

Abstract
The protein phosphatase SHP2/PTPN11 has been reported to be a key modulator of proliferative pathways in a wide range of malignancies. Intriguingly, SHP2 has also been described as a critical regulator of the tumor microenvironment. Based on this evidence SHP2 is considered a multifaceted target in cancer, spurring the notion that development of direct inhibitors of SHP2 would provide the two-fold benefit of tumor intrinsic and extrinsic inhibition. In this review, we will discuss the role of SHP2 in cancer and the tumor microenvironment, and the clinical strategies where SHP2 inhibitors are leveraged as combination agents to improve therapeutic response.
Keywords: SHP2, PTPN11, RAS/MAPK pathway, combination therapy, SHP2 allosteric inhibitors
Introduction
The SHP (Src homology region 2 (SH2)-containing protein tyrosine phosphatase) family of phosphatases are represented by two members; SHP1 (encoded by PTPN6) and SHP2 (encoded by PTPN11). SHP1 and SHP2 share similarities in primary sequence (61% sequence identity) and structure, but they differ in terms of expression and function. SHP1 is expressed in a more limited fashion in the hematopoietic system and in subsets of epithelial cells(1), whereas SHP2 is ubiquitously expressed(2). Importantly, SHP1 and SHP2 play different regulatory roles, with SHP1 playing a negative regulatory role in downregulating receptor signaling. SHP2, by contrast, is a pleiotropic molecule playing both positive and negative regulatory roles across multiple signaling nodes. Importantly, SHP2 plays a positive regulatory role in mediating growth factor receptor tyrosine kinase (RTK) signaling leading to activation of RAS and its effectors(3), therefore rationalizing a great deal of attention to therapeutic targeting of SHP2.
SHP2: a key regulator of the MAPK pathway in cancer
Growing evidence links the activity of SHP2 to cell transformation and cancer development particularly in the RAS/MAPK pathway even if a detailed molecular mechanism of action has not been completely refined yet. SHP2 modulates the signaling downstream of oncogenic RTKs and is found hyperactivated in gastric carcinoma(4), anaplastic large cell lymphoma(5), glioblastoma(6), breast cancer(7), and mediates the oncogenic signals of EGFR- and KRAS-driven cancers(8,9). SHP2 acts as a rheostat or regulaor of several growth-factor, chemokine, cytokine and integrin receptors(2), best exemplified by its critical role to modulate pathways downstream of RTKs. Active RTKs such as EGFR, MET, and HER2 expose phosphotyrosine residues recognized by SHP2 as docking sites(10,11). SHP2 is in turn phosphorylated at the tyrosine residues 542 and 580 in the PTP domain and undergoes a conformational change enhancing its phosphatase activity(12). Phosphorylated SHP2 binds to the Grb2/SOS1 complex via Gab1(13) and increases active RAS-GTP loading by interfering with its inactivation mediated by p120 RasGAP (Ras GTPase-activating protein)(14). SHP2 dephosphorylates RasGAP blocking its interaction with RAS-GTP at the plasma membrane, thus prolonging RAS activation. Mutagenesis studies have shown that the SHP2 mutant Y580F but not Y542F can bind Grb2 even if neither can activate the downstream MAPK cascade, suggesting specific roles for each tyrosine residue(15). Intriguingly, SHP2 also modulates MAPK activity through other substrates. For example, SHP2 dephosphorylates and inactivates Sprouty(16,17), a negative regulator of RAS(18), thus contributing to sustained MAPK activation. In addition, some studies have shown that SHP2 promotes the activation of SFKs (Src family kinases) through the dephosphorylation of two substrates: CBP (Csk binding protein) and Paxilin(19). Once dephosphorylated, these proteins cannot bind and activate CSK, a negative regulator of SFK, preventing downregulation of the MAPK pathway signaling. The Src/Ras pathway plays a crucial role not only in cell homeostasis but also in embryonic development, where it has been shown that Shp2 deletion results in trophoblast stem cells via the pro-apoptotic factor Bim(20).
Structure and enzymatic activity of SHP2
Structurally, SHP2 consists of two tandem SH2 domains (N-SH2, and C-SH2) followed by a protein tyrosine phosphatase (PTP) domain and a C-terminal tail containing a bipartite tyrosine motif (Y542 and Y580), which may play a role in regulating SHP2 activation(21) (Figure 1A). In its inactive state, SHP2 assumes a closed conformation wherein the N-SH2 and C-SH2 domains bind to and auto-inhibit the PTP domain (Figure 1A, Suppl Video 1). Upon the binding of the SH2 domains to phospho-peptide ligands, the PTP domain is released from auto-inhibition, where essentially, the N-SH2 domain, that shields the active site of the PTP domain undergoes a large conformational change, allowing the PTP active site and the catalytic C459 residue access to substrates, resulting in their dephosphorylation (Suppl Video 2). Therein, this elegant mechanism enables three key features for SHP2, which include (a) specific recruitment into signaling complexes containing high affinity SHP2 phospho-peptide ligands, (b) catalytic activation, and (c) juxtaposition of SHP2 to target proteins. In this way SHP2 efficiently dephosphorylates phosphorylated substrates to enable optimal pathway activation or inhibition. SHP2 activity downstream of RTK activity is characterized by its recruitment to phospho-peptide ligands present in RTKs and/or signaling adaptors, such as Grb2 and Gab1. Therein SHP2 regulates protein complex assembly and protein function through its phosphatase activity, leading to the efficient activation of guanosine nucleotide exchange factors (GEFs), such as SOS1, which in turn activate RAS through exchange of GDP for GTP.
Figure 1. Structure and key mutations on SHP2.
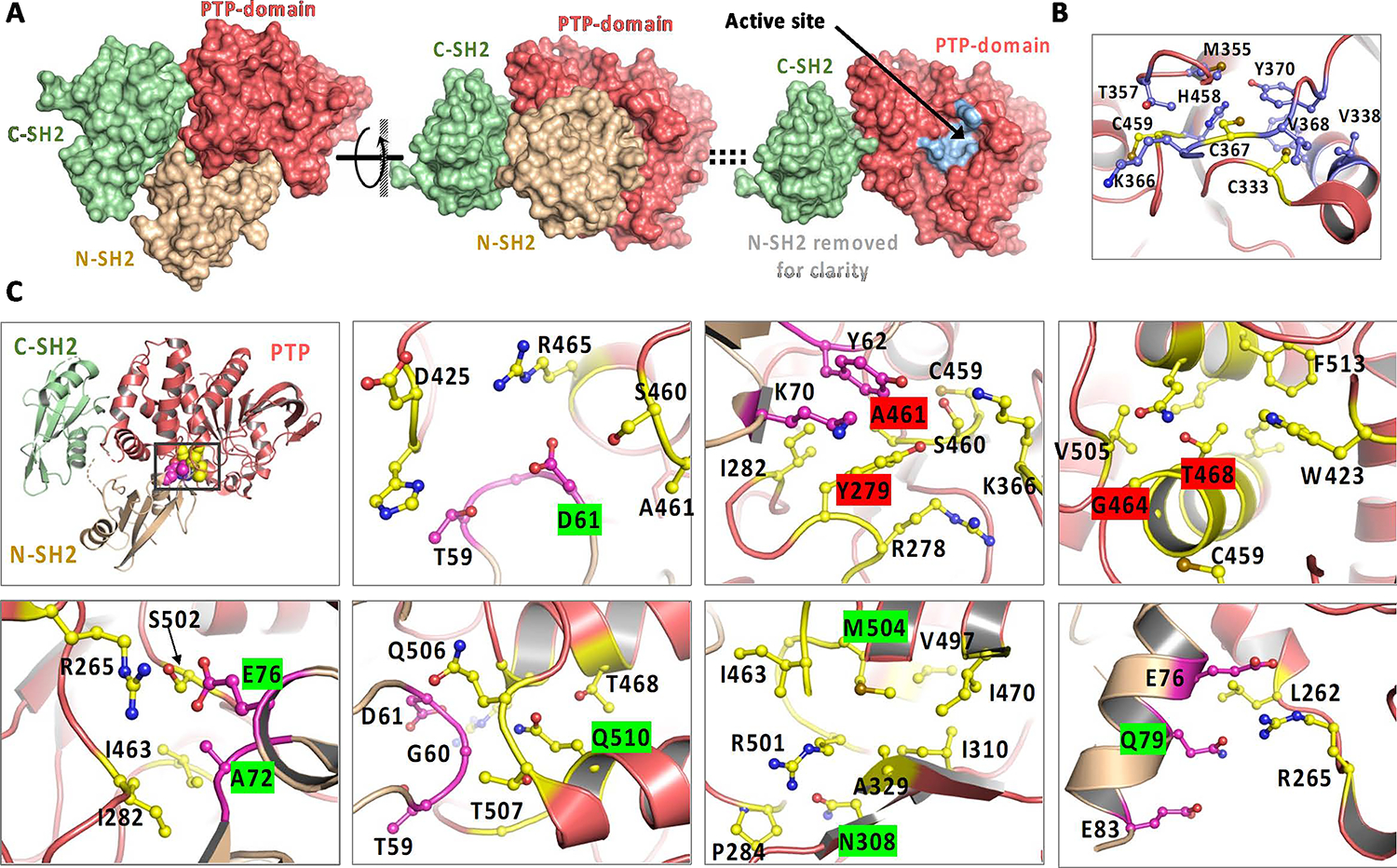
A. The overall structure of SHP2 contains 3 well-folded domains: two tandem SH2 domains (beige, light green) followed by the PTP (phospho-tyrosine phosphatase) domain (left); the structure with the N-SH2 domain facing out shows the N-SH2 domain bound on top of the PTP domain, in this view, and shields the active site from access to substrates (middle); (right) – Same view as (middle), with the N-SH2 domain removed for clarity, revealing the active site in the PTP domain, in light blue. B. Residues surrounding Catalytic C459 residue, and C367, C333, termed ‘backdoor cysteines’ are in a hydrophobic region of the protein. C. Molecular details of the regions in SHP2 around residues at which GOF (highlighted in green) hotspot mutations in Leukemias and Noonan syndrome and LOF (highlighted in red) hotspot mutations occur. Top, left: Hotspot mutations occur around the interaction site between N-SH2 and PTP domains. Residues in the PTP domain (Salmon color) that are around the site of the mutation are highlighted in yellow, and residues in the N-SH2 domain (light brown color) that are around the site of the mutation are highlighted in magenta.
In addition to the catalytic C459, the C333 residue has been termed the ‘backdoor cysteine” and reported to cause destabilizing effects compared to other PTPs, in which the corresponding residue is a conserved proline. C333, in addition to C367, surrounded by hydrophobic residues (Figure 1B) around the catalytic C459 have been suggested to provide protection to the C459 from inactivating oxidation(22).
Recurrent SHP2 mutations in cancer and other diseases
Germline variants on the PTPN11 gene converge on a set of developmental abnormalities defined as RASopathies. SHP2 variants have been associated with Noonan Syndrome (NS), with loss-of-function (LOF) or gain-of-function (GOF) leading to distinct pathologic outcomes(23). Typically, variants of SHP2 resulting in GOF lead to the impairment of its auto-inhibitory function, shifting the equilibrium towards a constitutively active conformation. Notably, germline LOF are associated with ~90% Noonan syndrome with multiple lentigines (NSML) cases(24). In contrast, GOF mutations associated with NS, are characterized by facial dysmorphia, shortened stature, bleeding irregularities, and heart and skeletal deformations. On the other hand, somatic GOF variants are associated with deregulation of SHP2 activity, leading to hyperactive signaling through the RAS/MAPK pathway. This has been associated with an increased risk of proliferation and tumorigenesis(25)(26), including myelodysplastic syndrome (MDS), juvenile myelomonocytic leukemia (JMML)(27), acute myeloid leukemia (AML)(28), acute lymphocytic leukemia (ALL)(29), and sporadic cases of lung, colon, and skin cancers (30,31).
