

Abstract
Ambient GABA in the brain activates GABAA receptors to produce tonic inhibition. Membrane potential influences both GABA transport and GABAA receptors and could thereby regulate tonic inhibition. We investigated the voltage-dependence of tonic currents in cultured rat hippocampal neurons using patch clamp techniques. Tonic GABAA conductance increased with depolarization from 15±3 pS/pF at −80 mV to 29±5 pS/pF at −40 mV. Inhibition of vesicular or nonvesicular GABA release did not prevent voltage-dependent increases of tonic conductance. Currents evoked with exogenous GABA (1 µM) were outwardly-rectifying, similar to tonic currents due to endogenous GABA. These results indicate that the voltage-dependent increase of tonic conductance was due to intrinsic GABAA receptor properties rather than an elevation of ambient GABA. Following transient depolarization to +40 mV, endogenous tonic currents measured at −60 mV were increased by 75±17%. This novel form of tonic current modulation, termed post-depolarization potentiation (PDP), recovered with a time constant of 63 s, was increased by exogenous GABA, and inhibited by GABAA receptor antagonists. Measurements of EGABA showed PDP was due to increased conductance and not a change in the anion gradient. To assess the functional significance of PDP, we used voltage-clamp waveforms that replicated epileptiform activity. PDP was produced by this pathophysiologic depolarization. These data show that depolarization produces prolonged potentiation of tonic conductance due to voltage-dependent properties of GABAA receptors. These properties are well suited to limit excitability during pathophysiologic depolarization accompanied by rises in ambient GABA, such as occur during seizures and ischemia.
Keywords: tonic inhibition, hippocampus, seizure, membrane potential, extrasynaptic
Introduction
In many areas of the brain ambient levels of extracellular GABA tonically activate GABAA receptors in extrasynaptic and perisynaptic locations (Semyanov et al., 2004; Farrant and Nusser, 2005). This tonic form of inhibition is mediated by GABAA receptors distinct from those underlying phasic inhibition at synapses and represents a potent inhibitory mechanism for neurons. For example, in mature cerebellar granule cells the total charge transfer from tonic inhibition is 3-fold larger than that produced by phasic inhibition (Hamann et al., 2002). Genetic or pharmacologic disruption of tonic inhibition increases firing rate of cerebellar granule cells in vitro and in vivo (Brickley et al., 2001; Chadderton et al., 2004), produces hippocampal hyperexcitability (Maguire et al., 2005; Glykys and Mody, 2006), promotes tonic firing of thalamocortical neurons (Cope et al., 2005), alters cellular and behavioral correlates of learning and memory (Cheng et al., 2006; Dawson et al., 2006), and influences anxiety-related behaviors (Shen et al., 2007). Additionally, the expression, localization, and function of GABAA receptor subunits underlying tonic inhibition is altered in experimental temporal lobe epilepsy (Houser and Esclapez, 2003; Peng et al., 2004; Scimemi et al., 2005; Zhang et al., 2007) suggesting a role for tonic inhibition in epileptogenesis. Defining the regulation of tonic inhibition is therefore highly relevant to brain function in health and disease.
Phasic inhibition is rapidly modulated over seconds to minutes through changes in presynaptic release, postsynaptic GABAA receptor modulation via second messenger cascades, and alterations in chloride gradients (Staley et al., 1995; Poisbeau et al., 1999; Radcliffe et al., 1999; Cai et al., 2002; Kullmann and Semyanov, 2002; Fujiwara-Tsukamoto et al., 2007; Wanaverbecq et al., 2007). In contrast, less is known about modulation of tonic inhibition over short time periods. The concentration of ambient GABA is influenced by vesicular GABA release at synapses, as well as both uptake and release of GABA by transporters (Wu et al., 2001; Richerson and Wu, 2003; Semyanov et al., 2003; Keros and Hablitz, 2005; Glykys and Mody, 2007; Wu et al., 2007). These mechanisms regulate tonic inhibition and are sensitive to changes in ongoing neural activity. Modulation of extrasynaptic GABAA receptor behavior could also regulate tonic inhibition. Because both GABA transporters and hippocampal GABAA receptors have voltage-dependent properties (Segal and Barker, 1984; Gray and Johnston, 1985; Yoon, 1994; Richerson and Wu, 2003; Wu et al., 2007), we investigated the effects of membrane depolarization on tonic GABA currents in rat hippocampal neurons. We find that membrane depolarization rapidly (within seconds) increases tonic GABA conductance. This increase of tonic current is independent of changes in ambient GABA or intracellular anion accumulation and is primarily due to intrinsic voltage-dependent properties of GABAA receptors. Finally, we demonstrate that potentiation of tonic currents by membrane depolarization persists following repolarization. This persistent effect is novel and is produced with pathophysiologic forms of depolarization. These properties are well suited to provide negative feedback to neurons during periods of robust depolarization.
Materials and Methods
Cell culture
Primary hippocampal cell cultures were prepared as previously described (Gaspary et al., 1998). In brief, 0–2 day old Sprague-Dawley rat pups of both sexes were decapitated and the hippocampi were dissected. The tissue was minced in sterile-filtered, HEPES-buffered solution and then treated with a digestion solution containing papain (10 U/ml), 0.5 mM EDTA, and cysteine (0.2 mg/ml) for 15 minutes. The enzyme-treated tissue was triturated in complete Minimum Essential Medium (MEM), trypsin inhibitor (1.5 mg/ml), and bovine serum albumin (1.5 mg/ml). Triturated cells were added to culture media (MEM/10%FBS, 100 U/ml penicillin, and 1.5 mg/ml streptomycin) and plated directly on polyornithine-coated round glass coverslips (Erie Scientific, Portsmouth, NH, USA) in 12 well culture dishes at a density of 2.5–5 × 105 cells/ml. After 1 hour, media was changed to 70% MEM/30% Neurobasal with B27 supplement (Gibco, Carlsbad, CA, USA). Cells were maintained in an incubator (Forma Scientific, model 3110, Marietta, OH) with a humidified environment containing 5% CO2 in room air at 37°C. Media was changed on day 5–6 to Neurobasal Medium with B27 supplement and 1 µM cytosine arabinoside (Ara-C), followed by half medium changes (without Ara-C) every 7 days.
Electrophysiology
Conventional whole-cell patch clamp techniques were used to record membrane currents from neurons aged 14–60 days in vitro. Cells were visualized with an Axiovert 200 inverted microscope with DIC optics (Carl Zeiss Inc., Thornwood, NY). Recordings were made using an Axopatch 1D amplifier, a Digidata 1440A A–D converter, and pClamp 10 software (Molecular Devices, Redwood City, CA). Data were acquired at 2–5 kHz and low-pass filtered at 1 kHz. Series resistance and whole-cell capacitance were determined and compensated 50–70% online; whole-cell capacitance was recorded as the compensation value from the front of the amplifier. Drug applications were made via a microperfusion device (SF-77B, Warner Instruments, Hamden, CT) connected to manifolds that allowed up to 5 test solutions to be applied in a single experiment. Flow through the manifolds was controlled with electronic solenoid valves (Parker Hannifin Corporation, General Valve Division, Cleveland, OH) and step application of test solutions was controlled by pClamp software. The microperfusion device produced 90% solution exchange times of 50–55 ms across open pipette tips. The recording chamber (RC-26, Warner Instruments, Hamden, CT) had a volume of ~0.3 ml and was continuously superfused at a rate of ~0.5 ml/min with bath solution that contained (in mM): 134 NaCl, 3 KCl, 1.4 NaH2PO4, 24 NaHCO3, 10 Dextrose, 2 MgCl2, 2 CaCl2, 1–3 Kynurenic acid, and 0.001 CGP 55845. The pH was 7.35–7.4 when bubbled with 95% O2/5% CO2. Osmolarity was adjusted to 305 mOsm with H2O. Kynurenic acid was omitted from solutions used to record spontaneous action potentials in the presence of 4-aminopyridine (4-AP). Patch electrodes were made with borosilicate glass without filament (593400, A–M Systems, Carlsborg, WA) using a micropipette puller (P-97; Sutter Instruments, Novato, CA). Pipettes had resistances of 2.5 to 3.5 MΩ when filled with an intracellular solution containing (in mM): 125 CsCl, 10 QX-314 chloride salt, 10 HEPES, 1 EGTA (pH corrected to 7.25 with CsOH). Osmolarity was adjusted to 275–285 mOsm with H2O as needed. In experiments with low intracellular Cl− this pipette solution was modified to include 120 mM Cs-methanesulfonic acid and 5 mM CsCl. To record action potentials in the presence of 4-AP, an intracellular solution was used that contained (in mM): 125 K-gluconate, 10 KCl, 10 HEPES, and 1 EGTA. All chemicals were purchased from Sigma except SKF-89976a, SNAP-5114 (Tocris-Cookson, Ellisville, MO), and QX-314 (Alomone Labs, Tel Aviv, Israel). Data acquisition was begun 3–5 minutes after establishing a whole-cell recording. Experiments were performed at room temperature, typically 23°C.
