

Abstract
BACKGROUND
Mitral valve (MV) elongation is a primary hypertrophic cardiomyopathy (HCM) phenotype and contributes to obstruction. The residual MV leaflet that protrudes past the coaptation point is especially susceptible to flow-drag and systolic anterior motion. Histopathological features of MVs in obstructive hypertrophic cardiomyopathy (OHCM), and of residual leaflets specifically, are unknown.
OBJECTIVES
The purpose of this study was to characterize gross, structural, and cellular histopathologic features of MV residual leaflets in OHCM. On a cellular-level, we assessed for developmental dysregulation of epicardium-derived cell (EPDC) differentiation, adaptive endocardial-to-mesenchymal transition and valvular interstitial cell proliferation, and genetically-driven persistence of cardiomyocytes in the valve.
METHODS
Structural and immunohistochemical staining were performed on 22 residual leaflets excised as ancillary procedures during myectomy, and compared with 11 control leaflets from deceased patients with normal hearts. Structural components were assessed with hematoxylin and eosin, trichrome, and elastic stains. We stained for EPDCs, EPDC paracrine signaling, valvular interstitial cells, endocardial-to-mesenchymal transition, and cardiomyocytes.
RESULTS
The residual leaflet was always at A2 segment and attached by slack, elongated and curlicued, myxoid chords. MV residual leaflets in OHCM were structurally disorganized, with expanded spongiosa and increased, fragmented elastic fibers compared with control leading edges. The internal collagenous fibrosa was attenuated and there was collagenous tissue overlying valve surfaces in HCM, with an overall trend toward decreased leaflet thickness (1.09 vs 1.47 mm, P = 0.08). No markers of primary cellular processes were identified.
CONCLUSIONS
MV residual leaflets in HCM were characterized by histologic findings that were likely secondary to chronic hemodynamic stress and may further increase susceptibility to systolic anterior motion.
Keywords: hypertrophic cardiomyopathy, immunohistochemistry, left ventricular outflow tract obstruction, mitral valve, pathology, surgery
Hypertrophic cardiomyopathy (HCM) is the most common heritable cardiomyopathy, characterized by hypertrophy not attributable to excess load. Left ventricular outflow tract (LVOT) obstruction is present in approximately two-thirds of patients with HCM and may cause symptoms including exercise intolerance, dyspnea, fatigue, and syncope.1 Obstruction is mediated by systolic anterior motion (SAM) of the mitral valve (MV) into a narrowed outflow tract. Mitral apparatus abnormalities, such as leaflet elongation and the presence of a residual leaflet that protrudes past the coaptation point, predispose patients to SAM, as valves with these abnormalities are pushed by even low-velocity flow-drag forces towards the interventricular septum.2
Approximately two-thirds of patients with HCM have elongated MVs.3,4 The initial impetus for this elongation is likely a primary, rather than secondary, process, as studies have found longer anterior and posterior mitral leaflets in genotype-positive, patients with phenotype-negative HCM compared with normal controls.4,5
Given the current understanding of HCM being caused by sarcomere mutations, the pathophysiological mechanism of MV elongation is not understood. Four hypotheses arise from the literature: dysregulated stem cell differentiation during valve development (developmental hypothesis), adaptive cell line activation in response to chronic tethering forces (adaptive hypothesis), genetic persistence of myocardial cells in the valve (genetic hypothesis), and mechanical stretch from chronic hemodynamic load (mechanical hypothesis).6–9 Only 1 study, by Klues et al in 1992, performed histopathological analysis of MVs from patients with HCM. They included valves from 10 obstructive and 12 non-obstructive patients and used Movat and hematoxylin and eosin (H&E) staining only, finding features they considered secondary in origin.3
In this study, we performed histopathological analysis of MV residual leaflets in patients with obstructive hypertrophic cardiomyopathy (OHCM), and compared them with cadaver valves from patients without structural heart disease. We also used cellular immunohistochemistry to investigate the aforementioned hypotheses and assess cellular composition.
METHODS
STUDY PARTICIPANTS.
Patients with confirmed OHCM scheduled for septal myectomy and possible MV repair or replacement provided consent for tissue excised during surgery to be stored and used for research.10 In addition to extended myectomy, the operating surgeon (D.G.S.) determined which patients would benefit from residual leaflet excision (ReLex) (n = 18) based on review of preoperative echocardiographic images with the referring cardiologist, transesophageal images at time of surgery, and direct intraoperative inspection.11 Of the 22 eligible patients consecutively enrolled, 18 had ReLex for mitral repair. Four (ages 63-82 years) were observed to have pliable residual leaflets on echocardiography and intraoperative visual inspection but were selected preoperatively for MV replacement because of severe posterior mitral annular calcification. Genotype analysis was offered to all patients and performed in 11.
Control autopsy valves were obtained either from eligible cadavers at our institution (n = 4, New York University [NYU]) or sent frozen from a collaborating institution (n = 10, Ohio State). At NYU, patients without pathologic evidence or clinical history of major cardiac or valvular disease were considered eligible. At our collaborating institution, valves were harvested from normal potential donor hearts that had been explanted but not transplanted. The exclusion criteria for controls are in the Supplemental Methods.
This study was approved by the institutional review board at NYU Grossman School of Medicine; informed consent was obtained from all subjects.
RESIDUAL LEAFLET EXCISION.
All patients considered for septal myectomy undergo rigorous analysis of their individual pathophysiology of obstruction, and a strategy for operative intervention is planned. The length of the anterior and posterior mitral leaflets is ascertained with echocardiography, with attention to the portion of leaflets extending beyond the area of coaptation–the residual leaflet.12,13 It is critical to establish a length of actual coaptation of 1 cm to qualify for ReLex. It is also vital to differentiate between leaflet tissues and what may be excessive chordal tissue; in this regard, 3-dimensional echocardiography is useful.11 During surgery, after myectomy is performed, it becomes easier to visualize the anterior mitral leaflet, then a nerve hook is used to bring the leading edge into the outflow tract. A video of the ReLex surgical technique for mitral shortening may be viewed.10
The residual leaflet of the anterior leaflet middle scallop (A2) is carefully excised after assuring adequate chordal support on either side (Supplemental Figure 1). Importantly, if the key characteristics of a residual leaflet (described in Results below) are not clearly apparent, it is critical to avoid excising the MV leading edge, regardless of preoperative echocardiographic impressions. Leading-edge excision in absence of characteristic findings leads to postoperative central mitral regurgitation.
TISSUE PROCESSING.
After excision, resected leaflet tissue was transported in saline, and then fixed in 10% Neutral Buffered Formalin for 48 hours. After pre-embedding processing, while in wax, the leaflet tissue was sectioned perpendicular to the free-edge and embedded in paraffin on-edge to enable visualization of leaflet layers, from atrial to ventricular surfaces. Septal myectomy samples from the same HCM subjects were similarly serially sectioned.
