

Abstract
Similar to dopamine (DA), cannabinoids strongly influence prefrontal cortical functions such as working memory, emotional learning, and sensory perception. While endogenous cannabinoid receptors (CB1Rs) are abundantly expressed in the prefrontal cortex (PFC), very little is known about endocannabinoid (eCB) signaling in this brain region. Recent behavioral and electrophysiological evidence has suggested a functional interplay between the dopamine and cannabinoid receptor systems, although the cellular mechanisms underlying this interaction remain to be elucidated. We examined this issue by combining neuroanatomical and electrophysiological techniques in PFC of rats and mice (both genders). Using immuno-electron microscopy, we show that CB1Rs and dopamine type 2 receptors (D2Rs) co-localize at terminals of symmetrical, presumably GABAergic, synapses in the PFC. Indeed, activation of either receptor can suppress GABA release onto layer 5 pyramidal cells. Furthermore, co-activation of both receptors via repetitive afferent stimulation triggers eCB-mediated long term depression of inhibitory transmission (I-LTD). This I-LTD is heterosynaptic in nature, requiring glutamate release to activate group I metabotropic glutamate receptors (mGluRs). D2Rs most likely facilitate eCB signaling at the presynaptic site as disrupting postsynaptic D2R signaling does not diminish I-LTD. Facilitation of eCB-LTD may be one mechanism by which DA modulates neuronal activity in the PFC and regulates PFC-mediated behavior in vivo.
Keywords: Neocortex, CB1 receptor, dopamine receptor, inhibition, LTD, presynaptic, transmitter release, schizophrenia
INTRODUCTION
Dopaminergic innervation of the prefrontal cortex (PFC) strongly modulates high-level cognitive processes (Le Moal and Simon, 1991; Goldman-Rakic, 1998; Seamans and Yang, 2004). Unlike fast ionotropic neurotransmitters, dopamine (DA) does not directly mediate synaptic transmission but affects it by altering the cellular or synaptic properties of target neurons (Greengard, 2001; Seamans and Yang, 2004; Iversen and Iversen, 2007). The recent link between DA and endocannabinoids (eCBs) (van der Stelt and Di Marzo, 2003; Laviolette and Grace, 2006) is particularly interesting in the PFC given the well-known hallucinogenic effects of exogenous cannabinoids and the potential eCB association with neuropsychiatric disorders that reflect PFC dysfunction (Ujike and Morita, 2004; Koethe et al., 2009; Sewell et al., 2009). However, it remains unclear exactly how eCBs regulate PFC function and whether DA plays a role in this regulation.
Short- and long-term synaptic plasticity mediated by eCBs has been reported in several brain regions (for recent reviews, see Chevaleyre et al., 2006; Lovinger, 2008; Heifets and Castillo, 2009; Kano et al., 2009). Recent evidence in the midbrain suggests that DA, via D2-like receptors (D2Rs), can facilitate eCB signaling and eCB-mediated plasticity (Melis et al., 2004; Kreitzer and Malenka, 2005; Yin and Lovinger, 2006; Kreitzer and Malenka, 2007; Shen et al., 2008). Consistent with the observation that D2R activation increases striatal levels of the eCB anandamide (AEA) in vivo (Giuffrida et al., 1999), it was suggested that DA acts via postsynaptic D2Rs by promoting the production of eCBs. However, like CB1Rs, D2Rs are coupled to Gi/o-proteins and their activation leads to the inhibition of adenylyl cyclase (AC), resulting in a reduction of cAMP levels and protein kinase A (PKA) activity. The observation that D2Rs may be present at presynaptic locations in the PFC (Fadda et al., 1984; Seamans et al., 2001) raises the possibility that CB1Rs and D2Rs may co-localize on the same axon terminals and perhaps be co-activated by physiological synaptic activity. It is unknown how co-incident activation of presynaptic CB1Rs and D2Rs in the PFC may affect transmitter release.
Most studies on eCB-mediated plasticity have concentrated on excitatory synapses and the PFC is no exception. Pharmacological activation of CB1Rs suppresses excitatory inputs to layer 5 pyramidal cells in rodent PFC (Auclair et al., 2000; Lafourcade et al., 2007) and repetitive stimulation in PFC can elicit long-term depression of excitatory transmission (E-LTD) (Lafourcade et al., 2007). However, given that a balance of excitation and inhibition shapes neuronal excitability and network firing patterns, knowledge of how eCBs influence inhibitory transmission in the PFC will be necessary to predict the functional impact of eCB signaling in this structure. Here, we investigate eCB signaling at inhibitory synapses in the PFC and demonstrate facilitation of inhibitory LTD (I-LTD) by the dopaminergic system. Our findings suggest a potential synaptic link between eCB and DA function in the PFC. Dysregulation of this crosstalk may play a role in neuropsychiatric disorders like schizophrenia.
MATERIALS AND METHODS
Double CB1R/D2R immunocytochemistry for electron microscopy (EM)
Female mice (n=3, 2 month old, C57 BL/6 strain) were deeply anesthetized with chloral hydrate (400 mg/kg body weight). CB1R −/− knockout mice (n=2; kindly provided by Dr. Giovanni Marsicano, Institute François Magendie, Bordeaux, France) (Marsicano et al., 2002) were used as controls of CB1R antibody specificity. All animals were transcardially perfused with phosphate-buffered solution (PB 0.1M, pH 7.4) and then fixed by 500 ml of 0.1% glutaraldehyde, 4% formaldehyde (freshly depolymerized from paraformaldehyde) and 0.2% picric acid in 0.1M PB (pH 7.4) prepared at 4°C. Tissue blocks were extensively rinsed in 0.1M PB (pH 7.4). Rostral vibrosections containing the PFC were cut at 50 μm and collected in 0.1M PB (pH 7.4) at room temperature (RT). Sections were preincubated in a blocking solution of 10% bovine serum albumin (BSA), 0.1% sodium azide and 0.02% saponin prepared in Tris-HCl buffered saline (TBS, pH 7.4) for 30 minutes at RT.
A preembedding silver-intensified immunogold method and an immunoperoxidase method were used for the co-localization of CB1R and D2R in mouse PFC sections. Two primary polyclonal antibodies were used in this study: goat CB1R raised against a 31 amino acid C-terminal sequence (NM007726) of the mouse CB1R (CB1-Go-Af450-1; Frontier Science Co. Ltd; 1-777-12, Shinko-nishi, Ishikari, Hokkaido, Japan), and rabbit D2R raised against a 28 amino acid sequence (aa 284-311) within the third cytoplasmic loop of the human D2R, which is common to both long and short forms of the receptor (AB5084P; Chemicon, Millipore, Temecula, CA, USA). The CB1R (2 μg/ml) and D2R (5 μg/ml) antibodies prepared in 10% BSA/TBS containing 0.1% sodium azide and 0.004% saponin were incubated simultaneously in PFC tissue on a shaker for 2 days at 4°C.
After several washes in 1% BSA/TBS, sections were incubated in a secondary 1.4 nm gold-labeled rabbit anti-goat IgG (Fab′ fragment, 1:100, Nanoprobes Inc., Yaphank, NY, USA) for the detection of CB1R, and in a biotinylated donkey anti-rabbit IgG (1:200, Jackson ImmunoResearch Inc., Baltimore, PA, USA) for the detection of D2R, both in 1% BSA/TBS with 0.004% saponin on a shaker for 4 hours at RT. PFC tissue was washed in 1% BSA/TBS and processed by a conventional avidin-biotin horseradish peroxidase complex method (ABC; Elite, Vector Laboratories Burlingame, CA, USA). The sections were washed in 1% BSA/TBS overnight at 4°C and postfixed in 1% glutaraldehyde in TBS for 10 minutes at RT. Following washes in double-distilled water, gold particles were silver-intensified with an HQ Silver kit (Nanoprobes Inc., Yaphank, NY, USA) for about 12 minutes in the dark and then washed in 0.1M PB (pH 7.4).
The tissue was preincubated subsequently with 0.05% DAB in 0.1M PB for 5 minutes, incubated by adding 0.01% hydrogen peroxide to the same solution for 5 minutes and washed in 0.1M PB for 2 hours at RT. Stained sections were osmicated (1% OsO4 in 0.1M PB, pH 7.4, 20 minutes), dehydrated in graded alcohols to propylene oxide and plastic-embedded flat in Epon 812. Ultrathin sections (80 nm) were collected on mesh nickel grids, stained with lead citrate, and examined in a PHILIPS EM2008S electron microscope. Tissue preparations were photographed by using a digital camera coupled to the electron microscope.
Specificity of the immunostainings was assessed as follows: CB1R antibodies were incubated in CB1R−/− PFC tissue in the same conditions as above. In this case, CB1R immunolabeling was not observed in cortical synapses of CB1R deficient mice (not shown). The D2R antibodies gave a similar faint cortical pattern as described in the light microscopy in previous publications using the same antiserum (Wang and Pickel, 2002; Lei et al., 2004). In addition, no specific staining was detected in sections in which the primary D2R antibodies were replaced by goat serum (not shown). Figure compositions were scanned at 300 dots per inch (dpi). Labeling and minor adjustments in contrast and brightness were made using Adobe Photoshop (CS, Adobe Systems, San Jose, CA, USA).
Quantitative analysis of electron microscopy images
The quantitative analysis was made in layer 5/6 of PFC sections obtained from 3 mice processed for CB1R/D2R co-localization with preembedding immunocytochemistry. 50-μm-thick PFC sections from each animal showing good and reproducible DAB immunoreaction (D2R) and silver-intensified gold particles (CB1R) were cut at 80 nm. Electron micrographs (18,000–28,000X) were taken from grids (132 μm side) containing DAB immunodeposits; all ultrathin sections showed a similar DAB labeling intensity indicating that selected areas were at the same depth. Positive CB1R labeling was determined by observation of at least one immunoparticle within approximately 30 nm of the plasmalemma of axon terminals forming symmetrical-type synapses. Presynaptic boutons with D2R immunodeposits making symmetrical synapses were also counted. Percentages of presynaptic inhibitory-like profiles positive for CB1R or D2R were analyzed and displayed using a statistical software package (GraphPad Prism 4, GraphPad Software Inc, San Diego, USA). Finally, the percentage of CB1R positive terminals at symmetrical synapses that co-localize with D2R as well as the percentage of immunoreactive D2R axonal boutons with CB1R immunolabeling that form symmetrical synapses were also analyzed. Data are presented as mean ± S.E.M. A total of 250 inhibitory-like presynaptic axon terminals in an area of 642.492 μm2 were measured and analyzed by Image-J (version 1.36).