Structurally, a large majority of all disease relevant mutations associated with SHP2 occur in the N-SH2 domain or the PTP domain(24), with the hotspot mutations clustering at the interface of the N-SH2:PTP domains (Figure 1C, Top, Left). A closer look at the residues around many of the GOF or LOF hotspot mutations helps us rationalize the impact of these modifications (Figure 1C). Molecular environments of hotspot mutations in Leukemias (at residues D61, A72, E76, Q510) and Noonan syndrome (Q79, N308, M504) that are activating mutations (highlighted in green), and hotspot mutations in NSML (Y279, A461, G464, T468) that are inactivating mutations (highlighted in red) are detailed in Figure 1B. An E76K mutation in the N-SH2 domain would cause a charge-charge repulsive interaction with R265 in the PTP-domain, destabilizing the inactive SHP2, resulting in the active, open conformation. An A72S mutation, while less activating than E76K, would cause a steric clash at the interface, shifting the equilibrium to the open-state, basally activating SHP2. A D61G mutation would remove a salt-bridge (positive-negative charge interaction) between R265 in the PTP-domain and D61 in N-SH2 domain, also shifting SHP2 to the open-state. A Q510P mutation would unfold the helix that Q510 is a part of and alter the interface between N-SH2 and the PTP domain. An N308D mutation would introduce a more polar side chain, presumably with a charge in the hydrophobic environment, and destabilize the protein folding enough to cause a disruption of interaction with N-SH2. The M504V mutation would be predicted to reduce the hydrophobicity at its location in PTP, altering conformation of I463, which contacts the N-SH2, destabilizing the closed state. The Q79R mutation in N-SH2 would directly cause a steric clash as well as a charge repulsion with R265 in the PTP domain, very similar to the E76K mutation, stabilizing the active conformation. For the inactivating mutations Y279C, A461T, G464A, T468M, these are located close to the active site residue, C459, and would be expected to alter the active site enough to interfere with substrate recognition and catalytic activity.
The ability of mutant SHP2 to drive oncogenesis in liquid tumors is supported by preclinical work leveraging GEMMs (Genetically Engineered Mouse Models)(32). Mice that express the gain-of-function (GOF) mutants D61G, D61Y or N308D - typically associated with leukemia or MDS - all develop a myeloproliferative syndrome(23,30,31). Likewise, mice expressing the E76K PTPN11 mutant, that is the most common and most active mutant found in JMML and acute leukemias, show aberrant activation of hematopoietic stem cells (HSCs) and myeloid progenitors, and eventually develop acute myeloid leukemia (AML), T or B cell acute lymphoblastic leukemia (T-ALL or B-ALL) (33). In addition to a cell-autonomous effect, germline PTPN11 activating mutations promote the development and progression of myeloproliferative syndromes through a perturbation of HSCs by an increased production of chemokines, such as CCL3, by the bone marrow microenvironment(34). Together with an amplification of the RTK and chemokine signaling, PTPN11 mutations can also promote genomic instability partly by causing centrosome amplification(33). The effect of PTPN11 GOF mutations on genomic instability was also confirmed by the D61G mutant, that causes chromosomal instability and increased susceptibility to tumors induced by DNA damage(35). SHP2 also localizes to the kinetochore and the centrosome and GOF mutants hyperactivates Polo-like kinase 1 (Plk1) by enhancing its phosphorylation mediated by c-Src kinase(35). In contrast to what is observed in liquid tumors, SHP2 activating mutations are relatively rare in solid cancers and are detected at low frequency in tumors such as neuroblastoma(28).
Other pathways regulated by SHP2
While this key role for SHP2 has been the focus for therapeutic intervention, SHP2 clearly plays a broader role in regulating the phospho-proteome. Recent proteomics studies have demonstrated that SHP2 impacts multiple substrates, resulting in complex regulatory effects which span both positive and negative regulatory roles, as well as roles in protecting phosphorylated peptides from dephosphorylation by other phosphatases(36)(37).
PI3K pathway.
SHP2 is also involved in the activation and sustained signaling of the PI3K pathway(38,39). Immunoprecipitation studies paired with mass spectrometry have shown the direct interaction across species between SHP2 and the regulatory subunit p85 but not the catalytic subunit p110(40,41). The SHP2/p85 complex also includes Gab1(42) or Gab2(41) and it has been shown that it is essential for KIT signaling in myeloproliferative disease (MPD)(43). Consistently it has also been observed that co-inhibition of SHP2 and PI3K can correct MPD by disrupting p85/SHP2/Gab2 interaction. Moreover, it has been demonstrated that SHP2 plays also a critical role in the development of resistance to PI3K inhibitors, as shown in preclinical models of metastatic breast cancer where combination of PI3K and SHP2 inhibition results not only in synergistic tumor growth inhibition but also inhibits metastasis formation(44). The role of the PI3K/SHP2 axis in the regulation of metastatic progression has been also demonstrated by overexpression experiments in ovarian tumor models(45). Similarly, SHP2 overexpression has been shown to increase cell migration in HeLa and SiHa cells while SHP2 knockdown reduces cell motility though a mechanism of action involving AKT.(46)
JAK/STAT pathway.
SHP2 both enhances and inhibits signaling in the JAK/STAT signaling pathway and different JAK/STAT pathways may be differentially regulated by SHP2 depending on the nature of the extracellular signals(47). The JAK1/STAT1 and the STAT3 pathways can be suppressed by SHP2. Suppression and inhibition of the STAT3 activity pathway by SHP2 in hepatocellular carcinoma highlights a tumor suppressive role of SHP2 in certain tissues(47,48). Conversely, some SHP2 mutants may enhance JAK2/STAT5 activation in hematopoietic cells and can activate the JAK-STAT pathway in other contexts(49). Besides mutations and receptor-mediated activation, additional mechanisms can enhance SHP2 activity in some cancers. An example is given in pancreatic cancer, where a germline variation of the long intergenic noncoding RNA LINC00673 can increase SHP2 stability by interfering with its degradation mediated by the PRPF19 ubiquitin ligase(50).
Hippo pathway.
SHP2 can also modulate the activity of the transcription factor YAP/TAZ revealing a distinct role of the phosphatase in the nucleus (defined here as nuclear SHP2, nSHP2). For example, the nuclear co-localization of SHP2 and YAP has been correlated with poor patient survival in non-small cell lung cancer(51), strongly suggesting the pro-oncogenic function of the SHP2/YAP axis. In addition, it has been shown that SHP2 binds YAP/TAZ and that this interaction modulates its translocation into the nucleus(52). Mechanistically, nSHP2 dephosphorylates parafibromin, and stimulates TCF/LEF- and TEAD-regulated genes. Intriguingly, the SHP2/YAP axis seems to play opposite roles depending on the cellular context. For example, in liver it has been shown that SHP2 can inhibit YAP-mediated organ regeneration(53) as well as cholangiocarcinoma progression(54), thus functioning as tumor suppressor. More detailed studies are certainly needed to better understand the interplay between SHP2 and YAP/TAZ even if a partial explanation to these conflicting reports is probably due to the complexity of the cross-talk, the variability in experimental conditions such as cell density, and other players not yet completely characterized. We can however speculate that this axis indicates that the role and localization of SHP2 are not limited to the membrane and that its phosphatase activity can modulate transcription factor activity. These observations and the recent development of TEAD inhibitors as combination agents(55) could provide the rationale for additional preclinical studies to investigate how concomitant inhibition of both pathways could benefit efficacy.
SHP2 at the interface between cancer and its microenvironment
In addition to its vital role in altering the function and fate of cancer cells themselves, SHP2 has multiple immuno-suppressive properties in the tumor microenvironment (TME, Figure 2). This is largely mediated via its modulation of various signaling pathways in immune cells such as macrophages and T lymphocytes and triggered through a complex mix of tumor intrinsic and extrinsic effects(56). Immune phenotypes associated with selective deletion or suppression of SHP2 in tumor cells and/or immune microenvironment using various genetic approaches have been well described. However, the relatively recent success in developing selective, allosteric inhibitors of SHP2, tested alone or in combination with various targeted therapies such as immune checkpoint blockade, has further elucidated and advanced our understanding of the complex role and mechanism(s) of action of SHP2 in the context of tumor immuno-regulation and established SHP2 as a promising immune-modulatory target.
Figure 2.
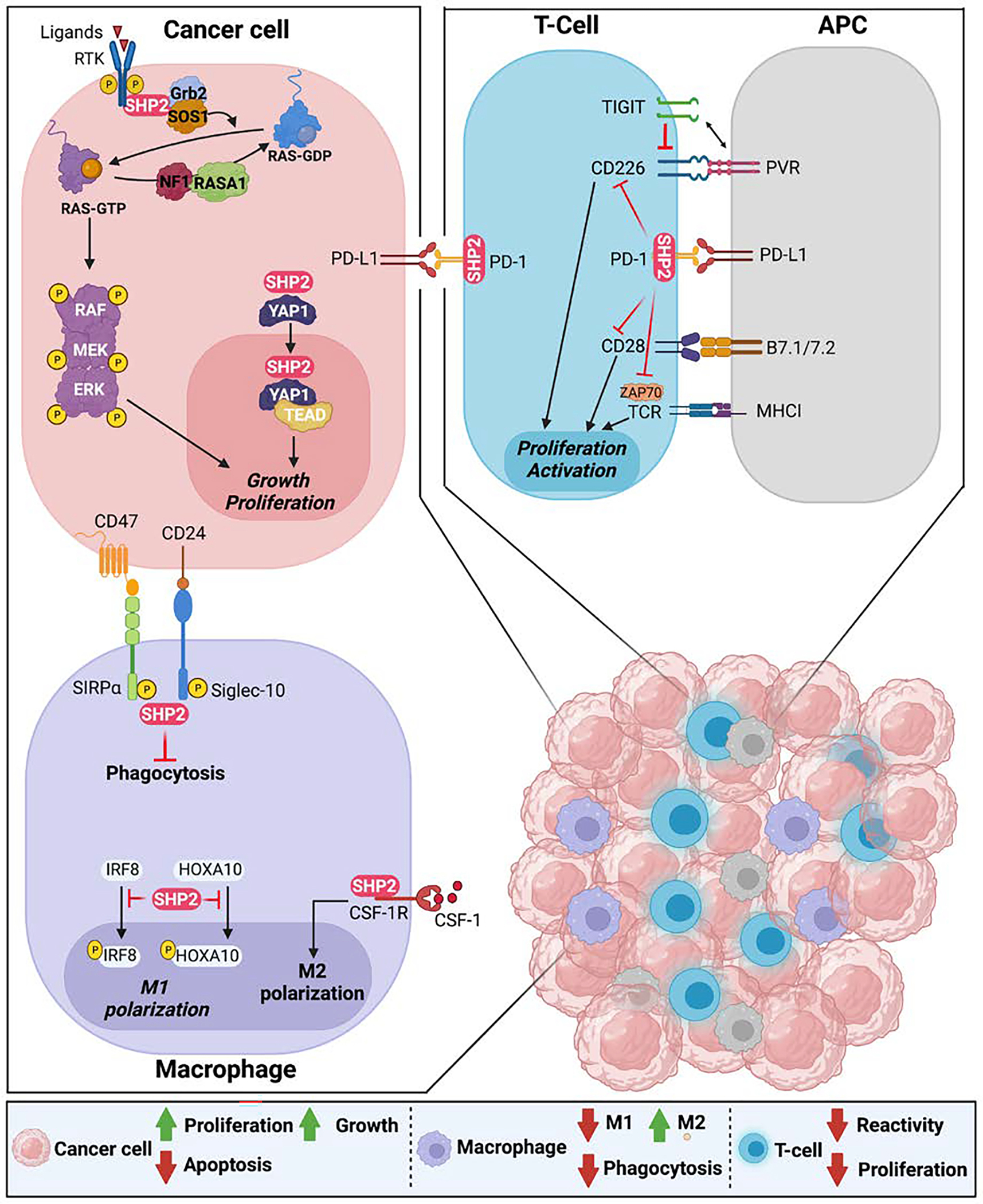
SHP2 is protein tyrosine phosphatase with pleiotropic function. It is a critical regulator of the RAS-MAPK pathway leading to cancer cell proliferation and growth. In T-cell, it is an integral downstream effector of the PD-1 cascade to halt activation and proliferation. In macrophages it promotes M2 polarization and proliferation and inhibits phagocytosis.
SHP2 in macrophages.