Analysis
Data analysis was performed with Clampfit (pClamp 10, Molecular Devices, Mountain View CA) and Origin (v6.1, Microcal Software, Northhampton, MA) software. Tonic current amplitudes were measured as the difference in mean holding current in the absence and presence of a GABAA receptor antagonist (bicuculline methiodide or SR95531). The mean current values were obtained from Gaussian fits of all-point amplitude histograms of current (e.g. Fig. 1B). Histograms were constructed from 2–10 seconds of data with a bin width of 1 pA. Gaussian fits were performed using a Levenberg-Marquardt curve-fitting algorithm provided in Clampfit software. Time constants for recovery of current potentiation were determined by fitting normalized current data with a single exponential function using a least squares curve-fitting routine provided in Origin software. The slope of conductance vs. voltage plots and I–V curves were determined by fitting data with a linear equation. Chord conductance, g, was calculated from the following equation:
where I is current amplitude, Em is membrane potential, and EGABA is the reversal potential for GABA currents (a composite of the equilibrium potential for permeant anions including Cl− and HCO3−). The theoretical Nernst potential for chloride was 0 mV and this value was used for the calculations of chord conductance presented in figure 1 & figure 2. In some experiments the reversal potential of tonic currents and currents evoked with exogenous GABA were measured directly and used as EGABA. The above equation was rearranged for the calculations of current presented in Fig. 10C. Data in Fig. 5E were fit with a Boltzmann equation of the following form:
where I/Icontrol is normalized current, q is the effective gating charge, V is membrane potential, V0.5 is the apparent half-maximal voltage, k is the Boltzmann constant, and T is temperature in Kelvin. Under our conditions the term kT was ~25.6 V*C. We calculated the percent inhibition for post-depolarization potentiation (PDP) of tonic currents produced by bicuculline as follows:
where %PDPBic and %PDPCon is the % potentiation of tonic currents seen following depolarization in the presence of bicuculline or under control conditions, respectively.
Figure 1.

Tonic GABAA conductance in rat hippocampal neurons increased with depolarization. A: Tonic GABAA receptor-mediated currents at −40 mV and +40 mV. Horizontal bars indicate period of bicuculline application (10 µM, Bic) in this and subsequent panels. B: Quantification of tonic current amplitude. All-points histograms were created from current data before and after Bic application (gray bars in right hand panel). The peaks of these histograms were fit with a Gaussian equation (solid lines in right hand panel) to determine mean current amplitudes (dashed lines). Tonic current was defined as change in mean holding current produced by bicuculline. C: Current-voltage (I–V) plot from the neuron in (A). Values are means of 2–3 measurements at each potential. Tonic currents were outwardly-rectifying. D: Tonic chord conductance as a function of voltage from data illustrated in panels A–C. Tonic conductance increased in a near-linear fashion with membrane depolarization. Solid line represents a linear fit of the data (slope = 13 pS/mV). E: Mean current density as a function of voltage for all neurons tested (n=12). F: Mean capacitance-specific conductance as a function of voltage for all neurons tested (n=12). Linear fit of the data had a slope of 0.32 pS*pF−1*mV−1.
Figure 2.

Voltage-dependent increases in tonic conductance were insensitive to inhibition of transporter-mediated or vesicular GABA release. A: Current responses to bicuculline application (Bic, 10 µM) under control conditions (left panel) and in the presence of SKF89976A (SKF, 40 µM) (right panel). Tonic current was increased by SKF. Holding potential was −40 mV. B: Mean tonic current density (pA/pF) vs. voltage under control conditions and in presence of SKF (n=6). Outward rectification was unaffected by SKF. C: Mean capacitance-specific conductance vs. voltage (n=6). There was a shift in the conductance curve, but the voltage-dependence of tonic conductance was unaffected by SKF, the slopes of the solid lines are 0.31 pS/pF*mV−1 and 0.33 pS/pF*mV−1 for control and SKF, respectively. Symbols are same as panel B. D: Current responses to Bic under control conditions (left panel) and with zero extracellular Ca2+/1 mM EGTA (right panel). As seen previously (Wu et al., 2006), phasic currents were reduced by zero extracellular Ca2+ but tonic currents were not (amplitudes of illustrated tonic currents were −31 pA for control and −38 pA for zero Ca2+). Holding potential was −60 mV. E: Tonic current density vs. voltage for experiments with zero Ca2+/1 mM EGTA (n=6). F: Capacitance-specific conductance vs. voltage for all neurons tested in zero Ca2+/1 mM EGTA. Same symbols as E. The voltage-dependent increase in tonic conductance was independent of extracellular Ca2+. Slopes of the solid lines are 0.30 pS/pF*mV−1 and 0.23 pS/pF*mV−1 for control and zero Ca2+, respectively.
Figure 10.

PDP was independent of intracellular Cl− accumulation. A: GABA currents measured every 30 s before and after depolarization to +40 mV. At t=0 GABA was co-applied to the depolarized neuron in a low extracellular [Cl−] bath solution (Na-methanesulfonate substitution for NaCl, [Cl−]o=3 mM). The net charge movement induced by GABA in low [Cl−] solution was −2.37 nC, indicating a net outward Cl− flux. PDP was still seen despite this reduction in intracellular Cl− concentration. No baseline adjustment was made to the displayed records. B: Mean normalized current from 7 experiments as in A (open circles). Filled circle represents the mean PDP produced by transient depolarization alone without GABA application. Solid line is a single exponential function.
Figure 5.

PDP of GABA current was time- and voltage-dependent. A: Current responses to GABA (1 µM) with step depolarizations of increasing duration. Duration of initial depolarization was 1 s and increased by 0.5 s with each subsequent trial (not all traces shown, illustrated traces are for 0, 1.5, 2.5, 3.5, and 4.5 s of depolarization). A progressive increase in current amplitude was seen upon repolarization as the duration of the preceding depolarization increased. Lower panel shows command potentials. Note that the current after 2.5 s of depolarization was larger than the peak current produced by GABA alone. B: Mean potentiation of current as a function of depolarization duration. Current was measured 1.5 s after repolarization and normalized to current values at corresponding time point of control trace (−60 mV) from experiments as in panel A (n=4). C: GABA-evoked currents (3 µM) with step depolarization to a range of potentials from −30 to +30 mV. Current at −60 mV was potentiated by transient depolarization to −30 mV (compared to control current recorded at −60 mV) and increased progressively as the magnitude of the preceding depolarization increased. D: Data from panel C on expanded time scale illustrating the current increase seen upon repolarization. Labels refer to value of preceding depolarization (−60 mV is control). E: Mean normalized current at −60 mV as a function of prepulse potential. Current was measured 1.5 s after repolarization and normalized to corresponding time point of control trace (n=4). Data from experiments as in panel C. Solid line represents a Boltzmann equation fit to the data; the apparent half-maximal voltage (V0.5) for PDP of GABA current was +10 mV.
Data values are presented as mean ± standard error of mean (SEM) and all error bars represent SEM. Statistical analyses were performed using Microsoft Excel (Bellevue, WA). A two-tailed, paired or homoscedastic Students t-test was used with a p-value of 0.05 considered as significant. Where appropriate the actual p-values are reported.
