

Abstract
GABAA receptors mediate the majority of inhibitory neurotransmission in the CNS. Genetic deletion of the α1 subunit of GABAA receptors results in a loss of α1-mediated fast inhibitory currents and a marked reduction in density of GABAA receptors. A grossly normal phenotype of α1-deficient mice suggests the presence of neuronal adaptation to these drastic changes at the GABA synapse. We used cDNA microarrays to identify transcriptional fingerprints of cellular plasticity in response to altered GABAergic inhibition in the cerebral cortex and cerebellum of α1 mutants. In silico analysis of 982 mutation-regulated transcripts highlighted genes and functional groups involved in regulation of neuronal excitability and synaptic transmission, suggesting an adaptive response of the brain to an altered inhibitory tone. Public gene expression databases permitted identification of subsets of transcripts enriched in excitatory and inhibitory neurons as well as some glial cells, providing evidence for cellular plasticity in individual cell types. Additional analysis linked some transcriptional changes to cellular phenotypes observed in the knock-out mice and suggested several genes, such as the early growth response 1 (Egr1), small GTP binding protein Rac1 (Rac1), neurogranin (Nrgn), sodium channel β4 subunit (Scn4b), and potassium voltage-gated Kv4.2 channel (Kcnd2) as cell type-specific markers of neuronal plasticity. Furthermore, transcriptional activation of genes enriched in Bergman glia suggests an active role of these astrocytes in synaptic plasticity. Overall, our results suggest that the loss of α1-mediated fast inhibition produces diverse transcriptional responses that act to regulate neuronal excitability of individual neurons and stabilize neuronal networks, which may account for the lack of severe abnormalities in α1 null mutants.
Keywords: knock-out, null mutant, microarray, gene expression, neuroadaptation, synapse, neuron, glia
Introduction
Null mutant mice are widely used to study effects of a single gene on complex traits. Several mouse lines with genetically altered GABAA receptor subunits have been generated to investigate the involvement of GABAergic system at cellular and behavioral levels (Boehm et al., 2004; Vicini and Ortinski, 2004). GABAA receptors mediate the majority of inhibitory neurotransmission in the CNS, and the GABAA α1 subunit is a constituent of the most abundant subunit combination in the brain: α1β2γ2. Mice lacking this subunit fail to switch from α2/α3 to the α1 subunit during early development, thus retaining long-lasting postsynaptic currents conferred by α2/α3 subunits (Vicini et al., 2001; Bosman et al., 2002; Goldstein et al., 2002; Ortinski et al., 2004). This increased efficacy at the GABA synapse does not result in gross phenotypic abnormalities or any apparent changes in breeding or development (Sur et al., 2001; Vicini et al., 2001).
Several studies suggest that compensatory changes could maintain the balance between inhibition and excitation within a range of normal functioning in the brain of α1 mutants. For example, an increase in decay time of GABA-mediated IPSCs recorded from cerebellar or hippocampal neurons of α1 knockout animals was accompanied by a decrease in amplitude (Vicini et al., 2001; Goldstein et al., 2002; Ortinski et al., 2004), frequency (Goldstein et al., 2002; Ortinski et al., 2004), or both (Goldstein et al., 2002; Ortinski et al., 2004), implying the presence of counter-balancing forces that regulate the efficacy of the GABA synapse. The loss of ~50% of GABAA receptors throughout the brain of α1 knock-outs (Sur et al., 2001; Kralic et al., 2002a) may also contribute to homeostatic compensation. Pharmacological studies suggest that neuronal plasticity is not limited to changes at the GABA synapse and that other neurotransmitter systems, such as glutamate and dopamine may also contribute to the maintenance of neuronal activity within physiologically relevant limits (Kralic et al., 2003; Reynolds et al., 2003).
An emerging approach in neuroscience is to investigate brain adaptation by describing transcriptional profiles associated with various genetic, pharmacological, and pathological perturbations (Pongrac et al., 2002; Tabakoff et al., 2003; Hitzemann et al., 2004; Ponomarev et al., 2004; Chesler et al., 2005; Sugino et al., 2006). The objective of the present study was to define transcriptional signatures of neuronal plasticity in response to altered inhibition in the mutant brain. We used cDNA microarrays and the α1 null mutants to describe mutation-driven transcriptional changes in the cerebral cortex and cerebellum, two brain regions that normally exhibit an abundant expression of the α1 subunit. Analysis of data from two independently created knock-out lines (Sur et al., 2001; Vicini et al., 2001) revealed genes and cellular pathways implicated in neuronal plasticity and synaptic homeostasis and allowed us to make predictions about their contribution to cellular phenotypes previously described in the α1 mutants (see supplemental Table 1, available at www.jneurosci.org as supplemental material). In addition, the availability of published expression databases permitted identification of plasticity-related transcriptional signatures in individual cell types.
Materials and Methods
Animals
All animal-related procedures were performed in accordance with the rules of the University of Texas at Austin and University of Pittsburgh Institutional Animal Use and Care Committees, which abide by the published guidelines of the United States Department of Agriculture and the United States Public Health Service. Male mice were used for microarray and immunohistochemistry experiments, and both males and females were used for Western blot validation and electrophysiology. Littermates were used in all experiments. Two mouse lines lacking the α1 subunit of the GABAA receptor have been independently created, one by Dr. Whiting’s group at the Merck Sharp and Dohme company (Sur et al., 2001) and the other by Dr. Homanics’ group at the University of Pittsburgh (Vicini et al., 2001). Both the Merck and Pittsburgh groups used a homologous recombination approach to generate knock-out animals with deleted exons 4 and 8, respectively. The Pittsburgh line had the “Neo” cassette removed, whereas the Merck mice retained it. Both lines were maintained by heterozygous mating on mixed genetic backgrounds of either C57BL/6J × 129/SvEv (Merck) or C57BL/6J × 129Sv/SvJ × FVB/N (Pittsburgh) inbred strains. Merck mice were obtained from a local colony, whereas brains of Pittsburgh null mutant and wild-type animals were shipped from Pittsburgh. All animals were housed three to five mice per cage under a 12 h light/dark cycle with food and water available ad libitum. All mice were killed by cervical dislocation and were 6–12 weeks of age at the time of testing.
Brain tissue collection and RNA isolation
Whole brains were removed from drug-naive mice, cut in half, and stored in RNAlater (Ambion, Austin, TX) solution at 4°C before additional dissection. Brains were later dissected into cerebral cortex, cerebellum, and the remaining brain tissue. Total RNA was extracted from the cortex and cerebellum of individual animals using TRIzol reagent (Invitrogen, Carlsbad, CA). The resulting total RNA samples were quantified on a NanoDrop ND-1000 Spectrophotometer (NanoDrop Technologies, Wilmington, DE), and RNA quality was determined on an Agilent 2100 Bioanalyzer using the RNA 6000 Nano LabChip kit (Agilent Technologies, Palo Alto, CA). The RNA samples were then stored at −80°C until use for microarray analysis.
Microarray analysis
Spotted cDNA microarrays were prepared and printed at the University of Texas at Austin according to the process described previously (Schena et al., 1995). Two independent mouse cDNA clone sets, Brain Molecular Anatomy Project and Sequence Verified (ResGen; Invitrogen), were printed on poly-L-lysine-coated microscope slides using a robotic arrayer. The resulting microarrays consisted of 16,950 clones representing ~11,000 unique mouse genes. Microarray hybridizations were performed using the Array 50 kit purchased from Genisphere (Hatfield, PA). The Genisphere protocol uses an end-labeling procedure that results in the attachment of an oligodendrimer labeled with ~50 fluorescent dye molecules to the cDNA of interest. This process allows for a high degree of sensitivity, which reduces the amount of total RNA necessary for differential signal resolution and allows for reduction in sequence length and composition-based intensity variance associated with other fluorescent labeling techniques. All microarrays used in this study were hybridized using ~15 μg of a common reference sample in channel 1 (Cy3) and 15 μg of total RNA from either the experimental or control samples in channel 2 (Cy5). This common reference sample was created by extracting total RNA from the whole brains of 100 male C57BL/6J mice (The Jackson Laboratory, Bar Harbor, ME) at 72 d of age. These RNA samples were combined and thoroughly mixed and then aliquoted and stored at −80°C. Microarray hybridizations were performed at 51°C by the two-step procedure outlined by the Genisphere protocol. The hybridized arrays were immediately scanned on an Axon GenePix 4000B dual channel laser scanner (Molecular Devices, Union City, CA.). The microarrays were then gridded and the image data translated to intensity values using the software package GenePix version 4.2 (Molecular Devices).
Normalization, filtering, and standardization of microarray data
Microarray normalization was performed using the R package, Statistics for Microarray Analysis (Yang et al., 2002). Specifically, the Lowess within-print-tip-group function contained in this package was used for normalization. The normalized data were then uploaded and stored in the Longhorn Array Database (Killion et al., 2003), which allowed for the compilation of multiple arrays for data retrieval. Log2 Cy5/Cy3 ratio of median spot intensity of background-subtracted data were used as the dependent variable. Poor quality spots detected by GenePix software were removed before statistical analysis. Only transcripts that were reliably detected on at least four microarray slides per genotype (or eight per mutant line) were selected for additional analysis. After initial analysis revealed similar global patterns of gene expression for the two mutant lines, individual data sets were collapsed across the lines to increase statistical power and specificity of mutation-driven transcriptional changes. Before doing so, data were subjected to a standardization procedure to reduce print tip-related variability. This procedure preserves numeric differences between knock-out and wild-type animals within each line and reduced variability introduced by different printing parameters. Finally, we removed statistical outliers from a combined data set by excluding values deviating from the genotype mean by >2 SDs.
Statistical methods
For a combined data set, each transcript was represented by 8–17 subjects per genotype per brain region. For each transcript element on the array, a two-tailed t test for independent samples (MS Excel 2002; Microsoft, Seattle, WA) was used to compare gene expression values between knock-out and wild-type groups. This analysis was followed by a false discovery rate analysis to control for multiple testing. We used a free-access Q-value software (Storey, 2002) to calculate a Q value for each transcript. Default program parameters (λ ranged from 0 to 0.95; smoother p0 method) were used.