SHP2 is involved in multiple signaling pathways in tumor- associated macrophages (TAMs), with a tumor-promoting role. Upon stimulation by the colony-stimulating factor-1 (CSF-1), SHP2 binds to the CSF receptor (CSF-1R)/Grb2/Gab2 complex on the inner membrane of TAMs and consequently activates the downstream RAS-ERK signaling pathway promoting the proliferation and the polarization of TAMs to immunosuppressive M2-like phenotype, thus favoring the survival, proliferation, and migration of tumor cells(57). SHP2 activity also modulates the function of the surface proteins CD47 and CD24. Known as “don’t eat me signal”, CD47 is expressed on the surface of tumor cells and forms a signaling complex with the signal regulatory protein α (SIRPα), an adaptor protein that is highly expressed on the membrane of myeloid cells. Consequently, SIRPα recruits specific substrates for dephosphorylation by SHP2 to inhibit intracellular signaling thus enabling tumor evasion from macrophage-mediated phagocytosis(58,59). Known as heat stable antigen or small-cell lung carcinoma cluster 4 antigen, the tumor-expressed CD24 binds to the inhibitory receptor sialic-acid-binding Ig-like lectin 10 (Siglec-10) on the surface of TAMs, eliciting an inhibitory signaling cascade and tumor evasion from phagocytosis triggered by the recruitment of SHP2 (and/or SHP1) to the immunoreceptor tyrosine-based inhibition motif (ITIM) of the cytoplasmic tail of Siglec-10(60). Myeloid-selective ablation of SHP2 inhibited B16 melanoma growth in mice through potentiating CXCL9 production in macrophages and facilitating CXCL9-mediated attraction of effector T cells, promoting antitumor adaptive immunity(61). In a recent study, myeloid-specific depletion of Shp2 diminished B16-F10 melanoma and MC17–51 fibrosarcoma growth in mice and the immuno-suppressive capacity of MDSC; further RNA-seq analysis of PMN-MDSC or TAMs from C17–51 tumors demonstrated the presence of enriched gene expression profiles of enhanced differentiation, activation and expression of immunostimulatory molecules(62). Myeloid specific deletion of SHP2 alone or in combination with PD-1 depletion enhanced bone marrow GM-CSF-mediated phosphorylation of the transcription factors HOXA10 and IRF8 which induce myeloid differentiation and monocyte/DC lineage commitment, respectively(62). Moreover, Shp2 deficiency in macrophages confers protection from colitis and colitis-driven colon carcinogenesis in mice through promoting the IL-10/STAT3 signaling and its dependent anti-inflammatory response(63). Dual inhibition of CSF1-R and SHP2 using self-assembled dual-inhibitor-loaded nanoparticles (DNTs) skewed the activation states of TAMs towards a more M1 phenotype, while simultaneously enhancing the phagocytic index(64). Together, these findings demonstrate that SHP2 promotes macrophage proliferation and M2 type polarization and that targeting SHP2 in macrophages increase anti-tumor immunity, providing evidence for the non-autonomous role of SHP2 on tumor progression.
SHP2 in T lymphocytes.
SHP2 plays a crucial role in the regulation of T cell function via binding to regulatory receptors that contain tandem phosphorylated immunoreceptor tyrosine-based inhibition motif (ITIM) and immunoreceptor tyrosine-based switch motif (ITSM) domains, including immune suppressive receptors such as PD-1. Upon stimulation by its PD-L1 ligand, PD-1 becomes phosphorylated by Src kinases at both ITIM and ITSM motifs located in its cytoplasmic tail. Phosphorylated ITSM interacts with C-terminal SH2 domain of SHP2, thereby recruiting SHP2 to PD-1 with high affinity while phosphorylated ITIM binds the N-terminal SH2 domain, displacing it from the catalytic pocket and activating its phosphatase activity(65). When recruited by PD-1, SHP2 directly dephosphorylates the costimulatory molecules CD28 and CD226, and consequently limits T-cell activation(66,67). In addition, SHP2 promotes the dephosphorylation of critical proteins in the T cell receptor (TCR) signaling complex such as ZAP70 kinase, CD3ζ, PKC-θ, and PLCγ2, leading to inhibition of the downstream PI3K-AKT and RAS-ERK signaling and ultimately reduction in TCR-mediated IL-2 production and T cell proliferation(68). However, in a T cell confined SHP2 knockout (KO) mouse model, SHP2 was found to be dispensable for PD-1 signaling in T cells and for establishing T cell exhaustion: the control of immunogenic tumors was not significantly improved and the response to anti-PD-1 checkpoint blockade therapy was not altered(69). This apparent discrepancy could be due to the emergence of compensatory mechanisms such as recruitment of alternative phosphatases to direct intracellular signaling. It was recently reported that PD-1 converges with the inhibitory receptor TIGIT through distinct mechanisms to disrupt the activation of the costimulatory receptor CD226 on T cells. While PD-1 inhibits the phosphorylation of CD226 via recruitment of SHP2, TIGIT restricts CD226 co-stimulation by blocking interaction with their shared ligand PVR (CD155) and impairs its function by directly disrupting CD226 homodimer formation. Since TIGIT and PD-1 can independently regulate CD226, dual blockade of both inhibitory receptors was required to fully restore CD226 signaling and to obtain an optimal CD8+ cytotoxic T cell response(70,71). These investigations will elucidate mechanistically how the phosphatase activity of SHP2 modulates T cell signaling and suggest unanticipated benefits of combining SHP-2 inhibition with novel targets for immune modulation.
SHP2 in Natural Killer Cells.
Although most studies focused on macrophages and T lymphocytes, the role of SHP2 in regulation of Natural Killer cells (NK cells) in the tumor microenvironment has been reported even if the exact mechanism of action has not been fully elucidated yet. SHP2 negatively regulates NK cell development and functions through its interaction with some inhibitory receptors on the surface of NK cells via the ITIM motif (72). More specifically, SHP2 inhibits activation of human NK cells upon recruitment to killer cell Ig-like receptors (KIRs), with overexpression of a catalytically inactive SHP2 mutant shown to reverse KIR-mediated inhibition of NK cell(73). In addition, SHP2 deficient NK cells had elevated cytolytic activity and IFN-γ production when targeting tumor cells but in a KIR- independent process(74). In a relatively more recent study, using mice conditionally deficient for SHP2 in the NK lineage, it was shown that SHP2 is largely dispensable for NK cell education. However, SHP2-deficient NK cells have reduced proliferation and survival when treated with high dose IL-15, elucidating an essential role for SHP2 in orchestrating NK cell metabolic activity following exposure to IL-15(75). More research is needed to further elucidate the role of SHP2 in modulating NK cells among other immune cells in the TME
SHP2 and other components of the TME.
In addition to its major role in altering the tumor immune microenvironment, SHP2 is shown to modulate other components of the TME such as endothelial cells and stromal fibroblasts. Studies have shown that SHP2 supports endothelial cell survival and growth in the remodeling tumor vasculature; SHP2 inhibition by SHP099, alone or in various combinations, impaired angiogenesis, reduced tumor vascularity and tumor growth and promoted remodeling of the stroma in various syngeneic and orthotopic tumor models(76,77). In addition, the selective SHP2 deletion in tumor endothelial cells through the use of inducible and endothelial-cell-specific SHP2 knockout mice inhibited tumor growth and angiogenesis while promoting the normalization of tumor vasculature(78). Studies have also shown that SHP2 promotes TGFβ1-induced epithelial-mesenchymal transition (EMT) in lung epithelial A549 cells(79) and is required for EMT stimulated by IL-6 in breast cancer cells(80). SHP2 also acts as a molecular checkpoint of TGFβ-induced JAK2/STAT3 signaling to regulate fibroblast activation and tissue fibrosis; genetic or pharmacologic inactivation of SHP2 in fibroblasts reduced JAK2/STAT3 signaling, inhibited TGFβ-induced pro-fibrotic effects manifested through various changes such as decrease in α-SMA levels, collagen release and formation of stress fibers, and ameliorated or protected from experimental fibrosis induced in various mouse models(81). All these studies provided compelling evidence on the involvement of SHP2 in stroma remodeling; however, this remains an immature area of research and more work is needed to dissect the mechanism of action of SHP2 in the TME, especially with its role in activating and reprogramming cancer associated fibroblasts (CAFs), a key component of the tumor stromal compartment with distinct molecular and functional subtypes across various malignancies such as PDAC (Pancreatic ductal adenocarcinoma), and with potential pro- and anti-tumorigenic dichotomy(82).
SHP2 inhibitors to reshape the TME.
Given the complexity of the function of SHP2 in several components of the TME, studies utilizing targeted SHP2 allosteric inhibitors have been key to understand its pleiotropic activity. For example, it has been shown that the inhibition of SHP2 by SHP099 delayed tumor growth by triggering anti-tumor immunity mediated by cytotoxic T cells and synergized with PD-1 blockade in syngeneic models of colon cancer. Notably, SHP099 treatment resulted in a significant increase of the number of NK1.1+IFN-γ+ cells, indicating a role of SHP2 in tumor growth retardation(83). Studies involving other allosteric inhibitors RMC-4550 and TNO155 demonstrated that SHP2 inhibition reverses immune suppression in syngeneic tumor mouse models through modulation of both innate and adaptive immunity(68,84,85). More specifically, RMC-4550 caused an increase in CD8+ T-cell tumor infiltrates in addition to class I MHC and PD-L1 expression on tumor cells; it also drove a direct and selective depletion of pro-tumorigenic M2 macrophages via attenuation of CSF1R signaling and increased M1 macrophages. The anti-tumor effects of RMC-4550 were additive with either anti-PD-L1 and anti-CTLA4 immune checkpoint inhibitors or an anti–CSF1R antibody, consistent with the pleiotropic function of SHP2(84). Similar to RMC-4550, TNO155 effectively inhibited immunosuppressive M2 macrophages, and showed combination activity with PD-1 blockade(85). A recent study leveraging KRas- and EGFR-mutant NSCLC GEMMs showed that SHP2 inhibition by SHP099 depleted alveolar and M2-like macrophage populations, induced tumor intrinsic secretion of CCL5 (RANTES) and CXCL10 (IP-10) chemokines with key roles in T cell recruitment and activation, and increased B and T lymphocyte infiltration in tumors. However, it also promoted the accumulation of immunosuppressive granulocytic MDSCs (gMDSCs) that was attributable to NF-κB-induced secretion of CXCR2 ligands largely in tumor cell, but with some expression in cancer-associated fibroblasts (CAFs)(77). Combined treatment of SHP099 with the CXCR1/2 inhibitor SX682 blocked the infiltration of a specific cluster of S100a8/9hi gMDSCs, generated Klrg1+ CD8+ effector T cells with high cytotoxic and proliferative capability, and improved survival of KRas- and Egfr-mutant models(77). This asserts the beneficial effect of testing the combinations SHP2 and CXCR1/2 inhibitors in the clinic. It would also be fair to speculate that SHP2 inhibition could impact not only immune cells but also stroma survival and that combination with emerging inhibitors of KRAS G12D in PDAC, where stroma plays a key supportive role, may turn out to be beneficial even if no experimental evidence has been generated yet.
Tumor stroma is highly dynamic and plays an essential role in providing a supportive microenvironment for tumor epithelial cell growth and progression. Gaining a comprehensive view and a deeper understanding of the complexity, functionality, heterogeneity and robustness of the immune context together with other non-immune components of the tumor stroma could identify new therapeutic vulnerabilities and combinatorial strategies with SHP2 inhibition; this would also necessitate more detailed studies to evaluate long term efficacy, safety and tolerability of such combination therapies. Developing methods and tools that allow the identification and quantification of variability and perturbations in the microenvironment in real-time remain a challenge. Most pre-clinical studies of SHP2 have used syngeneic tumor models implanted in the subcutaneous space. Although proven invaluable, these models provide an incomplete or even ambiguous picture on SHP2 inhibitor mode of action since, as shown in various studies, vascularization and sensitivity to anti-tumoral treatments are dependent upon the site of implantation. Efforts in ascertaining the best ways of generating pre-clinical models that can faithfully unveil the effect of targeting of SHP2 in combination with specific functional nodes in the complex cross-talk between cancer cells and TME components and are thus good indicators of therapeutic efficacy in humans are vital for medical advancement.
SHP2: A Therapeutic Target in Cancer
Because of its pleiotropic role, SHP2 has emerged as an appealing target in cancer therapy. Molecules targeting SHP2 can be distinguished between catalytic and allosteric inhibitors based on their mechanism of action. Catalytic inhibitors have been designed to target the PTP domain and despite the significant activity in vitro, their poor selectivity against SHP1 has limited their clinical development(86). Allosteric inhibition of SHP2 was first reported by Novartis in 2015(87) with the identification of a hitherto unknown pocket formed between the N-SH2, C-SH2 and PTP domains when SHP2 is in the auto-inhibited state and demonstrated that selective and potent allosteric inhibition of SHP2 was possible (Supplementary Figure 1). This finding unlocked a wealth of subsequent allosteric inhibitors inspired by SHP099 (1, Supplementary Figure 1). This original molecule and all subsequent allosteric inhibitors share the same general pharmacophore (2, Supplementary Figure 1) of an elaborated amino piperidine ring (green) attached to a heterocyclic ring (blue, commonly amino pyrazine or pyrazolopyrazine) which is connected to an aromatic moiety, either directly or via a heteroatom (red). Novartis’ ultimate clinical candidate, TNO155(88) (3, Supplementary Figure 1), maintains the aminopyrazine core of SHP099, with additional elaboration of the piperidine ring to a highly potent spirocyclic amine. Variations of this novel motif are contained in many subsequent reported SHP2 allosteric inhibitors, including RMC-4550 (4, Supplementary Figure 1)(89), clinical molecule GDC-1971(5, Supplementary Figure 1)(90), and patented molecules from Jacobio (6, Supplementary Figure 1)(91). All SHP2 allosteric inhibitors to date bind the same pocket, trapping SHP2 in an inactive configuration (Supplementary Figure 2)
Targeting of SHP2 in cancer has paved a new way in cancer therapy for at least three reasons. First, SHP2 has been one of the first examples of a non-kinase target within the RAS pathway in cancer and was the first tyrosine phosphatase for which an oncogenic role was demonstrated(27) and function inhibited. Second, SHP2 is usually not a driver in cancer, rather it co-operates with other key factors for pathway modulation. Third, while SHP2 inhibitors have very limited activity as single agents, they are expected to improve efficacy of a wide range of inhibitors within the MAPK pathway and beyond.
