

Abstract
Expression of the costimulatory molecule CD40 on both B cells and DCs is required for induction of experimental autoimmune encephalomyelitis (EAE), and cell-autonomous CD40 expression on B cells is required for primary T-dependent (TD) antibody responses.We now ask whether the function of CD40 expressed by different cell types in these responses is mediated by the same or different cytoplasmic domains. CD40 has been reported to possess multiple cytoplasmic domains, including distinct TRAF6 and TRAF2/3 binding motifs. To elucidate the in vivo function of these motifs in B cells and dendritic cells (DCs) involved in EAE and TD germinal center responses, we have generated knock-in mice containing distinct CD40 cytoplasmic domain TRAF-binding site mutations and have used these animals, together with bone marrow chimeric mice, to assess the roles that these motifs play in CD40 function. We found that both TRAF2/3 and TRAF6 motifs of CD40 are critically involved in EAE induction and demonstrated that this is mediated by a role of both motifs for priming of pathogenic T cells by DCs. In contrast, the TRAF2/3 binding motif, but not the TRAF6 binding motif, is required for B cell CD40 function in TD high affinity antibody responses. These data demonstrate that the requirements for expression of specific TRAF-binding CD40 motifs differ for B cells or DCs that function in specific immune responses and thus identify targets for intervention to modulate these responses.
Introduction
The costimulatory molecule CD40 is widely expressed on multiple antigen presenting cell (APC) types, and has been shown to play an important role in diverse immune responses, including induction/maintenance of autoimmune diseases and primary T-dependent (TD) antibody responses (1). These observations have led us to analyze in our previous studies the identity of the cell populations in which CD40 functions to mediate the autoimmune disease model experimental autoimmune encephalomyelitis (EAE) (2) and the TD germinal center (GC) response that generates class-switched high affinity antibody production (3, 4).
Multiple sclerosis (MS) is a disease of the central nervous system (CNS) that has an autoimmune component and that leads to progressive neurological deficits and disability (5). Analysis of the immune mechanisms underlying MS has been informed by the murine EAE model, which approximates the pathological features of MS. EAE can be induced by immunization with CNS antigens, including the myelin oligodendrocyte (MOG) protein and peptide (6, 7). In EAE, antigen-specific T cells, including differentiated IFN-γ-producing T helper (Th)1 and IL-17-producing Th17 cells, are primed by APCs in peripheral lymphoid organs. Th17 cells can also produce IFN-γ (IFN-γ and IL-17 double positive cells) mediated by T-bet and Runx1 or Runx3, and this developmental flexibility has been linked to the pathogenicity of Th17 cells in multiple autoimmune diseases, including EAE (8). Pathogenic T cells cross the blood brain barrier, are re-stimulated by local APCs in the CNS, and recruit additional immune cells that mediate tissue damage (9). In addition to the role of pathogenic Th cells, B cells are also required for EAE induced by recombinant human myelin oligodendrocytes glycoprotein (rhMOG) (10, 11), paralleling the observation that B cell infiltration, antibodies to myelin components, and activated complement are routinely detected in MS patients (12, 13). Our recent study showed that CD40 expression on both B cells and dendritic cells (DCs) is required for induction of EAE by rhMOG and that CD40 on these two cell types functions through distinct and complementary mechanisms, with CD40 on DC functioning through priming of pathogenic Th17 cells, while the role of CD40 on B cells is mediated by production of MOG-specific IgG (2).
In addition to a critical function in EAE, CD40 also play an essential role in TD antibody production, which is a critical aspect of the adaptive immune response to pathogens and vaccine development. To generate class-switched, high-affinity antigen-specific antibodies, antigen-activated B cells enter GC where cognate interaction with T follicular helper cells (Tfh) results in class switch recombination, somatic mutation of B cell antigen receptor (BCR) genes, and clonal expansion with selection of B cells expressing high affinity antigen-specific BCRs (14, 15). GC B cell responses and TD antibody responses are dependent on costimulatory interactions with T cells, including a requirement for CD40, as demonstrated by the inhibitory effects of blocking anti-CD40 or CD154 antibodies and studies of CD40 or CD154-deficient mice (16–19). Using conditional knockouts and bone marrow (BM) chimeras we have demonstrated that signals provided by CD40 expressed on B cells, but not on DC, are required for primary GC responses (3, 4).
It has been reported that the CD40 cytoplasmic tail contains multiple functional domains including two distinct intracellular motifs that regulate the binding to either TRAF6 or TRAF2/3, adapter proteins that are involved in the CD40 signaling cascade (20–22). It was therefore of interest to determine whether the same or distinct CD40 TRAF binding-motifs are important for CD40 function of B cells and DCs in immune responses. To address this question, we have generated mice expressing CD40 TRAF binding motif mutations by CRISPR/Cas9 technology. Using these mice, together with BM chimeras, we have identified distinct requirements for TRAF6 and TRAF2/3 binding motifs of CD40 on DC and B cells during both primary GC responses and the autoimmune pathogenesis of EAE.
Materials and methods.
Mice
CD40 cytoplasmic domain mutant mice were generated using CRISPR/Cas9 technology (Genome Modification Core, NCI, NIH). C57BL/6 (WT) mice were purchased from Charles River Laboratories. CD40−/− (B6.129P2-Cd40tm1Kik/J) and μMT (B6.129S2-Ighmtm1Cgn/J) mice were purchased from the Jackson Laboratory. CD40 fl/fl mice were provided by Dr. David Wagner (University of Colorado Denver - Anschutz Medical, Aurora, CO) as described previously (2). Mixed BM chimera mice were generated by reconstitution of 6×106 total T cell-depleted BM cells from μMT and CD40 WT/mutant donor mice at a 1:1 ratio into irradiated (950 rad) WT host mice by intravenous injection and were used after 6 weeks. Mice were maintained in accordance with National Institutes of Health guidelines. All animal experiments were approved by the National Cancer Institute Animal Care and Use Committee.
Immunoprecipitation and Western blotting
A combined stimulation/ immunoprecipitation protocol was used to isolate CD40 as previously described (23). Briefly, Protein G magnetic beads (Dynal) were coated with goat-anti-mouse IgG (5 μg/10 μl beads) (Jackson ImmunoResearch) followed by anti-mouse CD40 mAb 3/23 (10 μg/10 μl beads). After stimulation of splenocytes with CpG ODN1826 (0.2 μg/ml) overnight, the cells were washed and incubated with anti-CD40 coated magnetic beads for 20 min at 37°C. Following incubation, beads and cells were pelleted by centrifugation (2 min 300×g) and the medium discarded. Cells were lysed and beads washed as described (24). Bead-bound proteins were eluted in SDS-PAGE sample loading buffer by boiling and analyzed by SDS-PAGE and immunobloting using anti-TRAF2 (MBL M112–3) or anti-CD40 mAb (Abcam ab65853).
Immunizations and adoptive transfer studies
Mice were immunized i.p. with 100 μg NP-KLH or NP-OVA (Biosearch Technologies) mixed 1:1 with Imject Alum (Thermo) in a total volume of 200 μl. For adoptive transfer experiments, CD45.1 OT-II Tg CD4+ T cells were purified using a MACS CD4+ T cell isolation kit (Miltenyi Biotec) and 5×105 cells were transferred by intravenous injection into host mice 1 day before immunization as described previously (4).
EAE induction and scoring
EAE induction was performed by mixing 150 μg rhMOG (Protein Expression Laboratory, Leidos Biomedical Research, Inc., Frederick National Laboratory for Cancer Research, Frederick, MD) or 200 μg of MOG35–55 peptide (MEVGWYRSPFSRVVHLYRNGK) (GenScript) in complete Freund’s adjuvant containing Mycobacterium tuberculosis H37Ra (Difco Laboratories). Pertussis toxin (120 ng) (List Biological Laboratories) diluted in PBS was administered by intraperitoneal injection on days 0 and 2 post-immunization. The clinical severity of EAE was scored by an observer who was unaware of mouse genotypes using a grading scale of 0–5 as previously described (2, 7).
Isolation of mononuclear cells from peripheral lymph nodes and CNS parenchyma
Inguinal and axillary DLN were mixed and digested with 2.5 mg/ml collagenase D (Roche) and 1 mg/ml DNase I (Roche) for 30 min at 37°C. The samples were then filtered through 70 μm mesh strainers to generate single cell suspensions. For isolation of CNS-infiltrating cells, brain and spinal-cord tissues were minced and digested with collagenase D and DNase I as above. The samples were then filtered through 70 μm mesh strainers and centrifuged through a discontinuous Percoll (Sigma) density gradient (38% and 70%). Mononuclear cells at the interphase were removed, washed, and resuspended in culture medium for analysis by flow cytometry.
