LLC, TL015HD) was utilized for staining aortic root sections with CD68 antibody according to the manufacturer’s instructions. Images were acquired using an Olympus BX43 phase-contrast microscope. For ORO, H & E, Masson trichrome, and CD68 immunostaining, four sections per mouse spaced 100 μm apart were stained and analyzed, and mean area of four sections were shown. All image analyses were performed utilizing the Image-Pro Plus software (Media Cybernetics, Bethesda, MD).
2.6. Statistical analysis
All statistical analyses were performed using the GraphPad Prism 9 (La Jolla, CA, USA). The data are represented as mean ± SEM. The sample number (n) for each experiment/group is mentioned in the figure legends. The normality of data was investigated employing the Shapiro-Wilk normality test. Two-tailed Student’s t-test (parametric data) or Mann-Whitney test (nonparametric data) were used to compare to two groups, while more than two groups were analyzed using one- or two-way ANOVA followed by appropriate post hoc test for multiple comparisons where appropriate. Kruskal-Wallis test was used to compare more than two groups of non-parametric data. While using parametric tests to compare groups, it was assumed that all groups have the same standard deviation. A P-value < 0.05 was considered statistically significant.
Results
3.1. TSP1 protein expression is elevated in atherosclerotic arteries
Previous human studies have associated polymorphism in the TSP1 gene and TSP1 protein expression with the increased risk of myocardial infarction and atherosclerosis development, respectively 12,13. Despite these associations, the role of TSP1 in atherosclerosis development is largely unknown. To confirm whether TSP1 expression correlates with atherosclerosis development in mice, we collected murine atherosclerotic and non-atherosclerotic aortic tissue and quantified TSP1 expression using immunoblotting. The presence and absence of atherosclerotic lesions were confirmed with Oil red O (ORO) staining. Western blot experiments showed increased TSP1 protein expression in atherosclerotic inner curvature (IC) of 12-week Western diet-fed Apoe−/− mice compared with plaque-free descending aorta (DA) region (Fig. 1A and 1B).
Fig. 1. TSP1 protein levels are upregulated in murine atherosclerotic arteries.
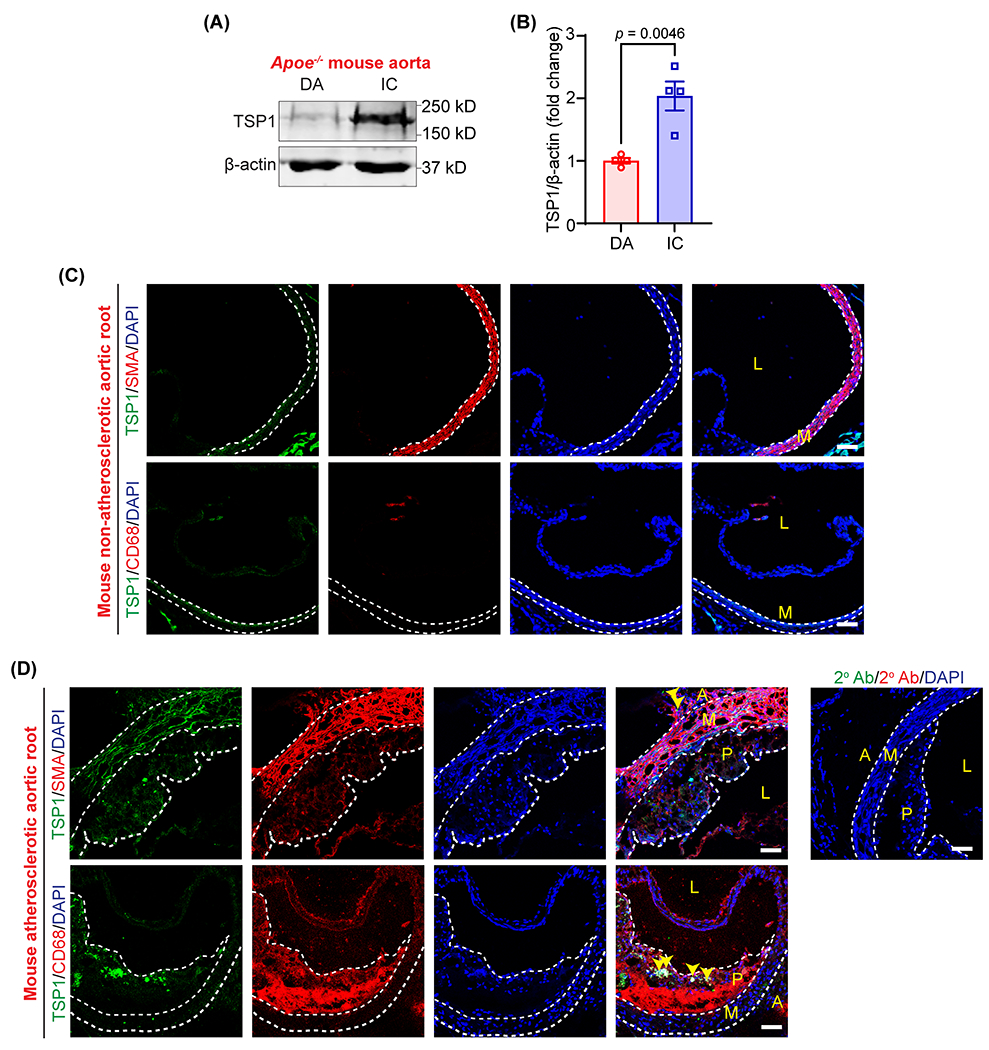
(A) Representative western blot images of TSP1 and β-actin expression in plaque-free DA segments and atherosclerotic IC of male Apoe−/− mice fed with a Western diet for 12 weeks. (B) Bar graph shows mean TSP1 protein levels (n = 4). (C & D) Wild-type and Apoe−/− mice (12 weeks Western diet) aortic sinus cross-sections were immunostained to investigate TSP1 (green), SMA (red), CD68 (macrophage marker, red), expression. Yellow arrowheads indicate TSP1 expression in adventitia and intraplaque areas (n = 3). Scale bar 50 μm. Statistical analyses were performed using a two-tailed unpaired student t-test (B). Data represent mean ± SEM. DA, descending aorta; IC, inner curvature; L, lumen; P, plaque; M, media and A, adventitia.
Next, immunostaining experiments were performed utilizing aortic root cross-sections from non-atherosclerotic wild-type mice and atherosclerotic Apoe−/− mice (12 weeks Western diet) to determine the layer(s) of the atherosclerotic arterial wall expressing TSP1. Immunostaining data demonstrated elevated TSP1 expression in Apoe−/− mice compared with wild-type mice (Fig. 1C and 1D). In particular, TSP1 expression was detected in some intraplaque CD68 (macrophage marker)-positive areas and adventitial layer (Fig. 1G). Taken together, these results demonstrate that TSP1 expression is upregulated in atherosclerotic arteries, and it localizes in the plaque area and adventitial layer (where lymphatic vessels are primarily localized). However, the role of TSP1 in regulating arterial lymphangiogenesis and atherosclerosis development is not known.
3.2. TSP1 suppresses both in vitro and in vivo lymphangiogenesis
Lymphatic vessels play an important role in the removal of cholesterol from atherosclerotic blood vessels via mediating RCT, and disruption of arterial lymphatic drainage induces adventitial inflammation and aggravates atherosclerosis development in hypercholesterolemic mice 2–4. We identified elevated TSP1 expression (Fig. 1C and D) in the adventitial layer of atherosclerotic arteries where LV are mainly present and TSP1 is a potent inhibitor of angiogenesis 7, yet surprisingly, its direct effect on lymphangiogenesis has not been investigated. Therefore, we sought to investigate the role of TSP1 in regulating lymphangiogenesis. First, we performed WST-1 assay to examine the effect of treatment with different TSP1 concentrations on LEC proliferation. As shown in Fig. 2A and S1A, TSP1 at 11 nM and 22 nM concentrations remarkably reduced VEGF-C-induced (100 ng/mL) LEC proliferation. Therefore, we chose 11 nM and 22 nM TSP1 concentrations for further experiments investigating TSP1’s effects on LEC migration and tube formation. Plasma levels of TSP1 in patients with vascular pathologies have been shown elevated (117 to 6,500 ng/mL) compared with control individuals (13 - 137 ng/mL) 25. Therefore, TSP1 concentrations used in the present study are pathologically relevant (5,000 ng/mL = 11 nM). However, exact TSP1 concentrations in the atherosclerotic and non-atherosclerotic arteries are not known. Next, immunostaining experiments were executed to determine Ki67-positive nuclei as a measure of cell proliferation and demonstrated a significantly decreased number of Ki67-positive nuclei in the TSP1-treated group compared with the control group (Fig. 2B). The results of scratch wound healing assay indicated that VEGF-C-stimulated LEC migration was inhibited by TSP1 treatment (Fig. 2C and S1B). Then, we performed Matrigel tube formation assay to examine the effect of TSP1 on the vessel-forming ability of LEC in vitro. Consistent with the findings of proliferation and migration assays, TSP1-challenged cells had reduced ability of tube formation as suggested by decreased tube length and lower number of branching points compared to vehicle-treated cells (Fig. 2D–F and S1C). Additional cell cycle analysis experiments showed increased percentage of cells in the G1 phase and reduced cell percentage in the S phase with TSP1 treatment, suggesting cell cycle inhibition in TSP1-treated cells (Fig. S2). Collectively, these data suggest that TSP1 inhibits lymphangiogenesis in vitro.
