

Abstract
The recurrence of tumors after years of disease-free survival has spurred interest in the concept that cancers may have a stem cell basis. Current speculation holds that as few as 0.1% of the tumor mass may be chemoresistant and radioresistant, harboring stem-like properties that drive tumor survival, development, and metastasis. There are intense investigations to characterize cancer stem cells on the basis of self-renewal and multi-lineage differentiation. Thus far, no successful targeted therapies have been developed and reached the clinic, but as these cells are isolated and characterized, insights may be unraveled. In this review, we discuss the controversy over the origins of the cancer stem cell hypothesis and the unforeseen factors that may facilitate breast cancer stem cell survival and metastasis. We discuss the role of tumor microenvironment, including carcinoma-associated fibroblasts, epigenetic factors, and the Th1/Th2 balance, in supporting breast cancer stem cells. In addition, we have incorporated ideas on the epithelial-to-mesenchymal transition in metastatic dissemination of epithelial malignancies. This area is relevant since breast cancer stem cells have been suggested to revert to a mesenchymal phenotype during the progression of cancer. Finally we discuss prospects on developing targeted therapy including novel treatment modalities such as oncolytic viral therapy, differentiation therapy, and nanotechnology.
Keywords: cancer stem cells, EMT, oncolytic viral therapy, microenvironment, side population, crosstalk
Genesis of breast cancer stem cells (CSCs): Ambiguity in interpretations
An increase in the interest in the origin of cancer stem cells (CSCs) occurred in 1997 when malignant stem cells were identified in acute myelogenous leukemia.1 These seminal experiments paved the way for more in-depth studies on stem cells in leukemia as well as other malignancies, such as breast, lung, brain, prostate, colon.2 The CSC hypothesis holds that a subset of malignant cells can give rise to the majority of other cells of a tumor and permit cancer maintenance and progression.2 Recent attention in the arena of breast cancer therapy has focused on targeting this subpopulation of cells with the hope of eliminating unnecessary cycles of relapse and remission. The resilience of these cells to cytotoxic therapy is presumably due to their low mitotic rates, compared to mitotic rates of more differentiated progenitors.3
Traditional theories about the origins of breast cancer held that each cancer cell has equal tumorigenic potential. This stochastic model was based on cancer initiation via accumulation of genetic insults.4 However, in light of new evidence on the heterogeneity of tumors and the emergence of therapeutic resistance, a new model termed the hierarchical model has taken the foreground.4,5 This model holds that any given tumor may contain genetically and morphologically diverse populations of cells, including primitive stem cells, transient amplifying cells, and terminally differentiated cells.5 Each cell type within the tumor is characterized by its own unique rate of division and ability to generate independent tumors in vivo.5 The hierarchical model holds that only a small proportion of cells can sustain cancers, and the heterogeneity of breast cancer may be attributable to clonal evolution.6,7 Moreover, the inefficiency of cancer metastasis at the cellular level lends credit to the idea that only a subset of cells within heterogeneous populations can invade traverse basement membranes and invade blood vessels.8
A unified definition of a CSC is not conserved among scientists, but generally includes at least two characteristics: 1) self-renewal to generate another malignant stem cell and, 2) ability to show lineage-specific differentiation. These properties of CSCs are supported by recent reports showing evidence for the presence of both primitive and differentiated cells in tumors.5 Breast CSCs comprise a phenotypically distinct subset of cells with clonogenic potential in vivo.7,9,10
One of the most important questions in the field of breast CSC biology involves the origin of these resilient cells. The clonal origins of breast CSCs have been investigated, but generalizations cannot be made due to the heterogeneity of breast tumors, including luminal A, luminal B, basal-like, and claudinlow, and other phenotypes.11 Gene profiling using microarray technology has revealed that at least four different subtypes of breast cancer exist, and each confers different prognoses.12 Another recent study described three subsets of mammary cells – basal stem/progenitor cells, luminal progenitors, and non-stem luminal cells.13 Regardless of the classification, the most prevalent breast cancer type appears to be basal-like, triple-negative cancers, which is characterized by lack of hormone and EGF receptors.14,15 Basal-like breast cancers are less differentiated and confer worse prognosis than express luminal subtypes. They are thought to arise from mammary stem cells, and thus, some scientists believe that breast CSCs may be more likely to arise from basal-like cancers versus luminal cancers. Basal-like cancers express basal cytokeratin-5, -6, -14, and -17 and demonstrate gene expression patterns similar to myoepithelial elements of the breast.16,17 In contrast, luminal cancers express cytokeratin-7, -8, and -19 and originate from milk-producing ductal epithelium of the breast lumen.16,18 The majority of ductal carcinoma in situ (DCIS) cases are luminal, but high-grade DCIS can take on basal-like characteristics.19
In a recent finding, luminal progenitors of the breast, rather than basal stem/progenitors, have been shown to most closely resemble basal breast tumors.13 Based on these studies, basal-like breast cancers may arise from a luminal progenitor population.13 In summary, the heterogeneity of breast cancers is evident from the idea that different classes of breast tumors may arise from different subtypes of epithelia in the normal breast. Although no clear conclusions can be made about the origins of luminal versus basal-like cancers, knowledge of the normal breast tissues that correspond most closely with each breast cancer subtype can be important for targeted therapy for different subsets of cancers. Another subtype, known as claudinlow breast cancer, has not been traditionally grouped into the basal/luminal scheme but may be characterized by expression of EMT markers.20,21 Whether different subtypes of breast cancer harbor different types of CSCs remains to be determined.
Numerous groups currently believe that breast CSCs are CD44+/CD24−/low, although the dynamic nature of CD marker expression has brought into question the reliability of these markers as definitive for breast CSCs. For example, the expression of the CD44+/CD24−/low phenotype throughout breast cancer progression may be influenced by epigenetic factors, genomic integrity, and factors involved in the epithelial-to-mesenchymal transition (see EMT section below).22,23 Furthermore, the characterization of breast CSCs may not be complete because new phenotypes such as ALDH1 positivity have been linked to self-renewability in breast cancer.24 ALDH1 positivity is becoming an increasingly important means of isolation of breast CSCs, along with identification of side populations expressing multi-drug resistance proteins (see Side Populations section).24–26
The origins of the breast CSC remain somewhat elusive, as scientists disagree as the evidence accrues. It is unclear if CSCs arise from de-differentiation of transient amplifying cells or progenitors vs. acquisition of oncogenic mutations by normal resident stem cells.5,27,28 In the former case, transient amplifying cells in local niches can undergo programs that allow them to gain self-renewal ability and limitless division capacity, like the stem cells from which they originally arose.27 In the latter case, spontaneous genetic damage or environmental insults can lead to transformation of stem cells, which continue to self-renew and give rise to new transformed cells.6,7,27
One of the most challenging aspects of the breast cancer biology involves the molecular mechanisms underlying genesis of breast CSCs, which point to a variety of signaling pathways common to both stem cells and cancer biology. Activation of the PI3K/Akt pathway through PTEN knockdown can lead to enrichment of mammary stem cells and progenitors.29 Crosstalk with the canonical Wnt/β-catenin pathway is partly responsible for these effects.29 Both pathways have been implicated in breast cancer progression and poor prognosis.29 Notch signaling is known to regulate cell survival, division, and fate determination, and it can lead to alterations in mammary stem cell self-renewal and increased proliferation.30 It has been shown to regulate cell fate in hematopoietic, neural, and other stem cells.10 Dysregulation of self-renewal ability may account for genesis of mammospheres harboring breast CSCs.30 Finally, the Hedgehog signaling pathway and the polycomb gene Bmi-1 are critical players in mammosphere formation and growth.15 This pathway is important because the CD44+/CD24−/low subset has been shown to employ aberrant Hedgehog signaling.15 Pharmacological manipulation of this pathway through agonists and antagonists disrupts the balance between self-renewal and differentiation. These diverse molecular pathways are likely necessary for survival and can be interconnected. Given that they have all been implicated to some degree in breast CSC generation, targeted therapy may pose formidable challenges.15,24,29
Crosstalk between breast cancer stem cells (CSCs) and their microenvironment
Normal physiological processes and environmental conditions dictate the behavior of cells, tissues, and organ systems.31 Carcinomas, for instance, are generally comprised of a mixture of various cellular elements, including transformed cells within a milieu of stromal cells, fibroblasts, and immune cells.32 Therefore, the in vitro study of breast CSCs in isolated systems, such as cell and tissue culture, has its limitations. It has been suggested that breast CSCs require a particular niche in which to grow for maintenance.8 Specific details of factors that may influence this niche will be discussed here.