Gene expression databases
Two public gene expression databases that characterize transcriptional fingerprints of individual cell types have been used to identify cellular expression of α1 mutation-associated genes. The Mouse Neuronal Expression Database (MNED; http://mouse.bio.brandeis.edu) describes transcriptomes of 12 neuronal populations obtained from the cerebral cortex, amygdala, hippocampus, and thalamus (Sugino et al., 2006). The Allen Brain Atlas (ABA; http://www.brain-map.org), a project of Allen Institute for Brain Science, uses nonisotopic in situ hybridization and high-resolution imaging to map mRNA expression of known mouse genes. MNED and ABA were used for genes detected in the cortex and cerebellum, respectively.
MNED data (Sugino et al., 2006) were downloaded from National Center for Biotechnology Information GEO database (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE2882). Genes detected in cortex from the present study were identified in the database by gene symbols, and their values were compared across different cortical neuronal types by a two-tail t test. If multiple probe sets were found for a single gene, sequences were “BLAST”ed, and probe sets with higher specificity score were selected for additional analysis. For functional group analysis (Table 1 and supplemental Table 4, available at www.jneurosci.org as supplemental material), genes for glutamate- and GABA-enriched groups were selected based on comparisons between the two neuronal classes (t test; p < 0.05).
Table 1.
A subset of over-represented functional groups and pathways known to play a role in neuronal plasticity (all over-represented groups are listed in supplemental Table 4, available at www.jneurosci.org as supplemental material). Six brain populations were examined: genes regulated in cortex (Whole Cortex) and cerebellum (Whole Cerebellum) and also enriched in four neuronal populations: cortical glutamate neurons (Glutamate enriched), cortical GABA neurons (GABA enriched), cerebellar Purkinje neurons (Purkinje enriched), and cerebellar granule neurons (Granule enriched). Color indicates a significance level from the over-representation analysis. Empty cells indicate the absence of over-represented groups from a particular functional domain in a particular brain population. The over-represented groups shown include both upregulated and downregulated transcripts (distributions not shown).
| Functional Domains | Whole Cortex | Glutamate enriched | GABA enriched | Whole Cerebellum | Granule enriched | Purkinje enriched |
|---|---|---|---|---|---|---|
| Synaptic transmission | synapse | axon | synapse | glutamate receptor activity | neuropeptide signaling pathway | |
| synaptic vesicle | synaptic vesicle | neuropeptide signaling pathway | ||||
| Protein trafficking & metabolism | SNARE binding | SNARE binding | clathrin-coated vesicle | protein autoprocessing | protein ubiquitination | |
| clathrin-coated vesicle | protein metabolism | peroxisome | ||||
| peptidase activity | protein binding | |||||
| Signal transduction | signal transduction | MAPKKK cascade | calmodulin binding | calmodulin binding | MAPK signaling pathway | |
| calmodulin binding | second-messenger signaling | second-messenger signaling | second-messenger signaling | |||
| protein kinase activity | protein kinase activity | basal transcription factors | ||||
| Cytoskeleton- dependent processes | Rac 1 cell motility signaling pathway | axon cargo transport | actin binding | actin binding | actin binding | |
| cell motility | regulation of actin cytoskeleton | cellular morphogenesis | cell motility | |||
| Rac 1 cell motility signaling pathway | ||||||
| Ion channels & ion homeostasis | ion homeostasis | voltage-gated potassium channel activity | calcium ion transport | ligand-gated ion channel activity | ||
| magnesium ion binding | calcium channel activity | |||||
| calcium ion transport | ||||||
| Other | response to stress | cell adhesion | ||||
| citrate cycle | gap junction | |||||
| citrate cycle |
Genes were identified by gene symbols from ABA. Images of sagittal brain slices corresponding to lateral distances 0.60–2.52 mm (Paxinos and Franklin, 2001) were used for analysis. The ABA gene expression filter was used to quantify expression of each gene from 0 to 4. A corresponding chart of ABA filter and our quantification method (weighted expression) is shown in supplemental Figure 5 (available at www.jneurosci.org as supplemental material). Expression of each gene was averaged across two to three images with best quality. For functional group analysis (Table 1 and supplemental Table 4, available at www.jneurosci.org as supplemental material), genes for Purkinje- and granule cell-enriched groups were selected such that expression values of these genes in their corresponding cell group exceed expression in other parts of the cerebellar cortex by one. For preferential expression in Purkinje and granule cells (see Fig. 4), genes were selected such that expression values of these genes in their corresponding cell group exceed expression in other parts of the cerebellar cortex by two.
Figure 4.
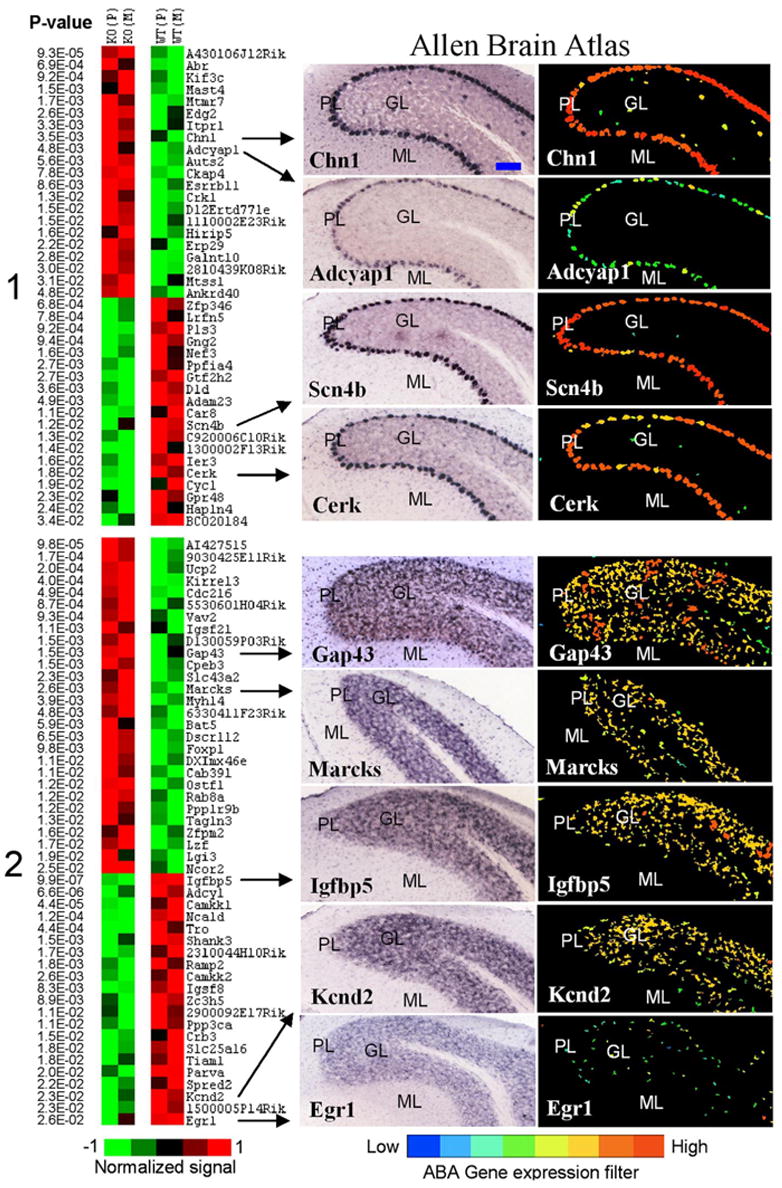
Cerebellum-specific genes with Purkinje and granule patterns of expression. Left, Differential expression between knock-out (KO) and wild-type (WT) lines is shown in pseudocolor with associated p values from a t test. Average values for Pittsburgh (P) and Merck (M) lines are shown for Purkinje-enriched (cluster 1) and granule-enriched (cluster 2) genes. Genes that show a distinct pattern of expression in Golgi cells of granule layer (for example, see Fig. 1) were excluded from cluster 2. Right, ABA images show regional distribution of nine selected transcripts. ML, Molecular layer; PL, Purkinje cell layer; GL, granule layer; DCN, deep cerebellar nuclei. Several genes with known patterns of expression in cerebellar tissue were present in their corresponding clusters: for example, IP3R (Itpr1) and Scn4b in Purkinje cells, and Igfbp5 and Kcnd2 in granule neurons. Most transcripts were also expressed to some extent in the DCN. The blue bar in the top left image represents ~100 μm. All images are copied and modified with permission from Allen Institute for Brain Science.
Other bioinformatics resources
Transcripts that passed a statistical threshold and had an assigned gene symbol were annotated into functional groups using a “WEB-based GEne SeT AnaLysis Toolkit” database, WebGestalt (http://genereg.ornl.gov/webgestalt). Functional annotations based on Gene Ontology Consortium (http://www.geneontology.org), BioCarta pathways (http://www.biocarta.com/genes/index.asp), and Kyoto Encyclopedia of Genes and Genomes database (http://www.genome.jp/kegg) were performed. The WebGestalt database was then used for functional group over-representation analysis. This software uses all genes detected on microarray as control and a hypergeometric test to estimate whether a biological function/pathway is perturbed (i.e., represented by more significantly regulated genes than expected by chance). Identification of over-represented (enriched) functional groups provides an additional level of statistical validity that mitigates effects of high false-positive rates for individual genes. An enrichment p value generated by a hypergeometric test is assigned to each group.