Flow cytometry
Cells were washed with FACS buffer (HBSS containing 0.2% BSA and 0.05% sodium azide), treated with anti-FcR (24G2), and then stained using specific antibodies. Anti-mouse CD4, CD8, B220, CD19, CD11c, MHC-II, and CD40 antibodies were purchased from Biolegend. For intracellular cytokine staining, mononuclear cells from DLN and CNS were stimulated with PMA/ionomycin for 2 hr at which time GolgiStop (BD Biosciences) was added for additional 2 hr. The cells were fixed and permeabilized with the BD Fix/Perm kit (BD Biosciences) according to the manufacturer’s instructions and stained with anti-IFN-γ, anti-IL-17A and isotype control antibody (BD Biosciences) for 30 min. Data were collected with a FACS LSR II or FACS Fortessa flow cytometer (BD Biosciences) and analyzed using FlowJo software.
Antibody ELISA
NP-specific and MOG-specific IgG was measured by ELISA. In brief, NP30-BSA (Biosearch Technologies) or rhMOG (10 μg/ml) was coated on ELISA plates (Immulon 4HBX; Thermo Fisher) at 4°C overnight. The high affinity IgG antibody titer was determined by coating plates with NP2-BSA overnight. The plates were washed with ELISA wash buffer (0.5% Tween in PBS), serially diluted sera were applied to the plates, and plates were incubated 2 hr at room temperature. After washing, anti-mouse IgG HRP (Southern Biotech) was used to detect bound serum IgG. After a wash step, 2,2’-Azino-di-(3-ethylbenzthiazoline-6-sulfonate) (ABTS) substrate (KPL) was added to the wells, and enzyme reaction was stopped after 20 mins using ABTS HRP Stop Solution (KPL). The optical density at 405 nm was measured using a SpectraMax iD3 plate reader.
Statistical analyses
Student’s t test with two-tailed distributions was performed for statistical analyses with single comparisons. For multiple comparisons, statistical analysis was performed with one-way ANOVA followed by Dunnett’s multiple comparison. P-values <0.05 were considered statistically significant.
Results
Generation of CD40 cytoplasmic domain mutant knock-in mice
In vitro studies have suggested that there are multiple functional motifs in the CD40 cytoplasmic domain that are involved in CD40 signaling. Among these are two cytoplasmic motifs that are thought to be important for the recruitment of the adaptor molecules TRAF6 or TRAF2/3 to CD40 (25, 26). We generated mice expressing cytoplasmic domain mutations at the endogenous CD40 locus by CRISPR/Cas9 as illustrated in Figure 1A. A proximal domain CD40 cytoplasmic tail mutant (termed CD40ΔT6) was generated by deletion of the sequence 234RQDPQE239, which is homologous to the TRAF6 binding motif in human CD40 (27). A distal domain CD40 cytoplasmic tail mutant (termed CD40ΔT2) was made by deletion of the sequence 251PVQET255, which corresponds to the TRAF2 binding motif 251PxQxT255sequence in human CD40 (27–29). We also established a third strain by truncation of sequence distal to amino acid 245 by inserting two stop codons (TAATAG), which preserves the proximal TRAF6 binding domain and deletes distal TRAF2/3 binding domain as well as CD40 cytoplasmic amino acids 246–278 (termed CD40ΔT2*). This region of the CD40 cytoplasmic domain is identical to that in human CD40 and also contains a putative second TRAF2 binding motif (29). All CD40 cytoplasmic domain mutants express levels of CD40 on the surface of B and DCs that are comparable to CD40 expressed in C57BL/6 (WT) mice both at steady state as well as after overnight stimulation with CpG (Fig. 1B). Co-immunoprecipitation studies using CD40-stimulated mouse splenocytes confirmed that TRAF2 bound to WT CD40 and CD40ΔT6 but was unable to bind to CD40 expressed in CD40ΔT2 and CD40ΔT2* mice (Fig 1C). Nearly identical results were obtained when examining the binding of epitope-tagged TRAF2 co-expressed with WT or muant CD40 molecules in transfected HeLa cells (Supplementary Fig 1). We were unable to reproducibly co-immunoprecipitate TRAF6 even in WT CD40 expressing mice, a finding that is consistent with other reports that failed to show direct TRAF6/CD40 interactions in vitro (27).
Figure 1. Generation of endogenous CD40 cytoplasmic domain mutant mice.
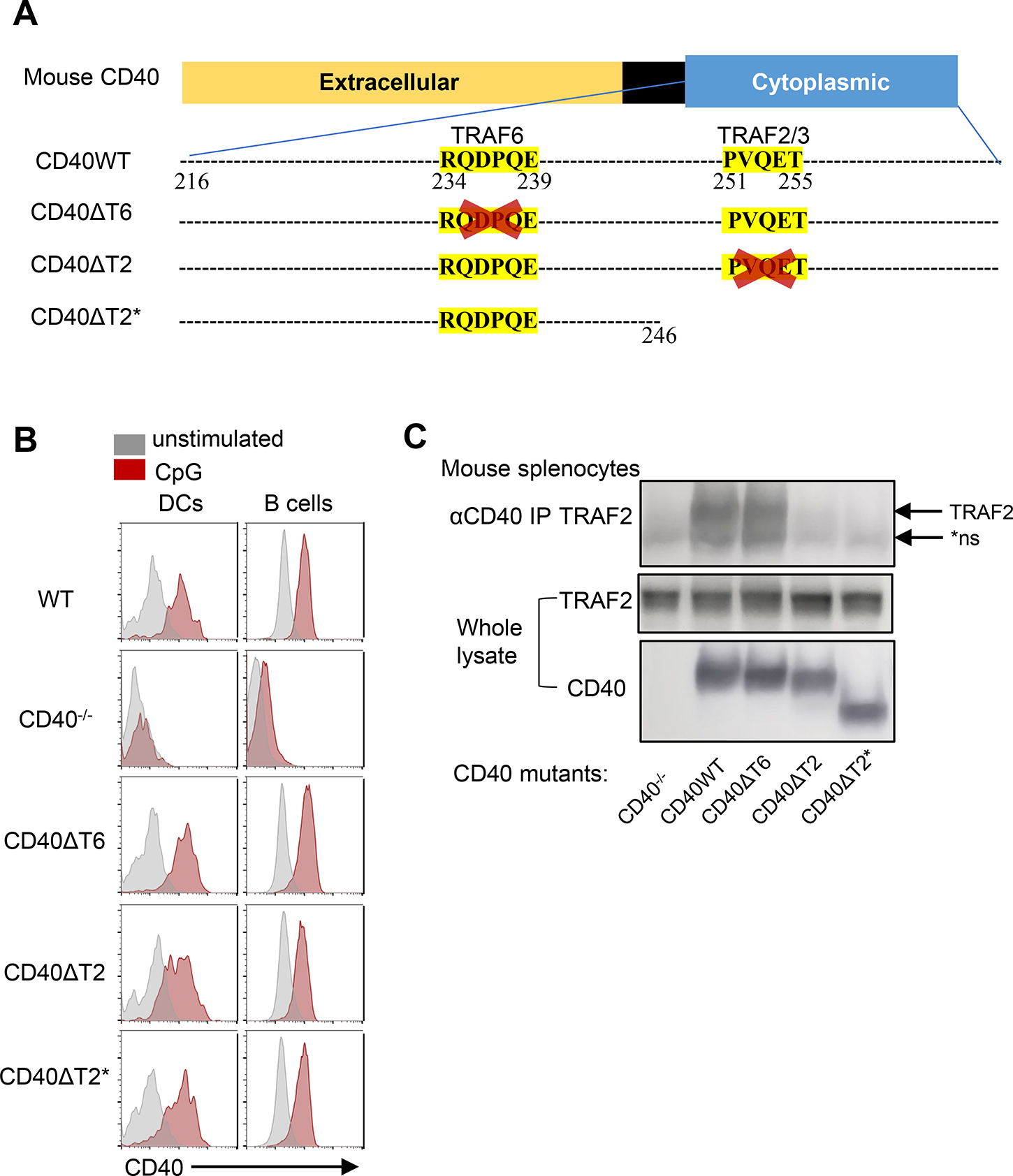
A. Schematic representation of WT CD40 and mutants. CD40ΔT6 was generated by deletion of the sequence 234RQDPQE239. CD40ΔT2 was made by deletion of the sequence 251PVQET255. CD40ΔT2* was made by truncation of sequence distal to amino acid 245 by inserting two stop codons (TAATAG). B. Representative FACS analysis of CD40 surface expression on the same mouse spleen DCs (PI−CD11c+B220−) and B cells (PI−CD11c−B220+) that were in 4°C overnight (gray) or incubated with CpG overnight (red). C. CD40 was immunoprecipitated from cell lysates of the indicated mice and analyzed bound proteins immunoblotting by using TRAF2 antibody. Lower two panels showed expresion of TRAF2 and CD40 in whole cell lysates. The arrow indicates the position of TRAF2 and the asterisk indicates the position of a non-specific band present even in CD40−/− mice (*ns).