As blocking SHP2 disrupts SOS1-mediated RAS-GTP loading, SHP2 inhibitors are expected to show an anti-proliferative activity in tumors where RAS cycling is at least partially intact and to have no effect when RAS is locked in an active state, like in the Q61 mutants(92). Based on this rationale, SHP2 inhibitors are combined with the recently discovered KRAS G12C inhibitors to enhance their activity as they covalently bind to the KRAS GDP-bound form. In addition to enhancing target alkylation, SHP2 inhibition represents a valid strategy in KRAS G12C patients where resistance to treatment has emerged through RTK-mediated bypass mechanisms facilitating the reactivation of MAPK signaling(93).
Moreover, SHP2 represents an appealing combination agent with RTK inhibitors to prevent and/or delay MAPK reactivation through an alternative RTK as mechanism of resistance. Notably, since this mechanism of resistance overlaps across all the RTKs such as EGFR(94), ALK(95), RET(96), ROS1(97), NTRK1(98) and FLT3(99), it suggests that SHP2 inhibitors can be in principle combined across receptors. Consistent with this hypothesis, inhibition of SHP2 is shown to overcome resistance mediated through alternative RTK-mediated bypass signaling(100,101).
Although an explicit co-targeting of individual RTKs conferring resistance to the inhibition of the driver RTKs is a rational therapeutic approach that is being explored(102,103), other RTKs may still serve as escape routes to any such combinations. Supporting this idea, Ryan and colleagues showed combined SHP2 and KRAS-G12C inhibition as a more effective approach to suppress RTK-mediated MAPK signaling rebound and in vivo tumor growth over a KRAS-G12Ci and anti-EGFR combination(104). This is consistent with the idea that it would be more effective blocking a central node of RTK signaling by inhibiting SHP2. In such case SHP2 can be considered a key at player of cancer plasticity and subsequently SHP2 inhibitors are ideal combination partners for all RTK blocking agents.
In addition, SHP2 inhibitors are expected to show anti-tumor activity in cancers where the RAS-MAPK pathway is generally deregulated either at the RTK level (through amplifications) or immediately downstream. In particular, SHP2 may be considered a key target for tumors that do not carry a specific mutation on a typical oncogene but rather show the loss of multiple tumor suppressor genes regulating MAPK activation. Some evidence is provided in studies leveraging an animal model of NSCLC driven by the concomitant loss of the two GTPase-activating proteins (GAPs) NF1 and RASA1(105), detected in roughly 2% of NSCLC patients with no known driver mutations(106). In addition, it has been shown in preclinical models of Small Cell Lung Cancer (SCLC) that the loss of the epigenetic modificatory KTMD2 activates ERBB2 and EGFR and that the combined inhibition of SHP2 and ERBB2 results in tumor regression(107). Moreover, SHP2 inhibition has shown to be an effective combination strategy in hepatocellular carcinoma in combination with an mTOR inhibitor (108). The studies collectively show that inhibition of SHP2 in cancer would represent a solid strategy to treat, prevent and delay resistance mechanisms to currently available targeted therapies.
Clinical development of SHP2 inhibitors
Currently, 15 small molecule SHP2 inhibitors have been reported to be in clinical development (Table 1), with all trials conducted in solid tumor oncology indications. While the earliest first-in-human studies generally enrolled all tumor genotypes, some of the later studies have taken a more targeted approach by enrolling mutant genotypes thought to be most sensitive to SHP2 inhibition, such as ALK- or ROS1-positive NSCLC, BRAF-V600E colorectal cancer (CRC), or NF1-mutant or BRAF class 3-mutant solid tumors (e.g. PF-07284892 in NCT04800822(109)). Another interesting strategy is exclusion of known activating mutations in RAS or BRAF which may render tumors resistant to SHP2 inhibition (e.g. TNO155 in NCT03114319). Yet despite the known activating mutations in PTPN11 found in JMML and Noonan syndrome, clinical studies of SHP2 inhibitors (SHP2i) in these disease areas have not been initiated to date.
Table 1:
Current clinical trials with SHP2 inhibitors
| Compound name | Sponsor(s) | Date of first clinical development | Phase of Development | Tumor types enrolled for FIH study | FIH study (ct.gov ID) |
|---|---|---|---|---|---|
| BBP-398 | Navire Pharma Inc., | Nov-20 | Ph1 / 1b | Advanced solid tumors followed by KRAS-G12C NSCLC & other, NF1 loss-of-function (LOF), and EGFR-mutant NSCLC | NCT04528836 |
| BPI-442096 | Betta Pharmaceuticals Co., Ltd. | Jun-22 | Ph1 | Advanced solid tumors followed by KRAS G12, Class-3 BRAF, NF1 LOF mutations, and RTK mutations, amplifications or rearrangements [excluding HCC] | NCT05369312 |
| BR790 | Jiangxi Qingfeng Pharmaceutical Co. Ltd. and Shanghai Gopherwood Biotech Co., Ltd. | May-21 | Ph1 | Advanced solid tumors | NCT04891653 |
| ERAS-601 | Erasca, Inc. | Dec-20 | Ph1 / Ph2 | Advanced or metastatic solid tumors followed by advanced or metastatic solid tumors with "specific molecular alterations" not enumerated. | NCT04670679 |
| ET0038 | Etern BioPharma (Shanghai) Co., Ltd | Oct-21 | Ph1 | Advanced solid tumors followed by advanced solid tumors with "specific mutations/rearrangements that result in hyperactivation of the RAS-MAPK pathway" - not enumerated. | NCT05354843 |
| GDC-1971 | Genentech Inc, / Relay Therapeutics | Feb-20 | Ph1 / 1b | Advanced or metastatic solid tumors; exclusions include KRAS G12D, G12V, G13X, and Q61X; BRAF V600E; or MEK mutations | NCT04252339 |
| GH21 | Suzhou Genhouse Bio Co., Ltd. | Feb-22 | Ph1 | Advanced or metastatic solid tumors | NCT05183243 |
| HBI-2376 | HUYABIO International, LLC. | Dec-21 | Ph1 | Advanced malignant solid tumors harboring KRAS or EGFR mutations | NCT05163028 |
| HS-10381 | Jiangsu Hansoh Pharmaceutical Co., Ltd. | Jun-22 | Ph1 | Advanced solid tumors | NCT05378178 |
| ICP-189 | Beijing InnoCare Pharma Tech Co., Ltd. | May-22 | Ph1 | Locally advanced unresectable or metastatic solid tumors | NCT05370755 |
| JAB-3068 | Jacobio Pharmaceuticals Co., Ltd. | Nov-18 | Ph1 / Ph2 | Advanced solid tumors | NCT03518554 |
| JAB-3312 | Jacobio Pharmaceuticals Co., Ltd. | Sep-19 | Ph1 / Ph2 | Advanced solid tumors | NCT04045496 |
| PF-07284892 | Pfizer Inc | Mar-21 | Ph1 / 1b | ALK- or ROS1-positive non-small cell lung cancer (NSCLC), BRAF-V600E colorectal cancer (CRC), or RAS- mutant, NF1-mutant or BRAF class 3-mutant solid tumors | NCT04800822 |
| RMC-4630 | Revolution Medicines, Inc. | Sep-18 | Ph1 / Ph2 | Advanced solid tumors followed by KRAS amplifications, KRASG12C (NSCLC), BRAF Class 3, or NF1 LOF (NSCLC and gynecological cancers) mutations | NCT03634982 |
| SH3809 | Nanjing Sanhome Pharmaceutical, Co., Ltd. | Apr-21 | Ph1 | Advanced solid tumors [except HCC] | NCT04843033 |
| TNO155 | Novartis Pharmaceuticals | May-17 | Ph1 / Ph2 | Advanced solid tumors, exclusing known activating KRAS, NRAS, HRAS, BRAF or PTPN11 (SHP2) mutations. (Exceptions are KRAS G12-mutant NSCLC's) | NCT03114319 |
Emerging efficacy data from the first SHP2 allosteric inhibitors to reach the clinic, RMC-4630 and TNO155, have shown that this mechanism of action is not highly effective on its own as an anti-tumor agent (110). Aside from one complete response (NF1 LOF uterine carcinoma) and one partial response (KRAS-G12C mutant NSCLC), both treated with RMC-4630, the best tumor responses reported have stable disease(111). This lack of tumor shrinkage was seen despite pharmacodynamic evidence of MAPK pathway suppression, despite the single-agent efficacy of SHP2i in preclinical xenograft models of KRAS-G12C and EGFR-amplified tumors. These human clinical data confirm the biological role of SHP2 as a signal integration node rather than a driver oncogene in most tumor types.
Encouraging preclinical combination data, informed by biological and mechanistic insights and a growing understanding of the tumor and its microenvironment, have warranted the rapid clinical evaluation of SHP2 inhibitors in treatment combinations (Table 2 and Figures 3 and 4). The current treatment approaches in combination with SHP2 inhibitors can be grouped into 2 main strategies: a) combination of SHP2 with targeted therapy (Figure 3); b) combination of SHP2 with immunotherapy (Figure 4).
Table 2:
Current combinations with SHP2 inhibitors
| Compound name | Drug combination partner | Partner drug MoA | Trial ID |
|---|---|---|---|
| BBP-398 | Nivolumab | anti-PD1 mAb | NCT05375084 |
| BR790 | Tislelizumab | anti-PD1 mAb | NCT05505877 |
| ERAS-601 | Sotorasib | KRAS-G12C inhibitor | NCT04959981 |
| Cetuximab | anti-EGFR mAb | NCT04670679 | |
| GDC-1971 | GDC-6036 | KRAS-G12C inhibitor | NCT04449874 |
| atezolizumab | anti-PDL1 mAb | not yet | |
| JAB-3068 | JS001 (toripalimab) | anti-PD1 mAb | NCT04721223 |
| JAB-3312 | JAB-21822 | KRAS-G12C inhibitor | NCT05288205 |
| Sotorasib | KRAS-G12C inhibitor | NCT04720976 | |
| Osimertinib | mutant EGFR inhibitor | ||
| Pembrolizumab | anti-PD1 mAb | ||
| Binimetinib | MEK1/2 inhibitor | ||
| PF-07284892 | Lorlatinib | ALK inhibitor | NCT04800822 |
| Binimetinib | MEK1/2 inhibitor | ||
| Cetuximab + encorafenib | anti-EGFR mAb + BRAFV600mut | ||
| RMC-4630 | Sotorasib | KRAS-G12C inhibitor | NCT05054725 |
| Adagrasib | KRAS-G12C inhibitor | NCT04418661 | |
| Pembrolizumab | anti-PD1 mAb | ||
| Osimertinib | mutant EGFR inhibitor | NCT03989115 | |
| Cobimetinib | MEK1/2 inhibitor | ||
| LY3214996 | ERK1/2 inhibitor | NCT04916236 | |
| TNO155 | Adagrasib | KRAS-G12C inhibitor | NCT04330664 |
| JDQ443 | KRAS-G12C inhibitor | NCT04699188 | |
| JDQ443 + tislelizumab | KRAS-G12Ci + anti-PD1 mAb | ||
| Nazartinib | mutant EGFR inhibitor | NCT03114319 | |
| Spartalizumab | anti-PD1 mAb | NCT04000529 | |
| Ribociclib | CDK4/6 inhibitor | ||
| Lorlatinib | ALK inhibitor | NCT04292119 | |
| Sotorasib | KRAS-G12C inhibitor | NCT04185883 | |
| Dabrafinib + LTT462 | BRAF-V600mut inhibitor + ERK1/2 inhibitor | NCT04294160 | |
| Dabrafinib + Trametinib | BRAF-V600mut inhibitor + MEK1/2 inhibitor | ||
| LY3537982 | KRAS-G12C inhibitor | NCT04956640 |
Figure 3.