To outline a set of potential downstream targets of the transcription factor Egr1, we selected a subset of genes coregulated with Egr1 in the cortex and used promoter analysis in conjunction with three gene expression databases. First, data from MNED was used to calculate Pearson Product Moment correlations between Egr1 and other cortical transcripts using individual values from the 12 neuronal populations (total, n = 12 × 3 = 36). Genes identified by this method were further filtered by promoter analysis using MATCH public version 1.0 (http://www.gene-regulation.com/pub/programs.html#match) to find transcription factor binding sites for Egr1 in the promoter regions (1 kb upstream from transcription initiation site) of genes coregulated with Egr1 in the cortex. MATCH uses a library of mononucleotide weight matrices from TRANS-FAC 6.0. We used the Alcohol Research Integrator databases maintained locally by Dr. Susan Bergeson at the University of Texas at Austin (https://trip.icmb.utexas.edu/cgi-bin/genedb.pl). These databases contain cDNA-based microarray data for DBA/2J and C57BL/6J mouse strains after different treatments with ethanol. Administration of ethanol vapor for 72 h to DBA/2J mice or an acute injection of 4 g/kg in C57BL/6J animals result in consistent downregulation of the Egr1 transcript at 6–7 and 24 h after ethanol treatment. We identified those cortical transcripts that showed similar patterns of expression. Finally, GeneNetwork databases (http://www.genenetwork.org) were used to obtain genetic correlations between transcripts regulated in the cortex of α1 mutants and the Egr1 transcript. Brain transcript abundance values from 32 BXD RI mouse strains and their parental inbred strains [C57BL/6J (B) and DBA/2J (D)] were used. These measurements of mRNA expression were obtained in naive mice using the Affymetrix U74Av2 and M430 microarrays (Chesler et al., 2004, 2005) for a forebrain region (whole brain without cerebellum). Affy ID# 98579_at probe-set was used to obtain transcript abundance values of Egr1 [UTHSC Brain mRNA U74Av2 (Dec03) PDNN database].
Western blot analysis
The cerebral cortex was dissected from knockout and wild-type animals of the Merck line and homogenized in 1 ml of immunoprecipitation buffer (150 mM sodium chloride, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris, pH 8.0) containing protease and phosphatase inhibitors (Santa Cruz Biotechnology, Santa Cruz, CA) using a Polytron homogenizer at 4°C. The homogenate was incubated on ice for 30 min and transferred to microcentrifuge tubes and centrifuged at 10,000 × g for 10 min at 4°C. The supernatant was removed and centrifuged two additional times. The clear lysate was used for Western blots and protein estimation (DC kit; Bio-Rad, Hercules, CA). To ensure that the amount of protein used for Western blot analysis was in the linear range of detection, a standard curve was generated for each protein using 10–100 μg of protein. The accuracy of protein loadings was assured by measuring the amount of protein in each sample and the linearity of standard curves for Egr1 and neurogranin (regression, R2 >0.9). Based on this standard curve, 20 μg (for neurogranin), 50 μg, and 100 μg (for Egr1) of protein were used for Western blot analysis. The proteins were separated on precast 4–15% SDS polyacrylamide gels (Bio-Rad) and transferred onto nitrocellulose membranes. The membranes were probed with the polyclonal Egr1 antibody (1:250; Santa Cruz Bio-technology) or polyclonal neurogranin antibody (1:500; Chemicon, Temecula, CA) and appropriate secondary antibodies. Signal was detected using the Kodak gel documentation system after incubating the membranes with Western Lightning chemiluminescence kit (PerkinElmer, Boston, MA) as per the manufacturer instructions. Band densities (net band intensity) were quantified using the Kodak gel documentation system. The density values for wild type were averaged for each gel, and all values were expressed as percentage of wild-type averages for each gel. Genotypes were then compared using a two-tailed t test for independent samples using percentage change as the dependent variable.
Immunohistochemistry
Immunohistochemistry was performed according to previously published protocols (Ryabinin and Wang, 1998; Bachtell et al., 1999). Briefly, mouse brains from the Merck line were dissected and fixed overnight, cryoprotected, and sectioned into 40 μm coronal slices. Immunohistochemical analysis was performed on every seventh section. Blocking was performed with 3% goat serum. Egr1 antibodies were purchased from Santa Cruz Biotechnology (1:10,000 dilution). Immunoreaction was detected with avidin–biotin–peroxidase complex (Vectastain ABC kit; Vector Laboratories, Burlingame, CA), and enzymatic development was performed with 3,3′-diaminobenzidine (Metal Enhanced DAB kit; Pierce, Rockford, IL).
Quantitative real-time PCR
A standard protocol from Applied Biosystems (Foster City, CA), p/n 4333458, was used for reverse transcription (RT)-PCR. TaqMan Universal PCR master mix, No AmpErase UNG (2X) and 20X Assays-on-Demand Gene Expression Assay MIX, TaqMan MGB probe FAM dye-labeled and unlabeled PCR primers were used. The quantity of RNA was calculated by using the standard curve method. Glyceraldehyde-3-phosphate dehydrogenase was used as control (assay identification: Mm99999915_g1). Assay identifications for the validated genes are listed in supplemental Table 5 (available at www.jneurosci.org as supplemental material). Significance was assessed by a one-tailed t test.
Electrophysiology
Midbrain slices were prepared from wild-type and GABAA α1 knock-out mice of the Merck line as described previously (Morikawa et al., 2003). Briefly, mice were deeply anesthetized with halothane and killed. The brain was rapidly removed into ice-cold artificial CSF (aCSF) containing the following (in mM): 126 NaCl, 2.5 KCl, 2.4 CaCl2, 1.2 MgCl2, 1.2 NaH2PO4, 21.4 NaHCO3, 11.1 glucose bubbled with 95% O2/5% CO2. Horizontal slices (210 μm) of the ventral midbrain were cut with a vibratome (Leica, Nussloch, Germany). Slices were allowed to recover in 34°C aCSF for at least 1 h before recording. Slices were transferred to the stage of an upright microscope (BX51WI; Olympus, Melville, NY) equipped with infrared-differential interference contrast optics and continuously perfused (4 ml/min) with bubbled aCSF at 34°C. Dopamine neurons were identified by their large, oblong soma and by their 1–4 Hz pacemaker firing (monitored in cell-attached mode). Recordings were made using a Multiclamp 700B amplifier (Molecular Devices). Data were filtered at 1 kHz, digitized at 2 kHz, and collected using Axograph 4.9 (Molecular Devices). Neurons were voltage clamped at −65 mV, and spontaneous miniature GABAA IPSCs (mIPSCs) were recorded in the presence of 10 μM DNQX, 1 μM strychnine, and 200 nM TTX. Patch pipettes (1.4–1.7 MΩ) were filled with an internal solution containing the following (in mM): 125 CsCl, 1 MgCl2, 5 EGTA, 10 HEPES, 2 Mg-ATP, 0.2 Na-GTP, 10 phosphocreatine, pH 7.25 (280 mOsm). Series resistance (typically 5–9 MΩ) was left uncompensated. mIPSCs were detected with a sliding template algorithm using Axograph. At least 200 mIPSCs were collected from each neuron.
Results
Initial comparison of the two independently created mutant lines revealed striking similarities in global gene expression (see supplemental note, available at www.jneurosci.org as supplemental material), indicating the involvement of developmentally conserved downstream mechanisms after the deletion of α1 subunit gene. In view of these results, we combined data sets from the two mutant lines for additional analysis. Initial analysis of the combined data set identified 982 differentially expressed transcripts: 688 and 326 for the cerebellum and cortex, respectively (supplemental Fig. 1, supplemental Tables 2, 3, available at www.jneurosci.org as supplemental material). The majority of differences were unique for each brain region with only a few common transcripts for both the cortex and cerebellum. Our first goal was to select transcripts implicated in neuronal plasticity by previous studies. A publication analysis using the PubMed search engine and the commercially available PathwayAssist program (version 3.0; Stratagene, La Jolla, CA) revealed several individual genes and pathways involved in regulation of synaptic transmission both presynaptically and postsynaptically (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). We then used public gene expression databases to search for cellular identity of these genes.
Using gene expression databases to ascribe transcriptional plasticity to individual cell types
We used a novel approach to classify transcripts regulated between mutant and wild-type mice into cellular categories. Two gene expression databases that characterize transcriptional fingerprints of individual cell types recently became available. We used the MNED (Sugino et al., 2006) and ABA to define cell type-specific expression of α1 mutation-associated genes in the cortex and cerebellum, respectively. Expression profiles of 217 cortex- and 425 cerebellum-specific genes were available in the databases. The validity of this approach was examined by comparing cellular distributions of previously characterized genes to data from MNED and ABA. Data from both data sets were consistent with the literature (see figures for details). This analysis revealed subsets of transcripts enriched in different types of neurons and glial cells, thus providing evidence for transcriptional plasticity in individual cell types. Genes enriched in one glial and four neuronal populations were then classified into functional groups and pathways, and the importance of these categories in cell functions was assessed by functional group over-representation analysis (Table 1 and supplemental Table 4, available at www.jneurosci.org as supplemental material). Functional groups with known or presumed roles in neuronal plasticity are shown in Table 1. Mostly different subsets of these functional groups in individual neuronal populations suggest that different cells use mostly different strategies to adapt to altered balance between inhibition and excitation.
Transcriptional changes in GABA and glutamate neurotransmission
Previous studies showed a compensatory upregulation of several α subunit proteins in the cortex and cerebellum of α1 mutants (Sur et al., 2001; Kralic et al., 2002a, 2006; Ogris et al., 2006). Therefore, our goal was to examine whether the deletion of the α1 subunit results in transcriptional regulation of other GABAA subunits. Nineteen transcripts representing 10 GABAA receptor subunits (α1, α2, α4, α6, β2, β3, γ1, γ2, ε, and δ) were present on the arrays used in our study. Results showed that none of the subunits were consistently changed (data not shown), thus confirming previous reports showing the lack of compensation at the mRNA level (Heinen et al., 2003; Ogris et al., 2006). Regulation of several genes and functional categories involved in protein trafficking and metabolism (Table 1) is consistent with several studies that suggested a posttranslational regulation of such compensation (Kralic et al., 2006; Ogris et al., 2006).