An important determinant of CSC behavior in the microenvironment of breast cancer may be TGF-β, an immunosuppressive cytokine with diverse functions including wound healing, fibrosis, and cell cycle regulation.33 Local TGF-β in the microenvironment can enhance motility of breast cancer cell through epithelial-to-mesenchymal transition (see EMT section).33 The role of TGF-β could be difficult to understand due to its dual role in cancer progression. TGF-β is a known cell cycle inhibitor and has been shown to prevent de novo cancer formation in mice.33,34 However, TGF-β is a major player in promotion of cell motility and invasiveness.34 Furthermore, the estrogen receptor status of the patient can determine the role of TGF-β in cancer progression, as TGF-β appears to enhance growth of hormone-independent tumors, while hindering growth of hormone-positive cancers.34
Aside from the critical role of TGF-β, emerging evidence indicates that microenvironmental factors involved in breast CSC survival may include protective effects of mesenchymal stem cells (MSCs).35 Although data on immune protection of breast CSCs have not been thoroughly investigated thus far, ongoing studies in our laboratory suggest that MSCs play an important role in helping breast cancer cells evade the immune system (unpublished). MSCs can migrate to site of tumor and cause tumors to become unresponsive to hormonal treatment.35 Specifically, the chemokine receptor 4 (CXCR4)/stromal cell-derived factor-1 (SDF-1) axis may govern cancer cell homing to sites of distant metastasis.8 This chemokine-receptor pair may also regulate adhesion to resident cells in distant sites.8,36 Therapy directed at microenvironmental factors have been proposed but have has limited utility thus far. Details are discussed in Targeted Therapy section.
Once MSCs migrate to sites of tumor, they can integrate themselves in the stroma and facilitate breast cancer progression.36–38 MSCs have been shown to give rise to carcinoma-associated fibroblasts (CAFs), which comprise the tumor stroma and facilitate metastasis, angiogenesis, and other processes necessary for cancer progression.38 CAFs are characterized in part by the expression of SDF-1.38 A reciprocal interaction can exist between cancer cells and their surrounding stromal elements.32 For example, a plethora of cytokines and growth factors secreted by cancer cells can affect local fibroblasts, which themselves act on cancer cells to regulate proliferation and breast cancer progression.32 Furthermore, the response of breast cancer cells to chemotherapy treatment is regulated by CAFs and likely involves cellular adhesions to the underlying basement membrane.32
Other evidence implicating microenvironmental influences on breast cancer include epigenetic changes brought about by co-culture with CAFs.39 Downregulation of the tumor suppressor cystatin M via promoter hypermethylation has been reported to occur in normal breast epithelial in contact with CAFs.39 In summary, these findings are concurrent activation of the PI3K/Akt1 pathway and emphasize the importance of not only intercellular communications via soluble factors but also communications via physical contact between cells.39
The interaction of the immune system with breast cancer cells is complex but must be considered before targeted therapy for CSCs can be implemented. There is a dynamic interplay between breast tumor stroma and immunological factors to facilitate or hinder cancer growth. A T helper 1 (Th1) phenotype, for example, appears to exert more anti-cancer effects than a T helper 2 (Th2) phenotype, and Th1 and Th2 responses are counterbalanced by each other.40 Breast cancer micrometastasis to seminal lymph nodes can results in polarization towards a Th1 phenotype, perhaps in an effort to eradicate metastatic cells41. However, the challenging aspect of this arena is that the Th1 cytokine effects may be bimodal, as some scientists believe that the inflammatory nature of the Th1 response may allow for development of DNA damage that facilitates transformation.42 The effects of the Th2 response in cancer are slightly clearer, as most reports demonstrate a facilitative effect of Th2 processes in cancer. Release of IL-4 by a Th2 response, for example, may prevent chemosensitivity and allow for escape from immune detection.42,43 Members of the IL-10 family, which are secreted in the Th2 response, have been shown to inhibit cell proliferation in cancers.44 In addition to the Th1/Th2 balance, IL-24, a component of the IL-20 cytokine family, has recently gained attention based on its activity against tumors via induction of apoptosis.44,45
Microenvironmental effects can influence the induction of the epithelial-to-mesenchymal transition (EMT) in breast cancer cells. EMT is a phenomenon that shares similarities with the process of wound formation and healing, which are also influenced by local inflammation and microenvironmental factors (see EMT section).46 In basal-like breast cancers, epigenetic remodeling has shown to occur by loss of the cell cycle inhibitor p16/INK4A, and this process is susceptible to microenvironmental influences from EMT factors. In synergy with the oncogene ras, factors found in serum, such as TGF-β, favor EMT.46 High serum conditions have been associated with transition towards a mesenchymal-like phenotype with gene methylation and repression of E-cadherin.46 The process by which EMT induces de novo methylation may give insight into the origin of cancers.46
Along with these immunological and epigenetic influences from the microenvironment, crosstalk between differentiation programs and stem cell niches likely governs the aggressive behavior of breast cancer. The interaction between stem cells and their microenvironment applies to breast CSCs and may allow for further insights into the behavior of these cells in a physiological or pathological context.47
Epithelial-to-mesenchymal transition (EMT) and breast CSCs
The process of cancer metastasis is complex and appears to involve a variety of changes sometimes referred to as the invasion-metastasis cascade.48,49 It includes alterations in gene expression signatures and cellular morphology.48 It is well accepted that certain populations of cancer cells harbor primitive phenotypes, which confer worse prognosis. In addition to these primitive phenotypes, entire biological processes that occur in embryonic development may also govern metastasis and invasiveness in breast cancer. This phenomenon, termed epithelial-to-mesenchymal transition (EMT), occurs in morphogenesis, wound healing, tissue repair, and tissue remodeling.50 Specifically, EMT follows the loss of epithelial cell polarity and development of a fibroblast-like morphology.51,52 As with the breast CSC hypothesis, correlations have been established between drug resistance and EMT, a finding which is consistent with the idea that primitive cells are less susceptible to chemotherapy and radiotherapy.52 EMT may mediate both cancer initiation and cancer progression, and EMT markers have been associated with poor prognosis.53,54
The details of EMT in breast malignancies are unclear to date, but are presumed to involve a variety of changes at the genetic, molecular, and cellular levels. Paradoxically, EMT cannot be described as a simple transition from epithelial cells to mesenchymal cells.55 It is more accurately described as a switch from differentiated luminal epithelium to undifferentiated basal-like epithelium.55 Phenotypically, EMT is characterized by loss of epithelial markers like E-cadherin, which functions in calcium-dependent adhesions, and other proteins like γ-catenin, zonula occludens-1 (Zo-1), and a gain of vimentin, fibronectin, and other mesenchymal markers.52,53 N-cadherin, which is upregulated during EMT, facilitates motility and migration of cancer cells.53 The significance of mesenchymal and epithelial phenotypes is evident in that these two tissue types have different architectures and therefore distinct functions in cancer progression.54 For example, epithelial cells are organized in a tight manner with strong attachments to each other and to basement membranes, while mesenchymal cells are not as well anchored and harbor greater tendencies for motility.54 They are more likely to form mammospheres, consistent with the basal-like epithelial phenotype.55 Furthermore, the secretion of enzymes in mesenchymal tissues facilities the migration of cancer cells.54 Therefore, the differences in the properties of epithelial and mesenchymal elements at the cellular and tissue levels explain why the EMT process in cancers poses significant challenges to breast cancer therapy. It is important to understand that cells undergoing EMT are still mammary epithelia, and the mesenchymal designation refers to various states of stemness versus differentiation of epithelial cells.55 Phenotypic changes involved in EMT are summarized in Table 1.
Table 1.