For control MVs, the distal portion of A2, including a small portion of the body of the valve, was resected from cadaver hearts after formalin fixation. Valves were processed in the same fashion as HCM surgical specimens. Frozen control valves from Ohio State were placed in 10% Neutral Buffered Formalin for 72 hours, and processed as above. Five-micron tissue sections were collected onto Plus slides (Fisher Scientific, Cat #22-042-924), air-dried, and stored at room temperature before use.
HISTOCHEMICAL STAINING.
Leaflet architecture was assessed using H&E, Masson’s Trichrome (collagen), and Weigert Resorcin/Fuchsin (elastin) staining protocols. Septal myocardium architecture was similarly assessed using H&E and trichrome stains. In leaflets where multiple sections were present, length and thickness were measured in the best-oriented section.
IMMUNOHISTOCHEMICAL STAINING.
Chromogenic immunohistochemistry was performed using antibodies shown in Supplemental Table 1. Cells from the epicardium-derived cell (EPDC) lineage were identified using anti-t-box transcription factor 18 (Tbx18) antibody, a proepicardial transcription factor that is constitutively suppressed in healthy adult EPDCs, but becomes re-expressed when epicardial to mesenchymal transition occurs in disease.14,15 To evaluate for paracrine secretion of periostin, sections were stained with an anti-periostin antibody. Interventricular septal myocardium specimens from HCM subjects were also stained with anti-Tbx18 and anti-periostin. Valvular interstitial cells (VICs) were identified using an anti-vimentin antibody. Inflammatory cells in the valve were assessed using an antibody against leukocyte common antigen CD45. Valves were investigated for the presence of cardiomyocytes using an anti-cardiac troponin I antibody. See Supplemental Appendix for additional methods.
To assess for endocardial to mesenchymal transition, valves were sequentially stained with an endothelial marker, anti-CD31, then a fibroblast marker, anti-α-smooth muscle actin (SMA), using sequential elution-stripping multiplex assay.16 See the Supplemental Appendix for detailed immunohistochemical methods.
PATHOLOGICAL ANALYSIS.
Structural stains (H&E, trichrome, elastic) were evaluated subjectively, as structural disorganization precluded quantification of layer thickness. Anti-periostin was evaluated subjectively due to its extracellular, tissue-level staining, and anti-CD45 and anti-troponin were evaluated subjectively because the relative paucity of cells precluded digital quantitative and statistical analysis. All slides were reviewed by investigator N.N., a dedicated cardiac pathologist, with simultaneous review by a second investigator (A.T.). Both investigators then summarized subjective findings; N.N. edited all final descriptions. Thickness was measured at the mid-point of each HCM leaflet, and 4 mm from the distal edge (the mean mid-point for HCM leaflets) in control leaflets. For quantitative cellular analysis methodology, see the Supplemental Appendix.
ECHOCARDIOGRAPHIC ANALYSIS.
Preoperative and postoperative transthoracic echocardiograms were reviewed by 1 cardiologist (D.M.). Measurements included MV anterior and posterior leaflet lengths, MV protrusion height, basal anteroseptal thickness, mitral regurgitation grade, presence of mitral annular calcification, LV ejection fraction, as well as peak LVOT gradient at rest, with Valsalva, standing, and post-exercise, when available. Mitral regurgitation (MR) was graded on a 0 to 4 ordinal scale, increasing from 0 = none, to 1 = trace, 2 = mild, 3 = moderate, and 4 = severe.
STATISTICAL ANALYSIS.
Welch 2-sample t-tests, Wilcoxon rank sum tests, and Spearman rank correlation coefficients were performed using a 2-sided P value threshold of 0.05 for significance. Statistical testing was conducted using RStudio (Build 372, RStudio, PBC) and means and standard deviations were calculated using Excel (V16.16.27, Microsoft). For more details, see Supplemental Table 3.
RESULTS
CLINICAL CHARACTERISTICS.
Clinical and echocardiographic characteristics of all HCM subjects are reported in Table 1. There were 22 HCM MVs and 11 control valves. Subjects’ age at time of surgery was 59 ± 12 years, and 41% were female. They had high rates of HCM symptoms; 91% had exertional dyspnea, 50% presyncope, and 23% syncope. The anterior leaflet length in HCM subjects was 33 ± 5 mm pre-op and 29 ± 5 mm post-op. Basal anteroseptal thickness was 18 ± 2 mm pre-op and 12 ± 2 mm post-op. Pre-op resting LVOT gradient was 49 ± 41 mm Hg and provoked gradient was 99 ± 3 mm Hg. Post-op resting LVOT gradient was 11 ± 4 mm Hg and provoked gradient was 11 ± 3 mm Hg. One patient (4.5%) had moderate MR post-op, all others had either trace or mild MR. Of the 11 patients who elected to have genotype testing, 2 had HCM-associated variants. Five had variants of uncertain significance and 4 were negative for an HCM variant.
Table 1.
Clinical and Echocardiographic Characteristics of OHCM Subjects
| Age (y) | 59 ± 12 | |
|
| ||
| Female (%) | 41 | |
|
| ||
| Clinical | ||
| NYHA functional class | 2.4 ± 0.5 | |
| Exertional dyspnea | 91 | |
| Presyncope | 50 | |
| Syncope | 23 | |
| Atrial fibrillation | 32 | |
| On beta blockers | 82 | |
| On calcium-channel blockers | 45 | |
| On disopyramide | 64 | |
|
| ||
| Structural | Preoperative | Postoperative |
|---|---|---|
| MV anterior leaflet length (mm) | 33 ± 5 | 29 ± 5 |
| MV posterior leaflet length (mm) | 15 ± 2 | 14 ± 2 |
| Basal anteroseptal thickness (mm) | 18 ± 3 | 12 ± 2 |
| Peak LVOT gradient at rest (mm Hg) | 49 ± 41 | 11 ± 3 |
| Peak LVOT gradient with provocation (mm Hg) | 99 ± 23 | 11 ± 4 |
| Mitral regurgitation grade (0-4) | 2a | 2a |
| Left ventricular ejection fraction (%) | 73 ± 4 | 68 ± 4 |
Values are mean ± SD or %.
The medians are reported.
LVOT = left ventricular outflow tract; MV = mitral valve; NYHA = New York Heart Association; OHCM = obstructive hypertrophic cardiomyopathy.
PATHOLOGICAL FINDINGS IN THE MITRAL VALVE.
Surgical anatomy.