Fig. 2. TSP1 suppresses both in vitro and in vivo lymphangiogenesis.
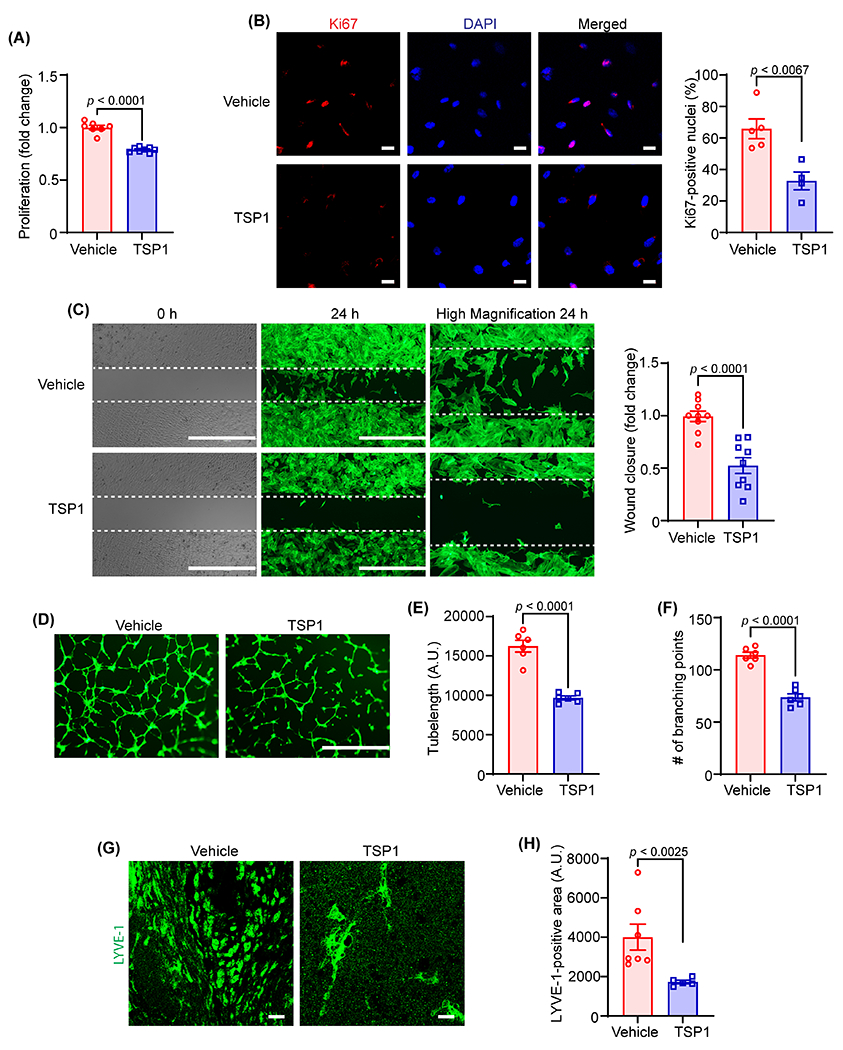
(A) Vehicle- and TSP1-pretreated (22 nM, 4 h) human LEC were stimulated with VEGF-C (100 ng/mL) and proliferation investigated after 48 h using WST-1 assay. Data are representative of three independent experiments performed at least in duplicate. (B) LEC-plated on coverslips were pretreated and stimulated as in (A) for 24 h. Cells were immunostained for Ki67 (red) and nuclei counterstained with DAPI (blue). Images of at least four random microscopic fields were captured. Representative images are shown. Scale bar 20 μm. Bar graph represents the percentage of Ki67-positive nuclei (n = 4-5). (C) LEC migration in response to vehicle (VEGF-C) and TSP1 (VEGF-C+TSP1) was investigated after 24 h using Culture-Insert 2 Well 24 (ibidi USA). Representative images of wounds at 0 h and 24 h are shown. Scale bar 1000 μm. Bar diagram shows quantification of wound closure (n = 9). (D) Vehicle- or TSP1-pretreated LEC were seeded in wells of a Matrigel-coated plate in basal medium containing VEGF-C ± TSP1 and tube formation determined after 6 h. Representative images of tube formation are shown. Scale bar 1000 μm. Images of random fields were captured, and tube length (E) and number of branching points (F) quantified (n = 6). (G-H) Wild-type male mice were injected s.c. with Matrigel solutions premixed with either VEGF-C or VEGF-C+TSP1. Plugs were isolated after 10 days, sectioned and immunostained for LYVE-1. (G) Representative images of LYVE-1 staining of cross-sections of the Matrigel plugs are shown. Scale bar 20 μm. (H) Quantification of LYVE-1-positive area (n = 5-7). Statistical analyses were performed using a two-tailed unpaired student t-test (A-C, E and F) and a Mann-Whitney test (H). Data represent mean ± SEM. LYVE-1, lymphatic vessel endothelial hyaluronan receptor-1.
To further investigate the anti-lymphangiogenic potential of TSP1 in vivo, a well-established in vivo lymphangiogenesis model 26,27, the Matrigel plug assay was employed. Matrigel solution containing VEGF-C with or without TSP1 was subcutaneously injected into wild-type mice on a C57BL/6 background. On day 10 after injection, Matrigel plugs were harvested, fixed, embedded, and processed for immunostaining with LYVE-1 antibody to determine LV density. As expected, there were remarkably reduced LYVE-1-positive areas in plugs containing VEGF-C + TSP1 compared with VEGF-C-comprising plugs (Fig. 2G and H). These results of in vivo and in vitro experiments demonstrate that TSP1 possesses an anti-lymphangiogenic potential.
3.3. CD47 mediates TSP1-induced inhibition of lymphangiogenesis
TSP1 is known to influence cellular responses via binding cell surface receptors mainly CD36 and CD47 28,29. Therefore, we next investigated the mRNA levels of CD47 and CD36 in LEC using quantitative RT-PCR (qRT-PCR) and compared the levels with human aortic endothelial cells. The qRT-PCR data revealed significantly higher expression of CD47 mRNA in LEC in comparison to CD36 (55-fold) (Fig. 3A). Additionally, LEC CD47 transcript levels were significantly higher than those in vascular endothelial cells (Fig. 3A). Consistently, higher expression of CD47 protein was observed in LEC compared with vascular endothelial cells (Fig. S3). These results suggest CD47 as a dominant TSP1 receptor in LEC. Then, to examine the engagement of CD47 in TSP1-stimulated inhibition of lymphangiogenesis, CD47 was silenced using a gene-specific siRNA pool (Fig. 3B). As expected, CD47-silenced LEC had a higher proliferation rate compared with control siRNA-treated cells with or without TSP1 challenge (Fig. 3C). Interestingly, CD47 silencing abrogated the effects of TSP1 on LEC migration (Fig. 3D) and tube formation (Fig. 3E–G), however, migration and tube formation ability of CD47 knockdown cells were comparable to control cells after vehicle treatment. The results of Matrigel plug assay showed increased LYVE-1 staining in plugs containing VEGF-C + TSP1+CD47-blocking antibody compared with VEGF-C + TSP1-containing plugs (Fig. 3H). However, no differences in CD45 (immune cell)-positive and CD34 (angiogenesis)-positive areas were observed in plugs among different groups (Fig. S4). Taken together, these data demonstrate that CD47 is involved in mediating anti-lymphangiogenic effects of TSP1.
Fig. 3. CD47 mediates TSP1-induced inhibition of lymphangiogenesis.
