

Summary
Meningococcal factor H-binding protein (fHbp) is a promising vaccine antigen that binds the human complement down-regulatory molecule factor H (fH), which enhances survival of the organism in serum. Based on sequence variability of the entire protein, fHbp has been divided into three variant groups or two sub-families. Here we present evidence based on phylogenic analysis of 70 unique fHbp amino acid sequences that the molecular architecture is modular. From sequences of natural chimeras we identified blocks of two to five invariant residues that flanked five modular variable segments. Although overall, 46% of the fHbp amino acids were invariant, based on a crystal structure, the invariant blocks that flanked the modular variable segments clustered on the membrane surface containing the amino-terminal lipid anchor, while the remaining invariant residues were located throughout the protein. Each of the five modular variable segments could be classified into one of two types, designated α or β, based on homology with segments encoded by variant 1 or 3 fHbp genes, respectively. Forty of the fHbps (57%) comprised only β (N=33) or β (N=7) type segments. The remaining 30 proteins (43%) were chimeras and could be classified into one of four modular groups. These included all 15 proteins assigned to the previously described variant 2 in sub-family A. The modular segments of one chimeric modular group had 96% amino acid identity with fHbp orthologs in Neisseria gonorrhoeae. Collectively the data suggest that recombination between N. meningitidis and N. gonorrhoeae progenitors generated a family of modular, antigenically diverse meningococcal factor H-binding proteins.
Keywords: GNA1870, Lp2086, vaccine, Neisseria meningitidis, complement
Introduction
Meningococcal factor H-binding protein (previously referred to as GNA1870 (Masignani et al., 2003) or LP2086 (Fletcher et al., 2004)) is a promising vaccine antigen for prevention of disease caused by Neisseria meningitidis. Two recombinant protein vaccines containing fHbp are in clinical development (Jansen et al., 2008; Rappuoli, 2008). Both vaccines recently were reported to elicit serum bactericidal responses in children and adults against genetically diverse meningococcal strains (Jansen et al., 2008; Marshall et al., 2008; Richmond et al., 2008; Snape et al., 2008){Plested, 2009 #2733}. An important function of fHbp is to bind human complement fH (Granoff et al., 2009; Madico et al., 2006), which down-regulates complement activation. Binding of fH to the bacterial surface is an important mechanism by which the pathogen survives in non-immune human serum or blood and evades innate host defenses (Madico et al., 2006; Schneider et al., 2006; Welsch et al., 2008). Serum antibodies directed at fHbp both activate classical complement-mediated bacteriolysis (Welsch et al., 2004), and block binding of fH (Madico et al., 2006; Welsch et al., 2008). Blocking binding of fH to the bacterial surface would be expected to increase susceptibility of the organism to complement-mediated killing. The main limitations of fHbp as a vaccine antigen are low density on the surface of some strains (Welsch et al., 2008) and variability (Beernink et al., 2007; Fletcher et al., 2004; Masignani et al., 2003).
In their original description of fHbp, Masignani and coworkers classified fHbp into three variant groups based on amino acid sequence diversity and lack of cross-reactive serum bactericidal antibody responses of immunized mice (Masignani et al., 2003). Using similar analyses, Fletcher and coworkers assigned fHbp variants into two sub-families, designated A and B (Fletcher et al., 2004). Sub-family B corresponded to variant group 1 of Masignani et al, and sub-family A included strains with fHbp in the variant 2 or 3 groups. Thus far, there has not been a consensus as to whether variant groups 2 and 3 are antigenically or phylogenically distinct.
In an effort to develop fHbp antigens capable of eliciting broad bactericidal antibody responses against strains with fHbp from different variant groups, our laboratory has conducted epitope mapping studies of anti-fHbp bactericidal mAbs (Beernink et al., 2008; Beernink et al., 2009b). Based on the locations of the epitopes, we prepared recombinant chimeric fHbp antigens containing domains from different variant groups (Beernink & Granoff, 2008). As part of these studies we noted that certain portions of the fHbp molecule shared amino acid sequence identity and cross-reactivity between variant 1 and 2 proteins (Beernink et al., 2009b), while other portions of the protein shared identity between variant 2 and 3 proteins (Beernink et al., 2008). Further, we noted examples of naturally occurring chimeric fHbps that all had in common certain blocks of invariant amino acids, which appeared to flank segments containing variable amino acids. The results of these preliminary analyses suggested the hypothesis that the fHbp architecture was “modular” with distinct variable segments derived from different progenitor sequences. This architecture had not been described by the previously published fHbp phylogenic studies (Fletcher et al., 2004; Jacobsson et al., 2009; Masignani et al., 2003), which were largely based on analyses of the variability of the overall sequences of the mature protein. To investigate the modular fHbp hypothesis, in the present study we performed phylogenic analyses of different segments of 70 distinct fHbps. Our goals were to increase our understanding of the evolution of fHbp variation and provide a basis for sub-classification of fHbps into different modular variant groups.
Methods
fHbp sequencing
We amplified the fHbp gene from genomic DNA prepared with the DNeasy Tissue kit (Qiagen, Valencia, CA) by polymerase chain reaction using primers A1 and B2 and cycling parameters described previously (Masignani et al., 2003). The PCR products were purified using QiaQuick PCR purification kit (Qiagen) and were eluted in 30 μl of sterile deionized H2O. The fHbp DNA sequences were determined by a commercial sequencing facility using the primers A1 and 22 described previously (Masignani et al., 2003).