Both CD40 TRAF6 and TRAF2/3 binding motifs on non-B cells are required for severe EAE
CD40 expression is required for mice to develop EAE (2, 30, 31). We therefore analyzed the requirement for the distinct TRAF6 and TRAF2/3 binding-motifs in EAE. Immunization of mice with rhMOG results in a form of EAE that requires both T and B cells, mirroring the known roles of these cell types in human MS (5, 10, 11, 32). All CD40 TRAF binding motif mutant mice showed a significant reduction of EAE severity relative to WT mice (Fig.2A), indicating that the CD40 TRAF6 and TRAF2/3 binding-motifs play important, and not completely redundant, roles in EAE. EAE severity in CD40−/− mice was significantly lower than in any single TRAF-binding motif mutant mouse, suggesting that the TRAF2/3 and TRAF6 binding motifs have partially overlapping functions and/or that additional CD40 domains function in EAE.
Figure 2. Both CD40 TRAF6 and TRAF2/3 binding motifs on non-B cells are required for EAE.
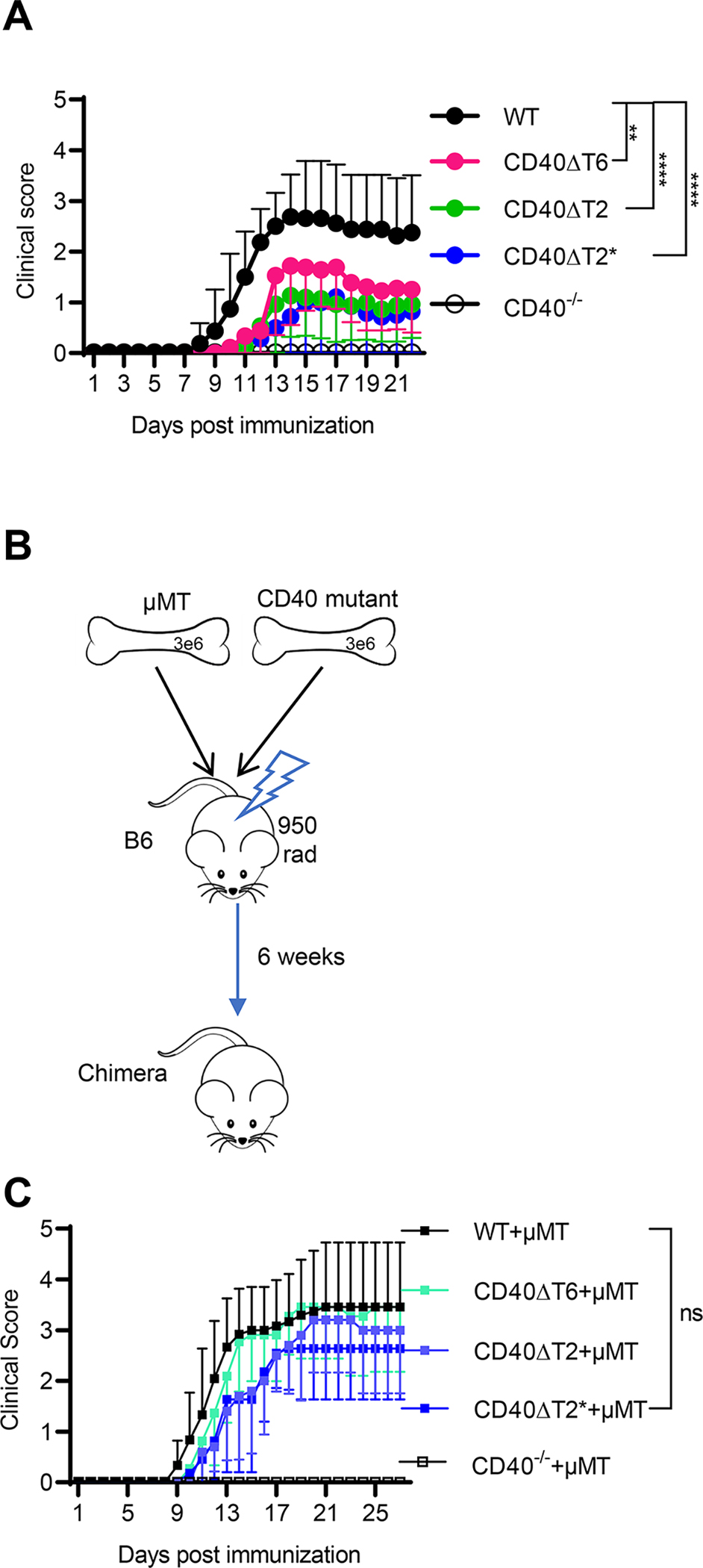
WT or CD40 mutant mice were immunized with rhMOG and the severity of EAE was examined over time. A. Clinical score of indicated mice immunized with rhMOG. B. Radiation BM chimeras were established by mixing BM from μMT mice with BM from CD40 mutant mice. C. EAE was induced in the indicated chimeric mice with rhMOG. EAE scores are combined from three independent experiments (mean ± S.E.M. using 10–18 mice per group). Median of the clinical score during day 11–27 for WT mice was compared to each CD40 mutant using a two-tailed non-parametric Mann-Whitney test. ** p<0.01, **** p<0.0001, ns not significant. Comparing clinical scores of CD40−/− mice to CD40ΔT6 (p<0.001), CD40ΔT2 (p<0.01), or CD40ΔT2* (p<0.01) mice also revealed statistically significant differences.
We have previously shown that CD40 expression on both DCs and B cells is required for the development of EAE (2). To address whether the CD40 TRAF-binding motifs on B cells or DCs (or both) play a role in EAE, we generated mixed bone marrow chimeras in which expression of mutant CD40 was restricted to B cells, while all other APCs in the mouse express WT CD40 (Fig. 2B). Chimeric mice with B cells derived from CD40−/− donors were completely protected from EAE, demonstrating a requirement for B cell CD40 in mediating EAE. By contrast, chimeras with B cells derived from CD40 TRAF-binding motif mutants all developed severe EAE similar to the WT group (Fig. 2C). Since EAE was less severe in mice expressing CD40 TRAF-binding motif mutations (in which all APC subtypes, including B cells, possess mutant CD40 proteins), these data show that the reduced EAE observed in CD40 TRAF-binding motif mutant mice was a consequence of impaired CD40 function on APCs other than B cells.
EAE can also be induced in mice by immunizing mice with MOG35–55 (7). In contrast to rhMOG-induced EAE, MOG35–55-induced EAE is a B cell-independent disease (10, 11). As was observed in rhMOG-based disease, each of the CD40 TRAF-binding motif mutant mice showed a significant reduction of EAE severity relative to WT mice when immunized with MOG35–55 (Supplementary Fig. 2). This result further supports our finding using chimeric mice and demonstrates that TRAF2/3 binding and TRAF6 binding motifs of CD40 APC other than B cells are important in EAE.
The CD40 TRAF6 and TRAF2/3 binding motifs function in EAE through optimal priming of Th17 cells
An early event following rhMOG immunization is the priming and differentiation of inflammatory cytokine-producing CD4+ T cells in lymph nodes, of which IL-17A-producing Th17 cells and IL-17A- and IFNγ-double-producing cells are the most pathogenic (33). Using conditional CD40−/− mice, we confirmed our previous observation (2) that CD40 expression on DCs, but not on B cells, is required for priming of pathogenic Th17 cells (Fig. 3A). To determine if the TRAF binding motifs on DC are involved in CD4 priming in draining lymph nodes (DLN), we measured the generation of cytokine-producing CD4+ T cells in DLN of WT and TRAF binding motif mutant mice following rhMOG immunization (Fig. 3B). All CD40 TRAF binding motif mutant mice showed lower numbers of CD4+ T cells, equivalent to the defective response of CD40−/− mice (Fig. 3C). The frequency of IL-17A-producing Th17 cells in DLN was markedly reduced in all CD40 TRAF binding motif mutant mice (Fig. 3D), demonstrating that the TRAF6 and TRAF2/3 binding motifs on DCs play a non-redundant role in the induction and differentiation of pathogenic Th17 cells in the periphery of rhMOG-immunized mice.
Figure 3. Both CD40 TRAF6 and TRAF2/3 binding motifs function in EAE by regulating Th17 cell priming.