We also examined other transcripts associated with GABA and glutamate neurotransmission. Ten genes were detected in the cerebellum, and cellular distribution of these transcripts was identified using ABA and other published literature (Fig. 1). Three genes involved in metabolism of GABA and glutamate (Abat, Got2, and Glud1) were upregulated in mutants suggesting an increased transmitter turnover. Regulation of ionotropic AMPA3 glutamate receptor (Gria3), metabotropic receptors mGluR2 (Grm2), mGluR7 (Grm7), and mGluR8 (Grm8) as well as glutamate vesicular transporter VGLUT2 (Slc17a6) and serine racemase (Srr), an enzyme for a modulator of NMDA receptors, D-serine, suggests participation of glutamate neurotransmission in regulation of inhibition-excitation balance in cerebellum. In addition, collybistin (Arhgef9), a protein involved in GABAA receptor clustering through deposition of gephyrin (Luscher and Keller, 2004), was downregulated, implicating this process as a part of homeostatic mechanisms at the GABA synapse. A wide cellular distribution of expression of these transcripts suggests a diverse cellular response to altered inhibition, which may result in a reorganization of neuronal networks in the cerebellum, a finding supported by other studies (Kralic et al., 2006; Ogris et al., 2006).
Figure 1.
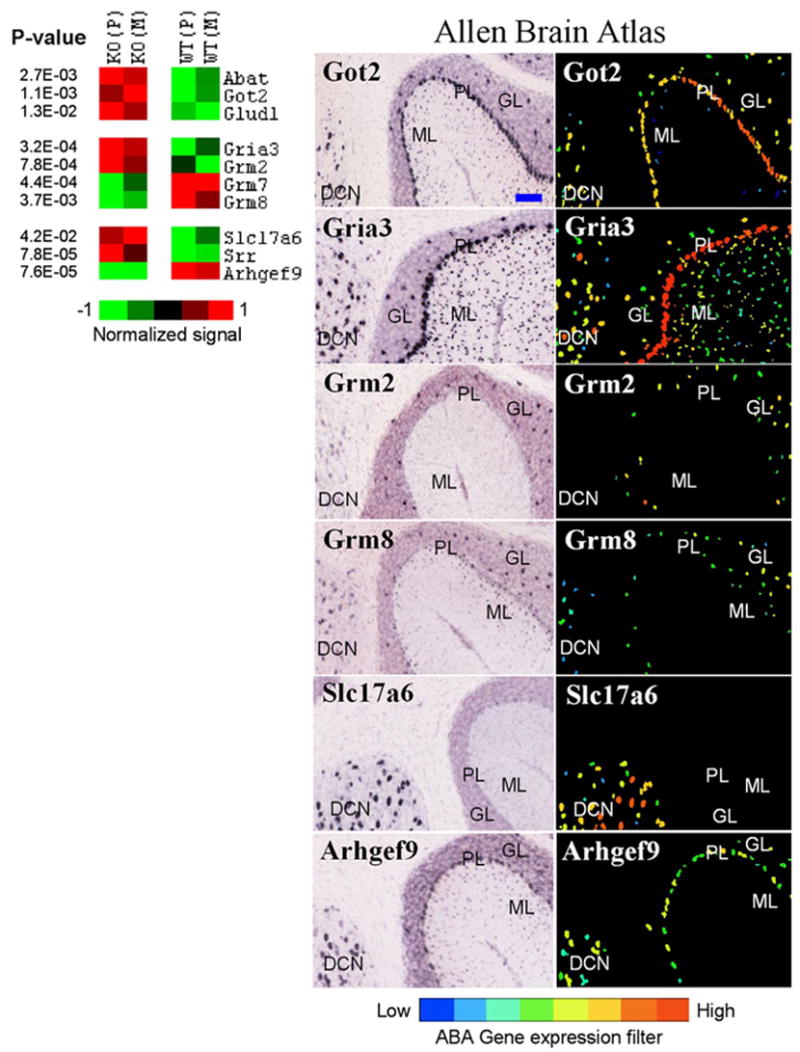
Genes differentially expressed in the cerebellum and involved in GABA and glutamate neurotransmission. Left, Differential expression between knock-out (KO) and wild-type (WT) lines is shown in pseudocolor with associated p values from a t test. Average values for Pittsburgh (P) and Merck (M) lines are shown. Right, ABA images show regional distribution of six selected transcripts. ML, Molecular layer; PL, Purkinje cell layer; GL, granule layer; DCN, deep cerebellar nuclei. Got2, Gria3, and Abat (not shown) are expressed in all four subregions examined. Grm2 is expressed in Golgi cells in the GL. Grm7 is predominantly expressed in Purkinje neurons, whereas Grm8 is detected in ML, DCN, and Golgi cells (Berthele et al., 1999). VGLUT2 (Slc17a6 ) was detected only in the DCN, and collybistin (Arhgef9) was present in PL, GL, and DCN. Glud1 and Srr are preferentially expressed in astrocytes (Zaganas et al., 2001; Ribeiro et al., 2002). The blue bar in the top left image represents ~100 μm. All images are copied and modified with permission from Allen Institute for Brain Science.
Disparate transcriptional profiles in cortical excitatory and inhibitory neurons
Expression of genes regulated in the cortex was examined across seven neuronal populations: three excitatory and four inhibitory. The four inhibitory populations in the cortex represent three subclasses of GABA interneurons: parvalbumin (Pvalb)-positive, somatostatin (Sst)-positive, and cholecystokinin (Cck)-positive (Sugino et al., 2006). From 217 genes that were matched between our data set and MNED, 145 were enriched in either all glutamate neurons or one of the three subclasses of GABA neurons (MNED; t test, p < 0.05). Interestingly, compared with the combined population of all GABA interneurons, the number of transcripts enriched in glutamate neurons greatly exceeded the chance level (supplemental Fig. 3, left panel, available at www.jneurosci.org as supplemental material; Fig. 2), suggesting that the loss of α1-mediated fast inhibition affected excitatory neurons to a larger degree than inhibitory neurons.
Figure 2.

Genes differentially expressed in cortex between knock-out (KO) and wild-type (WT) lines ( p < 0.01). Genes enriched in glutamate (cluster 1) and GABA (cluster 2) neurons (MNED; t test; p < 0.01). Cluster 3, Genes enriched in glia (detected by literature search and confirmed in MNED as overexpressed in whole tissue, compared with the 12 neuronal populations). Cluster 4, Genes found in MNED but not consistently different between glutamate and GABA neuronal classes (MNED; p > 0.01). Cluster 5, Genes not found in MNED. Several genes including Egr1 and neurogranin (Nrgn) were previously shown to be enriched in excitatory neurons (Chaudhuri et al., 1995; Singec et al., 2004).
Our MNED search also revealed that 80 genes showed a substantial heterogeneity in expression (difference of twofold and higher) across the seven cortical populations. A K-mean clustering identified eight different patterns of expression described in Figure 3. This analysis highlighted genes with cell-specific patterns of expression and suggested a foundation for neuronal plasticity in individual cell types. Results also suggested that although transcriptional responses in glutamate populations are mostly similar, different classes of inhibitory interneurons demonstrate substantial heterogeneity in cellular adaptation to α1 deletion. A quantitative assessment revealed that relative frequencies of observed changes were proportional to the abundance of α1 subunit; with over-representation of transcripts in higher expressing glutamate and Pvalb-positive GABA neurons and under-representation of transcripts in lower-expressing Sst-positive population (Fig. 3, bottom panel; supplemental Fig. 3, right panel, available at www.jneurosci.org as supplemental material).
Figure 3.

Left, Cortex-specific genes differentially expressed between knock-out (KO) and wild-type (WT) (t test; p values are shown on the left), which also show heterogeneous expression across seven types of cortical neurons (≥2-fold difference). Right, Corresponding data from MNED (Sugino et al., 2006). Fold change refers to the difference between lowest (black) and highest (bright yellow) expressing neurons. These genes are clustered according to their differential expression among three excitatory (Glut) and four inhibitory (GABA) neuronal types (for detailed description of neuronal types, see Sugino et al., 2006). The first four clusters represent genes enriched in all Glut populations but with different expression in GABA neurons. Clusters 5– 8 represent genes enriched in individual classes of GABA interneurons. Expression of three GABAA subunits across the seven neuronal populations is shown at the bottom.
Several genes that play a role in synaptic plasticity were enriched in either glutamate or GABA populations. For example, genes involved in presynaptic release machinery, such as Vamp1, Syn2, and Vat1 were enriched in different subclasses of GABA interneurons (Fig. 3), whereas Rac1, a small GTPase involved in regulation of actin cytoskeleton and formation of dendritic spines (Bolis et al., 2003; Tashiro and Yuste, 2004) was enriched in glutamate populations (Fig. 2). Other plasticity-related transcripts enriched in excitatory neurons included the transcription factor Egr1, neurogranin (Nrgn), prion protein (Prnp), and Cdk5.
Transcriptional signatures in cell populations of cerebellum
Cellular distribution of genes differentially expressed in the cerebellum was examined in four subregions: molecular layer, Purkinje cell layer, granule layer, and deep cerebellar nuclei. Cell populations and neuronal networks in the cerebellum are well characterized (Wang and Zoghbi, 2001; Carulli et al., 2004), and several patterns of cellular distribution could be identified. GABA-containing Purkinje neurons, the only output from the cerebellar cortex, were identified as large cells located in the Purkinje layer at the border between the molecular and granule layers. Bergman glia, astrocytes located in the Purkinje layer, can be identified as a signal scattered at the base of and between Purkinje neurons. Golgi inhibitory interneurons are relatively large cells dispersed in the granule layer (for example, see Grm2 in Fig. 1) and provide feedback inhibition to excitatory granule cells. Granule neurons are small cells that are located in the granule layer and constitute the most abundant neuronal population in the cerebellar cortex. They were identified by a relatively dense distribution of signal in the granule layer (for example, see expression of GABAA α6 subunit in supplemental Fig. 4, available at www.jneurosci.org as supplemental material). The ABA gene expression filter was used to quantify the relative abundance of 425 transcripts (supplemental Fig. 5, available at www.jneurosci.org as supplemental material).