Source of data
We analyzed protein sequences encoded by 76 fHbp genes determined in our laboratory from Neisseria meningitidis case isolates from the United States (Beernink et al., 2007), Europe (Beernink & Granoff, 2008; Beernink et al., 2008) and Africa (Beernink et al., 2009a). This data set included 48 sequences determined as part of our previous studies and 28 new sequences performed for the present study. We obtained 95 additional fHbp gene sequences from Genbank (http://www.ncbi.nlm.nih.gov) by performing translated BLAST (tblastn) searches with fHbp amino acid sequences from strains MC58 (variant 1/sub-family B) and M1239 (variant 3/sub-family A). Among the 171 nucleotide sequences from our collection and those in Genbank, we identified fHbp genes that encoded 64 unique protein sequences. These 64, plus 6 additional unique fHbp amino acid sequences obtained from the Neisseria.org fHbp peptide database (http://neisseria.org), were used for our analyses of 70 unique fHbp peptides. The respective Genbank accession numbers and/or peptide identification numbers and the characteristics of the source strains are listed in supplementary Table S1.
Thirty-eight (54.3%) of the 70 peptides were classified in the variant 1 group of Masignani et al (Masignani et al., 2003), 15 (21.4%) in the variant 2 group, and 17 (24.3%) in the variant 3 group. Of the 70 source strains, one was capsular group A, 57 were group B, seven were group C, two were group W-135, two were group X and one was group Y. Multi-locus sequence type (MLST) information was available for 59 of the strains of which 15 were from the ST-269 clonal complex, 12 were from the ST-11 complex, 10 were from the ST-41/44 complex, five each were from the ST-162 and ST-213 complexes, and three each were from the ST-8 and ST-32 complexes. Six strains were from other clonal complexes and had sequence types ST-4, ST-35, ST-751, ST-4821, ST-5403 and ST-6874. The 11 strains without MLST information were not available for testing.
Phylogenic analysis
The analysis of complete or partial protein sequences was performed on the platform at http://www.phylogeny.fr (Dereeper et al., 2008) and comprised the following steps. Sequences were aligned with MUSCLE (v3.7) (Edgar, 2004) configured for highest accuracy and the alignments were checked for accuracy using Gblocks (Castresana, 2000). The respective alignments, which contained up to three sites of insertions or deletions, were inspected and verified by adjacent invariant sequences. The phylogenic trees were reconstructed using the maximum likelihood method implemented in the PhyML program (v3.0 aLRT)(Guindon & Gascuel, 2003) Statistical tests for branch support were performed using the bootstrapping method (100 replicates). Phylograms were displayed with MEGA 4.0 (Tamura et al., 2007). The percent sequence identities within and between variable segment types of sequences were determined using ClustalW (Larkin et al., 2007). Additional information on the parameters used is provided in supplementary Table S2.
Results
Phylogenic analysis of mature proteins
We aligned the 70 unique fHbp amino acid sequences to generate a phylogram based on variability of the entire mature protein, which was arbitrarily rooted on peptide 1 (Figure 1). The analysis showed the two major branches previously designated as sub-families A and B (Fletcher et al., 2004). Sub-family A contained fHbp sequences in antigenic variant groups 2 and 3, and sub-family B corresponded to fHbps in the antigenic variant group 1 (Masignani et al., 2003). For some clonal complexes, there were examples of strains with fHbp in each of the variant groups (for example, for ST-11, peptide ID numbers 2, 3, 6, 9, 10, 11 and 78 in the variant 1 group, peptides 17, 22, 23 and 27 in the variant 2 group, and peptide 59 in the variant 3 group). The strains from the ST-32 clonal complex had fHbp in two of the variant groups (peptides 1 and 89 in the variant 1 group and peptide 76 in the variant 3 group). The ST-8 clonal complex had fHbp only in variant group 2 (peptides 16, 50, and 77). The distribution of fHbp variants in all of the observed clonal complexes is given in supplementary Table S3.
Figure 1.

Phylogram of fHbp based on 70 unique amino acid sequences. For each sequence, the peptide identification number assigned in the fHbp peptide database at http://Neisseria.org is shown and, if known, the multi-locus sequence type (MLST) clonal complex is shown in parentheses. The lower left branch shows variant group 1 as defined by Masignani et al (Masignani et al., 2003) (sub-family B of Fletcher et al (Fletcher et al., 2004)); Sub-family A contained two branches, variant groups 2 and 3. The phylogram was constructed by multiple sequence alignment as described in Methods and supplementary Table S2. The scale bar shown at the bottom indicates 5 amino acid changes per 100 residues.
Evidence of a modular architecture: invariant amino acids
Overall, 46% of the amino acids in the 70 fHbps were invariant. However, based on examples of naturally occurring chimeric fHbps (see below), we identified blocks of two to five invariant residues that flanked five modular variable segments. With the exception of the amino-terminal CSSG, each of the invariant blocks also was present in two N. gonorrhoeae orthologs (Genbank accession numbers AE004969 and CP001050) (Figure 2, Panel A). We mapped the variable and invariant residues onto a molecular model based on the published coordinates from the fHbp crystal structure (Figure 2, Panel B)(Schneider et al., 2009) The light gray residues represent the variable amino acids. The black residues represent the subset of invariant amino acids that flank the modular variable segments determined from inspection of sequences of natural chimeras and described below. The yellow residues represent the remaining invariant amino acids. The subset of invariant amino acids flanking each of the modular variable segments formed a distinct cluster on the surface of the protein predicted to be anchored to the cell wall (visible on the models in the center and on the right). This location suggests that there are structural constraints involving these invariant blocks, perhaps a requirement for a partner protein, for anchoring and/or orienting fHbp on the bacterial cell membrane.