The residual leaflet is located at the distal edge of A2. It appears as a tongue, with cowl-like excursion into the outflow tract. It is always attached to the papillary muscles by elongated, slack, and curlicued redundant myxoid chords (Supplemental Figure 1).
Structural histopathology.
The mean length of MV residual leaflets collected from patients with HCM was 8.0 ± 4.0 mm (Figure 1).
FIGURE 1. Residual Leaflet Histology.
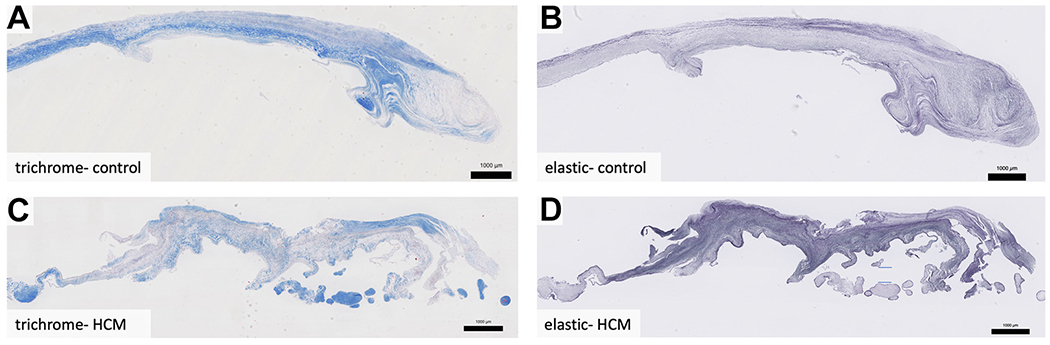
MVs from a cadaveric control (A, B) and a patient with hypertrophic cardiomyopathy (C, D), oriented with the proximal edge on left and free edge on right. Control valves have laminar structure, with trichrome staining (A) revealing a well-organized collagenous fibrosa layer that decreases in thickness in the distal leaflet (right), and elastic staining reveals a thin spongiosa layer that expands in the distal leaflet (B). Hypertrophic cardiomyopathy valves have attenuated fibrosa (C), expanded spongiosa (D), and are disorganized throughout. They also have superimposed collagenous tissue present at their distal portions (C, D) and an overall trend toward decreased thickness compared with controls. Weigert Resorcin/Fuchsin, elastic fibers = black. Trichrome, collagen = blue. HCM = hypertrophic cardiomyopathy.
The normal MV consists of the following layers from left atrial to left ventricular surfaces: atrialis, spongiosa, fibrosa, and ventricularis. The major histological findings in the substance of HCM valves were in the spongiosa and fibrosa layers. The spongiosa layer is normally composed of ground substance, including proteoglycans and glycosaminoglycans, and elastic fibers. In HCM valves, the spongiosa was expanded with disruption of the fibrosa (Figure 2). In the spongiosa of HCM valves, elastic fibers were increased in number, fragmented, and disorganized compared with their neat layering in controls (Figures 2B, 2D, 3B, and 3D). The collagen-rich fibrosa layer was attenuated in HCM valves (blue, Trichrome), featuring loose, fragmented collagen bundles that differed from its dense laminar nature in controls (Figures 2A, 2C, 3A, and 3C).
FIGURE 2. Structural Analysis.
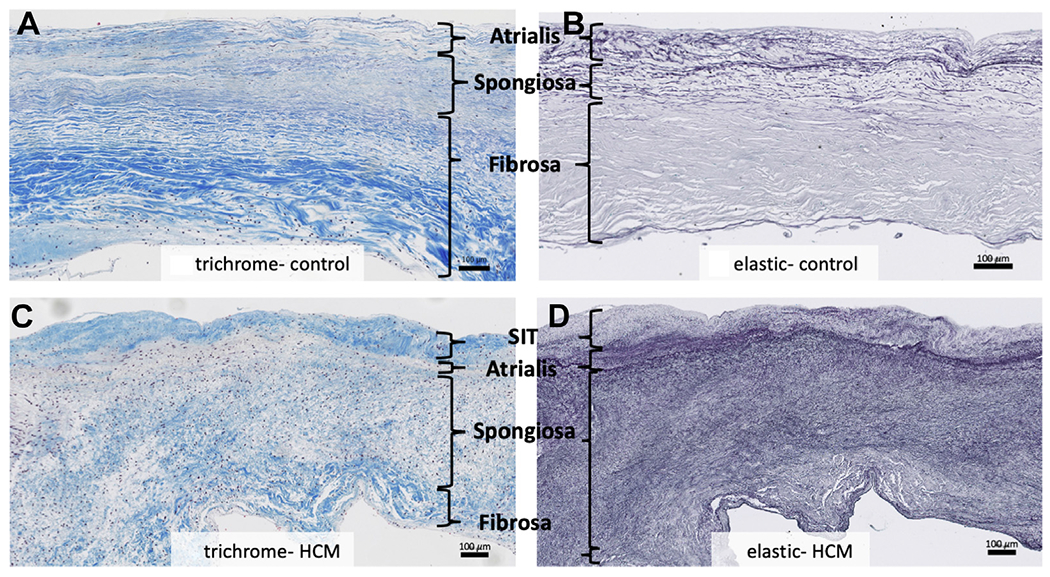
Compared to the Leading edge of normal control mitral valves (A, B), hypertrophic cardiomyopathy residual leaflets (C, D) are structurally disorganized. Trichrome staining of normal valves reveals a dense laminar fibrosa Layer (A). Trichrome staining of hypertrophic cardiomyopathy leaflets reveals attenuated fibrosa with loose, fragmented collagen bundles (C). Elastic staining of normal valves reveals a compact spongiosa Layer with Laminar elastic fibers (B). Elastic staining of hypertrophic cardiomyopathy leaflets reveals an expanded spongiosa Layer with fragmented, disorganized fibers (D). Weigert Resorcin/Fuchsin, elastic fibers = black. Trichrome, collagen = blue. HCM = hypertrophic cardiomyopathy; SIT = superimposed collagenous tissue.
FIGURE 3. Specific Structural Changes.
