

Abstract
Episodic ataxia type-2 (EA2) is an inherited movement disorder caused by mutations in the gene encoding the Cav2.1α1 subunit of the P/Q-type voltage-gated calcium channel that result in an overall reduction in the P/Q-type calcium current. A consequence of these mutations is loss of precision of pacemaking in cerebellar Purkinje cells. This diminished precision reduces the information encoded by Purkinje cells and is thought to contribute to symptoms associated with this disorder. The loss of the precision of pacemaking in EA2 is the consequence of reduced activation of calcium-dependant potassium channels (KCa) by the smaller calcium current and in vitro can be pharmacologically restored by KCa activators.
We used a well-established mouse model of EA2, the tottering (tg/tg) mouse, to examine the potential therapeutic utility of one such FDA-approved compound, chlorzoxazone (CHZ). Compared to wild-type Purkinje cells, we found the firing rate of tg/tg Purkinje cells in acutely prepared cerebellar slices to be very irregular. Bath application of CHZ successfully restored the precision of pacemaking in a dose-dependent manner. Oral administration of CHZ to tg/tg mice improved their baseline motor performance and reduced the severity, frequency and duration of episodes of dyskinesia without producing any adverse effects. We propose the use of CHZ, which is currently FDA-approved as a muscle relaxant, as a safe and novel treatment of EA2.
Introduction
Episodic ataxia type-2 (EA2) is a rare neurological disorder caused by mutations in the gene encoding the Cav2.1α1 subunit of the P/Q-type voltage-dependent calcium channel (Ophoff et al., 1996). The mutations associated with EA2 significantly reduce the P/Q-type calcium current (Barclay et al., 2001; Dove et al., 1998; Fletcher et al., 1996). Patients suffering from EA2 have transient attacks of ataxia, instability and dyskinesia with progressive inter-episode dystonia, general weakness and mild ataxia (Jen et al., 2007). These episodes are triggered by diverse stressors ranging from psychological stress and exercise to caffeine and ethanol (Jen et al., 2007). The symptoms are primarily cerebellar in origin (Jen et al., 2007).
Because of the reduction in the P/Q-type calcium current associated with EA2 mutations, the precision of Purkinje cell pacemaking is reduced in several animal models of this disorder (Walter et al., 2006). Consequently, the ability of Purkinje cells to encode motor-related information is significantly reduced (Walter et al., 2006). It is suggested that the loss in the precision of Purkinje cell pacemaking contributes to EA2 symptoms (Walter et al., 2006).
The mechanism by which P/Q-type calcium channels regulate pacemaking in Purkinje cells is well understood. With each action potential, the calcium that enters the Purkinje cell via these voltage-gated channels activates calcium-dependent potassium (KCa) channels (Womack et al., 2004). The increase in the KCa conductance, particularly of the small-conductance type (SK channels), sets the duration of individual interspike intervals in Purkinje cells (Walter et al., 2006; Womack et al., 2004). In support of this mechanism, in animal models of EA2 the irregular pacemaking of Purkinje cells can be restored to precision levels comparable with that of wild-type by pharmacological activation of SK channels with 1-EBIO (1-ethyl-2-benzimidazolinone) (Walter et al., 2006). Even more intriguing is the finding that chronic perfusion of 1-EBIO into the cerebellum of the ataxic mice reduced baseline ataxia and the episodes of dyskinesia (Walter et al., 2006). While these experiments serve as proof of concept and suggest that KCa channels might be an appropriate therapeutic target for treatment of EA2, it remains to be established whether systemic administration of such compounds is similarly effective. Because current therapeutic approaches for treatment of EA2 are limited (Jen et al., 2007), we explored the potential use of KCa activators by examining the efficacy of a structurally related (Cao et al., 2001; Syme et al., 2000), FDA-approved compound, chlorzoxazone (CHZ).
We find that the firing of Purkinje cells in a well established animal model of EA2, tottering (tg/tg) mice, is very irregular and that CHZ effectively restores the precision of the pacemaking. Behaviorally, oral administration of CHZ reduced the basal ataxia of tg/tg mice, and also lessened the probability that animals had episodes of stress-induced dyskinesia. At the concentrations used, no adverse effects of CHZ were noted. Our results strongly support the notion that SK channel activators are a promising group of therapeutic agents for treatment of the motor abnormalities associated with EA2.
Methods
Mutant mice (tottering, tg/tg) and wild-type mice (+/+) were originally purchased from Jackson Laboratories and subsequently bred, genotyped and kindly provided by Dr. Ellen Hess (Johns Hopkins University, Baltimore, MD). tottering mice are inbred on the C57Bl/6J strain for at least 25 generations. For control experiments, a different set of wild-type C57Bl/6J mice were obtained from Charles Rivers-Kingston (see Supplementary Material 1). All procedures employed were in accordance with the policies established by the Animal Institute Committee of the Albert Einstein College of Medicine.
Preparation of cerebellar slices
Mice (3-4 weeks old) were anesthetized with halothane and decapitated. The brain was quickly removed and placed on cold extracellular solution containing (in mM): 125 NaCl, 2.5 KCl, 26 NaHCO3, 1.25 NaH2PO4, 1 MgCl2, 2 CaCl2, and 10 glucose, pH= 7.4 when gassed with 95% O2:5% CO2. The cerebellum was dissected and mounted on a modified Oxford vibratome and 300-μm thick sagittal slices were made. The slices were kept in oxygenated extracellular solution at 34 °C for one hour, and then at room temperature until use (typically within 1-5 hours of preparation).
Extracellular recording
Slices were placed in a recording chamber on the stage of a Zeiss Axioskop microscope. Purkinje cells were visually identified using a 40× water-immersion objective with infrared optics. The slices were superfused with the recording solution and the temperature adjusted to 35±1°C. Measurements of spontaneous firing rate were made in the presence of 5 mM kynurenic acid (Spectrum Chemical MFG Corp., Gardena, CA), a broad spectrum ionotropic glutamate receptor antagonist (Stone, 1993), and 100 μM picrotoxin (Sigma, St. Louis, MO) a GABAA channel blocker (Yoon et al., 1993).
Extracellular recordings were obtained from single Purkinje cells using a home-made differential amplifier and glass pipette electrodes filled with extracellular solution. With this type of recording activity of Purkinje cells can be monitored for periods in excess of four to five hours without any noticeable change in the pattern or rate of their activity (Womack and Khodakhah, 2002). Data was sampled at 10 kHz using an analog-to-digital converter (PCI-MIO-16XE-10; National Instruments, Austin, TX), and acquired and analyzed using custom software written in LabView (National Instruments).
Chlorzoxazone and NS1619 were purchased from Sigma Aldrich (St. Louis, MO), and Iberiotoxin was obtained from Tocris Bioscience (Ellisville, MO, USA).
Behavioral analysis
We used the accelerating rotarod test as a paradigm to examine motor performance (Crawley, 2008; Walter et al., 2006). The apparatus consisted of a 3 cm diameter rotating rod (Rotamex-5, Columbus Instruments) elevated 55 cm above a covered platform. Each trial started from stationary position accelerating at a rate of 0.1 cm/s every second. Speed and latency to fall of the animals were automatically recorded by an interfaced computer. Every day, tg/tg and +/+ mice (4-5 months old) were tested in 10 consecutive trials.
