

Abstract
We present a combinatorial indexing method PerturbSci-Kinetics for capturing whole transcriptomes, nascent transcriptomes, and single guide RNA (sgRNA) identities across hundreds of genetic perturbations at the single-cell level. Profiling a pooled CRISPR screen targeting various biological processes, we show the gene expression regulation during RNA synthesis, processing, and degradation, miRNA biogenesis, and mitochondrial mRNA processing, systematically decoding the genome-wide regulatory network that underlies RNA temporal dynamics at scale.
Cellular functions are determined by the expression of millions of RNA molecules, which are tightly regulated by their synthesis, splicing, and degradation. However, understanding how key regulators impact genome-wide RNA kinetics is constrained by existing tools, which provide only snapshots of the transcriptome1–8. To resolve this challenge, we developed PerturbSci-Kinetics, combining CRISPR-based pooled genetic screen, single-cell RNA-seq by combinatorial indexing, and RNA metabolic labeling, to uncover single-cell transcriptome dynamics across extensive genetic perturbations.
PerturbSci-Kinetics features a combinatorial indexing strategy (‘PerturbSci’) for targeted capture of sgRNA transcripts that carries the same cellular barcode with the whole transcriptome (Fig 1a). In brief, we adopted the modified CROP-seq vector5, and developed a strategy for capturing sgRNA sequences6,7 through reverse transcription using a sgRNA-specific primer followed by targeted enrichment of sgRNA sequences via PCR (Extended Data Fig 1, Supplementary Note 1–2, Supplementary Table 1). With extensive optimizations (Extended Data Fig 2), PerturbSci achieves a high knockdown efficacy with a potent dual-repressor dCas9 (i.e., dCas9-KRAB-MeCP29), a high capture rate of sgRNA (i.e., up to 99.7% of cells), and can readily scale up for profiling a large number of cells using the three-level combinatorial indexing approach10 (Fig 1b, Supplementary Note 3).
Figure 1. PerturbSci-Kinetics enables joint profiling of transcriptome dynamics and high-throughput gene perturbations.
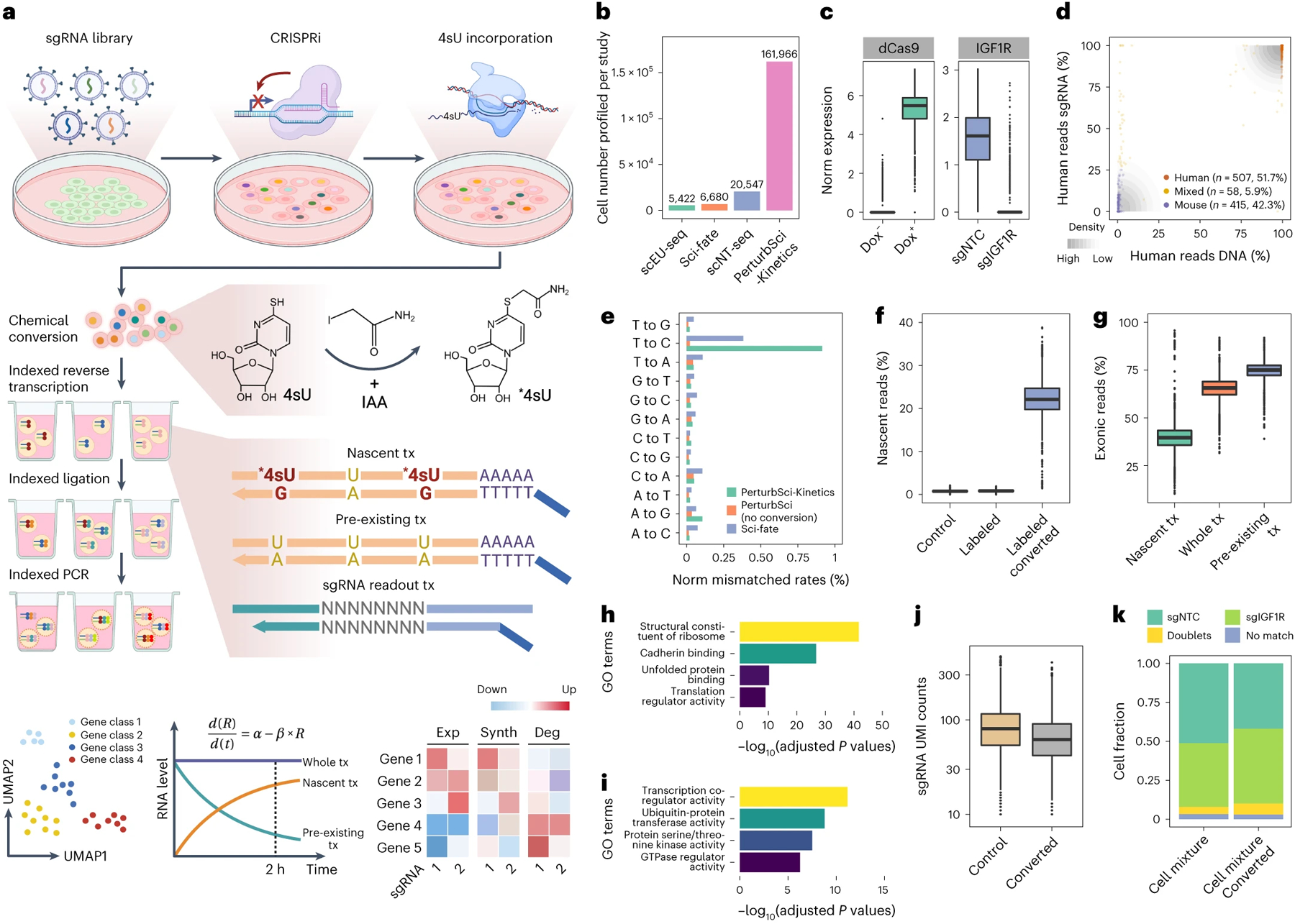
a. Scheme of PerturbSci-Kinetics. IAA, iodoacetamide. *4sU, chemically modified 4sU. R and Exp, steady-state expression. α and Synth, RNA synthesis rate. β and Deg, RNA degradation rate. b. Barplot showing the cell numbers profiled in this study and those from published single-cell RNA-seq coupled with metabolic labeling20–22. c. Left: the log-transformed normalized expression of dCas9-KRAB-MeCP2 in untreated (n = 3,344 cells) or Dox-induced HEK293-idCas9 cells (n = 1,419 cells). Right: the normalized expression of IGF1R in Dox-induced HEK293-idCas9 cells transduced with sgNTC (n = 688 cells) or sgIGF1R (n = 820 cells). d. An equal number of induced HEK293-idCas9-sgIGF1R cells and 3T3-CRISPRi-sgFto cells were mixed and were profiled using PerturbSci. Scatterplot showed the concordance between percentage of transcriptome and sgRNA reads mapping to human and mouse genomes and human and mouse sgRNA, respectively, for each cell. e. Barplot showing the sequencing-depth-normalized percentages of single-base mismatches in reads from sci-fate20, and PerturbSci-Kinetics on chemically converted or unconverted cells. f. Boxplot showing the fraction of nascent reads recovered from single cells without 4sU labeling and chemical conversion (n = 1,498 cells), 4sU-labeled cells without chemical conversion (n = 1,008 cells), and 4sU-labeled/converted cells (n = 2,568 cells). g. Boxplot showing the proportion of nascent, pre-existing, and whole-transcriptome reads mapped to exons of the genome across single cells (n=4,115 cells). h-i. Barplots showing the enriched Gene Ontology (GO) terms in genes with low (h) or high (i) nascent reads fractions. One-sided Fisher’s Exact Tests were conducted with the alternative hypothesis that the true odds ratio is greater than 1. j. Boxplot showing the sgRNA UMI counts/cell in cells with (n = 2,568 cells) or without the chemical conversion (n = 2,506 cells). k. Stacked barplot showing the fraction of converted/unconverted cells identified as sgNTC/sgIGF1R singlets, doublets, and cells with no sgRNA detected. Boxes in boxplots indicate the median and interquartile range (IQR) with whiskers indicating 1.5× IQR.
By incorporating 4-thiouridine (4sU) labeling11–17, PerturbSci-Kinetics retrieves time-resolved nascent transcriptomes at the single-cell resolution, distinguishing newly-synthesized transcripts from whole transcriptomes. The kinetic rates of mRNA such as RNA synthesis and degradation in each genetically-perturbed cell population were then inferred (Fig 1a, Methods). Our method incorporates several optimizations to reduce the cell loss (Extended Data Fig 2) and enhance the accuracy of nascent reads calling (Extended Data Fig 3). With three levels of combinatorial indexing, PerturbSci-Kinetics demonstrates orders of magnitude higher throughput than previous approaches coupling metabolic labeling and single-cell RNA-seq (e.g., scEU-seq, sci-fate, scNT-seq)18–22 (Fig 1b).
As a proof of concept, we established a human HEK293 cell line with inducible dCas9-KRAB-MeCP29 expression (HEK293-idCas9). We thoroughly validated the potent knockdown of target gene expression following Doxycycline (Dox) treatment (Fig 1c, Extended Data Fig 4a–c). Furthermore, we demonstrated the purity of the single-cell transcriptome and sgRNA capture of PerturbSci by profiling mixed human and mouse cells transduced with human and mouse-specific sgRNAs, respectively (Fig 1d).
We proceeded to validate the capability of PerturbSci-Kinetics in capturing the three-layer readout at the single-cell level. After 4sU labeling and chemical conversion, we observed a significant enrichment of T to C mismatches in the mapped reads, which is consistent with findings from our previous study20 (Fig 1e). A median of 22.1% of newly synthesized reads were recovered, in contrast to only 0.8% in control cells (Fig 1f). The proportion of reads mapped to exonic regions was also significantly lower in nascent reads compared with pre-existing reads (p-value < 1e-20, Tukey’s test after ANOVA) (Fig 1g). Moreover, genes with a higher fraction of nascent reads were significantly enriched in highly dynamic biological processes23 while housekeeping genes were strongly enriched in genes with a lower fraction of nascent reads (Fig 1h–i). Notably, the chemical conversion step is fully compatible with sgRNA detection. We recovered sgRNAs from 97% of chemically converted cells (a median of 62 sgRNA UMIs per cell), in which 92.6% were annotated as sgRNA singlets (Fig 1j–k).
To dissect the impact of genetic perturbations on transcriptome kinetics, we performed a PerturbSci-Kinetics screening on HEK293-idCas9 cells. These cells were transduced with a library of 699 sgRNAs, which included 15 no-target controls (NTC), targeting a total of 228 genes involved in diverse biological processes (Fig 2a, Supplementary Table 2). Following a 5-day puromycin selection, we harvested a proportion of cells for bulk library preparation (referred to as ‘day 0’ samples) and induced dCas9-KRAB-MeCP2 expression with Dox for seven more days. The screening window was carefully chosen to maximize gene knockdown efficiency, minimize population dropout8, and allow cells to attain transcriptomic steady states24 (Extended Data Fig 4d). We performed 200uM 4sU labeling for two hours at the end of the screening and harvested samples for both bulk and PerturbSci-Kinetics library preparation. As a quality control, the activation of CRISPRi significantly altered the abundance of sgRNAs in the pool, which was consistent across replicates and aligned with previous studies25. For example, genes involved in essential functions (e.g., DNA replication, ribosome assembly) were strongly depleted after the screening (Extended Data Fig 4e–g). Reassuringly, the number of sgRNA singlets recovered by PerturbSci-Kinetics correlated well with read counts of bulk screen libraries (Pearson correlation r = 0.988, p-value < 2.2e-16) (Fig 2b).
Figure 2. Characterizing the impact of genetic perturbations on gene-specific transcriptional and degradation dynamics with PerturbSci-Kinetics.
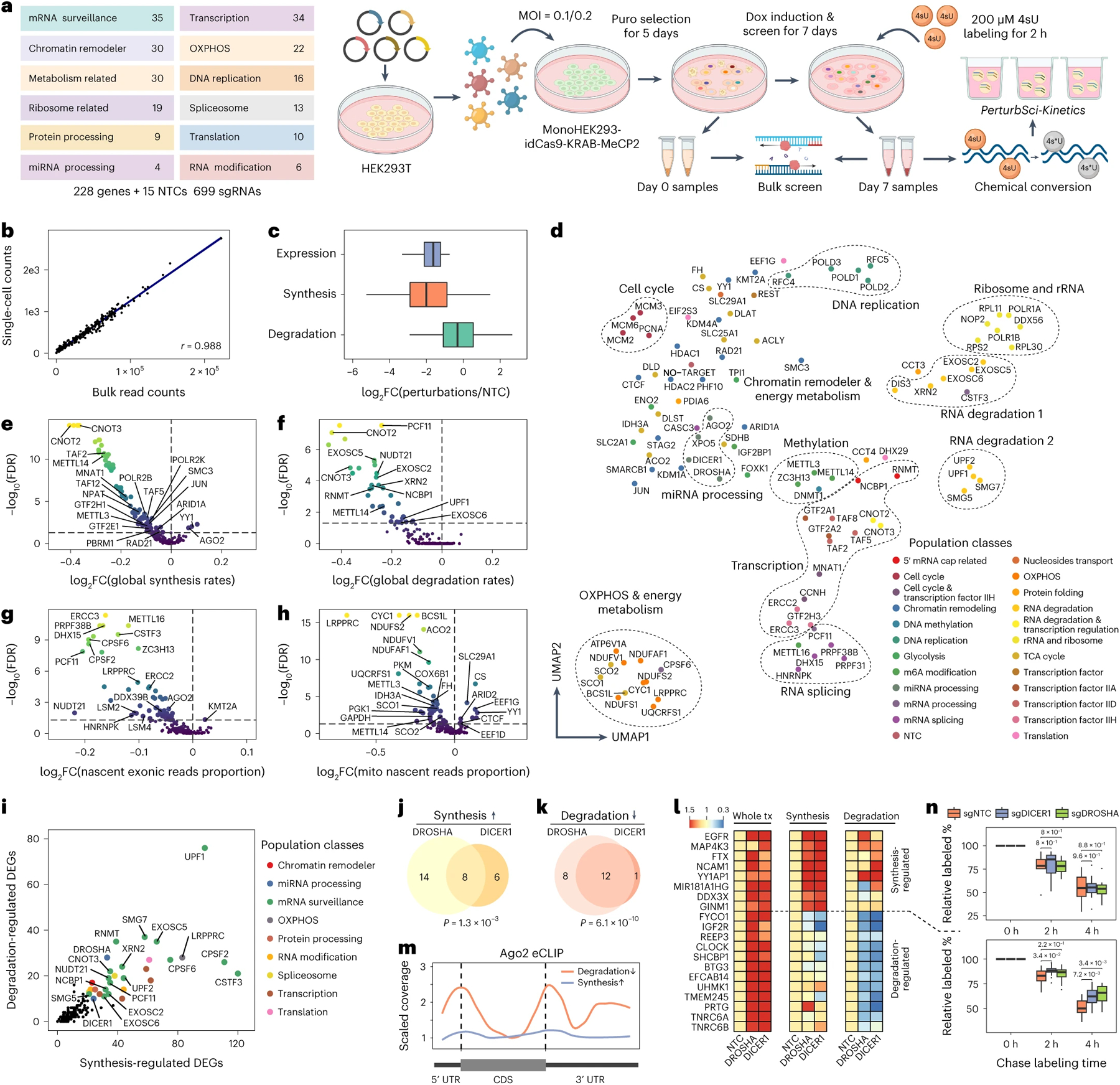
a. Scheme of the experimental design. b. The scatterplot shows the correlation between perturbation-associated cell count from PerturbSci-Kinetics and sgRNA read counts from bulk screen libraries. c. Boxplot showing the log2-transformed fold changes of gene expression, synthesis rates, and degradation rates of sgRNA-targeted genes (n = 203 genes) in perturbed cells expressing the corresponding sgRNA, compared to NTC. d. UMAP visualization of perturbed pseudobulk whole transcriptomes profiled by PerturbSci-Kinetics. We aggregated single-cell transcriptomes in each perturbation, followed by dimension reduction using PCA and visualization using UMAP. Population classes, the functional categories of genes targeted in different perturbations. e-h. Scatterplots showing the extent and the significance of changes on the distributions of global synthesis (e), degradation (f), proportions of exonic reads in the nascent transcriptome (g), and proportions of mitochondrial nascent reads (h) upon perturbations compared to NTC cells. The fold changes were calculated by dividing the median values of each perturbation with that of NTC cells and were log2 transformed. i. Scatterplot showing the number of synthesis/degradation-regulated DEGs from different perturbations. nDEGs: the number of DEGs. j-k. Venn diagrams showing the number of merged DEGs with significantly enhanced synthesis (j) or impaired degradation (k) between DROSHA and DICER1. One-sided Fisher’s Exact Tests were conducted with the alternative hypothesis that the true odds ratio is greater than 1. l. Heatmaps showing the steady-state expression, synthesis and degradation rate changes of genes included in j-k. Tiles of each row were colored by fold changes of values of perturbations relative to NTC. m. Lineplot showing the AGO2 binding patterns on transcripts of protein-coding genes in j-k revealed by eCLIP signal intensity. Data was obtained from a prior study42. n. Boxplots showing the relative proportion of labeled mRNA of transcription- (n = 8 genes) and degradation-regulated genes (n = 12 genes) after chase labeling for different time in HEK293-idCas9-sgNTC, sgDROSHA, and sgDICER1 cells. Two-sided student t-tests were performed between knockdown groups and the NTC group. Boxes in boxplots indicate the median and IQR with whiskers indicating 1.5× IQR.