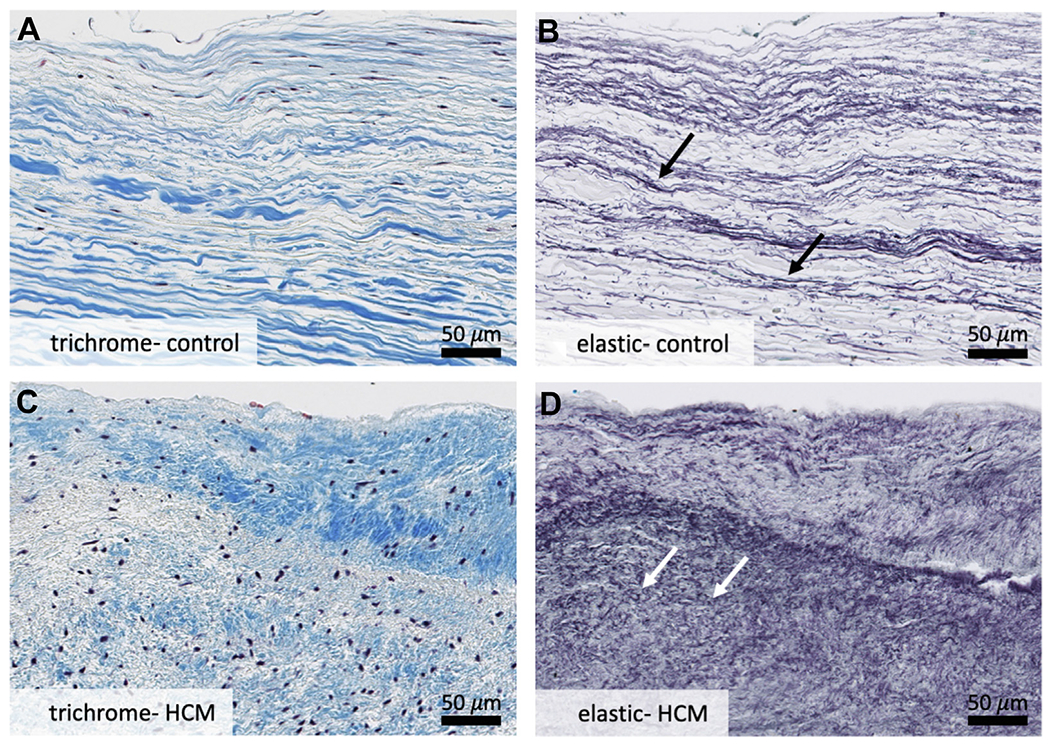
Higher-powered analysis of leading edge of control mitral valves (A, B) and hypertrophic cardiomyopathy residual leaflets (C, D) reveals disruption of the collagenous fibrosa layer and increased proliferation and fragmentation of elastic fibers in hypertrophic cardiomyopathy valves (D). A layer of superimposed collagenous tissue is also noted on surface of hypertrophic cardiomyopathy valves (C) that is not present on control valves (A). White arrows point to short, truncated elastic fibers as compared to normal fibers indicated by black arrows above. Overall, these changes are consistent with chronic hemodynamic stress on the hypertrophic cardiomyopathy residual leaflet. Weigert Resorcin/Fuchsin, elastic fibers = black. Trichrome, collagen = blue. HCM = hypertrophic cardiomyopathy.
Both the atrialis and ventricularis are normally composed of thin layers of elastic and collagen fibers (Figures 2A and 2B). In HCM valves, there was a layer of fibrosis super-imposed on both the atrial and ventricular surfaces of HCM valves that was beyond the atrialis and ventricularis (Figures 2C, 2D, 3C, and 3D) and best seen on trichrome and elastic stains. The super-imposed tissue was present on both valve surfaces, with slight atrial predominance, often encasing the chordae tendineae at their insertion sites (Figure 4A).
FIGURE 4. Notable Histologic Features.
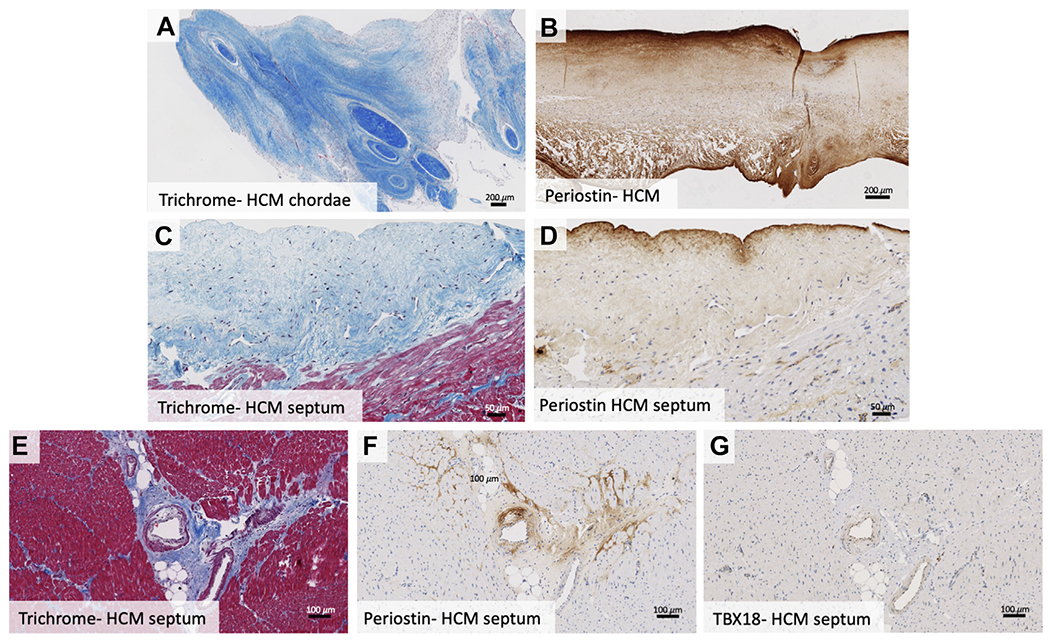
In some hypertrophic cardiomyopathy subjects, superimposed tissue extended beyond the valve, encasing the chordae tendineae at their insertion sites (A). Hypertrophic cardiomyopathy leaflets featured patchy periostin staining of the atrial and ventricular valve surfaces and with areas of superimposed tissue staining strongly (B). Endocardial surface fibrosis with neovascularization, lymphocyte proliferation, and periostin staining is noted in septal myocardium samples (C, D). Septal myocardial samples in hypertrophic cardiomyopathy patients showed interstitial and replacement fibrosis, with abnormal intramyocardial vessels with perivascular fibrosis that stained positive for periostin (E, F). Many cells comprising abnormal arteries exhibited Tbx18 positivity (G). HCM = hypertrophic cardiomyopathy; Tbx18 = t-box transcription factor 18.
With all layers included, there was a trend toward decreased thickness in HCM leaflets compared with controls (1.09 vs 1.47 mm, P = 0.08, 95% CI: [−0.06, 0.80]) (Supplemental Figure 2A).
Immunohistochemical findings.
Developmental markers.
Immunohistochemical staining with anti-Tbx18 for EPDCs showed no differences in the number of positive cells between HCM and control valves (309 vs 269 cells/high-power field, P = 0.26 [95% CI: −114 to 32]) (Supplemental Figure 2B, Supplemental Figures 3A and 3B).