To quantify the frequency, severity and duration of episodes of dyskinesia observed in tg/tg mice, the overall motor behavior was scored every 10 min before and after each rotarod session for up to 2 hours. The scoring followed a previously published scale (Weisz et al., 2005) as follows: 0=normal motor behavior; 1=slightly slowed or abnormal movements; 2=mild impairments, limited ambulation unless disturbed; 3=moderate impairment, limited ambulation even when disturbed, frequent abnormal postures; 4=severe impairment, almost no ambulation, sustained abnormal postures; 5=prolonged immobility in abnormal postures.
The stress and exertion associated with the rotarod trials resulted in long lasting episodes of paroxysmal dyskinesia. The severe attacks (4-5) typically last for 30-60 minutes and are very stereotyped. These attacks start with the involuntary extension of hind limbs followed by lowering of the hips and extending the knees, ankles and paws. Throughout these movements the back is abnormally arched. This posturing then spreads to the rest of the body, with particularly severe contractions of the neck and face muscles (Campbell et al., 1999). Most of these characteristics are notable every time a tg/tg mouse has a severe attack making the severe episodes (4-5) unambiguously distinguishable from milder motor impairments (levels 2-3).
Scoring of the severity of the symptoms was carried out by one of the authors with prior knowledge of the treatment implemented. To ascertain that this scoring was not biased, a few colleagues who were blind to the treatment were also asked to provide dyskinesia scores by reviewing a number of video-taped episodes. There were no significant or consistent differences between the scores assigned by five such observers blind to the treatment of the mice and the scores obtained by ourselves.
Chlorzoxazone (CHZ) was orally administrated to tg/tg and +/+ mice by adding it to their drinking water. The CHZ solution was prepared fresh every day by adding CHZ to a 0.1% solution of hydroxypropyl-β-cyclodextrin (Tocris Bioscience, Ellisville, MO, USA), and then adding a few drops of 1 N NaOH until CHZ was fully dissolved. The solution was supplemented with 10% sucrose to improve its taste and thus to ensure its consumption. To improve the accuracy of the water consumption measurements, the large water bottle in the cages were replaced with graduated 15 ml plastic tubes. The weight of the animals and the extent of their water intake were monitored daily throughout the experiment.
Because rodents are nocturnal, all behavioral tests were carried out during their dark cycle. All data are reported as Mean ± S.E.M. Data were analyzed using one-way ANOVA followed by Bonferroni's multiple comparison test, and considered to be statistically significant if p<0.05.
Results
Pacemaking of Purkinje cells of the tg/tg mice is very irregular
To investigate the efficacy of CHZ we used a well-established animal model of EA2, namely the tottering mice (tg/tg) (Jen et al., 2007). Because of a spontaneous mutation in the P/Q-type calcium channel, these mice suffer from a movement disorder which has remarkable resemblance to EA2 in humans (Jen et al., 2007; Wakamori et al., 1998). tg/tg mice have basal ataxia that becomes more severe with age and, like humans with EA2, manifest episodes of imbalance and dyskinesia triggered by a diverse set of stressors (Pietrobon, 2005) although the severity of the dyskinesia in these mice is more severe than that typically seen in patients.
We monitored the activity of wild-type (+/+) and tg/tg Purkinje cells by extracellularly recording their firing rate in acutely prepared cerebellar slices. To ensure that the recorded activity was solely intrinsic and not affected by synaptic inputs, fast excitatory and inhibitory synaptic transmissions were pharmacologically blocked. Consistent with that seen in other animals models of EA2 (Walter et al., 2006) and in tg/tg mice in vivo (Hoebeek et al., 2005), the pacemaking of Purkinje cells in slices made from tg/tg mice was significantly more irregular compared to that seen in Purkinje cells of +/+ mice (Figure 1). Raw traces of the spontaneous activity of +/+ and tg/tg Purkinje cell are shown in Figure 1A. The irregularity of the pacemaking of the tg/tg Purkinje cell compared with the regular pacemaking of the +/+ is evident from their corresponding interspike interval autocorrelograms shown in Figure 1B. As can be noted, compared to that of the +/+ Purkinje cells, the tg/tg autocorrelogram shows diminished peaks and wider distributions.
Figure 1. Chlorzoxazone decreases the irregularity of the intrinsic pacemaking of tottering Purkinje cells.
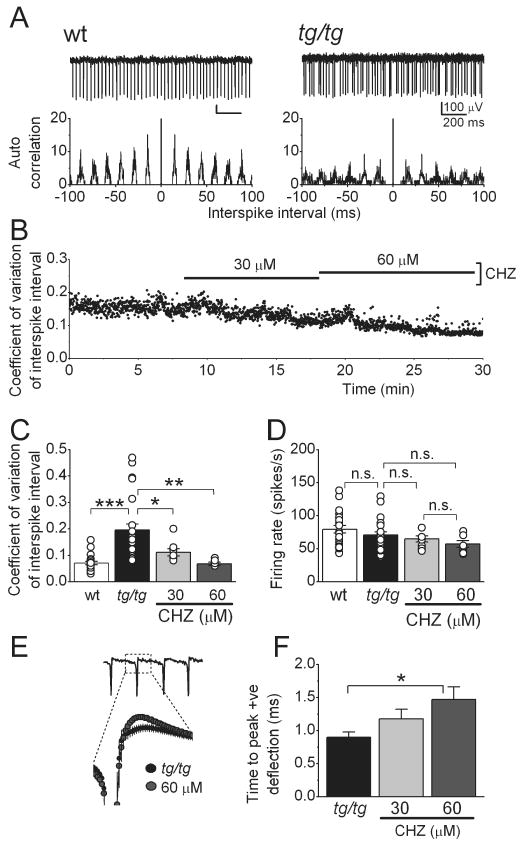
(A) Raw traces from extracellular recordings from wild-type (+/+) and tottering (tg/tg) Purkinje cells, with their interspike interval autocorrelograms below. Note the irregular firing of the tg/tg cell.
(B) Chlorzoxazone (CHZ) dose-dependently decreased the variation of the interspike interval in a tg/tg Purkinje cell whose spontaneous activity was extracellularly recorded.
(C) Average and individual values (circles) of the coefficient of variation of the interspike intervals in +/+ and tg/tg Purkinje cells. Note the higher values and the wider distribution of coefficient of variation in the mutant cells. (*) p<0.05, (**) p<0.01, (***) p<0.001 (One-way ANOVA, followed by Bonferroni correction), n=24 cells for tg/tg mice and n=19 cells for +/+ mice.
(D) Average and individual (circles) firing rates of the +/+ and tg/tg Purkinje cells shown in C, “n.s.” denotes not significant.
(E) CHZ increased the magnitude of the positive deflection of the spikes recorded extracellularly in tg/tg Purkinje cells. The expanded figure shows an averaged trace of the activity of 7 cells, in control and after adding CHZ 60 μM.
(F) Average magnitude of the positive deflection in control and after adding CHZ in increasing concentrations. (*) p<0.05 (One-way ANOVA, followed by Bonferroni correction).
To quantify the irregularity, for each cell studied we calculated the coefficient of variation (CV) of the interspike interval (Figure 1 B and C). The average CV in +/+ Purkinje cells was 0.07±0.01 (Figure 1C, n=22 cells, 3 mice) whereas in tg/tg it was 0.19±0.02 (n=24 cells, 3 mice, p<0.001). In addition, the spread of CV values in tg/tg cells was higher than in the +/+ (Figure 1C). Despite the higher irregularity, the firing rate was not different between +/+ and tg/tg Purkinje cells (Figure 1D), the average predominant firing rate was 79.4±5.6 spikes/s in +/+ compared to 70.6±4.7 spikes/s in tg/tg Purkinje cells (p>0.1).
