

Abstract
Purpose
To evaluate morphologic and functional contributions of Arrestin 1 (Arr1) and Arrestin 4 (Arr4) in cone photoreceptors, the authors examined the phenotypes of visual arrestin knockout mice (Arr1−/−, Arr4−/−, Arr1−/− Arr4−/− [Arr-DKO]) reared in darkness.
Methods
Retinal rods and cones were evaluated in wild-type (WT), Arr1−/−, Arr4−/−, and Arr-DKO mice using quantitative morphologic analysis, immunoblot, immunohistochemistry, TUNEL, and electroretinographic (ERG) techniques.
Results
Compared with either Arr4−/− or WT, Arr1−/− and Arr-DKO mice had increased apoptotic nuclei in their retinal outer nuclear layer (ONL) at postnatal day (P) 22. By P60, cone density was significantly diminished, but the ONL appeared normal. After 1 minute of background illumination, cone ERG b-wave amplitudes were similar in WT and all Arr KO mice. However, by 3 minutes and continuing through 15 minutes of light adaptation, the cone b-wave amplitudes of WT and Arr4−/− mice increased significantly over those of the Arr1−/− and Arr-DKO mice, which demonstrated no cone b-wave amplitude increase. In contrast, ERG flicker analysis after the 15-minute light adaptation period demonstrated no loss in amplitude for either Arr1−/− or Arr4−/− mice, whereas Arr-DKO demonstrated significantly lower amplitudes. When Arr1 expression was restored in Arr1−/− mice (p48Arr1−/−), normal cone density and light-adapted ERG b-wave amplitudes were observed.
Conclusions
In the adult dark-reared Arr1−/− and Arr-DKO mice, viable cones diminish over time. Arr1 expression is essential for cone photoreceptor survival and light adaptation, whereas either Arr1 or Arr4 is necessary for maintaining normal flicker responses.
Arrestin 1 (Arr1) was initially named either retinal S-antigen, which referred to the soluble fraction in the retina causing uveitis,1,2 or the 48-kDa (its molecular weight) protein.3,4 In the rod phototransduction cascade, Arr1 has an essential recovery role in “arresting” light-activated, phosphorylated rhodopsin.5 When dark-adapted mice are subjected to light, Arr1 translocates from the synapse and rod inner segments to the rod outer segments.6–8
Based on the molecular discovery of Arr1,9 three other homologues were later identified: two ubiquitously expressed β-arrestins (β-arrestin 1 and 2 [Arr2, Arr3])10 and cone arrestin or X-arrestin (Arrestin 4 [Arr4]), which is expressed in cones and a subpopulation of pinealocytes.11–14 Subsequently, Arr1 and Arr4 were shown to be coexpressed in mouse cones, with the concentration of Arr1 expression in dark-reared mice 50-fold higher (1.7 × 108 molecules/cone) than that of Arr4 (3. 3 × 106 molecules/cone).15 This Arr1 concentration in cones even exceeds its reported concentration in rods (~4.5 × 107 molecules/rod).8,16 A role for Arr4 in cone phototransduction has yet to be fully elucidated17; however, electrophysiological measurements from isolated cones indicate that S- and M-opsins require at least one visual arrestin (Arr1 or Arr4) for normal recovery and inactivation of phototransduction.15
Arr1 null mice (Arr1−/−) reared in cyclic light (164 lux) for at least 100 days or exposed to constant bright light (1250–1640 lux) for 1 week develop rod degeneration typified by the loss of photoreceptor nuclei and a decrease in outer nuclear layer (ONL) thickness.18 Partial rescue of this light-dependent degeneration in Arr1−/− mouse rods and its recovery function occurs in transgenic mice when either Arr1 (p48)19 or Arr420 is expressed on the Arr1 null background.
While investigating the cone function of visual arrestins in retinas of Arr1−/− mice, we observed a light-independent cone dystrophy phenotype depicted by an increase in apoptosis and cone photoreceptor cell loss (Brown BM, et al. IOVS 2007;48:ARVO E-Abstract 4644). An earlier report using Drosophila also described a light-independent photoreceptor degeneration that was accelerated by light when Arr1 was absent.21 Other mouse cone-specific degenerations have been reported, including knockout of the genes CNG3,22 Rpe65,23 Lrat,23 and Gnat2 (Cpfl3).24
In addition to a light-independent cone dystrophy phenotype in retinas of Arr1−/− mice, we observed a photopic ERG phenotype associated with cone light adaptation. A documented result of cone light adaptation, characterized by a b-wave amplitude increase of a dark-adapted subject during the first 15 minutes of exposure to a rod-saturating background light, has been observed in human, monkey, rat, and mouse.25–33 When cone b-wave responses to a bright light stimuli were measured over 15-minute exposure to a rod-saturating background light, we observed that wild-type (WT) and Arr4−/− mice exhibited similar b-wave amplitude increases though Arr1−/− and Arr-DKO did not. Before our discovery of Arr1 expression in cones, Arr1−/− was used as a model for a functionally rodless mouse.34 In their detailed study, Lyubarsky et al.34 reported no differences in the cone b-wave amplitudes of WT and Arr1−/− mice; however, they did not evaluate whether any progressive changes took place over their light-adapting period.
In this report, we present three observations related to Arr1 and Arr4 expression and function in mouse cones: light-independent cone degeneration, photopic ERG b-wave phenotype related to light adaptation resulting from the loss of Arr1 expression, and photopic ERG flicker phenotype when both visual arrestins were absent.