Patchy periostin staining was present on the surface of both control and HCM valves, with some extension into the valve substance. Strongest periostin staining was present in areas of fibrosis, including the super-imposed collagenous tissue on the HCM valve surfaces and around VIC clusters near the valve surface (Figure 4B). HCM valves tended toward stronger periostin staining on the ventricular surface, while controls tended to have stronger staining on the atrial surface.
Adaptive markers.
There was no significant difference in quantity of VICs between HCM and control valves (13.4% vs 7.7% vimentin positivity per high-power field, P = 0.29) (Supplemental Figures 2C, 3C, and 3D). However, in HCM valves, VICs were present throughout the valve thickness and disorganized, while in control valves they were more neatly layered, accentuating normal valves’ layered structure.
CD31-positive endothelial cells were diffusely present on the valvular surfaces and rarely present in the deeper layers of both HCM and control valves (Supplemental Figures 3E and 3F). α-SMA positive VICs were present in both HCM and control valves, predominantly scattered or clustered in the spongiosa and fibrosa (Supplemental Figures 3G and 3H). Contiguous bands of smooth muscle could be seen in the middle portions some valves, proximal to the free edge. CD31+/α-SMA+ cell colocalization was absent.
The number of CD45-positive inflammatory cells varied from rare small clusters to complete lack of inflammatory cells in the valves. Nearly all clusters contained fewer than 50 cells, with the majority consisting of 3 to 10 cells (Supplemental Figures 3I and 3J). There was no difference between the number of cells present in the HCM and control valves.
Genetic markers.
No troponin-positive cells were present in any HCM valves or normal controls, indicating absence of cardiomyocytes in the valve (Supplemental Figures 3K and 3L). Summary of all pathologic analyses, organized by mechanistic hypothesis, can be found in Table 2.
Table 2.
Pathologic Analysis by Mechanistic Hypothesis
| Hypothesis | Analysis (Stain) | Hypothesis | Findings |
|---|---|---|---|
| Mechanical | Elastic fibers (Weigert Resorcin/Fuschin) Collagen fibers (trichrome) Leaflet thickness |
Chronic flow drag forces and friction on the valve cause strain, stretch, and fibrotic tissue deposition | Compared with control valves, HCM residual leaflets showed: 1. An expanded spongiosa layer with increased, fragmented, and disorganized elastic fibers 2. An attenuated, disrupted, and disorganized collagenous fibrosa layer 3. An added layer of super-imposed collagenous tissue overlying both atrial and ventricular surfaces 4. Elongated, slack and curlicued chordae and a trend to decreased leaflet thickness |
| Developmental | Epicardium derived cells (TBX-18) Paracrine signaling from epicardium-derived cells (periostin) |
Dysregulated stem cell differentiation during valve development | No difference in number of epicardium-derived cells No significant difference in amount of paracrine signaling |
| Adaptive | Endocardial-to-mesenchymal transition (CD31-αSMA) Valvular interstitial cells (vimentin) Inflammatory cells (CD45) |
Adaptive cell lines activate in response to chronic tethering forces | No evidence of endocardial-to-mesenchymal transition in HCM or control valves No difference in valvular interstitial or inflammatory cells |
| Genetic | Cardiomyocytes (cardiac troponin I) | Persistence of mutated cardiomyocytes in the valve | No cardiomyocytes in HCM or control valves |
HCM = hypertrophic cardiomyopathy; Tbx-18 = t-box transcription factor 18.
Pathological findings in OHCM septal myocardium.
Moderate-to-marked myocyte hypertrophy was present in all HCM septal myocardial samples, characterized by irregularly shaped, hyperchromatic–or dark-staining–nuclei. Some samples showed myocyte disarray. Trichrome staining revealed endocardial fibrosis with neovascularization and small clusters of lymphocytes (Figure 4C). Multifocal interstitial and perivascular fibrosis were present in all samples, and replacement fibrosis in some samples (Figure 4E). Immunohistochemical staining for periostin was positive in the areas of fibrosis (Figures 4D and 4F). The endocardial surface in areas of endocardial fibrosis showed the strongest periostin staining (Figure 4D).
Among penetrating arterioles in the septal myocardium, some had normal walls, some had segmental wall thickening, and some had diffuse circumferential thickening. The abnormally thickened arteries, and abnormally thickened wall segments, displayed stronger periostin staining than normal arterial walls (Figures 4E and 4F). Anti-Tbx18 staining was positive in the endothelial cells of abnormally thickened arteries (Figure 4G).
ECHOCARDIOGRAPHIC CORRELATIONS.
In HCM valves, there was no significant correlation between Tbx18 positive cells and anterior leaflet length (ρ = −0.11, P = 0.639), or between vimentin positive cells and anterior leaflet length (ρ = −0.07, P = 0.771).
DISCUSSION
There were multiple differences between the 22 surgically resected residual MV leaflets from patients with OHCM and normal cadaveric controls. The OHCM residual leaflets had expanded spongiosa with abundant, fragmented elastic fibers, attenuated collagenous fibrosa, and superimposed collagenous tissue overlying the valve surfaces. They were attached by elongated, curlicued chordae and had a trend toward decreased thickness.
The observed changes may represent the long-term impact of abnormal flow dynamics in OHCM. Chronic strain on the leaflets from increased flow drag forces may stretch and ultimately shear the normally laminar elastic fibers and induce reactive proliferation–namely, induce myxoid change. These are both pathologic features that have been documented to varying degrees in patients with MV prolapse.17,18 Indeed, SAM has been analogized as “anteriorly directed MV prolapse,” as both conditions feature enlarged leaflets displaced from their normal position by the force of flow, leading to mitral regurgitation. Regardless, without the resilience provided by an internal collagenous fibrosa layer and organized, laminar elastic fibers, HCM residual leaflets are less stiff and robust than the normal distal MV. On direct inspection, the chordae attached to the residual leaflet are also curlicued and slack, stretched by the chronic hydrodynamic load with each beat of the heart. All these changes further increase the leaflet’s susceptibility to flow drag forces that push the residual leaflet anteriorly during early systole (Central Illustration). Chronic friction between the distal anterior and posterior leaflets, turbulence in the obstructed proximal LVOT, and years of mitral-septal contact may lead to the reactive fibrous tissue deposition overlying the leaflets.17
CENTRAL ILLUSTRATION. The Mitral Valve Residual Leaflet in Hypertrophic Cardiomyopathy.