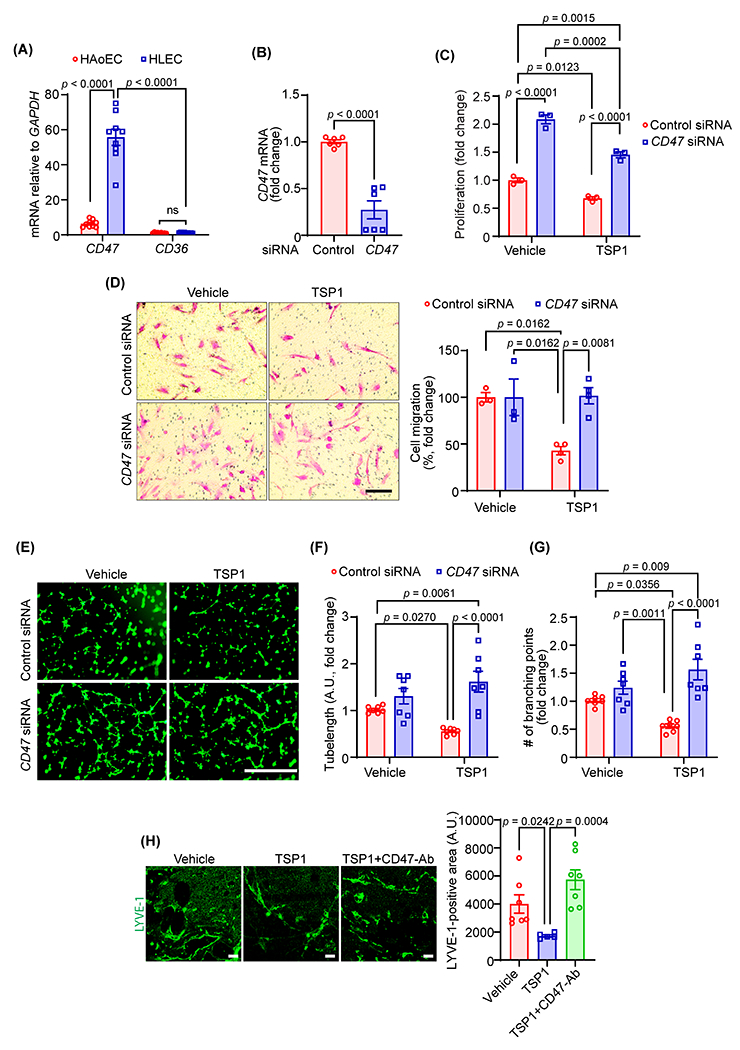
(A) Quantitative real-time PCR was performed to determine the relative mRNA levels of CD47 and CD36 in human LEC (HLEC) and human aortic endothelial cells (HAoEC). Bar graph represents mRNA levels in comparison to CD36 (n = 9). (B) Control and CD47 siRNA-treated LEC (48 h) were utilized to quantify CD47 transcript expression using qRT-PCR (n = 6). (C) WST-1 assay was conducted to investigate the effects of CD47-silencing on LEC proliferation in response to TSP1 treatment as described in Fig. 2A. Data are representative of three independent experiments performed at least in quadruplicate. (D) Control and CD47-silenced LEC were used to evaluate cell migration. Scale bar 200 μm. Bar graph represents the percentage of migrated cells (n = 3-4). (E-G) Control and CD47-silenced cells were pretreated as in Fig. 2D and seeded in wells of a Matrigel-coated plate in basal medium containing VEGF-C ± TSP1 and tube formation determined. Representative images of tube formation are shown (E). Scale bar 1000 μm. Tube length (F) and number of branching points (G) quantified (n = 7). (H) Wild-type male mice were injected s.c. with Matrigel solutions premixed with either VEGF-C, VEGF-C+TSP1+IgG or VEGF-C+TSP1+CD47-blocking antibody. Plugs were isolated after 10 days, sectioned and immunostained for LYVE-1. Representative images of LYVE-1 staining of the cross-sections of the Matrigel plugs and quantification of LYVE-1-positive area are shown (n = 5-7). Scale bar 20 μm. Statistical analyses were performed using two-way ANOVA (A, C, D, F and G) with Bonferroni’s (A), Tukey’s (C, F and G) and Sidak’s (D) multiple comparisons test, two-tailed unpaired student t-test (B) and Kruskal-Wallis test for multiple comparisons (H). Data represent mean ± SEM.
3.4. TSP1-induced CD47 activation blocks VEGF-C-stimulated lymphangiogenic signaling
Activation of VEGFR3 by its ligand VEGF-C is critical for lymphatic vessel formation and leads to downstream phosphorylation of molecules including AKT, ERK, and eNOS 30. Therefore, we examined whether TSP1 regulates VEGF-C-stimulated pro-lymphangiogenic signaling in LEC. Briefly, we treated human LEC with TSP1 (22 nM, 16 h), stimulated with VEGF-C (100 ng/mL, 15 min), and subjected cell lysates to immunoblotting. Cells were induced with VEGF-C for 15 min because maximum phosphorylation of AKT and ERK1/2 was observed after 15 min of incubation with VEGF-C (Fig. S5). As expected, VEGF-C treatment induced phosphorylation of AKT (Ser-473), eNOS (Ser-1177), and ERK1/2 (Thr-202/Tyr-204) in human LEC (Fig. 4A and D). However, TSP1-treated cells had no increase in AKT and eNOS activation following VEGF-C stimulation. No differences in ERK1/2 phosphorylation were observed between VEGF-C and VEGF-C+TSP1-treated cells (Fig. 4A–D). Further, TSP1 treatment suppressed cell proliferation and migration following stimulation with VEGFR3-specific mutant VEGF-C (C156S) (Fig. S6A and S6B). TSP1 also reduced VEGF-C (C156S)-stimulated AKT and eNOS phosphorylation compared with control cells (Fig. S6C). In separate experiments, we determined the role of TSP1 in regulating the expression of VEGFR3. As shown Fig. S7A, TSP1 treatment significantly reduced VEGFR3 protein levels in LEC, suggesting that TSP1 may inhibit lymphangiogenesis via downregulating VEGFR3 expression. However, no differences in FLT4 (VEGFR3) mRNA levels were observed between control and TSP-1 treated cells (Fig. S7B).
Fig. 4. TSP1-induced CD47 activation blocks VEGF-C-stimulated lymphangiogenic signaling.
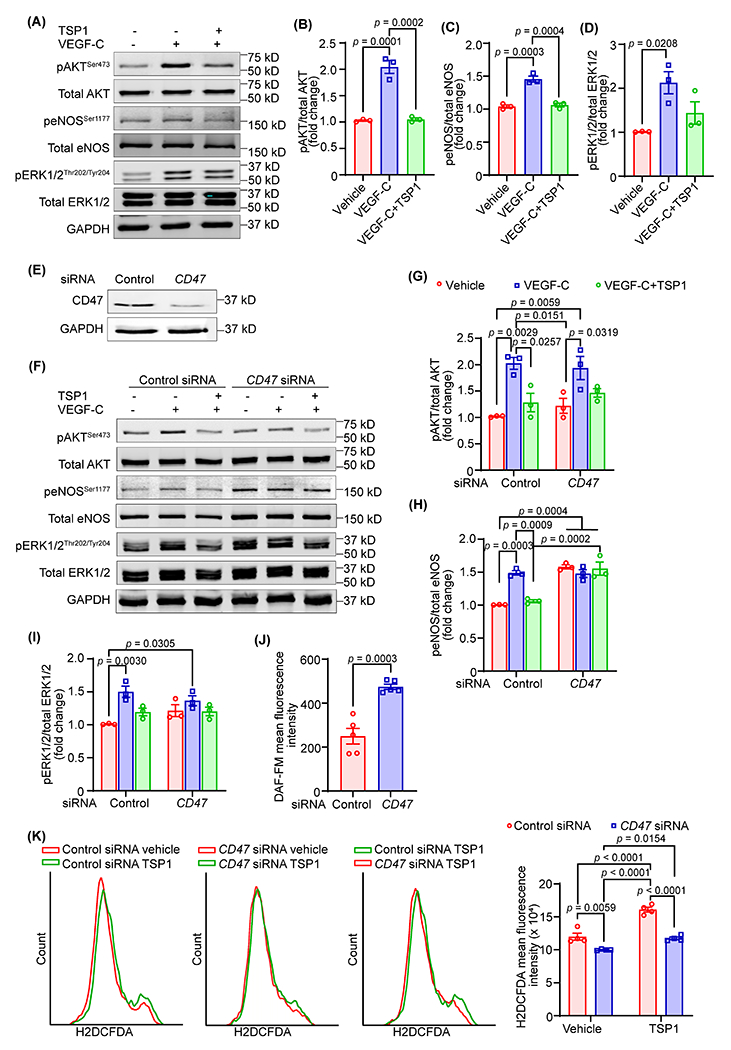
(A-D) LEC were pretreated with TSP1 (22 nM, 16 h) in 0.5% FBS containing basal media MV2, stimulated with VEGF-C (15 min), and cell lysates subjected to western blot analysis. (A) Representative western blot images are shown. (B-D) Bar diagrams represent mean protein levels expressed as a ratio of phospho to total proteins, AKT (B), eNOS (C), and ERK1/2 (D) (n = 3). (E) LEC were treated with control or CD47-siRNA (48 h) and immunoblotting done to determine CD47 expression (ab175388). (F-I) Control or CD47-siRNA treated cells were treated as in Fig. 4A and Western blot experiments executed. (F) Representative western blot images are shown. (G-I) Bar diagrams represent pAKT/total AKT (G), peNOS/total eNOS (H), and pERK1/2/total ERK1/2 (I) (n = 3). (J) Control or CD47-silenced LEC were pretreated with TSP1 (22 nM, 4 h), stimulated with VEGF-C (100 ng/mL, 1 h) and analyzed for nitric oxide (NO) production using DAF-FM diacetate. (K) Control or CD47-silenced LEC were pretreated with vehicle or TSP1 (1 h), incubated with H2DCFDA solution and fluorescence analyzed using flow cytometry. Statistical analyses were performed using one-way ANOVA (B-D), two-way ANOVA with Tukey’s test for multiple comparisons (G-I and K), and two-tailed unpaired student t-test (J). Data represent mean ± SEM.