Results
Voltage dependence of tonic GABA conductance
The voltage-dependence of tonic inhibition was evaluated in cultured hippocampal neurons by measuring tonic current amplitude over a range of membrane potentials from −100 mV to +40 mV. Attention was focused on larger neurons (whole-cell capacitance range = 43–100 pF) and recording conditions were designed to isolate GABAA receptor currents. Representative current traces recorded at −40 mV and +40 mV during bicuculline application are presented in Fig. 1A. Tonic current amplitude was defined as the change in mean holding current induced by application of 10 µM bicuculline. Mean holding current was determined from Gaussian fits to all-points amplitude histograms constructed from segments of current data (Fig. 1B). Bicuculline was applied within 30 seconds of changing membrane potential. As shown previously, there was a significant amount of tonic current in cultured neurons (Wu et al., 2001). Because neurons were continuously superfused by bath solution, this presumably resulted from activation of GABAA receptors by ambient GABA that was trapped between the recorded neurons and other neurons, glia, or the culture dish. Tonic currents were outwardly-rectifying, seen by comparing currents measured at +40 mV and −40 mV (Fig 1A,B; EGABA ~ 0 mV) and in the I–V curve for tonic current (Fig.1C). Tonic current amplitudes in Fig. 1C are the mean of 1–3 measurements at each potential. Calculation of chord conductance (using the theoretical ECl value of 0 mV as EGABA) showed that tonic GABA conductance increased in a near-linear manner with membrane depolarization (Fig. 1D). Summary data for current and conductance (normalized to membrane capacitance) also showed outward rectification and a near-linear increase in conductance (Fig 1E,F; n=12 cells). The mean specific conductance was 15±3 pS/pF at −80 mV and 29±5 pS/pF at −40 mV (p<0.01). On average, tonic GABA conductance of hippocampal neurons increased with depolarization by 0.32 pS*pF−1*mV−1 (Fig. 1F).
The prolonged membrane depolarization used above to measure tonic current could potentially produce changes in intracellular Cl− and HCO3− concentrations. This could increase current amplitude through changes in electrochemical driving force rather than an actual increase in conductance. To evaluate this possibility, cells were held at −60, −20, and +40 mV for 2–3 minutes and then given voltage ramps (−60 mV to +40 mV over 2.5 s) before and during bicuculline exposure. Point-by-point subtraction of these records yielded the “bicuculline-sensitive” ramp current and allowed determination of tonic current reversal potential (i.e. EGABA) (data not shown). The reversal potential for tonic current was not significantly different when measured from holding potentials of −60 mV, −20 mV, or +40 mV (−0.1±0.8 mV, 0.1±1.0 mV, and 2.0±0.9 mV, respectively; n=8, p-values of 0.81 and 0.13). Thus, changes in EGABA cannot account for the increased tonic current amplitudes seen with depolarization. For changes in driving force alone to account for the increased tonic current amplitude seen with depolarization from −40 mV to +40 mV in the neuron in Fig. 1A–B, EGABA would have needed to shift by +24 mV (much larger than the insignificant shift of +2 mV seen experimentally).
Similar results were obtained using SR95531 (SR, 100 µM) as the GABAA receptor antagonist to measure tonic current. Tonic currents measured with SR were outwardly-rectifying and the capacitance-specific conductance increased during depolarization with a slope of 0.21 pS*pF−1*mV−1 (n=4). The mean capacitance-specific tonic conductance seen with SR was 8±2 pS/pF and 15±4 pS/pF at −80 mV and −40 mV, respectively. Although the capacitance-specific conductance seen in experiments with SR was lower than values obtained with bicuculline, with either antagonist there was a ≈2-fold increase in tonic conductance when cells were depolarized from −80 to −40 mV. Because these data with SR were obtained from a different subset of neurons than those with bicuculline, it is not possible to directly compare the absolute conductance between the two datasets. The lower conductance seen with SR may be due to variability between hippocampal cultures.
Effect of reducing vesicular and non-vesicular GABA release on the voltage-dependence of tonic currents
The increase in tonic conductance seen with depolarization could be due to elevated ambient [GABA]. Ambient GABA may increase during membrane depolarization through reduction of driving force for GABA uptake into the recorded neuron or nonvesicular release of GABA by reversal of GABA transport (Richerson and Wu, 2003; Wu et al., 2007). Because there was no GABA in our intracellular recording solution, nonvesicular release from the depolarized neuron was not expected to occur, but it is possible that depolarization would reduce GABA uptake. To assess the contribution of GABA transporters to the voltage-dependence of tonic inhibition, we measured tonic current in the presence of the GAT1 inhibitor SKF-89976A (SKF). SKF inhibits GABA transport in these neurons with an IC50 of 1.3 µM and >80% inhibition is produced by 10 µM (Wu et al., 2007). To ensure maximal block of GABA transport we used a high concentration of SKF (40 µM). The current response to exogenous GABA (0.5 µM) was not affected by 40 µM SKF (104±8% of control, n=7, p=0.65; data not shown), indicating that SKF did not act directly on GABAA receptors. As previously reported (Rossi et al., 2003; Wu et al., 2003), application of SKF induced an inward current that reached a peak within 15–20 seconds (average peak amplitude of −283±68 pA, n=6) and decayed to a steady-state within 60 seconds. This inward current was completely blocked by bicuculline (10 µM) indicating that it was due to elevated ambient GABA acting on GABAA receptors (Fig. 2A). At −60 mV, tonic current density was reversibly increased by SKF from −0.91±0.11 pA/pF to −2.05±0.15 pA/pF (n=6, p-value <0.01). Mean current density and capacitance-specific conductance from cells studied with SKF are plotted against voltage in Fig. 2B–C. In the presence of SKF, tonic currents displayed outward rectification and the near-linear relationship between tonic conductance and membrane potential seen under control conditions persisted. The specific conductance in the presence of SKF increased from 29±3 pS/pF to 46±9 pS/pF when neurons were depolarized from −80 mV to −40 mV. The solid lines in Fig 2C are linear fits to the data with slopes of 0.31 pS*pF−1*mV−1 and 0.33 pS*pF−1*mV−1 for control and SKF, respectively. Thus, there was a shift in conductance to higher levels due to increased ambient [GABA], without a change in the voltage dependence. The GAT-3 inhibitor SNAP-5114 (100 µM) alone or in combination with SKF did not affect the voltage dependence of tonic current (n=3, data not shown).
Vesicular release of GABA also regulates tonic current amplitude (Glykys and Mody, 2007). Depolarization of a recorded neuron could increase vesicular release from the same neuron or from neighboring cells. The inclusion of QX-314 in our pipette solution would limit action potential mediated vesicular release of GABA from recorded neurons. Furthermore, recordings were made from large, pyramidal-appearing neurons so most neurons studied were probably not GABAergic. In hippocampal neurons, evoked IPSCs between neuron pairs are completely blocked by zero extracellular Ca2+ (Wu et al., 2006). The frequency and amplitude of miniature IPSCs are also greatly reduced in the absence of extracellular Ca2+ to 25% and 18% of control, respectively (Wu et al., 2006), and the residual vesicular release is not increased by depolarization. To ensure that tonic conductance was not increased by depolarization as a result of evoked vesicular GABA release, tonic current was measured in bath solution containing 1 mM EGTA and no Ca2+ (Fig. 2D). In contrast to the reduction of phasic currents, tonic current amplitude was unchanged in zero Ca2+/1 mM EGTA (Fig. 2E–F) nor was the outward rectification and voltage-dependent increase of tonic conductance, implying that these phenomena were not caused by depolarization-evoked vesicular release of GABA. These experiments also excluded a contribution of Ca2+ entry via voltage-gated calcium channels to the voltage-dependence of tonic currents.
Voltage-dependence of current evoked with exogenous GABA
Inhibition of GABA transport or vesicular GABA release did not affect the increase in tonic current with depolarization, suggesting that this was not due to an elevation of ambient [GABA]. To assess the voltage-dependence of GABAA receptors, exogenous GABA (1 µM) was applied to neurons held at a range of membrane potentials (−100 mV to +40 mV). Current amplitude was measured early during GABA application (near peak current at −60 mV) and near steady state. As previously reported for hippocampal neurons (Segal and Barker, 1984; Ashwood et al., 1987; Yoon, 1994; Pytel et al., 2006), GABA-evoked currents were outwardly-rectifying (Fig. 3). The outward rectification was most pronounced when currents were measured at the end of a 10 s GABA application (Fig. 3B–C). A GABA concentration of 1 µM is not selective for extrasynaptic/perisynaptic receptors that mediate tonic currents, but the steady-state currents induced by this level of GABA are most relevant to tonic activation of these receptors. These data also show an acceleration of desensitization with hyperpolarization and slow, continuous activation of current at +40 mV (Fig. 3A) (Yoon, 1994). This latter feature was seen with GABA concentrations of 1–30 µM.
Figure 3.