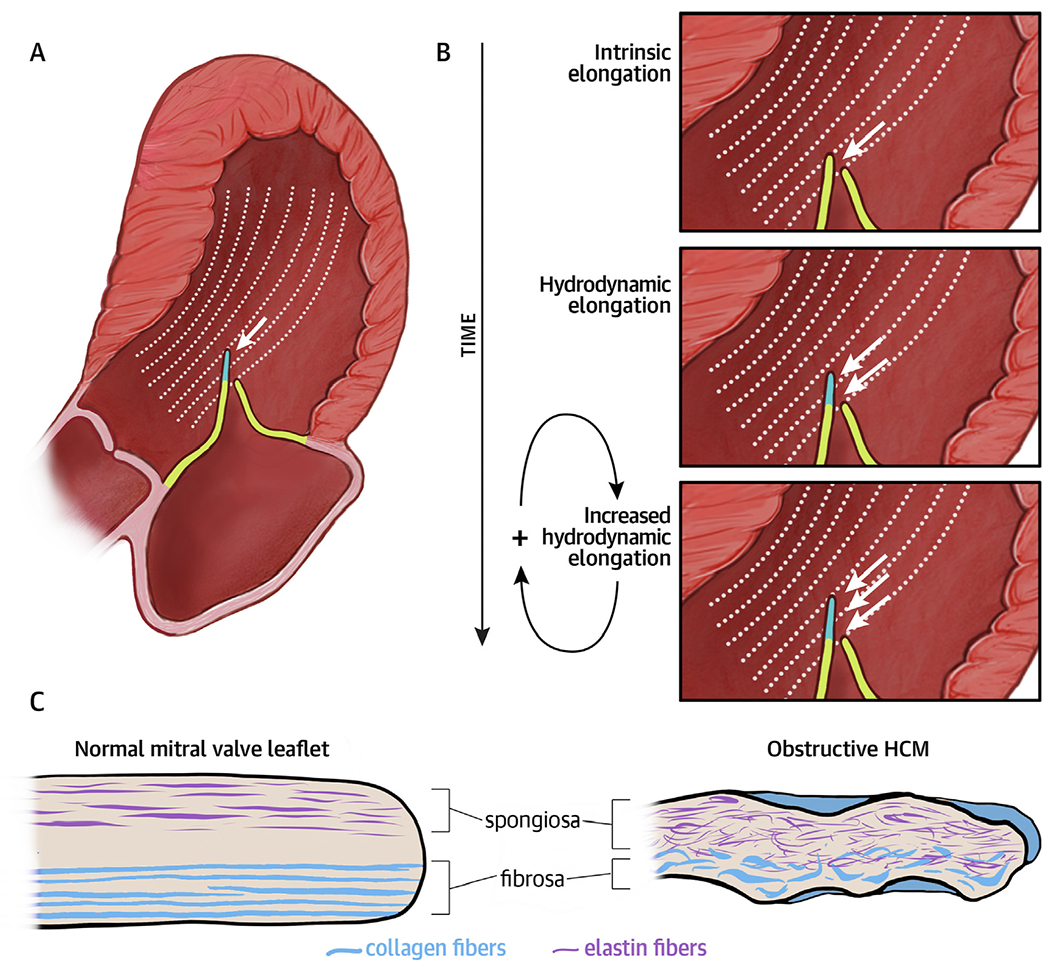
In obstructive hypertrophic cardiomyopathy, the residual leaflet extends beyond the coaptation point and is susceptible to flow drag forces that push it anteriorly, driving outflow tract obstruction (A). Our results suggest a hypothetical mechanism contributing to leaflet elongation: a feed-forward cycle in which chronic hydrodynamic forces over time cause myxoid changes that make the leaflet more susceptible to hydrodynamic elongation (B). Pushing hydrodynamic force on the residual leaflet (white arrows). Histologic analysis of resected residual leaflets showed structural disorganization with expanded spongiosa featuring increased fragmented elastic fibers, attenuated collagenous fibrosa, and superimposed collagenous tissue overlying the valve (C).
When combined with the understanding of MV elongation as a process present in non-obstructive as well as obstructive HCM patients, a novel 2-step conception of this process arises.19 First, we posit that a cellular process, not identified here, causes early leaflet elongation of the MV apparatus.2,4,20 Once the anterior leaflet is even slightly elongated or displaced, it may experience chronic hemodynamic stress from flow drag, turbulence, and increased friction against the posterior leaflet. This could then lead to the structural changes in the residual leaflet we describe, including disrupted spongiosa and fibrosa and superimposed collagen deposition, which further increase the residual leaflets’ susceptibility to these forces. A positive feedback cycle would then ensue, with the residual leaflet’s higher susceptibility to flow drag leading to increased stress and friction on the valve, further increasing susceptibility to these forces, and so on (Central Illustration). This cycle may cause greater SAM and LVOT obstruction over time. In sum, gross and pathologic examination supports the hypothesis that chronic SAM stretches and thins the residual anterior leaflet and its chordae tendinea. In rare cases, this can be complicated by chordal rupture.21
We aimed to elucidate the primary cellular pathway responsible for initial valve elongation through immunohistochemical analysis, albeit only in the residual leaflet. We tested 3 hypotheses for the etiology of primary MV elongation. We investigated a developmental hypothesis: in utero, mutated cardiomyocytes dysregulate the development of pluripotent EPDCs, causing abnormal differentiation and increased migration of these cells into the developing MV.6 This mechanism could explain other extended HCM phenotypes like fibrosis, disarray, and microvascular remodeling.
In our analysis, we found no significant difference in number of EPDCs, and paracrine periostin signaling was similar in HCM and control residual leaflets. This is consistent with experiments in a mouse model of HCM showing absence of MV elongation in embryos.22
Second, we investigated an adaptive hypothesis: that cellular pathways activate in response to chronic tethering forces. This process has been identified in the post-myocardial infarction period and is mediated by endocardial to mesenchymal transition, VIC activation, and fibrosis.7,8 We did not find large numbers of activated VICs in either HCM or control leaflets and found no areas of endocardial to mesenchymal transition. We also found few scattered clusters of inflammatory cells that did not appear increased in HCM valves. Altogether, lack of evidence for these cellular pathways and the quiescence of VICs suggest that residual leaflet elongation in HCM is a secondary stretch process due to hydrodynamic forces rather than cellularly mediated adaptive elongation.
And, finally, we explored a genetic hypothesis, that cardiomyocytes–the cells most directly affected by this disease–are present in the valve, either through persistence from early cardiac development or through abnormal migration.9 However, cardiomyocytes were absent in both control and HCM valves.
STUDY LIMITATIONS.