Postulated phenotypic changes undergone by breast cancer cells during EMT
| Loss of epithelial markers | Gain of mesenchymal markers |
|---|---|
| E-cadherin (CDH1) | vimentin |
| γ-cadherin | fibronectin |
| zonula occludens-1 (ZO1) | N-cadherin (CDH2) |
| Occluding | α-smooth muscle actin (α-SMA) |
| cytokeratin 8, cytokeratin 18 |
References: Li et al., 2009, Moen et al., 2009
The triggers of EMT are diverse and have been suggested to demonstrate some overlap with factors involved in CSC generation. TGF-β is one of the most widely recognized inducers of EMT.50 It has been suggested that, during this process, epithelial de-differentiation occurs, rather than simple phenotypical alterations.52,56 The gene signature of EMT has recently been explored and involves a variety of transcription factors. Snail and TWIST family members are found at high levels in certain epithelial cancers and negatively regulate E-cadherin expression, preventing intercellular adhesion while promoting cellular motility and dissemination.51,53
Associations between the breast CSC hypothesis and EMT hypothesis of cancer were established only recently as similarities in these two ideas were noted. Breast cancer cells undergoing EMT demonstrated a CD44+/CD24−/low phenotype typical of breast CSCs.56 The process of EMT has been linked to characteristics of stemness in normal and transformed epithelia.54 A more obvious link between breast CSCs and EMT is demonstrated in studies where overexpression of Snail or TWIST resulted in the development of CD44+/CD24−/low breast CSCs. Treatment with TGF-β can also lead to the breast CSC phenotype and enhanced ability for mammosphere formation.55
Like other biological processes, EMT in breast cancer is affected by immunological mediators.56 For breast CSCs, CD8+ T lymphocytes can induce EMT.56 Thus, the process of EMT in breast cancer should not be considered in isolation because cancer progression occurs within a physiological context that necessarily involves normal tumor responses by the host.56 Since immunological factors are involved in these processes, one can surmise that MSCs in the bone marrow likely play a crucial role via their immunomodulatory properties.
Identification of side populations in breast cancer
Advances in the identification and isolation of breast CSCs was accelerated by the identification of side population (SP) cells, due to lack of dye retention and chemotherapy efflux.57 The method is based on cells incubated with Hoechst dye 33342 or rhodamine, after which the cells are analyzed by flow cytometry for dye exclusion and size. Small SP cells would not retain dye and are considered to be primitive cells. Samples of different tissue types revealed that SP cells only represent a very small percentage of the cells (0.05–5%).58–60 Analyses based on efflux of Hoechst dye have identified putative SPs in various tissues such as skeletal muscle, lung, liver and mammary glands.60–65 Rare SP cells have also been seen in both the estrogen receptor (ER)-positive cell line MCF-7 and the ER-negative cell line Cal-51.58,66,67
The transporters responsible for dye efflux are diverse but appear to involve adenosine triphosphate (ATP)-dependent processes. When SPs are treated with the calcium-channel blocker verapamil, the low-staining SPs are lost, thus leading to the assumption that an ATP-binding cassette (ABC) transporter is responsible for the efflux of the dye.57 More specifically, studies indicate that ABCG2 (also known as breast cancer resistance protein-1) is a major mediator of dye efflux in various stem cells and can be used as a molecular marker for the SP phenotype.68,69 Furthermore, the degree of efflux activity seems to correlate with the maturation state, such that cells exhibiting the highest efflux activity are the most primitive.57
The use of SP in the identification of breast CSCs is supported in studies showing dye efflux in highly chemo-resistant breast cancer cells.70 The ability to efflux substrates is particularly important for the protection of CSCs. ABCG2 seems to protect stem cells from toxins. This is evident in ABCG2 knockout mice that are more sensitive to compounds such as vinblastine, ivermectin, topotecan, and mitoxantrone.71–73 Additionally, ABCG2 was identified in drug-selected MCF-7 breast cancer cells and found to efflux multiple chemotherapeutic drugs and xenobiotics.67,74 The expression of the full-length ABCG2 cDNA in MCF-7 breast cancer cells provided resistance to mitoxantrone, doxorubicin, and daunorubicin, and reduced daunorubicin accumulation and retention.74 These findings imply that SP cells have the ability to efflux toxicologically harmful substances out of breast cancer cells through an ABCG2-mediated cytoprotective mechanism and seem to confer chemo-resistance. The use of ABCG2 as a breast CSC marker seems theoretically plausible but is currently in question as evident on this idea is not well supported. A study demonstrated that highly purified ABCG2+ cells were not any more tumorigenic than ABCG2− cells.75 This suggests that even an ABCG2− population contains primitive stem-like cancer cells and correlates with the earlier fact that ABCG2 is not critical for normal stem cell growth and maintenance.
SP cancer cells exhibit stem cell-like ability by self-renewal and higher tumorigenicity as compared to non-SP (NSP) cells. The ability of self-renewal was seen when single purified SP cells from the ER-negative cell line Cal-51 produced a heterogenous mixture of SP and NSP cells.58 Cal-51 NSP cells failed to sustain proliferation beyond one week of culture.58 Similar to Cal-51, purified SP cells taken from U373 glioma and MCF-7 breast cancer generated NSP cells in vivo and preferentially expressed “stemness” genes, including Notch-1 and β-catenin.75 More importantly, SP cells purified from U373 and MCF-7 were found to be more tumorigenic than NSP cells.75
SP cells could be heterogenous, and include a subset that expresses ABCG2.25,26 In addition, it has been shown that both SPs and known stem cells also express other ABC transporterssuch as ABCB1 (also known as P-glycoprotein or MDR-1), ABCC1 (also known as MRP-1), and ABCA3.76,77 Although ABCG2 expression alone may not serve as a stem cell marker, there is evidence that SP cells isolated from various tissues are enriched in adult tissue stem cells.60,69,70,78,79 In the future, a better understanding of the subsets within the SP population and their surface markers would be critical to the identification of putative CSC, with implications for cancer treatment. Furthermore, it is important to consider that identification of SP cells is not limited to malignancies of the breast, as other solid organ cancers have been shown to harbor these populations.80 These include the ovary, bladder, pancreas, and stomach.80,81
Prospects on developing targeted therapy
The effectiveness of current chemotherapeutic agents is posed with challenges. These problems could be partly explained by the resistance of a small subset of chemo-resistant CSCs despite the clearance of the major bulk of cancer cells. As would be expected of stem cells, CSCs would be able to resurge by developing into new tumors. Thus, chemotherapy has limited effectiveness over the long-term. Thus far, there is no defined drug that specifically targets CSCs in the clinic. However, as the scientific evidence unravels, this would open avenues for new drugs. The particular targets that may allow for selective elimination of CSCs are unclear but have been speculated to include angiogenesis, ATP-binding cassette transporters, and the Patch pathway.82 Since decreased activity of Numb, a Notch inhibitor, has been found in nearly 50% of breast cancers, Notch and γ-secretase inhibitors have been proposed for breast CSC targeting and are currently in clinical trials.83 In any case, they have experienced little clinical success.84 The reasons for this may include unforeseen crosstalk with other molecular pathways and different levels of pathway activity among different tumors.84 The challenges inherent to targeting pathways such as Hedgehog, Wnt, and Notch, is that, although they may inhibit breast CSC propagation, they may also have adverse effects on normal adult stem cells as these pathways are vital in development.85
Technologies employing automated screening of compounds have been used, and reports have shown in vivo efficacy of an agent, salinomycin, that targets CSCs of epithelial origin.86 This compound has been shown to decrease breast CSC fraction, identified as CD44+/CD24−/low, by two orders of magnitude. These findings are based on reduced expression of CSC-specific genes.86 Paclitaxel has been shown to increase the proportion of the CD44+/CD24−/low population, a finding that demonstrates possible untoward effects of conventional chemotherapy for epithelial malignancies.86 Another agent, cyclopamine, inhibits the SMO protein and prostate and gastric cancer progression by acting through the Hedgehog signaling pathway, which facilitates maintenance of CD44+/CD24−/low breast cancer cells.82,87 In addition to these pharmacological agents, a recent finding on the involvement of PI3K/Akt signaling in breast cancer had led to the identification of perifosine as putative therapy for breast CSCs in vivo.29 Perifosine, an Akt inhibitor, reduces serial mammosphere formation.29 The challenges that face these types of agents revolve around the idea that their selective toxicities towards breast CSC, in comparison with normal stem cells and cancer cells, may be limited.