Methods
Animals
All mice were dark reared and treated according to the guidelines established by the Institute for Laboratory Animal Research (Guide for the Care and Use of Laboratory Animals), conformed to the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research, and were approved by the appropriate animal committees of the University of Southern California. Arr4−/− mice were created in our laboratory, the details of which have been published.15 The original Arr1−/− and p48Arr1−/− mice were generously provided by Jeannie Chen (University of Southern California).5 All WT and visual arrestins knockout (Arr KO) mice used in this study were on a mixed C57/Bl6J: 129SVJ background and resulted from breeding homozygous F2 littermates from the crossing of the original Arr1−/− and Arr4−/− mice: Arr1+/+ Arr4+/+ (designated colony control WT), Arr1−/−Arr4+/+ (Arr1−/−), Arr1+/+Arr4−/− (Arr4−/−), and Arr1−/−Arr4−/− (Arr-DKO). The p48Arr1−/− mice were produced by interbreeding a +p48 transgenic mouse with the Arr1−/− mice used in this study. The F1 offspring with the transgene are designated +p48Arr1−/−, whereas those without the transgene are designated p48Arr1−/−. Rod α-transducin (Tα−/−) mice were generously provided by Janice Lem (Tufts University).35
Polymerase Chain Reaction Genotype Analysis
PCR analysis of genomic DNA, isolated from tail clips, used specific primers for each visual arrestin to identify the genotype of all breeders and their offspring. Because the mice were derived from the 129SVJ background, the genomic DNA of all breeders was also tested to verify that the rd1 mutation was not present with primers specific for β-phosphodiesterase.36 Additional details of the visual arrestins knockout characterization and PCR conditions for the listed primer pairs have been published (http://www.cell.com/neuron/supplemental/S0896-6273(08)00,528-X).15
SDS-PAGE and Immunoblot Analysis
Standard procedures for protein analysis using polyacrylamide gel electrophoresis with sodium dodecyl sulfate (SDS-PAGE), followed by immunoblot analysis, were performed as previously described.37 Fifty micrograms of retinal homogenate (50 mM Tris-HCl, pH 7.6; 10 mM EDTA; 4 mM MgCl2; 40 μg/mL leupeptin, pepstatin, and aprotinin; and 0.5 mM phenylmethylsulfonyl fluoride) were mixed with Laemmli buffer, boiled, resolved on 11.5% SDS-PAGE, and either stained with Coomassie blue or transferred to polyvinylidene fluoride (PVDF) membranes. The membranes were blocked with 1% bovine serum albumin, incubated in primary antibody overnight and horseradish peroxidase (HRP)-conjugated secondary antibody, and visualized by enhanced chemiluminescence (ECL) detection. Specific primary polyclonal antibodies (pAbs) were used at the following dilutions: anti-rabbit Arrestin1 pAb C10C10 with peptide, AA288–295 (1:50,000; RERRGIALD), developed and characterized in our laboratory from previously published data of the bovine S-antigen monoclonal antibody (mAb) C10C10,38 and anti-rabbit pAb mouse cone arrestin-Luminaire juniors (mCAR-LUMIj) AA369–381 (CEEFMQHNSQTQS) at the carboxyl terminus of the mouse cone arrestin (1:10,000; Arr4) protein.39 The secondary antibody was HRP-conjugated anti-rabbit (1:10,000; Bio-Rad Laboratories, Hercules, CA).
Retinal Tissue Preparation
Dark-reared mice were killed by CO2 asphyxiation, and an orientation mark was made on the right eye at the limbus. The eye was enucleated and the cornea was removed, leaving the orientation mark. The eyecup was fixed for 3 hours at 4°C with 4% paraformaldehyde in phosphate-buffered saline (PBS), washed 2 × 15 minutes with PBS, and incubated in 30% sucrose overnight at 4°C. The lens was removed, and the eyecup was embedded in optimal cutting temperature compound (Tissue-Tek 4583; Sakura Finetek USA., Inc., Torrance, CA) and frozen in liquid nitrogen. Midsagittal 7-μm-thick frozen sections were cut through the optic nerve (Cryocut 1800 cryostat; Leica, Wetzlar, Germany) and mounted on slides.
Immunohistochemistry
The details of our immunohistochemistry (IHC) protocol have been published.17,39 Briefly, frozen retinal tissue sections were dried for 30 minutes and washed 2 × 5 minutes in PBS. The slides were blocked in 10% normal goat serum and 0.2% Triton X-100 in PBS in a humidified chamber at room temperature (RT) for 30 minutes and then incubated with 100 μL diluted primary antibody in 2% normal goat serum and 0.2% Triton X-100 in PBS in the chamber at 4°C overnight. The slides were washed 3 × 15 minutes with 100 mL PBS at RT with gentle shaking, and then 100 μL fluorescence-labeled secondary antibody (Vector Laboratories, Burlingame, CA) was diluted in 2% normal goat serum and 0.2% Triton X-100 in PBS, added to the slides, and incubated for 1 hour at RT in the chamber. The slides were washed 3 × 15 minutes with 100 mL PBS at RT and vacuum-dried. Coverslips were applied over mounting medium containing DAPI (Vector Laboratories) to stain the nuclei of all cells, and nail polish was used to eliminate evaporation. The slides were then photographed with a camera (SPOT SP401–115, software version 3.5; Diagnostic Instruments, Inc., Sterling Heights, MI) mounted on a fluorescence microscope (DMF; Leica Microsystems). Selected slides were also viewed on a confocal microscope (LSM 510; Zeiss MicroImaging, Thornwood, NY). Primary and secondary antibodies were used at the following dilutions: 1:1000, pAb mCar-LUMIj; 1:20,000, mAb S-antigen D9F2, AA360–369 (PEDPDTAKE,40 generously provided by Larry Donoso [Wills Eye Hospital]); 1:500, Alexa Fluor 488 (green) conjugated goat anti-rabbit or Alexa Fluor 568 (red) conjugated goat anti-mouse (Invitrogen, Carlsbad, CA).
Quantitative Morphometric Analysis
To measure the number of nuclear layers in the ONL, frozen retinal tissue sections were stained with Harris hematoxylin and eosin (Fisher Scientific, Pittsburgh, PA), placed in mounting medium (Vectamount; Fisher Scientific), and examined under a Leica microscope (40× objective) with a camera lucida attached.18 Each retinal hemisphere was divided into 12 equal segments, six on the superior and six on the inferior side of the optic nerve. The number of layers of nuclei in the ONL was counted at three locations in each of the 12 sections, averaged, and quantified with a graphic tablet (Wacom Technology, Vancouver, WA) and imaging software (AxioVision LE Rel. 4.1; Carl Zeiss Inc., Oberkochen, Germany).41
To quantitate the number of cones, frozen retinal sections were stained immunohistochemically with pAb mCar-LUMIj and the appropriate secondary fluorescent antibody and DAPI to stain the nuclei of all cells, and then they were examined under a Leica microscope. Each retinal hemisphere was divided into six 290-μm segments, three on the superior and three on the inferior side of the optic nerve, and the number of immunologically labeled cones in each segment was quantified as described previously.42,43
For both ONL and cone density measurements, two-way ANOVA with Bonferroni posttests were used to analyze the data from at least five mice of each genotype (WT, Arr1−/−, p48Arr1−/−) with a statistical program (Prism 5; GraphPad, San Diego, CA).44
TUNEL Analysis
Cell apoptosis analysis in the ONL of retinas from postnatal day (P) 22 WT and Arr1−/−, Arr4−/−, and Arr-DKO mice was performed on frozen retinal sections using a terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) system (DeadEnd Fluorometric; Promega, Madison, WI). After labeling with fluorescein-12-dUTP, slides were rinsed twice with sodium citrate buffer. To identify cones, the sections were stained immunologically with pAb mCAR-LUMIj with the appropriate secondary fluorescent antibody, as detailed in the IHC protocol, and then mounted with mounting medium containing DAPI to stain the nuclei of all cells. Apoptotic nuclei in three adjacent midsagittal whole retina sections cut through the optic nerve along the vertical meridian were counted and averaged. Two-way ANOVA with Bonferroni posttests was performed on data from at least four mice of each strain (WT, Arr1−/−, Arr4−/−, Arr-DKO), using a statistical program (Prism 5; GraphPad).