Figure 2.

Panel A. Schematic representation of fHbp showing positions of blocks of invariant residues (shown as black vertical rectangles). The top three panels show representative architectures of three N. meningitidis fHbp variants in groups 1, 2 and 3 (peptide ID numbers, 1, 16 and 28, respectively). The amino acid positions of the last residue in each variable segment are shown. With the exception of a longer, unrelated amino-terminal element, two N. gonorrhoeae orthologs (Ng, Genbank accession numbers AE004969 and CP001050) had the identical six invariant blocks of residues that flanked segments VA through VE. Panel B. Space-filling structural models of factor H binding protein based on the coordinates of fHbp in a complex with a fragment of human factor H (Schneider et al., 2009). The light gray residues represent the variable amino acids located within the modular variable segments. The invariant blocks of residues separating each of the variable segments are shown in black. The yellow residues represent the invariant amino acids outside of these blocks. The model on the left is the surface predicted to be anchored to the cell wall. The model in the center has been rotated 180 degrees on the Y-axis from the corresponding model on the left, while the model on the right has been rotated 90 degrees on the X-axis as compared with the model in the middle. The figure was constructed with PyMol (http://www.pymol.org).
Modular variable segments
Based on the positions of the blocks of invariant amino acids, the overall architecture could be divided into an amino-terminal repetitive element and five modular variable segments, which we designated VA-VE (Figure 2, Panel A). Segments VB and VD contained 15 and 19 amino acids, respectively, while segments VA, VC and VE contained 69, 62 and 71 amino acids, respectively. Within each of the modular variable segments, there were both invariant and variable amino acids. The percentages of variable amino acids in segments VA and VD were 49 and 47%, respectively, while the percentages of variable amino acids in segments VB, VC and VE ranged from 60% to 68%.
The amino-terminal repetitive element
For all 70 sequences, the mature fHbp began with a cysteine residue that is lipidated by signal peptidase II, which was followed by three invariant amino acid residues, SSG. This invariant sequence was followed by a repetitive element consisting of 1 to 6 glycine and/or serine residues and then by two invariant glycine residues (Figure 2, Panel A). The variable portion of the amino-terminal element consisted of a single glycine residue for 34 of the 70 proteins (nearly all in the previously described variant 1 group, Table 1), or GG residues for 12 of the proteins (all in the variant 2 group). The other common repetitive element sequence was GGGSGG (8 in the variant 1 group and 7 in the variant 3 group).
Table 1.
Sequence and distribution of amino-terminal repetitive element
| Variable Sequence 1 | Number of Peptides in Variant Group |
||
|---|---|---|---|
| Variant 1 | Variant 2 | Variant 3 | |
| G | 29 | 3 | 2 |
| GG | 0 | 12 | 0 |
| SGG | 0 | 0 | 1 |
| GGGS | 0 | 0 | 2 |
| SGSGG | 0 | 0 | 1 |
| GGGSGG | 8 | 0 | 7 |
| GGGSGS | 1 | 0 | 4 |
The mature fHbp begins with an invariant tetrapeptide, CSSG, which is followed by a repetitive variable sequence consisting of 1 to 6 glycine and/or serine residues, which is then followed by two invariant glycine residues.
Downstream modular variable segments
We analyzed separately the phylogeny of each the five modular variable segments of the 70 distinctive meningococcal fHbp peptide variants. In these analyses, the numbering of the amino acid residues was based on the mature fHbp peptide 1 encoded by a gene from MC58 in the previously assigned variant 1 group (Masignani et al., 2003). The lengths of the respective variant 2 and 3 proteins differed from that of the variant 1 protein by -1 and +7, respectively (Figure 2).
Segment VA began at amino acid residue 8 immediately after the invariant GG sequence and extended to position 73 while segment VB began at position 79 immediately after an invariant SRFDF sequence and extended to position 93 (Figure 2, Panel A). Based on the phylogram of the VA segments (Figure 3, top Panel), 33 of the 38 fHbps assigned to Masignani variant 1 group (shown in black) clustered together with those of the 15 fHbps in the variant 2 group (red), whereas all 17 fHbps in the variant 3 group (aqua) were located in a separate cluster. In the phylogram of the VB segments (bottom Panel), there was similar clustering of the variant 1 and 2 proteins, which were separate from those of the variant 3 proteins.
Figure 3.

Phylograms of unique fHbp amino acid sequences in modular variable segments VA (residues 8-73) and VB (residues 79-93). Segments derived from fHbp in the variant groups 1 are shown as black circles; variant 2 group as red circles, and variant 3 group as aqua circles. Where multiple proteins possessed an identical sequence in a segment, the number of proteins is given in parentheses. A.β.9 and B.α.3 refer to proteins with exceptional junctional points (See text). The scale bar indicates 2 amino acid changes per 100 residues. The histogram (Segment VA) shows the mean number of protein sequences (Y-axis) and percent amino acid identity (X-axis) generated by comparing each of the 48 α A segments to the corresponding α and β types A segments of all 70 proteins. The corresponding histograms comparing each of the α segments to the corresponding α and β types, and each of the β segments to the corresponding α and β types, for all five modular variable segments, VA-VE, are shown in supplementary Figure S1.