Chlorzoxazone restores the precision of pacemaking of tg/tg Purkinje cells
In the ducky mouse, an animal model of EA2 with a less severe phenotype (Brodbeck et al., 2002), we have previously shown that the SK channel activator 1-EBIO reduces the irregularity of Purkinje cell pacemaking (Walter et al., 2006). We thus examined whether the FDA-approved compound Chlorzoxazone (CHZ) was similarly efficacious in restoring the precision of pacemaking in tg/tg Purkinje cells. We monitored the activity of Purkinje cells in slices as described earlier, and then added CHZ to the bathing solution (Figure 1B). We found that CHZ effectively and dose-dependently made the firing of tg/tg Purkinje cells more regular (Figure 1B), a finding that is quantitatively reflected in the CV of interspike intervals (Figure 1C). In the 7 cells examined (3 mice), the average CV of the tg/tg Purkinje cells under control conditions was 0.21±0.03. Application of 30 μM CHZ reduced the CV to 0.11±0.01 (p<0.05 vs. control) and 60 μM reduced it further to 0.08±0.004 (p<0.01 vs. control), a value that was not significantly different from the CV obtained in +/+ Purkinje cells (p>0.1). At a concentration of 30 μM, CHZ did not significantly affect the baseline firing rate of tg/tg Purkinje cells (the firing rate was 70.1±4.5 spikes/s in control vs. 64.8±4.6 spikes/s in 30 μM CHZ, p>0.1), whereas 60 μM CHZ marginally reduced it to 59.6±3.6 spikes/s (p>0.1 vs. control; Figure 1D). The prominent effect of CHZ in reducing the CV of interspike intervals, however, was produced mainly by reducing the irregularity because 30 and 60 μM CHZ decreased the interspike interval standard deviations by 74% and 176% respectively, whereas the average firing rates were only reduced by 21% and 33%. Thus 30-60 μM CHZ can be effectively used to restore the precision of pacemaking in tg/tg Purkinje cells. It is intriguing that this concentration range is equivalent to the plasma concentration of CHZ found in rats after a clinically relevant dose (Kwon, 2003).
To restore the precision of pacemaking, consistent with its efficacy in activating SK channels, 1-EBIO increases the amplitude of the AHP in Purkinje cells (Walter et al., 2006). To determine the effects of CHZ on the action potential waveform, we examined the extracellularly recorded spikes which represent the derivate of the membrane potential. In this context, the negative deflection corresponds to the upstroke of the action potential, and the positive deflection to the down stroke of the action potential and its AHP. CHZ dose-dependently increased both the magnitude (Figure 1E) and the time from start of the negative deflection to peak positive deflection (0.86±0.06 ms in control, 1.01±0.05 ms in 30 μM CHZ, p<0.1, and, 1.22±0.11 ms in 60 μM CHZ p<0.05; n=9; Figure 1F). These data are consistent with the actions of 1-EBIO on the AHP reported previously (Walter et al., 2006), and the hypothesis that CHZ restores the precision of tottering Purkinje cell pacemaking by increasing the magnitude of the current carried by the calcium-dependent potassium channels.
In summary, these data demonstrate that CHZ effectively restores the precision of pacemaking in the mutant Purkinje cells in cerebellar slices in vitro. In agreement with such a mechanism of action, we note that systemic in vivo administration of a related compound, zoxazolamine, has been shown to transform the irregular firing of adult rat nigrostriatal dopaminergic neurons into a stable regular firing pattern (Matthews et al., 1984).
Modulators of BK channels do not alter the pacemaking of adult Purkinje cells
While selective for KCa channels, CHZ activates both SK and BK channels (Cao et al., 2001; Liu et al., 2003; Syme et al., 2000) and thus its efficacy in restoring the precision of pacemaking in the tg/tg mouse Purkinje cells might be the consequence of activation of both SK and BK channels. Because prior experiments have shown that SK specific activators such as 1-EBIO are effective in restoring the precision of pacemaking (Walter et al., 2006), here we examined the potential efficacy of BK channels in this process. We did so using two complementary approaches. In the first we explored whether in the presence of BK channel blockers CHZ remained efficacious in imposing regularity in the erratic activity of Purkinje cells in which calcium channels were partially blocked by low concentrations of cadmium (Walter et al., 2006). In the second we directly tested the efficacy of a BK-selective compound in restoring the precision of pacemaking.
The activity of Purkinje cells in acutely prepared cerebellar slices of adult C57Bl/6J mice was recorded extracellularly. Addition of blockers of fast synaptic transmission (kynurenic acid and picrotoxin) ensured that the recorded activity represented the intrinsic pacemaking of these neurons. To block calcium channels (which in Purkinje cells are primarily of P/Q-type), low concentrations of cadmium chloride (5-15 μM) were sequentially bath-applied until the very regular pacemaking of Purkinje cells became erratic (Walter et al., 2006). This was done quantitatively by measuring the coefficient of variation of their interspike intervals online (Figure 2A). We first tested whether CHZ remained effective in restoring the precision of pacemaking when BK channels were blocked with iberiotoxin (Ibtx) (Candia et al., 1992). As can be seen low concentrations of cadmium increased the CV from 0.06±0.01 to 0.13±0.02 (p<0.05; n=5 cells, 3 mice; Figure 2A and B) a value close to that seen in the tottering mice. At these low concentrations even though addition of cadmium made firing of Purkinje cells erratic it did not significantly alter their firing rate (54.0±4.0 and 57.8±6.8 spikes per second, in control and cadmium respectively, p>0.4, Figure 2C). In none of the adult Purkinje cells examined did application of 100 nM Iberiotoxin change the firing rate (63.2±11.2 spikes per second, p>0.4 compared to cadmium, Figure 2C), or the coefficient of variation of interspike intervals (0.13±0.02, p>0.1 vs. cadmium, Figure 2A and B). However, subsequent addition of 30 μM CHZ in the presence of Ibtx reduced the CV of interspike intervals to levels similar to control conditions (0.07±0.01, p<0.05 vs. Ibtx and cadmium; p>0.1 vs. control conditions, Figure 2A and B).
Figure 2. Chlorzoxazone decreases the irregularity pacemaking through SK channels.
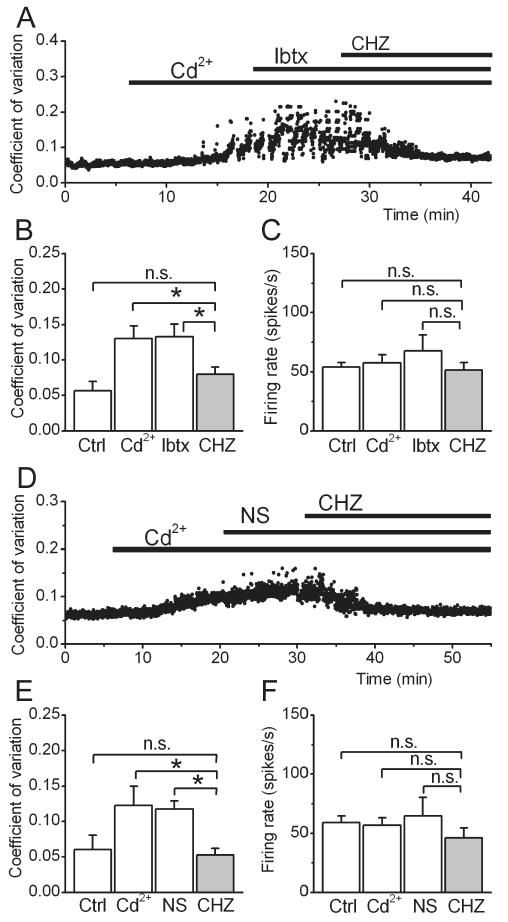
(A) The precision of pacemaking of a wild type cerebellar Purkinje cell was monitored by following coefficient of variation of its interspike intervals. Addition of low concentration of cadmium to partially block voltage-gated calcium channels made the firing of the cell irregular. 100 nM iberiotoxin was then added to block BK channels. Despite the blockade of BK channels, 30 μM CHZ restored the precision of pacemaking to control conditions.