In addition to these outlooks on targeted therapy, the use of oncolytic viruses has been investigated based on the ideas that (1) many viruses replicate more efficiently in tumors than in normal tissue, and (2) viruses can be loaded with transgenes that ultimately lead to toxicity against tumors.88 Oncolytic viral therapy has reached clinical trials.88–90 Among the administration methods employed are intra-arterial, intravenous, intraperitoneal, and/or intratumoral delivery.88–90 Advantage for viral therapy that specifically targets cancer is that they evade host resistance mechanisms. This is significant because, with chemotherapy and radiation treatment, cellular resistance frequently develops and these treatment modalities become ineffective.91 Oncolytic viruses appear to exert their effects on both the CSC and the non-CSC population.90 Two general classes of oncolytic viruses include wild-type viruses and attenuated viruses, both of which demonstrate toxicity towards human cells.91 DNA and RNA viruses have been explored and have been shown to alter various oncogenic signaling pathways and cell cycle checkpoints.92
Oncolytic viral therapy has demonstrated favorable results for breast malignancies.93 Reoviruses, which are in Phase II clinical trials, have been shown to alter ras signaling in breast cancer with reduction in the CSC population.93,94 Adenoviruses, in particular, may be important for targeting breast CSCs due to preference for epithelial tissue, and conditionally replicative adenoviral vectors are in Phase I clinical trials.90 In fact, studies have demonstrated a panel of oncoviruses with specificity towards the CD44+/CD24−/low population of breast cancer cells.95 Also, viruses can be armed with transgenes that target CSCs. For example, infection of invasive cancer breast cells using an adenovirus loaded with the receptor for TGF-β type II resulted in significant tumor regression in nude mice.96 These findings are consistent with the pan-suppressive effects of TGF-β on cell cycling.34
A comprehensive review of oncolytic viral therapy for breast cancer is incomplete without a discussion of the drawbacks. These include low efficacy in serum, adverse effects, potential toxicity to non-target cells, and the need for epidemiological control. Firstly, with regards to efficacy, complement-depleted plasma and IgM in serum can inactivate viruses via neutralization.89 Pre-treatment with cyclophosphamide has been shown to abolish immune-mediated viral destruction and enhance efficacy of oncolytic therapy.89 However, cyclophosphamide treatment can result in immune suppression, posing severe challenges to this method of cancer therapy.89 Another factor limiting the efficacy is the route of administration. Intravenous delivery, for example, is challenging because viruses can be subject to hepatic sequestration.90 Poor penetration of viruses into tumors presents a challenge to delivery methods.97 Adverse effects of adenoviral therapy include systemic toxicity, especially hepatotoxicity based on adenoviral tropism for the liver.98 Epidemiological control must be considered when employed these viruses, and attenuated viral strains are preferable. Thus, the challenges to oncolytic viral therapy are significant, and both safety and efficacy must be addressed. The ongoing clinical trials will provide answers to these pressing questions.
Targeted therapy involving the SDF-1/CXCR4 axis has been considered by many groups. This is rooted in the idea that the interaction between components of immune chemotaxis such as SDF-1 and CXCR4 may regulate breast cancer metastasis to the bone marrow.36,43 The use of CXCR4 antagonists such as AMD3100 and TN14003 has been suggested as a method to hinder breast tumor growth.8,99 Thus, targeting the microenvironment in the context of soluble factors surrounding tumor stroma has potential in future breast cancer therapy and may be necessary for elimination of resistant breast cancer cells in the bone marrow. Ongoing studies in our laboratory on targeting this interaction show favorable results in vitro.
The concept of CSC differentiation therapy has been proposed based on evidence that de-differentiation is associated with increased aggressiveness of cancers.5 This concept is strongly rooted in current clinical hematology and oncology, as all-trans-retinoic acid is used as therapy for acute myelogenous leukemia M3.100 Among the most well-recognized agents for differentiation therapy are the retinoids, which have been suggested for breast cancer differentiation.101,102 Quinoline compounds have been shown to cause breast cancer cells to reduce G1-S phase protein expression and induce breast cancer cells to enter G0 phase.103 Novel findings on the role of BRCA1 in breast cancer have shown that BRCA1 is necessary for differentiation of triple-negative cancers.11 BRCA1 knockdown leads to decreased luminal epithelial cells and increase the formation of secondary and tertiary mammospheres.11 BRCA1 knockdown also resulted in increased the proportion of ALDH1-positive cells and expansion of stem and progenitor cells.11 Although BRCA1 differentiation therapy is not widely accepted to date, prospects may be favorable with further investigations.
Recent investigations into differences between breast CSCs have highlighted the role of microRNAs (miRNAs), which have been implicated in regulation of self-renewal and cell fate of breast cancer cells.104 Although the detailed role of miRNAs in breast CSCs is out of the scope of this review, it is important to emphasize that non-coding molecules can have profound implications in cancer survival, maintenance, and progression.104 let-7 levels, for instance, are increased in differentiated cells and decreased in breast CSCs.104 Ectopic delivery of let-7 to breast CSCs can hinder proliferation and sphere formation and maintenance. As let-7 is a key regulator of stem cell differentiation, it may hold therapeutic potential in differentiation therapy.104 Other miRNAs may also be involved in breast CSC differentiation and maintenance of self-renewal. In a recent study, miRNA expression profiles of breast cancer cells and breast CSCs were determined. Both normal stem cells of the breast and breast CSCs were noted to downregulation of miR-200c, which functions in suppressing tumor progression through inhibition of clonal expansion.105 It has been suggested that normal stem cells and breast CSCs are subject to similar regulation by miRNAs.105 Therapy that targets breast CSCs may therefore lead to normal breast stem cell toxicity. The similarities between miRNA profiles and the complexity of miRNA networks that govern both normal stem cell and breast CSCs survival may pose challenges to developing targeted therapy. These hurdles can be overcome through first understanding the basic science prior to implementation of potential therapy in the clinic.
Prospects on differentiation therapy as a treatment modality for cancer is not limited to breast cancer, as vitamin D has been used in differentiation therapy for cancers of the colon, bone, and blood.106 For instance, miRNAs have been proposed as a method to induce differentiation in rhabdomyosarcoma.37 In addition, cell differentiation agent 2 (Cda-2) has been shown to inhibit glioblastoma proliferation through differentiation.107
Nanotechnology is emerging as a new tool for localizing and targeting breast cancer and as an addendum to current genomics and proteomic methods.108 Nanoparticles are comprises of entities such as quantum dots, which can be beneficial to cancer cell detection.109 Immunoglobulin-conjugated quantum dots have been used to identify HER2(+) breast cancer cells.109 Quantum dots have been shown to be more stable and more fluorescent than traditional organic dyes used in cell biology.109 Breast cancer biomarkers such as erbB2 and Ki-67 have been shown to be detected by nanoprobes.110 Nanoparticle vectors can also effectively target breast cancer cells and aid in delivery of anti-cancer agents.111 These findings represent only a few examples that emphasize in the increasingly important role of emerging technology in breast cancer biology. Further investigations into biotechnology are needed before nanotechnology becomes a key player in breast cancer cell tracking.
In order to achieve successful targeted therapy, a thorough investigation into the differences between breast CSC and normal mammary cells must be performed. Robust analysis of the basic science must precede clinical trials. Current treatment modalities such as chemotherapy, radiation, immunotherapy, and surgery for breast cancer are somewhat effective, but when one considers their limitations, treatments can be improved based on their current toxicity and the development of resistance.5,92 The introduction of oncolytic adenoviral therapy for breast cancer represents a new modality that may hold the answer to these challenges, and current positive findings merit further investigation as this may be an effective modality. The idea of viral oncolysis is currently in its infancy but, given that this mode of therapy is now in clinical trials, it may have promising potential for treatment of malignancy.91 Differentiation therapy holds promise based on its success in hematological malignancies and in vitro demonstrations of its effectiveness in breast cancer.103 Another interesting but weakly supported treatment modality for breast cancer is the use of peptide-derived vaccines.112 The basis for this treatment is that breast tumors are immunogenic and express various surface antigens.112 Nonetheless, the evidence is limited in this arena of treatment.112 The implementation of effective targeted therapy for breast cancer is complicated by the requirement that both the CSC and non-CSC populations must be effectively eliminated. The challenge is that signal transduction mechanisms that maintain the cellular survival could be distinct between the two subsets. Dynamics of gene networks and microenvironmental factors that regulate tumor progression in breast cancer must be considered while investigating therapeutic targets and developing new modalities of therapy.