Electrophysiological Recordings
All animal experiments were carried out in accordance with the National Institutes of Health guide for the care and use of laboratory animals. Acute prefrontal cortical slices were prepared from postnatal day (P) 16–25 Wistar rats and P24-46 C57 BL/6 mice from Charles River Laboratories (Wilmington, MA, USA) or from our colony of Zimmer line CB1R−/− mice (Zimmer et al., 1999). The CB1R−/− mouse colony was maintained as heterozygotes and genotyped before use as described previously (Takahashi and Castillo, 2006). Male and female animals were anesthetized with isofluorane, decapitated and their brains removed to a chilled cutting solution consisting of (in mM): 215 sucrose, 2.5 KCl, 26 NaHCO3, 1.6 NaH2PO4, 1 CaCl2, 4 MgCl2, 4 MgSO4 and 20 glucose. The posterior end of the brain was truncated flat with a razor blade and then the brain was positioned in the cutting chamber with the cut end on the bottom. Coronal slices (350μm thick) were cut with a DTK-2000 (Dosaka EM Co., Ltd., Japan) or Leica VT1200 S (Leica Microsystems, Germany) vibrating microslicer. Rostral sections containing the prelimbic-infralimbic region of the PFC were stored at room temperature in holding solution containing (in mM) 63 NaCl, 2.5 KCl, 22 NaHCO3, 1.4 NaH2PO4, 1.1 CaCl2, 3.2 MgCl2, 2 MgSO4, 15.5 glucose and 107.5 sucrose. Thirty minutes post sectioning, slices were placed in extracellular recording solution containing: 126 NaCl, 2.5 KCl, 18 NaHCO3, 1.2 NaH2PO4, 1.2 CaCl2, 2.4 MgCl2 and 11 glucose. All solutions were saturated with 95% O2 and 5% CO2 (pH 7.4). Slices were incubated for at least 30 minutes in the recording solution prior to experiments.
All experiments were conducted in a submersion-type recording chamber perfused at ~2 ml/min and the recording temperature was maintained at 30 ± 1°C using a TC-344B dual channel heater controller (Warner Instruments, Hamden, CT, USA). Whole-cell patch clamp experiments were performed in 10 μM NBQX or CNQX (AMPAR/KAR antagonists) and 25 μM APV (NMDAR antagonist). In recordings of miniature IPSCs (mIPSCs), 1 μM tetrodotoxin was additionally applied in the bath. To record GABAAR-mediated IPSCs, layer 5 pyramidal cells were voltage clamped at 0 mV using patch-type pipettes filled with intracellular solution containing (in mM): 131 Cs-Gluconate, 8 NaCl, 1 CaCl2, 10 EGTA, 10 glucose, 10 HEPES, 5 MgATP, 0.4 Na3GTP; pH = 7.2, 285 mmol/kg. In experiments with postsynaptic calcium chelators, 20 mM Cs and 10 mM EGTA was replaced with 20 mM BAPTA. In some experiments, we assessed the electrophysiological properties of the patched pyramidal cells using a K-based internal solution containing (in mM): 130 K-Gluconate, 10 NaCl, 0.2 EGTA, 10 HEPES, 5 MgATP, 0.4 Na3GTP; pH = 7.2, 285 mmol/kg. Series resistance (typically 5–15 MΩ) was monitored throughout the experiment with a −4 mV, 80 ms voltage step, and recordings with a greater than 10 % change in series resistance were excluded from analysis.
Stimulating electrodes were filled with extracellular recording solution and placed in layer 2/3 to stimulate distal inhibitory inputs and layer 5 to stimulate proximal inhibitory inputs. Patched cells were typically no more than 50 μm lateral from the tips of stimulating electrodes. Each pathway was stimulated every 20 s and synaptic responses typically ranged from 200 to 800 pA. I-LTD in the rat was triggered in layer 2/3 inputs by a 5 Hz-train of synaptic stimulation lasting 10 minutes, at twice the test stimulus intensity. In the mouse PFC, a shorter 5 Hz-train (5 minutes) was required to induce I-LTD. Stimulation was triggered by a Master-8 pulse generator (A.M.P.I., Jerusalem, Israel) and acquisition was controlled by custom written software in Igor Pro 4.09A (Wavemetrics, Inc., Lake Oswego, OR, USA). Whole-cell patch clamp recordings were performed using a Multiclamp 700B amplifier (Axon Instruments, Union City, CA, USA).
To assess the efficacy of loading PKI 6-22 peptide in the patch pipette to inhibit postsynaptic PKA activity, experiments were performed in the CA1 region to monitor slow after hyperpolarization potential currents (IAHP), which is modulated by PKA activation (Pedarzani and Storm, 1993). Transverse hippocampal slices (400 μm) were prepared from P23-25 Wistar rats. Slow IAHP were elicited by a depolarizing step (between 60 and 70 mV) for 100 ms in CA1 pyramidal cells voltage clamped between −50 to −55 mV with intracellular solution containing (in mM): 120 K-Gluconate, 20 KCl, 15 HEPES, 2 Mg-ATP, 0.2 GTP, 1 MgCl2 and 0.1 EGTA. 50 μM Sp-cAMPS was used to inhibit the slow IAHP component, and the IAHP amplitude was measured 15–20 minutes post Sp-cAMPS application under control conditions or in cells loaded with 2.5 μM or 100 μM PKI 6-22.
Quantitative Analysis of Electrophysiological Data
All values are provided as mean ± s.e.m. The paired-pulse ratio (PPR) was defined as the ratio of the amplitude of the second IPSC over the amplitude of the first IPSC. The magnitude of drug effects was calculated as the percentage change between baseline (averaged responses for the 10 minutes before drug application) and 20–30 minutes after start of WIN or H89 application, 10–15 minutes after start of quinpirole application or 20–30 minutes after the start of bromocriptine application. The magnitude of I-LTD was calculated as the percentage change between baseline and 20–30 minutes after complete delivery of tetanus. Statistical analysis was performed using Student’s independent t-test at the p<0.05 significance level in OriginPro 7.0 software (OriginLab Corporation, Northampton, MA, USA), unless otherwise stated.
Drugs
WIN 55,212-2, (−)-quinpirole hydrochloride, bromocriptine mesylate, (S)-(−)-sulpiride, SCH23390, LY367385, OR 486, and picrotoxin were purchased from Tocris-Cookson Inc. (Ellisville, MO, USA). PKI 6-22 and PKI 14-22 peptides were either obtained from Tocris-Cookson Inc. (Ellisville, MO, USA) or BIOMOL International (Plymouth Meeting, PA, USA). Sp-cAMPS was acquired from BIOMOL International (Plymouth Meeting, PA, USA). D-APV, NBQX, CNQX, MPEP and AM 251 were obtained from Ascent Scientific (North Sumerset, UK). H89 was procured from LC Labs (Woburn, MA, USA). Total DMSO in the bath solution was maintained at 0.1% or below in all experiments.
RESULTS
Anatomical evidence for CB1R and D2R presynaptic co-localization in the mouse PFC
We first assessed anatomically whether CB1Rs and D2Rs co-localize at inhibitory synaptic terminals in the PFC. Double immuno-electron microscopy was used to precisely determine the localization of CB1Rs and D2Rs in layers 2/3 (Fig. 1A,B) and 5/6 (Fig. 1C–F) of the mouse PFC. Immunoperoxidase labeling of D2Rs (thick arrows) and immunogold labeling of CB1Rs (thin arrows) was frequently observed within axonal profiles in both layers (Fig. 1A–F), which formed symmetrical synapses onto small postsynaptic dendrites and somatic membranes. CB1R immunoparticles were consistently associated with portions of presynaptic membranes away from the active zones, as expected for its known perisynaptic localization. To assess the degree of co-localization between CB1Rs and D2Rs, quantitative measurements were performed in layer 5/6 of PFC sections (Fig. 1G). Notably, about 50% of relatively small presynaptic boutons with restricted immunoreaction for the D2R co-localized with CB1R immunoparticles along their membranes. A similarly high proportion of inhibitory presynaptic boutons with CB1R immunolabeling also contained D2R immunoreactivity. Although this number is probably an underestimation, about 30% of presynaptic boutons with pleomorphic synaptic vesicles and symmetrical synapses were immunonegative for both CB1R and D2R in layers 5/6. The high number of symmetrical synapses expressing either CB1R or D2R suggests that eCBs and DA play significant roles in modulating inhibitory transmission in the PFC. The superficial layer 2/3 also exhibited the same pattern of CB1R/D2R co-localization in presynaptic terminals impinging symmetrically on postsynaptic membranes of cell bodies and small diameter dendrites (Fig. 1A, B). Thus, our EM images reveal a strong pattern of co-localization between CB1Rs and D2Rs at terminals of symmetrical, presumably inhibitory, synapses in both layers 2/3 and 5/6 of the mouse PFC.
Figure 1. Co-localization of presynaptic CB1R and D2R at symmetrical synapses in the mouse PFC.