The average coefficient of variation of interspike intervals (B) and firing rate (C) of 5 Purkinje cells following the experimental procedure described in panel (A). (*) p<0.05 (One-way ANOVA, followed by Bonferroni correction), “n.s.” denotes not significant.
(D) The pacemaking of a Purkinje cell was made irregular with addition of cadmium, and the selective BK channel activator NS1619 was bath applied at a concentration of μM. While NS1619 was ineffective in restoring the precision of pacemaking, subsequent addition of 30 μM CHZ returned the coefficient of variation of interspike intervals to pre-cadmium levels.
The average coefficient of variation of interspike intervals (E) and firing rate (F) of all the Purkinje cells examined following the experimental procedure described in panel (D). (*) p<0.05, “n.s.” denotes not significant.
As a complementary approach we then examined the efficacy of a specific BK channel activator NS1619 (Olesen et al., 1994) in restoring the precision of pacemaking. Even at a relatively high concentration of 30 μM, activation of BK channels did not reduce the irregularity of the Purkinje cell spontaneous activity (CV after NS1619 = 0.12±0.01, p>0.1 compared to cadmium, Figure 2D, E). This finding is in agreement with the observation that despite having a prominent role in juveniles, BK channels play a minor role in regulation of pacemaking in adult Purkinje cells (Womack et al., 2009). In the same cells, addition of 30 μM CHZ in the presence of NS1619 restored the precision of pacemaking as indicated by the reduction of interspike interval CV to control levels (0.05±0.01, p<0.05 vs. NS1619 and cadmium, and p>0.1 compared to control, Figure 2E).
Ideally, it would be desirable to demonstrate that CHZ can no longer affect the precision of pacemaking of Purkinje cells when SK channels are blocked. However, block of SK channels results in high frequency (>300 spikes per second) avid bursting in Purkinje cells (Womack and Khodakhah, 2003) and thus it is not possible to perform this experiment. Nonetheless, given the pharmacology of CHZ, the data presented strongly suggest that CHZ restores the precision of pacemaking in Purkinje cells primarily by activating SK channels.
Chlorzoxazone significantly improves the baseline motor performance of tg/tg mice
Given the efficacy of CHZ in restoring the precision of pacemaking of the tg/tg Purkinje cells, we explored whether it can reduce the motor deficits in these mice. Our main goal was to investigate the potential use of CHZ as a therapeutic agent. Thus, while we have previously shown that direct perfusion of a similar compound (1-EBIO) into the cerebellum reduces motor deficits in tg/tg mice (Walter et al., 2006), here we tested the efficacy of CHZ when it was administered orally by adding it to the drinking water of the mice. Based on the data described in the prior section, our target CSF concentration of CHZ was between 30-60 μM. To achieve this we measured the daily water intake of the mice and found it to be ≈2.5 ml per day. We next took into consideration the fact that in rats, orally administered CHZ has a half-life of ≈60 min, and a single dose of 50 mg/kg results in a plasma concentration of ≈90 μM (oral half-life = 57.8±7.3 min (Wan et al., 2006). Based on this, we estimated that if the mice maintain their normal fluid intake replacement of their drinking water with one which contains ≈33 mM CHZ is likely to yield a plasma concentration of about 30 μM. However, at this concentration the taste of the solution was significantly changed such that even with addition of sucrose to sweeten it, tg/tg mice did not drink it. We thus reduced the concentration of CHZ to 15 mM to reduce the bitterness of the resulting solution. Although this results in a lower plasma concentration than that ideally needed, at this concentration the daily fluid consumption of mice was comparable to their consumption of normal water.
To examine motor function, a group of adult tg/tg and adult +/+ mice were trained on an accelerating rotarod (Crawley, 2008) to proficiency (Figure 3A). 15 mM CHZ was then added to the drinking water of both groups. Within a couple days of receiving CHZ, the performance of tg/tg mice significantly increased (Figure 3A) whereas the performance of the +/+ mice was not affected. On average, the maximum speed reached by tg/tg mice was 8.7±0.2 RPM (n=10 mice), which increased to 13.8±0.2 RPM (p<0.001) during CHZ treatment (Figure 3B). The performance of the +/+ was 34.9±0.2 RPM before and 35.5±0.4 RPM during administration of CHZ (p>0.4). It is important to note that the attacks of dyskinesia typically started after the last rotarod trial and thus the higher performance of the mice with CHZ reported above mainly reflect improvements in baseline motor performance rather than reductions in the episodes of dyskinesia detailed in the next section.
Figure 3. Oral administration of CHZ improves motor performance in tottering mice.
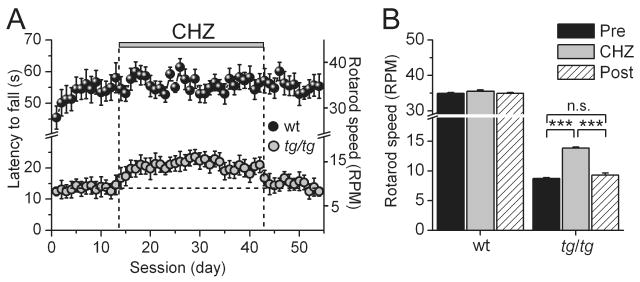
(A) The performance of tg/tg (n=10, orange symbols) and +/+ mice (n=11, black symbols) was evaluated daily using an accelerating rotarod paradigm. CHZ treatment is denoted with the vertical bar. CHZ improved the motor performance of tg/tg, but not wild type mice.
(B) Maximum speed achieved before CHZ treatment (average of 5 days), during treatment (CHZ, average of 2 weeks) and after terminating treatment (Post, average of the last 5 days) for +/+ and tg/tg mice. (***) p<0.001 (One-way ANOVA, followed by Bonferroni correction), “n.s.” denotes not significant.
We found no difference in the efficacy of CHZ in reducing ataxia in the tg/tg mice throughout the four-week period of treatment (Figure 3A). However, after the four-week CHZ treatment when the mice were returned to normal drinking water, the motor performance of tg/tg mice on the rotarod rapidly decreased to levels comparable to that prior to CHZ administration (9.3±0.3 RPM, p<0.001 vs. performance during CHZ treatment; p>0.1 vs. performance previous to CHZ treatment; Figure 3A and B).
Chlorzoxazone reduces the severity, frequency and duration of stress-induced attacks of dyskinesia in tg/tg mice
In addition to their baseline ataxia, various stressors cause severe episodes of dyskinesia in the tg/tg mice (Jen et al., 2007; Weisz et al., 2005). In our experimental group of mice, the exercise and the stress associated with the rotarod paradigm routinely triggered such episodes. We used these events to examine the efficacy of CHZ in reducing the probability that stress resulted in an episode. Moreover, using a published semi-quantitative scoring system (Weisz et al., 2005), we also evaluated the severity of motor disturbances observed. With this evaluation method a score of 0 is considered normal and that of 5 correlates with severe motor dysfunction (see Methods section for details).