We recovered 161,966 labeled cells with matched sgRNAs (88% of cells recovered in total), and 126,271 cells were annotated as sgRNA singlets (Extended Data Fig 4j). Despite the shallow sequencing depth (~8000 reads per cell), we achieved a median of 2,155 UMIs per cell. 698 out of 699 sgRNAs were successfully recovered, with a median of 28 sgRNA UMIs per cell. Subsequently, we excluded cells containing sgRNAs that demonstrated low knockdown efficiencies (<= 40% gene expression reduction compared to NTC) were excluded. The RT-qPCR validation on several individual sgRNAs corroborated the accuracy of our knockdown efficiency estimates (Extended Data Fig 4h–l). Ultimately, 98,315 cells were retained for downstream analysis, corresponding to a median of 484 cells per gene perturbation and a median knockdown efficiency of target genes at 67.7% (Fig 2c).
We next quantified gene-specific synthesis and degradation rates in each perturbation using an ordinary differential equation approach26 (Methods). As expected, genes targeted by the CRISPRi demonstrated substantially reduced synthesis rates, while their degradation rates exhibited only mild alterations (Fig 2c). As another validation, we observed significantly higher correlations of transcriptomes among sgRNAs targeting the same genes across multiple layers (e.g., whole/nascent transcriptome, synthesis/degradation rates, Extended Data Fig 5a). We then performed dimension reduction and Uniform Manifold Approximation and Projection (UMAP) visualization27 on aggregated whole transcriptomes of each perturbation. Perturbations targeting paralogous genes (e.g., EXOSC5 and EXOSC6) or related biological processes (e.g., RNA degradation, energy metabolism) were readily clustered together (Fig 2d). Similar analyses on gene-specific synthesis/degradation rates managed to group perturbations by their functions (Extended Data Fig 5b–c). Furthermore, by aggregating profiles of single cells carrying sgRNAs that target the same gene, we achieved robust estimations for both whole/nascent transcriptomes, as well as transcriptome kinetic rates (Extended Data Fig 5d).
We then investigated how genetic perturbations influence global transcriptome dynamics (Fig 2e–g, Extended Data Fig 6a–c, 6e–g, Supplementary Table 5–7). As expected, the knockdown of genes encoding proteins involved in transcription initiation (e.g., GTF2E1, TAF2), mRNA synthesis (e.g., POLR2B, POLR2K), and chromatin remodeling (e.g., SMC3, RAD21) significantly downregulated the global synthesis rates but not the degradation rates. Conversely, perturbations targeting critical biological processes such as DNA replication (e.g., POLA2, POLD1), ribosome synthesis, and rRNA processing (e.g., POLR1A, POLR1B, RPL11, RPS15A), mRNA and protein processing (e.g., CNOT2, CNOT3, CCT3, CCT4) reduced both global RNA synthesis and degradation, indicating a compensatory mechanism for maintaining transcriptome homeostasis28 (Fig 2e–f). Moreover, we noted significant reductions in exonic read fractions in nascent transcriptomes following perturbations related to RNA processing (e.g., NCBP1, LSM2, LSM4, CPSF2, CPSF6) and energy metabolism (e.g., GAPDH, NDUFS2), signifying dysregulated splicing dynamics (Fig 2g).
Interestingly, the knockdown of AGO2, a recognized post-transcriptional regulator29, led to an increase in global synthesis, suggesting its potential role in transcriptional repression (Fig 2e). The re-analysis of public datasets30,31 corroborated our observation. Specifically, genes exhibiting enriched AGO2 binding at transcription start sites (TSS) were markedly upregulated following AGO2 silencing (Extended Data Fig 7a–b). Additionally, the enrichment of AGO2 binding was observed immediately downstream of TSS, and was positively correlated with transcriptional pausing (Extended Data Fig 7a–d). For validation, we employed SLAM-seq32 to examine the transcriptomic response after AGO2 knockdown, identifying 78 highly-paused genes significantly upregulated. Notably, the nascent RNA of these genes showed increased 3’ end coverages compared to NTC, indicative of more efficient transcriptional elongation (Extended Data Fig 7e–f). Collectively, our integrated analyses robustly support the unconventional function of AGO2 in transcriptional repression.
We next investigated regulators of mitochondrial RNA dynamics by quantifying the fraction of nascent reads in single-cell mitochondrial transcriptomes. A significant reduction in mitochondrial transcriptome turnover was observed after perturbing metabolism-associated genes, including those encoding proteins involved in glycolysis (e.g., GAPDH, FH, PKM), the TCA cycle (e.g., ACO2, IDH3A), and oxidative phosphorylation (e.g., NDUFS2, COX6B1) (Fig 2h, Extended Data Fig 6d, 6h, Supplementary Table 8). Notably, LRPPRC emerged as a key mitochondrial RNA dynamics regulator, as its knockdown led to substantial reduction in both turnover rates and expression levels across the majority of mitochondrial protein-coding genes, and mitochondrial functional defect (Extended Data Fig 8a–c, Supplementary Table 9). In contrast, nuclear-encoded genes were primarily regulated at the transcriptional level upon LRPPRC knockdown (Extended Data Fig 8d–f). These kinetic changes in mitochondrial mRNA were validated through an independent PerturbSci-Kinetic experiment that profiled with LRPPRC knockdown (Extended Data Fig 8g–i). Recent studies have reported similar findings, observing impaired mitochondrial gene expression and mitochondrial functional defects in the hearts of LRPPRC knockout mice33 and in brown adipocyte-specific LRPPRC knockout mice34. This further corroborates the essential role of LRPPRC in maintaining mitochondrial mRNA homeostasis.
To further demonstrate the unique capacity of PerturbSci-Kinetics in unraveling the regulatory mechanisms that govern gene expression control, we identified 14,618 differentially expressed genes (DEGs) across perturbations, with 22.9% of them exhibited significant changes in their synthesis or degradation rates (Supplementary Table 10–11, Methods). Among these, DEGs regulated by RNA degradation were associated with perturbations in mRNA surveillance/processing genes (Fig 2i). For instance, our study revealed a set of significantly overlapped DEGs upon knockdown of DROSHA and DICER135,36, genes encoding two crucial RNases in the miRNA biogenesis pathway37 (Extended Data Fig 9a–c). These DEGs were regulated through distinct mechanisms: some genes were regulated by decreased degradation (e.g., genes encoding miRNA-mediated silencing complex (RISC) components: TNRC6A and TNRC6B), while others are regulated through increased transcription (e.g., miRNA host genes: MIR181A1HG, FTX; genes encoding protein involved in miRNA biogenesis: DDX3X) (Fig 2j–l, Supplementary Table 12). The RNA binding pattern of AGO2, a core component of RISC for miRNA-mediated mRNA degradation38, further validated our findings, exhibiting a strong enrichment in the UTRs of transcripts from degradation-regulated genes but not in synthesis-regulated genes (Fig 2m). This finding was further substantiated through PerturbSci-Kinetics profiling on individual sgRNA knockdown clones and SLAM-seq following 4sU chase labeling32 (Fig 2n, Extended Data Fig 9d–g).
Finally, we delved into the effects of genetic perturbations on RNA dynamics during cell cycle progression. Using our validation dataset, we separated cells into five clusters representing different cell cycle stages using cell cycle-related genes39 (Extended Data Fig 10a–c), and then calculated stage-specific kinetic rates of genes. Employing mfuzz clustering40, we identified four gene clusters displaying discrepant cell cycle time-course synthesis dynamics patterns. Among these, only genes in cluster 1 exhibited evident steady-state expression fluctuations (Extended Data Fig 10d). While their synthesis and degradation rates both increased along the cell cycle, the synthesis rates outpaced degradation rates, leading to an increase in steady-state mRNA levels from the S to the G2M stage. GO term analysis further supported the crucial roles of proteins encoded by these genes in cell cycle (Extended Data Fig 10e). Interestingly, in cells with DROSHA and DICER1 knockdown, we observed a similar steady-state expression pattern for genes in cluster 1, but with unresponsive degradation and compensated synthesis during cell cycle progression (Extended Data Fig 10f), suggesting the existence of synthesis-degradation feedback loops for gene regulation. In contrast, LRPPRC knockdown did not impact cell cycle-dependent RNA degradation dynamics (Extended Data Fig 10g), aligning with our results that it specifically affects mitochondrial mRNA stability. Together, our study emphasizes the coordinated regulation of gene expression throughout the cell cycle progression and highlights the presence of intricate feedback loops between RNA synthesis and degradation.
In summary, PerturbSci-Kinetics allows for the quantitative analysis of the genome-wide mRNA kinetics across genetic perturbations in a massively-parallel manner. Of note, there are several potential limitations to consider: First, extended 4sU labeling might impact cell states and potentially hinder the identification of sgRNA sequences. To mitigate this, we opted for a relatively short-term (2 hours) treatment to minimize such effects. Second, RNA dynamics identified by PerturbSci-Kinetics may not directly reflect causality in gene regulation, partly due to the gradual nature of CRISPRi-based gene knockdown. This limitation could be mitigated by coupling the technique with large-scale chemical perturbations. Third, the perturbation of essential genes might lead to significant dropout, affecting dynamic rate estimations due to limited cells and reads. Moreover, apoptosis-triggered mRNA decay might further complicate the analysis41. Therefore, we recommend excluding genetic perturbations that either lead to strong dropout effects or substantial disruption of cell cycle distribution during RNA dynamics analysis.
In spite of these limitations, our findings illuminate the distinct advantages of PerturbSci-Kinetics over conventional assays. Its multi-layer readout provides a comprehensive perspective on gene expression and RNA dynamics in response to genetic perturbations, facilitating high-throughput and parallel characterization of elements that govern gene-specific RNA dynamics. Moreover, given the low cost and high sensitivity of PerturbSci, we envision the potential to systematically dissect cell-type-specific gene regulatory networks across various biological contexts with an unparalleled scale and resolution.
Methods:
Cell culture
The 3T3-L1-CRISPRi cell line was obtained from the Tissue Culture facility at the University of California, Berkeley. The HEK293 cell line was a gift from the Scott Keeney Lab at Memorial Sloan Kettering Cancer Center. The HEK293T cell line and the NIH/3T3 cell line were obtained from ATCC. All cells were maintained at 37 °C and 5% CO2 in high glucose DMEM medium supplemented with L-Glutamine and Sodium Pyruvate (Gibco 11995065) and 10% Fetal Bovine Serum (FBS; Sigma F4135).
Cell lines generation
To generate HEK293 cells with Dox-inducible dCas9-KRAB-MeCP2 expression, the lentiviral plasmid Lenti-idCas9-KRAB-MeCP2-T2A-mCherry-Neo was constructed. After sequencing validation, the lentivirus was produced by co-transfecting Lenti-idCas9-KRAB-MeCP2-T2A-mCherry-Neo with psPAX2 (Addgene #12260) and pMD2.G (Addgene #12259) into low-passage HEK293T cells in a 10cm dish using Polyjet (SignaGen SL100688). After lentiviral titration, HEK293 cells were transduced at MOI = 0.2 for 48 hours. Cells were treated with 1ug/ml Dox (Sigma D5207) for 48 hours, and single cells with strong mCherry fluorescence were sorted for monoclonal generation.
The polyclone 3T3-CRISPRi cell line was generated in a similar way. pHR-SFFV-dCas9-BFP-KRAB (Addgenes #46911) was co-transfected with psPAX2 and pMD2.G to generate dCas9-expressing lentivirus, and the transduction at MOI=0.2 was performed on 3T3 cells. BFPhi cells (top 35% in BFP+ population) were sorted and the sorting was repeated twice more after cell expansion to enrich cells with strong dCas9 expression.
Single gene Knockdown and efficacy examination
CROP-seq-opti-Puro-T2A-GFP was assembled by adding a T2A-GFP downstream of Puromycin resistant protein coding sequence on the CROP-seq-opti plasmid (Addgene #106280). Oligos for individual guides cloning were ordered from IDT with the following design:
Plus strand: 5’-CACCG[20bp sgRNA plus strand sequence]-3’
Minus strand: 5’-AAAC[20bp sgRNA minus strand sequence]C-3’
Oligos were phosphorylated using T4 PNK (NEB M0201S) and were annealed. The CROP-seq-opti-Puro-T2A-GFP was digested by Esp3I (NEB R0734L), then the linearized backbone and the annealed duplex were ligated using the Blunt/TA Ligase Master Mix (NEB M0367S). Transformation, clone amplification, sequencing validation, lentivirus generation, and titer measurement were done as stated above.
Mouse 3T3-L1-CRISPRi cells and 3T3-CRISPRi cells were transduced with the lentivirus expressing non-target control (NTC) sgRNA or sgRNA targeting Fto. Human HEK293-idCas9 cells were transduced with lentivirus expressing NTC sgRNA or sgRNA targeting IGF1R during technique development, and HEK293-idCas9-sgXPO5, sgAGO2, sgDROSHA, sgDICER1, sgLRPPRC cell lines were later established for validating significant hits from the screen. Transduction was carried out at MOI = 0.2 with 8ug/ml of Polybrene for 48 hours. Transduced cells were then selected by either FACS or Puromycin treatment.
For RT-qPCR validation, primer pairs were selected from PrimerBank (https://pga.mgh.harvard.edu/primerbank/) and were synthesized from IDT. Total RNA of each sample was extracted using the RNeasy Mini kit (QIAGEN 74104). 1ug total RNA was then reverse-transcribed, and PowerUp™ SYBR™ Green Master Mix (Thermo A25742) was used for RT-qPCR following the manufacturer’s instructions. The data was analyzed and visualized by Graphpad Prism (9.2.0).
For flow cytometry validation, 1e6 cells of each sample were harvested and resuspended in 100ul of PBS-0.1% sodium azide-2% FBS. BV421 Mouse Anti-Human CD221 (BD 565966) and BV421 Mouse IgG1 k Isotype Control (BD 562438) at the final concentration of 10 ug/ml were added, and reactions were incubated at 4 °C in the dark with rotation for 30 minutes. Cells were then washed twice using PBS-0.1% sodium azide-2% FBS, and fluorescence signals were recorded. The data was analyzed and visualized by FlowJo (10.8.1).
Construction of the pooled sgRNA library
Genes to be included in our sgRNA library were selected based on following considerations: 1) essential and non-essential genes were identified using the bulk CRISPR screen data from a previous report25 and Depmap43, and both were included in the gene set. 2) To validate the ability of PerturbSci-kinetics to characterize gene-specific RNA dynamics, we selected genes involved in transcription, chromatin remodeling, RNA processing, and mRNA decay based on Gene Ontology terms44 and KEGG pathways45. 3) We ensured that all selected genes were expressed in the cell line to be used in our study. An in-house HEK293 EasySci-RNA dataset was used to select expressing genes that met criteria 1 and 2.
sgRNA sequences targeting genes of interest were obtained from an established optimized CRISPRi sgRNA library (set A)25. Finally, 684 sgRNAs targeting 228 genes (3 sgRNAs/gene) and 15 non-targeting controls were included in the present study.