Currents evoked with exogenous GABA were outwardly-rectifying. A: Current traces during GABA application (1 µM) at different holding potentials (as indicated). Baselines are adjusted to allow comparison of currents. Note the acceleration of desensitization at hyperpolarized potentials and the slow, continuous activation of currents at +40 mV. B: I–V plot of current from neuron in panel A. Currents were measured early during GABA application (near “peak” current at −100 mV; closed circle) and prior to offset of GABA (“steady-state”; open circle). Outward rectification is most pronounced for currents measured near steady-state. C: Mean current density (pA/pF) as a function of voltage from 5 cells studied as in panel A. D: Rectification ratio for endogenous tonic currents and currents evoked with exogenous GABA. Rectification ratio with exogenous GABA is for steady-state currents. No significant difference in rectification ratio was seen for currents evoked with exogenous GABA and endogenous tonic currents under control conditions (p=0.38). Rectification ratios of tonic currents with SKF or zero Ca2+ were not significantly different than control conditions (p=0.11 and p=0.90, respectively). Numbers in parentheses indicate number of cells in this and subsequent figures.
To compare the voltage-dependence of endogenous tonic currents and currents evoked with exogenous GABA we calculated the ratio of current at +40 mV to current at −40 mV (i.e. rectification ratio; Fig. 3D). The rectification ratio of currents evoked with exogenous GABA (1 µM) ranged between 1.93±0.2 for currents measured early during GABA application (near peak current at −60 mV) to 3.35±0.5 for currents measured near steady-state (n=5). These values were not significantly different from endogenous tonic currents under control conditions (rectification ratio 2.56±0.3, range=1.63–4.37, n=12). No significant changes in rectification ratio of tonic currents were seen in the presence of SKF (1.89±0.1, range=1.58–2.48, n=6; p=0.11) or with zero Ca2+/1 mM EGTA (2.78±0.5, range=1.70–4.73, n=6; p=0.46) compared to control conditions. The similar outward rectification of endogenous tonic currents and currents evoked with exogenous GABA is consistent with the voltage-dependence of tonic currents being primarily due to intrinsic properties of extrasynaptic GABAA receptors rather than to elevation of ambient GABA.
Post-depolarization potentiation (PDP) of GABA currents
While examining the voltage-dependence of GABA current, it was found that after GABA was applied at +40 mV, subsequent GABA currents at −60 mV were increased. To characterize this effect, repeated applications of GABA (1 µM) were made every 30 s at a holding potential of −60 mV. After a stable baseline was established, GABA was applied while neurons were depolarized to +40 mV (total depolarization time was 15 s). Neurons were then repolarized to −60 mV and the GABA response was measured again (Fig. 4A). On average, the peak and steady-state current amplitudes measured 30 s after repolarization were increased by 55±7% and 52±9%, respectively (n=5). This post-depolarization potentiation (PDP) of GABA current persisted for up to 120 s in some cells (see Fig 4A). Recovery from PDP followed an exponential time course. The time constant for recovery of PDP was estimated by normalizing current at −60 mV to baseline values and fitting a single exponential function to these data (Fig. 4B). The time constant for recovery of PDP was 25 s for the experiment illustrated (Fig. 4A–B) and was the same for summary data from 5 cells (Fig. 4C). PDP was concentration-dependent. With 3 µM GABA, steady-state currents measured 30 s after repolarization were potentiated by 131±9% (n=4) compared to 52±9% with 1 µM GABA (n=5; p<0.05). PDP was also seen with low intracellular Cl− (15 mM) and HCO3-free (HEPES buffer) bath solution (Supplemental Figure 1). Experiments with low intracellular Cl− were performed with 3 µM GABA. PDP with low intracellular Cl− averaged 161±35% (n=5) which was not different from PDP seen with 3 µM GABA and 135 mM intracellular Cl− (131±9%, n=4, p=0.48) (Supplemental Figure 1).
Figure 4.

Post-depolarization potentiation (PDP) of GABA currents. A: Current recordings during repeated GABA applications (1 µM, 5 s) at indicated times (holding potential −60 mV). At t=0 GABA was applied at +40 mV (note the slow activation of the GABA response similar to that seen in Fig. 3). Both peak and “steady-state” current at −60 mV were increased when measured 30 s after GABAA receptor activation at +40 mV. PDP of “steady-state” currents was 46% in this neuron. Baselines are adjusted to allow comparison of currents. B: Time course of normalized “steady-state” current amplitudes for data in panel A. Solid line represents a single exponential function fit to the data. Time constant for recovery of PDP was 25 s. C: Mean data from experiments on 5 cells as in panel A. The time constant for recovery was also 25 s.
Time- and voltage-dependence of PDP
To determine the minimum duration of depolarization required to potentiate GABA currents, neurons were depolarized to +40 mV for 1–4.5 s during GABA application (Fig 5A). Current was measured 1.5 s after repolarization to −60 mV and normalized to current at the corresponding time point during GABA application without depolarization. With 1 µM GABA the mean potentiation was 11±9% and 82±19% following depolarization of 1 s and 4.5 s, respectively (n= 4; Fig. 5B). For the cell shown in Fig. 5A, current after a 2.5 s depolarization was larger than the peak current produced by GABA application alone, suggesting that PDP was not simply reversal of desensitization.
The voltage-dependence of PDP was examined by depolarizing neurons from a holding potential of −60 mV to voltages between −30 mV and +30 mV (10 mV steps, 6.5 s duration) during GABA application (3 µM; Fig. 5C–D). Current was measured 1.5 s after repolarization to −60 mV and normalized to control current. Current potentiation was 13±3% (n=4) after depolarization to −30 mV even though the current continued to desensitize, suggesting that potentiation represents a different process than reversal of desensitization. Current potentiation increased progressively with greater depolarization reaching 99±19% at +30 mV (Fig. 5E). PDP was seen after depolarization to voltages at which little net current flow occurred (i.e. −10 mV to +10 mV), implying that PDP of GABA current was independent of intracellular anion accumulation during depolarization (see below).
PDP of tonic currents
PDP of tonic currents was investigated with continuous application of a low concentration of exogenous GABA (0.3 µM) to provide more selective activation of high-affinity extrasynaptic/perisynaptic GABAA receptors. The use of exogenous GABA would also minimize any contribution of voltage-dependent alterations in ambient [GABA] to PDP. Application of 0.3 µM GABA increased tonic current by −314±42 pA (n=11). Tonic current was measured at −60 mV with repeated application of bicuculline (10 µM, ~5 s) (Fig. 6A). After establishing a baseline, neurons were depolarized to +40 mV for 30 s, and tonic current was again measured after repolarization (Fig. 6A). Tonic current at −60 mV was increased by transient depolarization in all cells tested (n=11). Over the duration of a typical experiment (15–20 minutes), there was a progressive reduction in tonic current amplitude to −159±18 pA, apparently due to rundown and/or desensitization of extrasynaptic GABAA receptors (Fig. 6A–B). To account for this rundown/desensitization, tonic currents were normalized to the value preceding each depolarization. Peak potentiation under control conditions (measured 3–8 s after repolarization) averaged 122±13% (range 57–240%, n=11). Current rundown/desensitization was not associated with a reduction in PDP (p=0.44 for comparison of 1st and 2nd control depolarizations, n=11). PDP of tonic current recovered with a time course well described by a single exponential function (Fig 6B). The time constant for recovery averaged 45±4 s (range 23–78 s, n=11). PDP of tonic currents also occurred with smaller depolarizations to −20 mV (37±12%, n=4, data not shown).
Figure 6.