The motor performance of each mouse was scored for a total observation time of 130 minutes; 10 minutes prior to, and every 10 minutes for 2 hours after the rotarod test. In the graph shown in Figure 4A the width and shape of each bar reflects the average daily score of all tg/tg mice during the 130 minutes observation period. As can be noted in this and the average dyskinesia score shown in Figure 4B, CHZ treatment significantly reduced the average severity and duration of the stress-induced dyskinesia in the mice (see also the supplementary video). This was partly because with CHZ the probability that stress induced an attack was reduced. We first estimated the probability that the rotarod paradigm resulted in an episode of dyskinesia independent of the severity of the attack (anything above the baseline score). In the absence of CHZ the rotarod task resulted in an attack in all the mice, although not in all cases these attacks were highly severe (Figure 4C). During CHZ treatment, the probability that stress induced an attack was reduced to 74±5.0% (p<0.001). Moreover, the average severity (Figure 4D) and duration of these attacks (Figure 4E) were also significantly reduced. The maximum severity of the episodes of dyskinesia decreased from 3.80±0.09 to 2.78±0.11 (p<0.001, averaging the first 40 min after the rotarod session) and the average duration reduced from 56.5±1.3 min to 29.9±2.2 min (p<0.001). The main reason for the reduction in the average severity of the episodes of dyskinesia was the large decrease in the probability that the rotarod session triggered a very severe attack (those with a score of ≥3.5) (Figure 4F). In fact, CHZ treatment reduced the frequency of these highly severe attacks from an average of 68.9±3.0% to 23.6±5.2% (p<0.001). Further, the frequency of severe attacks returned to their pre-treatment values after removal of CHZ (72.9±2.9%, p<0.001 vs. frequency during CHZ; p>0.1 vs. frequency of attacks before CHZ). When the mice showed a very severe attack however, these attacks had comparable severity scores to the ones triggered previous to CHZ treatment (Figure 4G), although their duration was significantly shorter (Figure 4H), decreasing from an average of 53.9±1.5 min to 34.8±2.1 min during CHZ treatment (p<0.001, Figure 3H). When the CHZ treatment was terminated, the duration of these attacks returned to their pre-treatment levels (50.1±2.0 min, p<0.001 vs. CHZ; p>0.1 vs. pre-treatment). What is noteworthy is that CHZ had comparable efficacy in all mice. In other words, we did not find that some mice did not have any attacks whereas some did; the frequency of attacks was reduced to a similar extent in all treated mice. The pie charts in Figure 4I summarize the efficacy of CHZ in reducing the frequency of stress-evoked attacks of dyskinesia in tg/tg mice
Figure 4. CHZ treatment reduces the frequency and severity of the stress-induced episodes of dyskinesia in tottering mice.
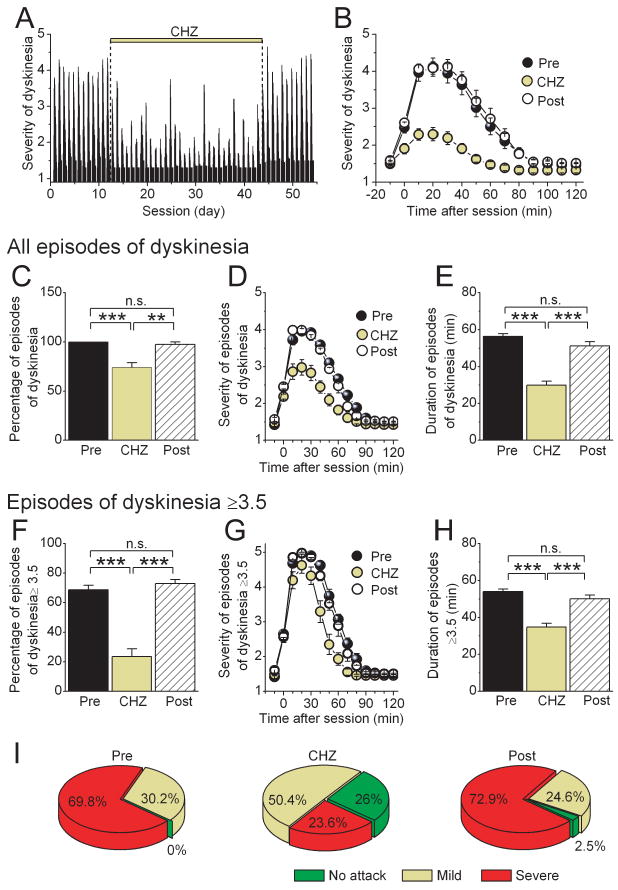
(A) The attacks of dyskinesia triggered in tg/tg mice as a consequence of the stress associated with the daily rotarod sessions were quantified in 10 minute intervals for each session. A score of 0 denotes normal behavior whereas a score of 5 corresponds to severe dyskinesia. During the treatment with CHZ, the average score was significantly reduced.
(B) The average severity of dyskinesia before, during, and after treatment with CHZ in the tottering mice shown in panel (A).
(C) The average probability that the rotarod paradigm resulted in an episode of dyskinesia irrespective of the severity of the attack. (***) p<0.001, (**) p<0.01 (One-way ANOVA, followed by Bonferroni correction), “n.s.” denotes not significant.
(D) The average severity of all attacks of dyskinesia before, during, and after treatment with CHZ.
(E) The duration of the attacks of dyskinesia shown in panel (D). (***) p<0.001, “n.s.” denotes not significant.
(F) The average probability that the rotarod paradigm resulted in severe episodes of dyskinesia (dyskinesia score ≥3.5). (***) p<0.001, “n.s.” denotes not significant.
(G) The average of only severe attacks revealed a significant shortening of the duration of such episodes during the treatment with CHZ. The graph depicts the progression in severity from before the rotarod session up to 120 min after.
(H) Average duration of severe episodes of dyskinesia. (***) p<0.001, “n.s.” denotes not significant.
(I) Summary results on the efficacy of CHZ in increasing the frequency of post-stress sessions in which tottering mice were free of symptoms (no attack), and correspondingly increasing the frequency of sessions in which they had an episode of dyskinesia with a score <3.5 (mild) or one with a score ≥3.5 (severe).
Lastly, we examined the consequence of long term treatment of mice with CHZ using a battery of behavioral tests. As detailed in the Supplementary Material 1, we found that even at higher concentrations oral administration of CHZ did not affect the cognitive function, muscle strength, or the overall gross motor performance of wild-type mice.
Collectively, the data presented indicate that systemic administration of CHZ is safe and that treatment of tg/tg mice with CHZ improves their baseline motor performance and also reduces the overall severity, frequency and duration of stress-induced episodes of dyskinesia.
Discussion
Episodic ataxia type2 and present therapeutic options
One of the most common forms of episodic ataxia is episodic ataxia type-2 (Jen et al., 2007). Patients affected by this condition not only have episodic attacks of dyskinesia, but also show mild baseline ataxia that progress in severity with time (Jen et al., 2004).
There are presently few therapeutic options and the only two viable drugs are acetazolamide (ACTZ) and 4-aminopyridine (4-AP) (Jen et al., 2007). While many patients respond well to ACTZ which both improves both some of the baseline symptoms and reduces the frequency of episodes of dyskinesia (Strupp et al., 2007), with time, many become non-responsive (Jen et al., 2007). 4-AP has been used recently to reduce the symptoms of EA2 (Strupp et al., 2004). While it is effective in improving baseline motor coordination and in reducing the frequency and severity of the episodic attacks (Glasauer et al., 2005; Lohle et al., 2008; Strupp et al., 2007), 4-AP has to be used with caution since as a potassium channel blocker it can be epileptogenic (Bever, Jr. et al., 1994; Judge and Bever, Jr., 2006). The mechanism of action of neither of these two drugs in the treatment of EA2 is fully understood although it is suggested that ACTZ prevents elevations in the intracellular pH (Strupp et al., 2007), whereas it is proposed that 4-AP increases the excitability of Purkinje cells (Strupp et al., 2008).