Electroretinography
Photopic cone ERG responses were recorded from mice in the presence of a steady, white, rod-suppressing, background light, as previously described.44 A bifurcated glass fiber optic delivered both maximum intensity (log 2.01 cd · s/m2) 10-μs bright flashes from a visual stimulator (model PS33; Grass Instruments, Braintree, MA) and the background light (200 cd/m2), with spectral peaks at 485, 530, and 543 nm and minimal transmission below 400 nm, to a level 1 cm from the cornea. Responses were captured on an electrovisual diagnostic system (Nicolet; Thermo Scientific, Pittsburgh, PA), at a half-amplitude bandwidth of 0.01 to 100 Hz. During and after the recording sessions, anesthetized mice were maintained at physiologic temperature on a 37°C water-filled heating pad connected to a heat therapy pump (Gaymar; Harvard Apparatus, Hollister, MA).
Mice were dark-reared, dark-adapted overnight, and anesthetized by intraperitoneal injection of ketamine (100 mg/kg body weight) and xylazine (10 mg/kg body weight). Pupils were dilated with topical phenylephrine (2.5%) and tropicamide (0.5%), and corneas were anesthetized with topical 0.5% proparacaine. Mice were placed in lateral recumbence with the visual axis of the right eye centered under the fiber optic. A modified carbon fiber-recording electrode was placed on the cornea. A reference 27-gauge subdermal platinum needle electrode was positioned in the lower eyelid, and a similar ground needle electrode was placed in the ipsilateral ear. The background light was turned on, and, beginning at 1 minute of light adaptation and continuing every 2 minutes afterward, retinal responses to 20 maximum intensity flashes delivered at 2 Hz were recorded and averaged. After 15 minutes of recording, the mice were maintained with the background light for 1 additional minute, at which time they were subjected to a 10-sweep, 10-Hz flicker stimulus, that was recorded and averaged. Responses from at least six mice from each genotype were averaged, and two-way ANOVA with Bonferroni posttests were performed on the data at each time point during recording.
Results
Expression of Visual Arrestins
Immunoblot analysis from an SDS-PAGE of retinal homogenates (Fig. 1, left) revealed the visual arrestins expression pattern of light-exposed P40 Arr1−/−, Arr4−/−, Arr-DKO, and WT mice. The 48-kDa Arr1 protein, immunoreactive with the Arr1-specific pAb C10C10, is visible in the WT and Arr4−/− mice but is absent in Arr1−/− and Arr-DKO homogenates (Fig. 1, middle). In contrast, a 43-kDa Arr4 protein, visualized with the specific pAb mCar-LUMIj, is expressed in the WT and Arr1−/− retinas but is absent in the Arr4−/− and Arr-DKO mice (Fig. 1, right). In Figure 2, IHC analysis of frozen retinal sections from each genotype exposed to light shows the same visual arrestin expression pattern as in Figure 1. WT retinas (Fig. 2A) have both visual arrestins, Arr4 (green) and Arr1 (red). Arr1−/− retinas (Fig. 2B) have only Arr4 expression (green), whereas Arr4−/− retinas (Fig. 2C) have only Arr1 expression (red). The retinas of Arr-DKO mice (Fig. 2D) express neither visual arrestin.
Figure 1.

Immunoblot analysis at P40 of visual arrestin expression in mice retinas from control (WT) and visual arrestin knockouts (Arr1−/−, Arr4−/−, Arr-DKO). Each lane represents ~50 μg retinal proteins from each genotype applied to 11.5% SDS-PAGE and either Coomassie Blue stained (left) or transferred to PVDF membrane and incubated with pAb C10C10 (Arr1) (middle) or pAb mCAR-LUMIj (Arr4) (right), followed by the appropriate HRP-conjugated secondary antibody and processed for ECL. Molecular weight markers (in kDa) are identified on the left.
Figure 2.
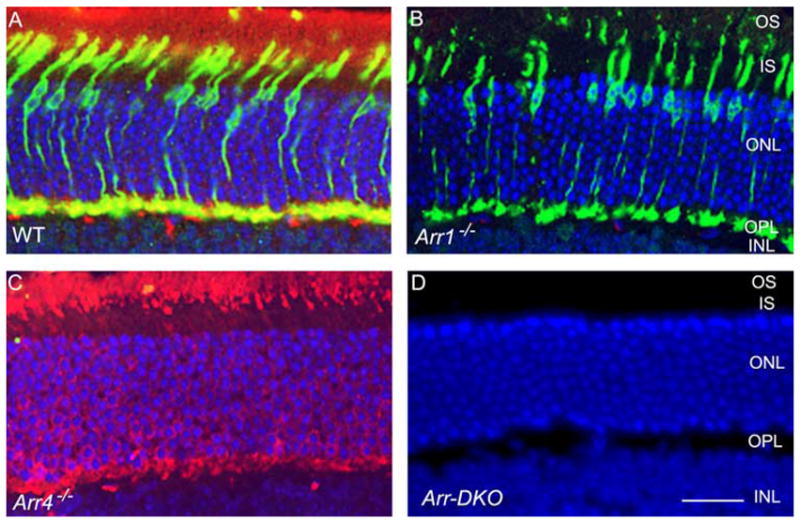
IHC analysis at P60 of visual arrestin expression in mice retinas from control (WT) and visual arrestin knockouts (Arr1−/−, Arr4−/−, Arr-DKO). Retinal frozen sections of WT (A), Arr1−/− (B), Arr4−/−(C), and Arr-DKO (D) were processed with Arr1 (mAb D9F2) and Arr4 (pAb mCAR-LUMIj) antibodies, followed by the appropriate secondary antibodies. Immunoreactive Arr1 is red (Texas red) and Arr4 is green (fluorescein). Dual immunolocalization of Arr1 and Arr4 is yellow. Nuclei are stained blue with DAPI. IS, inner segment; OS, outer segment. Original magnification, 40×. Scale bar, 20 μm.
In the light-exposed WT mice, Arr4 immunoreactive protein (green) is expressed throughout the cone photoreceptor, whereas most of the Arr1 protein (red) appears in the outer segment (OS), with some in the cone photoreceptor pedicles in the outer plexiform layer (OPL). This demonstrates an overlap in expression of the two visual arrestins (yellow) in the cones of these two layers of the light-exposed WT retina (Fig. 2A).