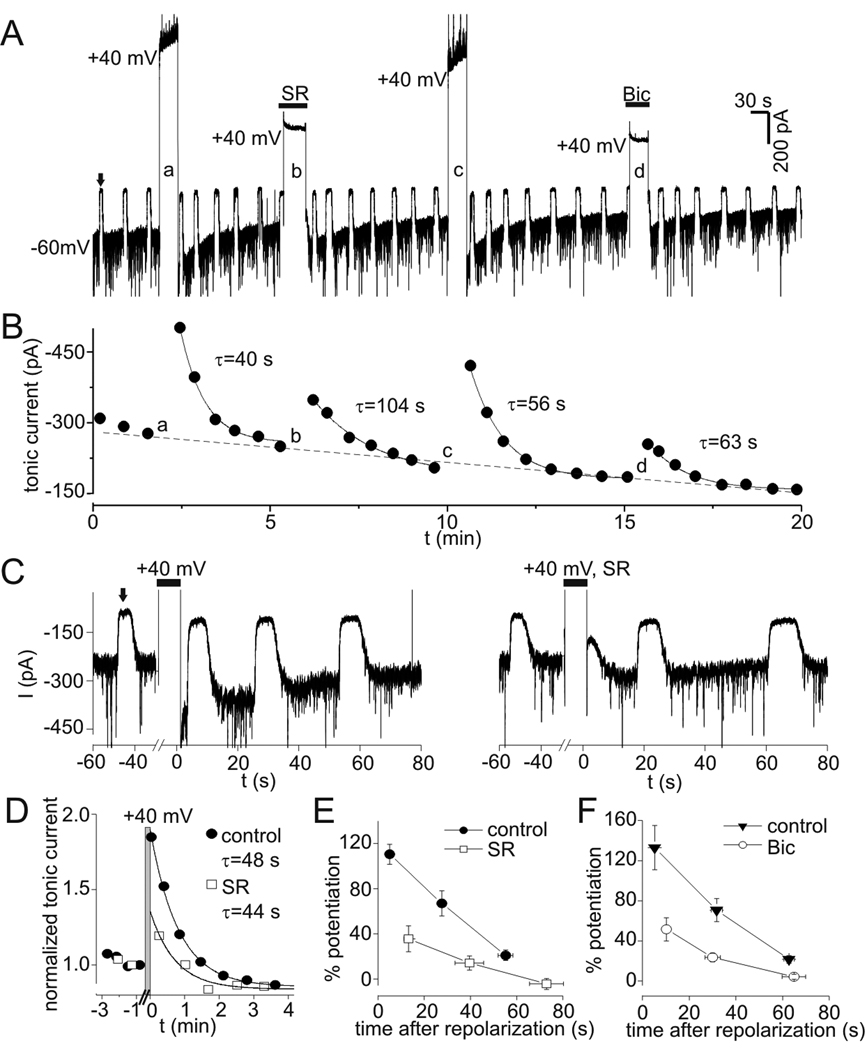
PDP of tonic currents. A: Tonic currents in the presence of low levels of exogenous GABA (0.3 µM). Upward deflections (e.g. the one indicated by solid arrow) represent response to 10 µM bicuculline (Bic) used to measure tonic current amplitude. Tonic current at −60 mV was increased by transient depolarization to +40 mV (large upward deflections). This increase was attenuated when depolarization occurred in the presence of the GABAA receptor antagonists SR95531 (SR, 100 µM) or Bic (10 µM). B: Tonic current amplitude from experiment in panel A. Solid lines represent single exponential fits, time constants (τ) for recovery after each depolarization are indicated. Lower case letters refer to sequentially labeled depolarizations in panel A. Note the progressive rundown of tonic current over duration of experiment (dashed line), which was seen in 11/11 cells studied with 0.3 µM GABA. C: PDP of tonic currents under control conditions (left panel) and when depolarization occurred in the presence of SR (right panel) on expanded time scale (different neuron than in A–B). Wash off of SR occurred in <10 s and was complete prior to measuring tonic current with Bic. D: Normalized tonic current from data in C. Data was normalized to value immediately preceding each depolarization and aligned so that repolarization occurred at t=0. Gray bar indicates period of depolarization to +40 mV. PDP in the presence of SR is reduced compared to control for 1 min after repolarization, much longer than the time required for wash of SR. Solid lines are single exponential fits to data with indicated time constants. E: Potentiation of first three measurements of tonic current after repolarization for experiments with and without SR. Peak PDP with SR was significantly reduced compared to both the first and second measurements under control conditions (p<0.01, n=5). F: Potentiation of tonic current vs. time after repolarization for experiments with and without Bic. PDP induced in Bic was significantly reduced compared to each corresponding control measurement (p<0.01, n=6).
PDP was attenuated when neurons were depolarized in the presence of the GABAA receptor antagonists SR or bicuculline (Fig. 6A–C). Wash of these antagonists began 1–3 s before repolarization and care was taken to allow complete wash of the antagonists prior to measuring tonic current (for example, see the right panel in Fig. 6C). Wash of SR was slower than bicuculline but was still complete within 10 s. Because of this, PDP was measured at variable times after depolarization with antagonists. To compare PDP under control conditions and after depolarization in the presence of antagonists, normalized tonic currents were aligned so that repolarization occurred at t=0 (Fig. 6D). With this method, it can be seen that SR application during depolarization reduced PDP for more than 60 s after repolarization (Fig. 6D), much longer than the time required for wash of SR. Mean potentiation is plotted as a function of time after repolarization in Fig. 6E and 6F. The first measurement of PDP after depolarization in the presence of SR was significantly reduced compared to both the first and second measurements under control conditions (n=5, p<0.01) (Fig. 6E). With bicuculline, PDP was also significantly reduced at all three time points compared to the corresponding control measurements (p<0.01, n=6). If incomplete wash of bicuculline after the prolonged application was the cause of PDP antagonism then the percent inhibition of PDP by bicuculline is predicted to decrease with time (as drug washed off). The percent inhibition produced by bicuculline for the 1st, 2nd, and 3rd measurements of tonic current after repolarization was 62±12%, 66±12%, and 83±16% (n=6). The trend for greater inhibition at the 3rd measurement was not significant (p=0.11). Thus, the reduction in PDP during application of antagonists was not due to incomplete washout of the drugs. The time constants for recovery of PDP following depolarization in the presence of SR (66±17 s, n=5) or bicuculline (47±8 s, n=6) were not significantly different from control (45±4 s, n=11, p=0.34 and 0.89, respectively).
Tonic current due to endogenous GABA was also persistently potentiated by depolarization (Fig. 7). In the absence of any exogenous GABA, tonic current was increased by >10% in response to transient depolarization in 67% (8/12) of cells tested, compared to 100% (11/11) of cells tested with 0.3 µM exogenous GABA. In cells that displayed PDP with endogenous GABA (defined as a >10% increase in current), the peak potentiation (75±17%, range 32–165, n=8) was smaller than seen with 0.3 µM GABA (121±13%, n=11, p=0.04). PDP with endogenous GABA recovered with a mean time constant of 63±15 s (Fig. 7B; range 25–135 s, n=8) and was antagonized by SR (100 µM) exposure during depolarization (Fig. 7C). Transient application of exogenous GABA (1 µM) for 5 s during neuronal depolarization only enhanced the subsequent PDP of endogenous tonic currents that occurred upon repolarization (Fig 7C). PDP of endogenous tonic current was also seen in the presence of SKF (40 µM) (153±53% potentiation, n=5) and in solutions with zero Ca2+/1 mM EGTA (52±2% potentiation, n=4) (data not shown).
Figure 7.

PDP of tonic currents due to endogenous GABA. A: Tonic currents due to endogenous GABA before and after depolarization to +40 mV. Period of depolarization is clipped to display the PDP of tonic current. Upward deflections (e.g. the one indicated by arrow) are the response to 10 µM bicuculline (Bic). B: Time course of normalized tonic current from panel A. Solid line is a single exponential fit with indicated time constant. C: Summary of peak PDP for tonic current due to endogenous GABA. Depolarization in the presence of 100 µM SR95531 (SR) attenuated PDP of endogenous tonic currents. PDP of endogenous tonic currents was increased by a 5 s application of exogenous GABA (1 µM) during the period of depolarization only (GABA). ** - p<0.01, * - p<0.05.
PDP was due to increased GABA conductance
The outward current produced by GABAA receptor activation at +40 mV represents an inward movement of anions and could lead to intracellular anion accumulation. The contribution of intracellular anion accumulation (and changes in electrochemical driving force) to PDP was evaluated by recording membrane currents in response to voltage ramps in the presence and absence of exogenous GABA (1–3 µM) (Fig. 8A). Difference currents were obtained by subtracting these records and used to determine the reversal potential of GABA-evoked currents (i.e. EGABA). To reproduce the timing previously used to measure PDP (Fig. 4A), difference currents were determined at baseline (t =−30 s) and then 30 s after GABA application at +40 mV (t=30 s) (Fig. 8B). Chord conductance at −60 mV was calculated using the measured value of EGABA. We also determined slope conductance from ramp currents using a linear fit to data between −60 mV and −10 mV. For the experiment illustrated in Fig. 8C, EGABA was −3.0 mV prior to depolarization. Thirty seconds after GABA was applied at +40 mV, EGABA was shifted to +3.6 mV indicating intracellular anion accumulation. The chord conductance was 17.2 nS under baseline conditions and 32.2 nS after depolarization, an 87% increase. Similar values were obtained from the slope conductance (a parameter that is independent of EGABA, values indicated in Fig. 8C). On average, EGABA shifted from −4.9±0.9 mV at baseline to +2.9±0.9 mV post-depolarization (n=5). Thus, the increased current during PDP cannot be accounted for simply by changes in driving force due to anion accumulation.
Figure 8.