Summary and Conclusions
The field of breast cancer biology is rapidly developing as scientists gain knowledge about the molecular underpinnings of breast CSCs. Although a consensus on the origins of the breast CSCs remains to be achieved, it is important to entertain the possibility that different types of breast cancer may originate from different elements of the breast. Establishment of a uniform definition of the breast CSC may not be in the best interest of the field. Further complicating the picture is that the interplay among various molecular pathways, including Wnt, PI3K/Akt, Hedgehog, and Notch, may be responsible for survival and propagation of breast cancer cells. Microenvironmental factors can adversely influence breast cancer progression by facilitating escape from immune surveillance, providing support via soluble factors and physical contact, and regulating epigenetic changes that lead to enhanced growth. The link between EMT, the CSC hypothesis, and side populations is stronger as commonalities are discovered between both ideas. Although many new treatment modalities such as oncolytic viral therapy, differentiation therapy, and nanotechnology have been designed, they are all met with challenges that currently prevent clinical success. Given the complexity of breast cancer biology, further investigations into the basic science of these cells will be necessary prior to the development of successful therapy.
Fig. 1.
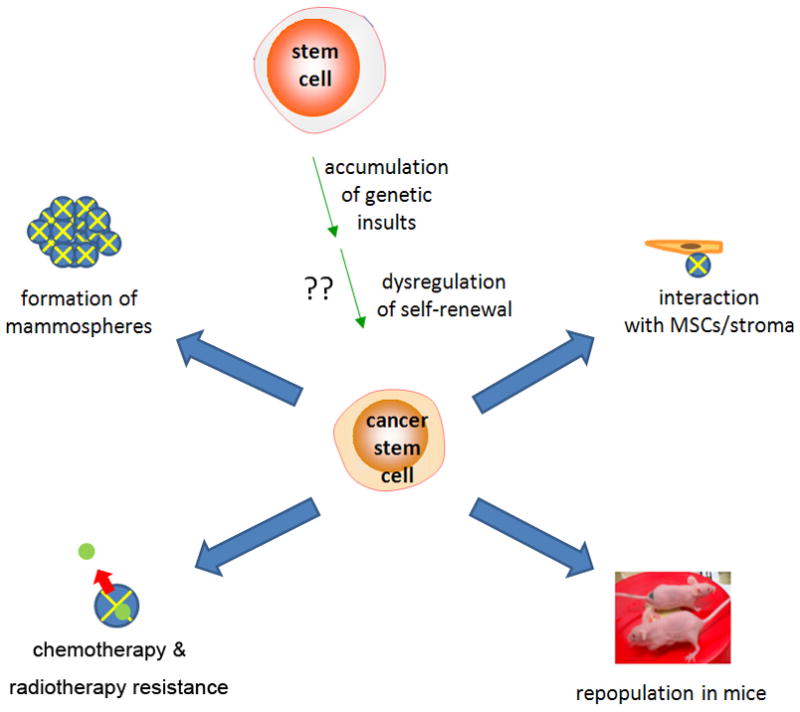
Origins and properties of breast cancer stem cells (CSCs). Origins of CSCs are unclear but may involve accumulation of genetic damage and dysregulation of normal self-renewal. Defining properties include resistance to chemotherapy, mammosphere formation, and in vivo tumor generation in immunodeficient mice. (Al-Hajj et al., 2003).
References
- 1.Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3(7):730–7. doi: 10.1038/nm0797-730. [DOI] [PubMed] [Google Scholar]
- 2.Schatton T, Frank NY, Frank MH. Identification and targeting of cancer stem cells. Bioessays. 2009;31(10):1038–1049. doi: 10.1002/bies.200900058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sakariassen PO, Immervoll H, Chekenya M. Cancer stem cells as mediators of treatment resistance in brain tumors: status and controversies. Neoplasia. 2007;9(11):882–92. doi: 10.1593/neo.07658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Odoux C, Fohrer H, Hoppo T, et al. A stochastic model for cancer stem cell origin in metastatic colon cancer. Cancer Res. 2008;68(17):6932–41. doi: 10.1158/0008-5472.CAN-07-5779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kang SK, Park JB, Cha SH. Multipotent, dedifferentiated cancer stem-like cells from brain gliomas. Stem Cells Dev. 2006;15(3):423–35. doi: 10.1089/scd.2006.15.423. [DOI] [PubMed] [Google Scholar]
- 6.Polyak K, Haviv I, Campbell IG. Co-evolution of tumor cells and their microenvironment. Trends Genet. 2009;25(1):30–8. doi: 10.1016/j.tig.2008.10.012. [DOI] [PubMed] [Google Scholar]
- 7.Polyak K. Breast cancer: origins and evolution. J Clin Invest. 2007;117(11):3155–63. doi: 10.1172/JCI33295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Croker AK, Allan AL. Cancer stem cells: implications for the progression and treatment of metastatic disease. J Cell Mol Med. 2008;12(2):374–90. doi: 10.1111/j.1582-4934.2007.00211.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100(7):3983–8. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kakarala M, Wicha MS. Implications of the cancer stem-cell hypothesis for breast cancer prevention and therapy. J Clin Oncol. 2008;26(17):2813–20. doi: 10.1200/JCO.2008.16.3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu S, Ginestier C, Charafe-Jauffret E, et al. BRCA1 regulates human mammary stem/progenitor cell fate. Proc Natl Acad Sci U S A. 2008;105(5):1680–5. doi: 10.1073/pnas.0711613105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gonzalez LO, Corte MD, Junquera S, et al. Expression and prognostic significance of metalloproteases and their inhibitors in luminal A and basal-like phenotypes of breast carcinoma. Hum Pathol. 2009;40(9):1224–33. doi: 10.1016/j.humpath.2008.12.022. [DOI] [PubMed] [Google Scholar]
- 13.Lim E, Vaillant F, Wu D, et al. Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nat Med. 2009;15(8):907–13. doi: 10.1038/nm.2000. [DOI] [PubMed] [Google Scholar]
- 14.Linn SC, Van’t Veer LJ. Clinical relevance of the triple-negative breast cancer concept: genetic basis and clinical utility of the concept. Eur J Cancer. 2009;45 (Suppl 1):11–26. doi: 10.1016/S0959-8049(09)70012-7. [DOI] [PubMed] [Google Scholar]
- 15.Liu S, Dontu G, Mantle ID, Patel S, Ahn NS, Jackson KW, Suri P, Wicha MS. Hedgehog signaling and Bmi-1 regulate self-renewal of normal and malignant human mammary stem cells. Cancer Res. 2006;66(12):6063–71. doi: 10.1158/0008-5472.CAN-06-0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ge Y, Sneige N, Eltorky MA, Wang Z, Lin E, Gong Y, Guo M. Immunohistochemical characterization of subtypes of male breast carcinoma. Breast Cancer Res. 2009;11(3):R28. doi: 10.1186/bcr2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ohta T, Wu W, Koike A, Asakawa H, Koizumi H, Fukuda M. Contemplating chemosensitivity of basal-like breast cancer based on BRCA1 dysfunction. Breast Cancer. 2009;16(4):268–74. doi: 10.1007/s12282-009-0115-y. [DOI] [PubMed] [Google Scholar]
- 18.MacDougall JR, Matrisian LM. Targets of extinction: identification of genes whose expression is repressed as a consequence of somatic fusion between cells representing basal and luminal mammary epithelial phenotypes. J Cell Sci. 2000;113(Pt 3):409–23. doi: 10.1242/jcs.113.3.409. [DOI] [PubMed] [Google Scholar]
- 19.Tang P, Wang X, Schiffhauer L, Wang J, Bourne P, Yang Q, Quinn A, Hajdu SI. Relationship between nuclear grade of ductal carcinoma in situ and cell origin markers. Ann Clin Lab Sci. 2006;36(1):16–22. [PubMed] [Google Scholar]
- 20.Creighton CJ, Li X, Landis M, et al. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc Natl Acad Sci U S A. 2009;106(33):13820–5. doi: 10.1073/pnas.0905718106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hennessy BT, Gonzalez-Angulo AM, Stemke-Hale K, et al. Characterization of a naturally occurring breast cancer subset enriched in epithelial-to-mesenchymal transition and stem cell characteristics. Cancer Res. 2009;69(10):4116–24. doi: 10.1158/0008-5472.CAN-08-3441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shipitsin M, Campbell LL, Argani P, et al. Molecular definition of breast tumor heterogeneity. Cancer Cell. 2007;11(3):259–73. doi: 10.1016/j.ccr.2007.01.013. [DOI] [PubMed] [Google Scholar]
- 23.Morel AP, Lièvre M, Thomas C, Hinkal G, Ansieau S, Puisieux A. Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS One. 2008;3(8):e2888. doi: 10.1371/journal.pone.0002888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ginestier C, Hur MH, Charafe-Jauffret E, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. 2007;1:555–67. doi: 10.1016/j.stem.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nadin BM, Goodell MA, Hirschi KK. Phenotype and hematopoietic potential of side population cells throughout embryonic development. Blood. 2003;102(7):2436–43. doi: 10.1182/blood-2003-01-0118. [DOI] [PubMed] [Google Scholar]
