

Abstract
Lineage plasticity has long been documented in both small cell lung cancer (SCLC) and neuroblastoma (NBL), two clinically distinct neuroendocrine (NE) cancers. In this study, we quantified the NE features of cancer as NE scores and performed a systematic comparison of SCLC and NBL. We found NBL and SCLC cell lines have highly similar molecular profiles and shared therapeutic sensitivity. In addition, NE heterogeneity was observed at both the inter- and intra-cell line levels. Surprisingly, we did not find a significant association between NE scores and overall survival in SCLC or NBL. We described many shared and unique NE score-associated features between SCLC and NBL, including dysregulation of Myc oncogenes, alterations in protein expression, metabolism, drug resistance, and selective gene dependencies.
Introduction
Small cell lung cancer (SCLC) and neuroblastoma (NBL) are two very different cancer types with respect to their etiology, mutation spectrum/load, classification scheme, therapeutic strategy, and prognosis. SCLC, accounting for 13% of lung cancers, is predominantly found in heavy smokers, with almost ubiquitous co-mutation of RB1 and TP53, and has a five-year survival rate of 7% as the disease is highly metastatic. SCLC staging typically follows the two-stage classification convention established by the Veterans Affairs Lung Study Group (VALSG) in the 1980s. In the United States, two-thirds of SCLC patients are diagnosed at the extensive stage, with cancers that have spread beyond the lung and nearby lymph nodes to other distant parts of the body. SCLC bears much resemblance to pulmonary neuroendocrine cells in their morphology and expression of NE markers [1], but studies from genetically engineered mouse models suggest that some SCLC may also arise from other lung cell types [2]. NBL, accounting for 6% of childhood cancers in the US, is derived from sympathoadrenal progenitor cells within the neural crest[3], often develops in and around the adrenal gland, exhibits frequent genetic alterations in MYCN or ALK, and has a five-year survival rate of 81%. Despite these differences, both SCLC and NBL are neuroendocrine (NE) tumors, and NE markers are routinely used in immunohistochemistry (IHC) to facilitate the clinical diagnosis of both cancer types. As one of the “small round blue cell tumors” of childhood, undifferentiated NBL also highly resembles SCLC histologically.
Interestingly, the ability to transdifferentiate from the NE to non-NE lineage has been documented for both SCLC and NBL. Over 35 years ago, “classic“ (NE) and “variant” (non-NE) SCLC were reported based on distinct cellular morphologies and biochemical properties [4]. In the recent decade, studies have shown that transdifferentiation of SCLC gives rise to intratumoral heterogeneity and mediates chemoresistance [5, 6]. More recently, it was shown that REST, YAP, and NOTCH mediate NE transition in both SCLC and normal lung [7]. For NBL, morphologically distinct cell types from cell lines established from the same patient tumor were observed over 50 years ago [8]. Distinct biochemical properties and the ability to interconvert have been reported for isogenic cell subclones [9]. In two more recent studies, the “sympathetic noradrenergic”(NE) and “neural crest cell-like” (non-NE) [10], or “adrenergic” (NE) and “mesenchymal” (non-NE) [11] NBL cell states have been shown to exhibit distinct epigenetic and transcriptomic profiles. It has also been shown that NOTCH regulates TF networks to drive NE transition in NBL and contribute to the development of chemoresistance in NBL[12]. These independent studies converged on similar NOTCH-mediated mechanisms in NE lineage switch and suggest shared NE-associated properties across different cancer types. However, the extent of such similarity is still unclear. In this study, we re-analyzed the molecular and clinical data generated from SCLC and NBL cell lines and tumors to compare their associations with NE heterogeneity side-by-side, to reveal the concordance and idiosyncrasy in the landscape of NE state-associated features in both cancer types.
Material and Methods
Clustering of cell lines by multiomics, drug sensitivity, and dependency data
For the clustering of cell lines based on RNA-seq data, we first conducted a principal component analysis for genes with a standard deviation larger than 0.4. The top ten principal components accounted for 41% of the total variance and were used for hierarchical clustering. For the clustering of RPPA and metabolomics data, we used all available features and did not filter the input features or perform principal component analysis.
Compared to the molecular profiling data, functional screening data tend to be noisier due to variations in experimental design or lack of differential sensitivity among the cell lines [13]. Therefore, for clustering dependency and drug data, we filter the input drug and dependency features by their consistency across multiple datasets. We collected nine compound screening datasets and three functional genomics datasets and conducted all possible combinations of pairwise correlations within the drug datasets and the dependency datasets respectively. For example, for a specific drug profiled by 4 datasets, C(4,2) = 6 inter-study pairwise correlation would be available, with each inter-study pairwise correlation assessing the measurement consistency for the same set of cell lines in two datasets. We then summarized these inter-study pairwise Pearson correlations by meta-analysis to generate consistency measures for each compound and gene [13]. For the clustering analysis in this study, we selected consistent dependency data features with r > 0.4, and for drug data, we selected consistent features with multiple comparison-adjusted p-values < 0.05. All hierarchical clustering was performed using Ward’s minimum variance method.
NE score computation
The original SCLC NE signature based on microarray gene expression data was described by Zhang et al. [14]. Here, we used the updated signature generated from RNA-seq expression[15]. A quantitative NE score can be generated from an NE signature using the formula NE score = (correl NE – correl non-NE)/2, where correl NE (or non-NE) is the Pearson correlation between the expression of the 50 genes in the test sample and expression/weight of these genes in the NE (or non-NE) cell line group. This score ranges from −1 to +1, where a positive score predicts NE and a negative score predicts non-NE cell types. The higher the score in absolute value, the better the prediction.
Comparison between bulk RNA-seq and scRNA-seq data
Bulk RNA-seq data (CCLE_depMap_19Q1_TPM.csv) from CCLE and scRNA-seq data (GSE157220_CPM_data.txt.gz) [16] downloaded from GEO were used to compute the NE score for cell lines as well as single cells within cell lines using the above approach. For the scRNA-seq data, the average NE score per cell line was calculated. A total of 191 cell lines were shared between the two datasets, including four SCLC and two NBL cell lines. Pearson correlation between the bulk RNA-seq NE scores and average scRNA-seq NE scores was used as a measure of agreement between the two profiling approaches (Figure 2C).
Data Availability
Cell line datasets
Copy number, RNA-seq, miRNA, histone PTM, metabolomics, and RPPA data were downloaded from DepMap. Compound sensitivity data for “CCLE” [17], “CTRP”[18], “GDSC1” and “GDSC2” [19], “PRISM_1st” and “PRISM_2nd” [20], and gene dependency data for demeter (RNAi) [21] and achilles [22] (CRISPR) were downloaded from DepMap and processed as previously described [13]. The cell line names and compound names were unified, and the datasets were processed to ensure that the lower value in each dataset always corresponded to a higher sensitivity. The processed data, lists of consistent compounds, and dependencies were downloaded from https://lccl.shinyapps.io/FDCE/. The scRNA-seq data for cell lines were downloaded from the Gene Expression Omnibus (GEO) repository GSE157220 [16].
Additional SCLC datasets
The following SCLC transcriptomic datasets “UTSW SCLC cell line,” “Drapkin_2018” (PDX) [23], tumor datasets “Rudin_2012” [24], “George_2015” [25] , “Jiang_2016” [26], and “Cai_2021” [15] were processed as previously described [15]. The processed data are available in our previous publication [15]. SCLC scRNA-seq data were downloaded from the HTAN portal [27].
Additional NBL datasets
In addition to the CCLE RNA-seq data, additional NBL cell line transcriptomic and associated sample phenotype data were downloaded from GEO using R package GEOquery [28] with the following accession numbers: GSE28019, GSE89413 [29], and GSE90683 [10]. For NBL patient tumor datasets, we included two partially overlapped NBL datasets from Therapeutically Applicable Research to Generate Effective Treatments (TARGET) (https://ocg.cancer.gov/programs/target) initiative, phs000467 [30]. “TARGET_microarray” was downloaded from the TARGET Data Matrix, whereas “TARGET_RNA-seq” was downloaded from the UCSC Toil RNAseq Recompute Compendium [31]. Additional NBL tumor datasets were downloaded from GEO with the following accession numbers: GSE120572 [32], GSE3446[33], GSE19274[34], GSE73517[35], GSE85047[36], GSE62564[37], GSE16476[38], and GSE3960[39].
Results
NBL and SCLC cell lines are molecularly similar
We have previously established an NE score calculation method for SCLC samples based on a gene expression signature generated from SCLC cell line transcriptomic data [14, 15]. This method takes the expression data of 25 NE genes and 25 non-NE genes as inputs and assigns a score ranging from −1 (non-NE) to 1 (NE) to each sample. From the pan-cancer study cancer cell line encyclopedia (CCLE)/dependency map (DepMap) [40] RNA-seq dataset, we averaged the expression of these 50 genes by different cancer lineages and performed hierarchical clustering (Figure 1A). Among cancer types in the sub-cluster with high expression of NE genes, SCLC and NBL had the highest number of cell lines in the CCLE collection. This allowed us to leverage the multi-dimensional profiling data from CCLE and DepMap for an in-depth comparison between SCLC and NBL. We computed NE scores for the pan-cancer cell lines and clustered the cell lines based on transcriptomic, functional proteomic (based on Reverse Phase Protein Arrays, RPPA), metabolomic, gene dependency, and drug sensitivity features (Figures 1B-H). We observed tight clusters of SCLC and NBL cell lines with high NE scores in each clustering analysis. These results suggest that SCLC and NBL cell lines are highly similar in these molecular aspects.
Figure 1. The molecular similarity between NBL and SCLC.