Light-Independent Cone Dystrophy in Arr1−/− Mice
The number of cones (green) in the P60 Arr1−/− retina (Fig. 2B) is lower than in the WT retina (Fig. 2A). DAPI staining (blue) of the nuclei in the ONL shows that all four genotypes (Figs. 2A–D) have similar number of rows of nuclei, suggesting that overall total photoreceptor cell number is maintained in the dark-reared mice. Quantitative spider plot analysis supports this initial observation: retinal sections through the optic nerve, showing the number of layers of nuclei in the ONL of WT, Arr1−/−, and p48Arr1−/− mice, indicate there is no statistically significant loss of photoreceptor outer nuclear layers in either the superior or the inferior retina of these dark-reared P60 mice (P > 0.05; Fig. 3A).
Figure 3.

Quantitative spider plot analysis of mouse retinas at P60 from control (WT), Arr1−/−, or p48Arr1−/−. (A) The total number of layers of nuclei in the ONL of hematoxylin and eosin–stained retinal sections through the optic nerve (0) was measured at 12 locations around the retina, six each in the superior and inferior hemispheres. At least four mice of each genotype were counted. Two-way ANOVA with Bonferroni posttests compared Arr1−/− or +p48Arr1−/− transgenic mice with WT at each segment measured. (B) The number of cones in the photoreceptor layer of mCAR-LUMIj immunologically positively stained retinal sections through the optic nerve (0) was counted in six 290-μm segments, three each in the superior and inferior hemispheres 100 μm from the optic nerve. At least four retinas of each genotype were counted. Two-way ANOVA with Bonferroni posttests compared KO mice with WT in all segments (***P < 0.001).
Figure 3B illustrates quantitative calculations of cone cell numbers from adjacent sections of the retinas of these mice and shows a significant reduction (24.2%; P < 0.001) in the number of cones in both the superior and the inferior regions of the Arr1−/− compared with WT mice, except in the extreme peripheral superior region (P > 0.05). By restoring Arr1 protein expression to photoreceptors of the Arr1−/− mice (p48Arr1−/−), cone numbers appeared normal in the superior region but were reduced in the inferior region of the retina (Fig. 3B).
At P22, positive TUNEL staining resulted in fewer apoptotic nuclei in WT and Arr4−/− mice than in Arr1−/− and Arr-DKO mice (Figs. 4A–D). Figure 4E shows quantitative TUNEL data analysis averaged from three sequential midsagittal retinal sections from the retinas of at least four mice in each group and shows a highly significant difference (P < 0.001) in the number of TUNEL-positive nuclei per retinal section when comparing WT mice with both Arr1−/− and Arr-DKO mice. Figure 4E also highlights that exposing the mice to 4 hours of room light (1400 lux) before killing increased the number of TUNEL-positive nuclei seen in Arr1−/− and Arr-DKO mice.
Figure 4.

Fluorometric TUNEL analysis of mice retinas at P22 from control (WT) and visual arrestin knockouts (Arr1−/−, Arr4−/−, Arr-DKO). Representative images illustrating cell apoptosis in the ONL from frozen retinal sections from P22. (A) WT, (B) Arr1−/−, (C) Arr4−/−, (D) Arr-DKO. Mice were killed in the dark and analyzed using the TUNEL system, followed by immunologic staining with Arr4 (pAb mCAR-LUMIj antibody) followed by the appropriate secondary antibody. Immunoreactive Arr4 is red (Texas red), and nuclei are stained blue with DAPI. Original magnification, 40×. Scale bar, 20 μm. (E) Histogram analysis summarizes these data from cell counting of all TUNEL-positive nuclei on averaging the total from three adjacent midsagittal whole retinal sections from at least four mice for each genotype killed either in the dark or after 4 hours of 1400 lux illumination. Bars represent ±SEM. Two-way ANOVA with Bonferroni posttests compared visual arrestin knockout mice with WT mice (***P < 0.001).
Electroretinography
Representative photopic ERG responses of WT and visual arrestin knockout (Arr KO) mice (P35–60) to bright light (log 2.0 cd · s/m2) flashes (stimulus) administered after 1 minute (left tracings) or 15 minutes (right tracings) of light adaptation to a rod-saturating background light (200 cd/m2) are presented in Figure 5A.
Figure 5.

ERG analysis of P35 to P60 control (WT), visual arrestin knockouts (Arr1−/−, Arr4−/−, Arr-DKO), p48Arr−/−, and Tα−/− mice. A bifurcated glass fiber optic delivered both maximum intensity (2.01 log cd · s/m2) 10-μs bright flashes and a 200 cd/m2 steady background white light to a level 1 cm from the cornea. (A) Representative responses to bright flashes (stimulus) of WT and visual arrestin KO mice at 1 minute (left waveform data) and after 15 minutes of continuous background illumination (right waveform data). Average photopic b-wave amplitudes (μV) recorded every 2 minutes during 15 minutes of light adaptation of the (B) WT and visual arrestin knockout mice, (C) transgenic p48 mice bred onto an Arr1 null background (Arr1−/−) with confirmed Arr1 (+p48Arr1−/−) or negative littermate controls (−p48Arr1−/−), and (D) rod alpha transducin (Tα−/−) mice. Two-way ANOVA with Bonferroni post-tests used for statistical comparisons with controls (***P < 0.001; **P < 0.01; *P < 0.05).
Averaged b-wave amplitudes recorded from WT and Arr KO mice every 2 minutes from 1 to 15 minutes during light adaptation are graphed in Figure 5B. The photopic b-wave amplitudes for these four genotypes are similar at 1 minute. For both WT and Arr4−/− mice, the amplitudes continued to increase from approximately 200 μV at 1 minute to approximately 500 μV at 15 minutes. In contrast, amplitudes of both the Arr1−/− and the Arr-DKO mice did not increase appreciably. Figure 5C shows b-wave responses of the p48Arr1−/− mice (+p48Arr1−/−, dark blue square) and their negative littermate controls (−p48Arr1−/−, light blue triangle). It shows that +p48Arr1−/− retinas expressing the Arr1 protein delivered from the expression of the transgene are rescued in their ability to light adapt compared with retinas without Arr1 expression in the −p48Arr1−/−. As seen in Figure 5D, rod α-transducin mice (Tα−/−, magenta triangle) light adapted in a manner similar to that of WT mice (black square).
For cone flicker analysis, light adaptation was continued to 16 minutes, at which time mice were subjected to a 10-sweep, 10-Hz flicker stimulus. Representative tracings are shown in Figure 6A. The average flicker response of the four genotypes and the p48 transgenic mice are summarized in Figure 6B. There is a significant decrease (P < 0.05) in the flicker response of Arr-DKO compared with WT. Unexpectedly, we observed a slight but significant increase (P < 0.05) in the Arr4−/− flicker response.
Figure 6.