In contrast to the VA and VB segments, in the phylograms of the respective VC (residues 98-159) and VE (residues 186-253) segments, all of the fHbps assigned to Masignani's variant 2 and 3 groups clustered together and were separated from those of the variant 1 group 1 (Figure 4). The phylogram of the VD segments (residues 162-180) had a different pattern from those of the other segments: All of the variant 1 proteins clustered with a subset of ten of the 15 variant 2 proteins and ten of the 17 variant 3 proteins (Figure 5). The remaining five variant 2 proteins and seven variant 3 proteins were located together in a separate cluster.
Figure 4.

Phylograms of unique fHbp amino acid sequences in variable segments VC (residues 98-159) and VE (residues 186-253). The colors of the circles in for segments in each variant group correspond to those described in the legend of Figure 3. E.β.10 refers to peptide ID no. 82 with an exceptional junctional point (See text). The scale bars indicate 2 amino acid changes per 100 residues.
Figure 5.

Phylogram of unique fHbp amino acid sequences in variable segment VD (residues 162-180). The colors of the circles in for peptides in each variant group correspond to those described in the legend of Figure 3. The scale bar represents 2 amino acid changes per 100 residues.
Each of the five modular variable segments segregated into one of two types (Table 2). One of the types had signature amino acid residues and sequence similarity to proteins in the antigenic variant 1 group. The other type had signature amino acid residues and sequence similarity to proteins in the antigenic variant 3 group. The latter also were similar to those of the gonococcal ortholog (see Figure 2, and Discussion section). For purposes of classification, we designated the first group of segments as α types and the second group as β types.
Table 2.
Amino acid identity within and between fHbp sequence lineages by segment.
| α Sequence Types |
β Sequence Types |
|||||||
|---|---|---|---|---|---|---|---|---|
| Variable Segment | Residue Range 1 | Signature residues 2 | No. 3 | Identity Range (%) | Signature residues 2 | No. 3 | Identity Range (%) | Identity between α and β Types (%) |
| VA | 8-73 | QSV | 48 | 89-100 | DSI and KDN | 21 | 87-100 | 69-83 |
| VB | 79-93 | IRQ | 51 | 86-100 | VQK | 17 | 100 | 60-66 |
| VC | 98-159 | QDS | 38 | 83-100 | NNP | 32 | 93-100 | 37-45 |
| VD | 162-180 | AG or AS | 58 | 89-100 | PN | 12 | 84-100 | 52-63 |
| VE | 186-253 | SLGI | 38 | 83-100 | HLAL | 31 | 92-1005 | 48-575 |
Amino acid numbering based on the mature fHbp from strain MC58 (peptide ID no.
See supplementary Table S3 for complete sequences of each variable segment.
For each segment, we calculated the percentages of the amino acid identity of each of the α type segments with each of the corresponding α or β type segments of the 70 fHbps. We performed a second calculation of the percentages of the amino acid identity of each of the β type segments with the corresponding α and β types. We generated histograms showing the respective mean frequencies of peptide variants with different percentages of amino acid identity. The inset panel on Figure 2 shows a representative histogram comparing each of the 48 α VA segments to the corresponding α and β type VA segments of the 70 proteins. The corresponding histograms comparing each of the α segments, and each of the β segments, to the corresponding α and β types for all five modular variable segments, VA-VE, are shown in supplementary Figure S1. For each of the modular variable segments, there was clear separation betweeen the percent amino acid identities of the respective the α and β types.
For each modular variable segment, distinct sequence variants were assigned a unique identifier beginning with a letter, A through E, to represent the modular variable segment; followed by an α or β to indicate the presence of residues with the respective types described above, followed by a number for each distinct sequence variant (listed in supplementary Table S4). For example, the VA segment contained 15 distinct α variants and nine β variants. The most common α variant (N=14) was designated A.α.1. The most common β variant of segment VA was designated A.β.1. Using this identifier system, each distinct fHbp can be described by a combination of five specific peptides (for example, peptide 1, A.α.2, B.α.1, C.α.5, D.α.5, E.α.8). These designations are provided for each of the 70 proteins in supplemental Table S1.
Exceptional chimeric fHbps
Four of the 70 fHbp sequences had junctional points between two of the segments that utilized alternative invariant sequences than those described in Figure 2. The VA segment of peptide ID no. 55 switched from a β type sequence to an α type sequence at an invariant AQGAE sequence starting at residue 50 rather than at SRFDF, which started at residue 74. This exceptional VA segment was designated A.β.9 (β because of its higher sequence identity to other β type A segments than to the α type A segments). The VB segment of two other fHbp peptide ID nos. 24 and 25, switched from an α type to a β type sequence at an invariant IEV sequence beginning at residue 82 instead of GEFQ at position 94. This exceptional VB segment, designated B.α.3, was categorized as an α type because of its higher sequence identity to other α type VB segments than to the β type VB segments. Finally, the VE segment of peptide ID no. 82 had an exceptional cross-over point from an α type sequence to a β type at residue A196 instead of at IEHLK starting at position 181. This segment was designated E.β.10
fHbp modular groups
Based on the phylogenic analysis of the five modular variable segments described above, we could categorize each of the 70 different fHbp variants into one of six distinct fHbp modular groups (Figure 6). Forty of the 70 fHbps (57%) comprised only α (N=33) or β (N=7) type segments, which were designated fHbp modular groups I and II, respectively (Figure 6). The remaining 30 (43%), which included the four with exceptional junctional points described above, could be classified into one of four chimeric groups derived from recombination of different α or β segments (designated fHbp modular groups III, IV, V or VI).
Figure 6.