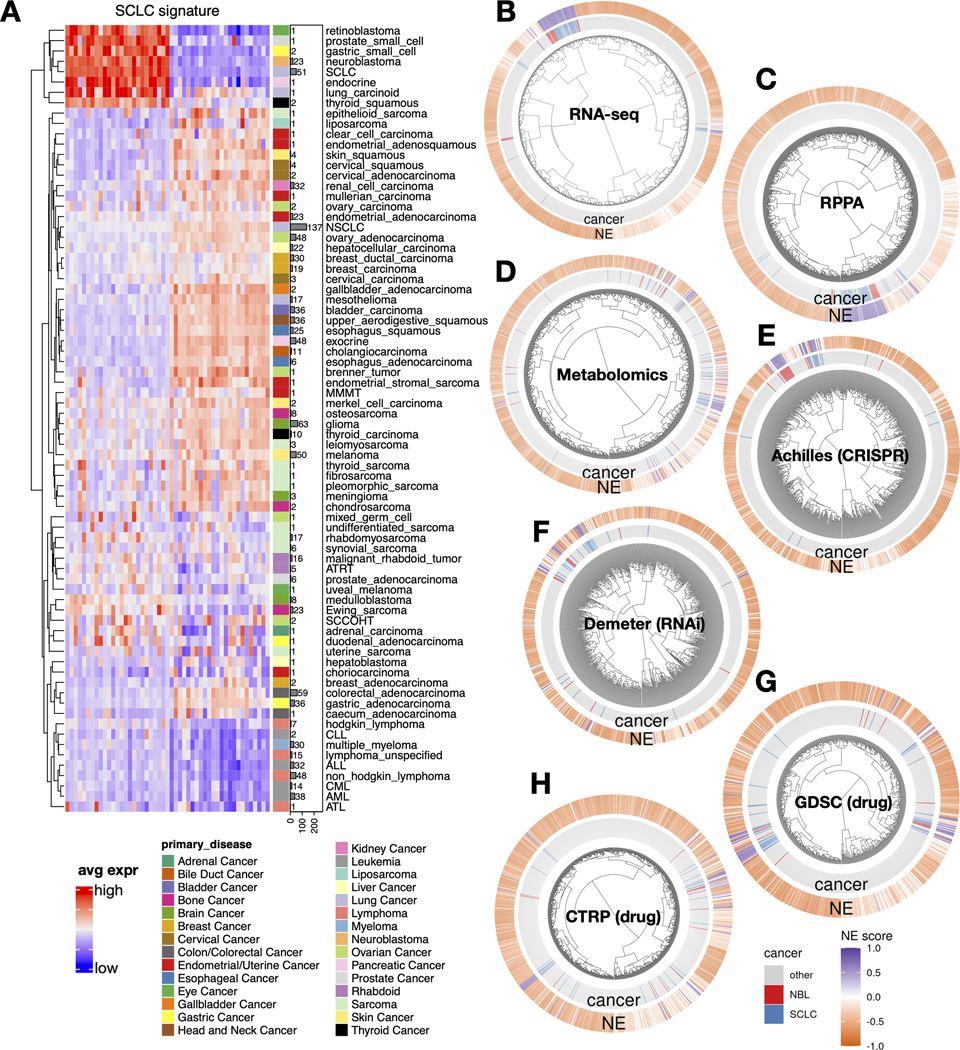
A. SCLC NE signature gene expression across different cancer lineages. The expression of NE (left half) and non-NE (right half) genes were averaged by cancer lineages and plotted as a heatmap. The number of cell lines per lineage are visualized as bars plotted right to the heatmap. Note that SCLC and NBL are the two cancer types with the highest number of cell lines in the cluster with high expression of NE genes. B-H, hierarchical clustering of cell lines by omics and functional screening datasets. 1165 cancer cell lines by expression of 19159 genes (B), 897 lines by 214 RPPA features (C), 926 lines by 225 metabolites (D), 688 lines by CRISPR effect score of 509 genes (E), 648 lines by RNAi effect score of 375 genes (F), 624 lines by 208 compounds from GDSC (G), and 794 lines by 168 compounds from CTRP (H). Note the clustering for RNA-seq data was based on the top 10 principal components, RPPA and metabolomics clusterings were based on all available features; dependency and drug clusterings were based on selected consistent features as previously summarized. Each leaf on the dendrogram represents a cell line. The inner rim right outside the dendrogram signifies the cancer lineage and the outer rim indicates the NE score of the cell line.
NE heterogeneity can be observed at inter- and intra-cell line levels for both SCLC and NBL
We assessed NE heterogeneity in SCLC and NBL cell lines by ranking the cell lines in the CCLE panel based on their NE scores. While most of the SCLC and NBL cell lines had positive NE scores and were enriched in the top, a few cell lines had negative NE scores, revealing inter-cell line NE heterogeneity (Figure 2A). We also examined the expression of SCLC NE score signature genes in SCLC and NBL cell lines. Although the signature was established in SCLC cell lines, it was also highly differentially expressed in NBL cell lines (Figure 2B). Four key transcription factors (ASCL1, NEUROD1, POU2F3, and YAP1) have been proposed to define the four molecular subtypes of SCLC. We described their relationship with SCLC NE scores in our previous study [15]. Although these transcription factors have not been used to classify NBL samples, when we examined their expression in NBL cell lines, we observed a pattern of segregation by NE score similar to SCLC - while high-NE-score NBL lines were found to have high expression of ASCL1 or NEUROD1, low-NE-score NBL lines had high expression of YAP1. However, no NBL line was found to express high levels of POU2F3, a tuft cell regulator [41] (Figure 2B). These results suggest that similar transcriptional regulations are involved in driving NE heterogeneity in SCLC and NBL cell lines.
Figure 2. Inter- and intra- cell line NE heterogeneity.

A. Inter-cell line NE heterogeneity. NE scores for CCLE pan-cancer cell lines were ranked from high to low. SCLC and NBL lines were highlighted by colors. Although most of the SCLC and NBL lines have high NE scores, a few of them also have low NE scores. B. Consistent gene expression pattern for SCLC NE signature genes observed for SCLC and NBL cell lines. Cell lines are in columns. Red/blue column left to the heatmap annotates the correlation between the gene expression and NE score; the expression of SCLC driver transcription factors (TFs) and NE scores was annotated above the heatmap. SCLC lines were further classified into four transcription factor (TF) classes. C. Average NE scores from scRNA-seq data align well with NE scores from bulk RNA-seq data for pan-cancer cell lines. D. Distribution of NE scores for lung cancer and neuroblastoma cell lines based scRNA-seq data. E-F. Intra-cell line NE heterogeneity. High- and low- NE score cells are found to coexist within the same SCLC cell line NCI-H1048 (E) or NBL cell line SKNAS (F). Single cells are in columns. Due to the high dropout rate of scRNA-seq data, only the top abundantly expressed genes are visualized.
To explore NE heterogeneity at the intra-cell line level, we compared scRNA-seq-based average NE scores to bulk RNA-seq-based NE scores for 191 cell lines (using scRNA-seq data available for a panel of pan-cancer cell lines [16]) and found a strong correlation (Figure 2C). We also observed that some SCLC and NBL cell lines had broader NE score distributions than others (Figure 2D). Using scRNA-seq data from SCLC patient tumors, we further observed the coexistence of high- and low-NE-score SCLC cells within tumors that exhibited highly variable NE scores (Figure S1A-C). Similarly, upon examining NE and non-NE gene expression across single cells in the SCLC cell line NCI-H1048 and NBL cell line SKNAS, we also observed the coexistence of high- and low-NE-score cells within the same cell line (Figures 2E-F). Notably, within these cell lines, cells with lower NE scores also had higher mesenchymal and IFN-response program scores, as previously annotated [16]. Additionally, low-NE-score cells from SCLC cell line NCIH1048 had higher epithelial senescence-associated program scores, although this association was not statistically significant in NBL cell line SKNAS because NBL is not epithelial (Figure S1D). These findings indicate that the lineage heterogeneity observed in patient tumors is preserved both among cell lines and within individual cells from the same cell line.
NE scores do not associate with overall survival in SCLC or NBL
Next, we tested whether NE scores were associated with disease outcomes in SCLC and NBL. As most patients with SCLC are diagnosed at an extensive stage, surgical resection of SCLC primary tumors is rare in practice. A recent study that profiled biopsied metastatic SCLC samples found no association between NE score and outcome [42]. We also investigated the prognosis association in a previously published dataset generated from 81 surgically resected SCLC tumors, of which 30 are stage III-IV samples [25]. We also did not find a significant association between the NE scores and overall patient survival (Figure 3A). With multiple NBL tumor datasets available, we performed a meta-analysis to assess the association between NE scores and overall survival in NBL. We also did not observe a consistent and significant result (Figure 3B). In the NBL datasets we investigated, the previously reported prognostic factors – age, MYCN amplification, and INSS stage 4 disease–were consistently associated with worse overall survival (Figure S2A-C), but we did not observe a significant difference in NE scores in groups stratified by these factors (Figures S2D-F). A small effect size was observed for NE score difference by relapse/progression status (Figure S2G); however, when comparing paired naïve and relapse samples from the same patient in two independent NBL studies, we did not identify a statistically significant difference in NE scores (Figure 3C). These findings suggest NE scores are not associated with prognosis in SCLC or NBL.
Figure 3. NE score is not associated with overall survival in SCLC or NBL.

A. Survival association analysis for SCLC based on 79 patients from the George_2015 study. NE score is not significantly associated with overall survival in univariate Cox regression or a multivariate model controlling for Sex and TNM stage. B. Meta-analysis for NBL based on seven studies and 1,531 patients. The result is also not statistically significant although significant results could be observed for individual studies, the trend was different. C. NE scores are not significantly altered in NBL relapsed samples. Paired samples from the same patients in two independent studies were compared.