ERG Flicker analysis of control (WT), visual arrestin knockouts (Arr4−/−, Arr1−/−, Arr-DKO), +p48Arr1−/−, or −p48Arr1−/− mice. (A) Representative 10-Hz flicker responses of WT and visual arrestin knockout mice measured after 16 minutes of background illumination. (B) 10-Hz flicker responses of WT, Arr4−/−, Arr1−/−, Arr-DKO, and p48 transgenic mice (+p48Arr1−/−) and their negative littermate controls (−p48Arr1−/−). Two-way ANOVA with Bonferroni posttests compared transgenic mice with WT mice (***P < 0.001; **P < 0.01; *P < 0.05).
Discussion
Lack of visual Arr1 expression in mice, or abnormal expression caused by a genetic defect in the ARR1 gene in humans, has a profound deleterious effect on rod electrophysiological function.5,18 Because of their increased susceptibility to light damage, the Arr1−/− mouse is a good animal model for the study of the light damage associated with autosomal recessive retinitis pigmentosa and Oguchi’s disease, a nonprogressive form of congenital stationary night blindness. Retinal damage reported in the Arr1 knockout mouse occurs when the mouse is subjected to a constant or cycling light environment, which leads to a constitutive signal flow arising from a defective opsin shut-off and abnormal recovery.18
Unexpectedly, in dark-reared Arr1−/− mice, we observed progressive light-independent cone dystrophy despite which the rod population appeared healthy. By P60, the Arr1−/− mice had approximately 75% the cone cell numbers that WT mice at this age (Fig. 3B). This dystrophy was specific for Arr1 because Arr4−/− mice had normal cone cell numbers (data not shown).
At P22, retinas of dark-reared Arr1−/− mice had increased numbers of apoptotic (TUNEL-positive) nuclei in the ONL, and the number of apoptotic nuclei increased with 4-hour exposure to room light (Fig. 4E). It has been reported that by P12, healthy cone nuclei migrate to line up adjacent to the outer limiting membrane (OLM); however, in the rd1 mouse, this cone nuclei migration is delayed.45 In our study, the TUNEL-positive nuclei at P22 did not line up near the OLM but were scattered throughout the ONL (Figs. 4B–D), possibly because they were in a late stage of apoptosis and either had never lined up or had lost their orientation during the process of cell death. Although it is not possible to positively identify the increased TUNEL-positive nuclei observed in the P22 Arr1−/− mice as apoptotic cones, we did observe that by P60 25% of the cones were absent (Fig. 3B).
In transgenic +p48Arr−/− mice, the rhodopsin promoter restores the 48-kDa rod Arr1 protein expression to the rods; however, the promoter may be “leaky”,46 resulting in the potential restoration of Arr1 expression in cones. Arr1 transgene expression, however, was only partially successful in rescuing the cone dystrophy. The cone cell numbers of these mice were indistinguishable from those of WT mice in the superior but not the inferior retina (Fig. 3B).
Given that Arr1 is not expressed in either rods or cones in Arr1−/− mice, why are cones not surviving in dark-reared mice? Compared with rods, cones have been shown to be particularly vulnerable to apoptotic cell death from calcium overload and oxidative stress, possibly because their metabolic rate of ATP demand and production is higher.47–49 In Stargardt macular dystrophy, associated with the accumulation of A2E resulting in subsequent oxidative stress, cones in the macula are selectively damaged.50 In models of rd1, cone cell death has been attributed to oxidative damage resulting from increased oxygen levels after rod death in the outer retina and reduced levels of insulin.51,52 If knocking out Arr1 results in increased reactive oxygen species, even when dark reared, the result may be the light-independent cone dystrophy we observed (Fig. 3B). We are investigating oxidative stress in this model.
Based on our electrophysiological data (Fig. 6B), Arr1 contributes to a cone-driven, photopic ERG b-wave amplitude increase observed in WT and Arr4−/− mice during light adaptation. This b-wave increase is not present in Arr1−/− and Arr-DKO mice. This phenomenon is Arr1 specific because the cone b-wave amplitude of Arr4−/− mice response increased normally during light exposure. When the 48-kDa Arr1 protein was restored to Arr1−/− mice, in +p48Arr1−/−, the photopic b-wave increase was also restored (Fig. 6C).
It has been reported that functional rods may be necessary for the photopic ERG amplitude increase during light adaptation.26 However, Peachey29 studied the light adaptation of the human rod system as a possible cause of cone ERG amplitude increases and concluded that rods were not a major contributor to this cone light adaptation. Our investigations, using the rod Tα−/− mouse, support this conclusion. The rod Tα−/− mouse has normal rod morphology but lacks rod function and has been used as a model for functionally rodless mice34; however, the b-wave amplitude increase during light exposure was similar to that in WT mice (Fig 5D).
In rods, the movement of Tα, recoverin, and Arr1 within the photoreceptor has been implicated in light adaptation (i.e., the desensitization of rods in response to increased light intensity.6,7,53,54 In the rods of a dark-adapted WT mouse, Arr1 expression is located predominantly in the inner segments and in the OPL containing the synaptic terminals. When an adapting background light is turned on, Arr1 slowly translocates from the OPL to the rod photoreceptor outer segments, arriving there after several minutes of constant illumination.55 Prolonged illumination and light adaptation also upregulate Arr1 gene expression and its translocation.56 Other investigators have seen a correlation between arrestin translocation and light adaptation. Lee57 showed that mutant Drosophila, with defective phosphoinositide metabolism resulting in delayed visual arrestin translocation, did not light adapt to continuous light. The flies had normal amounts of visual arrestin, but the arrestin did not translocate. Arshavsky58 proposed that, in rods, Arr1 translocation may be necessary for light adaptation because, in continuous light, the supply of Arr1 in the outer segment may be bound to activated rhodopsin, and additional Arr1 is needed to bind to newly activated receptors. Without this additional Arr1, the signal may overload the cell.
In cones, however, one form of light adaptation may be observed as an increase in b-wave response to bright light stimuli.59 A mechanism for Arr1 involvement in this cone light adaptation is unclear but likely involves protein movement. We have shown that Arr4 directionally translocates in cones as Arr1 does in rods.39 In dark-adapted mice, both Arr4 and Arr1 are highly expressed in the cone pedicles.15 McGinnis et al.60 showed intense Arr1 staining in the OPL and photoreceptor inner segments of dark-adapted WT mice. In the OPL, this probably reflected the presence of Arr1 in both rod spherules and the larger cone pedicles, which occupy a significant part of the OPL.60,61 By 6 minutes of light exposure, most of the immunologic reactive product was translocated from the photoreceptor inner segments, and, by 10 minutes, approximately 100% was absent from the OPL. The absence of detectable immunologic reactive staining in the OPL or photoreceptor inner segments after 6 to 10 minutes of light exposure suggests that Arr1 also translocates in cones with a time course similar to that in rods. Our ERG studies demonstrate a similar time course for light adaptation. In WT mice, photopic b-wave amplitude increased steadily after the background light was turned on. In Figure 5B, it can be seen that, by 7 minutes of continuous background light exposure, almost 80% of the b-wave amplitude increase has occurred.