As TSP1 suppressed LEC proliferation and caused cell cycle arrest, we next evaluated the expression of proteins including p21 and p53, which inhibit the cell cycle. Elevated levels of p53 and p21 have been shown to induce cell cycle arrest via inhibiting cyclin-dependent kinase (CDK) activity 31. Treatment with TSP1 stimulated the expression of CDK inhibitors p53 and p21 in LEC after 24 h (Fig. S7C). Further, we investigated the effects of TSP1 treatment on the expression of epsin1, which degrades VEGFR3 in LEC 26. Immunoblotting data did not show any differences in epsin 1 levels after TSP1 treatment (Fig. S7D).
Since CD47 is the highest expressing TSP1’s receptor in LEC and silencing of CD47 in LEC abrogated the effects of TSP1 on lymphangiogenesis, we next investigated the role of CD47 in TSP1-mediated inhibition of AKT and eNOS activation. CD47 silencing prevented TSP1-induced suppression (16 h) of AKT and eNOS phosphorylation in LEC (Fig. 4E–I). The time course TSP1 treatment experiments with control and CD47-knockdown cells showed improved AKT and ERK1/2 phosphorylation in CD47-siRNA treated cells after 8 h, however no differences were observed after 4 h TSP1 treatment (Fig. S8). Interestingly, basal eNOS activation was significantly higher in CD47-silenced cells compared with control siRNA-treated cells (Fig. 4F and H). Further, we noticed increased NO production by CD47-silenced cells following TSP1 treatment in comparison to TSP1-treated control cells (Fig. 4J). As NO bioavailability can be decreased due to enhanced ROS production, additional experiments were performed to measure ROS generation by control and CD47-silenced LEC. The data demonstrated reduced ROS production with CD47 silencing (Fig. 4K). Altogether, these results suggest that TSP1 reduces lymphangiogenesis via CD47-mediated suppression of AKT and eNOS activation.
3.5. LEC-specific deletion of Cd47 suppresses atherosclerosis
The lymphatic vasculature represents the primary route of cholesterol removal from atherosclerotic vessels and blockade of arterial lymphatic drainage aggravates atherosclerosis, and knockdown of CD47 from human LEC prevents TSP1-induced inhibition of lymphangiogenesis (Fig. 3). Therefore, to investigate the involvement of LEC Cd47 in the formation of atherosclerotic lesions, we generated LEC-specific Cd47 knockout mice (Cd47f/f Lyve-1 Cre+/−, Cd47ΔLEC) by breeding Cd47f/f mice with Lyve-1 Cre+ mice (Fig. S9A). Deletion of Cd47 protein from LYVE-1+ lymphatic vessel was confirmed using immunostaining experiments (Fig. S9B and S9C). Further, Cd47 mRNA levels were reduced >90% in LEC isolated from Cd47ΔLEC mice compared with those from control mice (Fig. S9D and S9E). Eight- to ten-weeks old Cd47ΔLEC and control Cd47WT mice were injected with AAV8-PCSK9 and fed a Western diet for 16 weeks. En face ORO staining of aortas collected from these mice showed substantially reduced atherosclerosis in Cd47ΔLEC mice compared with Cd47WT control mice (Fig. 5A–C). However, no significant differences in plasma total cholesterol, plasma triglycerides, fasting blood glucose levels, weight gain, and body composition (fat, lean and fluid mass) were observed between Cd47WT and Cd47ΔLEC mice (Fig. 5D–G and S10A). Additional hematoxylin-eosin and ORO staining performed on aortic sinus cross-sections from these mice demonstrated smaller lesion areas and reduced lipid accumulation in Cd47ΔLEC mice compared with control mice (Fig. 5H–J). Further histological staining conducted to characterize the aortic root atherosclerotic lesions exhibited increased collagen content and reduced CD68-positive area in Cd47ΔLEC mice in comparison to Cd47WT mice (Fig. 5H, 5K and 5L). Interaction of CD47 with phagocyte SIRPα negatively regulates efferocytic clearance of apoptotic cells, which contributes to necrotic core formation in atherosclerotic arteries 18. Therefore, we quantified the necrotic area in aortic root cross-sections and found no differences between the groups, suggesting no effects of LEC Cd47 deletion on necrotic core formation and efferocytosis (Fig. 5H, 5M and S10B). Next, to determine the effects of LEC-specific Cd47 deletion on systemic inflammation in hypercholesterolemic atherosclerotic mice, 13 cytokines of commercially available mouse inflammation panel (BioLegend) were measured. Plasma levels of pro-/anti-inflammatory cytokines including Il-12p70, Il-23, Il-1α, Ifn-γ, Tnf-α, Mcp-1, Il-6, Il-27, Il-17A, Ifn-β, Gm-csf and Il-1β, except Il-10, were not different between Cd47WT and Cd47ΔLEC mice (Fig. S11). Altogether, these results indicate that deletion of Cd47 selectively in LEC attenuates the development of atherosclerosis in hypercholesterolemic mice.
Fig. 5. LEC-specific Cd47 deficiency reduces atherosclerotic lesion formation.
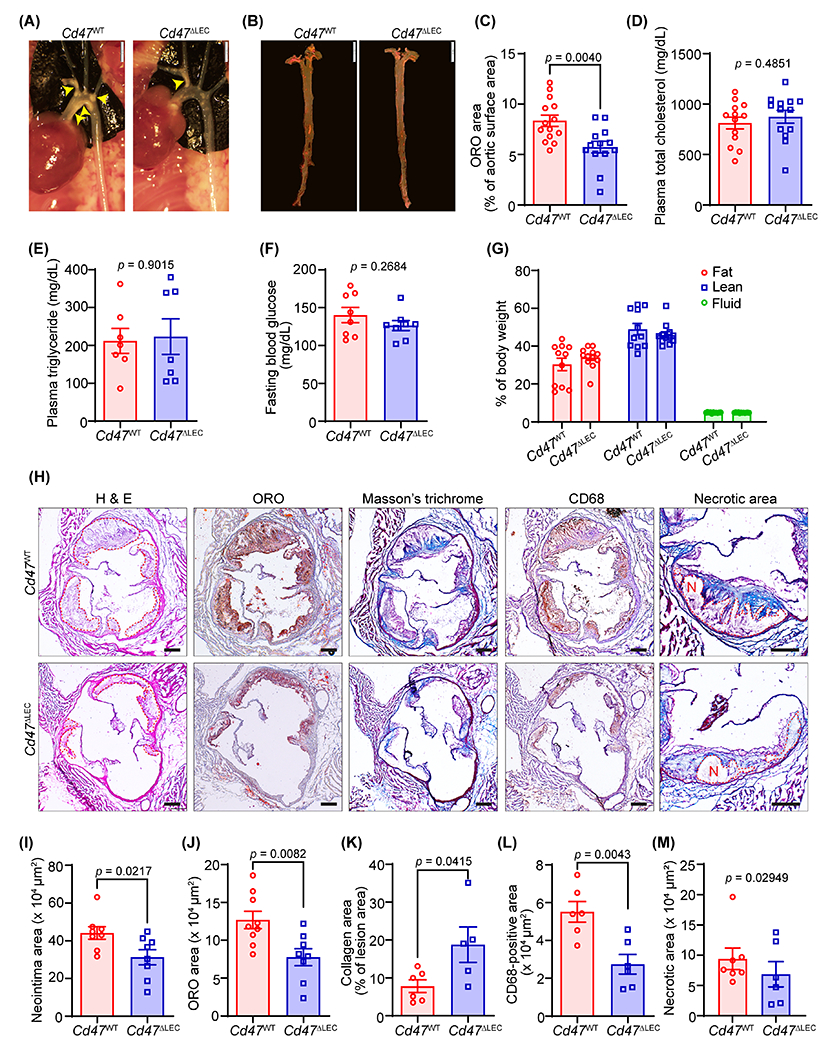
(A-M) Male Cd47WT and Cd47ΔLEC mice were injected with AAV8-PCSK9 i.p., fed a Western diet for 16 weeks and atherosclerosis analyzed. (A) Representative in situ images of aortic arch (yellow arrowheads point to atherosclerotic lesions). Scale bar 2 mm. (B) Representative en face ORO staining of aorta. Scale bar 5 mm. (C) Quantification of plaque area in aorta (n = 13-14). (D-G) Bar diagrams show plasma total cholesterol (D), plasma triglycerides (E), fasting blood glucose levels (F), and body composition (fat, lean, and fluid mass, G) (n = 8-13). (H) Representative images of aortic root cross-sections stained with H & E (neointima area), ORO (lipid accumulation), Masson’s trichrome (collagen content), CD68 (macrophage burden) and necrotic area (encircled in red). Scale bar 200 μm. (I-M) Bar diagrams show neointima area (I), lipid deposition (J), collagen content (K), macrophage accumulation (L), and necrotic area (M) in aortic root sections (n = 5-9). Statistical analyses were performed using a two-tailed unpaired Mann-Whitney test (C, E and M), two-tailed unpaired student t-test (D, F and I-L), and two-way ANOVA followed by Sidak’s post hoc test (G). Data represent mean ± SEM.