This analysis highlighted three patterns with relatively confined cellular distributions: (1) preferential expression in Purkinje neurons, (2) preferential expression in granule cells, and (3) enrichment in Bergman astrocytes and white matter glia. Transcripts that follow these patterns are shown in Figures 4 and 5. Several genes with known or presumed roles in neuronal plasticity were mapped to either Purkinje or granule neurons. For example, the sodium channel β4 subunit (Scn4b) is, in part, responsible for high-frequency firing of Purkinje neurons (Grieco et al., 2005). Because α1 subunit-containing GABAA receptors mediate majority of inhibition in Purkinje cells, the loss of this subunit drastically decreases GABA currents, which, surprisingly, did not affect firing frequency in null mutants (Kralic et al., 2005). A downregulation of Scn4b in knock-out mice may represent a homeostatic mechanism directed to restore firing in response to altered inhibition. Another example is regulation of Kcnd2, a potassium voltage-gated Kv4.2 channel prominent in the repolarization phase of the action potential in granule cells (Shibasaki et al., 2004; Strassle et al., 2005). Deletion of the α1 subunit resulted in a compensatory upregulation of other α subunits (Kralic et al., 2006; Ogris et al., 2006) and longer-lasting inhibitory currents in these neurons (Vicini et al., 2001; Ortinski et al., 2004). Downregulation of Kcnd2 may shorten the refractory period between action potentials and compensate for the increased inhibitory tone by increasing excitability of granule cells.
Figure 5.

Genes regulated in the cerebellum and detected in Bergman glia (BG) (clusters 1, 2) or white matter (clusters 3). Left, Differential expression between knock-out (KO) and wild-type (WT) lines is shown in pseudocolor with associated p values from a t test. Average values for Pittsburgh (P) and Merck (M) lines are shown. Right, ABA images show regional distribution of three selected transcripts. ML, Molecular layer; PL, Purkinje cell layer; GL, granule layer; WM, white matter. Genes from cluster 1 were predominantly expressed in BG, whereas cluster 2 and 3 genes were also detected in other cell populations. Several genes including Clud1, Srr, S100a1, Mobp, and Mbp are known glial markers. Asterisks show the position of Purkinje neurons. Arrows point to detectable signal. The blue bar in the top left image represents ~100 μm. All images are copied and modified with permission from Allen Institute for Brain Science.
Several transcripts were abundantly expressed in glia (Fig. 5). Interestingly, all transcripts detected in Bergman glia were up-regulated in knock-out mice, suggesting an activation of cerebellar astrocytes. Previous reports indicated that Bergman cells can detect and respond to glutamate signal from climbing and parallel fibers by regulating intracellular calcium (Bezzi and Volterra, 2001; Beierlein and Regehr, 2005). Upregulation of glutamate dehydrogenase 1 (Glud1) and serine racemase (Srr) suggests an active role of these cells in adaptation of neuronal networks to altered inhibition in general and in modulation of glutamate transmission in particular.
The immediate early gene, Egr1, is implicated in neuronal plasticity
Transcription factors regulate expression of other genes in response to specific stimuli. Of particular interest was the inducible transcription factor early growth response 1 (Egr1 also known as Zif268, Krox-24, and NGFI-A). The Egr1 transcript, enriched in excitatory neurons of the cortex and cerebellum, was downregulated in both of these tissues of knock-out mice (Figs. 2, 4). Egr1 has been widely used as a marker of neuronal activity (Knapska and Kaczmarek, 2004; Simon et al., 2004) and has been implicated in some forms of synaptic plasticity, associative learning, and development (Jones et al., 2001; Patra et al., 2004). These observations suggest that Egr1 can stabilize neuronal activity in response to destabilizing inhibition in α1 mutants by regulation of genes involved in activity-dependent plasticity.
To identify potential neuronal targets of Egr1, we used data from the MNED as well as other public databases. Our assumption was that genes effectively regulated by Egr1 should have correlated patterns of expression across different cell types. Data from MNED were used to calculate Pearson Product Moment correlations between Egr1 and other cortical transcripts using individual values from the 12 neuronal populations. Genes identified by this method were further filtered by selecting genes with at least one putative Egr1 binding site in their promoter region (1 kb). Genes shown in supplemental Figure 6 (available at www.jneurosci.org as supplemental material) are potential Egr1 targets, which may mediate its effects on neuronal activity. Some of them, such as Junb, Nrgn, Prnp, Rac1, and Syn2, have been implicated in neuronal plasticity by previous studies (Comb et al., 1992; Huang et al., 2004; Tashiro and Yuste, 2004; Papassotiropoulos et al., 2005; Sun et al., 2006). We further characterized one of the genes with expression highly correlated with Egr1, neurogranin (Nrgn). Neurogranin, a calcium/calmodulin-regulated protein is expressed postsynaptically and has been implicated in LTP induction and dendritic maintenance (Huang et al., 2004). Western blot analysis of Egr1 and Nrgn and an immunohistochemical analysis of Egr1 in cortical tissue corroborated our microarray findings (Fig. 6A, B; supplemental Fig. 7, available at www.jneurosci.org as supplemental material). Expression data from ABA also support correlated expression of these two genes (Fig. 6C). Egr1 can have both inducing and suppressing effects on its downstream targets. The latter can be mediated by competing with the structurally similar transcription factor Sp1 for binding sites (Zhang and Liu, 2003). A neurogranin promoter analysis revealed at least one sequence containing overlapping binding sites for Egr1 and Sp1 (data not shown), indicating that downregulation of Egr1 can result in upregulation of neurogranin. These observations are supported by a study that suggested neurogranin as a downstream target of Egr1 (Svaren et al., 2000) and another that reported an Sp1 binding site in promoter region of neurogranin (Sato et al., 1995). Expression of several transcripts enriched in different neuronal populations, including Bet1l, a potential target of Egr1, was validated with RT-PCR (supplemental Fig. 5, available at www.jneurosci.org as supplemental material).
Figure 6.
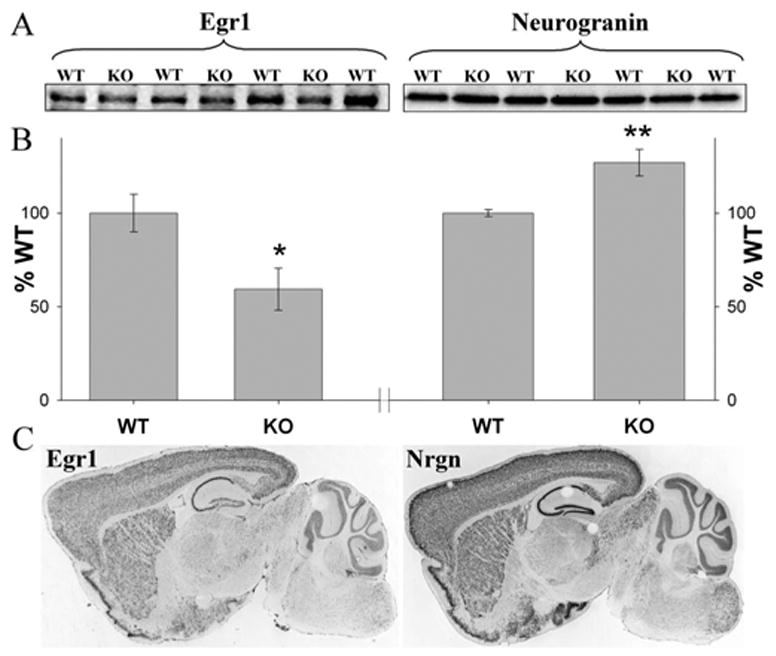
Validation of microarray results. A, B, Western blot results for two proteins from cortical tissue: Egr1 and neurogranin (Nrgn). A, Representative gels are shown. B, Comparison of knock-out (KO) and wild-type (WT) mice (*p < 0.05; **p < 0.01). C, ABA images showing similar expression patterns (mRNA) of the two genes in the brain.
Duration of inhibition: a driving force for neuronal plasticity
We also asked whether the deletion of the α1 subunit affected neurons in which this subunit is not expressed or expressed with low abundance. One study reported the detection of GABAA α1 subunit protein in dopamine neurons in humans (Petri et al., 2002), whereas two studies using single-cell RT-PCR in rodents failed to confirm this finding (Guyon et al., 1999; Okada et al., 2004), suggesting a minimal, if any, expression of α1 subunit in these cells. We investigated GABA neurotransmission in dopamine neurons using patch-clamp electrophysiology in acute slices. The decay time was longer and the frequency of GABA-mediated mIPSCs was lower in GABAA α1 knock-out mice compared with wild-type animals with no difference in mIPSC amplitude between mutant and control mice (Fig. 7). A small but consistent increase in duration of GABA currents in mutant mice suggests two possibilities. First, dopamine neurons may express α1 subunit in low abundance and lose a portion of fast inhibition with its deletion. Second, dopamine neurons may not express α1 subunit but regulate other α subunits to increase the proportion of GABAA receptors containing other α subunits in an activity-dependent manner. Partial support for the first possibility is provided by one study showing that even sparsely expressed α1 subunit affects kinetics of GABAA receptors (Bosman et al., 2005b). Another report supports the second possibility, showing an up-regulation of other α subunits in some cell types of α1 mutants, which do not normally express the α1 subunit and suggesting that neuroadaptation does not simply replace the α1 subunit but leads to reorganization of neuronal circuits (Kralic et al., 2006). In addition, in agreement with other reports (Goldstein et al., 2002; Ortinski et al., 2004), our results support the hypothesis that prolonged inhibitory currents in GABAA α1 knock-out mice is offset by a decrease in GABA release probability.
Figure 7.