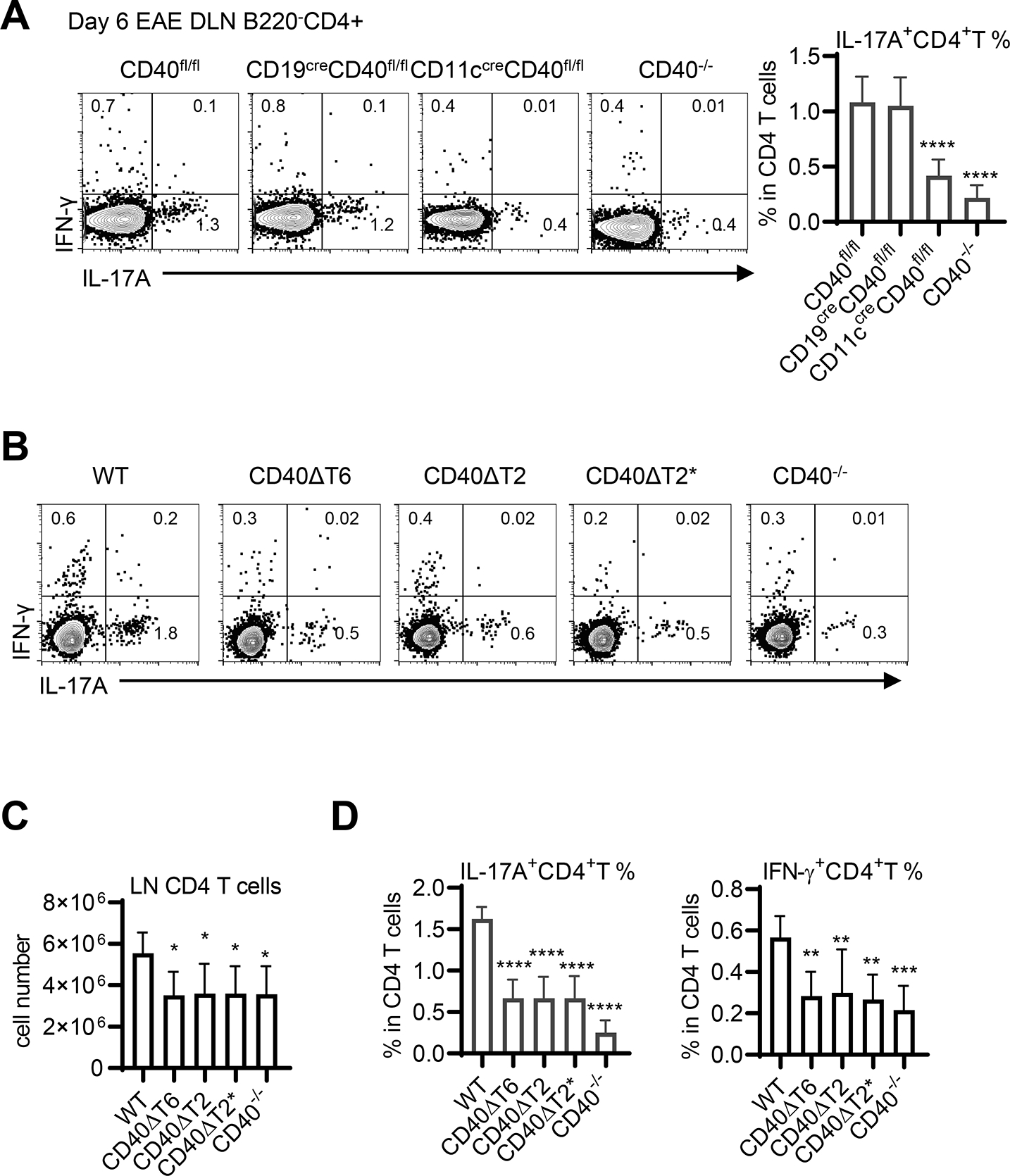
WT or CD40 mutant mice were immunized with rhMOG and 6 days after EAE induction CD4+ T cells from the DLN of the indicated mice were analyzed by FACS analysis. A. DLN cells from rhMOG-immunized CD40fl/fl mice, CD40−/− mice, or CD40fl/fl mice crossed with CD19-Cre or CD11c-Cre (to delete CD40 only on B cells or DCs, respectively) were analyzed for cytokine production by FACS analysis (in the CD4+ B220− gate) and the percentage of CD4 T cells expressing IL-17A was determined. B. DLN CD4+ T cells from rhMOG-immunized WT or CD40 TRAF binding-motif mutant mice were analyzed for cytokine production by FACS analysis (in the CD4+ B220− gate). The number of CD4 T cells in the DLN (C) and the percentage of all DLN CD4 T cells expressing either IL-17A or IFN- g was determined (D). Data are combined from two independent experiments (mean ± S.D. using 6 mice per group). One-way ANOVA followed by Dunnett’s test against the immunized WT group was performed for multiple comparisons. * p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001.
The CD40 TRAF6 and TRAF2/3 binding motifs function in EAE by promoting the accumulation of pathogenic Th17 cells in CNS
We also monitored the presence of CD4+ T cells in the CNS (brain and spinal cord) of rhMOG-immunized WT and CD40 TRAF binding motif mutant mice. Immunized CD40−/− mice, which did not develop disease, had very few CNS mononuclear cells and even fewer CD4+ T cells recoverable from brain and spinal cord (Fig. 4A). All CD40 TRAF binding motif mutant mice, which were significantly protected from EAE, had comparably reduced numbers of CD4+ T cells in the CNS when compared to WT mice. Substantial proportions of CD4+ T cells in the CNS of immunized CD40 WT mice were IL-17A+ and IL-17A+/IFN-γ+ (Fig. 4B), whereas the small numbers of CD4+ T cells isolated from the CNS of CD40−/− mice had significantly lower frequencies of cytokine-positive populations. All CD40 TRAF binding motif mutant mice had reduced frequencies of these pathogenic Th cells in the CNS, a finding that is consistent with reduced EAE severity and reduced frequencies of Th17 cells in the DLN in these mice. As was observed for clinical EAE severity, the reduction in Th17 cells in CD40−/− mice was even more profound than in any single TRAF binding motif mutant, suggesting that the functions of these two motifs are not completely redundant and/or that other domains of the CD40 molecule play a role in Th17 generation. Taken together with our conditional CD40 deletion data, these findings demonstrate the importance of the CD40 TRAF6 and TRAF2/3 binding motifs on DCs in the differentiation of pathogenic Th17 cells the periphery of rhMOG-immunized mice and in the pathogenesis of EAE.
Figure 4. The CD40 TRAF6 and TRAF2/3 binding-motifs function in EAE by mediating the accumulation of pathogenic Th17 cells in CNS.
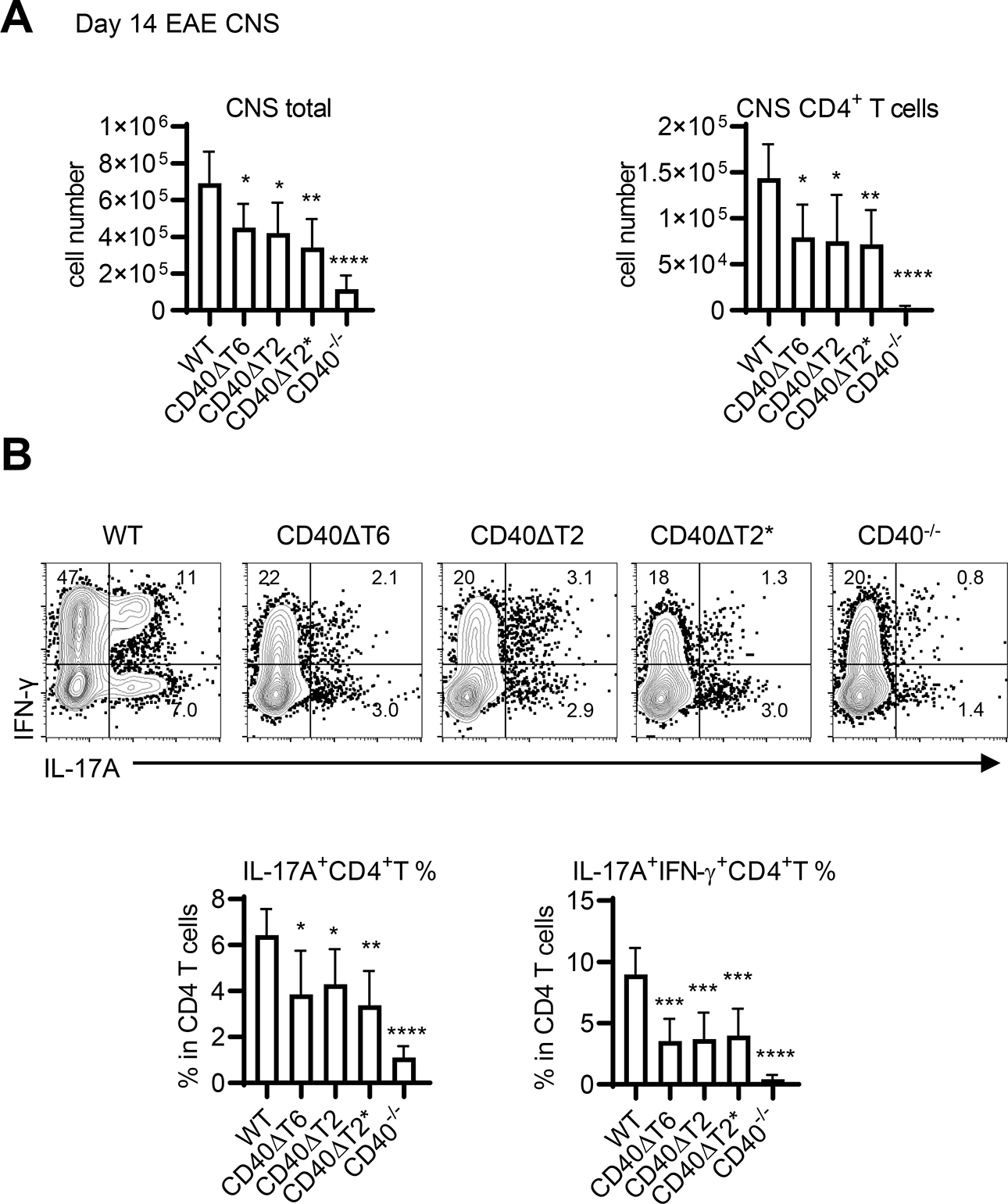
A. Day 14 post EAE induction, total mononuclear cells were recovered from the CNS and the total number of mononuclear cells as well as CD4+ T cells present in the CNS was determined by FACS analysis. B. CNS CD45.2+CD11b−CD4+ T cells were analyzed for cytokine production by FACS analysis and the percentage of all CNS CD4 T cells expressing either IL-17A alone, or both IL-17A and IFN-γ was determined. Data are combined from two independent experiments (mean ± S.D. using 6 mice per group). One-way ANOVA followed by Dunnett’s test comparing the immunized WT group to each CD40 mutant was performed for multiple comparisons. * p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001. Comparison of the immunized CD40−/− group to each CD40 mutant revealed statistically significant differences: for IL-17A+ CD4 T cells p<0.01 compared to ΔT2 and ΔT6, p<0.05 compared to ΔT2*; for IL17A+/IFN-g+ CD4 T cells p<0.05 compared to ΔT2 and ΔT6, p<0.01 compared to ΔT2*.