3.6. LEC-specific Cd47 knockout mice have increased arterial LV density
To investigate the pro- and anti-inflammatory nature of lesional macrophages, aortic root cross-sections of Cd47WT and Cd47ΔLEC mice were immunostained for CD68 and inducible nitric oxide synthase (iNOS, M1 marker) or arginase 1 (Arg1, M2 marker). Immunostaining analysis demonstrated elevated Arg1 expression in plaque CD68+ cells of Cd47ΔLEC mice compared with Cd47WT control mice, however, similar expression of iNOS was observed (Fig. 6A and 6B). These data suggest that Cd47 deficiency in LEC promotes M2 (pro-resolving) polarization of lesional macrophages. To determine whether decreased atherosclerotic lesion formation in LEC-specific Cd47 knockout mice associates with increased adventitial lymphangiogenesis, immunostaining was executed on aortic sinus cross-sections of hypercholesterolemic Cd47WT and Cd47ΔLEC mice (16 weeks Western diet). Consistent with increased lymphangiogenesis in in vitro and in vivo Matrigel plug assays with inhibition of CD47-mediated signaling, quantitation of LYVE-1-positive vessel-like structures revealed increased adventitial LV density in Cd47ΔLEC mice compared with Cd47WT mice (Fig. 6C, 6D and S10C). However, LV densities in hepatic tissue and ear dermal sheets were comparable (Fig. S12). We further examined the accumulation of CD11c+ dendritic cells and CD3+ T cells in atherosclerotic lesions, however, no significant differences were observed between the two groups of mice (Fig. S13). Collectively, these data suggest that LEC-specific Cd47 deletion promotes pro-resolving macrophage phenotype and augments adventitial LV density in hypercholesterolemic mice.
Fig. 6. LEC-specific deletion of Cd47 in mice increases arterial LV density.
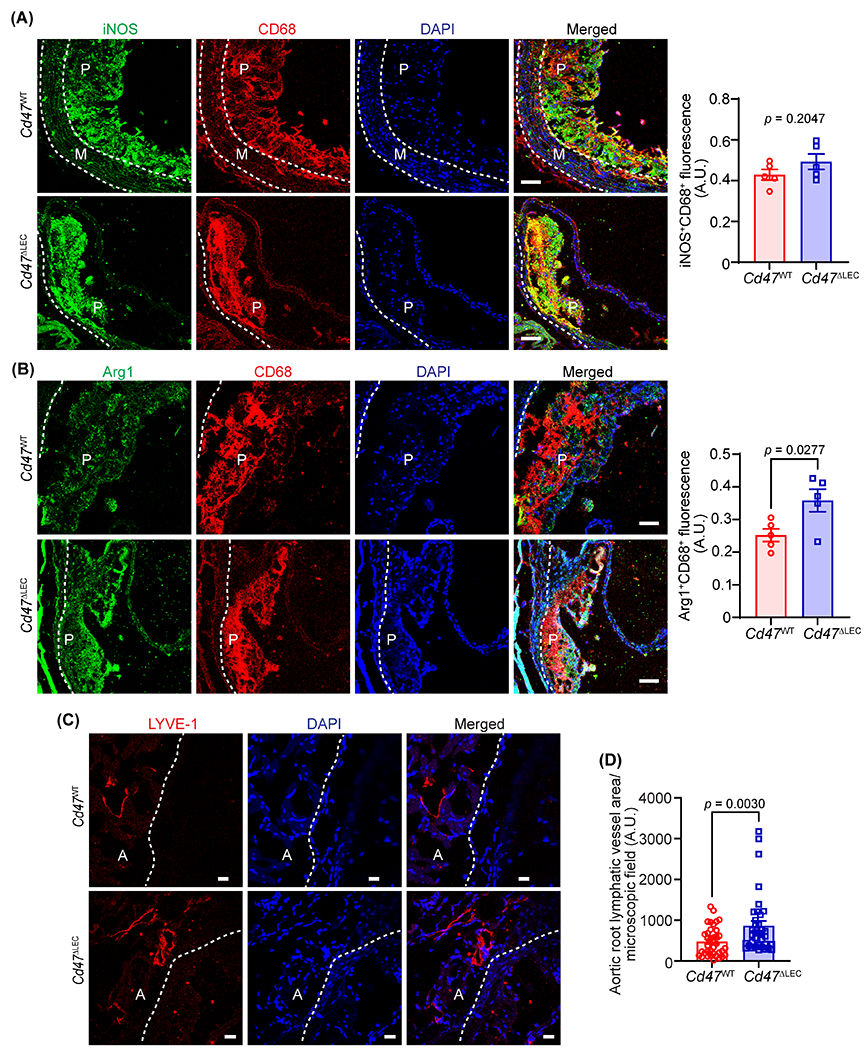
(A-D) Aortic root cross-sections from male AAV8-PCSK9-injected Cd47WT and Cd47ΔLEC mice (16 weeks Western diet) were immunostained for CD68, iNOS, Arg1 and LYVE-1. Nuclei were counterstained with DAPI (blue). Representative confocal images of iNOS (green, A) and CD68 (red); Arg1 (green, B) and CD68 (red) Scale bar 50 μm; LYVE-1 staining (red, C) Scale bar 20 μm, are shown (n = 5-6). Statistical analyses were performed using a two-tailed unpaired student t-test (A and B) and a Mann-Whitney test (C). Data represent mean ± SEM. P: plaque, A: adventitia and M: media.
3. Discussion
The lymphatic vasculature is indispensable for the removal of extracellular fluid, extravasated leukocytes, and inflammatory cytokines/lipids from the inflamed tissue like arterial wall to the surrounding lymph nodes and back to the systemic circulation, thereby contributing to the attenuation of inflammatory processes, resolution of inflammation and reduced atherosclerosis 2,3,32. In addition, hypercholesterolemia, a causative factor for atherosclerosis development, has been shown to degenerate LV and impair lymphatic drainage, which consequently leads to the accumulation of inflammatory cells in peripheral tissue 33,34. In the present study, for the first time, we explored the role of the TSP1-CD47 signaling axis in regulating lymphangiogenesis and atherosclerosis development. TSP1 expression was observed remarkably elevated in atherosclerotic arteries, particularly in the plaque area and adventitial layer. We found that TSP1 inhibits lymphangiogenesis via CD47-mediated blockade of lymphangiogenic signaling. Moreover, utilizing the novel LEC-specific Cd47-deficient mice, we report that deletion of Cd47 from LEC in mice suppresses atherosclerosis which is accompanied by increased adventitial lymphatic vessel density. Collectively, these findings suggest that LEC CD47 activation limits lymphangiogenesis and promotes atherosclerosis.
Various types of vascular cells and immune cells including VSMC, endothelial cells, adventitial fibroblasts, macrophages, and dendritic cells, which are present in the arterial and periarterial regions, express TSP1 35–37. Arterial TSP1 levels upregulate in various vascular pathologies such as abdominal aortic aneurysm, injury-induced neointima formation, and atherosclerosis 10–12,38 and conflicting roles of TSP1 in atherosclerosis development have been reported 14,15. Further, TSP1 is a potent endogenous inhibitor of angiogenesis 39 and yet knowledge of its ability to inhibit arterial lymphangiogenesis and the signaling mechanisms involved remains scant. To dissect the role of TSP1 in atherosclerotic lesion formation and manipulating arterial lymphangiogenesis, we first investigated the expression and localization of TSP1 protein in murine atherosclerotic vessels. Consistent with increased TSP1 expression in other vascular diseases, upregulated TSP1 protein expression was observed in mouse atherosclerotic arteries compared with non-atherosclerotic vascular tissue 11,40,41. We noted elevated TSP1 expression in the intraplaque CD68-positive areas and the adventitial layer of the arterial wall. Increased TSP1 expression has a pathophysiological significance as TSP1 treatment directly inhibited VEGF-C-stimulated LEC proliferation, migration, and both in vitro and in vivo lymphangiogenesis. A previous study reported that TSP1 via binding to CD36 present on macrophages suppresses the expression of lymphangiogenic factors, VEGF-C and VEGF-D, and consequently, reduces inflammation-induced corneal lymphangiogenesis 42. However, the physiological importance of TSP1-CD36 signaling in lymphangiogenesis has not been explored. Cifarelli et al. demonstrated reduced VEGF-C-induced lymphangiogenesis by CD36-silenced LEC compared with control cells 43. Conversely, the deletion of CD36 abrogated oxidized LDL-induced inhibition of lymphangiogenesis 34. These findings predict the differential effects of LEC CD36 deficiency depending on the presence and concentration of various pro- and anti-lymphangiogenic factors in the microenvironment.