PDP of GABA current was due to increased conductance. A: Membrane currents in response to voltage ramps before (control) and after GABA application (1 µM). The GABA-evoked ramp current (“difference current”) was obtained from point-by-point subtraction of these data for determination of reversal potential (i.e. EGABA) and slope conductance. B: Schematic of experimental protocol. Difference currents were obtained with voltage ramps 30 s before (t =−30 s) GABA was applied to a neuron depolarized to +40 mV (t=0) and then 30 s after this depolarization (t=30 s). C: GABA-evoked ramp currents during PDP. Difference currents are plotted as a function of voltage in the left- (t=−30 s) and right-hand (t=30 s) panels. Middle panel shows GABA response at +40 mV (t=0). Slope conductance was determined from a linear fit to current between −60 and −10 mV (solid lines in left-hand and right-hand panels). Chord conductance was calculated using the measured EGABA (indicated in figure) and current at −60 mV. Voltage ramps were given ~2 s into GABA applications. Slope and chord conductance were both increased by depolarization (values in figure). D: GABA-evoked (3 µM) ramp currents from a neuron recorded with pipette solution containing 15 mM Cl−. Same protocol as B-C. EGABA shifted from −57 mV at baseline (t=−30 s) to −32 mV after depolarization (t=30 s). Slope conductances are indicated in figure. E: Mean values of slope and chord conductance for experiments with [Cl−]in = 135 mM. The slope and chord conductances were increased by 60% and 56%, respectively (n=5, p<0.05). Legend applies to this panel and panel F. Data with [Cl−]in = 135 mM were pooled from experiments with 1–3 µM GABA. F: Mean values of slope and chord conductance for experiments with [Cl−]in = 15 mM. During PDP with [Cl−]in = 15 mM, conductance was increased by 91–132% (n=5, p<0.05). Data with [Cl−]in = 15 mM were obtained with 3 µM GABA.
PDP of conductance was also seen with physiological intracellular Cl− concentration ([Cl−]in=15 mM; Fig. 8D). With this lower [Cl−]in, EGABA was −59.8±2 mV at baseline and shifted to −28.5±5 mV 30 s after GABA (3 µM) was applied to depolarized neurons (n=5). Slope conductance of the neuron in Fig. 8D increased from 13 nS at baseline to 33 nS at t=30. Both slope and chord conductances were significantly increased after GABAA receptor activation at +40 mV with high or low intracellular Cl− concentrations (p<0.05, n=5 cells at each Cl− concentration) (Fig. 8E–F). In experiments with low [Cl−]in, chord conductance was calculated at −70 mV and slope conductance was determined with linear fits to data from −70 to −20 mV. PDP of GABA conductance was also seen in HCO3−-free bath solution (HEPES). The average chord conductance at −70 mV increased from 16.6 nS at baseline to 33.1 nS after depolarization in HEPES (n=2). There was no significant difference between the potentiation of slope conductance with low [Cl−]in (mean 86±26%, range 23–154%, n=5) and high [Cl−]in (mean 51±13%, range 20–94%, n=5, p-value=0.34).
Because PDP of GABA current was observed within seconds of repolarization (see Fig. 5A), it is possible that anion accumulation at these time points could be larger than that measured 30 s after repolarization. To evaluate this, chord conductance was measured before and after step depolarization in the presence of GABA (Supplemental Fig. 2). These data indicate that 1 s after repolarization, when anion accumulation should be maximal, PDP was associated with absolute increases in conductance.
Time course for PDP of tonic conductance
To determine the time course of tonic conductance changes produced by depolarization, we simultaneously measured reversal potential and tonic current amplitude in the presence of 0.3 µM exogenous GABA. A voltage ramp protocol (−30 to +30 mV over 500 ms) was repeated every 10 s from a holding potential of −60 mV. Bicuculline was applied at 30–50 s intervals and traces recorded in the presence of bicuculline were averaged and subtracted from control sweeps to yield tonic current amplitudes at −60 mV and bicuculline-sensitive ramp currents. Figure 9A illustrates bicuculline-sensitive ramp currents recorded during a baseline period at −60 mV, during depolarization to +40 mV, and then following repolarization to −60 mV (times of ramp currents indicated in figure). During depolarization, there was a progressive shift of reversal potential (i.e. EGABA) towards positive voltages that stabilized near +6 mV after 110 s of depolarization to +40 mV (experiment time of 260 s) (Fig 9A–B). Upon return to −60 mV, EGABA recovered towards control values over several minutes. Tonic conductance increased in parallel to EGABA during depolarization to +40 mV and continued to increase after EGABA approached a plateau (Fig. 9B). Tonic conductance of the cell illustrated in Fig. 9 peaked at 19.1 nS during depolarization and underwent a rapid drop to 8.7 nS upon return to −60 mV, but remained above the baseline value of 3.2 nS. PDP of tonic conductance following this prolonged (150 s) depolarization recovered to baseline with a time constant of 104 s (Fig. 9B). Similar results were obtained from other cells studied in this manner (n=4).
Figure 9.

Time course for PDP of tonic conductance. A: Bicuculline-sensitive ramp currents. Ramp currents were recorded every 10 s, first at a holding potential of −60 mV, during depolarization to +40 mV, and then after repolarization to −60 mV. Voltage ramps were from −30 to +30 mV over 0.5 s. Illustrated currents are difference currents obtained by subtracting ramp currents recorded in the presence of bicuculline from control currents. Bicuculline (10 µM) was applied every 30–50 s, the mean holding current at −60 mV in bicuculline was subtracted from control holding current prior to each voltage ramp to yield tonic current amplitude. Times at which currents were measured are indicated. Reversal potentials (i.e. EGABA) shifted towards more positive values during depolarization and recovered slowly after repolarization. Currents were recorded in the presence of 0.3 µM exogenous GABA. B: Double y-axis plot of tonic conductance (open circles) and EGABA (solid line) showing time course of changes produced by depolarization to +40 mV (horizontal bar). Chord conductance was calculated with EGABA and tonic current measured as in panel A. Conductance continued to increase during depolarization to +40 mV even after shifts in EGABA approached a plateau. Conductance dropped to 8.7 nS immediately upon repolarization to −60 mV, prior to substantial recovery of EGABA, but remained above baseline values. Solid line represents a single exponential fit to the recovery portion of conductance data. C: Time course for tonic current (open circles) from experiment in panels A–B. Theoretical changes in tonic current that would occur solely due to changes in driving force produced by shifts in EGABA, assuming no change in conductance from baseline, are plotted as a solid line for comparison to experimental data. PDP of tonic current in this situation would be only 27%, which is small compared to the 257% potentiation observed experimentally. Conversely, the theoretical current amplitude using the conductance values from panel B but assuming EGABA remained constant at −7 mV throughout the experiment is illustrated as triangles. In this case PDP would be 178% at its peak. Based on these calculations, changes in electromotive driving force from anion redistribution contributed no more than 10–30% to PDP of tonic current.
To assess the contribution of anion accumulation to PDP, we calculated the theoretical changes in tonic current that would result solely from shifts in EGABA for comparison to the observed changes in tonic current. Without any change in conductance, tonic current would decrease during depolarization as intracellular Cl−/HCO3− accumulated (Fig. 9C, solid line). This is in contrast to the slow increase in tonic current seen experimentally (Fig. 9C, open circles). Immediately following repolarization, tonic current would be increased by only 45 pA if there was only a shift of EGABA without conductance changes, a small increase compared to the observed increase in tonic current of 425 pA. Conversely, using experimentally determined chord conductance values we calculated the theoretical tonic current that would have resulted if EGABA remained constant (Fig. 9C, solid triangles). Given a chord conductance of 8.7 nS at the peak of PDP (Itonic = −590 pA, EGABA = +7.6 mV), the peak tonic current would be −463 pA if EGABA had remained constant at −7 mV. These calculations indicate that an increase in conductance was the dominant reason for the increase in current, and changes in electrochemical driving force due to anion accumulation contributed no more than 10–30% to PDP of tonic current.