Double labeling of receptors by combining a pre-embedding immunogold (CB1R) and an immunoperoxidase (D2R) method for electron microscopy (A–F). D2R immunoreactive (thick arrows) presynaptic axon terminals (ter) forming symmetrical synapses (arrowheads) with small dendrites (den) and cell bodies (soma) co-localize with CB1R immunoparticles (thin arrows) at their perisynaptic and extrasynaptic membranes. Note in A, CB1R immunolabeling (thin arrows) in a D2R immunonegative synaptic bouton making a somatic symmetrical synapse (arrowheads). Scale bars: 0.5 μm. (G) Left, percentages (mean ± standard error, S.E.M.) of the total number of symmetrical synapses analyzed (n=280) containing CB1R alone, D2R alone, both CB1R+D2R and neither CB1R nor D2R in layers 5/6 of the mouse PFC. Right, about 50% of CB1R immunopositive inhibitory-like synaptic terminals have D2R immunoreactivity and 50% of D2R immunoreactive symmetrical boutons are also equipped with CB1R immunometals.
Pharmacological activation of CB1Rs or D2Rs suppresses GABAergic transmission in mouse and rat PFC, most likely via a presynaptic mechanism
Because both CB1Rs and D2Rs are present at terminals of symmetrical presumably GABAergic synapses, we next tested whether activation of these receptors has any functional effect on GABAergic synaptic transmission. Whole-cell recordings of pharmacologically isolated IPSCs were obtained from layer 5 pyramidal cells in acute mouse PFC slices. The identity of pyramidal cells was confirmed by assessment of electrophysiological properties and pyramidal cell-like morphology under infrared-differential interference contrast (IR-DIC) microscopy (Fig. 2A). Exogenous bath application of the CB1R agonist WIN 55,212-2 (5 μM) suppressed IPSCs evoked by focal stimulation via electrodes placed distally in layer 2/3 as well as proximally in layer 5 (Fig. 2B). IPSC amplitudes following WIN application were 49.4 ± 7.9 % and 56.3 ± 9.9 % of baseline in layer 2/3 and layer 5, respectively (n=4). Consistent with a reduction in GABA release, paired-pulse ratio (PPR) increased from 0.83 ± 0.03 to 0.94 ± 0.04 in layer 2/3 (p<0.05) and from 0.84 ± 0.03 to 0.92 ± 0.04 in layer 5 (p<0.05). Similarly, activation of D2Rs by the agonist quinpirole (1 μM) also depressed IPSCs evoked in both layers (Fig. 2C). IPSC amplitudes following quinpirole application were 61.3 ± 8.6 % and 59.3 ± 8.9 % of baseline in layer 2/3 and layer 5, respectively (n=6). This reduction was associated with a change in PPR in both layers (layer 2/3: from 0.84 ± 0.06 to 0.94 ± 0.05; layer 5: from 0.76 ± 0.03 to 0.93 ± 0.06; p<0.05). Thus, activation of CB1Rs or D2Rs decreases GABAergic transmission in the mouse PFC, most likely via a reduction in GABA release.
Figure 2. Pharmacological activation of CB1Rs or D2Rs suppresses GABAergic responses evoked by stimulation in layers (L) 2/3 and 5 in the mouse PFC.
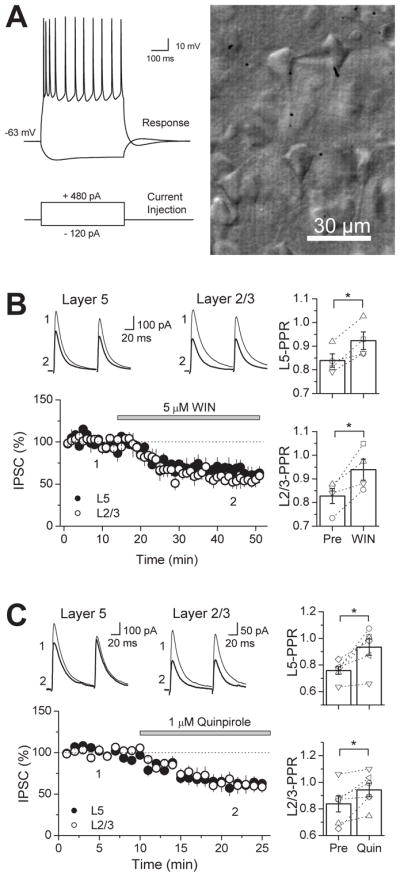
(A) Left, representative response of whole-cell patched L5 pyramidal cell in current clamp (above) while depolarizing or hyperpolarizing current injections were delivered (below) to show that targeting cells in L5 by their shape is a reliable way of patching pyramidal cells. Note the initial spike-adaption, followed by a slow regular spiking behavior. Right, photograph of image under IR-DIC microscopy of a patched cell in L5 of the PFC exhibiting a pyramidal cell-like morphology. (B) Time course of WIN 55,212-2 suppression on both proximal (black circles) and distal (white circles) GABAergic inputs in the mouse PFC (n=4 cells). Top, representative average IPSC traces from a single experiment, obtained at the time points in the time-course graph. Right, bar graph of the average PPR before and after WIN application. Open symbols represent the PPR of individual experiments. (c) Time course of quinpirole suppression on both proximal (black circles) and distal (white circles) GABAergic inputs in the mouse PFC (n=6 cells). Top, representative average IPSC traces from an individual experiment, obtained at the time points indicated. Right, bar graph of the average PPR before and after quinpirole application. Open symbols represent the PPR of individual experiments. * p<0.05.
WIN and quinpirole inhibition of GABAergic transmission is not exclusive to the mouse PFC. In rat PFC slices, IPSCs evoked by stimulating in layer 2/3 and recording from layer 5 pyramidal cells were also suppressed by 5 μM WIN to 41.4 ± 4.5 % of baseline (n=5) after WIN application and PPR increased from 0.77 ± 0.09 to 0.89 ± 0.09 (p<0.01, paired t-test) (Fig. 3A). The suppression was indeed mediated by CB1Rs as preincubation with the antagonist AM 251 (4 μM) abolished the WIN effect (91.8 ± 3.9 % of baseline; n=5, p<0.005). GABAergic inputs in the rat were also sensitive to quinpirole as bath application of this D2R agonist reduced IPSCs to 66.9 ± 4.6 % of baseline (n=8), an effect that is accompanied by a change in PPR from 0.71 ± 0.07 to 0.81 ± 0.05 (p<0.05) (Fig. 3B). Preincubation with the D2R antagonist sulpiride (10 μM) blocked quinpirole-induced suppression of GABAergic transmission (99.0 ± 3.8 % of baseline, n=6, p<0.005), supporting a role for D2Rs in this quinpirole effect. Moreover, a different and selective D2R agonist bromocriptine (2 μM) also suppressed IPSCs, reducing the amplitude to 72.5 ± 4.2 % of baseline (n=6; p<0.005) along with a change in PPR from 0.85 ± 0.04 to 0.99 ± 0.07 (p<0.05; paired t-test). Sulpiride also blocked the suppression by bromocriptine (102.5 ± 3.6 % of baseline, n=6, p<0.005). In the striatum, D2R activation may lead to eCB mobilization (Giuffrida et al., 1999; Yin and Lovinger, 2006), leading to a reduction in neurotransmitter release via CB1R activation (Yin and Lovinger, 2006). To explore this possibility in the PFC, we examined the role of CB1Rs in quinpirole-suppression of IPSCs; however, AM 251 did not block suppression by quinpirole (60.2 ± 9.5 % of baseline, n=5, p<0.05). Taken together with the EM data showing presynaptic localization of CB1Rs and D2Rs, the PPR changes following WIN or quinpirole application support a functional role for CB1Rs and D2Rs in modulating GABA release in both mouse and rat PFC.
Figure 3. Activation of CB1R or D2R suppresses GABAergic transmission in the rat PFC.

(A) Time course of WIN-suppression of IPSC amplitudes in the rat PFC (black circles) which is abolished in CB1R blockade with 4 μM AM 251 (white circles). Top left, representative average IPSC traces from a single control experiment, obtained at time points indicated. Top right, PPR plot before and after WIN application. PPR of L2/3-IPSCs significantly increased. Open symbols represent the PPR of individual experiments. (B) Time course of quinpirole-suppression of IPSC amplitudes in the rat PFC in control (black circles), under D2R antagonism with 10 μM sulpiride (white circles) and under CB1R block with 4 μM AM 251 (gray circles). Top left, representative average IPSC traces from a single control experiment. Top right, PPR plot before and after quinpirole application. PPR of L2/3-IPSCs significantly increased under control conditions and in AM 251. Open symbols represent the PPR of individual experiments. Bottom right, summary plot of suppression mediated by D2R agonists quinpirole (1 μM) and bromocriptine (2 μM). Effects of both drugs were blocked by sulpiride. However, AM 251 had no effect on quinprole-suppression. The number of experiments for each condition is indicated in parenthesis. ** p<0.01, *** p<0.005.