The single-stranded sgRNA library was synthesized in a pooled manner by IDT in the following format:
5’-GGCTTTATATATCTTGTGGAAAGGACGAAACACCG[20bp sgRNA plus strand sequence]GTTTAAGAGCTATGCTGGAAACAGCATAGCAAGTT-3’
100ng of oligo pool was amplified by PCR using primers targeting 5’ homology arm (HA) and 3’ HA. The PCR product was purified and the insert was cloned into Esp3I-digested CROP-seq-opti-Puro-T2A-GFP by Gibson Assembly. In parallel, a control Gibson Assembly reaction containing only the backbone was set. Both reactions were cleaned up by 0.75x AMPURE beads (Beckman Coulter A63882) and eluted in 5uL EB buffer (QIAGEN 19086), then were transformed into Endura Electrocompetent Cells (Lucigen 602422) by electroporation (Gene Pulser Xcell Electroporation System, Bio-Rad 1652662). After recovery, cells of each reaction were spread onto an 245 mm Square agarose plate (Corning, 431111) with 100ug/ml of Carbenicillin (Thermo, 10177012) and was then grown at 32 °C for 13 hours. All colonies from each reaction were scraped from the plates and the CROP-seq-opti-Puro-T2A-GFP-sgRNA plasmid library was extracted using ZymoPURE II Plasmid Midiprep Kit (Zymo, D4200). The lentiviral library was generated as stated.
The pooled PerturbSci-Kinetics screen experiment
For each replicate, 7e6 uninduced HEK293-idCas9 cells were seeded. Two replicates were transduced at MOI=0.1 and another two replicates were transduced at MOI=0.2. At least 1000x coverage was kept throughout the cell culture. At the end of the Puro selection, we harvested 1.4e6 cells in each replicate (2000x coverage/sgRNA) as day0 samples of the bulk screen and pellet down at 500xg, 4 °C for 5 minutes for genomic DNA extraction. For the rest of cells, the dCas9-KRAB-MeCP2 expression was induced by adding Dox at the final concentration of 1ug/ml, and L-glutamine+, sodium pyruvate-, high glucose DMEM was used to sensitize cells to perturbations on energy metabolism genes. Cells were cultured for additional 7 days. On day7, 6ml of the original media from each plate was mixed with 6uL of 200mM 4sU (Sigma T4509–25MG) dissolved in DMSO (VWR 97063–136) and was put back for nascent RNA metabolic labeling. After 2 hours of treatment, 1.4e6 cells in each replicate were harvested as day7 samples of the bulk screen, and the rest of the cells were fixed for PerturbSci-Kinetics profiling (see the next section).
Genomic DNA of bulk screen samples was extracted using Quick-DNA Miniprep Plus Kit (Zymo D4068T) following the manufacturer’s instructions. The bulk screen libraries were amplified from genomic DNA extracted using custom primers (Supplemental Note 2) for sequencing.
Step-by-step protocols for PerturbSci-Kinetics library preparation are included in Supplemental Note 1.
4sU pulse/chase labeling and SLAM-seq
HEK293-idCas9-sgAGO2 and sgNTC cells were induced with Dox for 7 days in 10cm dishes, and cells were labeled with 600uM 4sU for 20 minutes before total RNA extraction. HEK293-idCas9-sgDROSHA, sgDICER1, and sgNTC cells were induced with Dox for 7 days, and were treated with Dox+ medium containing 100uM 4sU for 18h. The medium was refreshed every 6h. Then chase labeling was performed by using medium with 10mM uridine (Sigma U3750–1G). Following 2h and 4h incubation, total RNA was extracted.
2–5 ug of total RNA from each sample was used for chemical conversion. RNA was diluted into 15ul, and mixed with 5ul of 100mM IAA, 5ul of NaPO4 (pH 8.0, 500mM) buffer, and 25ul of DMSO. The reaction was incubated at 50 °C for 15 minutes and was then quenched with 1ul 1M DTT.After RNA purification using the Monarch RNA Cleanup Kit (NEB T2030L), samples were immediately used for library construction.
Full-length and 3’end bulk SLAM-seq were used for different experimental purposes. For full-length bulk SLAM-seq library construction, the CRISPRclean Stranded Total RNA Prep with rRNA Depletion Kit (Jumpcode Genomics KIT1014) was used. For 3’end bulk SLAM-seq library construction, an in-house 3’end library preparation workflow was used. In brief, 250–500ng total mRNA was mixed with 1ul 100uM oligodT primer (ACGACGCTCTTCCGATCTNNNNNNNNNNTTTTTTTTTTTTTTT), 1ul 10mM each dNTP mix, 0.5ul SUPERase In and the volume was adjusted to 15ul with water. After RNA priming at 55C for 5min, 4ul 5xRT buffer and 1ul Maxima H Minus Reverse Transcriptase (Thermo EP0753) were added to the reaction, and reverse transcription was performed as recommended by the manufacturer. After 0.6x AMPURE beads purification, Second strand synthesis (NEB E6111L) was carried out by 1h incubation at 16 °C, then cDNA was purified by 0.6x AMPURE beads. Following Read2 tagmentation on 10ng cDNA using 1:20 V/V Nextera Read2-Tn5, the reaction was quenched, and the final library was prepared as EasySci-RNA10.
Reads processing
For bulk CRISPR screen libraries, bcl files were demultiplexed into fastq files based on index 7 barcodes. Reads for each sample were further extracted by index 5 barcode matching. Every read pair was matched against two constant sequences (Read1: 11–25bp, Read2: 11–25bp) to remove artifacts. For all matching steps, a maximum of 1 mismatch was allowed. Finally, sgRNA sequences were extracted from filtered read pairs (at 26–45bp of R1), assigned to sgRNA identities with no mismatch allowed, and read counts matrices at sgRNA and gene levels were quantified using python (2.7).
For PerturbSci-Kinetics, after demultiplexing on index 7, Read1 were matched against a constant sequence on the sgRNA capture primer to remove unspecific priming, and cell barcodes and UMI sequences sequenced in Read1 were added to the headers of the fastq files of Read2, which were retained for further processing. After trimming polyA sequences and low-quality bases from Read2 by Trim_Galore (0.6.7)46, reads were aligned to a customized reference genome consisting of a complete hg38 reference genome (GRCh38.p13 from GENCODE) and the dCas9-KRAB-MeCP2 sequence using STAR (2.7.9a)47. Reads with mapping score >= 30 were selected by samtools (1.13)48. Then deduplication at the single-cell level was performed based on the UMI sequences and the alignment location, and retained reads were split into SAM files per cell. These single-cell sam files were converted into alignment tsv files using the sam2tsv function in jvarkit (d29b24f)49. After background SNP removal, we considered T>C mismatches with the CIGAR string “M” and quality scores > 45 as 4sU site. And only reads with > 30% of T>C mutations among all mismatches were identified as nascent reads, and the list of reads was extracted from single-cell whole transcriptome sam files by the Picard (2.27.4)50. Finally, single-cell whole/nascent transcriptome gene x cell count matrices were constructed by assigning reads to genes51.
Read1 and read2 of PerturbSci-Kinetics sgRNA libraries were matched against constant sequences respectively, allowing a maximum of 1 mismatch. For each filtered read pair, cell barcode, sgRNA sequence, and UMI were extracted from designed positions. Extracted sgRNA sequences with a maximum of 1 mismatch from the sgRNA library were accepted and corrected, and the corresponding UMI was used for deduplication. De-duplication was performed by collapsing identical UMI sequences of each individual corrected sgRNA under a unique cell barcode. Cells with overall sgRNA UMI counts higher than 10 were maintained and the sgRNA x cell count matrix was constructed.
SLAM-seq reads were processed similarly. In brief, for 3’end SLAM-seq, UMI sequences in Read1 were extracted and were attached to the headers of Read2 by UMI-tools (1.1.2)52, and only read2 were further processed. After polyA and low quality base trimming by Trim_Galore, reads were aligned to the hg38 reference genome by STAR. In the scenario of high-concentration 4sU labeling, more loose alignment parameters were used (--outFilterMatchNminOverLread 0.2 --outFilterScoreMinOverLread 0.2). Reads were filtered by samtools, and PCR duplicates in passed reads were further removed by UMI-tools. Nascent reads were identified and extracted, and gene counting on both whole transcriptome and nascent transcriptome were performed as mentioned above but at the sample level. For full-length SLAM-seq, reads were processed similarly but paired-end reads were retained.
sgRNA singlets identification and off-target sgRNA removal
Cells with at least 300 whole transcriptome UMIs, 200 genes, 10 sgRNA UMIs, and unannotated reads ratio < 40% were kept. sgRNA singlets were assigned based on the following criteria: the most abundant sgRNA in the cell took >= 60% of total sgRNA counts and was at least 3-fold of the second most abundant sgRNA.
Target genes with the number of cells perturbed >= 50 were kept. The knockdown efficiency was calculated at the individual sgRNA level to remove potential off-target or inefficient sgRNAs: whole transcriptomes of cells receiving the same sgRNA were merged, normalized by CPM, then the fold changes of the target gene expressions were calculated by comparing the normalized expression levels between corresponding perturbations and NTC. sgRNAs with >= 40% of target gene expression reduction relative to NTC were regarded as “effective sgRNAs”, and singlets receiving these sgRNAs were kept as “on-target cells”. Downstream analyses were done at the target gene level by analyzing all cells receiving different sgRNAs targeting the same gene.
UMAP embedding on pseudo-cells
The count matrix of the “on-target” cells described above was loaded into Seurat27, and DEGs of each perturbation (compared to NTC) were retrieved. Cells from perturbations with >= 1 DEG and cells from genetic perturbations involved in similar pathways of the top perturbations were kept. The FC of the normalized gene expression between perturbations and NTC were calculated, and were binned based on the gene-specific expression levels in NTC. The top 3% of genes showing the highest fold changes within each bin were selected and merged as features for Principal Component Analysis (PCA). The top 9 PCs were used as input for UMAP embedding.
Differential expression analysis
Pairwise differential expression analyses between each perturbation and NTC cells were performed by Monocle 253. We selected significant hits (FDR < 0.05) with a >= 1.5-fold expression difference and CPM >= 5 in at least one of the tested cell pairs. More stringent criteria were used to obtain DEGs with high confidence: significant hits (FDR < 0.05) with a >= 1.5-fold expression difference and CPM >= 50 in at least one of the tested cell pairs were kept. For bulk RNA-seq libraries, genes with a minimum of 10 raw counts in at least one sample and expressed in at least a half of samples were kept, and EdgeR54 was used for bulk RNAseq DEGs analysis. Significant hits were selected at FDR < 0.05 level.
Synthesis and degradation rates calculation
After the induction of CRISPRi for 7 days, we assumed new transcriptomic steady states had been established at the perturbation level before the 4sU labeling, and the labeling didn’t disturb these new transcriptomic steady states. The following RNA dynamics differential equation is used for synthesis and degradation rates calculation similar to the previous study26:
| (1) |
In which R is the mRNA abundance of each gene, α is the synthesis rate of this gene, and β is the degradation rate of this gene. Since the RNA synthesis follows the zero-order kinetics and RNA degradation follows the first-order kinetics in cells, is determined by α and R·β.
As steady states had been established, the mRNA level of each gene didn’t change. We can get:
| (2) |
| (3) |
Under the assumption that the labeling efficiency was 100%, all nascent RNA were labeled during the 4sU incubation, and pre-existing RNA would only degrade. So, for nascent RNA (Rn), Rn(t = 0) = 0 and αn = α. For pre-existing RNA (Rn), and αp = 0. Based on these boundary conditions, we could further solve the differential equation above on nascent RNA and pre-existing RNA of each gene.
| (4) |
| (5) |
As both R and Rn were directly measured in PerturbSci-Kinetics, and cells were labeled by 4sU for 2 hours (t = 2), β can be calculated from equation 3 and 4. Then α can be solved by equation 3.
Due to the shallow sequencing and the sparsity of the single cell expression data, synthesis and degradation rates of DEGs were calculated at the target-gene pseudo-cell level. DEGs with only nascent counts or degradation counts were excluded from further examination since their rates couldn’t be estimated.
To examine the significance of synthesis and degradation rate changes upon perturbation, regarding the different cell sizes across different perturbations and NTC, which could affect the robustness of rate calculation, randomization tests were adopted. Only perturbations with cell number >= 50 were examined. For each DEG belonging to each perturbation, background distributions of the synthesis and degradation rate were generated: a subset of cells with the same size as the corresponding perturbed cells was randomly sampled from a mixed pool consisting of corresponding perturbed cells and NTC cells, then these cells were aggregated into a background pseudo-cell, and synthesis and degradation rates of the gene for testing were calculated as stated above, and the process was repeated for 500 times. Rates = 0 were assigned if only nascent counts or degradation counts were sampled during the process (referred to as invalid samplings), but only genes with less than 50 (10%) “invalid samplings” were kept for p-value calculation. The two-sided empirical p-values for the synthesis and degradation rate changes were calculated respectively by examining the occurrence of extreme values in background distributions compared to the rates from perturbed pseudo-cell. Rate changes with p-value < 0.05 were regarded as significant, and the directions of the rate changes were determined by comparing the rates from the perturbed pseudo-cell with the background mean values.
Global changes of key statistics upon perturbations
For global synthesis and degradation rate changes, considering the noise from lowly-expressed genes, we selected top1000 highly-expressed genes from NTC cells, then calculated their synthesis rates and degradation rates in NTC cells and all perturbations with cell number >= 50. K-S tests were performed to compare rate distributions between each perturbation and NTC cells. The distributions of exonic reads percentage in nascent reads from cells with the same target gene knockdown and NTC cells were compared using the K-S tests to identify genes affecting RNA processing. The proportion of nascent mitochondrial read counts to total mitochondrial read counts was calculated in each single cell, and its distributions between cells with knockdown and NTC cells were compared by the K-S tests to identify the master regulator of mitochondrial mRNA dynamics. In all global statistics examinations, Benjamini–Hochberg multiple hypothesis correction was performed, and comparisons with FDR <= 0.05 were considered as significant. The median value from each perturbation and NTC cells were compared to determine the direction of significant changes.
Coverage analysis
We reprocessed the raw data of AGO2 eCLIP obtained from Hela cells from Zhang, K et, al42. After adapter trimming, UMI extraction, mapping, and UMI-based deduplication, bam files were transformed to the single-base coverage by BEDtools55. The transcript regions of genes-of-interest were assembled based on the hg38 genome annotation gtf file from GENCODE. Briefly, for each gene, the exonic regions were extracted and were redivided into 5’UTR, CDS, and 3’UTR by the 5’most start codon and the 3’most stop codon annotated in the gtf. The AGO2 binding coverages of these designated regions were obtained by intersection and were binned. The gene-specific signal in each bin was normalized by the number of bases in each bin, and the binned coverage of each gene was scaled to be within 0–1. After aggregating scaled coverages of synthesis/degradation-regulated genes respectively, the lowest point within CDS was used as the second scaling factor.
Meta-gene coverage analysis was conducted to visualize the gene body distribution of newly transcribed RNA in NTC and AGO2-knockdown samples. Genomic coordinates of protein coding genes on chromosome 1–22 and chromosome X were retrieved from the hg38 genome annotation gtf file from GENCODE. Gene bodies were binned into 50 bins, ordered bins were exported as bed files. For input reads, two nascent reads BAM files per group from the pulse-labeling full-coverage SLAM-seq were merged using samtools, then reads with FLAG = 83/163 were assigned to genes on the plus strand, and reads with FLAG = 99/147 were assigned to genes on the minus strand. The gene-specific binned coverages were counted using the bedtools intersect command. Binned counts of each gene were normalized by total counts in the gene body, and the coverage of any group of genes was finally drawn by averaging the normalized signals across genes.
Public ChIP-seq, shRNA RNA-seq, GRO-seq data analysis
Genes with detectable expression were identified from shControl/shAGO2 bulk RNA-seq in ENCODE. Processed gene counts quantification tables were downloaded from the ENCODE portal. Only genes with mean transcript per million (TPM) > 1 across 4 samples and with detected expression in at least 3 of 4 samples were included. Log2 fold changes of each gene upon AGO2 silencing were calculated by dividing the mean TPM in the shAGO2 group with the mean TPM in the shControl group.
AGO2 ChIP-seq bam and narrow peak files from ENCODE were merged for identifying TSS binding of AGO2. TSS regions of genes with detectable expression (defined as 4kb around TSS) were retrieved, and genes were classified into AGO2 TSS peak+/− genes based on the overlap between their TSS regions with merged AGO2 ChIP-seq narrow peaks. The binding patterns were then visualized using the computeMatrix function in deepTools (3.5.1)56.