PDP was not due to direct effects of elevated intracellular Cl− on GABAA receptors
The results presented above demonstrate an absolute increase in whole-cell conductance during PDP of GABA current, independent of driving force changes due to anion accumulation (Chabwine et al., 2004). In addition to effects on driving force, elevations of [Cl−] can directly increase the unitary conductance of GABAA receptors (Bormann et al., 1987; Fatima-Shad and Barry, 1993). The similarity between the time course for recovery of EGABA and conductance during PDP seen in Fig. 9B raised the possibility that modulation of single-channel conductance by Cl− ions contributed to PDP. To evaluate this possibility, we measured GABA currents (1 µM) at −60 mV every 30 s before and after GABA was applied during depolarization. In these experiments, however, GABA was co-applied to depolarized neurons with a low Cl− bath solution (Na-methanesulfonate substitution for NaCl, 3 mM Cl−) so that GABA would produce inward currents (outward movements of anions) even at +40 mV (Fig. 10). A transient outward current was seen at +40 mV as the low Cl− solution washed off while the receptors were still deactivating. The net charge movement induced by GABA in low Cl− solutions was determined by integrating baseline-adjusted traces recorded at +40 mV. The net charge movement produced by this procedure averaged −2.0±0.5 nC (n=7), indicating a net outward movement of anions despite the transient outward current seen with solution exchange during receptor deactivation (Fig. 10A). PDP still occurred after GABA receptor activation in low Cl− solution (39±6%, n=7), indicating that anion accumulation is unnecessary for the production of PDP (Fig. 10B). The amount of PDP seen with low Cl− solutions was not significantly different from that in cells on the same coverslips using normal bath solution (42±4%, n=3, p=0.80). Transient depolarization alone (without GABA application) also potentiated GABA currents at −60 mV (16±6%, n=3) but this was reduced compared to that seen with GABA application during depolarization with normal or low Cl− bath solution in cells from the same coverslips (p=0.02 and 0.05, respectively) (Fig. 10B). Depolarization alone did not affect EGABA (see above). These data support the conclusion that PDP resulted primarily from direct effects of membrane potential on GABAA receptor function rather than the effects of anion accumulation on driving force and single channel conductance. Macroscopic GABA currents in outside-out patches also underwent PDP, adding further support to the conclusion that PDP resulted from intrinsic receptor properties independent of anion redistribution or cellular factors (Supplemental Figure 3).
Potentiation of extrasynaptic GABAA receptors during epileptiform activity
To determine whether a physiological or pathophysiological pattern of depolarization can cause PDP of GABAA receptors, we recorded spontaneous epileptiform bursts during current-clamp experiments in the presence of 4-aminopyridine (100 µM) (Fig. 11A). We then used this waveform as a voltage clamp command. This waveform had a maximum action potential frequency of 40 Hz and mean membrane potential of −25 mV. When this stimulus was used during application of exogenous GABA (1–10 µM), current was potentiated following repolarization (Fig. 11B). Current was measured 0.2 s and 2 s following the depolarizing stimulus and compared to control current (with no depolarization). Two hundred milliseconds after the depolarizing stimulus, currents were increased by 40.5±9% and 125±25% with 1 µM and 10 µM GABA, respectively (Fig. 11C, n=4 cells at each concentration, p<0.01). Tonic currents induced by 0.3 µM exogenous GABA were also potentiated (defined as >10% increase) by the pathophysiologic depolarizing stimulus in 7/10 cells (Fig. 11D,E). Tonic currents were increased by 21±3% and 5±4% when measured 2 s and 20 s after the depolarizing stimulus, respectively (Fig.11F, n=7). Similar to results with step depolarizations, the frequency of PDP with epileptiform depolarization was greater in the presence of exogenous GABA. With endogenous GABA, PDP of tonic currents was seen in only 3/10 neurons following epileptiform depolarization, consistent with the smaller magnitude of PDP seen with lower GABA concentrations. While the potentiation seen with epileptiform bursts was smaller than that produced with step depolarization, these results demonstrate that persistent, voltage-dependent changes in the function of GABAA receptors underlying tonic conductance are likely to occur during pathophysiological neuronal discharges.
Figure 11.

GABA current was potentiated by an epileptiform burst. A: Membrane potential of a hippocampal neuron in the presence of 4-aminopyridine (100 µM). Bursts of high frequency action potentials occur spontaneously. Data enclosed in the dashed box was used to create the stimulus waveform used in voltage-clamp experiments. B: Responses to GABA (1 or 10 µM) at a holding potential of −60 mV with and without epileptiform depolarization. Current amplitude was measured 0.2 and 2 s after the stimulus (see dashed lines in right panel) and compared to control currents at the same time points. C: Mean potentiation of GABA current by epileptiform depolarization at 0.2 s and 2 s after repolarization (n=4). D: Tonic current before and after epileptiform depolarization. Illustrated currents are the average of 3 trials from this cell recorded in presence of 0.3 µM exogenous GABA. E: Normalized tonic current amplitude from experiment in panel D. F: Mean potentiation of tonic current by epileptiform depolarization (n=7).
Discussion
We describe substantial modulation of tonic GABAA receptor mediated current by membrane potential over a physiologic range, with ~2-fold increase in tonic conductance with depolarization from −80 mV to −40 mV. Voltage-dependent increases of tonic conductance were independent of GABA release and currents evoked with exogenous GABA were outwardly-rectifying, indicating that the voltage-dependence of tonic inhibition is primarily due to intrinsic GABAA receptor properties rather than increased ambient [GABA]. A novel finding is that augmentation of tonic currents by voltage persists upon repolarization, a phenomenon we have termed post-depolarization potentiation (PDP). This study adds to the mechanisms by which depolarization produces persistent changes in GABAA current, including alterations of Cl− gradients (Chabwine et al., 2004), internalization of receptors (Goodkin et al., 2008), and interactions between receptors and GABAAR-associated protein (GABARAP) (Kawaguchi and Hirano, 2007).
Contribution of anion shifts to PDP of GABAA receptors
GABAA receptor activation affects intracellular Cl− concentration (Staley et al., 1995; Isomura et al., 2003). Our conclusion that PDP results from direct effects of voltage on GABAA receptor function depends on assessing the consequences of altered electrochemical driving force for Cl−/HCO3−. If anion accumulation during depolarization was large enough to potentiate currents after repolarization, it should also reduce currents during the period of depolarization. In contrast, we observed a slow activation of current during depolarization. Calculation of chord conductance (using measured EGABA) directly revealed an increase in GABA conductance during PDP. Thus, PDP of tonic inhibition is independent of changes in electrochemical driving force due to anion accumulation.
In addition to effects on driving force, elevation of intracellular Cl− concentration increases the unitary conductance of GABAA receptors (Bormann et al., 1987; Fatima-Shad and Barry, 1993). This cannot entirely account for PDP for several reasons. First, the increase in current was similar in experiments with low and high intracellular [Cl−] (Supplemental Fig. 1), whereas the effect of intracellular [Cl−] on GABAA receptor conductance is greatest at low [Cl−] (Fatima-Shad and Barry, 1993). Second, PDP was seen with levels of depolarization at which there was little net current flow (−10 to +10 mV, Fig. 5C) and PDP was induced in low Cl− bath solutions that caused a reduction in intracellular Cl− (Supplemental Fig. 3). Third, tonic conductance continued to increase during depolarization after changes in EGABA approached a plateau (Fig. 9B). Finally, calculations of conductance do not support a large effect of intracellular [Cl−] on GABAA receptor conductance. In the extreme case of anion accumulation (Fig. 9B), intracellular [Cl−] was ~104 mM at baseline and approached a plateau near 180 mM during depolarization (assuming no change in extracellular Cl−/HCO3− or intracellular HCO3−). With these Cl− concentrations we estimate from the literature that unitary conductance at −60 mV would increase from ~26 pS to ~40 pS, a 53% increase (Fatima-Shad and Barry, 1993). This is small compared to the observed whole-cell conductance increase of 178% during PDP (measured before EGABA recovered). These considerations support the conclusion that PDP is primarily due to direct effects of voltage on GABAA receptor function that persist upon repolarization. Our estimates suggest that intracellular anion accumulation contributes no more than 30% to PDP of tonic conductance.
Mechanism for PDP of GABAA receptor function
Depolarization affects GABAA receptor function by slowing desensitization (Hablitz, 1992; Oh and Dichter, 1992; Yoon, 1994), increasing open probability (Weiss, 1988; Curmi et al., 1993), increasing single channel conductance (Gray and Johnston, 1985; Fatima-Shad and Barry, 1993), enhancing GABA affinity (Fisher, 2002), and slowing deactivation (Mellor and Randall, 1998; Pytel and Mozrzymas, 2006). One or more of these effects likely contribute to PDP. One possibility is that modifications of conformation and/or kinetic behavior that occur to favor slower deactivation, higher open probability, higher conductance states, and higher GABA affinity recover slowly upon repolarization. Cellular factors including kinases, phosphatases, and structural proteins (e.g. GABARAP/RAPSYN) that modulate GABAA receptor function and cellular localization may also influence PDP (Jones and Westbrook, 1997; Brandon et al., 2000; Chen et al., 2000; Everitt et al., 2004; Kawaguchi and Hirano, 2007; Marsden et al., 2007). However, these types of GABAA receptor modifications are manifest over longer time frames than that seen for PDP (which never lasted more than 3 minutes). We did not examine the participation of second messenger cascades, but calcium entry is unlikely to be involved because voltage-dependent increases of tonic conductance occurred in zero extracellular Ca2+/1 mM EGTA. PDP of tonic currents was also observed with 10 mM intracellular BAPTA (unpublished observations).