Endogenous PKA activity is critical for basal release of GABA and is most likely involved in the suppression of inhibitory transmission by WIN and quinpirole in the PFC
Both CB1Rs and D2Rs are coupled to Gi/o-proteins and their activation leads to inhibition of the AC/cAMP/PKA pathway (Howlett et al., 2004; Neve et al., 2004), which may be responsible for the suppression of IPSCs in the PFC by WIN (Fig. 2B, 3A) and by quinpirole (Fig. 2C, 3B). If this were the case, we expect that directly blocking PKA activity with the inhibitor H89 should also reduce basal GABA release in the PFC. As in the hippocampus (Chevaleyre et al., 2007), bath application of 10 μM H89 resulted in a reduction of IPSCs to layer 5 pyramidal cells (65.0 ± 7.4 % of baseline, n=6), which was accompanied by a change in PPR (from 0.72 ± 0.03 to 0.84 ± 0.03; p<0.05, paired t-test) (Fig. 4A). It is known that inhibition of VGCCs and PKA contribute to CB1R-mediated depression of inhibitory transmission (Chevaleyre et al., 2006; Lovinger, 2008; Heifets and Castillo, 2009; Kano et al., 2009); thus, we examined the contribution of PKA in WIN-suppression in the PFC by measuring spontaneous miniature IPSCs (mIPSCs) to reduce contamination by the VGCC-sensitive component (Fig. 4B). As previously reported (Vaughan et al., 1999; Takahashi and Linden, 2000; Trettel and Levine, 2002; Chevaleyre et al., 2007), WIN (5 μM) reduced mIPSC frequency (from 4.14 ± 0.99 Hz to 2.00 ± 0.41 Hz, n=4, p<0.05, paired t-test), but not amplitude (from 11.3 ± 1.8 pA to 10.5 ± 1.5 pA, p>0.05, paired t-test). Preincubating PFC slices in 10 μM H89 lowered mIPSC frequency (1.62 ± 0.29 Hz, n=5, p<0.05) and occluded further reduction by subsequent WIN application (1.50 ± 0.19 Hz, n=5, p>0.05, paired t-test). The mechanism by which D2Rs suppress GABA release is less clear. One study in the VTA suggests that activation of D2Rs at the presynaptic terminal may suppress inhibitory transmission solely via a decrease in PKA activity (Pan et al., 2008). Another study using two-photon calcium imaging of axonal varicosities of VTA medium spiny neurons also implicate calcium sources in D2R-mediated inhibition (Mizuno et al., 2007). We tested whether quinpirole-suppression is sensitive to PKA inhibition and found that, in slices preincubated in H89, evoked IPSC amplitudes were unchanged following quinpirole application (108.9 ± 9.4 % of baseline, n=4) (Fig. 4C). To confirm that the lack of quinpirole-suppression in H89 indeed resulted from inhibition of PKA activity, we also used the potent cell membrane-permeable PKA inhibitor peptide, PKI 14-22, and found similar block of the quinpirole effect (97.4 ± 6.1 % of baseline, n=6, p<0.005). Thus, our results strongly suggest that PKA activity is involved in basal GABA release and by inhibiting this activity, CB1Rs or D2Rs can suppress GABAergic transmission.
Figure 4. Inhibiting PKA activity with H89 reduces GABA release and occludes WIN- and quinpirole-suppression of GABAergic transmission in the rat PFC.

(A) Time course of H89-suppression of IPSC amplitudes in the rat PFC (n = 6 cells). Top left, representative average IPSC traces from a single experiment, obtained at time points indicated. Top right, PPR plot before and after H89 application. PPR of L2/3-IPSCs significantly increased. Open symbols represent the PPR of individual experiments. (B) WIN-suppression of miniature IPSC (mIPSC) frequency in control is absent in slices preincubated in 10 μM H89. Top left, three representative mIPSC traces from a single control experiment before and after WIN application. Top right, three representative mIPSC traces from a single H89 experiment before and after WIN application. (C) Quinpirole-suppression of IPSC amplitudes in control (black circles is absent in slices preincubated in either 10 μM H89 (white circles) or 2.5 μM PKI 14-22 (gray circles). Top, representative average IPSC traces from single experiments. Bottom right, summary plot of quinpirole-suppression in control and block under PKA inhibition by H89 and PKI 14-22. The number of experiments for each condition is indicated in parenthesis. * p<0.05, *** p<0.005.
Pharmacological activation of D2Rs facilitates induction of I-LTD
Our EM images show co-localization of CB1Rs and D2Rs at terminals of symmetrical synapses in the PFC and we have provided functional evidence that independent activation of either receptor suppresses GABA release. Given that both CB1Rs and D2Rs can signal via PKA inhibition (Howlett et al., 2004; Neve et al., 2004), we next wondered whether simultaneous activation of both receptors will reveal an interaction between D2R- and CB1R-mediated effects. To this aim, we applied a low dose of quinpirole (500 nM) in the absence or presence of a submaximal concentration of WIN (50 nM) (Fig. 5A). Interestingly, while by itself, 500 nM quinpirole had very little effect of basal IPSC amplitude (96.2 ± 6.2 % of baseline, n=10), it significantly decreased IPSCs when CB1Rs were co-activated (78.4 ± 10.7 % of baseline, n=8, p<0.05). We next tested whether D2R activation could enhance eCB signaling in the PFC by delivering a 5 Hz-train of synaptic stimulation (see methods) in the presence of 500 nM quinpirole. While on its own, the 5-Hz stimulation was below threshold to trigger I-LTD, it became suprathreshold when D2Rs were activated (Fig. 5B). IPSC amplitudes post train were 97.6 ± 11.0 % of baseline (n=5) and 60.6 ± 3.3 % of baseline (n=14) in the absence and presence of quinpirole, respectively (p<0.005). Consistent with a role for D2Rs, I-LTD was abolished in the presence of sulpiride (98.6 ± 2.2 % of baseline, n=4, p<0.01) (Fig. 5C). Importantly, I-LTD was dependent on CB1R activation as it was blocked in AM 251 (99.6 ± 11.9 % of baseline, n=5, p<0.01) (Fig. 5D) and was occluded by preincubation with 5 μM WIN (99.1 ± 7.6 % of baseline, n=6, p<0.01) (Fig. 5E).
Figure 5. I-LTD can be triggered by a 5 Hz-train of synaptic stimulation in the presence of low dose quinpirole.

(A) Time course of 500 nM quinpirole effect on IPSC amplitude in the presence (black circles) or absence (white circles) of 50 nM WIN. Preincubation with a submaximal dose of WIN (+WIN) reveals the suppressive effect of an otherwise ineffective dose of quinpirole (control). Top, representative average IPSC traces from single experiments obtained from the time points indicated. (B) Time course plot of average IPSC amplitudes before and after 5 Hz stimulation train in the absence (white circles) or presence (black circles) of quinpirole. Responses only persistently depressed as a result of 5-Hz stimulation when D2R agonist is in the bath (+Quin). Top left, Representative average IPSC traces from single experiments obtained from the time points indicated. Top right, plot of PPR before and after train in the presence of D2R agonist. Open symbols represent single experiments. (C) Time course of control I-LTD (black circles) and the absence of I-LTD under D2R antagonism with 10 μM sulpiride (white circles). Top, representative average IPSC traces from a single control and sulpiride block experiment. (D) Time course of control I-LTD (black circles) and the absence of I-LTD in the presence of 4 μM AM251 (white circles). Top, Representative average IPSC traces from a single control and AM 251 block experiment. (E) Time course of control I-LTD (black circles) and its absence in slices that were preincubated in 5 μM WIN (white circles). Maximal CB1R activation occluded I-LTD as depicted in the summary bar plot on the top right. Top, Representative average IPSC traces from a single control and WIN occlusion experiment. (F) Time course of control I-LTD (black circles) and the lack of I-LTD in the presence of group I mGluR antagonists (white circles). Top, Representative average IPSC traces from single control and block experiments. * p<0.05.
In many brain regions, eCB mobilization in LTD is triggered by activation of group I mGluRs (Chevaleyre et al., 2006; Heifets and Castillo, 2009; Kano et al., 2009). Given evidence of the expression of these receptors in cell bodies and dendritic bundles of PFC pyramidal cells in layer 5 (Defagot et al., 2002; Lafourcade et al., 2007), we next examined the involvement of group I mGluRs in I-LTD. Indeed, I-LTD was abolished by the group I mGluR antagonists LY367385 and MPEP (98.9 ± 8.9 % of baseline, n=6; p<0.005), whereas interleaved I-LTD controls were normal (55.7 ± 5.2 % of baseline, n=6) (Fig. 5F). Thus, despite the need for quinpirole, I-LTD in the PFC is similar to the more classical form of eCB-LTD in other brain regions in that its induction requires group I mGluR activation.
DA facilitation of I-LTD most likely occurs via synergism between presynaptic D2R and CB1R signaling
D2Rs may act at several sites to facilitate I-LTD. We investigated the different possibilities in the following experiments. First, we tested whether D2Rs postsynaptically enhance eCB production and release in the PFC as has been suggested in the striatum and VTA (Giuffrida et al., 1999; Melis et al., 2004; Yin and Lovinger, 2006). Given that the AC/cAMP/PKA pathway is a principal effector of D2R-mediated effects (Neve et al., 2004), we blocked PKA in the entire slice by bath application of H89 (10 μM) or PKI 14-22 (2.5 μM) and compared this effect to inhibiting PKA specifically in the postsynaptic cell (Fig. 6A). Consistent with a presynaptic site of action by D2Rs, bath application of H89 or PKI 14-22 abolished I-LTD (92.6 ± 4.9 % of baseline, n=6, p<0.01; and 97.0 ± 14.3 % of baseline, n=6, p<0.01, respectively), whereas loading the membrane-impermeable PKI 6-22 peptide (2.5 μM or 100 μM) into the patch pipette did not prevent I-LTD (57.9 ± 5.6 % of baseline, n=4; and 53.8 ± 5.1 % of baseline, n=5, respectively). We verified that this loading procedure could reliably inhibit postsynaptic PKA by assaying the ability of the PKI peptide to reduce the PKA-dependent inhibition of the slow AHP current (IAHP) in CA1 pyramidal cells. Interestingly, loading PKI 6-22 at 2.5 μM or 100 μM were equally effective at decreasing the inhibition of slow IAHP induced by bath application of the PKA activator Sp-cAMPS (Fig. 6B). Hence, while I-LTD in the PFC is dependent on PKA activity, it appears that postsynaptic PKA does not play a significant role.
Figure 6. Neither postsynaptic PKA nor intracellular calcium rise is required for I-LTD.