The TRAF2/3 binding motif of CD40 expressed by B cells is required in T-dependent GC responses
We have previously reported that CD40 expression on B cells, but not on DC, is required for TD germinal center responses and high affinity antibody production (3, 4). Having established that both TRAF2/3 and TRAF6 binding motifs of CD40 are required for DC-dependent T cell priming in EAE, we assessed the role of these two TRAF binding sites on CD40 for primary T-dependent GC responses. CD40 WT and mutant mice were immunized with NP-KLH in alum and spleen GC B cells were analyzed 8 days after immunization by flow cytometry. In comparison to the response of mice containing WT CD40, GC B cell numbers were significantly decreased in CD40ΔT2 and CD40ΔT2* mutant mice to levels approximating that in complete CD40−/− mice. By contrast, the response of CD40ΔT6 mice was comparable to that of WT mice (Fig. 5A). The total B cell number in CD40−/− mice was comparable to WT mice in unimmunized mice (16, 17) and the total spleen B cell numbers were also comparable among all strains after immunization (Fig 5B, left). Like GC B cells responses, antigen-specific Tfh differentiation was significantly reduced in spleens from CD40ΔT2 and CD40ΔT2* mice and was unaffected in CD40ΔT6 mice (Fig. 5C). These results indicate that whereas CD40 does not play a role in the maintenance of total spleen B cells numbers, the CD40 TRAF2 binding motif, but not the TRAF6 binding motif, is required for antigen-specific Tfh differentiation and GC B cell generation. Despite the significantly reduced numbers of GC B cells and Tfh cells, the antigen-specific class switched IgG response measured as binding to NP30-BSA was unaffected by mutations in any of CD40 cytoplasmic TRAF binding-motif mutants (Fig. 5D). A similar result was observed by measuring the anti-rhMOG IgG levels in mice induced with EAE (Supplementary Fig.3). Notably, however, high affinity anti-NP IgG, measured as binding to NP2-BSA, was substantially reduced in CD40ΔT2 and CD40ΔT2* strains relative to that observed in WT mice, although greater than that of CD40−/− mice. High affinity anti-NP2 IgG from CD40ΔT6 mice was comparable to that found in immunized WT mice, a finding that is consistent with the observation that GC B cell numbers in immunized WT and CD40ΔT6 were identical.
Figure 5. The CD40 TRAF2 binding motif but not the TRAF6 binding motif is required for T-dependent germinal center responses.
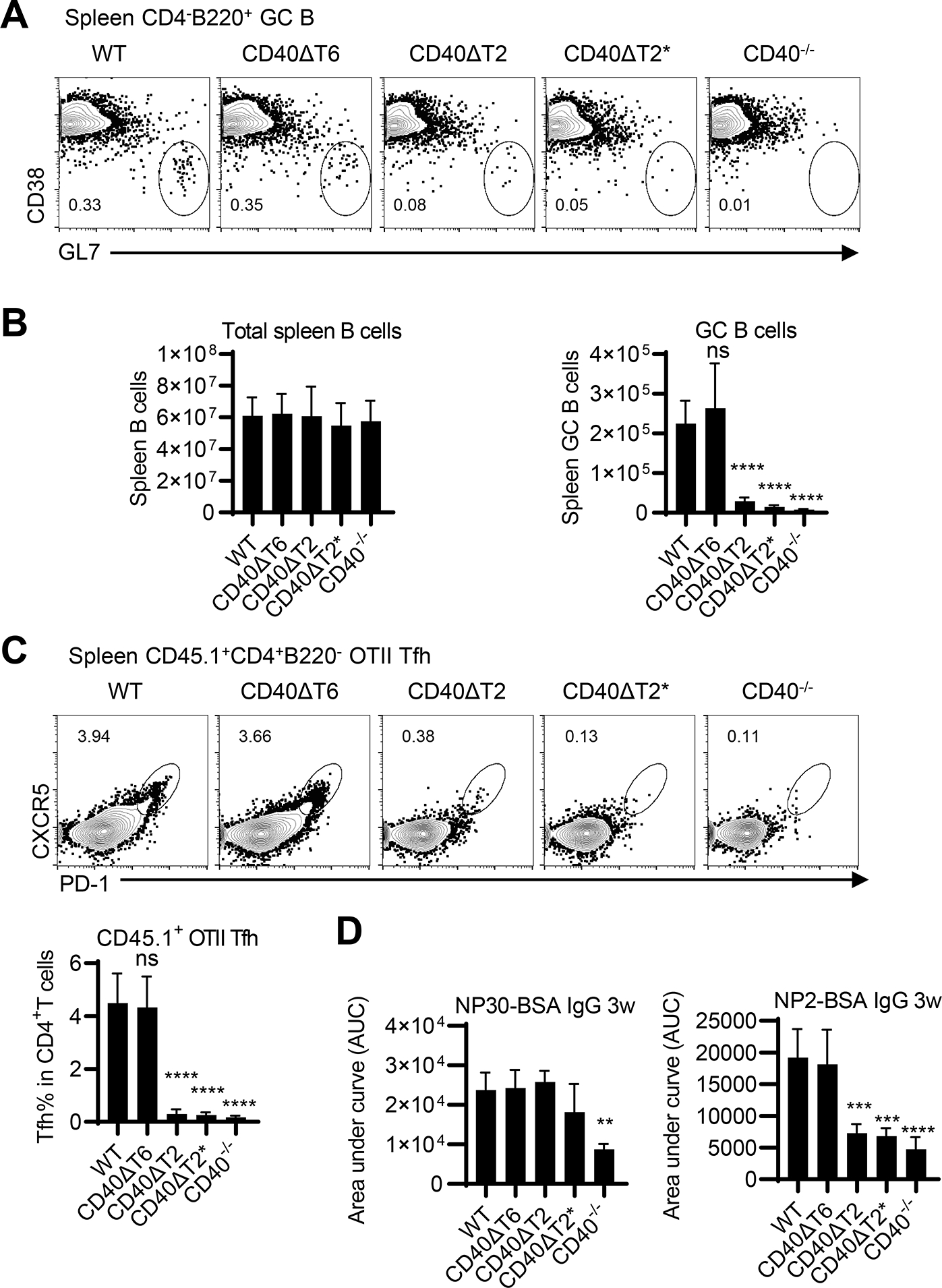
WT and CD40 mutant mice were immunized with NP-KLH in alum. After 8 days, GC B cells and total B cells were analyzed in the spleen. Representative FACS analysis (A) of CD38−GL7+ GC B cells in the CD4− B220+ B cell gate and statistical analyses of FACS data examining the total number of spleen B cells and GC B cells in each mouse strain (B). C. OT-II T cells (CD45.1) were transferred to the indicated recipient mice (CD45.2) and were immunized with NP-OVA/alum one day after cell injection. Transferred OT-II cells were analyzed at day 8 by FACS analysis. A representative flow cytometry profile of spleen OT-II cells (CD45.1+CD4+B220−) analyzed for the Tfh CXCR5high PD-1high phenotype and statistical analyses of FACS data showing the percentage of transferred OT-II cells expressing a Tfh phenotype were shown. D. WT and CD40 mutant mice were immunized with NP-KLH in alum. Serum was collected three weeks after immunization, and total NP-specific antibody (anti-NP30 IgG) and high-affinity NP antibody (anti-NP2 IgG) was determined by ELISA. Data are combined from two independent experiments (mean ± S.D. using 6 mice per group). One-way ANOVA followed by Dunnett’s test against immunized WT group was performed for multiple comparisons. ** p<0.01, *** p<0.001, **** p<0.0001.