GRO-seq data was downloaded from GEO and were reprocessed to depict the transcriptional pausing status of genes. 3’end of reads were trimmed against polyA by Cutadapt (3.4)57, and reads were then aligned to the hg38 reference genome using Bowtie2 (2.3.0)58. After filtering out unmapped reads using samtools, bam files were imported to R. TSS proximal regions and transcriptional elongation regions of protein coding genes with gene lengths >= 1kb were extracted, and the getPausingIndices() function from the BRGenomics package (3.17)59 was used to calculate the pausing indices of genes. Genes detected in both replicates were ranked by the pausing index within the replicate, and an averaged rank was used to study the association with AGO2 TSS binding.
Extended Data
Extended Data Fig. 1.
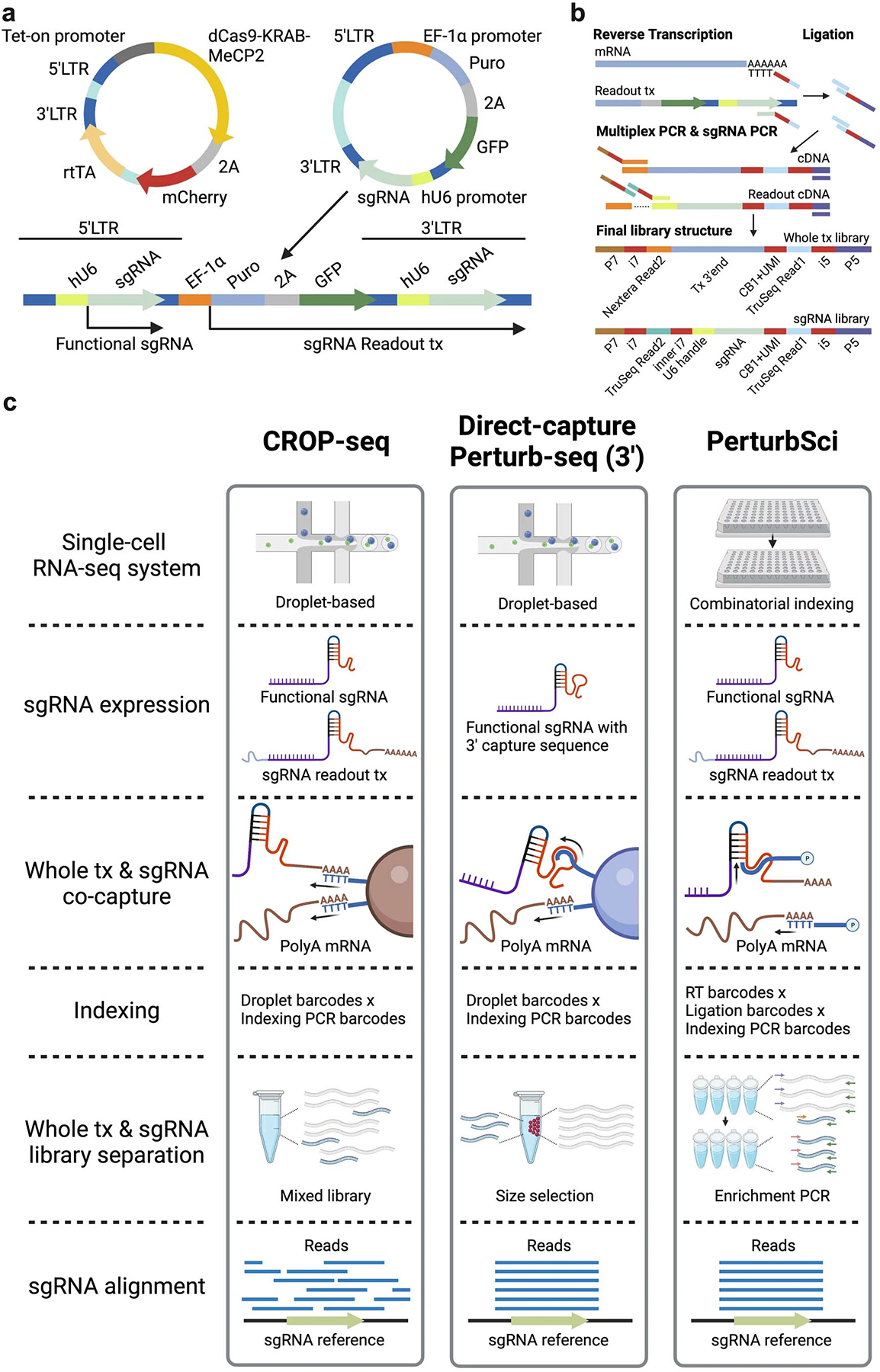
a. The vector system used in PerturbSci for dCas9 and sgRNA expression. The expression of the enhanced CRISPRi silencer dCas9-KRAB-MeCP29 was controlled by the tetracycline responsive (Tet-on) promoter. A GFP sequence was added to the original CROP-seq-opti plasmid6 as an indicator of successful sgRNA transduction and for the lentivirus titer measurement. The CROP-seq vector utilizes the self-replication mechanism of lentivirus during the integration for amplifying the sgRNA expression cassette. In this lentiviral plasmid, the sgRNA expression cassette replaced the U3 region of the 3’LTR5. During the lentiviral integration, the shortened 5’LTR of reverse-transcribed lentiviral genomic DNA was extended by using its 3’LTR as a template, and the sgRNA expression cassette is self-replicated to the 5’LTR62. The self-replicated sgRNA expression cassette at 5’LTR generates functional sgRNA for guiding dCas9, and the original expression cassette at 3’LTR is transcribed as a part of the Puro-GFP fusion transcript driven by the EF-1α promoter. b. The library preparation scheme and the final library structures of PerturbSci, including a scalable combinatorial indexing strategy with direct sgRNA capture and enrichment that reduced the library preparation cost, enhanced the sensitivity of the sgRNA capture compared to the original CROP-seq5, and avoided the extensive barcodes swapping detected in Perturb-seq6. c. A schematic comparison of chemistries between PerturbSci, CROP-seq5, and Direct-capture Perturb-seq7.
Extended Data Fig. 2.
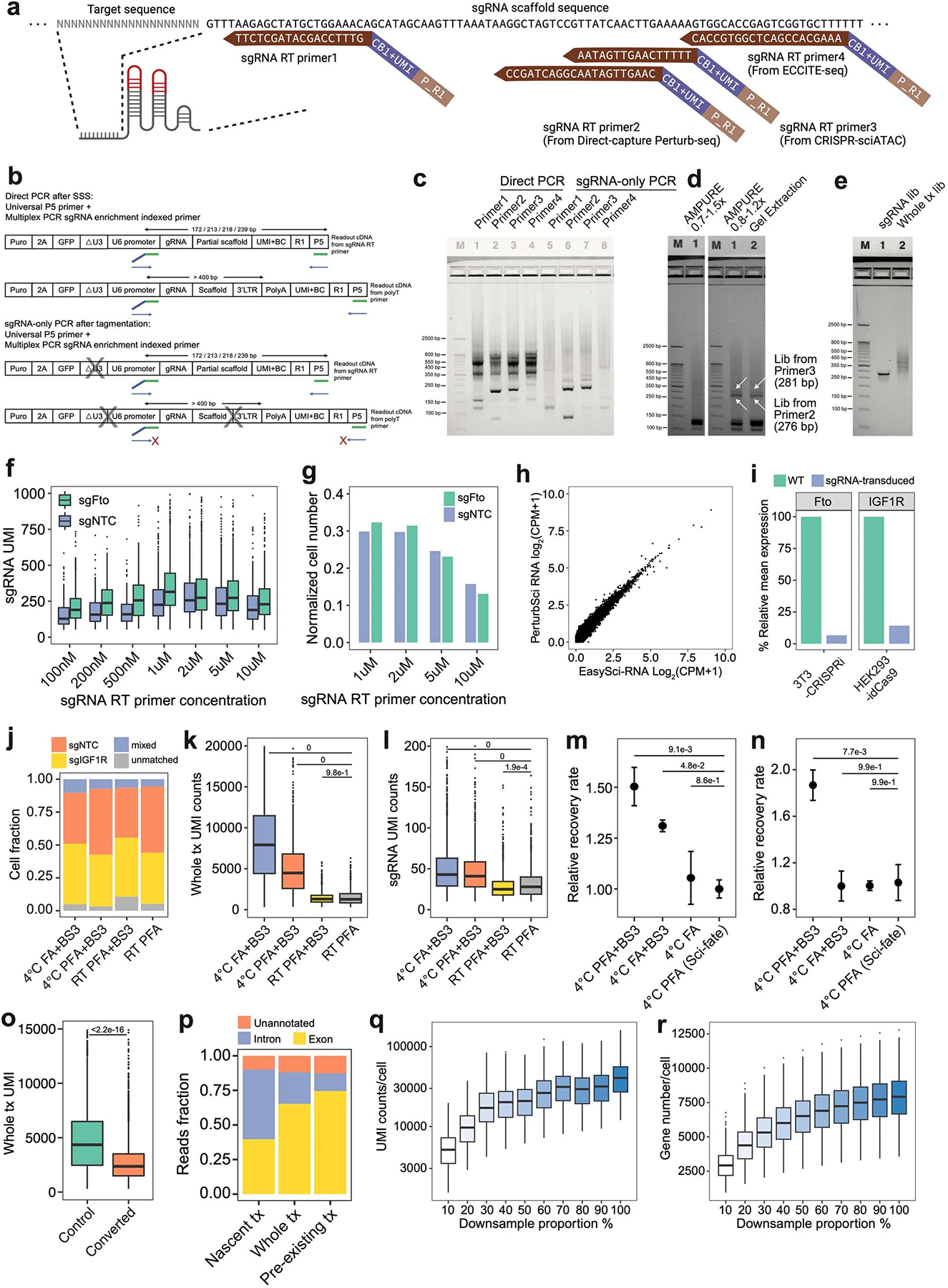
a. sgRNA primers of different designs were mixed with polyT primers respectively for RT. CB, cell barcode. P_R1, partial TruSeq read1 sequence. b-c. After RT, the capture efficiency of sgRNA by different RT primers was evaluated by “Direct PCR”, and the efficiency of by-product removal was examined by “sgRNA-only PCR”. 3 independent experiments were conducted. d. Different post-multiplex PCR purification strategies were tested. 3 independent experiments were conducted. e. A representative gel image of libraries of PerturbSci. 5 independent experiments were conducted. f-g. Boxplots showing sgRNA UMI counts (f) and the cell number recovered (g) from different sgRNA primer concentrations (n=230, 181, 149, 529, 512, 445, 299 cells from 100nM to 10uM groups for sgNTC cells, n = 499, 399, 246, 1237, 1215, 904, 537 cells from 100nM to 10uM groups for sgFto cells). h. Scatterplot showing the correlation between log2-transformed counts per million (CPM) profiled by PerturbSci and EasySci10 in the 3T3L1-CRISPRi cell line. i. Barplots showing effective knockdown in mouse 3T3-CRISPRi-sgFto cells and human HEK293-idCas9-sgIGF1R cells computationally assigned in the species-mixing experiment (Fig 1d). j-l. Barplots showing the cell identities fraction (j), whole transcriptome (k) and sgRNA UMI counts (l) detected per cell in different fixation conditions (n = 1508, 1132, 1247, 1084 cells for conditions from the left to the right). Tukey’s tests after one-way ANOVA were performed. m-n. Dotplots showing the relative recovery rate (n = 4, mean ± SEM) of HEK293-idCas9 cells in different fixation conditions following HCl permeabilization (m) and chemical conversion (n). Dunnett’s test after one-way ANOVA was performed. o. Boxplot showing the effect of chemical conversion on whole transcriptome UMI counts under 4°C PFA+BS3 fixation condition (n = 1988 cells in the control group, n = 4831 cells in the converted group). Two-sided Wilcoxon rank sum test was performed. p. Mapping statistics of reads from PerturbSci-Kinetics. q-r. Boxplots showing single-cell whole transcriptome UMI counts (q) and gene counts (r) under different sequencing depth (n = 500 cells in each subsampling group). Boxes in boxplots indicate the median and IQR with whiskers indicating 1.5× IQR.
Extended Data Fig. 3.
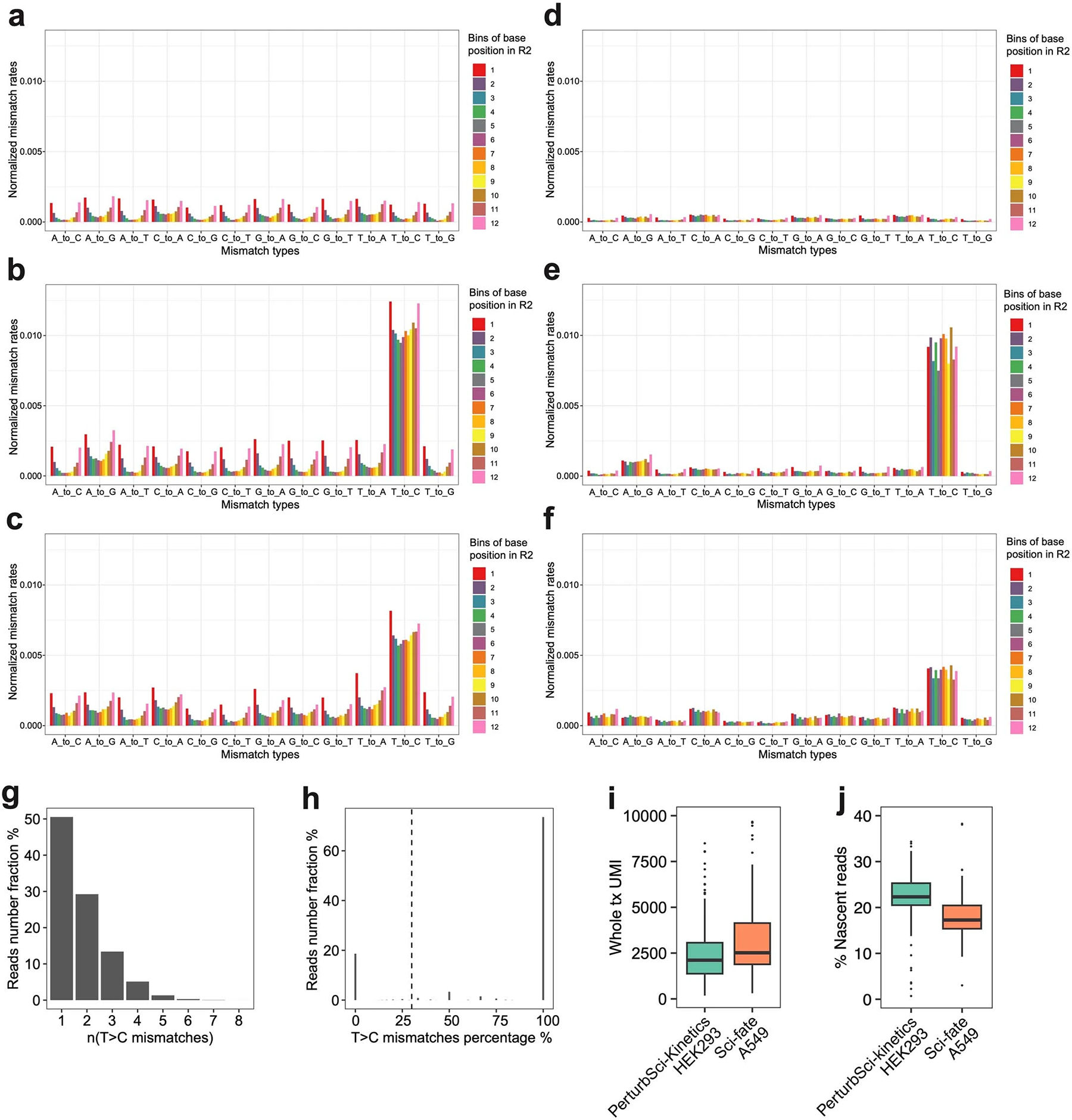
a-c. Barplots showing the normalized mismatch rates of all 12 mismatch types detected in unconverted cells (a), converted cells (b), and the original sci-fate A549 dataset20 (c) at different positions of the reads using the original sci-fate mutation calling pipeline20. d-f. Barplots showing the normalized mismatch rates of all 12 mismatch types detected in unconverted cells (d), converted cells (e), and the original sci-fate A549 dataset20 (f) at different positions of the reads using the updated mutation calling pipeline. Considering the different sequencing lengths between the present dataset and sci-fate, the Read2 from sci-fate were trimmed to the same length as the present dataset before processing. Compared to the original pipeline, the updated pipeline further filtered the mismatch based on the CIGAR string and only mismatches with “CIGAR = M” were kept. Normalized mismatch rates in each bin, the percentage of each type of mismatch in all sequencing bases within the bin. g-h. Statistics of T>C mutations in PerturbSci-Kinetics reads. Histogram showing the number of T>C mutations on reads that were identified to be from newly synthesized transcripts (g). For each read with high-quality mismatches identified, the fraction of mismatches from T>C mutations was calculated, which clearly separated the reads with background mutations and mutants introduced by 4sU in the plot (h). 30% was set as the cutoff to assign nascent reads as sci-fate20. i-j. Downsampling comparison between sci-fate20 and PerturbSci-Kinetics. A subset of raw reads in sci-fate A549 dataset20 were randomly selected to generate a downsampled dataset with the same single-cell raw reads number distribution with PerturbSci-Kinetics, and both datasets were processed using the same pipeline (n = 200 cells for each dataset). The single-cell whole transcriptome UMI counts (i) and the nascent reads proportions (j) between two datasets were compared. Boxes in boxplots indicate the median and IQR with whiskers indicating 1.5× IQR.