CD47 serves as a cognate receptor of TSP1, and its expression is upregulated in atherosclerotic arteries 18. Our results demonstrated CD47 as a major TSP1 receptor in LEC compared with CD36, and knockdown of CD47 in LEC and treatment with CD47-blocking antibody prevented the effects of TSP1 on lymphangiogenesis. Similarly, Hawighorst et al. showed little to no expression of CD36 receptor in LYVE-1+ LEC present in murine skin tumor tissue and human skin 44. However, we believe that LEC also have significant expression of CD36 as shown by us 34 and other groups 43,45 using both in vitro and in vivo studies. CD36 is a scavenger receptor, which helps in lipid internalization by various cell types including LEC. It has been shown by Wong et al that LEC mainly utilize fatty acid oxidation for their energy needs 45. LEC fatty acid oxidation promotes LEC proliferation as it provides acetyl-CoA, which helps to sustain the Krebs cycle and dNTP synthesis for cell division. Additional experiments performed using control- and CD47-siRNA-treated cells suggested that TSP1 suppresses VEGF-C-stimulated lymphangiogenic signaling involving AKT and eNOS phosphorylation in control cells, but not in CD47-silenced LEC. Exposure to TSP1 led to a significant increase in the expression of cell cycle inhibitors including p21 and p53. These data indicate that TSP1 is a potent lymphangiogenesis inhibitor and induces LEC senescence. Similarly, Gao et al. reported that TSP1 accelerates brain endothelial cell senescence in a CD47-dependent manner 46. It is possible that blockade of TSP1-CD47 signaling in LEC via CD47 deletion/blocking antibodies may contribute to LEC self-renewal via upregulating the expression of c-myc 47. However, we cannot exclude the engagement of other TSP1’s receptor(s) like CD36 or integrins in TSP1-mediated inhibition of LEC proliferation as a significant decrease in cell growth was observed in CD47-silenced cells following TSP1 treatment. TSP1 treatment downregulated VEGFR3 expression in LEC without affecting FLT4 (VEGFR3) mRNA and epsin 1 protein levels. TSP1 exposure may elevate the expression of other epsin proteins, which cause VEGFR3 degradation in the Golgi compartment 26 and/or promotes proteasomal- and autophagy-mediated VEGFR3 degradation 48. Oxidative stress via reducing NO bioavailability suppresses lymphangiogenesis 26. Moreover, CD47 deficiency in LEC improved NO production following TSP1 exposure and reduced TSP1-induced ROS production. Consistently, previous studies have shown that TSP1 reduces NO production in vascular endothelial cells and promotes ROS production in VSMC, endothelial cells, and macrophages 49–51. Besides, it has been shown that TSP1 via stimulating ROS production increases p53 levels and inhibits cell growth 52. However, it is not known whether treatment with antioxidants and/or NO donors can prevent TSP1-induced inhibition of lymphangiogenesis.
CD47 is a ubiquitously expressed protein and has pleiotropic effects in various cell types via interacting with multiple binding partners including TSP1, SIRPα, and integrins 53. Global CD47-deficient mice and atheroprone mice treated with CD47-blocking antibodies are protected from atherosclerosis, however, myeloid cell-specific CD47 loss augments lesion formation suggesting the significance of cell-specific CD47 blockade in atherosclerosis 18,20. We investigated the role of lymphatic endothelium Cd47 in atherosclerosis via generating novel LEC-specific Cd47 knockout mice and showed that LEC CD47 contributes to atherosclerotic lesion formation without affecting metabolic profile. Upregulated CD47 expression in atherosclerotic arteries inhibits phagocytosis, induces necrotic core formation, increase inflammation, and exacerbate atherosclerosis 18,54. Nonetheless, comparable necrotic core areas in Cd47WT and Cd47ΔLEC mice were observed, which demonstrates no role of LEC CD47 in regulating efferocytosis. Additionally, no differences in plasma proinflammatory cytokine levels were detected. Interestingly, we found reduced Il-10 levels in plasma of Cd47ΔLEC mice. Further experiments revealed reduced arterial inflammation in Cd47ΔLEC mice compared with control mice. CD47-SIRPα interaction plays important roles in regulating cell adhesion and migration. Deletion of Cd47 in LEC may affect transmigration of immune cells into LV.
Lymphatic vessels play important role in the maintenance of tissue homeostasis and are induced by inflammation to aid in the drainage of inflammatory cells, cytokines, and fluids, thereby promoting the resolution of inflammation. During atherogenesis, lymphangiogenesis is induced due to an increased concentration of lymphangiogenic factors like VEGF-C and VEGF-D 55, but elevated levels of anti-lymphangiogenic factors in the advanced plaques prevent lymphatic vessel formation and/or regress them 56. Our immunostaining analysis performed on aortic root cross-sections showed increased adventitial LV density with LEC-specific Cd47 deletion without affecting the accumulation of intraplaque dendritic cells and T-cells. Lymphatic vessels need to be functional to play their beneficial role in inflammatory diseases like atherosclerosis. None of the currently available methodologies allow the direct measurement of LV function in the adventitial layer of murine aortic arch inner curvature (area of increased atherosclerosis due to disturbed flow) and the aortic sinus. We speculate that there may be an improvement in lymphatic function with LEC Cd47 loss. It would be interesting to determine the number of immune cells in the proximity of adventitial LV as an indirect measure of lymphatic function. Most of the previous studies evaluated lymphatic function either in dermal tissue or the peritoneal cavity and associated it with RCT and atherosclerosis development. Martel et al. using aorta transplantation experiments showed reduced RCT in Apoe−/− mice treated with anti-lymphangiogenic VEGFR-3-blocking antibody compared with control antibody-treated mice, suggesting the critical role of lymphatics in macrophage RCT from the arterial wall 2. Yeo et al. recently devised a novel method to investigate the role of abdominal periaortic lymphatic drainage in descending aorta atherosclerosis and reported increased atherosclerosis with impaired drainage 3. Future functional studies using new innovative tools are required to investigate lymphatic function in the different regions of aortic arch and medium-sized arteries and determine its association with atherosclerosis development. Further, to fully understand the role of TSP1-CD47 signaling in regulating lymphatic function in mice, it will be important to quantify the levels of TSP1 in lymph fluid. As Lyve-1 is also expressed in a subset of M2-like aortic macrophages and Lyve-1+ macrophages affect arterial wall remodeling, it remains to be investigated whether deletion of Cd47 utilizing Lyve-1 Cre mice alters arterial stiffness and collagen deposition 57. It is possible that in vivo deletion of Cd47 from M2-like macrophages in the murine models utilized in the present study increased anti-inflammatory capacity of macrophages via blocking TSP1-CD47 signaling. Though we did not see any differences in the expression of CD47 in lesional macrophages between Cd47WT and Cd47ΔLEC mice, the role of macrophage CD47 may need to be ruled out using other LEC-specific Cre mouse line. Another limitation of the present study includes not determining the activation of LEC eNOS and AKT in our mouse models.
In summary, the present study suggests that TSP1 via activation of LEC CD47 restricts lymphangiogenesis and deletion of Cd47 in lymphatic endothelium increases arterial lymphangiogenesis and attenuates atherosclerosis. These findings demonstrate LEC CD47 inhibition as a potential therapeutic target to promote arterial lymphangiogenesis and reduce atherosclerosis.
Supplementary Material
Highlights.
Thrombospondin-1-induced lymphatic endothelial cell CD47 activation inhibits lymphangiogenesis.
CD47 deletion in lymphatic endothelial cells augments both in vitro and in vivo lymphangiogenesis.
Lymphatic endothelial cell-specific CD47 deletion suppresses atherosclerotic lesion formation.
Acknowledgments
We are very thankful to Dr. Gabor Csanyi, Vascular Biology Center, Augusta University for his feedback on the study design. In addition, we thank Esther Magdalena Marquez Wilkins, Neuroscience Imaging Center, UTHSC, for her help with confocal microscopy.
Sources of Funding
This work was supported by the National Institutes of Health (NIH) grants (K99HL146954 and R00HL146954), American Heart Association Postdoctoral Fellowship (17POST33661254) and UTHSC-College of Pharmacy Seed Research Grant awarded to BS, and NIH grant (R01AA028806) given to MG.
Nonstandard Abbreviations and Acronyms
- TSP1
thrombospondin-1
- LEC
lymphatic endothelial cell
- LV
lymphatic vessel
- IC
inner curvature of aortic arch
- DA
descending aorta
- AAV
adeno-associated virus
Footnotes
Disclosures
None.