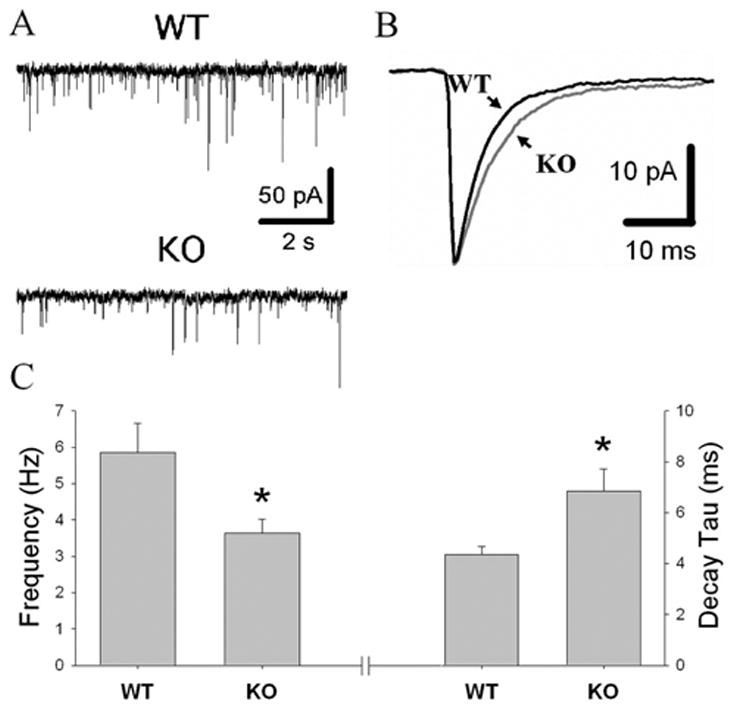
Electrophysiological results. A, Representative traces of GABAA mIPSCs in knock-out (KO) and wild-type (WT) mice. B, Average mIPSCs from representative WT and KO neurons. GABAA α1 KO mice exhibited mIPSCs with longer decay times (C, right) and a reduction in GABA release as measured by the frequency of mIPSCs (C, left) in midbrain DA neurons (n = 4 –5 for each genotype; *p < 0.05). Average mIPSC amplitude was similar for the KO and WT mice (not shown).
Patterns of neuronal activity play a major role in driving molecular changes underlying neuronal plasticity (Turrigiano, 1999; Ozaki, 2002; Turrigiano and Nelson, 2004). Therefore, it is of interest to determine whether patterns of neuronal activity control patterns of gene expression. As an initial step, we compared transcriptional changes associated with longer inhibition in α1 knock-out mice with published data on gene expression in the cortex after treatment with a high dose of diazepam (Huopaniemi et al., 2004). Diazepam is a benzodiazepine that binds to the GABAA/benzodiazepine receptor complex and increases the duration of inhibitory currents (Cohen et al., 2000; Cagetti et al., 2003), an effect similar to that produced by α1 gene deletion. Of 54 diazepam-regulated transcripts listed in Table 1 in the study by Huopaniemi et al. (2004), 18 were present on our array and detected in the cortex. From these 18 genes, five were differentially expressed, and six others showed a strong tendency to be differentially regulated in the mutant mice, with all 11 genes regulated in the same direction as diazepam-induced changes. The five significantly regulated genes were Egr1, Col4a1, Hivep1, Ier2, and Anp32e. This level of coincidence far exceeds a chance level (χ2; p < 0.01), which indicates that both pharmacological and genetic models of prolonged GABA inhibition are associated with some common transcriptional profiles, in part, driven by changes of the Egr1 transcription factor.
Discussion
Mutant mice lacking GABAA receptor subunits provide an opportunity to define molecular and neuronal plasticity produced by perturbation of this key inhibitory system. In particular, GABAA receptor α1 subunit null mutants are viable, fertile, and show only mild overt phenotypic alterations, despite a lack of α1-mediated fast inhibitory currents and a marked reduction in density of GABAA receptors. This suggests brain adaptation to altered inhibition. The present study used multiple approaches to detect genes and functional groups involved in regulation of mutation-driven downstream processes, providing a molecular basis for neuronal plasticity.
A novel aspect of our study is the use of public gene expression databases to define transcriptional patterns of neuronal plasticity with cellular resolution. This integrative strategy combined the sensitivity of whole-tissue microarrays with the specificity of single-cell transcriptomes. One basic assumption of this approach is that genes expressed at high levels in individual cell populations are important for cell functions and, therefore, are likely to also be important for cellular adaptation to external stimuli. Although this strategy does not assure cellular identity of plasticity changes, it allows for detection of patterns of gene expression changes that are characteristic of specific cellular populations. Using this approach, we were able to identify subsets of transcripts enriched in different neuronal and glial cell populations. Our results suggest that the transcriptional response to the loss of α1-mediated fast inhibition varies among different cell types and depends on several factors, including the abundance of the α1 subunit, cell-type specific transcriptomes, and signals from other neurons and glial cells. The high degree of heterogeneity of gene expression across neuronal cell types (Sugino et al., 2006) suggests that different cells may require different strategies to adapt to homeostatic perturbations. Functional group analysis supported this notion. Although major functional domains important for neuronal plasticity were represented to a similar degree across all neuronal types studied, specific pathways and components of these domains were different in different cell populations (Table 1).
Several transcriptional changes were consistent with expression of cellular phenotypes identified by previous studies of α1 null mutants. For example, downregulation of the transcription factor Egr1 was paralleled by decreased cortical activity (Bosman et al., 2005a), and a downregulation of Rac1 cell motility pathway (Tashiro and Yuste, 2004) was correlated with the reduction of mature mushroom-shaped spines (Heinen et al., 2003; Tashiro and Yuste, 2004). Regulation of several genes involved in protein trafficking and metabolism is also consistent with several reports that suggested the involvement of these processes in compensatory upregulation of other α subunit proteins in α1 knock-outs (Kralic et al., 2006; Ogris et al., 2006). Furthermore, transcriptional changes of Scn4b and Kcnd2 genes in Purkinje and granule cells, respectively, agree with compensatory regulation of neuronal excitability in these neurons (Shibasaki et al., 2004; Grieco et al., 2005). In addition, identification of cell type-specific genes not previously associated with synaptic plasticity provides a foundation for generating new hypotheses.
Overall, our molecular and electrophysiological data suggest that a shift in balance between inhibition and excitation produces diverse transcriptional responses that act to regulate neuronal excitability of individual neurons and stabilize neuronal networks, which may account for the lack of severe abnormalities in α1 null mutants. Other neurotransmitter systems, such as glutamate and dopamine, appear to be involved in such compensation (Kralic et al., 2003; Reynolds et al., 2003). Furthermore, transcriptional activation in Bergman astrocytes suggests an active role of glial cells in synaptic plasticity. These results support and extend previous findings implicating glia in modulation of neuronal circuits (Bezzi and Volterra, 2001; Beierlein and Regehr, 2005). It appears that the functional reorganization of neuronal networks does not restore homeostasis at its normal level. For example, at the behavioral level, the null mutants show an increased sensitivity to drug-induced motor activity. Compared with wild-type mice, mutant mice demonstrate a marked locomotor stimulation induced by small doses of ethanol (Blednov et al., 2003; Kralic et al., 2003), pronounced diazepam-induced sedation (Kralic et al., 2002b; Reynolds et al., 2003), and increased sensitivity to stereotypies induced by cocaine and amphetamine (Reynolds et al., 2003).
One important asset of our study is the use of two independently created knock-out lines. Our goal was to detect genes and pathways that are specifically affected by a deletion of GABAA α1 gene, regardless of the differences in genetic background, the method of producing the deletion or environmental effects. Integrating data from two knock-out lines allowed us to detect robust mutation-driven transcriptional changes present in both lines. Caution, however, should be exercised when considering results from genes located close to the α1 subunit gene on mouse chromosome 11. These genes are in linkage disequilibrium with the α1 gene and may differ between the mutant and wild-type genotypes because of possible differences between 129 and C57BL/6J background strains. Nevertheless, the increased statistical sensitivity of large sample sizes is advantageous for over-representation analysis that could now detect relevant functional groups even based on small transcriptional changes.
In conclusion, we propose that prolonged inhibition resulting from α1 subunit deficiency is offset by a shift in balance between excitation and inhibition. This adaptation results from a cascade of transcriptional changes which are distinct for excitatory and inhibitory neurons and act to restore neuronal firing within physiologically relevant limits. Transcription factors, such as Egr1, which controls plasticity-related secondary transcripts, may play an important role in regulation of neuronal activity. Latent effects of the adjustments achieved through transcriptional regulation may be revealed as altered sensitivity to drug-induced motor behaviors in null mutant mice.
Supplementary Material
Footnotes
This work was supported by National Institute on Alcohol Abuse and Alcoholism INIA Program Grants AA13520 and AA13518 and National Institutes of Health Grants AA06399 and AA10422. We thank Dr. Robert Williams for his valuable comments on initial drafts of this manuscript and Julie Owen, Carolyn Ferguson, and Ronjon Datta for their technical assistance.