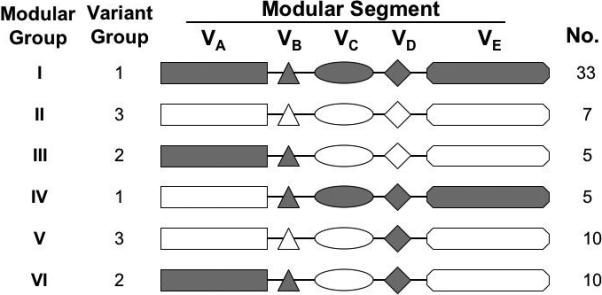
Schematic representation of six fHbp modular groups deduced from phylogenic analysis. Forty of the 70 proteins contained only α type segments or β type segments, and were designated as modular groups I and II, respectively. The remaining 30 proteins were chimeras with different combinations of α (shown in gray) and β segments (shown in white) and could be assigned to one of four modular groups (III-VI). The relationship between the modular group and Masignani variant group designation, and the number of unique sequences observed within each fHbp modular group, are shown.
Of the 38 fHbps in the Masignani variant 1 group, 33 were in modular group I and five were chimeras in modular group IV. Of the 17 proteins in the variant 3 group, seven were in modular group II and ten were chimeras in modular group V. All 15 variant 2 proteins were chimeras in modular groups III or VI.
Structural features of the modular variable fHbp segments
As described above for the invariant residues, we mapped the respective variable segments of fHbp onto a space-filled molecular model based on the published coordinates from the crystal structure the fHbp-fH complex (Figure 7) (Schneider et al., 2009). Each color on the model represents a specific modular variable segment and the white segments indicate the blocks of invariant residues that flank the variable segments.
Figure 7.

Space-filled structural models of factor H binding protein based on the coordinates of fHbp in a complex with a fragment of human factor H (Schneider et al., 2009). The five variable segments VA through VE are depicted in different colors (VA, blue; VB, orange; VC, green; VD, aqua; VE, violet) and the invariant blocks of residues separating each of the variable segments are shown in white. The model in the middle was generated by rotation of 180° around the Y-axis relative to the respective model on the far left. The model on the right was generated by a 90° rotation around the X-axis relative to the model in the middle. The factor H contact residues are depicted in black, and the residues affecting the epitopes of anti-fHbp mAbs are shown in yellow. The figure was constructed with PyMol (http://www.pymol.org).
The amino acid residues reported to be in contact with fH based are depicted in black. These residues formed clusters on variable segments VA (blue), VC (green) and VE (purple) and are visible in the models shown in the middle and far right of panel B. Since fH is known to bind to fHbp on live bacteria (Madico et al., 2006; Schneider et al., 2006), this binding site must be surface-exposed. Previous studies mapped the epitopes of ten bactericidal anti-fHbp mAbs (Beernink et al., 2008; Beernink et al., 2009b; Giuliani et al., 2005; Scarselli et al., 2009). The amino acids affecting expression of each of these epitopes are highlighted in yellow. With the exception of segment VB, all of the modular variable segments contained epitopes recognized by bactericidal mAbs. Note that the amino acids affecting epitope expression tended to be located on the periphery of the surface-exposed portion of the molecule (best visualized on the models in the middle and far right). In contrast, the residues in contact with fH were located in clusters in the central portions. The epitopes of certain mAbs such as JAR 3, 5 or 13, which were previously reported to inhibit binding of fH to fHbp (Beernink et al., 2008; Beernink et al., 2009b), involved amino acids located in proximity to some of the fH contact residues. However, there was no example of overlap between the two sets of contact residues. This finding suggests that even low affinity binding of mouse fH to fHbp may have affected the portion of fHbp recognized by the immune system.
Discussion
Based on previous analyses of the sequences of the mature protein, fHbp was classified into two sub-families (Fletcher et al., 2004) or three variant groups (Masignani et al., 2003) (Figure 1). However, neither of these classification systems, nor those used in subsequent studies (Bambini et al., 2009; Jacobsson et al., 2009) is entirely satisfactory since our analyses show that the overall architecture of fHbp is mosaic. The similar modular architectures of meningococcal and gonococcal fHbps (Figure 2, Panel A), suggest that during evolution the respective fHbp genes recombined to create antigenically diverse meningococcal factor H-binding proteins. The mosaic architecture of fHbp is not entirely surprising since Neisseria are known to transfer genes horizontally (Bentley et al., 2007; Hotopp et al., 2006), and other Neisserial proteins have been reported to have mosaic structures (Arreaza et al., 2003; Bilek et al., 2009; Callaghan et al., 2008; Ochiai et al., 2008; Rokbi et al., 1995; Szczesny & Lupas, 2008).
For each of the fHbp modular variable segments there was clear separation of the sequences into one of two types, designated α or β, based on the presence of “signature” amino acids and a high percentage of amino acid identity with variant 1 or variant 3 proteins (Table 2 and supplementary Figure S1). For a protein comprising five modular variable segments, each of which can be of one of two types, there are 25 = 32 theoretical independent modular combinations. That we identified only six modular groups (Figure 6) suggests that there are functional or structural constraints on the molecule that select for certain combinations of the variable modules.