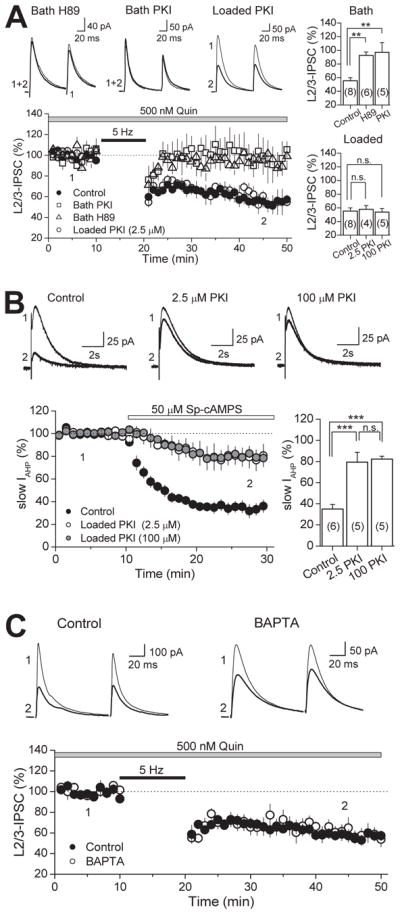
(A) Time course of I-LTD in interleaved controls (black circles), bath applied H89 (10 μM; white triangles), bath applied PKI 14-22 peptide (2.5 μM; white squares) and in cells loaded with 2.5μM PKI 6-22 peptide (white circles). Inhibiting PKA activity in the slice blocks I-LTD, but inclusion of PKI (at either 2.5 μM or 100 μM) in the postsynaptic cell does not, as depicted in the summary bar plots on the top and bottom right. Top, representative average IPSC traces from single H89 and PKI experiments obtained from time points indicated. (B) Verification that intracellular loading of the PKI 6-22 peptide effectively inhibits postsynaptic PKA activity. PKI loaded into CA1 pyramidal cells at either 2.5 μM or 100 μM significantly reduced the inhibition of slow AHP current (IAHP) induced by bath application of a specific PKA activator (Sp-cAMPS). Top, representative average AHP traces from single experiments at time points indicated. Bottom right, summary bar plot depicting the effect of loading PKI on slow IAHP inhibition. (C) Time course of I-LTD in control (black circles) and in cells loaded with 20 mM BAPTA (white circles). Chelating postsynaptic calcium has no effect on I-LTD. Top, Representative average IPSC traces from single experiments. The number of experiments for each condition is indicated in parenthesis. ** p<0.01, *** p<0.005.
Although inhibition of PKA activity is a main target of D2R signaling, alternative molecular players such as intracellular calcium stores have been linked to D2R-mediated effects (Nishi et al., 1997; Hernandez-Lopez et al., 2000). We tested whether intracellular calcium rise is necessary for eCB mobilization in I-LTD by loading the postsynaptic cell with the fast calcium chelator BAPTA (Fig. 6C). In the presence of quinpirole, the 5 Hz-train of synaptic stimulation still led to a long-lasting depression of IPSCs in the PFC (59.6 ± 6.6 % of baseline, n=5). Thus, similar to the hippocampus (Chevaleyre and Castillo, 2003) and amygdala (Azad et al., 2004), I-LTD in the PFC does not require postsynaptic calcium increases, further bolstering the idea that postsynaptic D2Rs are not involved. Our findings that D2Rs also localize to presynaptic inhibitory-like terminals, and that activation of these receptors may reduce GABA release via PKA inhibition, make presynaptic D2Rs the most likely mediators of I-LTD facilitation.
In the hippocampus, the level of spontaneous GABAergic activity can determine whether I-LTD occurs (Heifets et al., 2008). It was shown that I-LTD requires calcium increases at GABAergic terminals during the induction step, via the recruitment of calcium-dependent protein kinases and phosphatases. In the PFC, activation of D2Rs may increase interneuronal excitability and thus spontaneous inhibitory activity (Retaux et al., 1991; Grobin and Deutch, 1998). Although this effect is not seen by all (Gorelova and Yang, 2000; Seamans et al., 2001; Tseng and O’Donnell, 2007), it is formally possible that quinpirole may indirectly facilitate I-LTD in the PFC by enhancing the activity of interneurons whose terminals express CB1Rs. We examined whether sIPSC frequency increased in our hands following 500 nM quinpirole application but found no difference (20.8 ± 4.0 Hz before versus 17.7 ± 2.8 Hz after; p>0.05; n=9). Thus, an increase in spontaneous inhibitory activity cannot account for the facilitatory effect of quinpirole on eCB signaling.
Endogenous DA facilitates I-LTD when catechol-O-methyltransferase is inhibited
Thus far, we have activated D2Rs by applying the specific receptor agonist quinpirole. We next sought to determine whether endogenous DA can also facilitate eCB signaling. It is possible that DA deafferentation that is inherent to the slicing procedure prevented the 5 Hz-train from triggering I-LTD in the absence of quinpirole. To compensate for a reduction of DA levels that is highly expected in PFC slices, we pharmacologically blocked DA catabolism and tested whether synaptic stimulation will trigger I-LTD under this recording condition (Fig. 7). The major mammalian enzymes involved in the degradation of DA are catechol-O-methyltransferase (COMT) and monoamine oxidases, with relative contribution tilted towards COMT in the PFC (Karoum et al., 1994; Matsumoto et al., 2003; Yavich et al., 2007). Thus, to effectively increase DA levels in PFC slices, we pharmacologically blocked COMT activity with the inhibitor OR 486 (2 μM). 5 Hz-train failed to trigger I-LTD in the presence of OR 486 alone (100.4 ± 21.1 % of baseline, n=4; Fig. S1). Because DA can activate both D1Rs and D2Rs, with potentially opposing effects (Cepeda et al., 1993; Nishi et al., 1997; Seamans et al., 2001; Trantham-Davidson et al., 2004; Tseng and O’Donnell, 2004; Xu et al., 2009), an effect by D2Rs may potentially be masked by a conflicting D1R effect, depending on which receptor was more strongly activated. Therefore, we next tested I-LTD by co-application of OR 486 with the D1R antagonist SCH23390 (10 μM). Indeed, in the presence of OR 486 and SCH23390, I-LTD could be triggered by the 5 Hz synaptic stimulation (IPSC amplitude: 49.6 ± 4.6 % of baseline, n=10) (Fig. 7A). Consistent with a reduction in probability of GABA release, PPR in I-LTD experiments increased from 0.82 ± 0.03 to 0.92 ± 0.04 (p<0.05; paired t-test). A role for D2Rs was confirmed by the ability of sulpiride to reduce I-LTD in the presence of OR 486 (88.5 ± 8.4 % of baseline, n=8, p<0.005) (Fig. 7A). As was observed for the quinpirole-facilitated form, I-LTD in OR 486 and SCH23390 was also sensitive to CB1R blockade with AM 251 (93.0 ± 9.8 % of baseline, n=6, p<0.01) (Fig. 7B) and was not present in the CB1R knockout mice (KO: 92.2 ± 4.8 % of baseline, n=7, vs. WT: 70.7 ± 3.8 % of baseline, n=7, p<0.005) (Fig. 7C). Consistent with a role for PKA in D2R and CB1R-mediated effects on GABAergic transmission in the PFC, I-LTD in the presence of OR 486 and SCH23390 was reduced in H89 (91.0 ± 5.7 % of baseline, n=6, p<0.005) (Fig. 7D). Thus, taken together, our results strongly suggest that endogenous DA can activate D2Rs in the presynaptic GABAergic terminal to enhance eCB signaling and trigger I-LTD.
Figure 7. Increasing endogenous DA levels by inhibiting COMT also enables I-LTD.

(A) 5 Hz-stimulation in the presence of COMT inhibitor OR 486 and D1R antagonist SCH23390 elicited I-LTD (black circles) that was sensitive to D2R antagonist sulpiride (white circles). Top left, representative average traces from single experiments obtained at time points indicated. Top right, plot of PPR before and after delivery of 5 Hz-train, showing a change in PPR. (B) This I-LTD (black circles) was also sensitive to CB1R antagonist AM 251 (white circles). Top, representative average traces from single experiments obtained at time points indicated. (C) In the wild-type mouse (CB1+/+), a weaker 5 Hz-train (for 5 min) triggers I-LTD in the presence of OR 486 and SCH23390. This I-LTD is not present in the CB1 knockout littermates (CB1−/−). Top, representative average traces from single experiments obtained at time points indicated. (C) Time course of I-LTD in the presence of OR 486 and SCH23390 in control (black circles) and in slices bath applied with H89 (white circles). Top, representative average IPSC traces from single control and H89 block experiments in the rat PFC obtained at the time points indicated.
DISCUSSION
In this study, we combine anatomical and electrophysiological techniques to investigate the cellular mechanisms underlying the link between DA and eCB function in the PFC. Using EM, we first show immunolabeling of D2Rs and CB1Rs at terminals of symmetrical presumably GABAergic synapses in the PFC. Notably, quantitative analysis reveal a robust pattern of CB1R and D2R co-localization at these presynaptic sites. Consistent with this localization, activation of either receptor suppresses GABA release as indicated by an increase in PPR. Importantly, repetitive afferent stimulation in the presence of a D2R agonist induces eCB-dependent I-LTD that is insensitive to interference of postsynaptic D2R signaling. Taken together, our results strongly suggest a synergistic interaction between co-localized D2Rs and CB1Rs in regulating long-term depression of inhibitory transmission in the PFC.
Previous studies have shown that activation of CB1Rs in neocortex (Trettel and Levine, 2002; Bodor et al., 2005; Hill et al., 2007; Galarreta et al., 2008) or D2Rs in prefrontal cortex (Retaux et al., 1991; Seamans et al., 2001) can suppress inhibitory transmission, presumably by reducing GABA release. Our combined anatomical and physiological data support similar roles of CB1Rs and D2Rs on GABAergic transmission in the PFC. Furthermore, in keeping with a recent report in the nucleus accumbens (Pickel et al., 2006), we have found that D2Rs and CB1Rs can co-localize to sites presynaptic to symmetrical synapses in the PFC; this co-localization may provide an anatomical substrate for receptor interaction when co-activated. Co-activation of CB1Rs and D2Rs can have several potential effects on signal transduction of the respective individual receptors. In heterologous systems and striatal cultures, pharmacological co-activation of co-expressed CB1Rs and D2Rs has been shown to alter G-protein signaling, converting Gi/o-mediated pathways to Gs-coupled ones (Glass and Felder, 1997; Kearn et al., 2005), thereby enhancing rather than reducing transmitter release. There is also the possibility of synergistic or antagonistic crosstalk between the receptor systems in the presynapse (Kearn et al., 2005; Marcellino et al., 2008).