Potential mechanisms that contribute to ataxia in EA2
P/Q-type calcium channels which are affected in EA2 are highly expressed throughout the CNS (Evans and Zamponi, 2006) and are particularly enriched in axon terminals and in cerebellar Purkinje cells (Mori et al., 1991; Stea et al., 1994; Usowicz et al., 1992). Calcium influx primarily through these channels mediates synaptic transmission at CNS nerve endings (Evans and Zamponi, 2006). In cerebellar Purkinje cells, dendritic P/Q-type calcium channels generate calcium action potentials (Llinas and Sugimori, 1980), and somatic ones regulate the precision of pacemaking (Walter et al., 2006; Womack and Khodakhah, 2004). It has recently been suggested that irregular firing of cerebellar Purkinje cells contributes to motor symptoms associated with EA2 (Walter et al., 2006). The loss in the precision of pacemaking in affected Purkinje cells was found to be caused by reduced activation of KCa channels as a consequence of the smaller P/Q-type calcium current. On the basis of these findings it was postulated that KCa channels may constitute a novel therapeutic target in EA2. Consistent with this hypothesis, it was demonstrated that the symptoms of tottering mice whose cerebella were chronically perfused with the KCa channel activator 1-EBIO were appreciably reduced. The tottering mice are one of the most studied and best established models of EA2 (Jinnah et al., 2005). While the mutation in tottering mice results in a very large decrease in the P/Q calcium current density and more severe episodes of dyskinesia than typically seen in EA2 patients, these mice have served as a very useful and reliable model of EA2. At the concentrations used to improve the motor function of tottering mice, the most likely target of EBIO were cerebellar Purkinje cells and not other principal cells such as the neurons of the deep cerebellar nuclei (Alvina and Khodakhah, 2008; Walter et al., 2006; Womack et al., 2004; Womack and Khodakhah, 2003).
CHZ as a therapeutic agent for the treatment of EA2
Despite the efficacy of perfusion of cerebellum with EBIO in the tg/tg mice it is not practical to chronically perfuse agents into the cerebellum of patients. Moreover, the rarity of this disorder provides little incentive for pharmaceutical companies to explore novel agents. We thus explored the efficacy of systematic administration of CHZ, a FDA-approved analogue of 1-EBIO. In slice experiments we found that CHZ effectively restored the precision of pacemaking in tottering Purkinje cells. Although CHZ is presently FDA-approved as a centrally acting muscle relaxant (Chou et al., 2004), its mechanism of action of CHZ as a centrally acting muscle relaxant is not understood. It is interesting that the concentration range of CHZ that restores the precision of pacemaking in Purkinje cells is equivalent to the plasma concentration of CHZ found in rats after a clinically relevant dose (Kwon, 2003). Given recent findings that aberrant cerebellar activity can cause dyskinesia and even dystonia (LeDoux and Lorden, 1998; Pizoli et al., 2002; Richter and Loscher, 1998), it is possible that the therapeutic efficacy of CHZ as a centrally acting “muscle relaxant” might be as a consequence of its action on the activity of Purkinje cells.
When administered orally, CHZ improved the baseline motor performance and also significantly reduced the severity and frequency of stress-induced attacks of dyskinesia in the tottering mice. CHZ remained effective in reducing the motor symptoms for as long as it was administered and did not produce tolerance. It should be noted that our battery of tests did not reveal any adverse effects of CHZ on either motor function or cognitive tasks in wild type mice. In fact, in humans CHZ produces so few adverse effects that its rate of degradation by the liver is often used as a measure of liver function when other novel drugs are administered to human subjects (Ernstgard et al., 2004; Ernstgard et al., 2007). Unfortunately, the rarity of EA2 provides little incentive for pharmaceutical companies to explore novel agents. Our recent data (Alvina and Khodakhah, in review) also support the notion the therapeutic efficacy of 4-AP in EA2 might also be a consequence of 4-AP's ability to restore the precision of Purkinje cell pacemaking by broadening the duration of action potentials (thus permitting greater calcium influx per action potential). Since 4-AP can have neurologic side-effects in some patients, it might be of value to examine the efficacy of CHZ as an off-label prescription in patients affected with EA2.
Finally, one could cautiously suggest that CHZ might be effective in other hereditary ataxias and paroxysmal dyskinesias. For example, EA2 is allelic to Spinocerebellar ataxia type-6 (SCA6), which is the consequence of CAG repeats expansions within the P/Q-type calcium channel gene (Zhuchenko et al., 1997). Recently, it has been shown that, without affecting their intrinsic biophysical properties (gating and kinetics), this mutation results in an age-dependent decrease in the P/Q-type calcium current density in cerebellar Purkinje cells due to accumulation of mutated channels (Watase et al., 2008). It is therefore likely that the loss of precision of pacemaking seen in Purkinje cells affected with the EA2 mutation might also be present to some extent in SCA6 patients and may partially contribute to their symptoms. In contrast to EA2 there is significant cerebellar neurodegeneration in SCA6 and it is therefore unlikely that any pharmacological intervention would be able to rescue motor function to normalcy in this disorder. Nonetheless at face value, and pending further investigation, CHZ might be of some value in these patients and in other cerebellar disorders where the signal to noise ratio of the output of the cerebellum is reduced as a consequence of a reduction is the precision of encoded information in individual Purkinje cells, or collectively as a whole in the cerebellum due to degeneration and thus reduced averaging of the encoded information by the neurons of the deep cerebellar nuclei. In a recent clinical trial in Europe Riluzole, an activator of SK channels similar to CHZ, was prescribed to patients suffering from a diverse set of cerebellar ataxia. While the reasoning for prescription of a SK channel activator for these patients was entirely different to that presented here, it is encouraging to note that in this double blind placebo controlled study Riluzole was found to be extremely efficacious is reducing motor dysfunction (Ristori et al., 2010).
Supplementary Material
Acknowledgments
We thank members of Khodakhah laboratory for help and discussions. We are extremely grateful to Dr Palle Christophersen for his generous advice on SK channel activators.