Note that within a modular variable segment, only half to two-thirds of the amino acids were variable, and that within an α or β type, the respective sequences of a segment were conserved. If conservative amino acid substitutions are considered, the modular variable segments could have been defined at different boundary residues than those shown in Figure 2. However, the boundary residues were selected based on the invariant blocks observed in natural fHbp chimeras. Our hypothesis is that these invariant blocks represent junctional points for recombination that result in fit fHbp mutants. As additional data become available correlating fHbp variants with strain susceptibility to anti-fHbp bactericidal activity, the ability to bind complement fH, or changes in fHbp amino acid sequence over time, it may be possible to redefine the modular variable segments based on these functional or evolutionary considerations.
In previous studies, serum antibodies to recombinant fHbp in the variant 1 group were bactericidal primarily only against strains with variant 1 proteins (Beernink et al., 2007; Fletcher et al., 2004; Masignani et al., 2003), while antibodies to fHbp in the variant 2 or 3 groups had activity primarily against strains with homologous variant 2 or 3 proteins but had no activity against strains with fHbp in the variant 1 group. These observations suggest that epitopes in the VC and VE modular segments of fHbp, for which variant 1 proteins were phylogenically separated from those of variant 2 and 3 proteins (Figure 4), are more important for eliciting bactericidal antibodies than those in the VA, VB or VD modular segments, where variant 1 proteins clustered together with variant 2 and/or 3 proteins (Figures 3 and 5). However, all of the variable segments with the exception of segment VB contained residues previously identified as affecting epitopes recognized by murine bactericidal anti-fHbp mAbs (Beernink et al., 2008; Beernink et al., 2009b; Giuliani et al., 2005). Further, some of the mAbs inhibited binding of fH to the bacterial surface (Madico et al., 2006), and some combinations of mAbs that individually were not bactericidal elicited cooperative bactericidal activity (Beernink et al., 2008; Beernink et al., 2009b; Welsch et al., 2008). The latter included mAb JAR 4, which was specific for an epitope on variable segment VA (Beernink et al., 2009b). The implications of the different modular groups on susceptibility of strains to bactericidal activity of anti-fHbp antibodies elicited by different fHbp vaccines will require additional studies.
Approximately 40 percent of disease-causing isolates have fHbp in the previously described sub-family A (Fletcher et al., 2004), which includes both variant 2 and variant 3 as described by Masignani et al (Masignani et al., 2003). However, <3 percent of strains are in the fHbp variant 3 group, which means that the vast majority of sub-family A strains are in the variant 2 group, and these are all natural chimeras of variant 1 and 3 (The variant 2 group includes modular groups III and VI, Figure 6). The modular architecture of fHbp thus leads to difficulties in classification based on overall sequence. For epidemiologic and evolutionary purposes, classification of fHbp based on the six observed modular groups appears to be a more useful system. For complete annotation of an individual protein, its modular group can be supplemented by the peptide ID (from the Neisseria.org website) and the segment ID numbers of each of the five modular variable segments (supplementary Table S4). For vaccine development, additional data are needed correlating immunogenicity of fHbp from different modular groups and strain susceptibility to bactericidal activity.
The 70 fHbp sequences analyzed in the present study reflect the known amino acid diversity of fHbp reported to date. Since minor fHbp variants that are not widely transmitted in human populations were likely over-represented, our data do not reflect the prevalence of fHbp variants among disease-causing strains. Also, sequences from carriage isolates were not represented. In the future, population-based surveillance studies will be required to define the prevalence of fHbp from different modular groups among carriage and disease isolates, as well as changes over time, and the relationship between the fHbp modular group and other strain characteristics such as capsular group, clonal complex and PorA variable regions. Also, analyses at the DNA level are needed, particularly to determine which codons encoding fHbp may be under diversifying selective pressure.
In conclusion, our analyses showed that the overall architecture of fHbp is mosaic consisting of five modular variable segments. Collectively the data suggest that recombination occurred between N. meningitidis and N. gonorrhoeae progenitor sequences to generate an antigenically diverse family of meningococcal factor H-binding proteins. Our data thus provide insights into the evolution of fHbp variants, and may provide a rational basis for classification of fHbp variants that is more useful than previously described schemes based on overall amino acid identity of the entire mature protein.
Supplementary Material
{kind=link}
Supplementary Figure S1. For each of the five modular variable segments, VA to VE, the mean number of peptides (Y-axis) and percent amino acid identity (X axis) are shown. The histograms on the left were generated by comparing each of the α types to the corresponding α and β types for the other 69 peptides and calculating the respective mean frequency at each percent identity. The histograms on the right were generated by comparing each of the β types to the corresponding α and β types. Solid bars represent comparisons with sequences of α types and open bars represent comparisons with sequences of β types.
Acknowledgements
We thank Maurizio Comanducci, Novartis Vaccines, Siena Italy; Lee Harrison, University of Pittsburgh, PA; Jay Lucidarme, Health Protection Agency, UK; and Leonard Mayer, U.S. Centers for Disease Control and Prevention, Atlanta GA, for providing demographic and MLST information on some of the meningococcal strains described in this study. Drs. Rolando Feyt, University of Calgary, and Duccio Medini, Novartis Vaccines, Siena Italy, provided helpful comments on the manuscript. The work was supported by Public Health Service R01 grants AI 70955 (PTB) and R01 AI 46464 (DMG) from the National Institute of Allergy and Infectious Diseases, NIH. The work was performed in a facility funded by Research Facilities Improvement Program grant C06 RR 16226 from the National Center for Research Resources, NIH.