We hypothesize that Arr1 translocates in cones in a manner similar to that of rods, suggesting a functional role in the synapse that is inhibitory to amplitudes or a function in the OS that is stimulatory to amplitudes. In a mouse retina, it may be essential that Arr1 be present in the cone photoreceptor OS or be translocated away from the cone pedicle synapse to allow for light adaptation. Given that abnormal cone light adaptation and flicker response are observed, we are exploring alternative physiological roles and binding partners of the Arr1 in the photoreceptor synapses (Huang S-P, et al. IOVS 2009;50:ARVO E-Abstract 5430).
In conclusion, Arr1, but not Arr4, expression protects cones from light-independent dystrophy. This study also demonstrates for the first time that Arr1 plays a significant role in cone light adaptation reflected in the photopic ERG amplitude increase as the mouse transitions from dark to light. The 10-Hz flicker responses from the Arr1−/−, Arr4−/−, and Arr-DKO mice show that the presence of either visual arrestin is essential in the traditional role of resetting and restoring cone physiology after a light event. The significant reduction in flicker response after the removal of both visual arrestins demonstrates that one visual arrestin will partially substitute for the other in cone recovery.
Mutations in the human gene encoding ARR1 cause either an inherited recessive form of stationary night blindness known as Oguchi’s disease,62,63 associated with abnormal ERGs of reduced a-wave amplitudes and absent scotopic b-waves, or retinitis pigmentosa with retinal degeneration.64 Based on this work, Arr1−/− mice provide a model for better understanding of these retinal conditions. Future studies should closely examine photopic ERG and flicker responses in these patients.
Acknowledgments
Supported by National Institutes of Health Grants EY015851 (CMC) and EY03040 (DEI), the Mary D. Allen Endowment (CMC), a Research to Prevent Blindness Senior Scientific Investigator Award (CMC), Dorie Miller, and Patricia Beckman. CMC is the Mary D. Allen Chair in Vision Research, Doheny Eye Institute.
The authors thank Eric Tam, Freddi I. Zuniga, Lijuan Fu, Shun-Ping Huang, and Guey Shiang Wu for excellent technical assistance; Ernesto Barron for final figure preparation; Francis Concepcion for assistance in setting up the quantitative morphologic analysis; Larry Donoso for mAb D9F2; Jeannie Chen for Arr1−/− and p48Arr1−/− mice; and Janice Lem for rod αT−/− mice.
Footnotes
Disclosure: B.M. Brown, None; T. Ramirez, None; L. Rife, None; C.M. Craft, None
References
- 1.Wacker WB, Lipton MM. Experimental allergic uveitis: homologous retina as uveitogenic antigen. Nature. 1965;206:253–254. doi: 10.1038/206253a0. [DOI] [PubMed] [Google Scholar]
- 2.Wacker WB, Donoso LA, Kalsow CM, Yankeelov JA, Jr, Organisciak DT. Experimental allergic uveitis: isolation, characterization, and localization of a soluble uveitopathogenic antigen from bovine retina. J Immunol. 1977;119:1949–1958. [PubMed] [Google Scholar]
- 3.Kuhn H. Light-regulated binding of rhodopsin kinase and other proteins to cattle photoreceptor membranes. Biochemistry. 1978;17:4389–4395. doi: 10.1021/bi00614a006. [DOI] [PubMed] [Google Scholar]
- 4.Pfister C, Chabre M, Plouet J, et al. Retinal S antigen identified as the 48K protein regulating light-dependent phosphodiesterase in rods. Science. 1985;228:891–893. doi: 10.1126/science.2988124. [DOI] [PubMed] [Google Scholar]
- 5.Xu J, Dodd RL, Makino CL, Simon MI, Baylor DA, Chen J. Prolonged photoresponses in transgenic mouse rods lacking arrestin. Nature. 1997;389:505–509. doi: 10.1038/39068. [DOI] [PubMed] [Google Scholar]
- 6.Philp NJ, Chang W, Long K. Light-stimulated protein movement in rod photoreceptor cells of the rat retina. FEBS Lett. 1987;225:127–132. doi: 10.1016/0014-5793(87)81144-4. [DOI] [PubMed] [Google Scholar]
- 7.Whelan JP, McGinnis JF. Light-dependent subcellular movement of photoreceptor proteins. J Neurosci Res. 1988;20:263–270. doi: 10.1002/jnr.490200216. [DOI] [PubMed] [Google Scholar]
- 8.Strissel KJ, Sokolov M, Trieu LH, Arshavsky VY. Arrestin translocation is induced at a critical threshold of visual signaling and is superstoichiometric to bleached rhodopsin. J Neurosci. 2006;26:1146–1153. doi: 10.1523/JNEUROSCI.4289-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shinohara T, Dietzschold B, Craft CM, et al. Primary and secondary structure of bovine retinal S antigen (48-kDa protein) Proc Natl Acad Sci U S A. 1987;84:6975–6979. doi: 10.1073/pnas.84.20.6975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang J, Ferguson SS, Barak LS, et al. Molecular mechanisms of G protein-coupled receptor signaling: role of G protein-coupled receptor kinases and arrestins in receptor desensitization and resensitization. Receptors Channels. 1997;5:193–199. [PubMed] [Google Scholar]
- 11.Craft CM, Whitmore DH, Wiechmann AF. Cone arrestin identified by targeting expression of a functional family [published erratum appears in J Biol Chem. 1994;269:17756] J Biol Chem. 1994;269:4613–4619. [PubMed] [Google Scholar]
- 12.Murakami A, Yajima T, Sakuma H, McLaren MJ, Inana G. X-arrestin: a new retinal arrestin mapping to the X chromosome. FEBS Lett. 1993;334:203–209. doi: 10.1016/0014-5793(93)81712-9. [DOI] [PubMed] [Google Scholar]
- 13.Craft CM, Whitmore DH. The arrestin superfamily: cone arrestins are a fourth family. FEBS Lett. 1995;362:247–255. doi: 10.1016/0014-5793(95)00213-s. [DOI] [PubMed] [Google Scholar]
- 14.Sakuma H, Inana G, Murakami A, Higashide T, McLaren MJ. Immunolocalization of X-arrestin in human cone photoreceptors. FEBS Lett. 1996;382:105–110. doi: 10.1016/0014-5793(96)00163-9. [DOI] [PubMed] [Google Scholar]