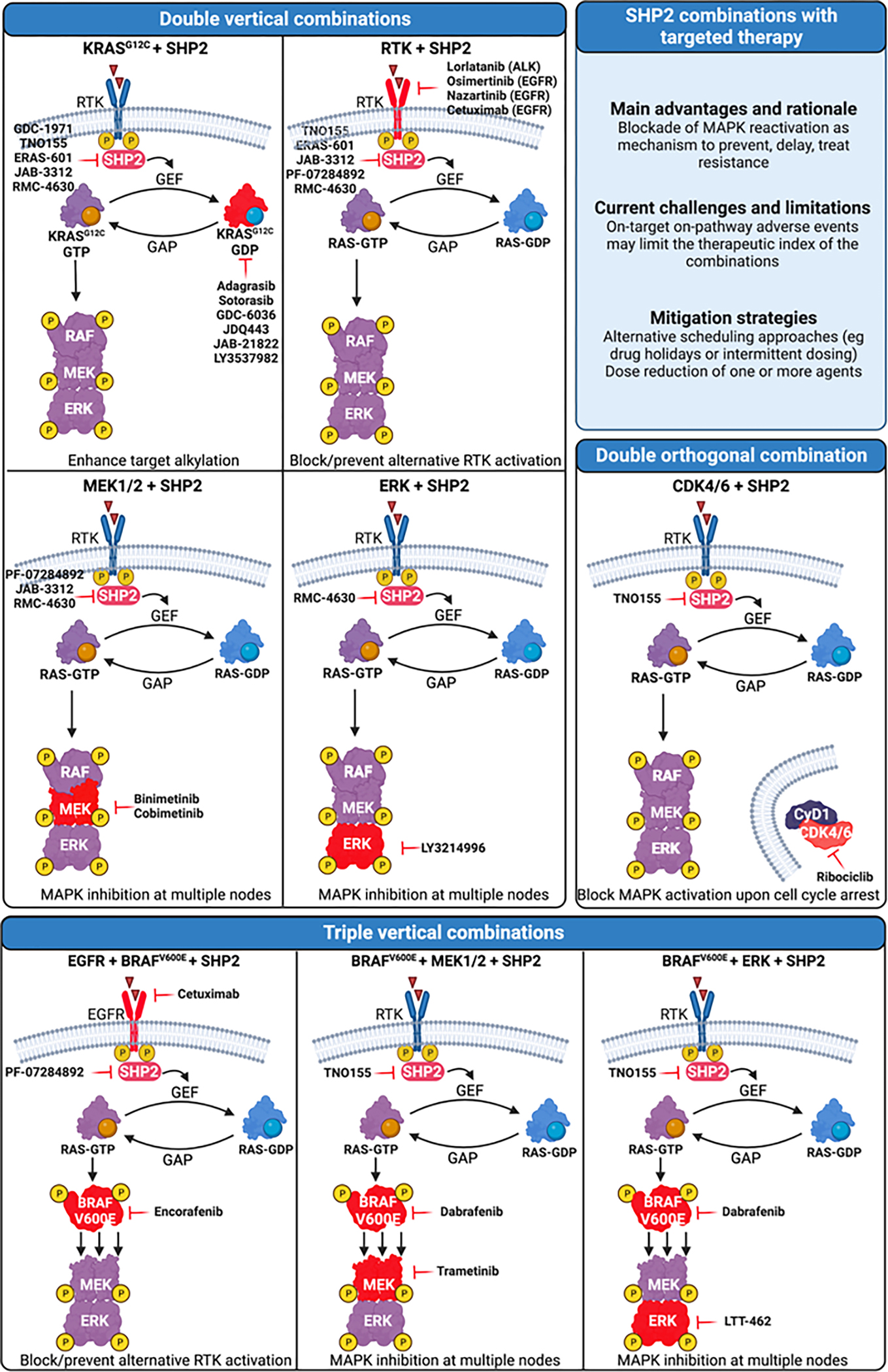
Combinations of SHP2 with targeted therapy currently tested in the clinic. Targets are highlighted in red. For the purpose of clarity, the location of RAS is depicted away from the membrane. Created with BioRender.com.
Figure 4.
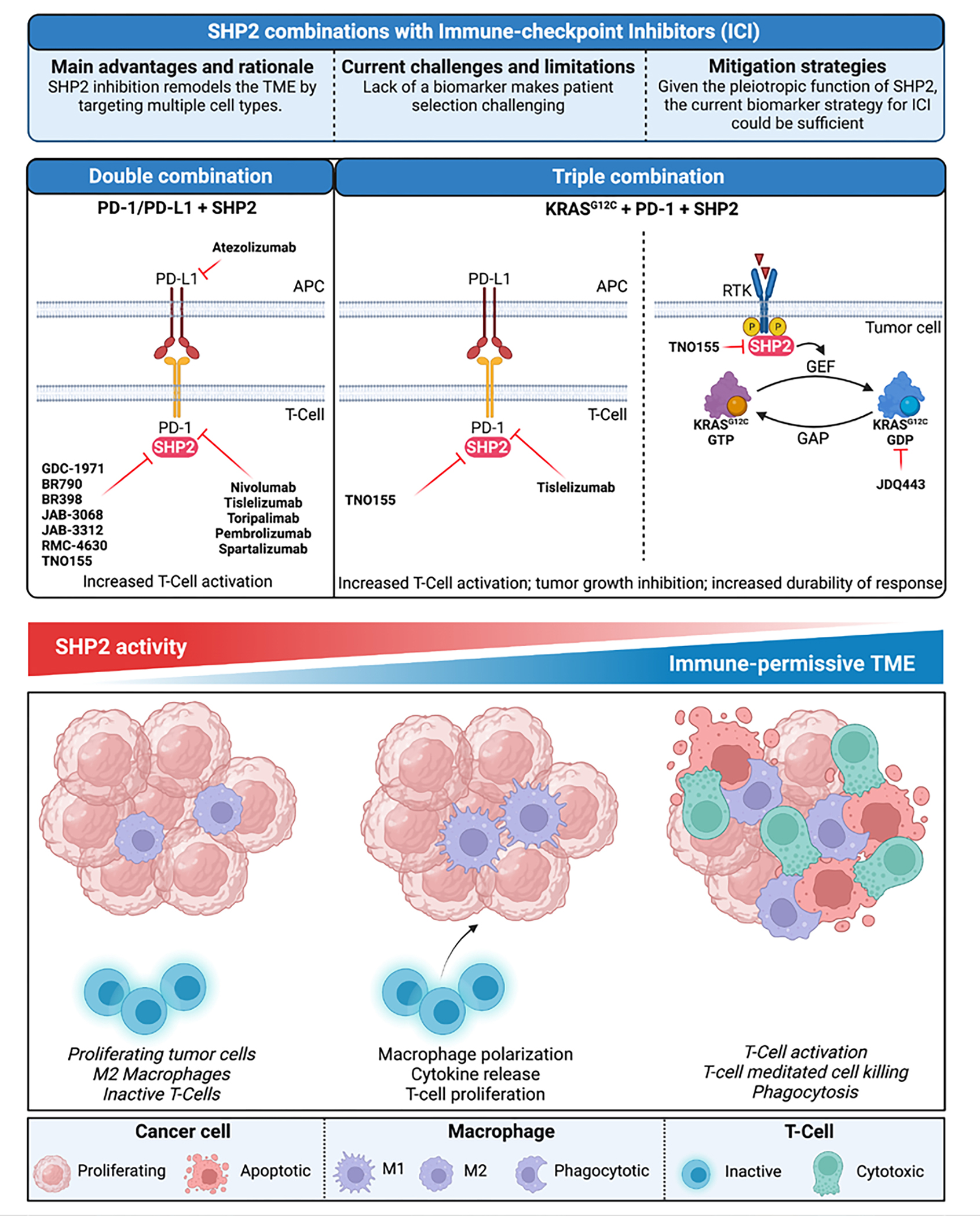
Combinations of SHP2 with immune checkpoint inhibitors currently tested in the clinic (upper panel). Schematic model of the changes occurring in the tumor microenvironment upon modulation of SHP2 activity (lower panel). Created with BioRender.com.
In the context of combinations with targeted therapy, the most common strategy aims at a vertical combination of the MAPK pathway to increase single agent activity, prevent MAPK-mediated resistance, and ultimately inhibit the pathway at multiple nodes.
KRAS-G12C inhibitors.
Small molecule compounds that target oncogenic KRAS-G12C via covalent reaction with the cysteine residue are only able to attack RAS in its inactive GDP-bound form. SHP2 inhibitors are a particularly attractive combination partner for these drugs, as SHP2 acts downstream of RTKs and upstream of SOS1/2 in the RAS signaling pathway and its phosphatase activity has been shown to inhibit several negative regulators of KRAS(112). SHP2 inhibition increases the occupancy of the KRAS-G12C-GDP state, enhancing the ability of KRAS-G12C inhibitors to couple to mutant KRAS. On the basis of this improved efficacy in preclinical studies, multiple KRAS-G12Ci / SHP2i pairs are being evaluated in clinical trials of G12C-mutated cancers (Table 2 and Figure 3). Indirect targeting of KRAS via SHP2 establishes a deeper target alkylation constitutes a form of vertical inhibition of this signaling pathway.
Tyrosine kinase inhibitors.
The mechanism of resistance to inhibitors targeting the RTK/RAS/MAPK pathway is commonly associated with bypass signaling at the level of RTKs. Combination strategies using SHP2 inhibitors acting downstream of these RTKs aim to block or prevent resistance and pathway reactivation by interrupting bypass RTK/RAS activation and combat tumor plasticity driving therapeutic adaptation. Preclinical data have shown synergy between SHP2 inhibitors and TKIs in tumors driven by RTK overexpression or activation, and suggested that SHP2 inhibitors can reverse some mechanisms of TKI resistance. This therapeutic hypothesis is being tested via combination of SHP2 inhibitors with both EGFR TKIs (osimertinib, nazartinib) and an ALK inhibitor (lorlatinib). In addition, multiple trials are investigating the combination of SHP2i with cetuximab, an anti-EGFR monoclonal antibody approved for treatment of RAS/RAF wild type colorectal carcinoma.
Additional vertical combinations.
Given the role of SHP2 as a key regulator that can orchestrate the initiation and progression of signaling cascades involved in cell proliferation, differentiation, and survival, it is not surprising that multiple signaling inhibitors of the MAPK pathway are being tested in combination with SHP2 inhibitors. These combination therapies are generally superior over single agents in prolonging responses to oncogenic pathway inhibition and in combating tumor heterogeneity and the emergence of pre-existing therapy-refractory tumor clones. Based on this rationale SHP2 inhibitors are currently tested in various malignancies with the MEK1/2 inhibitors Binimetinib or Cobimetinib(113) as well as in PDAC with the ERK inhibitor LY3214996(114). Similarly, SHP2 inhibitors have been integrated in already established combination therapies in colorectal cancer in the attempt of improving patient survival. In one instance SHP2 inhibitors are being tested together with the double combination Cetuximab/Encorafenib (EGFR/BRAFV600E) based on what has been shown by the BEACON trial(115,116). Another set of triple combination trials including SHP2 aim at preventing RTK-mediated resistance. In this case, instead of targeting only EGFR, the general RTK signaling is tackled by SHP2 inhibitors. Currently two trials are testing this hypothesis: the triple combination TNO155/Dabrafenib/Trametinib (SHP2/BRAFV600E/MEK1/2)(117) and the triple combination TNO155/Dabrafenib/LTT462 (SHP2/BRAFV600E/ERK).
Orthogonal combinations.
SHP2 inhibitors are also combined with targeted therapies beyond the MAPK pathway. An example is given by the combination of TNO155 with the CDK4/6 inhibitor Ribociclib. The rationale in this case is provided by the observation that CDK4/6 inhibitors impair G1 to S phase cell cycle progression driven by D-type cyclins, which are a convergent node of the MAPK pathway(118).
Combination of SHP2 with immunotherapy.