Myc oncogenes are differentially activated by NE states in SCLC and NBL
As members of the Myc oncogene family (MYC, MYCN, and MYCL) have been implicated in SCLC and NBL oncogenesis [43, 44], we attempted to dissect their relationship with the NE state. First, we examined copy number alterations of Myc oncogenes (Figure 4A). We found that MYC and MYCL were enriched in high-NE-score SCLC lines, whereas MYCN amplification was enriched in high-NE-score NBL lines. As MYCL is located on chromosome 1p, a frequently deleted region in NBL, MYCL loss appears to be frequent in NBL lines (Figure 4A). Examination of the gene expression data showed that the patterns for MYCL and MYCN agreed well with the copy number data (Figure 4B). Having made these observations in cell lines, we further examined the transcriptomic data from multiple SCLC and NBL studies. For SCLC, we included our in-house cell line RNA-seq data (UTSW cell lines), PDX dataset (Drapkin_2018), and four tumor datasets (Figures 4C). For NBL, we included three more cell line datasets along with the CCLE RNA-seq data (Figures 4D) and assembled 11 tumor datasets (Figures 4E). Meta-analyses with these datasets verified that the NE score associations with Myc oncogenes were consistent between multiple cell lines (Figure 4B) and patient tumor datasets (Figure 4F). Combined analysis of copy number, gene expression, and NE scores in the CCLE cell line dataset revealed upregulation of MYC expression in the low-NE-score lines without copy number gain, suggesting the transcriptional activation of MYC expression in the non-NE state for both SCLC and NBL (Figure 4G). We retrieved MYCN amplification status from eight NBL tumor datasets and assessed the association between NE score and MYCN expression while controlling for MYCN amplification (Figure 4H). Much stronger associations were observed across multiple studies in this multivariate linear model (Figure S3), suggesting the transcriptional activation of MYCN expression in the NE state NBL tumors. In summary, SCLC and NBL exhibit not only differential copy number gains but also differential transcriptional regulation for MYC family genes with regard to their NE status, with MYC transcriptionally upregulated in the non-NE state, MYCL preferentially amplified in high-NE score SCLC, and MYCN preferentially amplified and transcriptionally upregulated in high-NE score NBL.
Figure 4. NE score association with members of the Myc oncogene family in SCLC and NBL.

A-B. Copy number (A) and RNA expression (B) of Myc family genes in SCLC and NBL cell lines. Note that although MYC amplification was higher in the high-NE-score SCLC cell lines, its gene expression was higher in the low-NE-score cell lines for both SCLC and NBL lines. Frequent MYCL loss was found in NBL because MYCL is located in a frequently deleted region (chromosome 1p) in NBL. C. NE score vs. Myc gene member expression in SCLC studies. “UTSW cell line” is a cell line dataset; “Drapkin_2018” is a patient-derived xenograft dataset; “Rudin_2012”, “George_2015”, “Jiang_2016” and “Cai_2021” are all patient tumor datasets. D-E. NE score vs. Myc gene member expression in NBL cell line datasets (D) and tumor datasets (E). Note that some of the same cell lines were profiled in multiple studies. F. Forest plots visualizing meta-analysis of NE score association with Myc family genes. MYC expression is consistently associated with lower NE scores in SCLC and NBL samples (left). MYCL expression positively correlates with NE scores in SCLC but not NBL samples (middle). MYCN expression positively correlates with NE scores in NBL but not SCLC samples. G. Relationship between MYC copy number and gene expression in NBL and SCLC cell lines. Note that MYC amplification is only observed in SCLC cell lines. H. MYCN gene expression positively correlate with NE scores while controlling for MYCN amplification status in NBL patient tumors.
Consistent proteomic and metabolic changes are associated with NE-to-non-NE transition in SCLC and NBL
We performed NE score correlations with 12 sets of data from the CCLE/DepMap studies (Tables S1-S12). These include four sets of omics data (miRNA, histone PTM, RPPA, and metabolomics), six sets of compound screening data (CCLE, CTRP, GDSC1, GDSC2, PRISM_1ST, and PRISM_2nd), and two sets of gene dependency screening data (Demeter for RNAi and Achilles for CRISPR). The overall NE score association concordance was quite good for the omics datasets (Figure S4).
Upon close examination of the results of the RPPA associations (Figure 5A), we found most of the NE-score-associated features identified in SCLC cell lines could also be observed in NBL cell lines. Among the exceptions, Rb protein is decreased in the high-NE score SCLC, leading to an increase in cyclin E2 but this was not observed in NBL lines (Figure 5C), which could be explained by the frequent RB1 loss that occurs in SCLC but not NBL. Although a previous study suggests RB1 loss is highly enriched in YAPoff small-cell/neuro/neuroendocrine cancer lineages [45], the absence of RB1 mutation in NBL suggests the existence of an Rb-independent mechanism for YAP inactivation in NBL. Interestingly, in both SCLC and NBL, CDK-interacting protein/kinase inhibitory protein (CIP/KIP) p21 is upregulated in the low-NE-score lines whereas another CIP/KIP p27 is downregulated, suggesting that the NE state-specific cell cycle regulators are still consistent in these two cancer types despite differences in the upstream Rb loss. In the low-NE-score lines of both SCLC and NBL, we observed higher levels of receptor tyrosine kinases and their phosphorylation (EGFR, EGFR_pY1068, HER2_pY1248, and VEGFR2), higher levels of Hippo signaling components (YAP, YAP_pS127, and TAZ), pro-inflammatory proteins (p62, NF-kB-p65_pS536, PAI-1, and annexin 1), ribosome biogenesis markers (S6_pS240_S244 and S6_pS235_S236), and cell adhesion proteins (paxillin and CD49b). In the high-NE score lines of both SCLC and NBL, we found higher apoptotic machinery components (Smac, Bcl-2, Bim, and Bax), DNA repair proteins (MSH2 and MSH6), translation inhibitor 4E-BP1, and microtubule regulator Stathmin. Unique to SCLC, we observed higher epithelial junction proteins (Claudin-7 and E-cadherin) in the high-NE-score lines, these epithelial markers were however not expressed in the NBL lines (Figure 5A). We also examined the metabolomic associations and observed similar consistency between SCLC and NBL lines. (Figure 5B) In particular, many cholesteryl esters (CEs) were found to have higher levels in the low-NE-score SCLC lines; a weaker but similar trend was observed in NBL lines. We also found that both SCLC and NBL low-NE-score cell lines exhibited higher levels of citrate, aconitate, and isocitrate, three interconvertible metabolites, through the action of aconitase (Figures 5B and D).
Figure 5. NE score-associated protein and metabolic features are largely consistent in SCLC and NBL cell lines.

A-B. Heatmaps visualizing the relationship between NE scores and selected functional proteomic feature (A) or metabolites (B). In each heatmap, the left-side column denotes the Pearson correlation between the selected feature on the row and the NE score. The top colored rows denote NE scores and SCLC TF expression. The features were selected based on NE score correlation from the SCLC cell lines, adjusted p-value (p.adj) < 0.05 was used to select RPPA features and p.adj < 0.1 was used to select metabolic features. Note that although the selection was made from SCLC cell lines, a very similar pattern could be observed in NBL cell lines. C-D. Scatterplots visualizing the relationship between selected RPPA (C) and metabolic (D) features and NE scores in NBL and SCLC cell lines.
Consistent and unique therapeutic vulnerabilities in NE and non-NE subtypes of SCLC and NBL
The SCLC vs. NBL concordance for NE score – drug sensitivity associations was poorer than the omics data (Figure S4). We have previously demonstrated that drug screening data are more consistent for compounds directed against functionally important targets that are differentially expressed in a panel of cell lines [13]. For many of the compounds included in the screens, their targets may not be functionally important in the small panel of cell lines tested, which may explain the overall lower consistency. We reviewed the results (Tables S5-S10) to identify the most consistent associations across the multiple compound screens. Nine classes of compounds with different mechanisms of action (MOA) were selected. For each MOA class, we compared the NE score associations for different compounds in NBL and SCLC (Figure 6A). We also used meta-analysis to generate a summary correlation coefficient for each class of compounds from the SCLC and NBL assessments (Figures S5 and 6B). We found that in both SCLC and NBL, cell lines with higher NE scores were more resistant to drugs that target MEK, mTOR, XIAP, LCK, HSP90, and Abl but were more sensitive to BCL inhibitors. We also observed that higher NE scores were associated with resistance to microtubule inhibitors in SCLC, but not NBL cell lines, whereas higher NE scores were associated with resistance to BRD inhibitors in NBL, but not SCLC lines (Figure 6). Notably, although we identified differential therapeutic sensitivity within SCLC and NBL panels relative to their NE lineage, this does not tell us about the dynamic ranges of compound sensitivity in SCLC and NBL. In some cases, the dynamic range of compound sensitivity remains different between SCLC and NBL. For example, SCLC cell lines are the most resistant to MEK inhibitors, whereas NBL cell lines exhibit intermediate sensitivity over a broader range (Figures S6A-C). In other cases, we observed a similar overall sensitivity of SCLC and NBL cell lines. For example, both SCLC and NBL cell lines were more resistant to the HSP90 inhibitor 17-AAG, but more sensitive to the BCL inhibitor ABT-199, compared to the other cancer lineages (Figures S6D-E). In summary, our results revealed that the relative differential drug sensitivities associated with NE-to-non-NE transdifferentiation in SCLC and NBL were similar.
Figure 6. Similar and distinct NE score-associated therapeutic sensitivity in SCLC and NBL cell lines.

A. Correlation between NE scores and therapeutic sensitivity for drugs with selected targets. Therapeutic sensitivity data was previously harmonized such that a higher value represents more resistance in each study. For each of the nine selected targets, all compounds with the same target were identified from multiple studies. Pearson correlation coefficient r from correlating compound data with NE scores were calculated for NBL lines (x-axis values) and SCLC lines (y-axis values) respectively and visualized as a scatter plot, with colors annotating the source of data, and transparency annotating the statistical significance. B. Meta-analysis-summarized correlation between drug therapeutic sensitivity and NE scores in NBL (x-axis) and SCLC (y-axis) cell lines. Note that high NE scores are associated with resistance to inhibitors of LCK, MEK, XIAP, mTOR, HSP90, and Abl, and sensitivity to BCL inhibitors. NE scores are associated with resistance to BRD inhibitors in NBL but not SCLC whereas microtubule inhibitors resistance correlates with high NE scores in SCLC but not NBL cell lines.