- 15.Nikonov SS, Brown BM, Davis JA, et al. Mouse cones require an arrestin for normal inactivation of phototransduction. Neuron. 2008;59:462–474. doi: 10.1016/j.neuron.2008.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hanson SM, Van Eps N, Francis DJ, et al. Structure and function of the visual arrestin oligomer. EMBO J. 2007;26:1726–1736. doi: 10.1038/sj.emboj.7601614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhu X, Brown B, Li A, Mears AJ, Swaroop A, Craft CM. GRK1-dependent phosphorylation of S and M opsins and their binding to cone arrestin during cone phototransduction in the mouse retina. J Neurosci. 2003;23:6152–6160. doi: 10.1523/JNEUROSCI.23-14-06152.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen J, Simon MI, Matthes MT, Yasumura D, LaVail MM. Increased susceptibility to light damage in an arrestin knockout mouse model of Oguchi disease (stationary night blindness) Invest Ophthalmol Vis Sci. 1999;40:2978–2982. [PubMed] [Google Scholar]
- 19.Burns ME, Mendez A, Chen CK, et al. Deactivation of phosphorylated and nonphosphorylated rhodopsin by arrestin splice variants. J Neurosci. 2006;26:1036–1044. doi: 10.1523/JNEUROSCI.3301-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chan S, Rubin WW, Mendez A, et al. Functional comparisons of visual arrestins in rod photoreceptors of transgenic mice. Invest Ophthalmol Vis Sci. 2007;48:1968–1975. doi: 10.1167/iovs.06-1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Satoh AK, Ready DF. Arrestin1 mediates light-dependent rhodopsin endocytosis and cell survival. Curr Biol. 2005;15:1722–1733. doi: 10.1016/j.cub.2005.08.064. [DOI] [PubMed] [Google Scholar]
- 22.Biel M, Seeliger M, Pfeifer A, et al. Selective loss of cone function in mice lacking the cyclic nucleotide-gated channel CNG3. Proc Natl Acad Sci U S A. 1999;96:7553–7557. doi: 10.1073/pnas.96.13.7553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fan J, Rohrer B, Frederick JM, Baehr W, Crouch RK. Rpe65−/− and Lrat−/− mice: comparable models of Leber congenital amaurosis. Invest Ophthalmol Vis Sci. 2008;49:2384–2389. doi: 10.1167/iovs.08-1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chang B, Dacey MS, Hawes NL, et al. Cone photoreceptor function loss-3, a novel mouse model of achromatopsia due to a mutation in Gnat2. Invest Ophthalmol Vis Sci. 2006;47:5017–5021. doi: 10.1167/iovs.05-1468. [DOI] [PubMed] [Google Scholar]
- 25.Miyake Y, Horiguchi M, Ota I, Shiroyama N. Characteristic ERG-flicker anomaly in incomplete congenital stationary night blindness. Invest Ophthalmol Vis Sci. 1987;28:1816–1823. [PubMed] [Google Scholar]
- 26.Kondo M, Miyake Y, Piao CH, Tanikawa A, Horiguchi M, Terasaki H. Amplitude increase of the multifocal electroretinogram during light adaptation. Invest Ophthalmol Vis Sci. 1999;40:2633–2637. [PubMed] [Google Scholar]
- 27.Gouras P, Mackay CJ. Growth in amplitude of the human cone electroretinogram with light adaptation. Invest Ophthalmol Vis Sci. 1989;30:625–630. [PubMed] [Google Scholar]
- 28.Miller S, Sandberg MA. Cone electroretinographic change during light adaptation in retinitis pigmentosa. Invest Ophthalmol Vis Sci. 1991;32:2536–2541. [PubMed] [Google Scholar]
- 29.Peachey NS, Alexander KR, Derlacki DJ, Fishman GA. Light adaptation, rods, and the human cone flicker ERG. Vis Neurosci. 1992;8:145–150. doi: 10.1017/s0952523800009305. [DOI] [PubMed] [Google Scholar]
- 30.Peachey NS, Goto Y, al-Ubaidi MR, Naash MI. Properties of the mouse cone-mediated electroretinogram during light adaptation. Neurosci Lett. 1993;162:9–11. doi: 10.1016/0304-3940(93)90547-x. [DOI] [PubMed] [Google Scholar]
- 31.Moeller A, Eysteinsson T. Light adaptation of cones in rabbits and guinea pigs. Laeknabladid. 2001;87:221–226. [PubMed] [Google Scholar]
- 32.Alexander KR, Raghuram A, Rajagopalan AS. Cone phototransduction and growth of the ERG b-wave during light adaptation. Vision Res. 2006;46:3941–3948. doi: 10.1016/j.visres.2006.04.015. [DOI] [PubMed] [Google Scholar]
- 33.Bui BV, Fortune B. Origin of electroretinogram amplitude growth during light adaptation in pigmented rats. Vis Neurosci. 2006;23:155–167. doi: 10.1017/S0952523806232024. [DOI] [PubMed] [Google Scholar]
- 34.Lyubarsky AL, Lem J, Chen J, Falsini B, Iannaccone A, Pugh EN., Jr Functionally rodless mice: transgenic models for the investigation of cone function in retinal disease and therapy. Vision Res. 2002;42:401–415. doi: 10.1016/s0042-6989(01)00214-0. [DOI] [PubMed] [Google Scholar]
- 35.Calvert PD, Krasnoperova NV, Lyubarsky AL, et al. Phototransduction in transgenic mice after targeted deletion of the rod transducin alpha-subunit. Proc Natl Acad Sci U S A. 2000;97:13913–13918. doi: 10.1073/pnas.250478897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pittler SJ, Baehr W. Identification of a nonsense mutation in the rod photoreceptor cGMP phosphodiesterase β-subunit gene of the rd mouse. Proc Natl Acad Science U S A. 1991;88:8322–8326. doi: 10.1073/pnas.88.19.8322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brown BM, Carlson BL, Zhu X, Lolley RN, Craft CM. Light-driven translocation of the protein phosphatase 2A complex regulates light/dark dephosphorylation of phosducin and rhodopsin. Biochemistry. 2002;41:13526–13538. doi: 10.1021/bi0204490. [DOI] [PubMed] [Google Scholar]
- 38.Knospe V, Donoso LA, Banga JP, Yue S, Kasp E, Gregerson DS. Epitope mapping of bovine retinal S-antigen with monoclonal antibodies. Curr Eye Res. 1988;7:1137–1147. doi: 10.3109/02713688809001885. [DOI] [PubMed] [Google Scholar]
- 39.Zhu X, Li A, Brown B, Weiss ER, Osawa S, Craft CM. Mouse cone arrestin expression pattern: light-induced translocation in cone photoreceptors. Mol Vis. 2002;8:462–471. [PubMed] [Google Scholar]
- 40.Gregerson DS, Knospe V, Donoso LA. Selection of antibody epitopes in an immunopathogenic neural autoantigen. J Neuroimmunol. 1989;24:191–206. doi: 10.1016/0165-5728(89)90117-3. [DOI] [PubMed] [Google Scholar]