Anti-PD1/PD-L1 antibodies. In addition to its critical role in regulating the RAS-MAPK pathway, SHP2 is an integral downstream effector of immune signaling responses. Thus, SHP2 inhibitors can reshape the tumor microenvironment (TME) by inducing cytokine release, promoting T-cell activation and macrophage polarization to M2 (pro-inflammatory). These alterations in the TME present a therapeutic opportunity for targeting SHP2 in combination with anti-PD1/PD-L1. Preclinical studies supported the synergistic effect of SHP2 inhibition in modulating immune cell functions and demonstrated that SHP2i and anti-PD1/PD-L1combinations confer a substantial therapeutic benefit. Most of the SHP2 inhibitors brought to the clinic to date have initiated combination studies with anti-PD(L)1 antibodies (Table 2 and Figure 4). These trials are generally in indications where the checkpoint inhibitors have demonstrated efficacy, such as non-small cell lung carcinoma (NSCLC) and head and neck squamous cell carcinoma (HNSCC). These studies will test the hypothesis that SHP2 inhibition can reduce the immunosuppressive myeloid population and enhance cytotoxic T cell activity. Preliminary support for this hypothesis was provided by limited tumor biopsy data from patients receiving single-agent TNO155 or RMC-4630(119), showing an increase in tumor infiltrating T cells and a decrease in transcripts or cell markers associated with M2 macrophages. In addition, a triple combination approach to inhibit KRASG12C, SHP2, PD1 aims at synergizing cell-autonomous and non-cell autonomous activity of the SHP2 inhibitor to enhance durability of response.
Resistance to SHP2 inhibitors
While no mechanism of resistance to SHP2 inhibitors has emerged yet from the clinic, some experimental evidence has helped refining inclusion/exclusion criteria for patients with specific on-target and/or on-pathway mutations. For example, the mutations detected in leukemia on the SH2-domain E76K, D61Y and A72V keep SHP2 in an open, active conformation and are insensitive to SHP099, as opposite to the mutation E69K where the inhibitor is still effective(120) (121) (122). In addition, mutations on the PTP domain such as the double mutation T253M/Q257L (TM/QL)(87) or the mutation P491Q(113) confer drug resistance by interfering with the docking of the drug into the target while keeping the enzyme catalytically active. Moreover, on-pathway/off-target mutations may reduce significantly the activity of SHP2 inhibitors. A genome-wide CRISPR screen has identified that loss of INPPL1, MAP4K5, or LZTR1 genes results in resistance to SHP2 inhibition in vitro and in vivo by MAPK reactivation(123), as well as RAS/RAF mutant neuroblastoma cells do not respond to SHP099 as SHP2 activity is not required in this genetic context(124)
The future ahead
The ongoing and future trials with SHP2 inhibitors will help understand a few key questions. An important aspect is to dissect if SHP2 inhibition in combination with other targets would result in a deeper response rate compared to the single agent or in a general improvement of durability. Another open question is related to the chronic tolerability of such combination therapies. Many of the adverse events associated with most of the SHP2 inhibitors deriving from on-target off-cancer target inhibition include gastrointestinal tract toxicity such as diarrhea, decreased platelets and neutrophils, increased blood creatine phosphokinase, peripheral edema, and acneiform dermatitis (110). Since these side effects partially overlap with the typical toxicity related to inhibitors of the MAPK pathway, they may potentially impact the combinability of these agents. For this reason, combination strategies including alternate scheduling (on/off administration of the SHP2 inhibitor) or dosing regimens (acute high dose versus chronic lower dose) have been designed to balance efficacy and toxicity. The specific design as well as other features such as pharmacokinetics and potency will probably be a major differentiator among the trials mentioned and will refine the impact of SHP2 inhibitors in the clinic.
Conclusions
The multi-faceted role of the protein phosphatase SHP2 is a direct reflection of the complex cross-talk between cancer cells and the surrounding microenvironment. While the role of SHP2 has been extensively described in the context of the activation of the RAS-MAPK pathway, a clear idea of how SHP2 influences tumor extrinsic growth is still elusive and would require further investigation. Development of direct inhibitors of SHP2 and the clinical strategies associated to them will uncover additional aspects of this phosphatase in tumor biology
Supplementary Material
Statement of significance.
The SHP2 phosphatase functions as a pleiotropic factor and its inhibition not only hinders tumor growth, but also reshapes the tumor microenvironment. While their single agent activity may be limited, SHP2 inhibitors hold the potential of being key combination agents to enhance the depth and the durability of tumor response to therapy.
Acknowledgments
We thank Luca Gerosa and Eugene Chiang for the critical input while putting together this manuscript. The work is supported by grants R01 CA196703-01 and AIRC IG 2021 - ID. 2601 to Roberto Chiarle. Figure 3 and 4 have been generated with Biorender.
Footnotes
Conflict of interest: NMS, GP, EHK, JIA, JS, MM and DM are employees of Genentech Inc
References
- 1.Tsui HW, Hasselblatt K, Martin A, Mok SC, Tsui FWL. Molecular mechanisms underlying SHP-1 gene expression. Eur J Biochem. 2002;269:3057–64. [DOI] [PubMed] [Google Scholar]
- 2.Neel BG, Gu H, Pao L. The ‘Shp’ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem Sci. 2003;28:284–93. [DOI] [PubMed] [Google Scholar]
- 3.Shi Z-Q, Yu D-H, Park M, Marshall M, Feng G-S. Molecular Mechanism for the Shp-2 Tyrosine Phosphatase Function in Promoting Growth Factor Stimulation of Erk Activity. Mol Cell Biol. 2000;20:1526–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Higashi H, Tsutsumi R, Muto S, Sugiyama T, Azuma T, Asaka M, et al. SHP-2 Tyrosine Phosphatase as an Intracellular Target of Helicobacter pylori CagA Protein. Science. 2002;295:683–6. [DOI] [PubMed] [Google Scholar]
- 5.Voena C, Conte C, Ambrogio C, Erba EB, Boccalatte F, Mohammed S, et al. The Tyrosine Phosphatase Shp2 Interacts with NPM-ALK and Regulates Anaplastic Lymphoma Cell Growth and Migration. Cancer Res. 2007;67:4278–86. [DOI] [PubMed] [Google Scholar]
- 6.Zhan Y, Counelis GJ, O’Rourke DM. The protein tyrosine phosphatase SHP-2 is required for EGFRvIII oncogenic transformation in human glioblastoma cells. Exp Cell Res. 2009;315:2343–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aceto N, Sausgruber N, Brinkhaus H, Gaidatzis D, Martiny-Baron G, Mazzarol G, et al. Tyrosine phosphatase SHP2 promotes breast cancer progression and maintains tumor-initiating cells via activation of key transcription factors and a positive feedback signaling loop. Nat Med. 2012;18:529–37. [DOI] [PubMed] [Google Scholar]
- 8.Xu J, Zeng L-F, Shen W, Turchi JJ, Zhang Z-Y. Targeting SHP2 for EGFR inhibitor resistant non-small cell lung carcinoma. Biochem Bioph Res Co. 2013;439:586–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ruess DA, Heynen GJ, Ciecielski KJ, Ai J, Berninger A, Kabacaoglu D, et al. Mutant KRAS-driven cancers depend on PTPN11/SHP2 phosphatase. Nat Med. 2018;24:954–60. [DOI] [PubMed] [Google Scholar]
- 10.Vogel W, Lammers R, Huang J, Ullrich A. Activation of a Phosphotyrosine Phosphatase by Tyrosine Phosphorylation. Science. 1993;259:1611–4. [DOI] [PubMed] [Google Scholar]
- 11.Lechleider RJ, Sugimoto S, Bennett AM, Kashishian AS, Cooper JA, Shoelson SE, et al. Activation of the SH2-containing phosphotyrosine phosphatase SH-PTP2 by its binding site, phosphotyrosine 1009, on the human platelet-derived growth factor receptor. The J biological Chem. 1993;268:21478–81. [PubMed] [Google Scholar]
- 12.Barford D, Neel BG. Revealing mechanisms for SH2 domain mediated regulation of the protein tyrosine phosphatase SHP-2. Structure. 1998;6:249–54. [DOI] [PubMed] [Google Scholar]
- 13.Cunnick JM, Dorsey JF, Munoz-Antonia T, Mei L, Wu J. Requirement of SHP2 Binding to Grb2-associated Binder-1 for Mitogen-activated Protein Kinase Activation in Response to Lysophosphatidic Acid and Epidermal Growth Factor*. J Biol Chem. 2000;275:13842–8. [DOI] [PubMed] [Google Scholar]
- 14.Agazie YM, Hayman MJ. Molecular Mechanism for a Role of SHP2 in Epidermal Growth Factor Receptor Signaling. Mol Cell Biol. 2003;23:7875–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Araki T, Nawa H, Neel BG. Tyrosyl Phosphorylation of Shp2 Is Required for Normal ERK Activation in Response to Some, but Not All, Growth Factors*. J Biol Chem. 2003;278:41677–84. [DOI] [PubMed] [Google Scholar]
- 16.Hanafusa H, Torii S, Yasunaga T, Matsumoto K, Nishida E. Shp2, an SH2-containing Protein-tyrosine Phosphatase, Positively Regulates Receptor Tyrosine Kinase Signaling by Dephosphorylating and Inactivating the Inhibitor Sprouty*. J Biological Chem. 2004;279:22992–5. [DOI] [PubMed] [Google Scholar]
- 17.Jarvis LA, Toering SJ, Simon MA, Krasnow MA, Smith-Bolton RK. Sprouty proteins are in vivo targets of Corkscrew/SHP-2 tyrosine phosphatases. Development. 2006;133:1133–42. [DOI] [PubMed] [Google Scholar]
- 18.Hanafusa H, Torii S, Yasunaga T, Nishida E. Sprouty1 and Sprouty2 provide a control mechanism for the Ras/MAPK signalling pathway. Nat Cell Biol. 2002;4:850–8. [DOI] [PubMed] [Google Scholar]
- 19.Ren Y, Meng S, Mei L, Zhao ZJ, Jove R, Wu J. Roles of Gab1 and SHP2 in Paxillin Tyrosine Dephosphorylation and Src Activation in Response to Epidermal Growth Factor*. J Biological Chem. 2004;279:8497–505. [DOI] [PubMed] [Google Scholar]
- 20.Yang W, Klaman LD, Chen B, Araki T, Harada H, Thomas SM, et al. An Shp2/SFK/Ras/Erk Signaling Pathway Controls Trophoblast Stem Cell Survival. Dev Cell. 2006;10:317–27. [DOI] [PubMed] [Google Scholar]
- 21.Hof P, Pluskey S, Dhe-Paganon S, Eck MJ, Shoelson SE. Crystal Structure of the Tyrosine Phosphatase SHP-2. Cell. 1998;92:441–50. [DOI] [PubMed] [Google Scholar]
- 22.Yarnall MTN, Kim SH, Korntner S, Bishop AC. Destabilization of the SHP2 and SHP1 protein tyrosine phosphatase domains by a non-conserved “backdoor” cysteine. Biochem Biophys Reports. 2022;32:101370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Araki T, Chan G, Newbigging S, Morikawa L, Bronson RT, Neel BG. Noonan syndrome cardiac defects are caused by PTPN11 acting in endocardium to enhance endocardial-mesenchymal transformation. Proc National Acad Sci. 2009;106:4736–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kontaridis MI, Swanson KD, David FS, Barford D, Neel BG. PTPN11 (Shp2) Mutations in LEOPARD Syndrome Have Dominant Negative, Not Activating, Effects*. J Biol Chem. 2006;281:6785–92. [DOI] [PubMed] [Google Scholar]
- 25.Ney G, Gross A, Livinski A, Kratz CP, Stewart DR. Cancer incidence and surveillance strategies in individuals with RASopathies. Am J Medical Genetics Part C Seminars Medical Genetics. 2022;190:530–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chan G, Kalaitzidis D, Neel BG. The tyrosine phosphatase Shp2 (PTPN11) in cancer. Cancer Metast Rev. 2008;27:179–92. [DOI] [PubMed] [Google Scholar]
- 27.Tartaglia M, Niemeyer CM, Fragale A, Song X, Buechner J, Jung A, et al. Somatic mutations in PTPN11 in juvenile myelomonocytic leukemia, myelodysplastic syndromes and acute myeloid leukemia. Nat Genet. 2003;34:148–50. [DOI] [PubMed] [Google Scholar]