Funding: This work was supported by grants from NIH
Reference List
- Alvina K, Khodakhah K. Selective regulation of spontaneous activity of neurons of the deep cerebellar nuclei by N-type calcium channels in juvenile rats. J Physiol. 2008;586:2523–2538. doi: 10.1113/jphysiol.2007.148197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barclay J, Balaguero N, Mione M, Ackerman SL, Letts VA, Brodbeck J, Canti C, Meir A, Page KM, Kusumi K, Perez-Reyes E, Lander ES, Frankel WN, Gardiner RM, Dolphin AC, Rees M. Ducky mouse phenotype of epilepsy and ataxia is associated with mutations in the Cacna2d2 gene and decreased calcium channel current in cerebellar Purkinje cells. J Neurosci. 2001;21:6095–6104. doi: 10.1523/JNEUROSCI.21-16-06095.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bever CT, Jr, Young D, Anderson PA, Krumholz A, Conway K, Leslie J, Eddington N, Plaisance KI, Panitch HS, Dhib-Jalbut S. The effects of 4-aminopyridine in multiple sclerosis patients: results of a randomized, placebo-controlled, double-blind, concentration-controlled, crossover trial. Neurology. 1994;44:1054–1059. doi: 10.1212/wnl.44.6.1054. [DOI] [PubMed] [Google Scholar]
- Brodbeck J, Davies A, Courtney JM, Meir A, Balaguero N, Canti C, Moss FJ, Page KM, Pratt WS, Hunt SP, Barclay J, Rees M, Dolphin AC. The ducky mutation in Cacna2d2 results in altered Purkinje cell morphology and is associated with the expression of a truncated alpha 2 delta-2 protein with abnormal function. J Biol Chem. 2002;277:7684–7693. doi: 10.1074/jbc.M109404200. [DOI] [PubMed] [Google Scholar]
- Campbell DB, North JB, Hess EJ. Tottering mouse motor dysfunction is abolished on the Purkinje cell degeneration (pcd) mutant background. Exp Neurol. 1999;160:268–278. doi: 10.1006/exnr.1999.7171. [DOI] [PubMed] [Google Scholar]
- Candia S, Garcia ML, Latorre R. Mode of action of iberiotoxin, a potent blocker of the large conductance Ca(2+)-activated K+ channel. Biophys J. 1992;63:583–590. doi: 10.1016/S0006-3495(92)81630-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y, Dreixler JC, Roizen JD, Roberts MT, Houamed KM. Modulation of recombinant small-conductance Ca(2+)-activated K(+) channels by the muscle relaxant chlorzoxazone and structurally related compounds. J Pharmacol Exp Ther. 2001;296:683–689. [PubMed] [Google Scholar]
- Chou R, Peterson K, Helfand M. Comparative efficacy and safety of skeletal muscle relaxants for spasticity and musculoskeletal conditions: a systematic review. J Pain Symptom Manage. 2004;28:140–175. doi: 10.1016/j.jpainsymman.2004.05.002. [DOI] [PubMed] [Google Scholar]
- Crawley JN. Behavioral phenotyping strategies for mutant mice. Neuron. 2008;57:809–818. doi: 10.1016/j.neuron.2008.03.001. [DOI] [PubMed] [Google Scholar]
- Dove LS, Abbott LC, Griffith WH. Whole-cell and single-channel analysis of P-type calcium currents in cerebellar Purkinje cells of leaner mutant mice. J Neurosci. 1998;18:7687–7699. doi: 10.1523/JNEUROSCI.18-19-07687.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernstgard L, Johanson G, Karlsson AS, Warholm M. Phenotyping of cytochrome P450 2E1 in vitro and in vivo. Curr Drug Metab. 2007;8:493–498. doi: 10.2174/138920007780866843. [DOI] [PubMed] [Google Scholar]
- Ernstgard L, Warholm M, Johanson G. Robustness of chlorzoxazone as an in vivo measure of cytochrome P450 2E1 activity. Br J Clin Pharmacol. 2004;58:190–200. doi: 10.1111/j.1365-2125.2004.02132.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans RM, Zamponi GW. Presynaptic Ca2+ channels--integration centers for neuronal signaling pathways. Trends Neurosci. 2006;29:617–624. doi: 10.1016/j.tins.2006.08.006. [DOI] [PubMed] [Google Scholar]
- Fletcher CF, Lutz CM, O'Sullivan TN, Shaughnessy JD, Jr, Hawkes R, Frankel WN, Copeland NG, Jenkins NA. Absence epilepsy in tottering mutant mice is associated with calcium channel defects. Cell. 1996;87:607–617. doi: 10.1016/s0092-8674(00)81381-1. [DOI] [PubMed] [Google Scholar]
- Glasauer S, Kalla R, Buttner U, Strupp M, Brandt T. 4-aminopyridine restores visual ocular motor function in upbeat nystagmus. J Neurol Neurosurg Psychiatry. 2005;76:451–453. doi: 10.1136/jnnp.2004.045716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoebeek FE, Stahl JS, van Alphen AM, Schonewille M, Luo C, Rutteman M, van den Maagdenberg AM, Molenaar PC, Goossens HH, Frens MA, De Zeeuw CI. Increased noise level of purkinje cell activities minimizes impact of their modulation during sensorimotor control. Neuron. 2005;45:953–965. doi: 10.1016/j.neuron.2005.02.012. [DOI] [PubMed] [Google Scholar]
- Jen J, Kim GW, Baloh RW. Clinical spectrum of episodic ataxia type 2. Neurology. 2004;62:17–22. doi: 10.1212/01.wnl.0000101675.61074.50. [DOI] [PubMed] [Google Scholar]
- Jen JC, Graves TD, Hess EJ, Hanna MG, Griggs RC, Baloh RW. Primary episodic ataxias: diagnosis, pathogenesis and treatment. Brain. 2007;130:2484–2493. doi: 10.1093/brain/awm126. [DOI] [PubMed] [Google Scholar]
- Jinnah HA, Hess EJ, LeDoux MS, Sharma N, Baxter MG, DeLong MR. Rodent models for dystonia research: characteristics, evaluation, and utility. Mov Disord. 2005;20:283–292. doi: 10.1002/mds.20364. [DOI] [PubMed] [Google Scholar]
- Judge SI, Bever CT., Jr Potassium channel blockers in multiple sclerosis: neuronal Kv channels and effects of symptomatic treatment. Pharmacol Ther. 2006;111:224–259. doi: 10.1016/j.pharmthera.2005.10.006. [DOI] [PubMed] [Google Scholar]
- Kwon JW. Effects of glucose on the pharmacokinetics of intravenous chlorzoxazone in rats with acute renal failure induced by uranyl nitrate. J Pharm Sci. 2003;92:1604–1613. doi: 10.1002/jps.10426. [DOI] [PubMed] [Google Scholar]
- LeDoux MS, Lorden JF. Abnormal cerebellar output in the genetically dystonic rat. Adv Neurol. 1998;78:63–78. [PubMed] [Google Scholar]
- Liu YC, Lo YK, Wu SN. Stimulatory effects of chlorzoxazone, a centrally acting muscle relaxant, on large conductance calcium-activated potassium channels in pituitary GH3 cells. Brain Res. 2003;959:86–97. doi: 10.1016/s0006-8993(02)03730-7. [DOI] [PubMed] [Google Scholar]
- Llinas R, Sugimori M. Electrophysiological properties of in vitro Purkinje cell dendrites in mammalian cerebellar slices. J Physiol (Lond) 1980;305:197–213. doi: 10.1113/jphysiol.1980.sp013358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohle M, Schrempf W, Wolz M, Reichmann H, Storch A. Potassium channel blocker 4-aminopyridine is effective in interictal cerebellar symptoms in episodic ataxia type 2--a video case report. Mov Disord. 2008;23:1314–1316. doi: 10.1002/mds.22071. [DOI] [PubMed] [Google Scholar]
- Matthews RT, McMillen BA, Speciale SG, Jarrah H, Shore PA, Sanghera MK, Shepard PD, German DC. Effects of zoxazolamine and related centrally acting muscle relaxants on nigrostriatal dopaminergic neurons. Brain Res Bull. 1984;12:479–486. doi: 10.1016/0361-9230(84)90163-1. [DOI] [PubMed] [Google Scholar]