- 26.Montanaro F, Liadaki K, Schienda J, Flint A, Gussoni E, Kunkel LM. Demystifying SP cell purification: viability, yield, and phenotype are defined by isolation parameters. Exp Cell Res. 2004;298(1):144–54. doi: 10.1016/j.yexcr.2004.04.010. [DOI] [PubMed] [Google Scholar]
- 27.Oates JE, Grey BR, Addla SK, Samuel JD, Hart CA, Ramani V, Brown MD, Clarke NW. Hoechst 33342 side population identification is a conserved and unified mechanism in urological cancers. Stem Cells Dev. 2009 doi: 10.1089/scd.2008.0302. In press. [DOI] [PubMed] [Google Scholar]
- 28.Dittmar T, Nagler C, Schwitalla S, Reith G, Niggemann B, Zänker KS. Recurrence cancer stem cells--made by cell fusion? Med Hypotheses. 2009;73(4):542–7. doi: 10.1016/j.mehy.2009.05.044. [DOI] [PubMed] [Google Scholar]
- 29.Korkaya H, Paulson A, Charafe-Jauffret E, Ginestier C, Brown M, Dutcher J, Clouthier SG, Wicha MS. Regulation of mammary stem/progenitor cells by PTEN/Akt/beta-catenin signaling. PLoS Biol. 2009;7(6):e1000121. doi: 10.1371/journal.pbio.1000121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dontu G, Jackson KW, McNicholas E, Kawamura MJ, Abdallah WM, Wicha MS. Role of Notch signaling in cell-fate determination of human mammary stem/progenitor cells. Breast Cancer Res. 2004;6(6):R605–15. doi: 10.1186/bcr920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ischenko I, Seeliger H, Schaffer M, Jauch KW, Bruns CJ. Cancer stem cells: how can we target them? Curr Med Chem. 2008;15(30):3171–84. doi: 10.2174/092986708786848541. [DOI] [PubMed] [Google Scholar]
- 32.Sonnenberg M, van der Kuip H, Haubeis S, et al. Highly variable response to cytotoxic chemotherapy in carcinoma-associated fibroblasts (CAFs) from lung and breast. BMC Cancer. 2008;8:364. doi: 10.1186/1471-2407-8-364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tan AR, Alexe G, Reiss M. Transforming growth factor-beta signaling: emerging stem cell target in metastatic breast cancer? Breast Cancer Res Treat. 2009;115(3):453–95. doi: 10.1007/s10549-008-0184-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dabiri G, Tumbarello DA, Turner CE, Van de Water L. TGF-beta1 slows the growth of pathogenic myofibroblasts through a mechanism requiring the focal adhesion protein, Hic-5. J Invest Dermatol. 2008;128(2):280–91. doi: 10.1038/sj.jid.5700975. [DOI] [PubMed] [Google Scholar]
- 35.Rhodes LV, Muir SE, Elliott S, et al. Adult human mesenchymal stem cells enhance breast tumorigenesis and promote hormone independence. Breast Cancer Res Treat. 2009 doi: 10.1007/s10549-009-0458-2. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Corcoran KE, Trzaska KA, Fernandes H, et al. Mesenchymal stem cells in early entry of breast cancer into bone marrow. PLoS One. 2008;3(6):e2563. doi: 10.1371/journal.pone.0002563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mishra PJ, Merlino G. MicroRNA reexpression as differentiation therapy in cancer. J Clin Invest. 2009;119(8):2119–23. doi: 10.1172/JCI40107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mishra PJ, Mishra PJ, Humeniuk R, et al. Carcinoma-associated fibroblast-like differentiation of human mesenchymal stem cells. Cancer Res. 2008;68(11):4331–9. doi: 10.1158/0008-5472.CAN-08-0943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lin HJ, Zuo T, Lin CH, et al. Breast cancer-associated fibroblasts confer AKT1-mediated epigenetic silencing of Cystatin M in epithelial cells. Cancer Res. 2008;68(24):10257–66. doi: 10.1158/0008-5472.CAN-08-0288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Modolell M, Choi BS, Ryan RO, et al. Local suppression of T cell responses by arginase-induced L-arginine depletion in nonhealing leishmaniasis. PLoS Negl Trop Dis. 2009;3(7):e480. doi: 10.1371/journal.pntd.0000480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Matsuura K, Yamaguchi Y, Osaki A, et al. FOXP3 expression of micrometastasis-positive sentinel nodes in breast cancer patients. Oncol Rep. 2009;22(5):1181–7. doi: 10.3892/or_00000553. [DOI] [PubMed] [Google Scholar]
- 42.Scola L, Giacalone A, Marasà L, et al. Genetic determined downregulation of both type 1 and type 2 cytokine pathways might be protective against pancreatic cancer. Ann N Y Acad Sci. 2009;1155:284–8. doi: 10.1111/j.1749-6632.2008.03686.x. [DOI] [PubMed] [Google Scholar]
- 43.Winquist RJ, Boucher DM, Wood M, Furey BF. Targeting cancer stem cells for more effective therapies: Taking out cancer’s locomotive engine. Biochem Pharmacol. 2009;78(4):326–34. doi: 10.1016/j.bcp.2009.03.020. [DOI] [PubMed] [Google Scholar]
- 44.Dalloul A, Sainz-Perez A. Interleukin-24: A Molecule with Potential Anti-Cancer Activity and a Cytokine in Search of a Function. Endocr Metab Immune Disord Drug Targets. 2009 doi: 10.2174/187153009789839200. In press. [DOI] [PubMed] [Google Scholar]
- 45.Yang YJ, Chen DZ, Li LX, Sheng QS, Jin ZK, Zhao DF. Targeted IL-24 gene therapy inhibits cancer recurrence after liver tumor resection by inducing tumor cell apoptosis in nude mice. Hepatobiliary Pancreat Dis Int. 2009;8(2):174–8. [PubMed] [Google Scholar]
- 46.Dumont N, Wilson MB, Crawford YG, Reynolds PA, Sigaroudinia M, Tlsty TD. Sustained induction of epithelial to mesenchymal transition activates DNA methylation of genes silenced in basal-like breast cancers. Proc Natl Acad Sci U S A. 2008;105(39):14867–72. doi: 10.1073/pnas.0807146105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Charafe-Jauffret E, Monville F, Ginestier C, Dontu G, Birnbaum D, Wicha MS. Cancer stem cells in breast: current opinion and future challenges. Pathobiology. 2008;75(2):75–84. doi: 10.1159/000123845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hao-Xiang T, Qian W, Lian-Zhou C, et al. MicroRNA-9 reduces cell invasion and E-cadherin secretion in SK-Hep-1 cell. Med Oncol. 2009 doi: 10.1007/s12032-009-9264-2. In press. [DOI] [PubMed] [Google Scholar]
- 49.Fidler IJ. The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited. Nat Rev Cancer. 2003;3(6):453–8. doi: 10.1038/nrc1098. [DOI] [PubMed] [Google Scholar]
- 50.Kuroishi S, Suda T, Fujisawa T, et al. Epithelial-mesenchymal transition induced by transforming growth factor-beta1 in mouse tracheal epithelial cells. Respirology. 2009;14(6):828–37. doi: 10.1111/j.1440-1843.2009.01561.x. [DOI] [PubMed] [Google Scholar]
- 51.Tuhkanen H, Soini Y, Kosma VM, et al. Nuclear expression of Snail1 in borderline and malignant epithelial ovarian tumours is associated with tumour progression. BMC Cancer. 2009;9(1):289. doi: 10.1186/1471-2407-9-289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li Y, VandenBoom TG, 2nd, Kong D, et al. Up-regulation of miR-200 and let-7 by natural agents leads to the reversal of epithelial-to-mesenchymal transition in gemcitabine-resistant pancreatic cancer cells. Cancer Res. 2009;69(16):6704–12. doi: 10.1158/0008-5472.CAN-09-1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Moen I, Øyan AM, Kalland KH, et al. Hyperoxic treatment induces mesenchymal-to-epithelial transition in a rat adenocarcinoma model. PLoS One. 2009;4(7):e6381. doi: 10.1371/journal.pone.0006381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hollier BG, Evans K, Mani SA. The epithelial-to-mesenchymal transition and cancer stem cells: a coalition against cancer therapies. J Mammary Gland Biol Neoplasia. 2009;14(1):29–43. doi: 10.1007/s10911-009-9110-3. [DOI] [PubMed] [Google Scholar]