Identification and comparison of SCLC and NBL-specific gene dependencies
We observed very poor overall concordance between the RNAi and CRISPR dependency data for their association with NE scores in SCLC and NBL (Figure S4). We rationalized that this is because most genes were not selectively essential in the relatively small panel of SCLC or NBL cell lines assessed. Hence, we adopted a set of criteria for selecting cancer-specific gene dependencies. We looked for genes with RNAi vs. CRISPR gene effect scores positively correlated, as an indication of high reproducibility from independent dependency screening experiments, as well as negative correlations between RNAi or CRISPR gene effect scores and RNA-seq expression data on the premise that genes of selective functional importance are more highly expressed in the cells that depend on them, such that these cells also have more negative gene effect scores that indicate higher dependence. These measures from the SCLC and NBL panels were assembled to prioritize the SCLC-specific vulnerabilities (Table S13). Indeed, when we examined these correlations, the known SCLC subtype drivers and the most common NBL driver genes all met this set of criteria (Figure 7A-B). We further closely examined genes with high RNAi vs. CRISPR correlation, and high anti-correlations between RNA expression and the gene effect scores as selected vulnerabilities (Figure 7C). Among the SCLC-selected vulnerabilities, along with ASCL1, we found several other NE lineage transcription factors (SOX11, FOXA2, NKX2–1) were more selectively essential for high-NE-score cell lines, whereas several genes involved in cell adhesion and motility (VCL, PXN, ACTR3, and RAC1) were found to be more selectively essential for low-NE-score cell lines; we also found genes frequently amplified in SCLC (IRS2, CCNE1, and NFIB) [46] although these genes do not have gene effect scores significantly correlated with NE scores. Interestingly, among these SCLC-selected vulnerabilities, we also identified genes that are well characterized for their roles in NBL, such as the ciliary neurotrophic factor CNTF [47] and S-phase kinase-associated protein 2 (SKP2 [48]. Among the very few vulnerabilities selected from both SCLC and NBL, we identified BCL2, a well-characterized gene in both cancer types. Consistent with our observation in the therapeutic sensitivity analysis, high-NE-score cell lines from both SCLC and NBL were more sensitive to BCL2 depletion (Figures 7D-E). As only nine NBL cell lines were included in the RNAi dependency screen, the reliability of our NBL-selected vulnerabilities might have been undermined by the underpowered input datasets. Nevertheless, we were able to identify a few genes known to be important for NBL, such as GATA Binding protein 3 GATA3 [49], complement decay-accelerating factor CD55 [50], Forkhead Box R2 FOXR2 [51], and breast cancer anti-estrogen resistance protein 1 BCAR1(/p130Cas) [52]. Among these, selective essentiality for GATA3 was only observed for high-NE-score NBL cell lines but not SCLC cell lines (Figure 7F-G), whereas BCAR1 appears to be a shared vulnerability for low-NE-score cell lines in both NBL and SCLC (Figures 7H-I). Overall, we observed unique and shared gene dependencies between SCLC and NBL cell lines, some of which also exhibited NE/non-NE lineage-specific selectivity.
Figure 7. Similar and distinct NE score-associated gene dependencies in SCLC and NBL cell lines.

A-B. Selection of SCLC (A) and NBL (B) vulnerabilities based on the consistency (positive correlation) between CRISPR and RNAi data, and anti-correlation between dependency data and gene expression data. Pearson correlation coefficients from RNAi-CRISPR (left), RNAi-RNA expr (middle), and CRISPR-RNA expr (right) correlations were computed for all genes. The distributions of these coefficients are plotted as diagonal panels; pairwise correlations among these three sets of correlation coefficients were visualized as scatter plots in the lower triangular panels and the Pearson correlation coefficients are printed in the upper triangular panels. The four SCLC subtype driver TFs and the NBL oncogenic driver MYCN all have high consistency between CRISPR and RNAi data and high anti-correlation between dependency data and gene expression data. Areas with r > 0.4 from RNAi - CRISPR correlation, and r < −0.4 from RNAi - RNA expr and CRISPR - RNAi correlation were demarcated by light gray squares. C. Correlation between NE scores and effect scores of selected dependencies in SCLC and NBL. The upper part of the heatmap displays selected vulnerabilities for SCLC and was ordered by correlations between NE scores and the effect scores in SCLC cell lines; likewise, the lower part of the heatmap displays selected vulnerabilities for NBL. Genes with magenta arrows are showcased in D-I. Cell lines are ordered by their NE scores and annotated with NE score and SCLC driver TF expression. D-I, Comparison of selected gene dependencies in SCLC and NBL. In each plot, variable names are shown in the diagonal boxes, and scatter plots display relationships between each pairwise combination of variables. Lower triangular plots are colored by NE scores whereas upper triangular plots for SCLC figures are colored by TF classes. Pearson correlation coefficients are provided in lower triangular boxes for SCLC and upper triangular boxes for NBL. Refer to legends in C for color annotations.
Validations in additional cancer types
Given the distinct differences in etiology, risk factors, and molecular mechanisms between SCLC and NBL, we sought to validate our findings by analyzing cell lines from other cancer types. To this end, we identified lineage subtypes with at least two cell lines that exhibited positive neuroendocrine (NE) scores based on transcriptomic data (Figure S7A) and selected cell lines from medulloblastoma, prostate adenocarcinoma, Ewing sarcoma, and non-small cell lung cancer (NSCLC) for further investigation.
We confirmed that the original SCLC NE signature genes were differentially expressed by NE subtype in these cell lines (Figure S7B). We also examined the reverse-phase protein array (RPPA) and metabolomics features that were associated with NE scores in SCLC, finding good agreement with these features in the four additional cancer types (Figure S7C-D). Furthermore, we explored the relationship between Myc gene family members’ copy number and RNA expression and NE scores in the four cancer types. However, we observed no consistent pattern (Figure S7E). Notably, while c-Myc expression was anti-correlated with NE scores in SCLC and NBL, we found a strong positive correlation in medulloblastoma.
Due to the very limited number of cell lines available for drug sensitivity profiling in medulloblastoma, prostate adenocarcinoma, and Ewing sarcoma, we only compared SCLC and NSCLC for their NE score-associated drug sensitivity. Our results indicated agreement for MEK and BCL inhibitors (Figure S7F). However, this finding is not entirely robust, as NSCLC has few NE+ cell lines, and even fewer were profiled for drug sensitivity.
Discussion
Different cancers of the NE lineage have historically been investigated as separate entities, owing to their distinct clinical presentations. The common Notch-mediated NE lineage plasticity and the adjacency of SCLC and NBL in cancer cell line clustering by multi-omics datasets (Figure 1) prompted us to perform a systematic comparison of these two cancer types. In this paper, we identified numerous common molecular associations with NE states in both cancer types. Most of the proteomic and metabolic features observed to associate with NE states in SCLC could be validated in NBL (Figure 5). NE score-associated transcriptomes are also highly similar between SCLC and NBL. We previously reported cell-autonomous immune gene repression in SCLC and pulmonary neuroendocrine cells in the NE state and transdifferentiation into the non-NE lineage releases the repression of immune genes [15]. Recently, similar observations have been reported for NBL [53]. Besides immune genes, many other genes also are differentially expressed by NE status in both SCLC and NBL. Although our omics analyses in this study do not include large-scale transcriptomics comparison, this topic is explored in greater depth in a companion manuscript [54].
In the cell line cluster generated by RNA-seq, RPPA, metabolomics, drug sensitivity, and gene dependency data, we observed NBL and SCLC lines cluster tighter with each other in RNA-seq and RPPA data (Figure 1B-H). There may be several potential reasons. It is possible that while gene and protein expression patterns are hardwired by lineage specificity, the cell metabolism and functional liabilities are subjected to many additional feedback regulations. For example, chromosome instability may not be related to NE transdifferentiation, but the resulting replication stress could modulate nucleotide biosynthesis metabolism [55] and alter a cell’s response to DNA damaging drugs. Another contributing factor may be the insufficient coverage of metabolome and the drug targets of the drug screening panels may not adequately capture the functions differentially regulated by NE transdifferentiation. It is also worth noting that drug and gene dependency datasets are generally noisier than molecular profiling data (4), which may explain their poor NE score correlation agreement between SCLC and NBL (Figure S4), as well as their less robust clustering of the SCLC and NBL cell lines.
Our investigation of Myc family members in Figure 4 revealed that MYCN amplification is enriched in high-NE score NBL cell lines, MYCL amplification is enriched in high-NE score SCLC cell lines, whereas increased MYC gene expression is observed in low NE score cell lines and tumor samples of both SCLC and NBL. Interestingly, MYCN has been shown to drive NE prostate cancer initiation [56], as it can epigenetically activate neural lineage gene expression in prostate cancer [57]. A similar mechanism may also apply to neuroblastoma, where the dependence on MYCN to epigenetically sustain NE lineage could explain the high MYCN levels observed in samples with high NE scores. On the other hand, in a SCLC mouse model, it has been shown that c-Myc can activate Notch to drive the loss of NE fate [58]. Upon c-Myc activation, SCLC cells undergo a transition from Ascl1-positive state to Neurod1-positive state [59], and eventually to Yap1-positive state [58]. In another mouse model of SCLC, it has been shown that loss of Ascl1 can revert SCLC to a more neural crest like fate [60]. In NBL, while the ASCL1-high adrenergic-type (NE) cells are committed to the adrenergic lineage, the ASCL1-low mesenchymal-type (non-NE) neuroblastoma cells are also known to resemble neural crest derived precursor cells [12]. Therefore, the interplay between Myc family members, ASCL1, Notch signaling, and other factors in regulating NE plasticity in cancer may be reflecting their roles in driving cell fate towards NE or non-NE lineage during normal tissue development. However, whether these findings can be generalized to other cancer types remained to be verified. As shown in Figure S7E, some medulloblastoma cell lines with high MYC expression still exhibit high NE scores, indicating that c-Myc activation does not always result in the loss of NE fate. Additionally, loss of NE fate may occur upon Notch activation regardless of c-Myc status[7]. Furthermore, it is important to note that different types of cancer may involve distinct factors in the regulation of NE transdifferentiation. For example, NE prostate cancer driven by MYCN is highly dependent on Rb deletion [61], while high MYCN expression in NBL does not coincide with Rb deletion. Therefore, the specific factors that collaborate to regulate NE fate in different cancer types may impact the generalizability of MYC-dependent NE fate modulation. Further research is needed to fully understand the role of Myc genes and others in the complex regulation of NE transdifferentiation in different types of cancer.