References
- Bachtell RK, Wang YM, Freeman P, Risinger FO, Ryabinin AE. Alcohol drinking produces brain region-selective changes in expression of inducible transcription factors. Brain Res. 1999;847:157–165. doi: 10.1016/s0006-8993(99)02019-3. [DOI] [PubMed] [Google Scholar]
- Beierlein M, Regehr WG. Conventional synapses for unconventional cells. Neuron. 2005;46:694–696. doi: 10.1016/j.neuron.2005.05.017. [DOI] [PubMed] [Google Scholar]
- Berthele A, Platzer S, Laurie DJ, Weis S, Sommer B, Zieglgansberger W, Conrad B, Tolle TR. Expression of metabotropic glutamate receptor subtype mRNA (mGluR1–8) in human cerebellum. NeuroReport. 1999;10:3861–3867. doi: 10.1097/00001756-199912160-00026. [DOI] [PubMed] [Google Scholar]
- Bezzi P, Volterra A. A neuronglia signalling network in the active brain. Curr Opin Neurobiol. 2001;11:387–394. doi: 10.1016/s0959-4388(00)00223-3. [DOI] [PubMed] [Google Scholar]
- Blednov YA, Walker D, Alva H, Creech K, Findlay G, Harris RA. GABAA receptor alpha 1 and beta 2 subunit null mutant mice: behavioral responses to ethanol. J Pharmacol Exp Ther. 2003;305:854–863. doi: 10.1124/jpet.103.049478. [DOI] [PubMed] [Google Scholar]
- Boehm SL, Jr, Ponomarev I, Jennings AW, Whiting PJ, Rosahl TW, Garrett EM, Blednov YA, Harris RA. Gamma-aminobutyric acid A receptor subunit mutant mice: new perspectives on alcohol actions. Biochem Pharmacol. 2004;68:1581–1602. doi: 10.1016/j.bcp.2004.07.023. [DOI] [PubMed] [Google Scholar]
- Bolis A, Corbetta S, Cioce A, de Curtis I. Differential distribution of Rac1 and Rac3 GTPases in the developing mouse brain: implications for a role of Rac3 in Purkinje cell differentiation. Eur J Neurosci. 2003;18:2417–2424. doi: 10.1046/j.1460-9568.2003.02938.x. [DOI] [PubMed] [Google Scholar]
- Bosman LW, Rosahl TW, Brussaard AB. Neonatal development of the rat visual cortex: synaptic function of GABAA receptor alpha subunits. J Physiol (Lond) 2002;545:169–181. doi: 10.1113/jphysiol.2002.026534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosman L, Lodder JC, van Ooyen A, Brussaard AB. Role of synaptic inhibition in spatiotemporal patterning of cortical activity. Prog Brain Res. 2005a;147:201–204. doi: 10.1016/S0079-6123(04)47015-0. [DOI] [PubMed] [Google Scholar]
- Bosman LW, Heinen K, Spijker S, Brussaard AB. Mice lacking the major adult GABAA receptor subtype have normal number of synapses, but retain juvenile IPSC kinetics until adulthood. J Neurophysiol. 2005b;94:338–346. doi: 10.1152/jn.00084.2005. [DOI] [PubMed] [Google Scholar]
- Cagetti E, Liang J, Spigelman I, Olsen RW. Withdrawal from chronic intermittent ethanol treatment changes subunit composition, reduces synaptic function, and decreases behavioral responses to positive allosteric modulators of GABAA receptors. Mol Pharmacol. 2003;63:53–64. doi: 10.1124/mol.63.1.53. [DOI] [PubMed] [Google Scholar]
- Carulli D, Buffo A, Strata P. Reparative mechanisms in the cerebellar cortex. Prog Neurobiol. 2004;72:373–398. doi: 10.1016/j.pneurobio.2004.03.007. [DOI] [PubMed] [Google Scholar]
- Chaudhuri A, Matsubara JA, Cynader MS. Neuronal activity in primate visual cortex assessed by immunostaining for the transcription factor Zif268. Vis Neurosci. 1995;12:35–50. doi: 10.1017/s095252380000729x. [DOI] [PubMed] [Google Scholar]
- Chesler EJ, Lu L, Wang J, Williams RW, Manly KF. WebQTL: rapid exploratory analysis of gene expression and genetic networks for brain and behavior. Nat Neurosci. 2004;7:485–486. doi: 10.1038/nn0504-485. [DOI] [PubMed] [Google Scholar]
- Chesler EJ, Lu L, Shou S, Qu Y, Gu J, Wang J, Hsu HC, Mountz JD, Baldwin NE, Langston MA, Threadgill DW, Manly KF, Williams RW. Complex trait analysis of gene expression uncovers polygenic and pleiotropic networks that modulate nervous system function. Nat Genet. 2005;37:233–242. doi: 10.1038/ng1518. [DOI] [PubMed] [Google Scholar]
- Cohen AS, Lin DD, Coulter DA. Protracted postnatal development of inhibitory synaptic transmission in rat hippocampal area CA1 neurons. J Neurophysiol. 2000;84:2465–2476. doi: 10.1152/jn.2000.84.5.2465. [DOI] [PubMed] [Google Scholar]
- Comb MJ, Kobierski L, Chu HM, Tan Y, Borsook D, Herrup K, Hyman SE. Regulation of opioid gene expression: a model to understand neural plasticity. NIDA Res Monogr. 1992;126:98–112. [PubMed] [Google Scholar]
- Goldstein PA, Elsen FP, Ying SW, Ferguson C, Homanics GE, Harrison NL. Prolongation of hippocampal miniature inhibitory postsynaptic currents in mice lacking the GABA(A) receptor alpha1 subunit. J Neurophysiol. 2002;88:3208–3217. doi: 10.1152/jn.00885.2001. [DOI] [PubMed] [Google Scholar]
- Grieco TM, Malhotra JD, Chen C, Isom LL, Raman IM. Open-channel block by the cytoplasmic tail of sodium channel beta4 as a mechanism for resurgent sodium current. Neuron. 2005;45:233–244. doi: 10.1016/j.neuron.2004.12.035. [DOI] [PubMed] [Google Scholar]
- Guyon A, Laurent S, Paupardin-Tritsch D, Rossier J, Eugene D. Incremental conductance levels of GABAA receptors in dopaminergic neurones of the rat substantia nigra pars compacta. J Physiol (Lond) 1999;516:719–737. doi: 10.1111/j.1469-7793.1999.0719u.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinen K, Baker RE, Spijker S, Rosahl T, van Pelt J, Brussaard AB. Impaired dendritic spine maturation in GABAA receptor alpha1 subunit knock out mice. Neuroscience. 2003;122:699–705. doi: 10.1016/s0306-4522(03)00477-9. [DOI] [PubMed] [Google Scholar]
- Hitzemann R, Reed C, Malmanger B, Lawler M, Hitzemann B, Cunningham B, McWeeney S, Belknap J, Harrington C, Buck K, Phillips T, Crabbe J. On the integration of alcohol-related quantitative trait loci and gene expression analyses. Alcohol Clin Exp Res. 2004;28:1437–1448. doi: 10.1097/01.alc.0000139827.86749.da. [DOI] [PubMed] [Google Scholar]
- Huang KP, Huang FL, Jager T, Li J, Reymann KG, Balschun D. Neurogranin/RC3 enhances long-term potentiation and learning by promoting calcium-mediated signaling. J Neurosci. 2004;24:10660–10669. doi: 10.1523/JNEUROSCI.2213-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huopaniemi L, Keist R, Randolph A, Certa U, Rudolph U. Diazepam-induced adaptive plasticity revealed by alpha1 GABAA receptor-specific expression profiling. J Neurochem. 2004;88:1059–1067. doi: 10.1046/j.1471-4159.2003.02216.x. [DOI] [PubMed] [Google Scholar]
- Jones MW, Errington ML, French PJ, Fine A, Bliss TV, Garel S, Charnay P, Bozon B, Laroche S, Davis S. A requirement for the immediate early gene Zif268 in the expression of late LTP and long-term memories. Nat Neurosci. 2001;4:289–296. doi: 10.1038/85138. [DOI] [PubMed] [Google Scholar]
- Killion PJ, Sherlock G, Iyer VR. The Longhorn Array Database (LAD): an open-source, MIAME compliant implementation of the Stanford Microarray Database (SMD) BMC Bioinformatics. 2003;4:32. doi: 10.1186/1471-2105-4-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knapska E, Kaczmarek L. A gene for neuronal plasticity in the mammalian brain: Zif268/Egr-1/NGFI-A/Krox-24/TIS8/ZENK? Prog Neurobiol. 2004;74:183–211. doi: 10.1016/j.pneurobio.2004.05.007. [DOI] [PubMed] [Google Scholar]
- Kralic JE, Korpi ER, O’Buckley TK, Homanics GE, Morrow AL. Molecular and pharmacological characterization of GABA(A) receptor alpha1 subunit knockout mice. J Pharmacol Exp Ther. 2002a;302:1037–1045. doi: 10.1124/jpet.102.036665. [DOI] [PubMed] [Google Scholar]
- Kralic JE, O’Buckley TK, Khisti RT, Hodge CW, Homanics GE, Morrow AL. GABA(A) receptor alpha-1 subunit deletion alters receptor subtype assembly, pharmacological and behavioral responses to benzodiazepines and zolpidem. Neuropharmacology. 2002b;43:685–694. doi: 10.1016/s0028-3908(02)00174-0. [DOI] [PubMed] [Google Scholar]
- Kralic JE, Wheeler M, Renzi K, Ferguson C, O’Buckley TK, Grobin AC, Morrow AL, Homanics GE. Deletion of GABAA receptor alpha 1 subunit-containing receptors alters responses to ethanol and other anesthetics. J Pharmacol Exp Ther. 2003;305:600–607. doi: 10.1124/jpet.102.048124. [DOI] [PubMed] [Google Scholar]
- Kralic JE, Criswell HE, Osterman JL, O’Buckley TK, Wilkie ME, Matthews DB, Hamre K, Breese GR, Homanics GE, Morrow AL. Genetic essential tremor in gamma-aminobutyric acidA receptor alpha1 subunit knockout mice. J Clin Invest. 2005;115:774–779. doi: 10.1172/JCI23625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kralic JE, Sidler C, Parpan F, Homanics GE, Morrow AL, Fritschy JM. Compensatory alteration of inhibitory synaptic circuits in cerebellum and thalamus of gamma-aminobutyric acid type A receptor alpha1 subunit knockout mice. J Comp Neurol. 2006;495:408–421. doi: 10.1002/cne.20866. [DOI] [PubMed] [Google Scholar]