Although we have previously shown that CD40 on DCs is not required for TD antibody responses, we specifically examined the importance of the TRAF binding-motifs on B cells during this process. We constructed radiation BM chimeras in which BM from μMT mice was mixed with BM from CD40 cytoplasmic domain mutant mice and transferred into lethally irradiated WT recipients. The B cells in these chimeric mice possess either WT or mutant CD40, while all other cell types (including DCs) have intact CD40 (Fig 6A). The total numbers of spleen B cells in each chimera were comparable (Fig. 6B); however, chimeras with CD40ΔT2 and CD40ΔT2* B cells, but not CD40ΔT6 B cells, showed significant reduction of GC B cells following immunization with NP-KLH in alum. Taken together with our studies in CD40 TRAF binding motif mutant mice, these data demonstrate that the CD40 TRAF2 binding motif, but not the TRAF6 binding motif, in B cells is required for germinal center development and high affinity antibody production.
Figure 6. The CD40 TRAF2 binding motif on B cells is required for T-dependent germinal center responses.
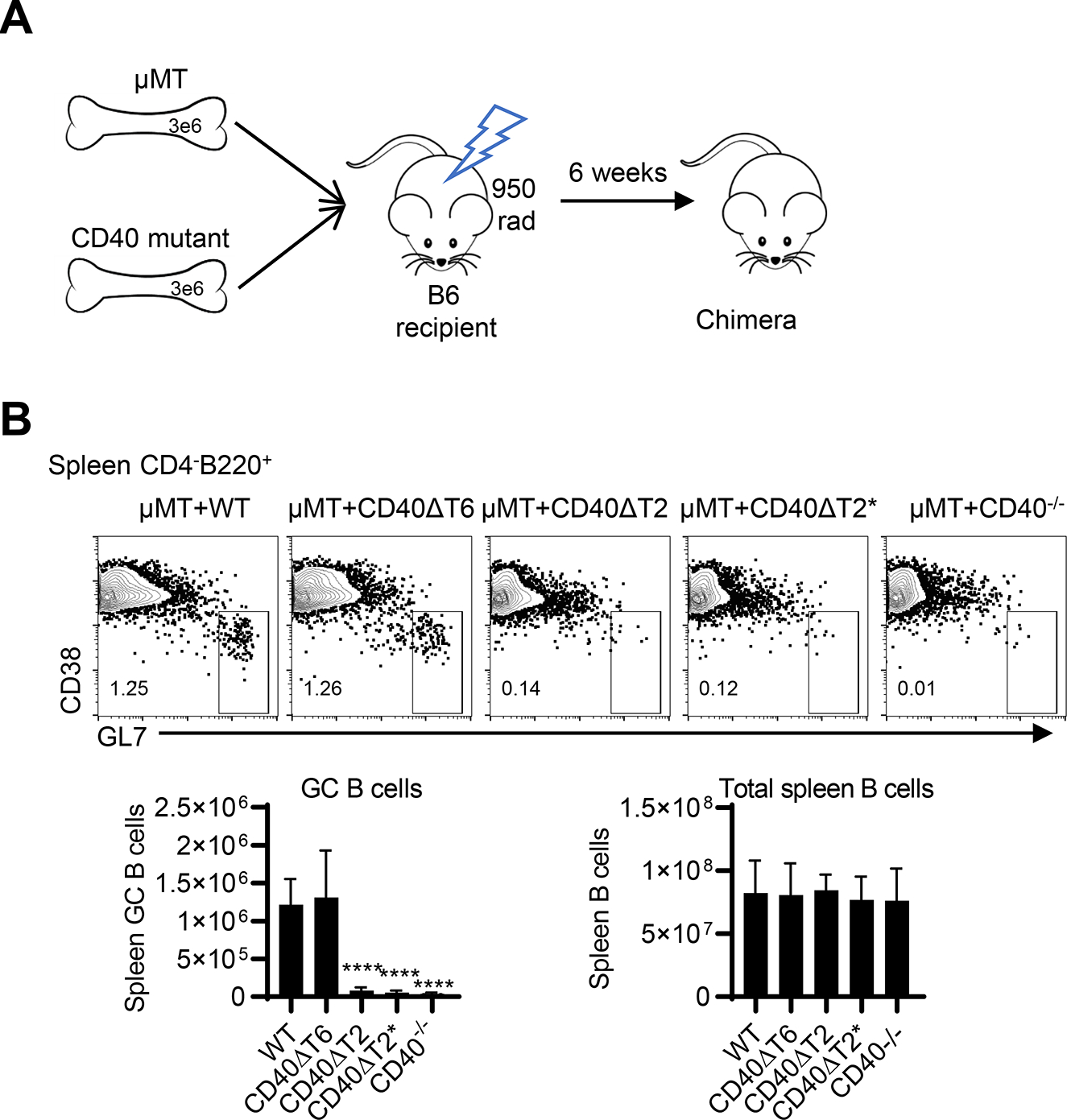
A. Radiation BM chimeras were established by mixing BM from μMT mice with BM from CD40 WT or mutant mice and transferring these BM cells into lethally irradiated B6 recipients. After 6 weeks, chimeric mice express WT or mutant CD40 only on B cells whereas all other APC cell types possess WT CD40. B. The indicated chimeric mice were immunized with NP-KLH in alum and after 8 days, GC B cells and total B cells were analyzed in the spleen. Representative FACS analysis of CD38−GL7+ GC B cells in CD4−B220+ B cell gate. Statistical analyses of FACS data examining the total number of GC B cells and total spleen B cells, respectively. Data are combined from two independent experiments (mean ± S.D. using 6 mice per group). One-way ANOVA followed by Dunnett’s test against immunized C57BL/6 group was performed for multiple comparisons. **** p<0.0001.
Discussion
CD40 is a cell surface receptor that belongs to the tumor necrosis factor receptor family and serves as a costimulatory molecule expressed on multiple APC lineages. The cytoplasmic domain of CD40 has been implicated as essential in CD40-CD154 costimulatory signal transduction in a variety of in vitro systems; however, understanding of the roles played by this domain in vivo is incomplete (21, 25, 34). The data presented here reveal cell type- and cytoplasmic domain-specific requirements for CD40 function in vivo. We show that both TRAF6- and TRAF2/3 binding motifs of CD40 on DCs plays an essential role in rhMOG-induced EAE by regulating pathogenic Th cell priming. In contrast, the cytoplasmic membrane distal TRAF2/3 binding motif, but not the membrane proximal TRAF6 binding motif, is required for CD40 expressed on B cells for TD humoral immune responses, including optimal GC B cell production, Tfh generation, and expression of high affinity antibodies.
Previous work in our lab, and others, highlighted the critical role of CD40 in mediating autoimmune and TD GC responses. The interaction between CD40 and TRAF molecules has been intensively investigated in cell lines, which suggested that TRAFs are involved in CD40 function (22, 25, 26, 35–37). Here, we have generated mutations in the proposed CD40 cytoplasmic TRAF2 binding motif and have found that endogenous TRAF2 does not bind to CD40 in spleen cells in which this motif has been mutated (CD40ΔT2 and CD40ΔT2*) or when TRAF2 is co-expressed with CD40ΔT2 or CD40ΔT2* in HeLa cells, consistent with previous reports (27, 38). Although we were able to reliably co-immunoprecipitate CD40 with TRAF2, we were unable to detect the binding of CD40 with TRAF6 in mouse spleen cells or in transfected HeLa cells. It is important to note that this does not mean that TRAF6 does not interact with CD40 in vivo, but simply shows that under the immunoprecipitation conditions used in our study the interaction is not preserved. This result is, however, consistent with a previous report that also failed to detect any binding between CD40 and TRAF6 in vitro (27). It is also possible that alterations in the CD40 cytoplasmic domains used in our and other studies act through CD40 interaction with as yet unidentified molecules. Despite deletion of the TRAF6 and TRAF2/3 binding motifs, both surface expression of CD40 as well as TLR-mediated upregulation of CD40 expression proceeds normally in these mice, demonstrating that some components of the machinery driving CD40 expression and upregulation are independent of the CD40 cytoplasmic TRAF binding motifs.