We have tested for a potential interaction between the eCB and DA systems in the PFC and found that nominal activation of D2Rs with a low dose of quinpirole can facilitate eCB signaling at GABAergic synapses to trigger I-LTD. D2Rs most likely act by enhancing a process downstream of eCB mobilization from the postsynaptic cell because blocking postsynaptic PKA activity has no effect on I-LTD, while global block of PKA in the slice does. D2Rs may signal via pathways other than PKA. In the striatum, activation of D2Rs has been shown to lead to stimulation of phospholipase Cβ isoforms and mobilization of intracellular calcium stores (Nishi et al., 1997; Hernandez-Lopez et al., 2000). However, two lines of evidence argue against a role for postsynaptic D2Rs in facilitating I-LTD in the PFC. First, quinpirole-suppression of IPSCs is completely insensitive to AM 251, indicating that D2R activation is insufficient to trigger eCB production in the PFC. Second, intracellular calcium rise is not required for I-LTD because loading the postsynaptic cell with BAPTA has no effect on plasticity. Because both CB1R and D2R signaling can lead to inhibition of the PKA pathway, we postulate that co-activation of CB1Rs and D2Rs enables I-LTD in the PFC by cooperatively lowering PKA activity below a threshold level as supported by our observation of an enhancement of quinpirole-mediated suppression by a submaximal concentration of WIN (Fig. 5A). A similar mechanism has recently been suggested for I-LTD in the VTA (Pan et al., 2008). The precise molecular PKA target underlying I-LTD remains to be identified.
It is widely accepted that DA modulates PFC function. Here, we show that DA may influence synaptic transmission and plasticity in the PFC by facilitating eCB signaling via presynaptic D2Rs. Interestingly, our results using the COMT inhibitor OR486 alone, in the absence of the D1R antagonist SCH23390 (Fig. S1), suggest that D1R activation may antagonize D2R-facilitated I-LTD. This raises the question of whether DA likely facilitates I-LTD in the intact brain. However, some evidence suggests that D1Rs and D2Rs may be selectively activated in vivo. D2Rs show comparatively higher affinity to DA than D1Rs (Creese et al., 1983), and thus D2R activation may require lower concentrations of DA. It has been proposed, at least in the nucleus accumbens, that basal DA release continuously activate D2Rs, whereas phasic DA release is needed to elevate DA levels enough to stimulate D1Rs (Grace, 1991). Moreover, tonic versus phasic DA transmission can be differentially evoked by distinct sets of afferents to DA neurons (Goto and Grace, 2005). It is possible that D2Rs may be preferentially activated during tonic DA release conditions in the PFC as well, making GABAergic synapses primed for eCB signaling and I-LTD.
It is unclear how activation of D1Rs opposes PFC I-LTD in our experiments; D1Rs may directly antagonize downstream D2R/CB1R signaling in the GABAergic terminal, and/or mediate I-LTP at the same or different GABAergic synapse. We have shown with EM that a high proportion of presynaptic D2Rs are co-localized with CB1Rs (Fig. 1), but the extent of overlap between presynaptic D1Rs and CB1Rs remains unknown. Furthermore, given the high expression of postsynaptic D1Rs in the PFC (Ariano and Sibley, 1994; Smiley et al., 1994; Lidow et al., 2003), it is possible that these receptors mask I-LTD by mediating a potentiation of GABAergic transmission via an increase in number or conductance of GABAA receptors as has been shown for D1R and AMPAR (Sun et al., 2005).
In the PFC, eCBs have been shown to depress excitatory transmission (Lafourcade et al., 2007) and we now demonstrate that inhibitory transmission is also under eCB regulation. What, then, is the overall effect of eCBs on the balance of excitation and inhibition in the PFC and how is PFC output modulated? The answers to these questions may lie in the differential induction of E-LTD and I-LTD. It is interesting to note that eCB-mediated E-LTD in the PFC can be triggered by a slightly stronger synaptic stimulation train (e.g. 10 Hz versus 5 Hz) and does not require DA transmission. Furthermore, postsynaptic calcium rise is necessary for E-LTD but not I-LTD, suggesting that multiple mechanisms underlying eCB production may exist. Future studies will be needed to examine whether DA can modulate this form of E-LTD. Interestingly, a form of E-LTD, induced by brief high frequency (50 Hz) stimulation, has previously been described in the PFC to depend on DA (Otani et al., 1998); the involvement of eCB signaling in this E-LTD is yet to be determined. Another provocative question is whether synaptic activity can simultaneously trigger both eCB-mediated E-LTD and I-LTD. If so, it will be important to assess changes in the firing properties of the output layer 5 pyramidal cells in PFC under conditions of intact excitation and inhibition.
In humans, cannabis consumption has dramatic effects on PFC-mediated function and can trigger schizophrenia-like states in normal individuals, exacerbate psychotic symptoms in schizophrenic patients and increase the risk of developing schizophrenia in predisposed individuals (Ujike and Morita, 2004; Koethe et al., 2009; Sewell et al., 2009). In addition, studies using CB1R-deficient mice have revealed an important role of eCBs in the extinction of learned behaviors such as fear (Marsicano et al., 2002; Varvel and Lichtman, 2002), which are partly mediated by the PFC (Morgan et al., 1993; Morrow et al., 1999). It is currently unknown how eCBs and cannabinoid agonists can lead to these effects. The involvement of DA in the central actions of cannabinoids has been suggested (van der Stelt and Di Marzo, 2003; Laviolette and Grace, 2006). Cannabinergic signaling may lead to DA release (Cadogan et al., 1997; van der Stelt and Di Marzo, 2003) and can modulate D2R agonist-induced behavior (Beltramo et al., 2000; Gorriti et al., 2005). Indeed, microdialysis measurements have revealed an increase of DA in the PFC following in vivo administration of the cannabinoid Delta(9)-tetrahydrocannabinol (Pistis et al., 2002). Notably, a decrease of GABA in the PFC was also observed in this study. Given that an imbalance of excitatory and inhibitory transmission in the PFC has been proposed to contribute to schizophrenia (Lewis et al., 2005; Gonzalez-Burgos and Lewis, 2008), these changes in neurotransmitter levels provide insight into how schizophrenic symptoms may emerge following cannabis consumption. We propose that a synergic interaction between DA and eCB function at the synaptic level in the PFC (i.e., to trigger I-LTD) may play a role in schizophrenia in predisposed cannabis users.
Supplementary Material
Acknowledgments
We especially thank Dr. Andrés Chávez for his work that forms the basis for Figure 6B, and Drs. Wade Regehr, David Lovinger and Giovanni Marsicano for providing CB1 knockout mice. We also thank all members of the Castillo Lab for critical reading of the manuscript. Funding for P.E. Castillo’s laboratory is provided by NIH/NIDA and by NARSAD. Funding for P. Grandes’ laboratory is provided by The Basque Country Government grant GIC07/70-IT-432-07, and by Red de Trastornos Adictivos, RETICS, Instituto de Salud Carlos III, MICINN, grant number: RD07/0001/2001. N. Puente is supported by The Basque Country University grant for PhD Researcher’s Specialization.
References
- Ariano MA, Sibley DR. Dopamine receptor distribution in the rat CNS: elucidation using anti-peptide antisera directed against D1A and D3 subtypes. Brain Res. 1994;649:95–110. doi: 10.1016/0006-8993(94)91052-9. [DOI] [PubMed] [Google Scholar]
- Auclair N, Otani S, Soubrie P, Crepel F. Cannabinoids modulate synaptic strength and plasticity at glutamatergic synapses of rat prefrontal cortex pyramidal neurons. Journal of Neurophysiology. 2000;83:3287–3293. doi: 10.1152/jn.2000.83.6.3287. [DOI] [PubMed] [Google Scholar]
- Azad SC, Monory K, Marsicano G, Cravatt BF, Lutz B, Zieglgansberger W, Rammes G. Circuitry for associative plasticity in the amygdala involves endocannabinoid signaling. J Neurosci. 2004;24:9953–9961. doi: 10.1523/JNEUROSCI.2134-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltramo M, de Fonseca FR, Navarro M, Calignano A, Gorriti MA, Grammatikopoulos G, Sadile AG, Giuffrida A, Piomelli D. Reversal of dopamine D(2) receptor responses by an anandamide transport inhibitor. J Neurosci. 2000;20:3401–3407. doi: 10.1523/JNEUROSCI.20-09-03401.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodor AL, Katona I, Nyiri G, Mackie K, Ledent C, Hajos N, Freund TF. Endocannabinoid signaling in rat somatosensory cortex: laminar differences and involvement of specific interneuron types. J Neurosci. 2005;25:6845–6856. doi: 10.1523/JNEUROSCI.0442-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadogan AK, Alexander SP, Boyd EA, Kendall DA. Influence of cannabinoids on electrically evoked dopamine release and cyclic AMP generation in the rat striatum. J Neurochem. 1997;69:1131–1137. doi: 10.1046/j.1471-4159.1997.69031131.x. [DOI] [PubMed] [Google Scholar]