- Luscher B, Keller CA. Regulation of GABAA receptor trafficking, channel activity, and functional plasticity of inhibitory synapses. Pharmacol Ther. 2004;102:195–221. doi: 10.1016/j.pharmthera.2004.04.003. [DOI] [PubMed] [Google Scholar]
- Morikawa H, Khodakhah K, Williams JT. Two intracellular pathways mediate metabotropic glutamate receptor-induced Ca 2+ mobilization in dopamine neurons. J Neurosci. 2003;23:149–157. doi: 10.1523/JNEUROSCI.23-01-00149.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogris W, Lehner R, Fuchs K, Furtmuller B, Hoger H, Homanics GE, Sieghart W. Investigation of the abundance and subunit composition of GABAA receptor subtypes in the cerebellum of alpha1-subunit-deficient mice. J Neurochem. 2006;96:136–147. doi: 10.1111/j.1471-4159.2005.03509.x. [DOI] [PubMed] [Google Scholar]
- Okada H, Matsushita N, Kobayashi K, Kobayashi K. Identification of GABAA receptor subunit variants in midbrain dopaminergic neurons. J Neurochem. 2004;89:7–14. doi: 10.1111/j.1471-4159.2004.02271.x. [DOI] [PubMed] [Google Scholar]
- Ortinski PI, Lu C, Takagaki K, Fu Z, Vicini S. Expression of distinct alpha subunits of GABAA receptor regulates inhibitory synaptic strength. J Neurophysiol. 2004;92:1718–1727. doi: 10.1152/jn.00243.2004. [DOI] [PubMed] [Google Scholar]
- Ozaki M. Analysis of patterned neuronal impulses and function of neuregulin. NeuroSignals. 2002;11:191–196. doi: 10.1159/000065434. [DOI] [PubMed] [Google Scholar]
- Papassotiropoulos A, Wollmer MA, Aguzzi A, Hock C, Nitsch RM, deq Uervain DJ. The prion gene is associated with human long-term memory. Hum Mol Genet. 2005;14:2241–2246. doi: 10.1093/hmg/ddi228. [DOI] [PubMed] [Google Scholar]
- Patra RC, Blue ME, Johnston MV, Bressler J, Wilson MA. Activity-dependent expression of Egr1 mRNA in somatosensory cortex of developing rats. J Neurosci Res. 2004;78:235–244. doi: 10.1002/jnr.20243. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. The mouse brain in stereotaxic coordinates. 2. San Diego, CA: Academic; 2001. [Google Scholar]
- Petri S, Krampfl K, Dengler R, Bufler J, Weindl A, Arzberger T. Human GABA A receptors on dopaminergic neurons in the pars compacta of the substantia nigra. J Comp Neurol. 2002;452:360–366. doi: 10.1002/cne.10379. [DOI] [PubMed] [Google Scholar]
- Pongrac J, Middleton FA, Lewis DA, Levitt P, Mirnics K. Gene expression profiling with DNA microarrays: advancing our understanding of psychiatric disorders. Neurochem Res. 2002;27:1049–1063. doi: 10.1023/a:1020904821237. [DOI] [PubMed] [Google Scholar]
- Ponomarev I, Schafer GL, Blednov YA, Williams RW, Iyer VR, Harris RA. Convergent analysis of cDNA and short oligomer microarrays, mouse null mutants and bioinformatics resources to study complex traits. Genes Brain Behav. 2004;3:360–368. doi: 10.1111/j.1601-183x.2004.00088.x. [DOI] [PubMed] [Google Scholar]
- Reynolds DS, O’Meara GF, Newman RJ, Bromidge FA, Atack JR, Whiting PJ, Rosahl TW, Dawson GR. GABA(A) alpha 1 subunit knock-out mice do not show a hyperlocomotor response following amphetamine or cocaine treatment. Neuropharmacology. 2003;44:190–198. doi: 10.1016/s0028-3908(02)00370-2. [DOI] [PubMed] [Google Scholar]
- Ribeiro CS, Reis M, Panizzutti R, de Miranda J, Wolosker H. Glial transport of the neuromodulator D-serine. Brain Res. 2002;929:202–209. doi: 10.1016/s0006-8993(01)03390-x. [DOI] [PubMed] [Google Scholar]
- Ryabinin AE, Wang YM. Repeated alcohol administration differentially affects c-Fos and FosB protein immunoreactivity in DBA/2J mice. Alcohol Clin Exp Res. 1998;22:1646–1654. [PubMed] [Google Scholar]
- Sato T, Xiao DM, Li H, Huang FL, Huang KP. Structure and regulation of the gene encoding the neuron-specific protein kinase C substrate neurogranin (RC3 protein) J Biol Chem. 1995;270:10314–10322. doi: 10.1074/jbc.270.17.10314. [DOI] [PubMed] [Google Scholar]
- Schena M, Shalon D, Davis RW, Brown PO. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science. 1995;270:467–470. doi: 10.1126/science.270.5235.467. [DOI] [PubMed] [Google Scholar]
- Shibasaki K, Nakahira K, Trimmer JS, Shibata R, Akita M, Watanabe S, Ikenaka K. Mossy fibre contact triggers the targeting of Kv4.2 potassium channels to dendrites and synapses in developing cerebellar granule neurons. J Neurochem. 2004;89:897–907. doi: 10.1111/j.1471-4159.2004.02368.x. [DOI] [PubMed] [Google Scholar]
- Simon P, Schott K, Williams RW, Schaeffel F. Posttranscriptional regulation of the immediate-early gene EGR1 by light in the mouse retina. Eur J Neurosci. 2004;20:3371–3377. doi: 10.1111/j.1460-9568.2004.03811.x. [DOI] [PubMed] [Google Scholar]
- Singec I, Knoth R, Ditter M, Volk B, Frotscher M. Neurogranin is expressed by principal cells but not interneurons in the rodent and monkey neocortex and hippocampus. J Comp Neurol. 2004;479:30–42. doi: 10.1002/cne.20302. [DOI] [PubMed] [Google Scholar]
- Storey JD. A direct approach to false discovery rates. J R Soc Lond B Biol Sci. 2002;64:479–498. [Google Scholar]
- Strassle BW, Menegola M, Rhodes KJ, Trimmer JS. Light and electron microscopic analysis of KChIP and Kv4 localization in rat cerebellar granule cells. J Comp Neurol. 2005;484:144–155. doi: 10.1002/cne.20443. [DOI] [PubMed] [Google Scholar]
- Sugino K, Hempel CM, Miller MN, Hattox AM, Shapiro P, Wu C, Huang ZJ, Nelson SB. Molecular taxonomy of major neuronal classes in the adult mouse forebrain. Nat Neurosci. 2006;9:99–107. doi: 10.1038/nn1618. [DOI] [PubMed] [Google Scholar]
- Sun J, Bronk P, Liu X, Han W, Sudhof TC. Synapsins regulate use-dependent synaptic plasticity in the calyx of Held by a Ca 2+/calmodulin-dependent pathway. Proc Natl Acad Sci USA. 2006;103:2880–2885. doi: 10.1073/pnas.0511300103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sur C, Wafford KA, Reynolds DS, Hadingham KL, Bromidge F, Macaulay A, Collinson N, O’Meara G, Howell O, Newman R, Myers J, Atack JR, Dawson GR, McKernan RM, Whiting PJ, Rosahl TW. Loss of the major GABAA receptor subtype in the brain is not lethal in mice. J Neurosci. 2001;21:3409–3418. doi: 10.1523/JNEUROSCI.21-10-03409.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svaren J, Ehrig T, Abdulkadir SA, Ehrengruber MU, Watson MA, Milbrandt J. EGR1 target genes in prostate carcinoma cells identified by microarray analysis. J Biol Chem. 2000;275:38524–38531. doi: 10.1074/jbc.M005220200. [DOI] [PubMed] [Google Scholar]
- Tabakoff B, Bhave SV, Hoffman PL. Selective breeding, quantitative trait locus analysis, and gene arrays identify candidate genes for complex drug-related behaviors. J Neurosci. 2003;23:4491–4498. doi: 10.1523/JNEUROSCI.23-11-04491.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tashiro A, Yuste R. Regulation of dendritic spine motility and stability by Rac1 and Rho kinase: evidence for two forms of spine motility. Mol Cell Neurosci. 2004;26:429–440. doi: 10.1016/j.mcn.2004.04.001. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG. Homeostatic plasticity in neuronal networks: the more things change, the more they stay the same. Trends Neurosci. 1999;22:221–227. doi: 10.1016/s0166-2236(98)01341-1. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG, Nelson SB. Homeostatic plasticity in the developing nervous system. Nat Rev Neurosci. 2004;5:97–107. doi: 10.1038/nrn1327. [DOI] [PubMed] [Google Scholar]
- Vicini S, Ortinski P. Genetic manipulations of GABAA receptor in mice make inhibition exciting. Pharmacol Ther. 2004;103:109–120. doi: 10.1016/j.pharmthera.2004.06.001. [DOI] [PubMed] [Google Scholar]
- Vicini S, Ferguson C, Prybylowski K, Kralic J, Morrow AL, Homanics GE. GABAA receptor alpha1 subunit deletion prevents developmental changes of inhibitory synaptic currents in cerebellar neurons. J Neurosci. 2001;21:3009–3016. doi: 10.1523/JNEUROSCI.21-09-03009.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang VY, Zoghbi HY. Genetic regulation of cerebellar development. Nat Rev Neurosci. 2001;2:484–491. doi: 10.1038/35081558. [DOI] [PubMed] [Google Scholar]
- Yang YH, Dudoit S, Luu P, Lin DM, Peng V, Ngai J, Speed TP. Normalization for cDNA microarray data: a robust composite method addressing single and multiple slide systematic variation. Nucleic Acids Res. 2002;30:e15. doi: 10.1093/nar/30.4.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaganas I, Waagepetersen HS, Georgopoulos P, Sonnewald U, Plaitakis A, Schousboe A. Differential expression of glutamate dehydrogenase in cultured neurons and astrocytes from mouse cerebellum and cerebral cortex. J Neurosci Res. 2001;66:909–913. doi: 10.1002/jnr.10058. [DOI] [PubMed] [Google Scholar]
- Zhang X, Liu Y. Suppression of HGF receptor gene expression by oxidative stress is mediated through the interplay between Sp1 and Egr-1. Am J Physiol Renal Physiol. 2003;284:F1216–F1225. doi: 10.1152/ajprenal.00426.2002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.