Abbreviations
- fH
factor H
- fHbp
factor H binding protein
- MLST
multilocus sequence type
- peptide ID no.
peptide identification number from http://neisseria.org
Footnotes
Four supplementary tables (with supplementary references), listing source strains and characteristics of unique fHbp variants sorted by modular group, parameters used to phylogenetic analyses, distribution of fHbp variants in each clonal complex, and unique sequences in fHbp variable segments and numbers from each variant group, and a supplementary figure, showing the mean number of peptides and percentage amino acid identity for each of the five modular variable segments, are available with the online version of this paper.
References
- Arreaza L, Alcala B, Salcedo C, de la Fuente L, Vazquez JA. Dynamics of the penA gene in serogroup C meningococcal strains. J Infect Dis. 2003;187:1010–1014. doi: 10.1086/368170. [DOI] [PubMed] [Google Scholar]
- Bambini S, Muzzi A, Olcen P, Rappuoli R, Pizza M, Comanducci M. Distribution and genetic variability of three vaccine components in a panel of strains representative of the diversity of serogroup B meningococcus. Vaccine. 2009;27:2794–2803. doi: 10.1016/j.vaccine.2009.02.098. [DOI] [PubMed] [Google Scholar]
- Beernink PT, Welsch JA, Harrison LH, Leipus A, Kaplan SL, Granoff DM. Prevalence of factor H-binding protein variants and NadA among meningococcal group B isolates from the United States: implications for the development of a multicomponent group B vaccine. J Infect Dis. 2007;195:1472–1479. doi: 10.1086/514821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beernink PT, Granoff DM. Bactericidal antibody responses induced by meningococcal recombinant chimeric factor H-binding protein vaccines. Infect Immun. 2008;76:2568–2575. doi: 10.1128/IAI.00033-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beernink PT, Welsch JA, Bar-Lev M, Koeberling O, Comanducci M, Granoff DM. Fine antigenic specificity and cooperative bactericidal activity of monoclonal antibodies directed at the meningococcal vaccine candidate, factor H-binding protein. Infect Immun. 2008;76:4232–4240. doi: 10.1128/IAI.00367-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beernink PT, Caugant DA, Welsch JA, Koeberling O, Granoff DM. Meningococcal factor H-binding protein variants expressed by epidemic capsular group A, W-135, and X strains from Africa. J Infect Dis. 2009a;199:1360–1368. doi: 10.1086/597806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beernink PT, Lopasso C, Angiolillo A, Felici F, Granoff D. A region of the N-terminal domain of meningococcal factor H-binding protein that elicits bactericidal antibody across antigenic variant groups. Mol Immunol. 2009b;46:1647–1653. doi: 10.1016/j.molimm.2009.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley SD, Vernikos GS, Snyder LA. Meningococcal genetic variation mechanisms viewed through comparative analysis of serogroup C strain FAM18. PLoS Genet. 2007;3:e23. doi: 10.1371/journal.pgen.0030023. other authors. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilek N, Ison CA, Spratt BG. Relative contributions of recombination and mutation to the diversification of the opa gene repertoire of Neisseria gonorrhoeae. J Bacteriol. 2009;191:1878–1890. doi: 10.1128/JB.01518-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callaghan MJ, Buckee CO, Jolley KA, Kriz P, Maiden MC, Gupta S. The effect of immune selection on the structure of the meningococcal opa protein repertoire. PLoS Pathog. 2008;4:e1000020. doi: 10.1371/journal.ppat.1000020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 2000;17:540–552. doi: 10.1093/oxfordjournals.molbev.a026334. [DOI] [PubMed] [Google Scholar]
- Dereeper A, Guignon V, Blanc G. Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 2008;36:W465–469. doi: 10.1093/nar/gkn180. other authors. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher LD, Bernfield L, Barniak V. Vaccine potential of the Neisseria meningitidis 2086 lipoprotein. Infect Immun. 2004;72:2088–2100. doi: 10.1128/IAI.72.4.2088-2100.2004. other authors. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giuliani MM, Santini L, Brunelli B. The region comprising amino acids 100 to 255 of Neisseria meningitidis lipoprotein GNA 1870 elicits bactericidal antibodies. Infect Immun. 2005;73:1151–1160. doi: 10.1128/IAI.73.2.1151-1160.2005. other authors. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granoff DM, Welsch JA, Ram S. Binding of complement factor H (fH) to Neisseria meningitidis is specific for human fH and inhibits complement activation by rat and rabbit sera. Infect Immun. 2009;77:764–769. doi: 10.1128/IAI.01191-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- Hotopp JC, Grifantini R, Kumar N. Comparative genomics of Neisseria meningitidis: core genome, islands of horizontal transfer and pathogen-specific genes. Microbiology. 2006;152:3733–3749. doi: 10.1099/mic.0.29261-0. other authors. [DOI] [PubMed] [Google Scholar]
- Jacobsson S, Hedberg ST, Molling P, Unemo M, Comanducci M, Rappuoli R, Olcen P. Prevalence and sequence variations of the genes encoding the five antigens included in the novel 5CVMB vaccine covering group B meningococcal disease. Vaccine. 2009;27:1579–1584. doi: 10.1016/j.vaccine.2008.12.052. [DOI] [PubMed] [Google Scholar]