Extended Data Fig. 4.
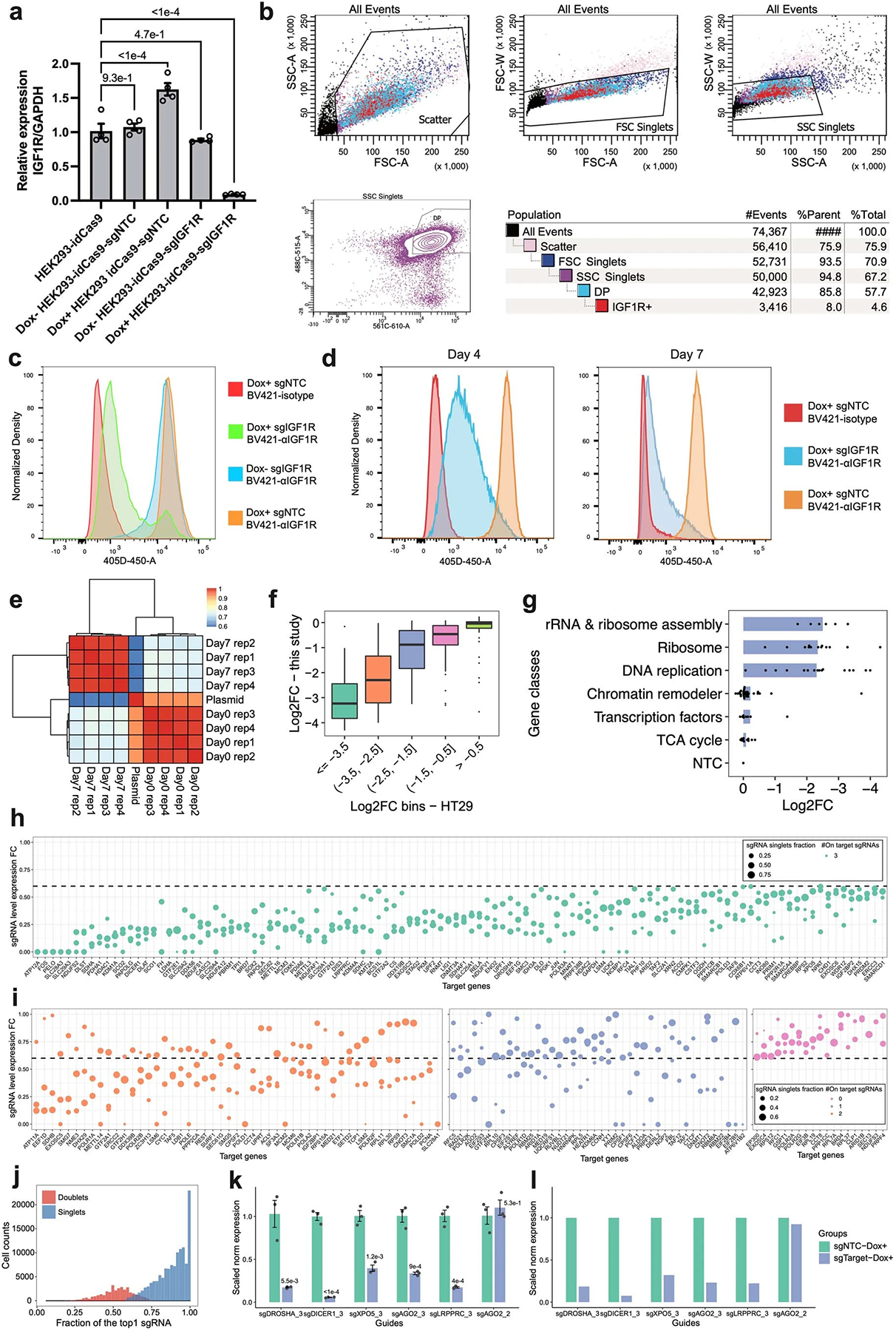
a-d. Inducible IGF1R mRNA and protein knockdown were further validated by RT-qPCR (a) after 3-day Dox induction (n=4 biologically independent samples, data are presented as mean ± SEM. Dunnett’s test after one-way ANOVA was performed.) and by flow cytometry (b-d). The representative gating strategy for flow cytometry is shown in (b). Cells were treated with Dox+/Dox- media for 7 days before the flow-cytometry assay (c). To find the minimal time of Dox induction with stable knockdown, sgIGF1R and sgNTC cells were induced for either 4 days or 7 days and the IGF1R abundance was examined. Isotype, isotype control. αIGFIR, anti-IGF1R. e. Heatmap showing the Pearson correlations of normalized sgRNA read counts between the plasmid library and bulk screen replicates. f. Boxplot showing the reproducible trends of deletion upon CRISPRi between the present study and a prior report25 (n = 10, 57, 45, 49, 68 genes in each bin from left to right). g. Barplot showing the knockdown of genes with higher essentiality resulted in stronger cell growth arrest. h-i. Dotplots showing the expression fold changes of target genes upon CRISPRi induction compared to NTC in the single-cell PerturbSci-Kinetics dataset. Each dot represents a sgRNA. Fold change < 0.6 was used for sgRNA filtering, and genes with 3, 2, 1, 0 on-target sgRNA(s) were visualized in b-e, respectively. FC, fold change. j. Histogram showing the distribution of the fraction of the most abundant sgRNA in singlets (78%) and doublet cells (22%). k-l. The accuracy of sgRNA targeting efficiency in PerturbSci-Kinetics was further confirmed by RT-qPCR. Individual HEK293-idCas9 clones expressing 5 sgRNAs with high efficiency and 1 off-target sgRNA were established. RT-qPCR was conducted after 3-day Dox induction (n=3 biologically independent samples). Data are presented as mean ± SEM, and two-sided Student’s t-test were performed (k). Mean expressions of target genes in NTC and corresponding cells in the original PerturbSci-Kinetics dataset were exhibited (l). Boxes in boxplots indicate the median and IQR with whiskers indicating 1.5× IQR.
Extended Data Fig. 5.
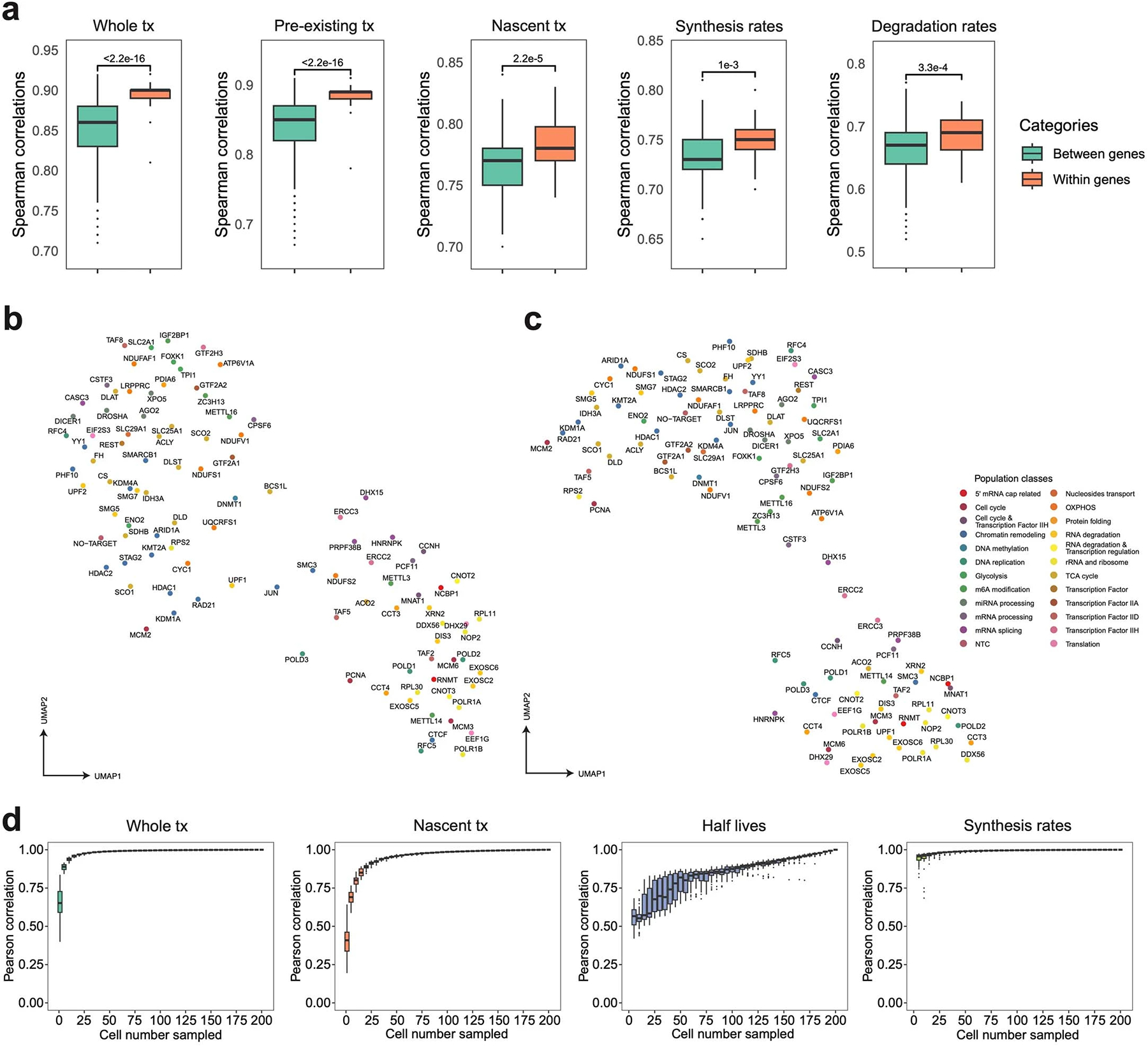
a. Boxplots showing the pairwise correlation coefficients of sgRNAs targeting the same/different genes, computed using aggregated whole transcriptomes, pre-existing transcriptomes, nascent transcriptomes, gene-specific synthesis rates and degradation rates. Considering the data sparsity and different cell numbers across perturbations, 150 cells per sgRNA were assembled into one pseudobulk for downstream analysis. Spearman correlation coefficients were calculated using DEGs between perturbations and NTC in the pooled screen. Two-sided Welch’s t-tests were performed. b-c. UMAP of pseudobulk perturbations by inferred synthesis rates (b) and degradation rates (c). DEGs between all perturbations-NTC pairs were combined, and their synthesis and degradation rates were calculated for each perturbation. Only genes with calculable synthesis or degradation rate in at least 75% of pseudobulk perturbations were used for dimension reduction. The top 12 and 15 principal components from the synthesis and degradation rates matrix were used for UMAP visualization, respectively. These UMAPs showed meaningful patterns. For example, RNA exosome genes (e.g., EXOSC2, EXOSC5, EXOSC6), nonsense-mediated mRNA decay pathway members (e.g., SMG5, SMG7), ribosomal biogenesis genes (e.g., NOP2, RPL30, RPL11, POLR1A, POLR1B), miRNA biogenesis pathway members (e.g., DICER1, DROSHA, XPO5, and AGO2) were in relative proximity in both UMAPs. Chromatin remodelers (e.g., HDAC1, HDAC2, STAG2, RAD21, KMT2A, KDM1A, ARID1A) were closely clustered in synthesis rates-derived UMAP, while m6A regulators (e.g., METTL3, METTL16, ZC3H13, IGF2BP1) and polyadenylation factors (e.g., CPSF6, CSTF3) were closer to each other in degradation rates-derived UMAP. d. Boxplots showing effects of cell number on the estimation of the pseudobulk whole/nascent transcriptome expression, gene-specific half-life, and synthesis rate. We conducted 50 random samplings for each cell number on sgDROSHA cells, then we aggregated profiles of sampled cells and retrieved pseudobulk expression levels and estimated RNA dynamics rates. We calculated the Pearson correlation coefficients between each downsampled pseudobulk group and unsampled pseudobulk sample. Boxes in boxplots indicate the median and IQR with whiskers indicating 1.5× IQR.
Extended Data Fig. 6.
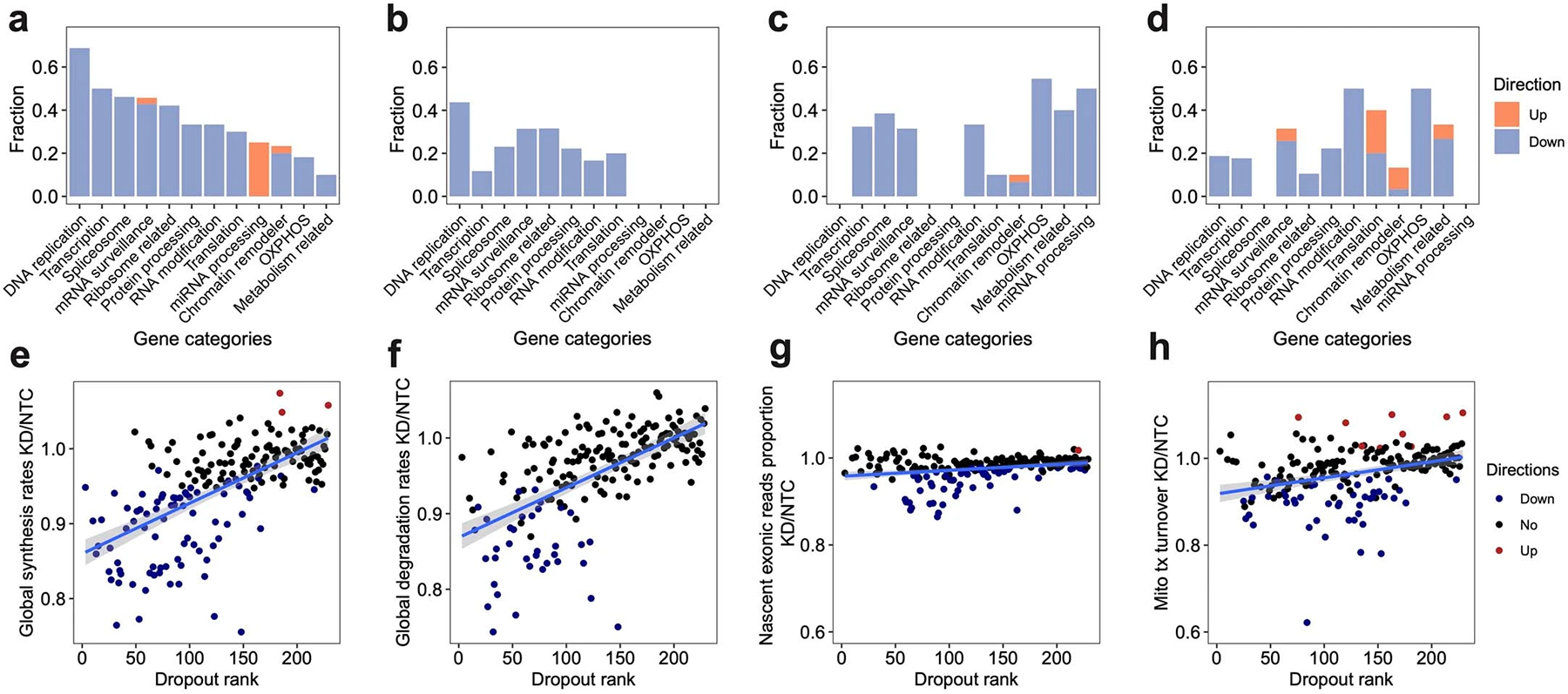
a-d. For each gene category, we calculated the fraction of genetic perturbations associated with significant changes in global synthesis rates (a), global degradation rates (b), proportions of exonic reads in the nascent transcriptome (c), and proportions of mitochondrial nascent reads (d). Overall global transcription could be affected by more genes than degradation. Perturbation on essential genes, such as DNA replication genes, could affect both global synthesis and degradation. Perturbations on chromatin remodelers only specifically impaired the global synthesis rates but not the degradation rates, supporting the established theory that gene expression is regulated by chromatin folding. In addition to the enrichment of genes in transcription, spliceosome and mRNA surveillance, perturbation on OXPHOS genes and metabolism-related genes also affected the RNA processing, consistent with the fact that 5’ capping, 3’ polyadenylation, and RNA splicing are highly energy-dependent processes. That knockdown of OXPHOS genes and metabolism-related genes could reduce the mitochondrial transcriptome dynamics and also supported the complex feedback mechanisms between energy metabolism and mitochondrial transcription63. e-h. Scatterplots showing the relationships between dropout effects and global synthesis rates (e), global degradation rates (f), proportions of exonic reads in the nascent transcriptome (g), and mitochondrial RNA turnover (h). A linear regression line was fitted and ±95% confidence intervals are visualized for each metric. Dropout rank, the ascending rank of gene-level sgRNA counts log2FC from the bulk screen. Directions were assigned as shown in Figure 2e–h. Both global synthesis and degradation rates showed strong negative correlations with dropout, indicating knocking out essential genes generally resulted in impaired global RNA synthesis and degradation. In contrast, proportions of exonic reads in the nascent transcriptome were much more stable across perturbations, and were only specifically affected by genes functioning in RNA processing. Proportions of mitochondrial nascent reads were also prone to be affected by genetic perturbation, but directions of changes depend more on the functions of perturbed genes than the essentiality of genes.