- 55.Mani SA, Guo W, Liao MJ, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133(4):704–15. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Santisteban M, Reiman JM, Asiedu MK, et al. Immune-induced epithelial to mesenchymal transition in vivo generates breast cancer stem cells. Cancer Res. 2009;69(7):2887–95. doi: 10.1158/0008-5472.CAN-08-3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Goodell MA, Rosenzweig M, Kim H, et al. Dye efflux studies suggest that hematopoietic stem cells expressing low or undetectable levels of CD34 antigen exist in multiple species. Nat Med. 1997;3(12):1337–45. doi: 10.1038/nm1297-1337. [DOI] [PubMed] [Google Scholar]
- 58.Christgen M, Ballmaier M, Bruchhardt H, von Wasielewski R, Kreipe H, Lehmann U. Identification of a distinct side population of cancer cells in the Cal-51 human breast carcinoma cell line. Mol Cell Biochem. 2007;306(1–2):201–12. doi: 10.1007/s11010-007-9570-y. [DOI] [PubMed] [Google Scholar]
- 59.Triel C, Vestergaard ME, Bolund L, Jensen TG, Jensen UB. Side population cells in human and mouse epidermis lack stem cell characteristics. Exp Cell Res. 2004;295(1):79–90. doi: 10.1016/j.yexcr.2003.11.032. [DOI] [PubMed] [Google Scholar]
- 60.Alvi AJ, Clayton H, Joshi C, et al. Functional and molecular characterisation of mammary side population cells. Breast Cancer Res. 2003;5(1):R1–8. doi: 10.1186/bcr547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Asakura A, Seale P, Girgis-Gabardo A, Rudnicki MA. Myogenic specification of side population cells in skeletal muscle. J Cell Biol. 2002;159(1):123–34. doi: 10.1083/jcb.200202092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Clayton H, Titley I, Vivanco M. Growth and differentiation of progenitor/stem cells derived from the human mammary gland. Exp Cell Res. 2004;297(2):444–60. doi: 10.1016/j.yexcr.2004.03.029. [DOI] [PubMed] [Google Scholar]
- 63.Summer R, Kotton DN, Sun X, Ma B, Fitzsimmons K, Fine A. Side population cells and Bcrp1 expression in lung. Am J Physiol Lung Cell Mol Physiol. 2003;285(1):L97–104. doi: 10.1152/ajplung.00009.2003. [DOI] [PubMed] [Google Scholar]
- 64.Giangreco A, Shen H, Reynolds SD, Stripp BR. Molecular phenotype of airway side population cells. Am J Physiol Lung Cell Mol Physiol. 2004;286(4):L624–30. doi: 10.1152/ajplung.00149.2003. [DOI] [PubMed] [Google Scholar]
- 65.Shimano K, Satake M, Okaya A, et al. Hepatic oval cells have the side population phenotype defined by expression of ATP-binding cassette transporter ABCG2/BCRP1. Am J Pathol. 2003;163(1):3–9. doi: 10.1016/S0002-9440(10)63624-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Litman T, Brangi M, Hudson E, et al. The multidrug-resistant phenotype associated with overexpression of the new ABC half-transporter, MXR (ABCG2) J Cell Sci. 2000;113(Pt 11):2011–21. doi: 10.1242/jcs.113.11.2011. [DOI] [PubMed] [Google Scholar]
- 67.Doyle LA, Ross DD. Multidrug resistance mediated by the breast cancer resistance protein BCRP (ABCG2) Oncogene. 2003;22(47):7340–58. doi: 10.1038/sj.onc.1206938. [DOI] [PubMed] [Google Scholar]
- 68.Zhou S, Schuetz JD, Bunting KD, Colapietro AM, Sampath J, Morris JJ, Lagutina I, Grosveld GC, Osawa M, Nakauchi H, Sorrentino BP. The ABC transporter Bcrp1/ABCG2 is expressed in a wide variety of stem cells and is a molecular determinant of the side-population phenotype. Nat Med. 2001;7(9):1028–34. doi: 10.1038/nm0901-1028. [DOI] [PubMed] [Google Scholar]
- 69.Scharenberg CW, Harkey MA, Torok-Storb B. The ABCG2 transporter is an efficient Hoechst 33342 efflux pump and is preferentially expressed by immature human hematopoietic progenitors. Blood. 2002;99(2):507–12. doi: 10.1182/blood.v99.2.507. [DOI] [PubMed] [Google Scholar]
- 70.Kim M, Turnquist H, Jackson J, et al. The multidrug resistance transporter ABCG2 (breast cancer resistance protein 1) effluxes Hoechst 33342 and is overexpressed in hematopoietic stem cells. Clin Cancer Res. 2002;8(1):22–8. [PubMed] [Google Scholar]
- 71.Zhou S, Morris JJ, Barnes Y, Lan L, Schuetz JD, Sorrentino BP. Bcrp1 gene expression is required for normal numbers of side population stem cells in mice, and confers relative protection to mitoxantrone in hematopoietic cells in vivo. Proc Natl Acad Sci U S A. 2002;99(19):12339–44. doi: 10.1073/pnas.192276999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jonker JW, Buitelaar M, Wagenaar E, et al. The breast cancer resistance protein protects against a major chlorophyll-derived dietary phototoxin and protoporphyria. Proc Natl Acad Sci U S A. 2002;99(24):15649–54. doi: 10.1073/pnas.202607599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Schinkel AH, Smit JJ, van Tellingen O, et al. Disruption of the mouse mdr1a P-glycoprotein gene leads to a deficiency in the blood-brain barrier and to increased sensitivity to drugs. Cell. 1994;77(4):491–502. doi: 10.1016/0092-8674(94)90212-7. [DOI] [PubMed] [Google Scholar]
- 74.Doyle LA, Yang W, Abruzzo LV, et al. A multidrug resistance transporter from human MCF-7 breast cancer cells. Proc Natl Acad Sci U S A. 1998;95(26):15665–70. doi: 10.1073/pnas.95.26.15665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Patrawala L, Calhoun T, Schneider-Broussard R, Zhou J, Claypool K, Tang DG. Side population is enriched in tumorigenic, stem-like cancer cells, whereas ABCG2+ and ABCG2- cancer cells are similarly tumorigenic. Cancer Res. 2005;65(14):6207–19. doi: 10.1158/0008-5472.CAN-05-0592. [DOI] [PubMed] [Google Scholar]
- 76.Lechner A, Leech CA, Abraham EJ, Nolan AL, Habener JF. Nestin-positive progenitor cells derived from adult human pancreatic islets of Langerhans contain side population (SP) cells defined by expression of the ABCG2 (BCRP1) ATP-binding cassette transporter. Biochem Biophys Res Commun. 2002;293(2):670–4. doi: 10.1016/S0006-291X(02)00275-9. [DOI] [PubMed] [Google Scholar]
- 77.Hirschmann-Jax C, Foster AE, Wulf GG, et al. A distinct “side population” of cells with high drug efflux capacity in human tumor cells. Proc Natl Acad Sci U S A. 2004;101(39):14228–33. doi: 10.1073/pnas.0400067101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Challen GA, Bertoncello I, Deane JA, Ricardo SD, Little MH. Kidney side population reveals multilineage potential and renal functional capacity but also cellular heterogeneity. J Am Soc Nephrol. 2006;17(7):1896–912. doi: 10.1681/ASN.2005111228. [DOI] [PubMed] [Google Scholar]
- 79.Dontu G, Abdallah WM, Foley JM, et al. In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev. 2003;17(10):1253–70. doi: 10.1101/gad.1061803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gao Q, Geng L, Kvalheim G, Gaudernack G, Suo Z. Identification of cancer stem-like side population cells in ovarian cancer cell line OVCAR-3. Ultrastruct Pathol. 2009;33(4):175–81. doi: 10.1080/01913120903086072. [DOI] [PubMed] [Google Scholar]