References
- 1.Lawton JS, Tamis-Holland JE, Bangalore S, Bates ER, Beckie TM, Bischoff JM, Bittl JA, Cohen MG, DiMaio JM, Don CW, et al. 2021 ACC/AHA/SCAI Guideline for Coronary Artery Revascularization: Executive Summary: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation. 2022;145:e4–e17. doi: 10.1161/CIR.0000000000001039 [DOI] [PubMed] [Google Scholar]
- 2.Martel C, Li W, Fulp B, Platt AM, Gautier EL, Westerterp M, Bittman R, Tall AR, Chen SH, Thomas MJ, et al. Lymphatic vasculature mediates macrophage reverse cholesterol transport in mice. J Clin Invest. 2013;123:1571–1579. doi: 10.1172/JCI63685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yeo KP, Lim HY, Thiam CH, Azhar SH, Tan C, Tang Y, See WQ, Koh XH, Zhao MH, Phua ML, et al. Efficient aortic lymphatic drainage is necessary for atherosclerosis regression induced by ezetimibe. Sci Adv. 2020;6. doi: 10.1126/sciadv.abc2697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rademakers T, van der Vorst EP, Daissormont IT, Otten JJ, Theodorou K, Theelen TL, Gijbels M, Anisimov A, Nurmi H, Lindeman JH, et al. Adventitial lymphatic capillary expansion impacts on plaque T cell accumulation in atherosclerosis. Sci Rep. 2017;7:45263. doi: 10.1038/srep45263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vuorio T, Nurmi H, Moulton K, Kurkipuro J, Robciuc MR, Ohman M, Heinonen SE, Samaranayake H, Heikura T, Alitalo K, et al. Lymphatic vessel insufficiency in hypercholesterolemic mice alters lipoprotein levels and promotes atherogenesis. Arterioscler Thromb Vasc Biol. 2014;34:1162–1170. doi: 10.1161/ATVBAHA.114.302528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Milasan A, Smaani A, Martel C. Early rescue of lymphatic function limits atherosclerosis progression in Ldlr(−/−) mice. Atherosclerosis. 2019;283:106–119. doi: 10.1016/j.atherosclerosis.2019.01.031 [DOI] [PubMed] [Google Scholar]
- 7.Bornstein P. Thrombospondins function as regulators of angiogenesis. J Cell Commun Signal. 2009;3:189–200. doi: 10.1007/s12079-009-0060-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bornstein P. Thrombospondins as matricellular modulators of cell function. J Clin Invest. 2001;107:929–934. doi: 10.1172/JCI12749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Isenberg JS, Romeo MJ, Abu-Asab M, Tsokos M, Oldenborg A, Pappan L, Wink DA, Frazier WA, Roberts DD. Increasing survival of ischemic tissue by targeting CD47. Circ Res. 2007;100:712–720. doi: 10.1161/01.RES.0000259579.35787.4e [DOI] [PubMed] [Google Scholar]
- 10.Raugi GJ, Mullen JS, Bark DH, Okada T, Mayberg MR. Thrombospondin deposition in rat carotid artery injury. Am J Pathol. 1990;137:179–185. [PMC free article] [PubMed] [Google Scholar]
- 11.Sajid M, Hu Z, Guo H, Li H, Stouffer GA. Vascular expression of integrin-associated protein and thrombospondin increase after mechanical injury. J Investig Med. 2001;49:398–406. doi: 10.2310/6650.2001.33784 [DOI] [PubMed] [Google Scholar]
- 12.Riessen R, Kearney M, Lawler J, Isner JM. Immunolocalization of thrombospondin-1 in human atherosclerotic and restenotic arteries. Am Heart J. 1998;135:357–364. doi: 10.1016/s0002-8703(98)70105-x [DOI] [PubMed] [Google Scholar]
- 13.Stenina OI, Topol EJ, Plow EF. Thrombospondins, their polymorphisms, and cardiovascular disease. Arterioscler Thromb Vasc Biol. 2007;27:1886–1894. doi: 10.1161/ATVBAHA.107.141713 [DOI] [PubMed] [Google Scholar]
- 14.Moura R, Tjwa M, Vandervoort P, Van Kerckhoven S, Holvoet P, Hoylaerts MF. Thrombospondin-1 deficiency accelerates atherosclerotic plaque maturation in ApoE−/− mice. Circ Res. 2008;103:1181–1189. doi: 10.1161/CIRCRESAHA.108.185645 [DOI] [PubMed] [Google Scholar]
- 15.Ganguly R, Khanal S, Mathias A, Gupta S, Lallo J, Sahu S, Ohanyan V, Patel A, Storm K, Datta S, et al. TSP-1 (Thrombospondin-1) Deficiency Protects ApoE(−/−) Mice Against Leptin-Induced Atherosclerosis. Arterioscler Thromb Vasc Biol. 2021;41:e112–e127. doi: 10.1161/ATVBAHA.120.314962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Isenberg JS, Ridnour LA, Perruccio EM, Espey MG, Wink DA, Roberts DD. Thrombospondin-1 inhibits endothelial cell responses to nitric oxide in a cGMP-dependent manner. Proc Natl Acad Sci U S A. 2005;102:13141–13146. doi: 10.1073/pnas.0502977102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lahdenranta J, Hagendoorn J, Padera TP, Hoshida T, Nelson G, Kashiwagi S, Jain RK, Fukumura D. Endothelial nitric oxide synthase mediates lymphangiogenesis and lymphatic metastasis. Cancer Res. 2009;69:2801–2808. doi: 10.1158/0008-5472.CAN-08-4051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kojima Y, Volkmer JP, McKenna K, Civelek M, Lusis AJ, Miller CL, Direnzo D, Nanda V, Ye J, Connolly AJ, et al. CD47-blocking antibodies restore phagocytosis and prevent atherosclerosis. Nature. 2016;536:86–90. doi: 10.1038/nature18935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jarr KU, Nakamoto R, Doan BH, Kojima Y, Weissman IL, Advani RH, Iagaru A, Leeper NJ. Effect of CD47 Blockade on Vascular Inflammation. N Engl J Med. 2021;384:382–383. doi: 10.1056/NEJMc2029834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Singla B, Lin HP, Ahn W, Xu J, Ma Q, Sghayyer M, Dong K, Cherian-Shaw M, Zhou J, Huo Y, et al. Loss of myeloid cell-specific SIRPalpha, but not CD47, attenuates inflammation and suppresses atherosclerosis. Cardiovasc Res. 2021. doi: 10.1093/cvr/cvab369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gubbels Bupp MR. Sex, the aging immune system, and chronic disease. Cell Immunol. 2015;294:102–110. doi: 10.1016/j.cellimm.2015.02.002 [DOI] [PubMed] [Google Scholar]
- 22.Bourassa PA, Milos PM, Gaynor BJ, Breslow JL, Aiello RJ. Estrogen reduces atherosclerotic lesion development in apolipoprotein E-deficient mice. Proc Natl Acad Sci U S A. 1996;93:10022–10027. doi: 10.1073/pnas.93.19.10022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kumar S, Kang DW, Rezvan A, Jo H. Accelerated atherosclerosis development in C57Bl6 mice by overexpressing AAV-mediated PCSK9 and partial carotid ligation. Lab Invest. 2017;97:935–945. doi: 10.1038/labinvest.2017.47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Singla B, Lin HP, Chen A, Ahn W, Ghoshal P, Cherian-Shaw M, White J, Stansfield BK, Csanyi G. Role of R-spondin 2 in arterial lymphangiogenesis and atherosclerosis. Cardiovasc Res. 2020. doi: 10.1093/cvr/cvaa244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McCrohan MB, Huang SW, Sleasman JW, Klein PA, Kao KJ. Plasma thrombospondin as an indicator of intravascular platelet activation in patients with vasculitis. Thromb Haemost. 1987;58:850–852. [PubMed] [Google Scholar]
- 26.Wu H, Rahman HNA, Dong Y, Liu X, Lee Y, Wen A, To KH, Xiao L, Birsner AE, Bazinet L, et al. Epsin deficiency promotes lymphangiogenesis through regulation of VEGFR3 degradation in diabetes. J Clin Invest. 2018;128:4025–4043. doi: 10.1172/JCI96063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Song L, Ding S, Ge Z, Zhu X, Qiu C, Wang Y, Lai E, Yang W, Sun Y, Chow SA, et al. Nucleoside/nucleotide reverse transcriptase inhibitors attenuate angiogenesis and lymphangiogenesis by impairing receptor tyrosine kinases signalling in endothelial cells. Br J Pharmacol. 2018;175:1241–1259. doi: 10.1111/bph.14036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roberts DD, Miller TW, Rogers NM, Yao M, Isenberg JS. The matricellular protein thrombospondin-1 globally regulates cardiovascular function and responses to stress via CD47. Matrix Biol. 2012;31:162–169. doi: 10.1016/j.matbio.2012.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Adams JC, Lawler J. The thrombospondins. Cold Spring Harb Perspect Biol. 2011;3:a009712. doi: 10.1101/cshperspect.a009712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Makinen T, Veikkola T, Mustjoki S, Karpanen T, Catimel B, Nice EC, Wise L, Mercer A, Kowalski H, Kerjaschki D, et al. Isolated lymphatic endothelial cells transduce growth, survival and migratory signals via the VEGF-C/D receptor VEGFR-3. EMBO J. 2001;20:4762–4773. doi: 10.1093/emboj/20.17.4762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dotto GP. p21(WAF1/Cip1): more than a break to the cell cycle? Biochim Biophys Acta. 2000;1471:M43–56. doi: 10.1016/s0304-419x(00)00019-6 [DOI] [PubMed] [Google Scholar]