Our lab recently demonstrated that CD40 on DCs, but not on B cells, is essential for peripheral Th1 and Th17 priming and CNS accumulation in rhMOG-induced EAE (2). In the present study we used the same rhMOG-induced EAE model to further examine the cellular mechanism mediating the function of CD40 in this autoimmune disease. Mice expressing either TRAF6 or TRAF2/3 binding motif mutations developed less severe EAE than did mice expressing WT CD40. The use of mixed bone marrow chimeras revealed that this was not a consequence of CD40 TRAF binding motif deletion on B cells, since EAE severity was not affected when expression of either TRAF6 or TRAF2/3 binding-motif mutant was restricted to B cells. These data point to a role of these domains of CD40 in a cell other than a B cell. Since expression of CD40 on DCs, but not on B cells, impairs peripheral Th17 priming, the inability of both CD40 TRAF binding motif mutant mice to generate pathogenic Th17 cells in the periphery of rhMOG-immunized mice demonstrates that these two motifs exert distinct and non-redundant roles of DC CD40 in priming of CD4 T cells. Analysis of the T cell subsets in CNS of EAE mice revealed that all CD40 TRAF binding motif mutant mice used in this study had impaired pathogenic Th17 CNS infiltration, a result that could be a consequence of simply reduced numbers of peripheral pathogenic Th17 cells primed by DCs or by another APC in the brain that also requires both TRAF binding-motifs for full activity. It is interesting to note that a recent study reported that myeloid cells plays a role in EAE (39), leading to the possibility that the non-APC expressing the CD40 TRAF binding motif-mutations used in this study could also be due to CD40 expression on one of these cell types. The observation that clinical EAE severity as well as Th17 priming in CD40−/− mice was even lower than in mice with single TRAF-binding motif mutations suggests that TRAF2/3 and TRAF6 binding motifs have partially overlapping functions or that additional CD40 domains function in EAE.
Previous studies using conditional deletion of TRAFs have addressed the role of specific TRAF proteins in humoral immune responses. Conditional deletion of B cell TRAF2, but not TRAF3, suppressed TD antibody responses but not T cell-independent antibody responses (40, 41). By contrast, conditional deletion of TRAF6 in B cells did not affect germinal center formation or antibody affinity maturation in mice immunized with TD antigens (42), unlike the profound defect in these responses in both CD40-deficient mice or in mice lacking CD40 on B cells (3, 4). Curiously, deletion of TRAF6 on B cells suppressed Ig class switching following immunization with either TD and T cell-independent antigens (42). Since T cell-independent antibody responses do not require CD40-CD40L signaling (16), these data reveal an additional function of TRAF6 in humor immune responses in B cells that is independent of CD40. CD40ΔT2 or CD40ΔT2* mutant mice had significantly reduced GC B cells and antigen-specific Tfh after TD protein antigen immunization, consistent with a role of CD40-TRAF2 interactions during the TD GC response. By generating mixed BM chimeras we also demonstrated a B cell-intrinsic effect of this process, confirming a B cell, but not DC, requirement for CD40 in TD immune responses as we have reported previously (4, 43). Interestingly, the significantly reduced GC B and Tfh cells did not result in decrease in total antigen specific anti-NP30 or anti-rhMOG IgG levels in the serum three weeks post immunization although high affinity anti-NP antibody measured by binding to NP2-BSA was substantially reduced in CD40ΔT2 or CD40ΔT2* mutant mice. In a previous report, transgenic mice expressing TRAF2 binding motif mutant of human CD40 also showed normal serum IgG levels in mice lacking GC after TD antigen immunization response (44). GC-independent serum IgG was also reported in lymphotoxin-alpha-deficient mice, which fail to form GC in the spleen but mount an anti-NP IgG1 response similar to WT mice after immunization with the TD antigen NP-OVA (45). Consistent with previous reports, our data suggest that normal extrafollicular plasma B cell differentiation may occur in the absence of GC response (44).
Two different reports have described CD40−/− mice expressing CD40 transgenes with cytoplasmic TRAF6 and TRAF2/3 binding-motif mutations with expression driven by either the I-Eα promoter (28) or the Ig heavy chain promoter (27). In the I-Eα driven-CD40 mutants, both TRAF6 and TRAF2/3 binding-motif mutations mount a normal GC response after immunization with sheep erythrocytes, however after immunization with NP-KLH/CFA antigen specific IgG1 was significantly reduced in the serum from TRAF6, but not TRAF2/3, binding motif mutant mice (28). By contrast, in Ig heavy chain driven-CD40 mutants, TRAF6 binding motif mutant mice mount a normal GC response and anti-KLH IgG in serum while TRAF2/3 binding motif mutant mice can not (27). Spleen B cells from our CD40 cytoplasmic TRAF6 binding motif mutant mice retain the ability to bind TRAF2 and are able to effectively mount a CD40-dependent GC response, demonstrating this motif is not involved in TRAF2 binding and is not important for TD immune responses. Thus our data, generated from CRISPR/Cas9 mutagenesis of endogenous CD40, and showing that the CD40 binding motif for TRAF2 is essential for CD40-mediated GC formation and high affinity anti-NP2 IgG but not total anti-NP30 IgG production, are most consistent with the findings of Jabara. et al (27).
In conclusion, our data reveal important new information regarding the functional roles of two distinct CD40 cytoplasmic domains expressed on distinct cell types in vivo. DCs require both TRAF2/3 and TRAF6-binding motifs for optimal ability to prime peripheral Th17 cells following immunization with rhMOG and in the development of EAE. In contrast, B cells require the CD40 TRAF2 binding motif, but not the TRAF6 binding motif, in the generation of TD antibody responses. These findings may have translational relevance for strategies specifically targeting pathogenic versus adaptive aspects of CD40-dependent immune response.
Study approval
Animal experiments were approved by the NCI Institute Animal Care and Use Committee. All mice were maintained in accordance with US National Institutes of Health guidelines.
Supplementary Material
Key points:
The TRAF2/3 and TRAF6 binding motifs of CD40 are critically involved in EAE induction
Both motifs are involved in priming of pathogenic T cells by DCs
The TRAF2/3 motif, but not the TRAF6 motif, is required for TD antibody responses
Acknowledgements
We thank Dr.Vanja Lazarevic and Dr. Gail Bishop for their thoughtful comments and review of this manuscript. We thank Nelson Cole for making CD40 mutant constructs for HeLa transfection; Bernardo Rosa, Elena Kuznetsova, and Tarra Dumas from the NCI animal facility for assistance, caring for, and scoring experimental mice.
Funding
This work was supported by the Intramural Research Program of the National Cancer Institute, National Institutes of Health.
Footnotes
This work was supported by the Intramural Research Program of the National Institutes of Health.
References
- 1.Zhang Q, and Vignali DA 2016. Co-stimulatory and Co-inhibitory Pathways in Autoimmunity. Immunity 44: 1034–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lu Y, Xu M, Dorrier CE, Zhang R, Mayer CT, Wagner D, McGavern DB, and Hodes RJ 2022. CD40 Drives Central Nervous System Autoimmune Disease by Inducing Complementary Effector Programs via B Cells and Dendritic Cells. J Immunol 209: 2083–2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lumsden JM, Williams JA, and Hodes RJ 2003. Differential requirements for expression of CD80/86 and CD40 on B cells for T-dependent antibody responses in vivo. J Immunol 170: 781–787. [DOI] [PubMed] [Google Scholar]
- 4.Watanabe M, Fujihara C, Radtke AJ, Chiang YJ, Bhatia S, Germain RN, and Hodes RJ 2017. Co-stimulatory function in primary germinal center responses: CD40 and B7 are required on distinct antigen-presenting cells. J Exp Med 214: 2795–2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ramaglia V, Rojas O, Naouar I, and Gommerman JL 2021. The Ins and Outs of Central Nervous System Inflammation-Lessons Learned from Multiple Sclerosis. Annu Rev Immunol 39: 199–226. [DOI] [PubMed] [Google Scholar]
- 6.Stromnes IM, and Goverman JM 2006. Passive induction of experimental allergic encephalomyelitis. Nat Protoc 1: 1952–1960. [DOI] [PubMed] [Google Scholar]
- 7.Stromnes IM, and Goverman JM 2006. Active induction of experimental allergic encephalomyelitis. Nat Protoc 1: 1810–1819. [DOI] [PubMed] [Google Scholar]