- 81.Kabashima A, Higuchi H, Takaishi H, et al. Side population of pancreatic cancer cells predominates in TGF-beta-mediated epithelial to mesenchymal transition and invasion. Int J Cancer. 2009;124(12):2771–9. doi: 10.1002/ijc.24349. [DOI] [PubMed] [Google Scholar]
- 82.Lou H, Dean M. Targeted therapy for cancer stem cells: the patched pathway and ABC transporters. Oncogene. 2007;26(9):1357–60. doi: 10.1038/sj.onc.1210200. [DOI] [PubMed] [Google Scholar]
- 83.Pece S, Serresi M, Santolini E, et al. Loss of negative regulation by Numb over Notch is relevant to human breast carcinogenesis. J Cell Biol. 2004;167(2):215–21. doi: 10.1083/jcb.200406140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rizzo P, Osipo C, Foreman K, Golde T, Osborne B, Miele L. Rational targeting of Notch signaling in cancer. Oncogene. 2008;27(38):5124–31. doi: 10.1038/onc.2008.226. [DOI] [PubMed] [Google Scholar]
- 85.Zardawi SJ, O’Toole SA, Sutherland RL, Musgrove EA. Dysregulation of Hedgehog, Wnt and Notch signalling pathways in breast cancer. Histol Histopathol. 2009;24(3):385–98. doi: 10.14670/HH-24.385. [DOI] [PubMed] [Google Scholar]
- 86.Gupta PB, Onder TT, Jiang G, et al. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell. 2009;138(4):645–59. doi: 10.1016/j.cell.2009.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tanaka H, Nakamura M, Kameda C, et al. The Hedgehog signaling pathway plays an essential role in maintaining the CD44+CD24−/low subpopulation and the side population of breast cancer cells. Anticancer Res. 2009;29(6):2147–57. [PubMed] [Google Scholar]
- 88.Crompton AM, Kirn DH. From ONYX-015 to armed vaccinia viruses: the education and evolution of oncolytic virus development. Curr Cancer Drug Targets. 2007;7(2):133–9. doi: 10.2174/156800907780058862. [DOI] [PubMed] [Google Scholar]
- 89.Ikeda K, Ichikawa T, Wakimoto H, et al. Oncolytic virus therapy of multiple tumors in the brain requires suppression of innate and elicited antiviral responses. Nat Med. 1999;5(8):881–7. doi: 10.1038/11320. [DOI] [PubMed] [Google Scholar]
- 90.Short JJ, Curiel DT. Oncolytic adenoviruses targeted to cancer stem cells. Mol Cancer Ther. 2009;8(8):2096–102. doi: 10.1158/1535-7163.MCT-09-0367. [DOI] [PubMed] [Google Scholar]
- 91.Cripe TP, Wang PY, Marcato P, Mahller YY, Lee PW. Targeting cancer-initiating cells with oncolytic viruses. Mol Ther. 2009 Oct;17(10):1677–82. doi: 10.1038/mt.2009.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Thirukkumaran C, Morris DG. Oncolytic viral therapy using reovirus. Methods Mol Biol. 2009;542:607–34. doi: 10.1007/978-1-59745-561-9_31. [DOI] [PubMed] [Google Scholar]
- 93.Marcato P, Dean CA, Giacomantonio CA, Lee PW. Oncolytic reovirus effectively targets breast cancer stem cells. Mol Ther. 2009;17(6):972–9. doi: 10.1038/mt.2009.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lal R, Harris D, Postel-Vinay S, de Bono J. Reovirus: Rationale and clinical trial update. Curr Opin Mol Ther. 2009;11(5):532–9. [PubMed] [Google Scholar]
- 95.Bauerschmitz GJ, Ranki T, Kangasniemi L, et al. Tissue-specific promoters active in CD44+CD24−/low breast cancer cells. Cancer Res. 2008;68(14):5533–9. doi: 10.1158/0008-5472.CAN-07-5288. [DOI] [PubMed] [Google Scholar]
- 96.Seth P, Wang ZG, Pister A, et al. Development of oncolytic adenovirus armed with a fusion of soluble transforming growth factor-beta receptor II and human immunoglobulin Fc for breast cancer therapy. Hum Gene Ther. 2006;17(11):1152–60. doi: 10.1089/hum.2006.17.1152. [DOI] [PubMed] [Google Scholar]
- 97.Nguyen TL, Tumilasci VF, Singhroy D, Arguello M, Hiscott J. The emergence of combinatorial strategies in the development of RNA oncolytic virus therapies. Cell Microbiol. 2009;11(6):889–97. doi: 10.1111/j.1462-5822.2009.01317.x. [DOI] [PubMed] [Google Scholar]
- 98.Waddington SN, McVey JH, Bhella D, et al. Adenovirus serotype 5 hexon mediates liver gene transfer. Cell. 2008;132(3):397–409. doi: 10.1016/j.cell.2008.01.016. [DOI] [PubMed] [Google Scholar]
- 99.Smith MC, Luker KE, Garbow JR, et al. CXCR4 regulates growth of both primary and metastatic breast cancer. Cancer Res. 2004;64(23):8604–12. doi: 10.1158/0008-5472.CAN-04-1844. [DOI] [PubMed] [Google Scholar]
- 100.Melinkeri SR, Gupta RK, Dabadghao S. A Sweet-like syndrome manifesting as gingival hyperplasia and myositis without cutaneous involvement. Ann Hematol. 2002;81(7):397–8. doi: 10.1007/s00277-002-0470-7. [DOI] [PubMed] [Google Scholar]
- 101.Baj G, Arnulfo A, Deaglio S, et al. Retinoids in breast cancer prevention and treatment. A review of the literature. Eur J Gynaecol Oncol. 2000;21(4):411–5. [PubMed] [Google Scholar]
- 102.Hansen LA, Sigman CC, Andreola F, Ross SA, Kelloff GJ, De Luca LM. Retinoids in chemoprevention and differentiation therapy. Carcinogenesis. 2000;21(7):1271–9. [PubMed] [Google Scholar]
- 103.Martirosyan AR, Rahim-Bata R, Freeman AB, Clarke CD, Howard RL, Strobl JS. Differentiation-inducing quinolines as experimental breast cancer agents in the MCF-7 human breast cancer cell model. Biochem Pharmacol. 2004;68(9):1729–38. doi: 10.1016/j.bcp.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 104.Yu F, Yao H, Zhu P, et al. let-7 regulates self renewal and tumorigenicity of breast cancer cells. Cell. 2007;131(6):1109–23. doi: 10.1016/j.cell.2007.10.054. [DOI] [PubMed] [Google Scholar]
- 105.Shimono Y, Zabala M, Cho RW, et al. Downregulation of miRNA-200c links breast cancer stem cells with normal stem cells. Cell. 2009;138(3):592–603. doi: 10.1016/j.cell.2009.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Gocek E, Studzinski GP. Vitamin D and differentiation in cancer. Crit Rev Clin Lab Sci. 2009;46(4):190–209. doi: 10.1080/10408360902982128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lin CL, Wang MH, Qin YF, Fang M, Xie BB, Zhong XY. Differentiation of SWO-38 glioma cells induced by CDA-2 is mediated by peroxisome proliferator-activated receptor gamma. J Neurooncol. 2009;95(1):29–36. doi: 10.1007/s11060-009-9900-x. [DOI] [PubMed] [Google Scholar]
- 108.Leporatti S, Vergara D, Zacheo A, et al. Cytomechanical and topological investigation of MCF-7 cells by scanning force microscopy. Nanotechnology. 2009;20(5):55103. doi: 10.1088/0957-4484/20/5/055103. [DOI] [PubMed] [Google Scholar]
- 109.Wu X, Liu H, Liu J, Haley KN, et al. Immunofluorescent labeling of cancer marker Her2 and other cellular targets with semiconductor quantum dots. Nat Biotechnol. 2003;21(1):41–6. doi: 10.1038/nbt764. [DOI] [PubMed] [Google Scholar]
- 110.Wang HN, Vo-Dinh T. Multiplex detection of breast cancer biomarkers using plasmonic molecular sentinel nanoprobes. Nanotechnology. 2009;20(6):65101. doi: 10.1088/0957-4484/20/6/065101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Mangipudi SS, Canine BF, Wang Y, Hatefi A. Development of a genetically engineered biomimetic vector for targeted gene transfer to breast cancer cells. Mol Pharm. 2009;6(4):1100–9. doi: 10.1021/mp800251x. [DOI] [PubMed] [Google Scholar]
- 112.Disis ML, Salazar LG, Knutson KL. Peptide-Based Vaccines in Breast Cancer. Breast Dis. 2009;20:3–11. doi: 10.3233/bd-2004-20102. [DOI] [PubMed] [Google Scholar]