- 28.Bentires-Alj M, Paez JG, David FS, Keilhack H, Halmos B, Naoki K, et al. Activating Mutations of the Noonan Syndrome-Associated SHP2/PTPN11 Gene in Human Solid Tumors and Adult Acute Myelogenous Leukemia. Cancer Res. 2004;64:8816–20. [DOI] [PubMed] [Google Scholar]
- 29.Kanumuri R, Pasupuleti SK, Burns SS, Ramdas B, Kapur R. Targeting SHP2 phosphatase in hematological malignancies. Expert Opin Ther Tar. 2022;26:319–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Araki T, Mohi MG, Ismat FA, Bronson RT, Williams IR, Kutok JL, et al. Mouse model of Noonan syndrome reveals cell type– and gene dosage–dependent effects of Ptpn11 mutation. Nat Med. 2004;10:849–57. [DOI] [PubMed] [Google Scholar]
- 31.Chan G, Kalaitzidis D, Usenko T, Kutok JL, Yang W, Mohi MG, et al. Leukemogenic Ptpn11 causes fatal myeloproliferative disorder via cell-autonomous effects on multiple stages of hematopoiesis. Blood. 2009;113:4414–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mohi MG, Williams IR, Dearolf CR, Chan G, Kutok JL, Cohen S, et al. Prognostic, therapeutic, and mechanistic implications of a mouse model of leukemia evoked by Shp2 (PTPN11) mutations. Cancer Cell. 2005;7:179–91. [DOI] [PubMed] [Google Scholar]
- 33.Xu D, Liu X, Yu W-M, Meyerson HJ, Guo C, Gerson SL, et al. Non–lineage/stage-restricted effects of a gain-of-function mutation in tyrosine phosphatase Ptpn11 (Shp2) on malignant transformation of hematopoietic cells. J Exp Med. 2011;208:1977–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dong L, Yu W-M, Zheng H, Loh ML, Bunting ST, Pauly M, et al. Leukaemogenic effects of Ptpn11 activating mutations in the stem cell microenvironment. Nature. 2016;539:304–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu X, Zheng H, Qu C-K. Protein Tyrosine Phosphatase Shp2 (Ptpn11) Plays an Important Role in Maintenance of Chromosome Stability. Cancer Res. 2012;72:5296–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vemulapalli V, Chylek LA, Erickson A, Pfeiffer A, Gabriel K-H, LaRochelle J, et al. Time-resolved phosphoproteomics reveals scaffolding and catalysis-responsive patterns of SHP2-dependent signaling. Elife. 2021;10:e64251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhu P, Wu X, Zhang R-Y, Hsu C-C, Zhang Z-Y, Tao WA. An Integrated Proteomic Strategy to Identify SHP2 Substrates. J Proteome Res. 2022;21:2515–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang SQ, Tsiaras WG, Araki T, Wen G, Minichiello L, Klein R, et al. Receptor-Specific Regulation of Phosphatidylinositol 3′-Kinase Activation by the Protein Tyrosine Phosphatase Shp2. Mol Cell Biol. 2002;22:4062–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goodwin CB, Yang Z, Yin F, Yu M, Chan RJ. Genetic disruption of the PI3K regulatory subunits, p85α, p55α, and p50α, normalizes mutant PTPN11-induced hypersensitivity to GM-CSF. Haematologica. 2012;97:1042–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Breitkopf SB, Yang X, Begley MJ, Kulkarni M, Chiu Y-H, Turke AB, et al. A Cross-Species Study of PI3K Protein-Protein Interactions Reveals the Direct Interaction of P85 and SHP2. Sci Reports. 2016;6:20471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu C-J, O’Rourke DM, Feng G-S, Johnson GR, Wang Q, Greene MI. The tyrosine phosphatase SHP-2 is required for mediating phosphatidylinositol 3-kinase/Akt activation by growth factors. Oncogene. 2001;20:6018–25. [DOI] [PubMed] [Google Scholar]
- 42.Mattoon DR, Lamothe B, Lax I, Schlessinger J. The docking protein Gab1 is the primary mediator of EGF-stimulated activation of the PI-3K/Akt cell survival pathway. BMC Biology. 2004;2:24–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mali RS, Ma P, Zeng L-F, Martin H, Ramdas B, He Y, et al. Role of SHP2 phosphatase in KIT-induced transformation: identification of SHP2 as a druggable target in diseases involving oncogenic KIT. Blood. 2012;120:2669–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Amante RJ, Jehanno C, Silva DD, Coissieux M-M, Ackerknecht M, Romanet V, et al. PI3K inhibition circumvents resistance to SHP2 blockade in metastatic triple-negative breast cancer. J Mammary Gland Biol Neoplasia. 28, 13 2023; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hu Z, Li J, Gao Q, Wei S, Yang B. SHP2 overexpression enhances the invasion and metastasis of ovarian cancer in vitro and in vivo. OncoTargets Ther. 2017;10:3881–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cao M, Gao D, Zhang N, Duan Y, Wang Y, Mujtaba H, et al. Shp2 expression is upregulated in cervical cancer, and Shp2 contributes to cell growth and migration and reduces sensitivity to cisplatin in cervical cancer cells. Pathol - Res Pract. 2019;215:152621. [DOI] [PubMed] [Google Scholar]
- 47.Xu D, Qu C-K. Protein tyrosine phosphatases in the JAK/STAT pathway. Front Biosci. 2008; 13:4925–4932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bard-Chapeau EA, Li S, Ding J, Zhang SS, Zhu HH, Princen F, et al. Ptpn11/Shp2 Acts as a Tumor Suppressor in Hepatocellular Carcinogenesis. Cancer Cell. 2011;19:629–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yu W-M, Daino H, Chen J, Bunting KD, Qu C-K. Effects of a Leukemia-associated Gain-of-Function Mutation of SHP-2 Phosphatase on Interleukin-3 Signaling*. J Biol Chem. 2006;281:5426–34. [DOI] [PubMed] [Google Scholar]
- 50.Zheng M, Liu Y, Wu C, Yang K, Wang Q, Zhou Y, et al. Novel PROTACs for degradation of SHP2 protein. Bioorg Chem. 2021;110:104788. [DOI] [PubMed] [Google Scholar]
- 51.Chen M-J, Wang Y-C, Wu D-W, Chen C-Y, Lee H Association of nuclear localization of SHP2 and YAP1 with unfavorable prognosis in non-small cell lung cancer. Pathology - Res Pract. 2019;215:801–6. [DOI] [PubMed] [Google Scholar]
- 52.Tsutsumi R, Masoudi M, Takahashi A, Fujii Y, Hayashi T, Kikuchi I, et al. YAP and TAZ, Hippo Signaling Targets, Act as a Rheostat for Nuclear SHP2 Function. Dev Cell. 2013;26:658–65. [DOI] [PubMed] [Google Scholar]
- 53.Watkins RD, Buckarma EH, Tomlinson JL, McCabe CE, Yonkus JA, Werneburg NW, et al. SHP2 inhibition enhances Yes-associated protein mediated liver regeneration in murine partial hepatectomy models. Jci Insight. 2022;7:e159930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Buckarma EH, Werneburg NW, Conboy CB, Kabashima A, O’Brien DR, Wang C, et al. The YAP-Interacting Phosphatase SHP2 Can Regulate Transcriptional Coactivity and Modulate Sensitivity to Chemotherapy in Cholangiocarcinoma. Mol Cancer Res. 2020;18:1574–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hagenbeek TJ, Zbieg JR, Hafner M, Mroue R, Lacap JA, Sodir NM, et al. An allosteric pan-TEAD inhibitor blocks oncogenic YAP/TAZ signaling and overcomes KRAS G12C inhibitor resistance. Nat Cancer. 2023; 4:812–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang Y, Mohseni M, Grauel A, Diez JE, Guan W, Liang S, et al. SHP2 blockade enhances anti-tumor immunity via tumor cell intrinsic and extrinsic mechanisms. Sci Rep-uk. 2021;11:1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Achkova D, Maher J. Role of the colony-stimulating factor (CSF)/CSF-1 receptor axis in cancer. Biochem Soc T. 2015;44:333–41. [DOI] [PubMed] [Google Scholar]
- 58.Zhang W, Huang Q, Xiao W, Zhao Y, Pi J, Xu H, et al. Advances in Anti-Tumor Treatments Targeting the CD47/SIRPα Axis. Front Immunol. 2020;11:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vonderheide RH. CD47 blockade as another immune checkpoint therapy for cancer. Nat Med. 2015;21:1122–3. [DOI] [PubMed] [Google Scholar]
- 60.Barkal AA, Brewer RE, Markovic M, Kowarsky M, Barkal SA, Zaro BW, et al. CD24 signalling through macrophage Siglec-10 is a target for cancer immunotherapy. Nature. 2019;572:392–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xiao P, Guo Y, Zhang H, Zhang X, Cheng H, Cao Q, et al. Myeloid-restricted ablation of Shp2 restrains melanoma growth by amplifying the reciprocal promotion of CXCL9 and IFN-γ production in tumor microenvironment. Oncogene. 2018;37:5088–100. [DOI] [PubMed] [Google Scholar]
- 62.Christofides A, Katopodi X-L, Cao C, Karagkouni D, Aliazis K, Yenyuwadee S, et al. SHP-2 and PD-1-SHP-2 signaling regulate myeloid cell differentiation and antitumor responses. Nat Immunol. 2023;24:55–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Xiao P, Zhang H, Zhang Y, Zheng M, Liu R, Zhao Y, et al. Phosphatase Shp2 exacerbates intestinal inflammation by disrupting macrophage responsiveness to interleukin-10. J Exp Med. 2019;216:337–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ramesh A, Kumar S, Nandi D, Kulkarni A. CSF1R- and SHP2-Inhibitor-Loaded Nanoparticles Enhance Cytotoxic Activity and Phagocytosis in Tumor-Associated Macrophages. Adv Mater. 2019;31:e1904364. [DOI] [PubMed] [Google Scholar]
- 65.Marasco M, Berteotti A, Weyershaeuser J, Thorausch N, Sikorska J, Krausze J, et al. Molecular mechanism of SHP2 activation by PD-1 stimulation. Sci Adv. 2020;6:eaay4458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hui E, Cheung J, Zhu J, Su X, Taylor MJ, Wallweber HA, et al. T cell costimulatory receptor CD28 is a primary target for PD-1–mediated inhibition. Science. 2017;355:1428–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang B, Zhang W, Jankovic V, Golubov J, Poon P, Oswald EM, et al. Combination cancer immunotherapy targeting PD-1 and GITR can rescue CD8+ T cell dysfunction and maintain memory phenotype. Sci Immunol. 2018; 3, eaat7061. [DOI] [PubMed] [Google Scholar]
- 68.Sheppard K-A, Fitz LJ, Lee JM, Benander C, George JA, Wooters J, et al. PD-1 inhibits T-cell receptor induced phosphorylation of the ZAP70/CD3zeta signalosome and downstream signaling to PKCtheta. Febs Lett. 2004;574:37–41. [DOI] [PubMed] [Google Scholar]
- 69.Rota G, Niogret C, Dang AT, Barros CR, Fonta NP, Alfei F, et al. Shp-2 Is Dispensable for Establishing T Cell Exhaustion and for PD-1 Signaling In Vivo. Cell Reports. 2018;23:39–49. [DOI] [PubMed] [Google Scholar]
- 70.Banta KL, Xu X, Chitre AS, Au-Yeung A, Takahashi C, O’Gorman WE, et al. Mechanistic convergence of the TIGIT and PD-1 inhibitory pathways necessitates co-blockade to optimize anti-tumor CD8+ T cell responses. Immunity. 2022;55:512–526.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chiang EY, Mellman I. TIGIT-CD226-PVR axis: advancing immune checkpoint blockade for cancer immunotherapy. J Immunother Cancer. 2022;10:e004711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vivier E, Nunès JA, Vély F. Natural Killer Cell Signaling Pathways. Science. 2004;306:1517–9. [DOI] [PubMed] [Google Scholar]
- 73.Yusa S, Campbell KS. Src Homology Region 2-Containing Protein Tyrosine Phosphatase-2 (SHP-2) Can Play a Direct Role in the Inhibitory Function of Killer Cell Ig-Like Receptors in Human NK Cells. J Immunol. 2003;170:4539–47. [DOI] [PubMed] [Google Scholar]