- Cepeda C, Buchwald NA, Levine MS. Neuromodulatory actions of dopamine in the neostriatum are dependent upon the excitatory amino acid receptor subtypes activated. Proc Natl Acad Sci U S A. 1993;90:9576–9580. doi: 10.1073/pnas.90.20.9576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevaleyre V, Castillo PE. Heterosynaptic LTD of hippocampal GABAergic synapses: a novel role of endocannabinoids in regulating excitability. Neuron. 2003;38:461–472. doi: 10.1016/s0896-6273(03)00235-6. [DOI] [PubMed] [Google Scholar]
- Chevaleyre V, Takahashi KA, Castillo PE. Endocannabinoid-mediated synaptic plasticity in the CNS. Annu Rev Neurosci. 2006;29:37–76. doi: 10.1146/annurev.neuro.29.051605.112834. [DOI] [PubMed] [Google Scholar]
- Chevaleyre V, Heifets BD, Kaeser PS, Sudhof TC, Castillo PE. Endocannabinoid-mediated long-term plasticity requires cAMP/PKA signaling and RIM1alpha. Neuron. 2007;54:801–812. doi: 10.1016/j.neuron.2007.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creese I, Sibley DR, Hamblin MW, Leff SE. The classification of dopamine receptors: relationship to radioligand binding. Annu Rev Neurosci. 1983;6:43–71. doi: 10.1146/annurev.ne.06.030183.000355. [DOI] [PubMed] [Google Scholar]
- Defagot MC, Villar MJ, Antonelli MC. Differential localization of metabotropic glutamate receptors during postnatal development. Dev Neurosci. 2002;24:272–282. doi: 10.1159/000066741. [DOI] [PubMed] [Google Scholar]
- Fadda F, Gessa GL, Marcou M, Mosca E, Rossetti Z. Evidence for dopamine autoreceptors in mesocortical dopamine neurons. Brain Res. 1984;293:67–72. doi: 10.1016/0006-8993(84)91453-7. [DOI] [PubMed] [Google Scholar]
- Galarreta M, Erdelyi F, Szabo G, Hestrin S. Cannabinoid sensitivity and synaptic properties of 2 GABAergic networks in the neocortex. Cereb Cortex. 2008;18:2296–2305. doi: 10.1093/cercor/bhm253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giuffrida A, Parsons LH, Kerr TM, Rodriguez de Fonseca F, Navarro M, Piomelli D. Dopamine activation of endogenous cannabinoid signaling in dorsal striatum. Nat Neurosci. 1999;2:358–363. doi: 10.1038/7268. [DOI] [PubMed] [Google Scholar]
- Glass M, Felder CC. Concurrent stimulation of cannabinoid CB1 and dopamine D2 receptors augments cAMP accumulation in striatal neurons: evidence for a Gs linkage to the CB1 receptor. J Neurosci. 1997;17:5327–5333. doi: 10.1523/JNEUROSCI.17-14-05327.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman-Rakic PS. The cortical dopamine system: role in memory and cognition. Adv Pharmacol. 1998;42:707–711. doi: 10.1016/s1054-3589(08)60846-7. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Burgos G, Lewis DA. GABA neurons and the mechanisms of network oscillations: implications for understanding cortical dysfunction in schizophrenia. Schizophr Bull. 2008;34:944–961. doi: 10.1093/schbul/sbn070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorelova NA, Yang CR. Dopamine D1/D5 receptor activation modulates a persistent sodium current in rat prefrontal cortical neurons in vitro. J Neurophysiol. 2000;84:75–87. doi: 10.1152/jn.2000.84.1.75. [DOI] [PubMed] [Google Scholar]
- Gorriti MA, Ferrer B, del Arco I, Bermudez-Silva FJ, de Diego Y, Fernandez-Espejo E, Navarro M, Rodriguez de Fonseca F. Acute delta9-tetrahydrocannabinol exposure facilitates quinpirole-induced hyperlocomotion. Pharmacol Biochem Behav. 2005;81:71–77. doi: 10.1016/j.pbb.2005.02.004. [DOI] [PubMed] [Google Scholar]
- Goto Y, Grace AA. Dopaminergic modulation of limbic and cortical drive of nucleus accumbens in goal-directed behavior. Nat Neurosci. 2005;8:805–812. doi: 10.1038/nn1471. [DOI] [PubMed] [Google Scholar]
- Grace AA. Phasic versus tonic dopamine release and the modulation of dopamine system responsivity: a hypothesis for the etiology of schizophrenia. Neuroscience. 1991;41:1–24. doi: 10.1016/0306-4522(91)90196-u. [DOI] [PubMed] [Google Scholar]
- Greengard P. The neurobiology of slow synaptic transmission. Science. 2001;294:1024–1030. doi: 10.1126/science.294.5544.1024. [DOI] [PubMed] [Google Scholar]
- Grobin AC, Deutch AY. Dopaminergic regulation of extracellular gamma-aminobutyric acid levels in the prefrontal cortex of the rat. J Pharmacol Exp Ther. 1998;285:350–357. [PubMed] [Google Scholar]
- Heifets BD, Castillo PE. Endocannabinoid signaling and long-term synaptic plasticity. Annu Rev Physiol. 2009;71:283–306. doi: 10.1146/annurev.physiol.010908.163149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heifets BD, Chevaleyre V, Castillo PE. Interneuron activity controls endocannabinoid-mediated presynaptic plasticity through calcineurin. Proc Natl Acad Sci U S A. 2008;105:10250–10255. doi: 10.1073/pnas.0711880105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez-Lopez S, Tkatch T, Perez-Garci E, Galarraga E, Bargas J, Hamm H, Surmeier DJ. D2 dopamine receptors in striatal medium spiny neurons reduce L-type Ca2+ currents and excitability via a novel PLC[beta]1-IP3-calcineurin-signaling cascade. J Neurosci. 2000;20:8987–8995. doi: 10.1523/JNEUROSCI.20-24-08987.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill EL, Gallopin T, Ferezou I, Cauli B, Rossier J, Schweitzer P, Lambolez B. Functional CB1 receptors are broadly expressed in neocortical GABAergic and glutamatergic neurons. J Neurophysiol. 2007;97:2580–2589. doi: 10.1152/jn.00603.2006. [DOI] [PubMed] [Google Scholar]
- Howlett AC, Breivogel CS, Childers SR, Deadwyler SA, Hampson RE, Porrino LJ. Cannabinoid physiology and pharmacology: 30 years of progress. Neuropharmacology. 2004;47(Suppl 1):345–358. doi: 10.1016/j.neuropharm.2004.07.030. [DOI] [PubMed] [Google Scholar]
- Iversen SD, Iversen LL. Dopamine: 50 years in perspective. Trends Neurosci. 2007;30:188–193. doi: 10.1016/j.tins.2007.03.002. [DOI] [PubMed] [Google Scholar]
- Kano M, Ohno-Shosaku T, Hashimotodani Y, Uchigashima M, Watanabe M. Endocannabinoid-mediated control of synaptic transmission. Physiol Rev. 2009;89:309–380. doi: 10.1152/physrev.00019.2008. [DOI] [PubMed] [Google Scholar]
- Karoum F, Chrapusta SJ, Egan MF. 3-Methoxytyramine is the major metabolite of released dopamine in the rat frontal cortex: reassessment of the effects of antipsychotics on the dynamics of dopamine release and metabolism in the frontal cortex, nucleus accumbens, and striatum by a simple two pool model. J Neurochem. 1994;63:972–979. doi: 10.1046/j.1471-4159.1994.63030972.x. [DOI] [PubMed] [Google Scholar]
- Kearn CS, Blake-Palmer K, Daniel E, Mackie K, Glass M. Concurrent stimulation of cannabinoid CB1 and dopamine D2 receptors enhances heterodimer formation: a mechanism for receptor cross-talk? Mol Pharmacol. 2005;67:1697–1704. doi: 10.1124/mol.104.006882. [DOI] [PubMed] [Google Scholar]
- Koethe D, Hoyer C, Leweke FM. The endocannabinoid system as a target for modelling psychosis. Psychopharmacology (Berl) 2009 doi: 10.1007/s00213-009-1591-7. [DOI] [PubMed] [Google Scholar]
- Kreitzer AC, Malenka RC. Dopamine modulation of state-dependent endocannabinoid release and long-term depression in the striatum. J Neurosci. 2005;25:10537–10545. doi: 10.1523/JNEUROSCI.2959-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, Malenka RC. Endocannabinoid-mediated rescue of striatal LTD and motor deficits in Parkinson’s disease models. Nature. 2007;445:643–647. doi: 10.1038/nature05506. [DOI] [PubMed] [Google Scholar]
- Lafourcade M, Elezgarai I, Mato S, Bakiri Y, Grandes P, Manzoni OJ. Molecular components and functions of the endocannabinoid system in mouse prefrontal cortex. PLoS ONE. 2007;2:e709. doi: 10.1371/journal.pone.0000709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laviolette SR, Grace AA. The roles of cannabinoid and dopamine receptor systems in neural emotional learning circuits: implications for schizophrenia and addiction. Cell Mol Life Sci. 2006;63:1597–1613. doi: 10.1007/s00018-006-6027-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Moal M, Simon H. Mesocorticolimbic dopaminergic network: functional and regulatory roles. Physiol Rev. 1991;71:155–234. doi: 10.1152/physrev.1991.71.1.155. [DOI] [PubMed] [Google Scholar]
- Lei W, Jiao Y, Del Mar N, Reiner A. Evidence for differential cortical input to direct pathway versus indirect pathway striatal projection neurons in rats. J Neurosci. 2004;24:8289–8299. doi: 10.1523/JNEUROSCI.1990-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis DA, Hashimoto T, Volk DW. Cortical inhibitory neurons and schizophrenia. Nat Rev Neurosci. 2005;6:312–324. doi: 10.1038/nrn1648. [DOI] [PubMed] [Google Scholar]
- Lidow MS, Koh PO, Arnsten AF. D1 dopamine receptors in the mouse prefrontal cortex: Immunocytochemical and cognitive neuropharmacological analyses. Synapse. 2003;47:101–108. doi: 10.1002/syn.10143. [DOI] [PubMed] [Google Scholar]
- Lovinger DM. Presynaptic modulation by endocannabinoids. Handb Exp Pharmacol. 2008:435–477. doi: 10.1007/978-3-540-74805-2_14. [DOI] [PubMed] [Google Scholar]
- Marcellino D, Carriba P, Filip M, Borgkvist A, Frankowska M, Bellido I, Tanganelli S, Muller CE, Fisone G, Lluis C, Agnati LF, Franco R, Fuxe K. Antagonistic cannabinoid CB1/dopamine D2 receptor interactions in striatal CB1/D2 heteromers. A combined neurochemical and behavioral analysis. Neuropharmacology. 2008;54:815–823. doi: 10.1016/j.neuropharm.2007.12.011. [DOI] [PubMed] [Google Scholar]