Our comparison of SCLC and NBL therapeutic vulnerabilities revealed similar NE score associations for several classes of compounds with shared MOA. Interestingly, individual studies in SCLC or NBL have also reported many of these associations, such as MEK inhibitors for NBL [62], HSP90 inhibitors for SCLC [63] and NBL [64], and BCL2 inhibitors for SCLC [65] and NBL [66]. We also identified cancer-unique vulnerabilities that have been previously reported, such as BRD inhibitors and GATA3 essentiality for NBL [67, 68]. Our findings suggest that NE plasticity may serve as a venue for therapy resistance in both SCLC and NBL for such drugs, as long-term monotherapy targeting cells of one lineage may create a selective pressure that shifts the population towards the other lineage. Importantly, our systematic investigation has mapped out the lineage-specific vulnerabilities in the NE and non-NE states. Coupled with our observation that high- and low- NE-score cells can co-exist within the same SCLC or NBL cell line (Figure 2), it would be interesting for future work to devise combinatorial therapies with drugs that target both NE and non-NE states and compare the efficacy to monotherapy using cell lines or cell line-derived xenografts as preclinical models. Although we did not experimentally validate the NE lineage-specific therapeutic vulnerabilities identified in this study, we have used five compound screening datasets and two gene essentiality screening datasets to choose features based on agreement across multiple datasets to maximize result reliability. Therefore, we believe that the generated results are credible and offer valuable insights for future work.
Compared to the omics analyses, relatively fewer similarities were observed at the functional liability levels. Several reasons might explain this discrepancy. First, functional data is much noisier than -omics data [13]. Second, fewer SCLC and NBL cell lines were included in the functional screening datasets and this compromised the statistical power for target discovery (Figure S4). As our concordance-based approach requires examining data from common cell lines between two datasets, this further reduces the available sample size for analysis. Lastly, the unique vulnerabilities for SCLC and NBL may stem from cancer drivers that act orthogonally to NE status. One such example is NFIB, which has been characterized as a metastatic promoter in SCLC [69].
In summary, our study provides a comprehensive molecular reference for features and vulnerabilities associated with NE-to-non-NE lineage transitions in SCLC and NBL. We also identified unique features that require further investigation in the context of each cancer type. While our focus was on SCLC and NBL, which are known for their NE-to-non-NE transitions, we discovered that many molecular features associated with NE scores in SCLC are also relevant to other cancers, such as prostate cancer and NSCLC, which exhibit non-NE to NE transitions to develop resistance to therapy (see Figure S7). Overall, our findings can guide the development of combinatorial therapies targeting lineage plasticity in SCLC, NBL, and other cancers that display NE heterogeneity.”
Supplementary Material
Implications.
Our work establishes a reference for molecular changes and vulnerabilities associated with NE to non-NE transdifferentiation through mutual validation of SCLC and NBL samples.
Acknowledgment
L.C. received support from UTSW ACS-IRG (IRG-21-142-16) and a Lung Cancer SPORE Career Enhancement Program award from P50CA70907. This study is supported by fundings from the National Institutes of Health [R01GM140012, R01GM141519, R01DE030656, U01CA249245, U01AI156189, R35CA22044901, P30CA142543, P50CA70907, U01CA213338, U24CA213274 and R35GM136375], and the Cancer Prevention and Research Institute of Texas [RP190107 and RP180805]. R.J.D. received funding from Howard Hughes Medical Institute. This article is subject to HHMI’s Open Access to Publications policy. HHMI lab heads have previously granted a nonexclusive CC BY 4.0, license to the public, and a sublicensable license to HHMI in their research articles. Pursuant to those licenses, the author-accepted manuscript of this article can be made freely available under a CC BY4.0 license immediately upon publication.
R.J.D. is an advisor for Agios Pharmaceuticals and Vida Ventures and a co-founder of Atavistik Bio. J.D.M receives licensing fees from the NIH and UTSW for distribution of human tumor cell lines.
Footnotes
Declaration of Interests
The other authors declare no competing interests.
Reference
- [1].Bensch KG, Corrin B, Pariente R, Spencer H, Oat-cell carcinoma of the lung. Its origin and relationship to bronchial carcinoid, Cancer, 22 (1968) 1163–1172. [DOI] [PubMed] [Google Scholar]
- [2].Yang D, Denny SK, Greenside PG, Chaikovsky AC, Brady JJ, Ouadah Y, Granja JM, Jahchan NS, Lim JS, Kwok S, Kong CS, Berghoff AS, Schmitt A, Reinhardt HC, Park KS, Preusser M, Kundaje A, Greenleaf WJ, Sage J, Winslow MM, Intertumoral Heterogeneity in SCLC Is Influenced by the Cell Type of Origin, Cancer Discov, 8 (2018) 1316–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Johnsen JI, Dyberg C, Wickstrom M, Neuroblastoma-A Neural Crest Derived Embryonal Malignancy, Front Mol Neurosci, 12 (2019) 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Gazdar AF, Carney DN, Nau MM, Minna JD, Characterization of variant subclasses of cell lines derived from small cell lung cancer having distinctive biochemical, morphological, and growth properties, Cancer Res, 45 (1985) 2924–2930. [PubMed] [Google Scholar]
- [5].Calbo J, van Montfort E, Proost N, van Drunen E, Beverloo HB, Meuwissen R, Berns A, A functional role for tumor cell heterogeneity in a mouse model of small cell lung cancer, Cancer Cell, 19 (2011) 244–256. [DOI] [PubMed] [Google Scholar]
- [6].Lim JS, Ibaseta A, Fischer MM, Cancilla B, O’Young G, Cristea S, Luca VC, Yang D, Jahchan NS, Hamard C, Antoine M, Wislez M, Kong C, Cain J, Liu YW, Kapoun AM, Garcia KC, Hoey T, Murriel CL, Sage J, Intratumoural heterogeneity generated by Notch signalling promotes small-cell lung cancer, Nature, 545 (2017) 360–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Shue YT, Drainas AP, Li NY, Pearsall SM, Morgan D, Sinnott-Armstrong N, Hipkins SQ, Coles GL, Lim JS, Oro AE, Simpson KL, Dive C, Sage J, A conserved YAP/Notch/REST network controls the neuroendocrine cell fate in the lungs, Nat Commun, 13 (2022) 2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Tumilowicz JJ, Nichols WW, Cholon JJ, Greene AE, Definition of a continuous human cell line derived from neuroblastoma, Cancer Res, 30 (1970) 2110–2118. [PubMed] [Google Scholar]
- [9].Ross RA, Spengler BA, Biedler JL, Coordinate morphological and biochemical interconversion of human neuroblastoma cells, J Natl Cancer Inst, 71 (1983) 741–747. [PubMed] [Google Scholar]
- [10].Boeva V, Louis-Brennetot C, Peltier A, Durand S, Pierre-Eugene C, Raynal V, Etchevers HC, Thomas S, Lermine A, Daudigeos-Dubus E, Geoerger B, Orth MF, Grunewald TGP, Diaz E, Ducos B, Surdez D, Carcaboso AM, Medvedeva I, Deller T, Combaret V, Lapouble E, Pierron G, Grossetete-Lalami S, Baulande S, Schleiermacher G, Barillot E, Rohrer H, Delattre O, Janoueix-Lerosey I, Heterogeneity of neuroblastoma cell identity defined by transcriptional circuitries, Nat Genet, 49 (2017) 1408–1413. [DOI] [PubMed] [Google Scholar]
- [11].van Groningen T, Koster J, Valentijn LJ, Zwijnenburg DA, Akogul N, Hasselt NE, Broekmans M, Haneveld F, Nowakowska NE, Bras J, van Noesel CJM, Jongejan A, van Kampen AH, Koster L, Baas F, van Dijk-Kerkhoven L, Huizer-Smit M, Lecca MC, Chan A, Lakeman A, Molenaar P, Volckmann R, Westerhout EM, Hamdi M, van Sluis PG, Ebus ME, Molenaar JJ, Tytgat GA, Westerman BA, van Nes J, Versteeg R, Neuroblastoma is composed of two super-enhancer-associated differentiation states, Nat Genet, 49 (2017) 1261–1266. [DOI] [PubMed] [Google Scholar]