Our immunohistochemical analysis did not show an active cellular pathway in the residual leaflet. Three limitations of our study may be responsible. First, we only studied the most distal portion of the MV, the region most strongly affected by hemodynamic forces and most directly contributing to obstruction, and therefore excised. Evidence for the cellular signaling pathway that triggers the valve’s primary elongation may lie in not-excised more proximal portions. Second, the briefly reported observations of Klues et al3 on whole excised MVs differed from ours, which may reflect differing hemodynamic impacts of obstruction on the residual leaflet compared with more proximal portions of the valve. Accessing proximal leaflets from humans may prove elusive as mitral repair is greatly preferred over replacement. Third, for our cellular analysis, we performed immunohistochemistry on formalin-fixed human valves rather than proteomics or transcriptomics on fresh tissue. However, our pathologic analysis enabled us to detect important structural abnormalities that would not have been illuminated by the aforementioned methods. Finally, the trend toward decreased thickness of HCM valves without reaching statistical significance may have been due to our study being underpowered to detect this difference, limited by access to normal cadaveric valves, and to the competing effects of superimposed collagen deposition and thinned fibrosa.
FUTURE DIRECTIONS.
Future studies should build on this newfound understanding of the MV residual leaflet in OHCM and work to identify cellular processes responsible for initial valve body elongation by performing pathologic, immunohistochemical, proteomic, and transcriptomic studies in animal models of HCM.
Additionally, in the septum, we found EPDCs to be present only in pathologically thickened penetrating vessels, and positive periostin staining in both abnormal vessels and areas of fibrosis. Future studies comparing periostin and Tbx18 staining in the septal myocardium of patients with and without HCM could shed light on the developmental hypothesis and potential of EPDCs as contributors to vascular narrowing and septal fibrosis.
Finally, future mixed-methods studies comparing MV changes in patients with obstructive and non-obstructive HCM that include longitudinal echocardiographic data would provide valuable insight into the changes attributable to the hemodynamic effects of obstruction. A prior echocardiographic study showed longer anterior leaflet lengths and longer residual leaflets in patients with obstructive vs non-obstructive HCM and both variables were longer than in normal controls.23 A cardiac magnetic resonance study showed no difference in anterior leaflet length between patients with and without obstruction.4 However, there was a large difference in the ratio of the anterior leaflet length/LVOT diameter, indicating that increased anterior leaflet lengths in relation to other LV geometric features are more common in obstruction. Residual leaflet length was not measured in that study.
CLINICAL IMPLICATIONS.
The histologic abnormalities and gross evidence of slack curlicued chordae support the concept of mitral leaflet stretch and damage associated with long-standing SAM. They suggest that SAM may beget SAM by further elongation, decreased resilience to abnormal flow dynamics, and the potential for impaired coaptation and increased regurgitation.
First, this hypothesis could explain the clinical observation that obstructive symptoms may worsen rapidly once they develop and, surprisingly, first occur in older individuals. Second, the observed mitral structural damage may contribute to complications such as endocarditis, and even chordal rupture, which has been observed in patients with elongated MVs and myxoid change.21
Finally, this self-perpetuating mechanism of lengthening and the noted histological degeneration support a hypothesis that earlier intervention to decrease SAM, obstruction, and structural damage to leaflets might benefit even patients with NYHA functional class I to II symptoms. Interventions such as negative inotropic pharmacotherapy or myectomy for class II patients could be fruitful areas for future study, as early alleviation of obstruction may help prevent damage to the leaflets, progression of SAM, and obviate post-myectomy MR, enhancing its ultimate benefit.24–28
CONCLUSIONS
The residual leaflet of the MV in OHCM is characterized by myxoid change, with fragmentation, proliferation, and disorganization of elastic fibers, and attenuation of the normally collagen-rich fibrosa layer. These changes both reflect the impact of chronic hemodynamic stress and likely increase leaflets’ susceptibility to the flow drag forces that drive obstruction. To the extent that these pathologic abnormalities suggest ongoing stretch and damage to the residual leaflet, they raise the hypothesis that early medical or surgical interventions to decrease or abolish obstruction might slow their development.
Supplementary Material
PERSPECTIVES.
COMPETENCY IN MEDICAL KNOWLEDGE:
The MV residual leaflet is damaged and stretched by chronic hemodynamic stress. These changes increase its susceptibility to SAM and, therefore, more obstruction.
COMPETENCY IN PATIENT CARE:
These data suggest the hypothesis that early medical or surgical intervention to decrease outflow tract obstruction might attenuate progression of obstruction and SAM.
TRANSLATIONAL OUTLOOK 1:
Proteomics and transcriptomics studies on whole MV leaflets explanted from patients with HCM or animal models could identify cellular pathways responsible for initiating MV body elongation.
TRANSLATIONAL OUTLOOK 2:
Studies should be undertaken that compare early vs standard-timing of medical/surgical intervention to decrease obstruction. The present study provides suggestive support for, but does not prove, the hypothesis that earlier intervention may improve outcomes.
ACKNOWLEDGMENT
The authors are grateful to Ms Eloise Sherrid for her contribution to the Central Illustration.
FUNDING SUPPORT AND AUTHOR DISCLOSURES
The NYU Langone Health Center for Biospecimen Research and Development, Histology and Immunohistochemistry Laboratory (RRID:SCR_018304), is supported in part by the Laura and Isaac Perlmutter Cancer Center Support Grant; NIH/NCI P30CA016087. The authors have reported that they have no relationships relevant to the contents of this paper to disclose.
ABBREVIATIONS AND ACRONYMS
- EPDC
epicardium-derived cell
- HCM
hypertrophic cardiomyopathy
- H&E
hematoxylin and eosin
- LVOT
left ventricular outflow tract
- MV
mitral valve
- OHCM
obstructive hypertrophic cardiomyopathy
- ReLex
residual leaflet excision
- SAM
systolic anterior motion
- SMA
smooth muscle actin
- Tbx18
t-box transcription factor 18
- VIC
valvular interstitial cell
Footnotes
The authors attest they are in compliance with human studies committees and animal welfare regulations of the authors’ institutions and Food and Drug Administration guidelines, including patient consent where appropriate. For more information, visit the Author Center.
APPENDIX For supplemental tables, figures, and methods, please see the online version of this paper.