- Jansen KU, McNeil LK, Dragalin V, Anderson AS, Hoiseth SK, Arora A, Emini EE, Zlotnick GW, Jones T. Bivalent recombinant LP2086 vaccine to provide broad protection against Neisseria meningitidis B disease: Immunological correlates of protection and how to assess coverage against invasive MnB strains.. In: van Alphen L, van Ley P, van den Dobbelsteen G, editors. 16th International Pathogenic Neisseria Conference; Rotterdam, The Netherlands. 2008. [Google Scholar]
- Larkin MA, Blackshields G, Brown NP. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. other authors. [DOI] [PubMed] [Google Scholar]
- Madico G, Welsch JA, Lewis LA. The meningococcal vaccine candidate GNA1870 binds the complement regulatory protein factor H and enhances serum resistance. J Immunol. 2006;177:501–510. doi: 10.4049/jimmunol.177.1.501. other authors. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall H, Nissen MD, Richmond P, Lambert SB, Roberton DM, Jones T, Lockhart S, Gruber W, Arona A. A randomized, placebo-controlled, double-blind, phase 1 trial of ascending doses of meningococcal group B rLP2086 vaccine in healthy adults.. In: van Alphen L, van Ley P, van den Dobbelsteen G, editors. 16th International Pathogenic Neisseria Conference.; Rotterdam, The Netherlands. 2008. [Google Scholar]
- Masignani V, Comanducci M, Giuliani MM. Vaccination against Neisseria meningitidis using three variants of the lipoprotein GNA1870. J Exp Med. 2003;197:789–799. doi: 10.1084/jem.20021911. other authors. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochiai S, Ishiko H, Yasuda M, Deguchi T. Rapid detection of the mosaic structure of the Neisseria gonorrhoeae penA Gene, which is associated with decreased susceptibilities to oral cephalosporins. J Clin Microbiol. 2008;46:1804–1810. doi: 10.1128/JCM.01800-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plested JS, Welsch JA, Granoff DM. Ex vivo model of meningococcal bacteremia using human blood for measuring vaccine-induced serum passive protective activity. Clin Vaccine Immunol. 2009;16:785–791. doi: 10.1128/CVI.00007-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappuoli R. The application of reverse vaccinology, Novartis MenB vaccine developed by design.. In: van Alphen L, van Ley P, van den Dobbelsteen G, editors. 16th International Pathogenic Neisseria Conference.; Rotterdam, The Netherlands. 2008. [Google Scholar]
- Richmond P, Marshall H, Nissen MD, Lambert S, Jones T, Gruber W, Arora A. A randomized, observer-blinded, active control, phase 1 trial of meningococcal serogroup B rLP2086 vaccine in healthy children and adolescents aged 8 to 14 years.. In: van Alphen L, van Ley P, van den Dobbelsteen G, editors. 16th International Pathogenic Neisseria Conference.; Rotterdam, The Netherlands. 2008. [Google Scholar]
- Rokbi B, Maitre-Wilmotte G, Mazarin V, Fourrichon L, Lissolo L, Quentin-Millet MJ. Variable sequences in a mosaic-like domain of meningococcal tbp2 encode immunoreactive epitopes. FEMS Microbiol Lett. 1995;132:277–283. doi: 10.1016/0378-1097(95)00326-z. [DOI] [PubMed] [Google Scholar]
- Scarselli M, Cantini F, Santini L. Epitope mapping of a bactericidal monoclonal antibody against the factor H binding protein of Neisseria meningitidis. J Mol Biol. 2009;386:97–108. doi: 10.1016/j.jmb.2008.12.005. other authors. [DOI] [PubMed] [Google Scholar]
- Schneider MC, Exley RM, Chan H, Feavers I, Kang YH, Sim RB, Tang CM. Functional significance of factor H binding to Neisseria meningitidis. J Immunol. 2006;176:7566–7575. doi: 10.4049/jimmunol.176.12.7566. [DOI] [PubMed] [Google Scholar]
- Schneider MC, Prosser BE, Caesar JJ. Neisseria meningitidis recruits factor H using protein mimicry of host carbohydrates. Nature. 2009;458:890–893. doi: 10.1038/nature07769. other authors. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snape MD, Dawson T, Morant A, John B, Ohene-Kena R, Borrow R, Oster P, Pollard AJ. Immunogenicity and reactogenicity of a novel serogroup B Neisseria meningitidis vaccine administered from 6 months of age.. In: Alphen L. v., Dobbelsteen G. v. d., Ley P. v. d., editors. 16th International Pathogenic Neisseria Conference.; Rotterdam, The Netherlands. 2008. [Google Scholar]
- Szczesny P, Lupas A. Domain annotation of trimeric autotransporter adhesins--daTAA. Bioinformatics. 2008;24:1251–1256. doi: 10.1093/bioinformatics/btn118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Dudley J, Nei M, Kumar S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24:1596–1599. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- Welsch JA, Rossi R, Comanducci M, Granoff DM. Protective activity of monoclonal antibodies to genome-derived neisserial antigen 1870, a Neisseria meningitidis candidate vaccine. J Immunol. 2004;172:5606–5615. doi: 10.4049/jimmunol.172.9.5606. [DOI] [PubMed] [Google Scholar]
- Welsch JA, Ram S, Koeberling O, Granoff DM. Complement-dependent synergistic bactericidal activity of antibodies against factor H-binding protein, a sparsely distributed meningococcal vaccine antigen. J Infect Dis. 2008;197:1053–1061. doi: 10.1086/528994. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1. For each of the five modular variable segments, VA to VE, the mean number of peptides (Y-axis) and percent amino acid identity (X axis) are shown. The histograms on the left were generated by comparing each of the α types to the corresponding α and β types for the other 69 peptides and calculating the respective mean frequency at each percent identity. The histograms on the right were generated by comparing each of the β types to the corresponding α and β types. Solid bars represent comparisons with sequences of α types and open bars represent comparisons with sequences of β types.