- [12].van Groningen T, Akogul N, Westerhout EM, Chan A, Hasselt NE, Zwijnenburg DA, Broekmans M, Stroeken P, Haneveld F, Hooijer GKJ, Savci-Heijink CD, Lakeman A, Volckmann R, van Sluis P, Valentijn LJ, Koster J, Versteeg R, van Nes J, A NOTCH feed-forward loop drives reprogramming from adrenergic to mesenchymal state in neuroblastoma, Nat Commun, 10 (2019) 1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Cai L, Liu H, Minna JD, DeBerardinis RJ, Xiao G, Xie Y, Assessing Consistency Across Functional Screening Datasets in Cancer Cells, Bioinformatics, (2021). [DOI] [PMC free article] [PubMed]
- [14].Zhang W, Girard L, Zhang YA, Haruki T, Papari-Zareei M, Stastny V, Ghayee HK, Pacak K, Oliver TG, Minna JD, Gazdar AF, Small cell lung cancer tumors and preclinical models display heterogeneity of neuroendocrine phenotypes, Transl Lung Cancer Res, 7 (2018) 32–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Cai L, Liu H, Huang F, Fujimoto J, Girard L, Chen J, Li Y, Zhang YA, Deb D, Stastny V, Pozo K, Kuo CS, Jia G, Yang C, Zou W, Alomar A, Huffman K, Papari-Zareei M, Yang L, Drapkin B, Akbay EA, Shames DS, Wistuba II, Wang T, Johnson JE, Xiao G, DeBerardinis RJ, Minna JD, Xie Y, Gazdar AF, Cell-autonomous immune gene expression is repressed in pulmonary neuroendocrine cells and small cell lung cancer, Commun Biol, 4 (2021) 314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Kinker GS, Greenwald AC, Tal R, Orlova Z, Cuoco MS, McFarland JM, Warren A, Rodman C, Roth JA, Bender SA, Kumar B, Rocco JW, Fernandes P, Mader CC, Keren-Shaul H, Plotnikov A, Barr H, Tsherniak A, Rozenblatt-Rosen O, Krizhanovsky V, Puram SV, Regev A, Tirosh I, Pan-cancer single-cell RNA-seq identifies recurring programs of cellular heterogeneity, Nat Genet, 52 (2020) 1208–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehar J, Kryukov GV, Sonkin D, Reddy A, Liu M, Murray L, Berger MF, Monahan JE, Morais P, Meltzer J, Korejwa A, Jane-Valbuena J, Mapa FA, Thibault J, Bric-Furlong E, Raman P, Shipway A, Engels IH, Cheng J, Yu GK, Yu J, Aspesi P Jr., de Silva M, Jagtap K, Jones MD, Wang L, Hatton C, Palescandolo E, Gupta S, Mahan S, Sougnez C, Onofrio RC, Liefeld T, MacConaill L, Winckler W, Reich M, Li N, Mesirov JP, Gabriel SB, Getz G, Ardlie K, Chan V, Myer VE, Weber BL, Porter J, Warmuth M, Finan P, Harris JL, Meyerson M, Golub TR, Morrissey MP, Sellers WR, Schlegel R, Garraway LA, The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity, Nature, 483 (2012) 603–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Basu A, Bodycombe NE, Cheah JH, Price EV, Liu K, Schaefer GI, Ebright RY, Stewart ML, Ito D, Wang S, Bracha AL, Liefeld T, Wawer M, Gilbert JC, Wilson AJ, Stransky N, Kryukov GV, Dancik V, Barretina J, Garraway LA, Hon CS, Munoz B, Bittker JA, Stockwell BR, Khabele D, Stern AM, Clemons PA, Shamji AF, Schreiber SL, An interactive resource to identify cancer genetic and lineage dependencies targeted by small molecules, Cell, 154 (2013) 1151–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Iorio F, Knijnenburg TA, Vis DJ, Bignell GR, Menden MP, Schubert M, Aben N, Goncalves E, Barthorpe S, Lightfoot H, Cokelaer T, Greninger P, van Dyk E, Chang H, de Silva H, Heyn H, Deng X, Egan RK, Liu Q, Mironenko T, Mitropoulos X, Richardson L, Wang J, Zhang T, Moran S, Sayols S, Soleimani M, Tamborero D, Lopez-Bigas N, Ross-Macdonald P, Esteller M, Gray NS, Haber DA, Stratton MR, Benes CH, Wessels LFA, Saez-Rodriguez J, McDermott U, Garnett MJ, A Landscape of Pharmacogenomic Interactions in Cancer, Cell, 166 (2016) 740–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Corsello SM, Nagari RT, Spangler RD, Rossen J, Kocak M, Bryan JG, Humeidi R, Peck D, Wu X, Tang AA, Wang VM, Bender SA, Lemire E, Narayan R, Montgomery P, Ben-David U, Garvie CW, Chen Y, Rees MG, Lyons NJ, McFarland JM, Wong BT, Wang L, Dumont N, O’Hearn PJ, Stefan E, Doench JG, Harrington CN, Greulich H, Meyerson M, Vazquez F, Subramanian A, Roth JA, Bittker JA, Boehm JS, Mader CC, Tsherniak A, Golub TR, Discovering the anti-cancer potential of non-oncology drugs by systematic viability profiling, Nat Cancer, 1 (2020) 235–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].McFarland JM, Ho ZV, Kugener G, Dempster JM, Montgomery PG, Bryan JG, Krill-Burger JM, Green TM, Vazquez F, Boehm JS, Golub TR, Hahn WC, Root DE, Tsherniak A, Improved estimation of cancer dependencies from large-scale RNAi screens using model-based normalization and data integration, Nat Commun, 9 (2018) 4610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].DepMap 20Q4 Public, 2020.
- [23].Drapkin BJ, George J, Christensen CL, Mino-Kenudson M, Dries R, Sundaresan T, Phat S, Myers DT, Zhong J, Igo P, Hazar-Rethinam MH, Licausi JA, Gomez-Caraballo M, Kem M, Jani KN, Azimi R, Abedpour N, Menon R, Lakis S, Heist RS, Buttner R, Haas S, Sequist LV, Shaw AT, Wong KK, Hata AN, Toner M, Maheswaran S, Haber DA, Peifer M, Dyson N, Thomas RK, Farago AF, Genomic and Functional Fidelity of Small Cell Lung Cancer Patient-Derived Xenografts, Cancer Discov, 8 (2018) 600–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Rudin CM, Durinck S, Stawiski EW, Poirier JT, Modrusan Z, Shames DS, Bergbower EA, Guan Y, Shin J, Guillory J, Rivers CS, Foo CK, Bhatt D, Stinson J, Gnad F, Haverty PM, Gentleman R, Chaudhuri S, Janakiraman V, Jaiswal BS, Parikh C, Yuan W, Zhang Z, Koeppen H, Wu TD, Stern HM, Yauch RL, Huffman KE, Paskulin DD, Illei PB, Varella-Garcia M, Gazdar AF, de Sauvage FJ, Bourgon R, Minna JD, Brock MV, Seshagiri S, Comprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small-cell lung cancer, Nat Genet, 44 (2012) 1111–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].George J, Lim JS, Jang SJ, Cun Y, Ozretic L, Kong G, Leenders F, Lu X, Fernandez-Cuesta L, Bosco G, Muller C, Dahmen I, Jahchan NS, Park KS, Yang D, Karnezis AN, Vaka D, Torres A, Wang MS, Korbel JO, Menon R, Chun SM, Kim D, Wilkerson M, Hayes N, Engelmann D, Putzer B, Bos M, Michels S, Vlasic I, Seidel D, Pinther B, Schaub P, Becker C, Altmuller J, Yokota J, Kohno T, Iwakawa R, Tsuta K, Noguchi M, Muley T, Hoffmann H, Schnabel PA, Petersen I, Chen Y, Soltermann A, Tischler V, Choi CM, Kim YH, Massion PP, Zou Y, Jovanovic D, Kontic M, Wright GM, Russell PA, Solomon B, Koch I, Lindner M, Muscarella LA, la Torre A, Field JK, Jakopovic M, Knezevic J, Castanos-Velez E, Roz L, Pastorino U, Brustugun OT, Lund-Iversen M, Thunnissen E, Kohler J, Schuler M, Botling J, Sandelin M, Sanchez-Cespedes M, Salvesen HB, Achter V, Lang U, Bogus M, Schneider PM, Zander T, Ansen S, Hallek M, Wolf J, Vingron M, Yatabe Y, Travis WD, Nurnberg P, Reinhardt C, Perner S, Heukamp L, Buttner R, Haas SA, Brambilla E, Peifer M, Sage J, Thomas RK, Comprehensive genomic profiles of small cell lung cancer, Nature, 524 (2015) 47–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Jiang L, Huang J, Higgs BW, Hu Z, Xiao Z, Yao X, Conley S, Zhong H, Liu Z, Brohawn P, Shen D, Wu S, Ge X, Jiang Y, Zhao Y, Lou Y, Morehouse C, Zhu W, Sebastian Y, Czapiga M, Oganesyan V, Fu H, Niu Y, Zhang W, Streicher K, Tice D, Zhao H, Zhu M, Xu L, Herbst R, Su X, Gu Y, Li S, Huang L, Gu J, Han B, Jallal B, Shen H, Yao Y, Genomic Landscape Survey Identifies SRSF1 as a Key Oncodriver in Small Cell Lung Cancer, PLoS Genet, 12 (2016) e1005895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Chan JM, Quintanal-Villalonga Á, Gao V, Xie Y, Allaj V, Chaudhary O, Masilionis I, Egger J, Chow A, Walle T, Mattar M, Yarlagadda DV, Wang JL, Uddin F, Offin M, Ciampricotti M, Bhanot UK, Lai WV, Bott MJ, Jones DR, Ruiz A, Hollmann T, Poirier JT, Nawy T, Mazutis L, Sen T, Pe’er D, Rudin CM, Single cell profiling reveals novel tumor and myeloid subpopulations in small cell lung cancer, bioRxiv, (2020).