- 8.Wang Y, Godec J, Ben-Aissa K, Cui K, Zhao K, Pucsek AB, Lee YK, Weaver CT, Yagi R, and Lazarevic V 2014. The transcription factors T-bet and Runx are required for the ontogeny of pathogenic interferon-gamma-producing T helper 17 cells. Immunity 40: 355–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Damsker JM, Hansen AM, and Caspi RR 2010. Th1 and Th17 cells: adversaries and collaborators. Ann N Y Acad Sci 1183: 211–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lyons JA, Ramsbottom MJ, and Cross AH 2002. Critical role of antigen-specific antibody in experimental autoimmune encephalomyelitis induced by recombinant myelin oligodendrocyte glycoprotein. European journal of immunology 32: 1905–1913. [DOI] [PubMed] [Google Scholar]
- 11.Lyons JA, San M, Happ MP, and Cross AH 1999. B cells are critical to induction of experimental allergic encephalomyelitis by protein but not by a short encephalitogenic peptide. European journal of immunology 29: 3432–3439. [DOI] [PubMed] [Google Scholar]
- 12.Hauser SL 2020. Progress in Multiple Sclerosis Research: An Example of Bedside to Bench. JAMA 324: 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hauser SL, Waubant E, Arnold DL, Vollmer T, Antel J, Fox RJ, Bar-Or A, Panzara M, Sarkar N, Agarwal S, Langer-Gould A, Smith CH, and Group HT 2008. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N Engl J Med 358: 676–688. [DOI] [PubMed] [Google Scholar]
- 14.Vinuesa CG, Linterman MA, Yu D, and MacLennan IC 2016. Follicular Helper T Cells. Annu Rev Immunol 34: 335–368. [DOI] [PubMed] [Google Scholar]
- 15.Mesin L, Ersching J, and Victora GD 2016. Germinal Center B Cell Dynamics. Immunity 45: 471–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kawabe T, Naka T, Yoshida K, Tanaka T, Fujiwara H, Suematsu S, Yoshida N, Kishimoto T, and Kikutani H 1994. The immune responses in CD40-deficient mice: impaired immunoglobulin class switching and germinal center formation. Immunity 1: 167–178. [DOI] [PubMed] [Google Scholar]
- 17.Castigli E, Alt FW, Davidson L, Bottaro A, Mizoguchi E, Bhan AK, and Geha RS 1994. CD40-deficient mice generated by recombination-activating gene-2-deficient blastocyst complementation. Proc Natl Acad Sci U S A 91: 12135–12139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Noelle RJ, Roy M, Shepherd DM, Stamenkovic I, Ledbetter JA, and Aruffo A 1992. A 39-kDa protein on activated helper T cells binds CD40 and transduces the signal for cognate activation of B cells. Proc Natl Acad Sci U S A 89: 6550–6554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Renshaw BR, Fanslow WC 3rd, Armitage RJ, Campbell KA, Liggitt D, Wright B, Davison BL, and Maliszewski CR 1994. Humoral immune responses in CD40 ligand-deficient mice. The Journal of experimental medicine 180: 1889–1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jalukar SV, Hostager BS, and Bishop GA 2000. Characterization of the roles of TNF receptor-associated factor 6 in CD40-mediated B lymphocyte effector functions. J Immunol 164: 623–630. [DOI] [PubMed] [Google Scholar]
- 21.Rothe M, Wong SC, Henzel WJ, and Goeddel DV 1994. A novel family of putative signal transducers associated with the cytoplasmic domain of the 75 kDa tumor necrosis factor receptor. Cell 78: 681–692. [DOI] [PubMed] [Google Scholar]
- 22.Ishida T, Mizushima S, Azuma S, Kobayashi N, Tojo T, Suzuki K, Aizawa S, Watanabe T, Mosialos G, Kieff E, Yamamoto T, and Inoue J 1996. Identification of TRAF6, a novel tumor necrosis factor receptor-associated factor protein that mediates signaling from an amino-terminal domain of the CD40 cytoplasmic region. J Biol Chem 271: 28745–28748. [DOI] [PubMed] [Google Scholar]
- 23.Hostager BS, Fox DK, Whitten D, Wilkerson CG, Eipper BA, Francone VP, Rothman PB, and Colgan JD 2010. HOIL-1L interacting protein (HOIP) as an NF-kappaB regulating component of the CD40 signaling complex. PLoS One 5: e11380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rowland SL, Tremblay MM, Ellison JM, Stunz LL, Bishop GA, and Hostager BS 2007. A novel mechanism for TNFR-associated factor 6-dependent CD40 signaling. J Immunol 179: 4645–4653. [DOI] [PubMed] [Google Scholar]
- 25.Xie P, Kraus ZJ, Stunz LL, and Bishop GA 2008. Roles of TRAF molecules in B lymphocyte function. Cytokine Growth Factor Rev 19: 199–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bishop GA, Moore CR, Xie P, Stunz LL, and Kraus ZJ 2007. TRAF proteins in CD40 signaling. Adv Exp Med Biol 597: 131–151. [DOI] [PubMed] [Google Scholar]
- 27.Jabara H, Laouini D, Tsitsikov E, Mizoguchi E, Bhan A, Castigli E, Dedeoglu F, Pivniouk V, Brodeur S, and Geha R 2002. The binding site for TRAF2 and TRAF3 but not for TRAF6 is essential for CD40-mediated immunoglobulin class switching. Immunity 17: 265–276. [DOI] [PubMed] [Google Scholar]
- 28.Ahonen C, Manning E, Erickson LD, O’Connor B, Lind EF, Pullen SS, Kehry MR, and Noelle RJ 2002. The CD40-TRAF6 axis controls affinity maturation and the generation of long-lived plasma cells. Nat Immunol 3: 451–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lu LF, Cook WJ, Lin LL, and Noelle RJ 2003. CD40 signaling through a newly identified tumor necrosis factor receptor-associated factor 2 (TRAF2) binding site. J Biol Chem 278: 45414–45418. [DOI] [PubMed] [Google Scholar]
- 30.Abromson-Leeman S, Maverakis E, Bronson R, and Dorf ME 2001. CD40-mediated activation of T cells accelerates, but is not required for, encephalitogenic potential of myelin basic protein-recognizing T cells in a model of progressive experimental autoimmune encephalomyelitis. European journal of immunology 31: 527–538. [DOI] [PubMed] [Google Scholar]
- 31.Becher B, Durell BG, Miga AV, Hickey WF, and Noelle RJ 2001. The clinical course of experimental autoimmune encephalomyelitis and inflammation is controlled by the expression of CD40 within the central nervous system. The Journal of experimental medicine 193: 967–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang A, Rojas O, Lee D, and Gommerman JL 2021. Regulation of neuroinflammation by B cells and plasma cells. Immunol Rev 299: 45–60. [DOI] [PubMed] [Google Scholar]
- 33.Wang Y, Godec J, Ben-Aissa K, Cui K, Zhao K, Pucsek AB, Lee YK, Weaver CT, Yagi R, and Lazarevic V 2014. The transcription factors T-bet and Runx are required for the ontogeny of pathogenic interferon-gamma-producing T helper 17 cells. Immunity 40: 355–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cao Z, Xiong J, Takeuchi M, Kurama T, and Goeddel DV 1996. TRAF6 is a signal transducer for interleukin-1. Nature 383: 443–446. [DOI] [PubMed] [Google Scholar]
- 35.Hu HM, O’Rourke K, Boguski MS, and Dixit VM 1994. A novel RING finger protein interacts with the cytoplasmic domain of CD40. J Biol Chem 269: 30069–30072. [PubMed] [Google Scholar]
- 36.Cheng G, Cleary AM, Ye ZS, Hong DI, Lederman S, and Baltimore D 1995. Involvement of CRAF1, a relative of TRAF, in CD40 signaling. Science 267: 1494–1498. [DOI] [PubMed] [Google Scholar]
- 37.Rothe M, Sarma V, Dixit VM, and Goeddel DV 1995. TRAF2-mediated activation of NF-kappa B by TNF receptor 2 and CD40. Science 269: 1424–1427. [DOI] [PubMed] [Google Scholar]
- 38.Hostager BS, and Bishop GA 1999. Cutting edge: contrasting roles of TNF receptor-associated factor 2 (TRAF2) and TRAF3 in CD40-activated B lymphocyte differentiation. J Immunol 162: 6307–6311. [PubMed] [Google Scholar]
- 39.Aarts SA, Seijkens TT, Kusters PJ, van Tiel CM, Reiche ME, den Toom M, Beckers L, van Roomen CP, de Winther MP, Kooij G, and Lutgens E 2019. Macrophage CD40 signaling drives experimental autoimmune encephalomyelitis. J Pathol 247: 471–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Woolaver RA, Wang X, Dollin Y, Xie P, Wang JH, and Chen Z 2018. TRAF2 Deficiency in B Cells Impairs CD40-Induced Isotype Switching That Can Be Rescued by Restoring NF-kappaB1 Activation. J Immunol 201: 3421–3430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xie P, Stunz LL, Larison KD, Yang B, and Bishop GA 2007. Tumor necrosis factor receptor-associated factor 3 is a critical regulator of B cell homeostasis in secondary lymphoid organs. Immunity 27: 253–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kobayashi T, Kim TS, Jacob A, Walsh MC, Kadono Y, Fuentes-Panana E, Yoshioka T, Yoshimura A, Yamamoto M, Kaisho T, Akira S, Monroe JG, and Choi Y 2009. TRAF6 is required for generation of the B-1a B cell compartment as well as T cell-dependent and -independent humoral immune responses. PLoS One 4: e4736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lumsden JM, Williams JA, and Hodes RJ 2003. Differential requirements for expression of CD80/86 and CD40 on B cells for T-dependent antibody responses in vivo. J Immunol 170: 781–787. [DOI] [PubMed] [Google Scholar]
- 44.Yasui T, Muraoka M, Takaoka-Shichijo Y, Ishida I, Takegahara N, Uchida J, Kumanogoh A, Suematsu S, Suzuki M, and Kikutani H 2002. Dissection of B cell differentiation during primary immune responses in mice with altered CD40 signals. Int Immunol 14: 319–329. [DOI] [PubMed] [Google Scholar]
- 45.Matsumoto M, Lo SF, Carruthers CJ, Min J, Mariathasan S, Huang G, Plas DR, Martin SM, Geha RS, Nahm MH, and Chaplin DD 1996. Affinity maturation without germinal centres in lymphotoxin-alpha-deficient mice. Nature 382: 462–466. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.