Extended Data Fig. 7.
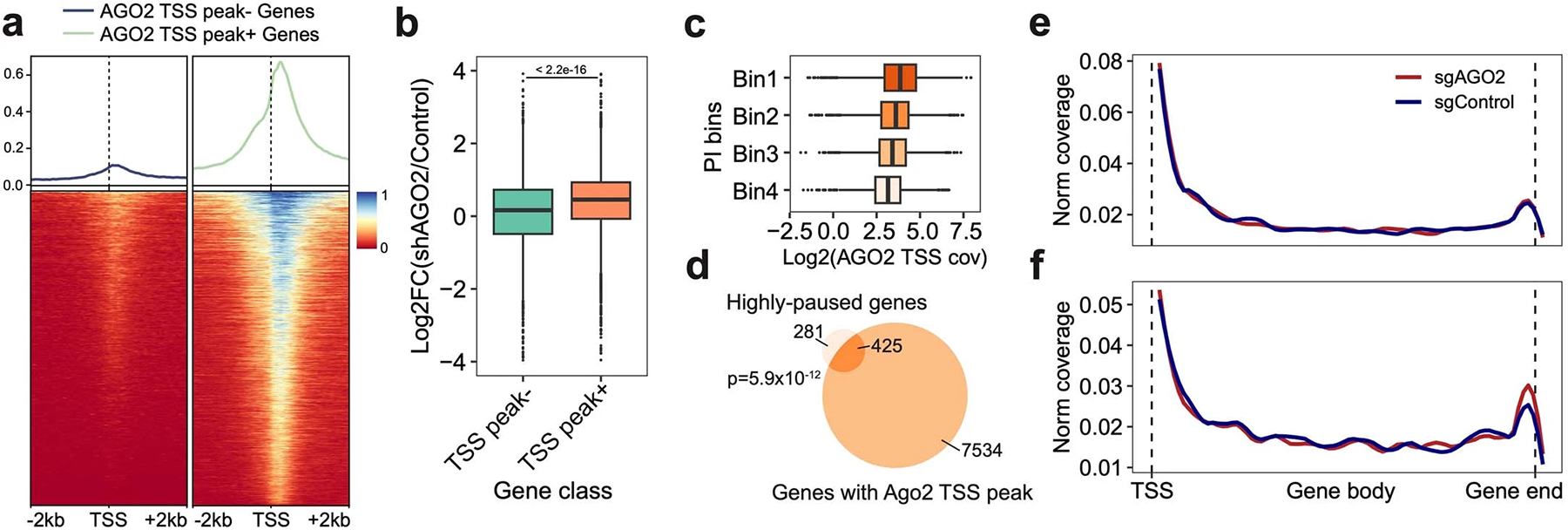
a. The density plot (top) and heatmap (bottom) show the density of AGO2 ChIP-seq reads around TSS of genes with or without enriched AGO2 TSS binding peaks. b. Boxplot showing the log2FC of gene expression between AGO2-silenced and control groups of genes with (n = 7315 genes) or without AGO2 TSS binding peaks (n = 3615 genes). Two-sided Wilcoxon rank sum test was performed. c. Boxplot displaying the positive correlation between PI of genes and normalized AGO2 ChIP-seq coverage within corresponding TSS regions. c-d. Genes were separated into 4 bins based on the average ranks of PI in two replicates (Methods). The Venn diagram highlights the significant association between AGO2 TSS binding and the strong pausing status of genes. One-sided Fisher’s Exact Test was conducted with the alternative hypothesis that the true odds ratio is greater than 1. Highly-paused genes, genes with top 10% of average PI ranks. e-f. Highly-paused genes were split into two groups, 1) significantly-upregulated genes upon AGO2 knockdown or 2) genes without significant expression changes. We then calculated the nascent RNA coverage of these two groups of genes in sgNTC and sgAGO2 cells. Notably, only genes in group 1 displayed increased 3’ end enrichment upon AGO2 knockdown (f). Boxes in boxplots indicate the median and IQR with whiskers indicating 1.5× IQR.
Extended Data Fig. 8.
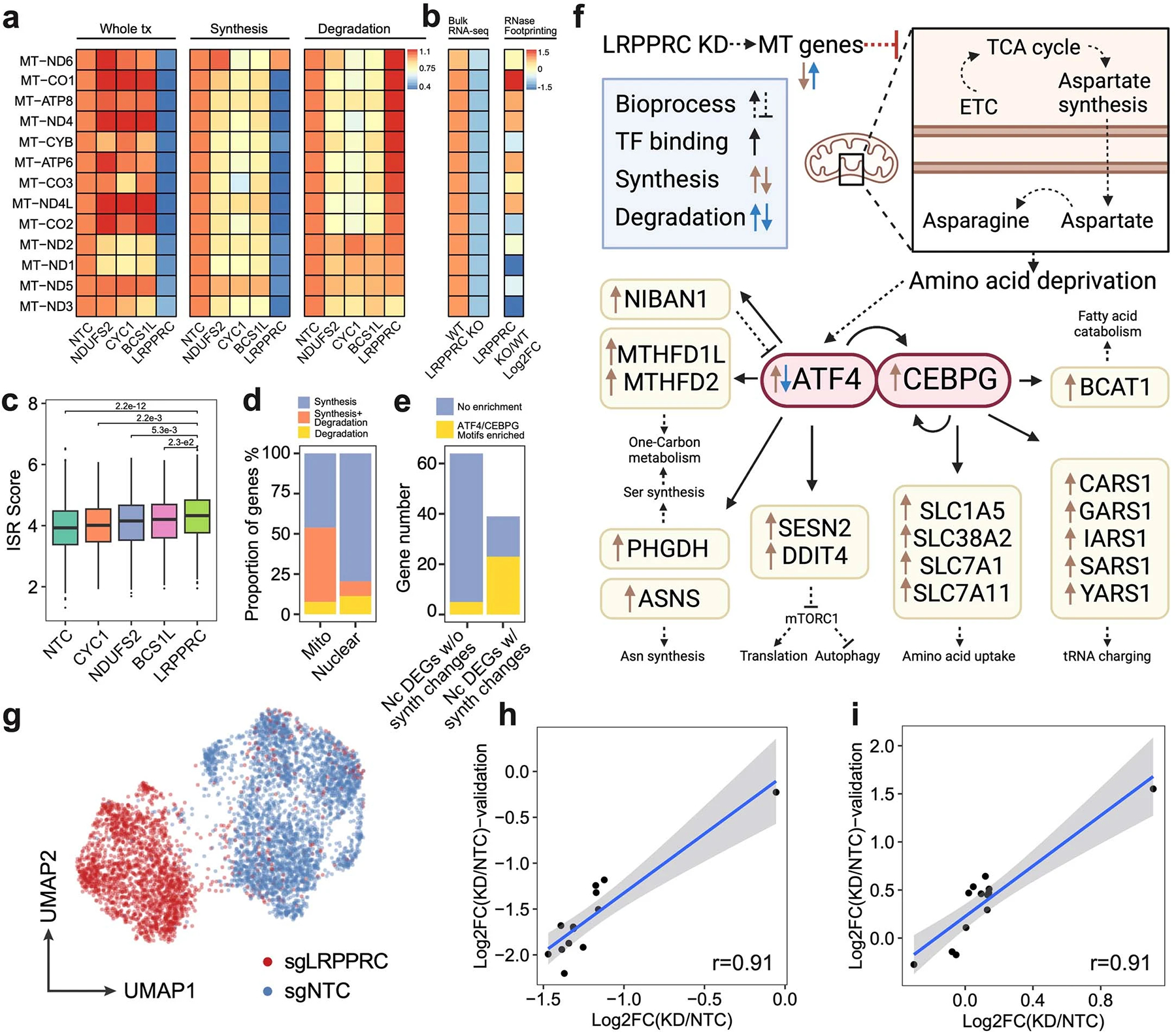
a. Heatmap showing the relative FC of gene expression, synthesis and degradation rates of mitochondrial protein-coding genes upon NDUFS2, CYC1, BCS1L and LRPPRC knockdown compared to NTC cells. b. The heatmap (left) showed mean z-scored mitochondrial gene expression changes between wild-type and LRPPRC-knockout mice heart tissue samples reported by Siira, S.J., et al.33. The DEG statistical examination was conducted by the original study. The heatmap (right) showed the FC of the mRNA secondary structure increase upon LRPPRC knockdown observed in the same prior report33, which positively correlated with the accelerated degradation of mitochondrial genes detected in our study (coefficient of Pearson correlation = 0.708, p-value = 6.8e-3). c. Boxplot showing the distribution of integrated stress response scores of single cells (n = 2758, 478, 768, 631, 504 cells in each group from left to right). Dunnett’s test after one-way ANOVA was performed. ISR, integrated stress response. ISR score, the average normalized expression of genes within the ISR transcription program identified by Genome-wide Perturb-seq8. d. Barplot showing the fraction of genes regulated by synthesis, degradation or both in mitochondrial/nuclear-encoded DEGs. e. Barplot showing the enrichment of ATF4/CEBPG motifs at promoter regions of DEGs with/without significant synthesis changes. We identified two transcription factors (ATF4 and CEBPG) that were significantly upregulated upon LRPPRC knockdown, and motifs of their protein product were significantly over-represented in TSS regions of the synthesis-regulated nuclear-encoded DEGs. Nc DEGs w/o synth changes, Nuclear-encoded DEGs without synthesis changes. Nc DEGs w/ synth changes, Nuclear-encoded DEGs with synthesis changes. f. The transcriptional regulatory network in LRPPRC perturbation inferred from our analysis. It was consistent with the prior study64 that ATF4 was regulated at both transcriptional and post-transcriptional levels. g. Single-cell UMAP of HEK293-idCas9-sgNTC/sgLRPPRC cells in the validation dataset. h-i. Correlations of synthesis and degradation rate changes of mitochondrial mRNA upon LRPPRC knockdown between the original screen and the validation dataset. A linear regression line was fitted and ±95% confidence intervals are visualized for each metric. r, coefficient of Pearson correlation. Boxes in boxplots indicate the median and IQR with whiskers indicating 1.5× IQR.
Extended Data Fig. 9.
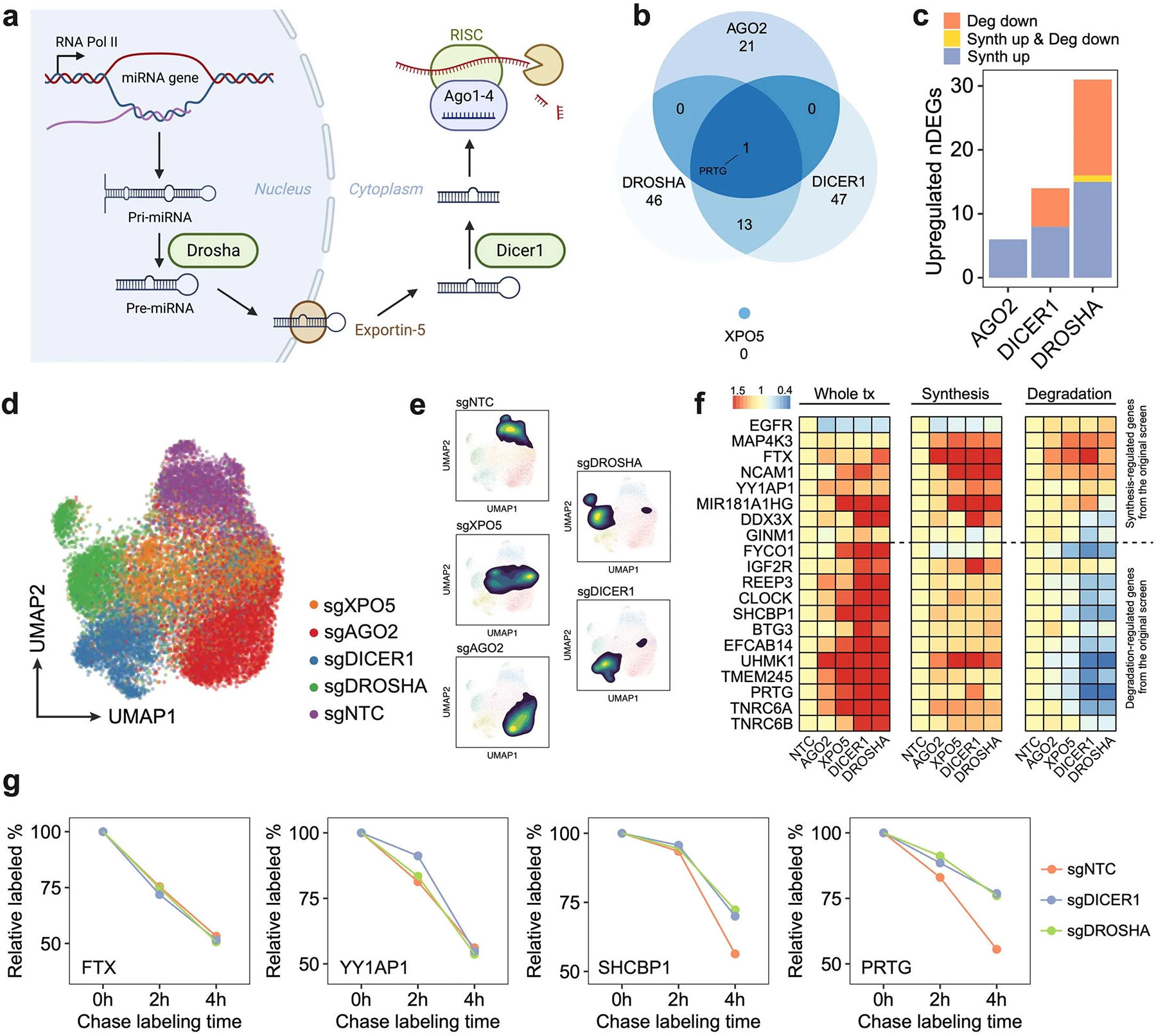
a. Illustration of the canonical miRNA biogenesis pathway. After the transcription of miRNA host genes, the primary miRNA (pri-miRNA) forms into a hairpin and is processed by DROSHA. Processed precursor miRNA (pre-miRNA) is transported to the cytoplasm by Exportin-5. The stem loop is cleaved by DICER1, and one strand of the double-stranded short RNA is selected and loaded into the RISC for targeting mRNA35. b. Venn diagram showing the overlap of upregulated DEGs across perturbations on four genes encoding main members of the miRNA pathway. The knockdown of DROSHA and DICER1 in this pathway resulted in significantly overlapped DEGs (p-value = 2.2e-16, one-sided Fisher’s exact test). In contrast, AGO2 knockdown resulted in more unique transcriptome features. XPO5 knockdown showed no upregulated DEGs, consistent with a previous report in which XPO5 silencing minimally perturbed the miRNA biogenesis36. c. Bar plot showing the fraction of upregulated DEGs driven by synthesis/degradation changes upon DROSHA, DICER1, and AGO2 perturbations. While DROSHA and DICER1 knockdown resulted in increased synthesis and reduced degradation, AGO2 knockdown only affected gene expression transcriptionally, consistent with our finding that AGO2 knockdown resulted in a global increase of synthesis rates (Fig 2e), and further supported its roles in nuclear transcription regulation65–67. As DROSHA is upstream of DICER1 in the pathway, we observed stronger effects of DROSHA knockdown than DICER1 knockdown, which was supported by the previous study36. d-e. UMAP of sgNTC cells and single cells with individual miRNA biogenesis pathway genes knockdown. f. Reproducible steady-state expression, synthesis rate, and degradation rate changes of synthesis/degradation-regulated genes in the validation dataset. g. Example genes showing unchanged (transcription-regulated genes: FTX, YY1AP1) and enhanced (degradation-regulated genes: SHCBP1, PRTG)
Extended Data Fig. 10.