- [28].Davis S, Meltzer PS, GEOquery: a bridge between the Gene Expression Omnibus (GEO) and BioConductor, Bioinformatics, 23 (2007) 1846–1847. [DOI] [PubMed] [Google Scholar]
- [29].Harenza JL, Diamond MA, Adams RN, Song MM, Davidson HL, Hart LS, Dent MH, Fortina P, Reynolds CP, Maris JM, Transcriptomic profiling of 39 commonly-used neuroblastoma cell lines, Sci Data, 4 (2017) 170033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Pugh TJ, Morozova O, Attiyeh EF, Asgharzadeh S, Wei JS, Auclair D, Carter SL, Cibulskis K, Hanna M, Kiezun A, Kim J, Lawrence MS, Lichenstein L, McKenna A, Pedamallu CS, Ramos AH, Shefler E, Sivachenko A, Sougnez C, Stewart C, Ally A, Birol I, Chiu R, Corbett RD, Hirst M, Jackman SD, Kamoh B, Khodabakshi AH, Krzywinski M, Lo A, Moore RA, Mungall KL, Qian J, Tam A, Thiessen N, Zhao Y, Cole KA, Diamond M, Diskin SJ, Mosse YP, Wood AC, Ji L, Sposto R, Badgett T, London WB, Moyer Y, Gastier-Foster JM, Smith MA, Guidry Auvil JM, Gerhard DS, Hogarty MD, Jones SJ, Lander ES, Gabriel SB, Getz G, Seeger RC, Khan J, Marra MA, Meyerson M, Maris JM, The genetic landscape of high-risk neuroblastoma, Nat Genet, 45 (2013) 279–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Vivian J, Rao AA, Nothaft FA, Ketchum C, Armstrong J, Novak A, Pfeil J, Narkizian J, Deran AD, Musselman-Brown A, Schmidt H, Amstutz P, Craft B, Goldman M, Rosenbloom K, Cline M, O’Connor B, Hanna M, Birger C, Kent WJ, Patterson DA, Joseph AD, Zhu J, Zaranek S, Getz G, Haussler D, Paten B, Toil enables reproducible, open source, big biomedical data analyses, Nat Biotechnol, 35 (2017) 314–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Ackermann S, Cartolano M, Hero B, Welte A, Kahlert Y, Roderwieser A, Bartenhagen C, Walter E, Gecht J, Kerschke L, Volland R, Menon R, Heuckmann JM, Gartlgruber M, Hartlieb S, Henrich KO, Okonechnikov K, Altmuller J, Nurnberg P, Lefever S, de Wilde B, Sand F, Ikram F, Rosswog C, Fischer J, Theissen J, Hertwig F, Singhi AD, Simon T, Vogel W, Perner S, Krug B, Schmidt M, Rahmann S, Achter V, Lang U, Vokuhl C, Ortmann M, Buttner R, Eggert A, Speleman F, O’Sullivan RJ, Thomas RK, Berthold F, Vandesompele J, Schramm A, Westermann F, Schulte JH, Peifer M, Fischer M, A mechanistic classification of clinical phenotypes in neuroblastoma, Science, 362 (2018) 1165–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Asgharzadeh S, Pique-Regi R, Sposto R, Wang H, Yang Y, Shimada H, Matthay K, Buckley J, Ortega A, Seeger RC, Prognostic significance of gene expression profiles of metastatic neuroblastomas lacking MYCN gene amplification, J Natl Cancer Inst, 98 (2006) 1193–1203. [DOI] [PubMed] [Google Scholar]
- [34].Cole KA, Huggins J, Laquaglia M, Hulderman CE, Russell MR, Bosse K, Diskin SJ, Attiyeh EF, Sennett R, Norris G, Laudenslager M, Wood AC, Mayes PA, Jagannathan J, Winter C, Mosse YP, Maris JM, RNAi screen of the protein kinome identifies checkpoint kinase 1 (CHK1) as a therapeutic target in neuroblastoma, Proc Natl Acad Sci U S A, 108 (2011) 3336–3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Henrich KO, Bender S, Saadati M, Dreidax D, Gartlgruber M, Shao C, Herrmann C, Wiesenfarth M, Parzonka M, Wehrmann L, Fischer M, Duffy DJ, Bell E, Torkov A, Schmezer P, Plass C, Hofer T, Benner A, Pfister SM, Westermann F, Integrative Genome-Scale Analysis Identifies Epigenetic Mechanisms of Transcriptional Deregulation in Unfavorable Neuroblastomas, Cancer Res, 76 (2016) 5523–5537. [DOI] [PubMed] [Google Scholar]
- [36].Rajbhandari P, Lopez G, Capdevila C, Salvatori B, Yu J, Rodriguez-Barrueco R, Martinez D, Yarmarkovich M, Weichert-Leahey N, Abraham BJ, Alvarez MJ, Iyer A, Harenza JL, Oldridge D, De Preter K, Koster J, Asgharzadeh S, Seeger RC, Wei JS, Khan J, Vandesompele J, Mestdagh P, Versteeg R, Look AT, Young RA, Iavarone A, Lasorella A, Silva JM, Maris JM, Califano A, Cross-Cohort Analysis Identifies a TEAD4-MYCN Positive Feedback Loop as the Core Regulatory Element of High-Risk Neuroblastoma, Cancer Discov, 8 (2018) 582–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Wang C, Gong B, Bushel PR, Thierry-Mieg J, Thierry-Mieg D, Xu J, Fang H, Hong H, Shen J, Su Z, Meehan J, Li X, Yang L, Li H, Labaj PP, Kreil DP, Megherbi D, Gaj S, Caiment F, van Delft J, Kleinjans J, Scherer A, Devanarayan V, Wang J, Yang Y, Qian HR, Lancashire LJ, Bessarabova M, Nikolsky Y, Furlanello C, Chierici M, Albanese D, Jurman G, Riccadonna S, Filosi M, Visintainer R, Zhang KK, Li J, Hsieh JH, Svoboda DL, Fuscoe JC, Deng Y, Shi L, Paules RS, Auerbach SS, Tong W, The concordance between RNA-seq and microarray data depends on chemical treatment and transcript abundance, Nat Biotechnol, 32 (2014) 926–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Molenaar JJ, Koster J, Zwijnenburg DA, van Sluis P, Valentijn LJ, van der Ploeg I, Hamdi M, van Nes J, Westerman BA, van Arkel J, Ebus ME, Haneveld F, Lakeman A, Schild L, Molenaar P, Stroeken P, van Noesel MM, Ora I, Santo EE, Caron HN, Westerhout EM, Versteeg R, Sequencing of neuroblastoma identifies chromothripsis and defects in neuritogenesis genes, Nature, 483 (2012) 589–593. [DOI] [PubMed] [Google Scholar]
- [39].Wang Q, Diskin S, Rappaport E, Attiyeh E, Mosse Y, Shue D, Seiser E, Jagannathan J, Shusterman S, Bansal M, Khazi D, Winter C, Okawa E, Grant G, Cnaan A, Zhao H, Cheung NK, Gerald W, London W, Matthay KK, Brodeur GM, Maris JM, Integrative genomics identifies distinct molecular classes of neuroblastoma and shows that multiple genes are targeted by regional alterations in DNA copy number, Cancer Res, 66 (2006) 6050–6062. [DOI] [PubMed] [Google Scholar]
- [40].Tsherniak A, Vazquez F, Montgomery PG, Weir BA, Kryukov G, Cowley GS, Gill S, Harrington WF, Pantel S, Krill-Burger JM, Meyers RM, Ali L, Goodale A, Lee Y, Jiang G, Hsiao J, Gerath WFJ, Howell S, Merkel E, Ghandi M, Garraway LA, Root DE, Golub TR, Boehm JS, Hahn WC, Defining a Cancer Dependency Map, Cell, 170 (2017) 564–576 e516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Huang YH, Klingbeil O, He XY, Wu XS, Arun G, Lu B, Somerville TDD, Milazzo JP, Wilkinson JE, Demerdash OE, Spector DL, Egeblad M, Shi J, Vakoc CR, POU2F3 is a master regulator of a tuft cell-like variant of small cell lung cancer, Genes Dev, 32 (2018) 915–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Lissa D, Takahashi N, Desai P, Manukyan I, Schultz CW, Rajapakse V, Velez MJ, Mulford D, Roper N, Nichols S, Vilimas R, Sciuto L, Chen Y, Guha U, Rajan A, Atkinson D, El Meskini R, Weaver Ohler Z, Thomas A, Heterogeneity of neuroendocrine transcriptional states in metastatic small cell lung cancers and patient-derived models, Nat Commun, 13 (2022) 2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Johnson BE, Ihde DC, Makuch RW, Gazdar AF, Carney DN, Oie H, Russell E, Nau MM, Minna JD, myc family oncogene amplification in tumor cell lines established from small cell lung cancer patients and its relationship to clinical status and course, J Clin Invest, 79 (1987) 1629–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Huang M, Weiss WA, Neuroblastoma and MYCN, Cold Spring Harb Perspect Med, 3 (2013) a014415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Pearson JD, Huang K, Pacal M, McCurdy SR, Lu S, Aubry A, Yu T, Wadosky KM, Zhang L, Wang T, Gregorieff A, Ahmad M, Dimaras H, Langille E, Cole SPC, Monnier PP, Lok BH, Tsao MS, Akeno N, Schramek D, Wikenheiser-Brokamp KA, Knudsen ES, Witkiewicz AK, Wrana JL, Goodrich DW, Bremner R, Binary pan-cancer classes with distinct vulnerabilities defined by pro- or anti-cancer YAP/TEAD activity, Cancer Cell, 39 (2021) 1115–1134 e1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Kim KB, Dunn CT, Park KS, Recent progress in mapping the emerging landscape of the small-cell lung cancer genome, Exp Mol Med, 51 (2019) 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Heymanns J, Unsicker K, Neuroblastoma cells contain a trophic factor sharing biological and molecular properties with ciliary neurotrophic factor, Proc Natl Acad Sci U S A, 84 (1987) 7758–7762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Muth D, Ghazaryan S, Eckerle I, Beckett E, Pohler C, Batzler J, Beisel C, Gogolin S, Fischer M, Henrich KO, Ehemann V, Gillespie P, Schwab M, Westermann F, Transcriptional repression of SKP2 is impaired in MYCN-amplified neuroblastoma, Cancer Res, 70 (2010) 3791–3802. [DOI] [PubMed] [Google Scholar]
- [49].Hoene V, Fischer M, Ivanova A, Wallach T, Berthold F, Dame C, GATA factors in human neuroblastoma: distinctive expression patterns in clinical subtypes, Br J Cancer, 101 (2009) 1481–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Weng Z, Lin J, He J, Gao L, Lin S, Tsang LL, Zhang H, He X, Wang G, Yang X, Zhou H, Zhao H, Li G, Zou L, Jiang X, Human embryonic stem cell-derived neural crest model unveils CD55 as a cancer stem cell regulator for therapeutic targeting in MYCN-amplified neuroblastoma, Neuro Oncol, 24 (2022) 872–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Schmitt-Hoffner F, van Rijn S, Toprak UH, Mauermann M, Rosemann F, Heit-Mondrzyk A, Hubner JM, Camgoz A, Hartlieb S, Pfister SM, Henrich KO, Westermann F, Kool M, FOXR2 Stabilizes MYCN Protein and Identifies Non-MYCN-Amplified Neuroblastoma Patients With Unfavorable Outcome, J Clin Oncol, 39 (2021) 3217–3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Fagerstrom S, Pahlman S, Nanberg E, Protein kinase C-dependent tyrosine phosphorylation of p130cas in differentiating neuroblastoma cells, J Biol Chem, 273 (1998) 2336–2343. [DOI] [PubMed] [Google Scholar]
- [53].Sengupta S, Das S, Crespo AC, Cornel AM, Patel AG, Mahadevan NR, Campisi M, Ali AK, Sharma B, Rowe JH, Huang H, Debruyne DN, Cerda ED, Krajewska M, Dries R, Chen M, Zhang S, Soriano L, Cohen MA, Versteeg R, Jaenisch R, Spranger S, Romee R, Miller BC, Barbie DA, Nierkens S, Dyer MA, Lieberman J, George RE, Mesenchymal and adrenergic cell lineage states in neuroblastoma possess distinct immunogenic phenotypes, Nat Cancer, 3 (2022) 1228–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Cai L, Sondhi V, Zhu M, Akbay E, DeBerardinis RJ, Xie Y, Minna JD, Xiao G, Gazdar A, The small cell lung cancer neuroendocrine transdifferentiation explorer, bioRxiv, (2022).