- Marsicano G, Wotjak CT, Azad SC, Bisogno T, Rammes G, Cascio MG, Hermann H, Tang J, Hofmann C, Zieglgansberger W, Di Marzo V, Lutz B. The endogenous cannabinoid system controls extinction of aversive memories. Nature. 2002;418:530–534. doi: 10.1038/nature00839. [DOI] [PubMed] [Google Scholar]
- Matsumoto M, Weickert CS, Akil M, Lipska BK, Hyde TM, Herman MM, Kleinman JE, Weinberger DR. Catechol O-methyltransferase mRNA expression in human and rat brain: evidence for a role in cortical neuronal function. Neuroscience. 2003;116:127–137. doi: 10.1016/s0306-4522(02)00556-0. [DOI] [PubMed] [Google Scholar]
- Melis M, Pistis M, Perra S, Muntoni AL, Pillolla G, Gessa GL. Endocannabinoids mediate presynaptic inhibition of glutamatergic transmission in rat ventral tegmental area dopamine neurons through activation of CB1 receptors. J Neurosci. 2004;24:53–62. doi: 10.1523/JNEUROSCI.4503-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno T, Schmauss C, Rayport S. Distinct roles of presynaptic dopamine receptors in the differential modulation of the intrinsic synapses of medium-spiny neurons in the nucleus accumbens. BMC Neurosci. 2007;8:8. doi: 10.1186/1471-2202-8-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan MA, Romanski LM, LeDoux JE. Extinction of emotional learning: contribution of medial prefrontal cortex. Neurosci Lett. 1993;163:109–113. doi: 10.1016/0304-3940(93)90241-c. [DOI] [PubMed] [Google Scholar]
- Morrow BA, Elsworth JD, Inglis FM, Roth RH. An antisense oligonucleotide reverses the footshock-induced expression of fos in the rat medial prefrontal cortex and the subsequent expression of conditioned fear-induced immobility. J Neurosci. 1999;19:5666–5673. doi: 10.1523/JNEUROSCI.19-13-05666.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neve KA, Seamans JK, Trantham-Davidson H. Dopamine receptor signaling. J Recept Signal Transduct Res. 2004;24:165–205. doi: 10.1081/rrs-200029981. [DOI] [PubMed] [Google Scholar]
- Nishi A, Snyder GL, Greengard P. Bidirectional regulation of DARPP-32 phosphorylation by dopamine. J Neurosci. 1997;17:8147–8155. doi: 10.1523/JNEUROSCI.17-21-08147.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otani S, Blond O, Desce JM, Crepel F. Dopamine facilitates long-term depression of glutamatergic transmission in rat prefrontal cortex. Neuroscience. 1998;85:669–676. doi: 10.1016/s0306-4522(97)00677-5. [DOI] [PubMed] [Google Scholar]
- Pan B, Hillard CJ, Liu QS. D2 dopamine receptor activation facilitates endocannabinoid-mediated long-term synaptic depression of GABAergic synaptic transmission in midbrain dopamine neurons via cAMP-protein kinase A signaling. J Neurosci. 2008;28:14018–14030. doi: 10.1523/JNEUROSCI.4035-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedarzani P, Storm JF. PKA mediates the effects of monoamine transmitters on the K+ current underlying the slow spike frequency adaptation in hippocampal neurons. Neuron. 1993;11:1023–1035. doi: 10.1016/0896-6273(93)90216-e. [DOI] [PubMed] [Google Scholar]
- Pickel VM, Chan J, Kearn CS, Mackie K. Targeting dopamine D2 and cannabinoid-1 (CB1) receptors in rat nucleus accumbens. J Comp Neurol. 2006;495:299–313. doi: 10.1002/cne.20881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pistis M, Ferraro L, Pira L, Flore G, Tanganelli S, Gessa GL, Devoto P. Delta(9)-tetrahydrocannabinol decreases extracellular GABA and increases extracellular glutamate and dopamine levels in the rat prefrontal cortex: an in vivo microdialysis study. Brain Res. 2002;948:155–158. doi: 10.1016/s0006-8993(02)03055-x. [DOI] [PubMed] [Google Scholar]
- Retaux S, Besson MJ, Penit-Soria J. Opposing effects of dopamine D2 receptor stimulation on the spontaneous and the electrically evoked release of [3H]GABA on rat prefrontal cortex slices. Neuroscience. 1991;42:61–71. doi: 10.1016/0306-4522(91)90150-m. [DOI] [PubMed] [Google Scholar]
- Seamans JK, Yang CR. The principal features and mechanisms of dopamine modulation in the prefrontal cortex. Prog Neurobiol. 2004;74:1–58. doi: 10.1016/j.pneurobio.2004.05.006. [DOI] [PubMed] [Google Scholar]
- Seamans JK, Gorelova N, Durstewitz D, Yang CR. Bidirectional dopamine modulation of GABAergic inhibition in prefrontal cortical pyramidal neurons. J Neurosci. 2001;21:3628–3638. doi: 10.1523/JNEUROSCI.21-10-03628.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sewell RA, Ranganathan M, D’Souza DC. Cannabinoids and psychosis. Int Rev Psychiatry. 2009;21:152–162. doi: 10.1080/09540260902782802. [DOI] [PubMed] [Google Scholar]
- Shen W, Flajolet M, Greengard P, Surmeier DJ. Dichotomous dopaminergic control of striatal synaptic plasticity. Science. 2008;321:848–851. doi: 10.1126/science.1160575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smiley JF, Levey AI, Ciliax BJ, Goldman-Rakic PS. D1 dopamine receptor immunoreactivity in human and monkey cerebral cortex: predominant and extrasynaptic localization in dendritic spines. Proc Natl Acad Sci U S A. 1994;91:5720–5724. doi: 10.1073/pnas.91.12.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Zhao Y, Wolf ME. Dopamine receptor stimulation modulates AMPA receptor synaptic insertion in prefrontal cortex neurons. J Neurosci. 2005;25:7342–7351. doi: 10.1523/JNEUROSCI.4603-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi KA, Linden DJ. Cannabinoid receptor modulation of synapses received by cerebellar Purkinje cells. J Neurophysiol. 2000;83:1167–1180. doi: 10.1152/jn.2000.83.3.1167. [DOI] [PubMed] [Google Scholar]
- Takahashi KA, Castillo PE. The CB1 cannabinoid receptor mediates glutamatergic synaptic suppression in the hippocampus. Neuroscience. 2006;139:795–802. doi: 10.1016/j.neuroscience.2006.01.024. [DOI] [PubMed] [Google Scholar]
- Trantham-Davidson H, Neely LC, Lavin A, Seamans JK. Mechanisms underlying differential D1 versus D2 dopamine receptor regulation of inhibition in prefrontal cortex. J Neurosci. 2004;24:10652–10659. doi: 10.1523/JNEUROSCI.3179-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trettel J, Levine ES. Cannabinoids depress inhibitory synaptic inputs received by layer 2/3 pyramidal neurons of the neocortex. J Neurophysiol. 2002;88:534–539. doi: 10.1152/jn.2002.88.1.534. [DOI] [PubMed] [Google Scholar]
- Tseng KY, O’Donnell P. Dopamine-glutamate interactions controlling prefrontal cortical pyramidal cell excitability involve multiple signaling mechanisms. Journal of Neuroscience. 2004;24:5131–5139. doi: 10.1523/JNEUROSCI.1021-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng KY, O’Donnell P. D2 dopamine receptors recruit a GABA component for their attenuation of excitatory synaptic transmission in the adult rat prefrontal cortex. Synapse. 2007;61:843–850. doi: 10.1002/syn.20432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ujike H, Morita Y. New perspectives in the studies on endocannabinoid and cannabis: cannabinoid receptors and schizophrenia. J Pharmacol Sci. 2004;96:376–381. doi: 10.1254/jphs.fmj04003x4. [DOI] [PubMed] [Google Scholar]
- van der Stelt M, Di Marzo V. The endocannabinoid system in the basal ganglia and in the mesolimbic reward system: implications for neurological and psychiatric disorders. Eur J Pharmacol. 2003;480:133–150. doi: 10.1016/j.ejphar.2003.08.101. [DOI] [PubMed] [Google Scholar]
- Varvel SA, Lichtman AH. Evaluation of CB1 receptor knockout mice in the Morris water maze. J Pharmacol Exp Ther. 2002;301:915–924. doi: 10.1124/jpet.301.3.915. [DOI] [PubMed] [Google Scholar]
- Vaughan CW, McGregor IS, Christie MJ. Cannabinoid receptor activation inhibits GABAergic neurotransmission in rostral ventromedial medulla neurons in vitro. Br J Pharmacol. 1999;127:935–940. doi: 10.1038/sj.bjp.0702636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Pickel VM. Dopamine D2 receptors are present in prefrontal cortical afferents and their targets in patches of the rat caudate-putamen nucleus. J Comp Neurol. 2002;442:392–404. doi: 10.1002/cne.10086. [DOI] [PubMed] [Google Scholar]
- Xu TX, Sotnikova TD, Liang C, Zhang J, Jung JU, Spealman RD, Gainetdinov RR, Yao WD. Hyperdopaminergic tone erodes prefrontal long-term potential via a D2 receptor-operated protein phosphatase gate. J Neurosci. 2009;29:14086–14099. doi: 10.1523/JNEUROSCI.0974-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yavich L, Forsberg MM, Karayiorgou M, Gogos JA, Mannisto PT. Site-specific role of catechol-O-methyltransferase in dopamine overflow within prefrontal cortex and dorsal striatum. J Neurosci. 2007;27:10196–10209. doi: 10.1523/JNEUROSCI.0665-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin HH, Lovinger DM. Frequency-specific and D2 receptor-mediated inhibition of glutamate release by retrograde endocannabinoid signaling. Proc Natl Acad Sci U S A. 2006;103:8251–8256. doi: 10.1073/pnas.0510797103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmer A, Zimmer AM, Hohmann AG, Herkenham M, Bonner TI. Increased mortality, hypoactivity, and hypoalgesia in cannabinoid CB1 receptor knockout mice. Proc Natl Acad Sci U S A. 1999;96:5780–5785. doi: 10.1073/pnas.96.10.5780. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.