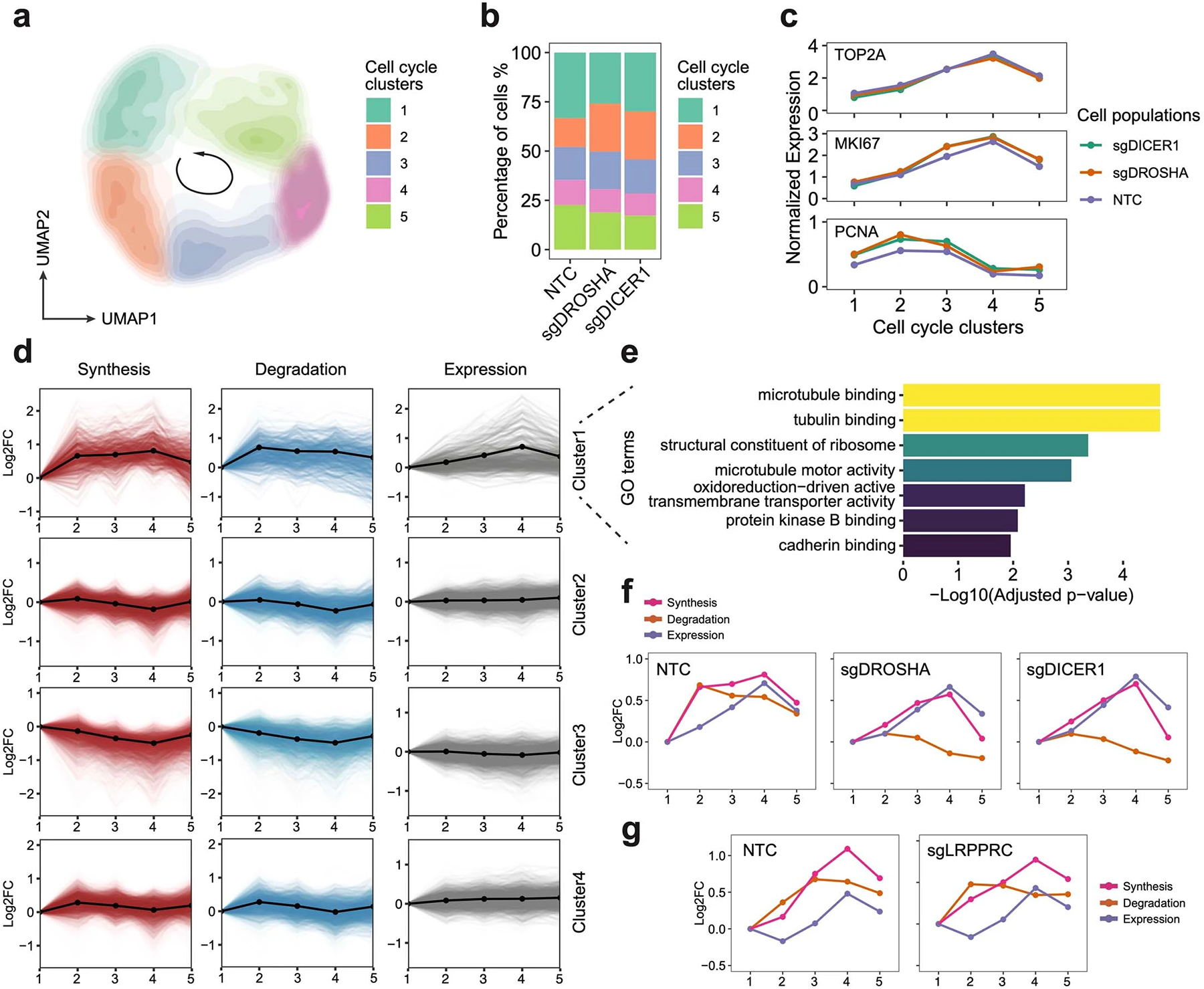
a. UMAP embedding of cells with miRNA pathway genes knockdowns and NTC cells reflected the cell-cycle progression. b. Stacked barplot showing the cell cycle distribution of cells from each perturbation. c. The expression changes of cell cycle marker genes in cell cycle clusters. d. The cell cycle time-course synthesis rates, degradation rates, and steady-state expression changes of 4 gene clusters. Solid lines with dots, the mean values and the average trend of all genes within the cluster. e. The top enriched GO terms of genes in the cluster 1 identified in GO enrichment analysis. f. Averaged trends of cell cycle time-course synthesis rates, degradation rates, and steady-state expression changes of cluster 1 genes in HEK293-idCas9-sgNTC, sgDROSHA, sgDICER1 cells. g. Averaged trends of cell cycle time-course synthesis rates, degradation rates, and steady-state expression changes of genes in cluster 1 in HEK293-idCas9-sgNTC and sgLRPPRC cells. Considering potential strong batch effects from distinct genetic perturbation, cell cycle clustering analysis in (g) was performed independently of (a), and cell cycle clusters in (g) were not fully synchronized with clusters in (f).
Supplementary Material
Supplementary Note 1: Detailed experiment protocols for PerturbSci-Kinetics, including all materials and equipment needed, step-by-step descriptions, and representative gel images.
Supplementary Note 2: The overall primer design of PerturbSci-Kinetics.
Supplementary Note 3: The overall costs for PerturbSci-Kinetics library preparation. Reagents used in each step were included, and the costs were calculated based on the scale of the real experiment.
Supplementary Table 1: Primer sequences used in PerturbSci-Kinetics. Sequences of the oligo pool library, bulk screen sequencing primer, shortdT RT primers, sgRNA capture primers, ligation primers, sgRNA inner i7 primers, and P5/P7 primers were included. The columns indicate the positions on the 96-well plate (Well positions), an identifier of the sequence (Names), the full primer sequence (Sequences), and the barcode sequence (Barcodes).
Supplementary Table 2: Genes and sgRNAs included in the study. Each gene (“gene_symbol”) has 3 sgRNAs, and they were named in the format “Gene_number” (“names”). sgRNA sequences were included in “sgRNA_seq”. The “gene_class” is the functional category of each gene.
Supplementary Table 3: Raw sgRNA counts of the bulk screen samples collected at different time points. Read counts of each sgRNA (“sgRNA_name”) from 4 replicates at day 0 and day 7 were included.
Supplementary Table 4: Relative sgRNA abundance fold changes between day 7 and day 0. The “Day7_vs_Day0_repX” is the fold changes of relative sgRNA abundance at the gene level.
Supplementary Table 5: Information about perturbations that showed significant global synthesis rate changes. Kolmogorov-Smirnov tests and Benjamini-Hochberg multiple hypothesis correction were performed. The “adj.p” is the false discovery rate adjusted for multiple comparisons. The “direction” is the direction of the changes on the global synthesis rates distributions comparing perturbed cells to the NTC cells, and the “KD_median/NTC_median” is the quantitative measurement of the changes. The “gene_class” is the functional category of target genes (“Perturbations”).
Supplementary Table 6: Information about perturbations that showed significant global degradation rate changes. Kolmogorov-Smirnov tests and Benjamini-Hochberg multiple hypothesis correction were performed. The “adj.p” is the false discovery rate adjusted for multiple comparisons. The “direction” is the direction of the changes on the global degradation rates distributions comparing perturbed cells to the NTC cells, and the “KD_median/NTC_median” is the quantitative measurement of the changes. The “gene_class” is the functional category of target genes (“Perturbations”).
Supplementary Table 7: Information about perturbations that showed significant nascent exonic reads ratio changes. Kolmogorov-Smirnov tests and Benjamini-Hochberg multiple hypothesis correction were performed. The “adj.p” is the false discovery rate adjusted for multiple comparisons. The “direction” is the direction of the changes on the nascent exonic reads ratio distributions comparing perturbed cells to the NTC cells, and the “KD_median/NTC_median” is the quantitative measurement of the changes. The “gene_class” is the functional category of target genes (“Perturbations”).
Supplementary Table 8: Information about perturbations that showed significant mitochondrial RNA turnover changes. Kolmogorov-Smirnov tests and Benjamini-Hochberg multiple hypothesis correction were performed. The “adj.p” is the false discovery rate adjusted for multiple comparisons. The “direction” is the direction of the changes in the distributions of mitochondrial nascent/total reads ratio comparing perturbed cells to the NTC cells, and the “KD_median/NTC_median” is the quantitative measurement of the changes. The “gene_class” is the functional category of target genes (“Perturbations”).
Supplementary Table 9: Steady-state expression and synthesis/degradation dynamics of mitochondrial genes upon LRPPRC, NDUFS2, CYC1, BCS1L perturbations. The “synth_rate”, “synth_FC”, “synth_pval”, “synth_direction” are the synthesis rate of the gene in the perturbed cells, the fold change of the synthesis rate of the gene in the perturbed cells compared to the NTC cells, the significance of the synthesis rate change, and the direction of the synthesis rate changes. DEG statistical examinations were performed using Monocle2, and the significance of rate changes was examined through randomization tests with empirical two-sided p-values. The “deg_rate”, “deg_FC”, “deg_pval”, “deg_direction” are the degradation rate of the gene in the perturbed cells, the fold change of the degradation rate of the gene in the perturbed cells compared to the NTC cells, the significance of the degradation rate change, and the direction of the degradation rate changes. The “DEG_qval” and “DEG_fold.change” are the multiple comparison-corrected FDR and the fold change of the steady-state gene expression change in perturbed cells compared to the NTC cells.
Supplementary Table 10: Filtered differentially expressed genes between perturbations with cell number >= 50 and NTC. For each gene (“Gene_symbol”), the “perturbation” is the target gene in perturbed cells. The “DEGs_direction” is the direction of gene expression changes comparing perturbed cells to the NTC cells, and the “DEGs_FC” is the fold change of the gene expression changes comparing perturbed cells to the NTC cells. The “max.CPM.between.KD.NTC” and “min.CPM.between.KD.NTC” are the pseudobulk expression levels of the gene that showed higher and lower expression in perturbed cells or the NTC cells. The expression level was quantified by counts per million. The “qval” is the false discovery rate (one-sided likelihood ratio test with adjustment for multiple comparisons).
Supplementary Table 11: Differentially expressed genes with significant synthesis and/or degradation changes. DEG statistical examinations were performed using Monocle2, and the significance of rate changes was examined through randomization tests with empirical two-sided p-values. The “perturbations” is the target gene of the perturbed cells, and the “Gene_symbols” is the symbols of DEGs with significant synthesis and/or degradation rate changes in corresponding perturbations. The type of significant rate change of each gene is included in the “Regulation_type”. The “Synth_deg_FC”, the “Synth_deg_direction”, and the “Synth_deg_pval” reflect the fold change, the direction of the change, and the randomization test p-value of the rate indicated in the “Regulation_type”. “DEGs_FC”, “DEGs_direction”, and “max.expr.between.KD.NTC” are the fold changes of gene expression, the direction of the change, and the maximum pseudobulk CPM between the corresponding perturbation and the NTC cells.
Supplementary Table 12: Steady-state expression and synthesis/degradation dynamics of merged DEGs upon DROSHA and DICER1 perturbations. DEG statistical examinations were performed using Monocle2, and the significance of rate changes was examined through randomization tests with empirical two-sided p-values. The “synth_rate”, “synth_FC”, “synth_pval”, “synth_direction” are the synthesis rate of the gene in the perturbed cells, the fold change of the synthesis rate of the gene in the perturbed cells compared to the NTC cells, the significance of the synthesis rate change, and the direction of the synthesis rate changes. The “deg_rate”, “deg_FC”, “deg_pval”, “deg_direction” are the degradation rate of the gene in the perturbed cells, the fold change of the degradation rate of the gene in the perturbed cells compared to the NTC cells, the significance of the degradation rate change, and the direction of the degradation rate changes. The “DEG_fold.change” and “DEG_qval” are the fold change of the steady-state gene expression change in perturbed cells compared to the NTC cells and the multiple comparison-corrected FDR.
Acknowledgments
We would like to express our gratitude to all members of the Cao lab for their helpful discussions and feedback. We thank Dr. Rahul Satija at New York Genome Center for his insightful feedback related to this work. We thank the Tissue Culture facility of the University of California, Berkeley for providing the 3T3L1 cell line, and Zhi Zheng at Memorial Sloan Kettering Cancer Center for providing the HEK293 cell line, and Shuyuan Cheng at Memorial Sloan Kettering Cancer Center for assisting with the supply of specific reagents. We thank members of the Rockefeller University Flow Cytometry Resource Center for their extensive help with FACS sorting. We also thank members of the Information Technology and HPC team at Rockefeller University, especially J. Banfelder and B. Jayaraman for their great support. The graphic illustrations in this study were generated using BioRender.com. We acknowledge that the research leading to this publication was partly supported by The G. Harold and Leila Y. Mathers Charitable Foundation. Additionally, the work received funding from grants provided by the NIH (1DP2HG012522, 1R01AG076932, and RM1HG011014) and the Mathers Foundation, awarded to J.C.
Footnotes
Code Availability
The computation scripts for processing PerturbSci-Kinetics were included as supplementary files. Scripts and the user manual are available for open access in GitHub: https://github.com/JunyueCaoLab/PerturbSci_Kinetics61.
Competing interests statement
J.C., W.Z., and Z.X. are listed as inventors on a patent related to PerturbSci-Kinetics (U.S. provisional patent application 63/385,479). Other authors declare no competing interests.
Data Availability
The data generated by this study can be downloaded in raw and processed forms from the NCBI Gene Expression Omnibus (GEO)60 (GSE218566). The AGO2 eCLIP data was obtained from the GEO database (GSE115146), and raw data from samples SRR7240709 and SRR7240710 were downloaded. Processed gene counts tables of RNA-seq on shControl/shAGO2 samples were downloaded from the ENCODE portal (ENCSR495YSS, ENCSR898NWE). The AGO2 ChIP-seq bam and narrow peak files were downloaded from the ENCODE portal (ENCSR151NQL). The GRO-seq data was obtained from the GEO database (GSE97072), and raw data from samples SRR5379790 and SRR5379791 were downloaded. The reference genome hg38 and corresponding genomic annotation gtf file were downloaded from the GENCODE database (Release 38, GRCh38.p13).