- [55].Mullen NJ, Singh PK, Nucleotide metabolism: a pan-cancer metabolic dependency, Nat Rev Cancer, (2023) 1–20. [DOI] [PMC free article] [PubMed]
- [56].Lee JK, Phillips JW, Smith BA, Park JW, Stoyanova T, McCaffrey EF, Baertsch R, Sokolov A, Meyerowitz JG, Mathis C, Cheng D, Stuart JM, Shokat KM, Gustafson WC, Huang J, Witte ON, N-Myc Drives Neuroendocrine Prostate Cancer Initiated from Human Prostate Epithelial Cells, Cancer Cell, 29 (2016) 536–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Berger A, Brady NJ, Bareja R, Robinson B, Conteduca V, Augello MA, Puca L, Ahmed A, Dardenne E, Lu X, Hwang I, Bagadion AM, Sboner A, Elemento O, Paik J, Yu J, Barbieri CE, Dephoure N, Beltran H, Rickman DS, N-Myc-mediated epigenetic reprogramming drives lineage plasticity in advanced prostate cancer, J Clin Invest, 129 (2019) 3924–3940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Ireland AS, Micinski AM, Kastner DW, Guo B, Wait SJ, Spainhower KB, Conley CC, Chen OS, Guthrie MR, Soltero D, Qiao Y, Huang X, Tarapcsak S, Devarakonda S, Chalishazar MD, Gertz J, Moser JC, Marth G, Puri S, Witt BL, Spike BT, Oliver TG, MYC Drives Temporal Evolution of Small Cell Lung Cancer Subtypes by Reprogramming Neuroendocrine Fate, Cancer Cell, (2020). [DOI] [PMC free article] [PubMed]
- [59].Mollaoglu G, Guthrie MR, Bohm S, Bragelmann J, Can I, Ballieu PM, Marx A, George J, Heinen C, Chalishazar MD, Cheng H, Ireland AS, Denning KE, Mukhopadhyay A, Vahrenkamp JM, Berrett KC, Mosbruger TL, Wang J, Kohan JL, Salama ME, Witt BL, Peifer M, Thomas RK, Gertz J, Johnson JE, Gazdar AF, Wechsler-Reya RJ, Sos ML, Oliver TG, MYC Drives Progression of Small Cell Lung Cancer to a Variant Neuroendocrine Subtype with Vulnerability to Aurora Kinase Inhibition, Cancer Cell, 31 (2017) 270–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Olsen RR, Ireland AS, Kastner DW, Groves SM, Spainhower KB, Pozo K, Kelenis DP, Whitney CP, Guthrie MR, Wait SJ, Soltero D, Witt BL, Quaranta V, Johnson JE, Oliver TG, ASCL1 represses a SOX9(+) neural crest stem-like state in small cell lung cancer, Genes Dev, 35 (2021) 847–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Brady NJ, Bagadion AM, Singh R, Conteduca V, Van Emmenis L, Arceci E, Pakula H, Carelli R, Khani F, Bakht M, Sigouros M, Bareja R, Sboner A, Elemento O, Tagawa S, Nanus DM, Loda M, Beltran H, Robinson B, Rickman DS, Temporal evolution of cellular heterogeneity during the progression to advanced AR-negative prostate cancer, Nat Commun, 12 (2021) 3372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Tanaka T, Higashi M, Kimura K, Wakao J, Fumino S, Iehara T, Hosoi H, Sakai T, Tajiri T, MEK inhibitors as a novel therapy for neuroblastoma: Their in vitro effects and predicting their efficacy, J Pediatr Surg, 51 (2016) 2074–2079. [DOI] [PubMed] [Google Scholar]
- [63].Workman P, Powers MV, Chaperoning cell death: a critical dual role for Hsp90 in small-cell lung cancer, Nat Chem Biol, 3 (2007) 455–457. [DOI] [PubMed] [Google Scholar]
- [64].Regan PL, Jacobs J, Wang G, Torres J, Edo R, Friedmann J, Tang XX, Hsp90 inhibition increases p53 expression and destabilizes MYCN and MYC in neuroblastoma, Int J Oncol, 38 (2011) 105–112. [PMC free article] [PubMed] [Google Scholar]
- [65].Fennell DA, Bcl-2 as a target for overcoming chemoresistance in small-cell lung cancer, Clin Lung Cancer, 4 (2003) 307–313. [DOI] [PubMed] [Google Scholar]
- [66].Lamers F, Schild L, den Hartog IJ, Ebus ME, Westerhout EM, Ora I, Koster J, Versteeg R, Caron HN, Molenaar JJ, Targeted BCL2 inhibition effectively inhibits neuroblastoma tumour growth, Eur J Cancer, 48 (2012) 3093–3103. [DOI] [PubMed] [Google Scholar]
- [67].Puissant A, Frumm SM, Alexe G, Bassil CF, Qi J, Chanthery YH, Nekritz EA, Zeid R, Gustafson WC, Greninger P, Garnett MJ, McDermott U, Benes CH, Kung AL, Weiss WA, Bradner JE, Stegmaier K, Targeting MYCN in neuroblastoma by BET bromodomain inhibition, Cancer Discov, 3 (2013) 308–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Peng H, Ke XX, Hu R, Yang L, Cui H, Wei Y, Essential role of GATA3 in regulation of differentiation and cell proliferation in SK-N-SH neuroblastoma cells, Mol Med Rep, 11 (2015) 881–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Semenova EA, Kwon MC, Monkhorst K, Song JY, Bhaskaran R, Krijgsman O, Kuilman T, Peters D, Buikhuisen WA, Smit EF, Pritchard C, Cozijnsen M, van der Vliet J, Zevenhoven J, Lambooij JP, Proost N, van Montfort E, Velds A, Huijbers IJ, Berns A, Transcription Factor NFIB Is a Driver of Small Cell Lung Cancer Progression in Mice and Marks Metastatic Disease in Patients, Cell Rep, 16 (2016) 631–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Cell line datasets
Copy number, RNA-seq, miRNA, histone PTM, metabolomics, and RPPA data were downloaded from DepMap. Compound sensitivity data for “CCLE” [17], “CTRP”[18], “GDSC1” and “GDSC2” [19], “PRISM_1st” and “PRISM_2nd” [20], and gene dependency data for demeter (RNAi) [21] and achilles [22] (CRISPR) were downloaded from DepMap and processed as previously described [13]. The cell line names and compound names were unified, and the datasets were processed to ensure that the lower value in each dataset always corresponded to a higher sensitivity. The processed data, lists of consistent compounds, and dependencies were downloaded from https://lccl.shinyapps.io/FDCE/. The scRNA-seq data for cell lines were downloaded from the Gene Expression Omnibus (GEO) repository GSE157220 [16].
Additional SCLC datasets
The following SCLC transcriptomic datasets “UTSW SCLC cell line,” “Drapkin_2018” (PDX) [23], tumor datasets “Rudin_2012” [24], “George_2015” [25] , “Jiang_2016” [26], and “Cai_2021” [15] were processed as previously described [15]. The processed data are available in our previous publication [15]. SCLC scRNA-seq data were downloaded from the HTAN portal [27].
Additional NBL datasets
In addition to the CCLE RNA-seq data, additional NBL cell line transcriptomic and associated sample phenotype data were downloaded from GEO using R package GEOquery [28] with the following accession numbers: GSE28019, GSE89413 [29], and GSE90683 [10]. For NBL patient tumor datasets, we included two partially overlapped NBL datasets from Therapeutically Applicable Research to Generate Effective Treatments (TARGET) (https://ocg.cancer.gov/programs/target) initiative, phs000467 [30]. “TARGET_microarray” was downloaded from the TARGET Data Matrix, whereas “TARGET_RNA-seq” was downloaded from the UCSC Toil RNAseq Recompute Compendium [31]. Additional NBL tumor datasets were downloaded from GEO with the following accession numbers: GSE120572 [32], GSE3446[33], GSE19274[34], GSE73517[35], GSE85047[36], GSE62564[37], GSE16476[38], and GSE3960[39].