- 74.Purdy AK, Campbell KS. SHP-2 Expression Negatively Regulates NK Cell Function. J Immunol. 2009;183:7234–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Niogret C, Miah SMS, Rota G, Fonta NP, Wang H, Held W, et al. Shp-2 is critical for ERK and metabolic engagement downstream of IL-15 receptor in NK cells. Nat Commun. 2019;10:1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang Y, Salvucci O, Ohnuki H, Tran AD, Ha T, Feng J, et al. Targeting the SHP2 phosphatase promotes vascular damage and inhibition of tumor growth. Embo Mol Med. 2021;13:e14089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tang KH, Li S, Khodadadi-Jamayran A, Jen J, Han H, Guidry K, et al. Combined Inhibition of SHP2 and CXCR1/2 Promotes Antitumor T-cell Response in NSCLC. Cancer Discov. 2021;12:47–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Xu Z, Guo C, Ye Q, Shi Y, Sun Y, Zhang J, et al. Endothelial deletion of SHP2 suppresses tumor angiogenesis and promotes vascular normalization. Nat Commun. 2021;12:6310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Li S, Wang L, Zhao Q, Liu Y, He L, Xu Q, et al. SHP2 Positively Regulates TGFβ1-induced Epithelial-Mesenchymal Transition Modulated by Its Novel Interacting Protein Hook1*. J Biological Chem. 2014;289:34152–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sun X, Zhang J, Wang Z, Ji W, Tian R, Zhang F, et al. Shp2 Plays a Critical Role in IL-6-Induced EMT in Breast Cancer Cells. Int J Mol Sci. 2017;18:395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zehender A, Huang J, Györfi A-H, Matei A-E, Trinh-Minh T, Xu X, et al. The tyrosine phosphatase SHP2 controls TGFβ-induced STAT3 signaling to regulate fibroblast activation and fibrosis. Nat Commun. 2018;9:3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Caligiuri G, Tuveson DA. Activated fibroblasts in cancer: Perspectives and challenges. Cancer Cell. 2023;41:434–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhao M, Guo W, Wu Y, Yang C, Zhong L, Deng G, et al. SHP2 inhibition triggers anti-tumor immunity and synergizes with PD-1 blockade. Acta Pharm Sinica B. 2019;9:304–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Quintana E, Schulze CJ, Myers DR, Choy TJ, Mordec K, Wildes D, et al. Allosteric Inhibition of SHP2 Stimulates Antitumor Immunity by Transforming the Immunosuppressive Environment. Cancer Res. 2020;80:2889–902. [DOI] [PubMed] [Google Scholar]
- 85.Liu C, Lu H, Wang H, Loo A, Zhang X, Yang G, et al. Combinations with Allosteric SHP2 Inhibitor TNO155 to Block Receptor Tyrosine Kinase Signaling. Clin Cancer Res. 2021;27:342–54. [DOI] [PubMed] [Google Scholar]
- 86.Blaskovich MAT. Drug discovery and protein tyrosine phosphatases. Curr Med Chem. 2009;16:2095–176. [DOI] [PubMed] [Google Scholar]
- 87.Chen Y-NP, LaMarche MJ, Chan HM, Fekkes P, Garcia-Fortanet J, Acker MG, et al. Allosteric inhibition of SHP2 phosphatase inhibits cancers driven by receptor tyrosine kinases. Nature. 2016;535:148–52. [DOI] [PubMed] [Google Scholar]
- 88.LaMarche MJ, Acker M, Argintaru A, Bauer D, Boisclair J, Chan H, et al. Identification of TNO155, an Allosteric SHP2 Inhibitor for the Treatment of Cancer. J Med Chem. 2020;63:13578–94. [DOI] [PubMed] [Google Scholar]
- 89.Nichols RJ, Haderk F, Stahlhut C, Schulze CJ, Hemmati G, Wildes D, et al. RAS nucleotide cycling underlies the SHP2 phosphatase dependence of mutant BRAF-, NF1- and RAS-driven cancers. Nat Cell Biol. 2018; 20: 1064–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Williams B, Taylor A, Orozco O, Owen C, Kelley E, Lescarbeau A, et al. Abstract 3327: Discovery and characterization of the potent, allosteric SHP2 inhibitor GDC-1971 for the treatment of RTK/RAS driven tumors. Cancer Res. 2022;82:3327–3327. [Google Scholar]
- 91.Huifeng H, Panliang G, Cunbo M, Kang D. Novel heterocyclic derivatives useful as SHP2 inhibitors. 2020. Worldwide patent WO2017211303A1 [Google Scholar]
- 92.Gebregiworgis T, Kano Y, St-Germain J, Radulovich N, Udaskin ML, Mentes A, et al. The Q61H mutation decouples KRAS from upstream regulation and renders cancer cells resistant to SHP2 inhibitors. Nat Commun. 2021;12:6274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Akhave NS, Biter AB, Hong DS. Mechanisms of Resistance to KRASG12C-Targeted Therapy. Cancer Discov. 2021;11:1345–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Leonetti A, Sharma S, Minari R, Perego P, Giovannetti E, Tiseo M. Resistance mechanisms to osimertinib in EGFR-mutated non-small cell lung cancer. Brit J Cancer. 2019;121:725–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Dardaei L, Wang HQ, Singh M, Fordjour P, Shaw KX, Yoda S, et al. SHP2 inhibition restores sensitivity in ALK-rearranged non-small-cell lung cancer resistant to ALK inhibitors. Nat Med. 2018;24:512–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Rosen EY, Won HH, Zheng Y, Cocco E, Selcuklu D, Gong Y, et al. The evolution of RET inhibitor resistance in RET-driven lung and thyroid cancers. Nat Commun. 2022;13:1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Charest A, Wilker EW, McLaughlin ME, Lane K, Gowda R, Coven S, et al. ROS Fusion Tyrosine Kinase Activates a SH2 Domain–Containing Phosphatase-2/Phosphatidylinositol 3-Kinase/Mammalian Target of Rapamycin Signaling Axis to Form Glioblastoma in Mice. Cancer Res. 2006;66:7473–81. [DOI] [PubMed] [Google Scholar]
- 98.Cocco E, Schram AM, Kulick A, Misale S, Won HH, Yaeger R, et al. Resistance to TRK inhibition mediated by convergent MAPK pathway activation. Nat Med. 2019;25:1422–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Alotaibi AS, Yilmaz M, Kanagal-Shamanna R, Loghavi S, Kadia TM, DiNardo CD, et al. Patterns of Resistance Differ in Patients with Acute Myeloid Leukemia Treated with Type I versus Type II FLT3-inhibitors. Blood Cancer Discov. 2020;2:125–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sun Y, Meyers BA, Czako B, Leonard P, Mseeh F, Harris AL, et al. Allosteric SHP2 Inhibitor, IACS-13909, Overcomes EGFR-Dependent and EGFR-Independent Resistance Mechanisms toward Osimertinib. Cancer Res. 2020;80:4840–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Deng Y, Ma G, Vallega KA, Wang D, Wang M, Wang C, et al. Therapeutic efficacy of the novel SHP2 degrader SHP2-D26, alone or in combination, against lung cancer is associated with modulation of p70S6K/S6, Bim and Mcl-1. Cancer Gene Ther. 2022;1–12. [DOI] [PubMed] [Google Scholar]
- 102.Sequist LV, Han J-Y, Ahn M-J, Cho BC, Yu H, Kim S-W, et al. Osimertinib plus savolitinib in patients with EGFR mutation-positive, MET-amplified, non-small-cell lung cancer after progression on EGFR tyrosine kinase inhibitors: interim results from a multicentre, open-label, phase 1b study. Lancet Oncol. 2020;21:373–86. [DOI] [PubMed] [Google Scholar]
- 103.Oxnard GR, Yang JC-H, Yu H, Kim S-W, Saka H, Horn L, et al. TATTON: a multi-arm, phase Ib trial of osimertinib combined with selumetinib, savolitinib, or durvalumab in EGFR-mutant lung cancer. Ann Oncol. 2020;31:507–16. [DOI] [PubMed] [Google Scholar]
- 104.Ryan MB, Coker O, Sorokin A, Fella K, Barnes H, Wong E, et al. KRASG12C-independent feedback activation of wild-type RAS constrains KRASG12C inhibitor efficacy. Cell Reports. 2021;39:110993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Yousefi M, Boross G, Weiss C, Murray CW, Hebert JD, Cai H, et al. Combinatorial Inactivation of Tumor Suppressors Efficiently Initiates Lung Adenocarcinoma with Therapeutic Vulnerabilities. Cancer Res. 2022;82:1589–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hayashi T, Desmeules P, Smith RS, Drilon A, Somwar R, Ladanyi M. RASA1 and NF1 are Preferentially Co-Mutated and Define A Distinct Genetic Subset of Smoking-Associated Non–Small Cell Lung Carcinomas Sensitive to MEK Inhibition. Clin Cancer Res. 2018;24:1436–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Pan Y, Han H, Hu H, Wang H, Song Y, Hao Y, et al. KMT2D deficiency drives lung squamous cell carcinoma and hypersensitivity to RTK-RAS inhibition. Cancer Cell. 2023;41:88–105.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Mulero-Sánchez A, Ramirez CF, Chatinier A du, Wang H, Koomen SJI, Song J, et al. Rational combination of SHP2 and mTOR inhibition for the treatment of hepatocellular carcinoma. Mol Oncol. 2023; 17:964–980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Drilon A, Sharma MR, Johnson ML, Yap TA, Gadgeel S, Nepert D, et al. SHP2 Inhibition Sensitizes Diverse Oncogene-Addicted Solid Tumors to Re-treatment with Targeted Therapy. Cancer Discov. 2023;OF1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Brana I, Shapiro G, Johnson ML, Yu HA, Robbrecht D, Tan DS-W, et al. Initial results from a dose finding study of TNO155, a SHP2 inhibitor, in adults with advanced solid tumors. J Clin Oncol. 2021;39:3005–3005. [Google Scholar]
- 111.Koczywas M, Haura E, Janne PA, Pacheco JM, Ulahannan S, Wang JS, et al. Abstract LB001: Anti-tumor activity and tolerability of the SHP2 inhibitor RMC-4630 as a single agent in patients with RAS-addicted solid cancers. Cancer Res. 2021;81:LB001–LB001. [Google Scholar]
- 112.Montagner A, Yart A, Dance M, Perret B, Salles J-P, Raynal P. A Novel Role for Gab1 and SHP2 in Epidermal Growth Factor-induced Ras Activation*. J Biol Chem. 2005;280:5350–60. [DOI] [PubMed] [Google Scholar]
- 113.Fedele C, Ran H, Diskin B, Wei W, Jen J, Geer MJ, et al. SHP2 Inhibition Prevents Adaptive Resistance to MEK inhibitors in Multiple Cancer Models. Cancer Discov. 2018;8:CD-18–0444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Frank KJ, Mulero-Sánchez A, Berninger A, Ruiz-Cañas L, Bosma A, Görgülü K, et al. Extensive preclinical validation of combined RMC-4550 and LY3214996 supports clinical investigation for KRAS mutant pancreatic cancer. Cell Reports Medicine. 2022;3:100815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Tabernero J, Grothey A, Cutsem EV, Yaeger R, Wasan H, Yoshino T, et al. Encorafenib Plus Cetuximab as a New Standard of Care for Previously Treated BRAF V600E–Mutant Metastatic Colorectal Cancer: Updated Survival Results and Subgroup Analyses from the BEACON Study. J Clin Oncol. 2021;39:273–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kopetz S, Grothey A, Yaeger R, Cutsem EV, Desai J, Yoshino T, et al. Encorafenib, Binimetinib, and Cetuximab in BRAF V600E–Mutated Colorectal Cancer. New Engl J Med. 2019;381:1632–43. [DOI] [PubMed] [Google Scholar]
- 117.Corcoran RB, Atreya CE, Falchook GS, Kwak EL, Ryan DP, Bendell JC, et al. Combined BRAF and MEK Inhibition With Dabrafenib and Trametinib in BRAF V600–Mutant Colorectal Cancer. J Clin Oncol. 2015;33:4023–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.O’Leary B, Finn RS, Turner NC. Treating cancer with selective CDK4/6 inhibitors. Nat Rev Clin Oncol. 2016;13:417–30. [DOI] [PubMed] [Google Scholar]
- 119.Chen AY-C, Haura E, Pacheco J, Koczywas M, Gordon M, Ulahannan S, et al. Abstract LB050: Modulation of innate and adaptive immunity in blood and tumor of patients receiving the SHP2 inhibitor RMC-4630. Cancer Res. 2021;81:LB050–LB050. [Google Scholar]
- 120.Sun X, Ren Y, Gunawan S, Teng P, Chen Z, Lawrence H, et al. Selective inhibition of leukemia-associated SHP2E69K mutant by the allosteric SHP2 inhibitor SHP099. Leukemia. 2018;32:1246–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Pádua RAP, Sun Y, Marko I, Pitsawong W, Stiller JB, Otten R, et al. Mechanism of activating mutations and allosteric drug inhibition of the phosphatase SHP2. Nat Commun. 2018;9:4507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wang R-R, Ma Y, Du S, Li W-Y, Sun Y-Z, Zhou H, et al. Exploring the reason for increased activity of SHP2 caused by D61Y mutation through molecular dynamics. Comput Biol Chem. 2019;78:133–43. [DOI] [PubMed] [Google Scholar]
- 123.Wei W, Geer MJ, Guo X, Dolgalev I, Sanjana NE, Neel BG. Genome-wide CRISPR/Cas9 screens reveal shared and cell-specific mechanisms of resistance to SHP2 inhibition. J Exp Med. 2023;220:e20221563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Cai J, Jacob S, Kurupi R, Dalton KM, Coon C, Greninger P, et al. High-risk neuroblastoma with NF1 loss of function is targetable using SHP2 inhibition. Cell Reports. 2022;40:111095. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.