REFERENCES
- 1.Maron MS, Olivotto I, Zenovich AG, et al. Hypertrophic cardiomyopathy is predominantly a disease of Left ventricular outflow tract obstruction. Circulation. 2006;114:2232–2239. [DOI] [PubMed] [Google Scholar]
- 2.Sherrid MV, Balaram S, Kim B, Axel L, Swistel DG. The mitral valve in obstructive hypertrophic cardiomyopathy: a test in context. J Am Coll Cardiol. 2016;67:1846–1858. [DOI] [PubMed] [Google Scholar]
- 3.Klues HG, Maron BJ, Dollar AL, Roberts WC. Diversity of structural mitral valve alterations in hypertrophic cardiomyopathy. Circulation. 1992;85:1651–1660. [DOI] [PubMed] [Google Scholar]
- 4.Maron MS, Olivotto I, Harrigan C, et al. Mitral valve abnormalities identified by cardiovascular magnetic resonance represent a primary phenotypic expression of hypertrophic cardiomyopathy. Circulation. 2011;124:40–47. [DOI] [PubMed] [Google Scholar]
- 5.Hagege AA, Dubourg O, Desnos M, et al. Familial hypertrophic cardiomyopathy. Cardiac ultrasonic abnormalities in genetically affected subjects without echocardiographic evidence of left ventricular hypertrophy. Eur Heart J. 1998;19:490–499. [DOI] [PubMed] [Google Scholar]
- 6.Olivotto I, Cecchi F, Poggesi C, Yacoub MH. Developmental origins of hypertrophic cardiomyopathy phenotypes: a unifying hypothesis. Nat Rev Cardiol. 2009;6:317–321. [DOI] [PubMed] [Google Scholar]
- 7.Levine RA, Hagege AA, Judge DP, et al. Mitral valve disease-morphology and mechanisms. Nat Rev Cardiol. 2015;12:689–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bartko PE, Dal-Bianco JP, Guerrero JL, et al. Effect of losartan on mitral valve changes after myocardial infarction. J Am Coll Cardiol. 2017;70:1232–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Lange FJ, Moorman AF, Anderson RH, et al. Lineage and morphogenetic analysis of the cardiac valves. Circ Res. 2004;95:645–654. [DOI] [PubMed] [Google Scholar]
- 10.Swistel DG, Sherrid MV. The surgical management of obstructive hypertrophic cardiomyopathy: the RPR procedure-resection, plication, release. Ann Cardiothorac Surg. 2017;6:423–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nampiaparampil RG, Swistel DG, Schlame M, Saric M, Sherrid MV. Intraoperative two- and three-dimensional transesophageal echocardiography in combined myectomy-mitral operations for hypertrophic cardiomyopathy. J Am Soc Echocardiogr. 2018;31:275–288. [DOI] [PubMed] [Google Scholar]
- 12.Swistel DG, DeRose JJ, Sherrid MV. Operative techniques in thoracic and cardiovascular surgery. In: Cohn LHA, Sundt TM, eds. Management of Patients With Complex Hypertrophic Cardiomyopathy: Resection/Plication/Release. WB Saunders; 2004:261–267. [Google Scholar]
- 13.Swistel DG, Balaram SK. Surgical myectomy for hypertrophic cardiomyopathy in the 21st century, the evolution of the “RPR” repair: resection, plication, and release. Prog Cardiovasc Dis. 2012;54:498–502. [DOI] [PubMed] [Google Scholar]
- 14.Zhou B, Honor LB, He H, et al. Adult mouse epicardium modulates myocardial injury by secreting paracrine factors. J Clin Invest. 2011;121:1894–1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Christoffels VM, Grieskamp T, Norden J, Mommersteeg MT, Rudat C, Kispert A. Tbx18 and the fate of epicardial progenitors. Nature. 2009;458:E8–E9. discussion E9-10. [DOI] [PubMed] [Google Scholar]
- 16.Paulsen JD, Zeck B, Sun K, Simoes C, Theise ND, Chiriboga L. Keratin 19 and mesenchymal markers for evaluation of epithelialmesenchymal transition and stem cell niche components in primary biliary cholangitis by sequential elution-stripping multiplex immunohistochemistry. J Histotechnol. 2020;43:163–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roberts WC, Vowels TJ, Ko JM, Hebeler RF Jr. Gross and histological features of excised portions of posterior mitral leaflet in patients having operative repair of mitral valve prolapse and comments on the concept of missing (=ruptured) chordae tendineae. J Am Coll Cardiol. 2014;63:1667–1674. [DOI] [PubMed] [Google Scholar]
- 18.Hjortnaes J, Keegan J, Bruneval P, et al. Comparative histopathological analysis of mitral valves in Barlow disease and fibroelastic deficiency. Semin Thorac Cardiovasc Surg. 2016;28:757–767. [DOI] [PubMed] [Google Scholar]
- 19.Hagege AA, Bruneval P, Levine RA, Desnos M, Neamatalla H, Judge DP. The mitral valve in hypertrophic cardiomyopathy: old versus new concepts. J Cardiovasc Transl Res. 2011;4:757–766. [DOI] [PubMed] [Google Scholar]
- 20.Groarke JD, Galazka PZ, Cirino AL, et al. Intrinsic mitral valve alterations in hypertrophic cardiomyopathy sarcomere mutation carriers. Eur Heart J Cardiovasc Imaging. 2018;19:1109–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boissier F, Achkouty G, Bruneval P, et al. Rupture of mitral valve chordae in hypertrophic cardiomyopathy. Arch Cardiovasc Dis. 2015;108:244–249. [DOI] [PubMed] [Google Scholar]
- 22.Captur G, Ho CY, Schlossarek S, et al. The embryoLogicaL basis of subcLinicaL hypertrophic cardiomyopathy. Sci Rep. 2016;6:27714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ro R, Halpern D, Sahn DJ, et al. Vector flow mapping in obstructive hypertrophic cardiomyopathy to assess the relationship of early systoLic left ventricular flow and the mitral valve. J Am Coll Cardiol. 2014;64:1984–1995. [DOI] [PubMed] [Google Scholar]
- 24.Dybro AM, Rasmussen TB, Nielsen RR, Andersen MJ, Jensen MK, Poulsen SH. Randomized trial of metoprolol in patients with obstructive hypertrophic cardiomyopathy. J Am Coll Cardiol. 2021;78:2505–2517. [DOI] [PubMed] [Google Scholar]
- 25.Nistri S, Olivotto I, Maron MS, et al. Beta blockers for prevention of exercise-induced left ventricuLar outflow tract obstruction in patients with hypertrophic cardiomyopathy. Am J Cardiol. 2012;110:715–719. [DOI] [PubMed] [Google Scholar]
- 26.Sherrid MV, Barac I, McKenna WJ, et al. Multicenter study of the efficacy and safety of disopyramide in obstructive hypertrophic cardiomyopathy. J Am Coll Cardiol. 2005;45:1251–1258. [DOI] [PubMed] [Google Scholar]
- 27.Olivotto I, Oreziak A, Barriales-Villa R, et al. Mavacamten for treatment of symptomatic obstructive hypertrophic cardiomyopathy (EX-PLORER-HCM): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2020;396:759–769. [DOI] [PubMed] [Google Scholar]
- 28.Alashi A, Smedira NG, Hodges K, et al. Outcomes in guideline-based class I indication versus earlier referral for surgical myectomy in hypertrophic obstructive cardiomyopathy. J Am Heart Assoc. 2021;10:e016210. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.