- 41.Concepcion FA. Essential Role of the Carboxyl-Terminus for Proper Rhodopsin Trafficking and “Enlightenment” to the Path-way(s) Causing Retinal Degeneration in a Mouse Model Expressing a Truncated Rhodopsin Mutant (dissertation) Los Angeles: University of Southern California; 2007. [Google Scholar]
- 42.Coleman JE, Zhang Y, Brown GA, Semple-Rowland SL. Cone cell survival and downregulation of GCAP1 protein in the retinas of GC1 knockout mice. Invest Ophthalmol Vis Sci. 2004;45:3397–3403. doi: 10.1167/iovs.04-0392. [DOI] [PubMed] [Google Scholar]
- 43.Haire SE, Pang J, Boye SL, et al. Light-driven cone arrestin translocation in cones of postnatal guanylate cyclase-1 knockout mouse retina treated with AAV-GC1. Invest Ophthalmol Vis Sci. 2006;47:3745–3753. doi: 10.1167/iovs.06-0086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhu X, Wu K, Rife L, et al. Carboxypeptidase E is required for normal synaptic transmission from photoreceptors to the inner retina. J Neurochem. 2005;95:1351–1362. doi: 10.1111/j.1471-4159.2005.03460.x. [DOI] [PubMed] [Google Scholar]
- 45.Rich KA, Zhan Y, Blanks JC. Migration and synaptogenesis of cone photoreceptors in the developing mouse retina. J Comp Neurol. 1997;388:47–63. [PubMed] [Google Scholar]
- 46.Woodford BJ, Chen J, Simon MI. Expression of rhodopsin promoter transgene product in both rods and cones. Exp Eye Res. 1994;58:631–635. doi: 10.1006/exer.1994.1058. [DOI] [PubMed] [Google Scholar]
- 47.Perkins GA, Ellisman MH, Fox DA. The structure-function correlates of mammalian rod and cone photoreceptor mitochondria: observations and unanswered questions. Mitochondrion. 2004;4:695–703. doi: 10.1016/j.mito.2004.07.020. [DOI] [PubMed] [Google Scholar]
- 48.Rogers BS, Symons RC, Komeima K, et al. Differential sensitivity of cones to iron-mediated oxidative damage. Invest Ophthalmol Vis Sci. 2007;48:438–445. doi: 10.1167/iovs.06-0528. [DOI] [PubMed] [Google Scholar]
- 49.Punzo C, Kornacker K, Cepko CL. Stimulation of the insulin/mTOR pathway delays cone death in a mouse model of retinitis pigmentosa. Nat Neurosci. 2009;12:44–52. doi: 10.1038/nn.2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sun H, Nathans J. Mechanistic studies of ABCR, the ABC transporter in photoreceptor outer segments responsible for autosomal recessive Stargardt disease. J Bioenerg Biomembr. 2001;33:523–530. doi: 10.1023/a:1012883306823. [DOI] [PubMed] [Google Scholar]
- 51.Punzo C, Kornacker K, Cepko CL. Stimulation of the insulin/mTOR pathway delays cone death in a mouse model of retinitis pigmentosa. Nat Neurosci. 2009;12:44–52. doi: 10.1038/nn.2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Komeima K, Rogers BS, Campochiaro PA. Antioxidants slow photoreceptor cell death in mouse models of retinitis pigmentosa. J Cell Physiol. 2007;213:809–815. doi: 10.1002/jcp.21152. [DOI] [PubMed] [Google Scholar]
- 53.Broekhuyse RM, Tolhuizen EF, Janssen AP, Winkens HJ. Light-induced shift and binding of S-antigen in retinal rods. Curr Eye Res. 1985;4:613–618. doi: 10.3109/02713688508999993. [DOI] [PubMed] [Google Scholar]
- 54.Sokolov M, Lyubarsky AL, Strissel KJ, et al. Massive light-driven translocation of transducin between the two major compartments of rod cells: a novel mechanism of light adaptation. Neuron. 2002;34:95–106. doi: 10.1016/s0896-6273(02)00636-0. [DOI] [PubMed] [Google Scholar]
- 55.Nair KS, Hanson SM, Mendez A, et al. Light-dependent redistribution of arrestin in vertebrate rods is an energy-independent process governed by protein-protein interactions. Neuron. 2005;46:555–567. doi: 10.1016/j.neuron.2005.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Codega P, Della Santina L, Gargini C, et al. Prolonged illumination up-regulates arrestin and two guanylate cyclase activating proteins: a novel mechanism for light adaptation. J Physiol. 2009;587:2457–2472. doi: 10.1113/jphysiol.2009.168609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lee SJ, Xu H, Kang LW, Amzel LM, Montell C. Light adaptation through phosphoinositide-regulated translocation of Drosophila visual arrestin. Neuron. 2003;39:121–132. doi: 10.1016/s0896-6273(03)00390-8. [DOI] [PubMed] [Google Scholar]
- 58.Arshavsky VY. Protein translocation in photoreceptor light adaptation: a common theme in vertebrate and invertebrate vision. Sci STKE. 2003:E43. doi: 10.1126/stke.2003.204.pe43. [DOI] [PubMed] [Google Scholar]
- 59.Stockman A, Langendorfer M, Smithson HE, Sharpe LT. Human cone light adaptation: from behavioral measurements to molecular mechanisms. J Vis. 2006;6:1194–1213. doi: 10.1167/6.11.5. [DOI] [PubMed] [Google Scholar]
- 60.Elias RV, Sezate SS, Cao W, McGinnis JF. Temporal kinetics of the light/dark translocation and compartmentation of arrestin and alpha-transducin in mouse photoreceptor cells. Mol Vis. 2004;10:672–681. [PubMed] [Google Scholar]
- 61.Carter-Dawson LD, LaVail MM, Sidman RL. Differential effect of rd mutation on rods and cones in mouse retina. Invest Ophthalmology Vis Sci. 1978;17:489–498. [PubMed] [Google Scholar]
- 62.Fuchs S, Nakazawa M, Maw M, Tamai M, Oguchi Y, Gal A. A homozygous 1-base pair deletion in the arrestin gene is a frequent cause of Oguchi disease in Japanese. Nat Genet. 1995;10:360–362. doi: 10.1038/ng0795-360. [DOI] [PubMed] [Google Scholar]
- 63.Nakazawa M, Wada Y, Fuchs S, Gal A, Tamai M. Oguchi disease: phenotypic characteristics of patients with the frequent 1147delA mutation in the arrestin gene. Retina. 1997;17:17–22. doi: 10.1097/00006982-199701000-00004. [DOI] [PubMed] [Google Scholar]
- 64.Nakamachi Y, Nakamura M, Fujii S, Yamamoto M, Okubo K. Oguchi disease with sectoral retinitis pigmentosa harboring adenine deletion at position 1147 in the arrestin gene. Am J Ophthalmol. 1998;125:249–251. doi: 10.1016/s0002-9394(99)80100-7. [DOI] [PubMed] [Google Scholar]