References:
- 1.Jaitin DA et al. Dissecting Immune Circuits by Linking CRISPR-Pooled Screens with Single-Cell RNA-Seq. Cell 167, 1883–1896.e15 (2016). [DOI] [PubMed] [Google Scholar]
- 2.Adamson B et al. A Multiplexed Single-Cell CRISPR Screening Platform Enables Systematic Dissection of the Unfolded Protein Response. Cell 167, 1867–1882.e21 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dixit A et al. Perturb-Seq: Dissecting Molecular Circuits with Scalable Single-Cell RNA Profiling of Pooled Genetic Screens. Cell 167, 1853–1866.e17 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xie S, Duan J, Li B, Zhou P & Hon GC Multiplexed Engineering and Analysis of Combinatorial Enhancer Activity in Single Cells. Mol. Cell 66, 285–299.e5 (2017). [DOI] [PubMed] [Google Scholar]
- 5.Datlinger P et al. Pooled CRISPR screening with single-cell transcriptome readout. Nat. Methods 14, 297–301 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hill AJ et al. On the design of CRISPR-based single-cell molecular screens. Nat. Methods 15, 271–274 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Replogle JM et al. Combinatorial single-cell CRISPR screens by direct guide RNA capture and targeted sequencing. Nat. Biotechnol 38, 954–961 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Replogle JM et al. Mapping information-rich genotype-phenotype landscapes with genome-scale Perturb-seq. Cell 185, 2559–2575.e28 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yeo NC et al. An enhanced CRISPR repressor for targeted mammalian gene regulation. Nat. Methods 15, 611–616 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sziraki A et al. A global view of aging and Alzheimer’s pathogenesis-associated cell population dynamics and molecular signatures in the human and mouse brains. Preprint at 10.1101/2022.09.28.509825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cleary MD, Meiering CD, Jan E, Guymon R & Boothroyd JC Biosynthetic labeling of RNA with uracil phosphoribosyltransferase allows cell-specific microarray analysis of mRNA synthesis and decay. Nat. Biotechnol 23, 232–237 (2005). [DOI] [PubMed] [Google Scholar]
- 12.Dolken L et al. High-resolution gene expression profiling for simultaneous kinetic parameter analysis of RNA synthesis and decay. RNA 14, 1959–1972 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miller C et al. Dynamic transcriptome analysis measures rates of mRNA synthesis and decay in yeast. Mol. Syst. Biol 7, 458–458 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Duffy EE et al. Tracking Distinct RNA Populations Using Efficient and Reversible Covalent Chemistry. Mol. Cell 59, 858–866 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schwalb B et al. TT-seq maps the human transient transcriptome. Science 352, 1225–1228 (2016). [DOI] [PubMed] [Google Scholar]
- 16.Rabani M et al. Metabolic labeling of RNA uncovers principles of RNA production and degradation dynamics in mammalian cells. Nat. Biotechnol 29, 436–442 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miller MR, Robinson KJ, Cleary MD & Doe CQ TU-tagging: cell type–specific RNA isolation from intact complex tissues. Nat. Methods 6, 439–441 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Erhard F et al. scSLAM-seq reveals core features of transcription dynamics in single cells. Nature 571, 419–423 (2019). [DOI] [PubMed] [Google Scholar]
- 19.Hendriks G-J et al. NASC-seq monitors RNA synthesis in single cells. Nat. Commun 10, 3138 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cao J, Zhou W, Steemers F, Trapnell C & Shendure J Sci-fate characterizes the dynamics of gene expression in single cells. Nat. Biotechnol 38, 980–988 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Qiu Q et al. Massively parallel and time-resolved RNA sequencing in single cells with scNT-seq. Nat. Methods 17, 991–1001 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Battich N et al. Sequencing metabolically labeled transcripts in single cells reveals mRNA turnover strategies. Science 367, 1151–1156 (2020). [DOI] [PubMed] [Google Scholar]
- 23.Kawata K et al. Metabolic labeling of RNA using multiple ribonucleoside analogs enables the simultaneous evaluation of RNA synthesis and degradation rates. Genome Res. 30, 1481–1491 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schwanhäusser B et al. Global quantification of mammalian gene expression control. Nature 473, 337–342 (2011). [DOI] [PubMed] [Google Scholar]
- 25.Sanson KR et al. Optimized libraries for CRISPR-Cas9 genetic screens with multiple modalities. Nat. Commun 9, 5416 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Qiu X et al. Mapping transcriptomic vector fields of single cells. Cell 185, 690–711.e45 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stuart T et al. Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902.e21 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sun M et al. Global analysis of eukaryotic mRNA degradation reveals Xrn1-dependent buffering of transcript levels. Mol. Cell 52, 52–62 (2013). [DOI] [PubMed] [Google Scholar]
- 29.Iwakawa H-O & Tomari Y Life of RISC: Formation, action, and degradation of RNA-induced silencing complex. Mol. Cell 82, 30–43 (2022). [DOI] [PubMed] [Google Scholar]
- 30.Van Nostrand EL et al. A large-scale binding and functional map of human RNA-binding proteins. Nature 583, 711–719 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.ENCODE Project Consortium et al. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 583, 699–710 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Herzog VA et al. Thiol-linked alkylation of RNA to assess expression dynamics. Nat. Methods 14, 1198–1204 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Siira SJ et al. LRPPRC-mediated folding of the mitochondrial transcriptome. Nat. Commun 8, 1532 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Paulo E et al. Brown adipocyte ATF4 activation improves thermoregulation and systemic metabolism. Cell Rep. 36, 109742 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Treiber T, Treiber N & Meister G Regulation of microRNA biogenesis and its crosstalk with other cellular pathways. Nat. Rev. Mol. Cell Biol 20, 5–20 (2019). [DOI] [PubMed] [Google Scholar]
- 36.Kim Y-K, Kim B & Kim VN Re-evaluation of the roles of DROSHA, Export in 5, and DICER in microRNA biogenesis. Proc. Natl. Acad. Sci. U. S. A 113, E1881–9 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chipman LB & Pasquinelli AE miRNA Targeting: Growing beyond the Seed. Trends Genet. 35, 215–222 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Heinrichs A A slice of the action. Nat. Rev. Mol. Cell Biol 5, 677–677 (2004). [Google Scholar]
- 39.Butler A, Hoffman P, Smibert P, Papalexi E & Satija R Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol 36, 411–420 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Futschik ME & Carlisle B Noise-robust soft clustering of gene expression time-course data. J. Bioinform. Comput. Biol 3, 965–988 (2005). [DOI] [PubMed] [Google Scholar]
- 41.Thomas MP et al. Apoptosis Triggers Specific, Rapid, and Global mRNA Decay with 3’ Uridylated Intermediates Degraded by DIS3L2. Cell Rep. 11, 1079–1089 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang K et al. A novel class of microRNA-recognition elements that function only within open reading frames. Nat. Struct. Mol. Biol 25, 1019–1027 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pacini C et al. Integrated cross-study datasets of genetic dependencies in cancer. Nat. Commun 12, 1661 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ashburner M et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet 25, 25–29 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kanehisa M & Goto S KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Krueger F A wrapper around Cutadapt and FastQC to consistently apply adapter and quality trimming to FastQ files, with extra functionality for RRBS data. TrimGalore (accessed on 27 August 2019). [Google Scholar]
- 47.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Danecek P et al. Twelve years of SAMtools and BCFtools. Gigascience 10, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lindenbaum P JVarkit: java-based utilities for Bioinformatics. (2015) doi: 10.6084/m9.figshare.1425030.v1. [DOI] [Google Scholar]
- 50.Picard. https://broadinstitute.github.io/picard/.
- 51.Putri GH, Anders S, Pyl PT, Pimanda JE & Zanini F Analysing high-throughput sequencing data in Python with HTSeq 2.0. Bioinformatics 38, 2943–2945 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Smith T, Heger A & Sudbery I UMI-tools: modeling sequencing errors in Unique Molecular Identifiers to improve quantification accuracy. Genome Research vol. 27 491–499 Preprint at 10.1101/gr.209601.116 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Qiu X et al. Reversed graph embedding resolves complex single-cell trajectories. Nat. Methods 14, 979–982 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Robinson MD, McCarthy DJ & Smyth GK edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Quinlan AR & Hall IM BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ramírez F et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–5 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Martin M Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17, 10–12 (2011). [Google Scholar]
- 58.Langmead B & Salzberg SL Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.DeBerardine M BRGenomics. (Bioconductor, 2020). doi: 10.18129/B9.BIOC.BRGENOMICS. [DOI] [Google Scholar]
- 60.Xu Z et al. PerturbSci-Kinetics: Dissecting key regulators of transcriptome kinetics through scalable single-cell RNA profiling of pooled CRISPR screens. Gene Expression Omnibus. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE218566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xu Z et al. PerturbSci_Kinetics. Github. https://github.com/JunyueCaoLab/PerturbSci_Kinetics. [Google Scholar]
- 62.Miyoshi H, Blömer U, Takahashi M, Gage FH & Verma IM Development of a self-inactivating lentivirus vector. J. Virol 72, 8150–8157 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Scarpulla RC, Vega RB & Kelly DP Transcriptional integration of mitochondrial biogenesis. Trends Endocrinol. Metab 23, 459–466 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pakos-Zebrucka K et al. The integrated stress response. EMBO Rep. 17, 1374–1395 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Janowski BA et al. Involvement of AGO1 and AGO2 in mammalian transcriptional silencing. Nat. Struct. Mol. Biol 13, 787–792 (2006). [DOI] [PubMed] [Google Scholar]
- 66.Griffin KN et al. Widespread association of the Argonaute protein AGO2 with meiotic chromatin suggests a distinct nuclear function in mammalian male reproduction. Genome Res. 32, 1655–1668 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Moshkovich N et al. RNAi-independent role for Argonaute2 in CTCF/CP190 chromatin insulator function. Genes Dev. 25, 1686–1701 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Note 1: Detailed experiment protocols for PerturbSci-Kinetics, including all materials and equipment needed, step-by-step descriptions, and representative gel images.
Supplementary Note 2: The overall primer design of PerturbSci-Kinetics.
Supplementary Note 3: The overall costs for PerturbSci-Kinetics library preparation. Reagents used in each step were included, and the costs were calculated based on the scale of the real experiment.
Supplementary Table 1: Primer sequences used in PerturbSci-Kinetics. Sequences of the oligo pool library, bulk screen sequencing primer, shortdT RT primers, sgRNA capture primers, ligation primers, sgRNA inner i7 primers, and P5/P7 primers were included. The columns indicate the positions on the 96-well plate (Well positions), an identifier of the sequence (Names), the full primer sequence (Sequences), and the barcode sequence (Barcodes).
Supplementary Table 2: Genes and sgRNAs included in the study. Each gene (“gene_symbol”) has 3 sgRNAs, and they were named in the format “Gene_number” (“names”). sgRNA sequences were included in “sgRNA_seq”. The “gene_class” is the functional category of each gene.
Supplementary Table 3: Raw sgRNA counts of the bulk screen samples collected at different time points. Read counts of each sgRNA (“sgRNA_name”) from 4 replicates at day 0 and day 7 were included.
Supplementary Table 4: Relative sgRNA abundance fold changes between day 7 and day 0. The “Day7_vs_Day0_repX” is the fold changes of relative sgRNA abundance at the gene level.
Supplementary Table 5: Information about perturbations that showed significant global synthesis rate changes. Kolmogorov-Smirnov tests and Benjamini-Hochberg multiple hypothesis correction were performed. The “adj.p” is the false discovery rate adjusted for multiple comparisons. The “direction” is the direction of the changes on the global synthesis rates distributions comparing perturbed cells to the NTC cells, and the “KD_median/NTC_median” is the quantitative measurement of the changes. The “gene_class” is the functional category of target genes (“Perturbations”).
Supplementary Table 6: Information about perturbations that showed significant global degradation rate changes. Kolmogorov-Smirnov tests and Benjamini-Hochberg multiple hypothesis correction were performed. The “adj.p” is the false discovery rate adjusted for multiple comparisons. The “direction” is the direction of the changes on the global degradation rates distributions comparing perturbed cells to the NTC cells, and the “KD_median/NTC_median” is the quantitative measurement of the changes. The “gene_class” is the functional category of target genes (“Perturbations”).
Supplementary Table 7: Information about perturbations that showed significant nascent exonic reads ratio changes. Kolmogorov-Smirnov tests and Benjamini-Hochberg multiple hypothesis correction were performed. The “adj.p” is the false discovery rate adjusted for multiple comparisons. The “direction” is the direction of the changes on the nascent exonic reads ratio distributions comparing perturbed cells to the NTC cells, and the “KD_median/NTC_median” is the quantitative measurement of the changes. The “gene_class” is the functional category of target genes (“Perturbations”).
Supplementary Table 8: Information about perturbations that showed significant mitochondrial RNA turnover changes. Kolmogorov-Smirnov tests and Benjamini-Hochberg multiple hypothesis correction were performed. The “adj.p” is the false discovery rate adjusted for multiple comparisons. The “direction” is the direction of the changes in the distributions of mitochondrial nascent/total reads ratio comparing perturbed cells to the NTC cells, and the “KD_median/NTC_median” is the quantitative measurement of the changes. The “gene_class” is the functional category of target genes (“Perturbations”).
Supplementary Table 9: Steady-state expression and synthesis/degradation dynamics of mitochondrial genes upon LRPPRC, NDUFS2, CYC1, BCS1L perturbations. The “synth_rate”, “synth_FC”, “synth_pval”, “synth_direction” are the synthesis rate of the gene in the perturbed cells, the fold change of the synthesis rate of the gene in the perturbed cells compared to the NTC cells, the significance of the synthesis rate change, and the direction of the synthesis rate changes. DEG statistical examinations were performed using Monocle2, and the significance of rate changes was examined through randomization tests with empirical two-sided p-values. The “deg_rate”, “deg_FC”, “deg_pval”, “deg_direction” are the degradation rate of the gene in the perturbed cells, the fold change of the degradation rate of the gene in the perturbed cells compared to the NTC cells, the significance of the degradation rate change, and the direction of the degradation rate changes. The “DEG_qval” and “DEG_fold.change” are the multiple comparison-corrected FDR and the fold change of the steady-state gene expression change in perturbed cells compared to the NTC cells.
Supplementary Table 10: Filtered differentially expressed genes between perturbations with cell number >= 50 and NTC. For each gene (“Gene_symbol”), the “perturbation” is the target gene in perturbed cells. The “DEGs_direction” is the direction of gene expression changes comparing perturbed cells to the NTC cells, and the “DEGs_FC” is the fold change of the gene expression changes comparing perturbed cells to the NTC cells. The “max.CPM.between.KD.NTC” and “min.CPM.between.KD.NTC” are the pseudobulk expression levels of the gene that showed higher and lower expression in perturbed cells or the NTC cells. The expression level was quantified by counts per million. The “qval” is the false discovery rate (one-sided likelihood ratio test with adjustment for multiple comparisons).
Supplementary Table 11: Differentially expressed genes with significant synthesis and/or degradation changes. DEG statistical examinations were performed using Monocle2, and the significance of rate changes was examined through randomization tests with empirical two-sided p-values. The “perturbations” is the target gene of the perturbed cells, and the “Gene_symbols” is the symbols of DEGs with significant synthesis and/or degradation rate changes in corresponding perturbations. The type of significant rate change of each gene is included in the “Regulation_type”. The “Synth_deg_FC”, the “Synth_deg_direction”, and the “Synth_deg_pval” reflect the fold change, the direction of the change, and the randomization test p-value of the rate indicated in the “Regulation_type”. “DEGs_FC”, “DEGs_direction”, and “max.expr.between.KD.NTC” are the fold changes of gene expression, the direction of the change, and the maximum pseudobulk CPM between the corresponding perturbation and the NTC cells.
Supplementary Table 12: Steady-state expression and synthesis/degradation dynamics of merged DEGs upon DROSHA and DICER1 perturbations. DEG statistical examinations were performed using Monocle2, and the significance of rate changes was examined through randomization tests with empirical two-sided p-values. The “synth_rate”, “synth_FC”, “synth_pval”, “synth_direction” are the synthesis rate of the gene in the perturbed cells, the fold change of the synthesis rate of the gene in the perturbed cells compared to the NTC cells, the significance of the synthesis rate change, and the direction of the synthesis rate changes. The “deg_rate”, “deg_FC”, “deg_pval”, “deg_direction” are the degradation rate of the gene in the perturbed cells, the fold change of the degradation rate of the gene in the perturbed cells compared to the NTC cells, the significance of the degradation rate change, and the direction of the degradation rate changes. The “DEG_fold.change” and “DEG_qval” are the fold change of the steady-state gene expression change in perturbed cells compared to the NTC cells and the multiple comparison-corrected FDR.
Data Availability Statement
The data generated by this study can be downloaded in raw and processed forms from the NCBI Gene Expression Omnibus (GEO)60 (GSE218566). The AGO2 eCLIP data was obtained from the GEO database (GSE115146), and raw data from samples SRR7240709 and SRR7240710 were downloaded. Processed gene counts tables of RNA-seq on shControl/shAGO2 samples were downloaded from the ENCODE portal (ENCSR495YSS, ENCSR898NWE). The AGO2 ChIP-seq bam and narrow peak files were downloaded from the ENCODE portal (ENCSR151NQL). The GRO-seq data was obtained from the GEO database (GSE97072), and raw data from samples SRR5379790 and SRR5379791 were downloaded. The reference genome hg38 and corresponding genomic annotation gtf file were downloaded from the GENCODE database (Release 38, GRCh38.p13).