- 32.Oliver G, Kipnis J, Randolph GJ, Harvey NL. The Lymphatic Vasculature in the 21(st) Century: Novel Functional Roles in Homeostasis and Disease. Cell. 2020;182:270–296. doi: 10.1016/j.cell.2020.06.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lim HY, Rutkowski JM, Helft J, Reddy ST, Swartz MA, Randolph GJ, Angeli V. Hypercholesterolemic mice exhibit lymphatic vessel dysfunction and degeneration. Am J Pathol. 2009;175:1328–1337. doi: 10.2353/ajpath.2009.080963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Singla B, Lin HP, Ahn W, White J, Csanyi G. Oxidatively Modified LDL Suppresses Lymphangiogenesis via CD36 Signaling. Antioxidants (Basel). 2021;10. doi: 10.3390/antiox10020331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sid B, Sartelet H, Bellon G, El Btaouri H, Rath G, Delorme N, Haye B, Martiny L. Thrombospondin 1: a multifunctional protein implicated in the regulation of tumor growth. Crit Rev Oncol Hematol. 2004;49:245–258. doi: 10.1016/j.critrevonc.2003.09.009 [DOI] [PubMed] [Google Scholar]
- 36.Armstrong LC, Bornstein P. Thrombospondins 1 and 2 function as inhibitors of angiogenesis. Matrix Biol. 2003;22:63–71. doi: 10.1016/s0945-053x(03)00005-2 [DOI] [PubMed] [Google Scholar]
- 37.Doyen V, Rubio M, Braun D, Nakajima T, Abe J, Saito H, Delespesse G, Sarfati M. Thrombospondin 1 is an autocrine negative regulator of human dendritic cell activation. J Exp Med. 2003;198:1277–1283. doi: 10.1084/jem.20030705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu Z, Morgan S, Ren J, Wang Q, Annis DS, Mosher DF, Zhang J, Sorenson CM, Sheibani N, Liu B. Thrombospondin-1 (TSP1) contributes to the development of vascular inflammation by regulating monocytic cell motility in mouse models of abdominal aortic aneurysm. Circ Res. 2015;117:129–141. doi: 10.1161/CIRCRESAHA.117.305262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Good DJ, Polverini PJ, Rastinejad F, Le Beau MM, Lemons RS, Frazier WA, Bouck NP. A tumor suppressor-dependent inhibitor of angiogenesis is immunologically and functionally indistinguishable from a fragment of thrombospondin. Proc Natl Acad Sci U S A. 1990;87:6624–6628. doi: 10.1073/pnas.87.17.6624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang H, Zhou T, Sorenson CM, Sheibani N, Liu B. Myeloid-Derived TSP1 (Thrombospondin-1) Contributes to Abdominal Aortic Aneurysm Through Suppressing Tissue Inhibitor of Metalloproteinases-1. Arterioscler Thromb Vasc Biol. 2020;40:e350–e366. doi: 10.1161/ATVBAHA.120.314913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rogers NM, Sharifi-Sanjani M, Yao M, Ghimire K, Bienes-Martinez R, Mutchler SM, Knupp HE, Baust J, Novelli EM, Ross M, et al. TSP1-CD47 signaling is upregulated in clinical pulmonary hypertension and contributes to pulmonary arterial vasculopathy and dysfunction. Cardiovasc Res. 2017;113:15–29. doi: 10.1093/cvr/cvw218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cursiefen C, Maruyama K, Bock F, Saban D, Sadrai Z, Lawler J, Dana R, Masli S. Thrombospondin 1 inhibits inflammatory lymphangiogenesis by CD36 ligation on monocytes. J Exp Med. 2011;208:1083–1092. doi: 10.1084/jem.20092277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cifarelli V, Appak-Baskoy S, Peche VS, Kluzak A, Shew T, Narendran R, Pietka KM, Cella M, Walls CW, Czepielewski R, et al. Visceral obesity and insulin resistance associate with CD36 deletion in lymphatic endothelial cells. Nat Commun. 2021;12:3350. doi: 10.1038/s41467-021-23808-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hawighorst T, Oura H, Streit M, Janes L, Nguyen L, Brown LF, Oliver G, Jackson DG, Detmar M. Thrombospondin-1 selectively inhibits early-stage carcinogenesis and angiogenesis but not tumor lymphangiogenesis and lymphatic metastasis in transgenic mice. Oncogene. 2002;21:7945–7956. doi: 10.1038/sj.onc.1205956 [DOI] [PubMed] [Google Scholar]
- 45.Wong BW, Wang X, Zecchin A, Thienpont B, Cornelissen I, Kalucka J, Garcia-Caballero M, Missiaen R, Huang H, Bruning U, et al. The role of fatty acid beta-oxidation in lymphangiogenesis. Nature. 2017;542:49–54. doi: 10.1038/nature21028 [DOI] [PubMed] [Google Scholar]
- 46.Gao Q, Chen K, Gao L, Zheng Y, Yang YG. Thrombospondin-1 signaling through CD47 inhibits cell cycle progression and induces senescence in endothelial cells. Cell Death Dis. 2016;7:e2368. doi: 10.1038/cddis.2016.155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kaur S, Soto-Pantoja DR, Stein EV, Liu C, Elkahloun AG, Pendrak ML, Nicolae A, Singh SP, Nie Z, Levens D, et al. Thrombospondin-1 signaling through CD47 inhibits self-renewal by regulating c-Myc and other stem cell transcription factors. Sci Rep. 2013;3:1673. doi: 10.1038/srep01673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lecker SH, Goldberg AL, Mitch WE. Protein degradation by the ubiquitin-proteasome pathway in normal and disease states. J Am Soc Nephrol. 2006;17:1807–1819. doi: 10.1681/ASN.2006010083 [DOI] [PubMed] [Google Scholar]
- 49.Isenberg JS, Frazier WA, Roberts DD. Thrombospondin-1: a physiological regulator of nitric oxide signaling. Cell Mol Life Sci. 2008;65:728–742. doi: 10.1007/s00018-007-7488-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Csanyi G, Yao M, Rodriguez AI, Al Ghouleh I, Sharifi-Sanjani M, Frazziano G, Huang X, Kelley EE, Isenberg JS, Pagano PJ. Thrombospondin-1 regulates blood flow via CD47 receptor-mediated activation of NADPH oxidase 1. Arterioscler Thromb Vasc Biol. 2012;32:2966–2973. doi: 10.1161/ATVBAHA.112.300031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Csanyi G, Feck DM, Ghoshal P, Singla B, Lin H, Nagarajan S, Meijles DN, Al Ghouleh I, Cantu-Medellin N, Kelley EE, et al. CD47 and Nox1 Mediate Dynamic Fluid-Phase Macropinocytosis of Native LDL. Antioxid Redox Signal. 2017;26:886–901. doi: 10.1089/ars.2016.6834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Meijles DN, Sahoo S, Al Ghouleh I, Amaral JH, Bienes-Martinez R, Knupp HE, Attaran S, Sembrat JC, Nouraie SM, Rojas MM, et al. The matricellular protein TSP1 promotes human and mouse endothelial cell senescence through CD47 and Nox1. Sci Signal. 2017;10. doi: 10.1126/scisignal.aaj1784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sick E, Jeanne A, Schneider C, Dedieu S, Takeda K, Martiny L. CD47 update: a multifaceted actor in the tumour microenvironment of potential therapeutic interest. Br J Pharmacol. 2012;167:1415–1430. doi: 10.1111/j.1476-5381.2012.02099.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hansson GK, Libby P, Tabas I. Inflammation and plaque vulnerability. J Intern Med. 2015;278:483–493. doi: 10.1111/joim.12406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Grzegorek I, Drozdz K, Chmielewska M, Gomulkiewicz A, Jablonska K, Piotrowska A, Karczewski M, Janczak D, Podhorska-Okolow M, Dziegiel P, et al. Arterial wall lymphangiogenesis is increased in the human iliac atherosclerotic arteries: involvement of CCR7 receptor. Lymphat Res Biol. 2014;12:222–231. doi: 10.1089/lrb.2013.0048 [DOI] [PubMed] [Google Scholar]
- 56.Taher M, Nakao S, Zandi S, Melhorn MI, Hayes KC, Hafezi-Moghadam A. Phenotypic transformation of intimal and adventitial lymphatics in atherosclerosis: a regulatory role for soluble VEGF receptor 2. FASEB J. 2016;30:2490–2499. doi: 10.1096/fj.201500112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lim HY, Lim SY, Tan CK, Thiam CH, Goh CC, Carbajo D, Chew SHS, See P, Chakarov S, Wang XN, et al. Hyaluronan Receptor LYVE-1-Expressing Macrophages Maintain Arterial Tone through Hyaluronan-Mediated Regulation of Smooth Muscle Cell Collagen. Immunity. 2018;49:326–341 e327. doi: 10.1016/j.immuni.2018.06.008 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.