- Mori Y, Friedrich T, Kim MS, Mikami A, Nakai J, Ruth P, Bosse E, Hofmann F, Flockerzi V, Furuichi T. Primary structure and functional expression from complementary DNA of a brain calcium channel. Nature. 1991;350:398–402. doi: 10.1038/350398a0. [DOI] [PubMed] [Google Scholar]
- Olesen SP, Munch E, Moldt P, Drejer J. Selective activation of Ca(2+)-dependent K+ channels by novel benzimidazolone. Eur J Pharmacol. 1994;251:53–59. doi: 10.1016/0014-2999(94)90442-1. [DOI] [PubMed] [Google Scholar]
- Ophoff RA, Terwindt GM, Vergouwe MN, van Eijk R, Oefner PJ, Hoffman SM, Lamerdin JE, Mohrenweiser HW, Bulman DE, Ferrari M, Haan J, Lindhout D, van Ommen GJ, Hofker MH, Ferrari MD, Frants RR. Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca2+ channel gene CACNL1A4. Cell. 1996;87:543–552. doi: 10.1016/s0092-8674(00)81373-2. [DOI] [PubMed] [Google Scholar]
- Pietrobon D. Function and dysfunction of synaptic calcium channels: insights from mouse models. Curr Opin Neurobiol. 2005;15:257–265. doi: 10.1016/j.conb.2005.05.010. [DOI] [PubMed] [Google Scholar]
- Pizoli CE, Jinnah HA, Billingsley ML, Hess EJ. Abnormal cerebellar signaling induces dystonia in mice. J Neurosci. 2002;22:7825–7833. doi: 10.1523/JNEUROSCI.22-17-07825.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter A, Loscher W. Pathology of idiopathic dystonia: findings from genetic animal models. Prog Neurobiol. 1998;54:633–677. doi: 10.1016/s0301-0082(97)00089-0. [DOI] [PubMed] [Google Scholar]
- Ristori G, Romano S, Visconti A, Cannoni S, Spadaro M, Frontali M, Pontieri FE, Vanacore N, Salvetti M. Riluzole in cerebellar ataxia: A randomized, double-blind, placebo-controlled pilot trial. Neurology. 2010;74:839–845. doi: 10.1212/WNL.0b013e3181d31e23. [DOI] [PubMed] [Google Scholar]
- Stea A, Tomlinson WJ, Soong TW, Bourinet E, Dubel SJ, Vincent SR, Snutch TP. Localization and functional properties of a rat brain alpha 1A calcium channel reflect similarities to neuronal Q- and P-type channels. Proc Natl Acad Sci U S A. 1994;91:10576–10580. doi: 10.1073/pnas.91.22.10576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone TW. Neuropharmacology of quinolinic and kynurenic acids. Pharmacol Rev. 1993;45:309–379. [PubMed] [Google Scholar]
- Strupp M, Kalla R, Dichgans M, Freilinger T, Glasauer S, Brandt T. Treatment of episodic ataxia type 2 with the potassium channel blocker 4-aminopyridine. Neurology. 2004;62:1623–1625. doi: 10.1212/01.wnl.0000125691.74109.53. [DOI] [PubMed] [Google Scholar]
- Strupp M, Kalla R, Glasauer S, Wagner J, Hufner K, Jahn K, Brandt T. Aminopyridines for the treatment of cerebellar and ocular motor disorders. Prog Brain Res. 2008;171:535–541. doi: 10.1016/S0079-6123(08)00676-6. [DOI] [PubMed] [Google Scholar]
- Strupp M, Zwergal A, Brandt T. Episodic ataxia type 2. Neurotherapeutics. 2007;4:267–273. doi: 10.1016/j.nurt.2007.01.014. [DOI] [PubMed] [Google Scholar]
- Syme CA, Gerlach AC, Singh AK, Devor DC. Pharmacological activation of cloned intermediate- and small-conductance Ca(2+)-activated K(+) channels. Am J Physiol Cell Physiol. 2000;278:C570–C581. doi: 10.1152/ajpcell.2000.278.3.C570. [DOI] [PubMed] [Google Scholar]
- Usowicz MM, Sugimori M, Cherksey B, Llinas R. P-type calcium channels in the somata and dendrites of adult cerebellar Purkinje cells. Neuron. 1992;9:1185–1199. doi: 10.1016/0896-6273(92)90076-p. [DOI] [PubMed] [Google Scholar]
- Wakamori M, Yamazaki K, Matsunodaira H, Teramoto T, Tanaka I, Niidome T, Sawada K, Nishizawa Y, Sekiguchi N, Mori E, Mori Y, Imoto K. Single tottering mutations responsible for the neuropathic phenotype of the P-type calcium channel. J Biol Chem. 1998;273:34857–34867. doi: 10.1074/jbc.273.52.34857. [DOI] [PubMed] [Google Scholar]
- Walter JT, Alvina K, Womack MD, Chevez C, Khodakhah K. Decreases in the precision of Purkinje cell pacemaking cause cerebellar dysfunction and ataxia. Nat Neurosci. 2006;9:389–397. doi: 10.1038/nn1648. [DOI] [PubMed] [Google Scholar]
- Wan J, Ernstgard L, Song BJ, Shoaf SE. Chlorzoxazone metabolism is increased in fasted Sprague-Dawley rats. J Pharm Pharmacol. 2006;58:51–61. doi: 10.1211/jpp.58.1.0007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watase K, Barrett CF, Miyazaki T, Ishiguro T, Ishikawa K, Hu Y, Unno T, Sun Y, Kasai S, Watanabe M, Gomez CM, Mizusawa H, Tsien RW, Zoghbi HY. Spinocerebellar ataxia type 6 knockin mice develop a progressive neuronal dysfunction with age-dependent accumulation of mutant CaV2.1 channels. Proc Natl Acad Sci U S A. 2008;105:11987–11992. doi: 10.1073/pnas.0804350105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisz CJ, Raike RS, Soria-Jasso LE, Hess EJ. Potassium channel blockers inhibit the triggers of attacks in the calcium channel mouse mutant tottering. J Neurosci. 2005;25:4141–4145. doi: 10.1523/JNEUROSCI.0098-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Womack M, Khodakhah K. Active contribution of dendrites to the tonic and trimodal patterns of activity in cerebellar purkinje neurons. J Neurosci. 2002;22:10603–10612. doi: 10.1523/JNEUROSCI.22-24-10603.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Womack MD, Chevez C, Khodakhah K. Calcium-activated potassium channels are selectively coupled to P/Q-type calcium channels in cerebellar Purkinje neurons. J Neurosci. 2004;24:8818–8822. doi: 10.1523/JNEUROSCI.2915-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Womack MD, Hoang C, Khodakhah K. Large conductance calcium-activated potassium channels affect both spontaneous firing and intracellular calcium concentration in cerebellar Purkinje neurons. Neuroscience. 2009;162:989–1000. doi: 10.1016/j.neuroscience.2009.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Womack MD, Khodakhah K. Somatic and dendritic small-conductance calcium-activated potassium channels regulate the output of cerebellar purkinje neurons. J Neurosci. 2003;23:2600–2607. doi: 10.1523/JNEUROSCI.23-07-02600.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Womack MD, Khodakhah K. Dendritic control of spontaneous bursting in cerebellar Purkinje cells. J Neurosci. 2004;24:3511–3521. doi: 10.1523/JNEUROSCI.0290-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon KW, Covey DF, Rothman SM. Multiple mechanisms of picrotoxin block of GABA-induced currents in rat hippocampal neurons. J Physiol. 1993;464:423–439. doi: 10.1113/jphysiol.1993.sp019643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuchenko O, Bailey J, Bonnen P, Ashizawa T, Stockton DW, Amos C, Dobyns WB, Subramony SH, Zoghbi HY, Lee CC. Autosomal dominant cerebellar ataxia (SCA6) associated with small polyglutamine expansions in the alpha 1A-voltage-dependent calcium channel. Nat Genet. 1997;15:62–69. doi: 10.1038/ng0197-62. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.