The voltage-dependence of GABAA receptors is dependent on subunit composition. The molecular substrates for the voltage-dependence of GABAA receptors are complex with interplay between expressed subunits. For example, α1β3δ receptors are outwardly-rectifying and α6β3δ receptors are inwardly-rectifying but this pattern switches for binary receptors without δ subunits, with marked outward rectification of α6β3 receptors compared to α1β3 (Bianchi et al., 2002). Basic amino acids in α and β subunits influence voltage-dependent properties (e.g. rectification, desensitization, and deactivation) (Fisher, 2002; Leidenheimer and Robinson, 2003). Because tonic inhibition in hippocampal neurons is mediated by both α5 subunit- and δ subunit-containing receptors, we speculate these contributed to the voltage-dependence and PDP of tonic inhibition described here (Glykys et al., 2008). Indeed, current mediated by α1β3δ and α5β3γ2L receptors display significant outward rectification, similar to tonic currents (Burgard et al., 1996; Bianchi et al., 2002; Semyanov et al., 2003).
Because PDP is independent of anion shifts, reduction of PDP with GABAA receptor antagonists must result from disruption of normal GABAA receptor gating during depolarization. Thus, modulation of extrasynaptic GABAA receptors by voltage is facilitated by agonist occupancy and gating (Weiss, 1988). Consistent with this, transient application of exogenous GABA during depolarization (to activate more receptors) enhanced PDP of endogenous tonic currents and currents evoked with periodic GABA application. In the case of tonic currents, exogenous GABA exposure during depolarization could enhance PDP by increasing the fraction of agonist-bound receptors. This is unlikely the only effect of depolarization because currents produced with periodic application of GABA also underwent PDP, indicating persistent augmentation of receptor function independent of an increased fraction of agonist-bound receptors. Variable activation of extrasynaptic receptors by ambient GABA in cultured neurons (due to differences in cell density or geometry of cell-cell contacts) may explain why PDP of endogenous tonic current was observed in only 67% of cells compared to 100% of cells with exogenous GABA. The activation of a greater number of receptors with exogenous GABA would lead to a greater number of receptors undergoing PDP, thereby increasing the magnitude of PDP and the frequency that PDP of tonic currents was observed.
Significance of voltage-dependent tonic conductance and PDP
Our results were obtained in cell culture at room temperature, very different conditions from the situation in vivo. However, because we are studying intrinsic biophysical properties of GABAA receptors, and ambient GABA is present both in vitro and in vivo, our basic observations are likely relevant to the in vivo situation. Temperature elevations do not affect tonic current in hippocampal slice (Semyanov et al., 2003) and GABA currents in CA1 pyramidal cells are outwardly-rectifying at near-physiologic temperatures (30–34°C) (Ashwood et al., 1987). These findings suggest that the voltage-dependent potentiation we report would also be seen at higher temperatures, although we have not tested this.
Tonic inhibition regulates neuronal excitability through effects on membrane potential and input resistance. The effect on membrane potential is dependent on the chloride gradient and could be hyperpolarizing or depolarizing. Independent of polarity, tonic GABA conductance provides a shunting form of inhibition, reducing the response to synaptic inputs. This alters the input-output relationship of neurons by offsetting action potential threshold and reducing action potential frequency (Brickley et al., 2001; Mitchell and Silver, 2003; Semyanov et al., 2003; Chadderton et al., 2004; Cope et al., 2005; Bonin et al., 2007). During high-frequency firing, when the mean membrane potential of hippocampal neurons can be −25 mV, we estimate that tonic conductance would increase by ~68% (for resting potential of −70 mV). Experimental and theoretical work predict this will further offset stimulus threshold and reduce the gain of action potential firing in response to excitatory synaptic inputs (Doiron et al., 2001; Mitchell and Silver, 2003). However, recent results indicate that the primary effect of tonic conductance in CA1 pyramidal cells is to offset action potential threshold (Pavlov et al., 2009). Voltage-dependent increases in tonic conductance may allow hippocampal neurons to rapidly adjust their input-output relationship in response to ongoing neural activity.
Voltage-dependent modulation of tonic conductance is magnified by its persistence following repolarization. We found that PDP occurred with patterns of depolarization similar to that experienced by neurons during ischemia and seizures. The magnitude of PDP was dependent on GABA concentration, making PDP highly relevant to these pathologic conditions that involve large depolarizations accompanied by increased extracellular GABA (During and Spencer, 1993; Allen et al., 2004). PDP of tonic conductance is well suited to provide negative feedback over seconds to minutes in these situations. Because membrane potential fluctuations do not occur uniformly throughout neurons, voltage-dependent modulation and PDP could occur within specific subcellular compartments. Potentiation of tonic inhibition in dendrites would alter synaptic integration and affect action potential propagation, similar to the effects of other ion channels, with implications for both firing rate and synaptic plasticity (Sjostrom et al., 2008).
Supplementary Material
Acknowledgements
The authors appreciate discussions with Dr. David S. Weiss during the course of experiments. The authors express special gratitude to the late Dr. Susan S. Spencer for her tireless commitment to colleagues, education, and science. This work was supported by the NIH (NS43288) (G.B.R. and Y.W.), the Bumpus Foundation (G.B.R.), the VAMC (G.B.R. & C.B.R.), and the National Epifellows Foundation (C.B.R.).
References
- Allen NJ, Rossi DJ, Attwell D. Sequential release of GABA by exocytosis and reversed uptake leads to neuronal swelling in simulated ischemia of hippocampal slices. Journal of Neuroscience. 2004;24:3837–3849. doi: 10.1523/JNEUROSCI.5539-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashwood TJ, Collingridge GL, Herron CE, Wheal HV. Voltage-clamp analysis of somatic gamma-aminobutyric acid responses in adult rat hippocampal CA1 neurones in vitro. Journal of Physiology. 1987;384:27–37. doi: 10.1113/jphysiol.1987.sp016441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi MT, Haas KF, Macdonald RL. a1 and a6 subunits specify distinct desensitization, deactivation and neurosteroid modulation of GABAA receptors containing the delta subunit. Neuropharmacology. 2002;43:492–502. doi: 10.1016/s0028-3908(02)00163-6. [DOI] [PubMed] [Google Scholar]
- Bonin RP, Martin LJ, MacDonald JF, Orser BA. α5 GABAA receptors regulate the intrinsic excitability of mouse hippocampal pyramidal neurons. Journal of Neurophysiology. 2007;98:2244–2254. doi: 10.1152/jn.00482.2007. [DOI] [PubMed] [Google Scholar]
- Bormann J, Hamill OP, Sakmann B. Mechanism of anion permeation through channels gated by glycine and gamma-aminobutyric acid in mouse cultured spinal neurones. Journal of Physiology. 1987;385:243–286. doi: 10.1113/jphysiol.1987.sp016493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandon NJ, Delmas P, Kittler JT, McDonald BJ, Sieghart W, Brown DA, Smart TG, Moss SJ. GABAA receptor phosphorylation and functional modulation in cortical neurons by a protein kinase C-dependent pathway. Journal of Biological Chemistry. 2000;275:38856–38862. doi: 10.1074/jbc.M004910200. [DOI] [PubMed] [Google Scholar]
- Brickley SG, Revilla V, Cull-Candy SG, Wisden W, Farrant M. Adaptive regulation of neuronal excitability by a voltage-independent potassium conductance. Nature. 2001;409:88–92. doi: 10.1038/35051086. [DOI] [PubMed] [Google Scholar]
- Burgard EC, Tietz EI, Neelands TR, Macdonald RL. Properties of recombinant gamma-aminobutyric acid A receptor isoforms containing the a5 subunit subtype. Molecular Pharmacology. 1996;50:119–127. [PubMed] [Google Scholar]
- Cai X, Flores-Hernandez J, Feng J, Yan Z. Activity-dependent bidirectional regulation of GABA(A) receptor channels by the 5-HT(4) receptor-mediated signalling in rat prefrontal cortical pyramidal neurons. Journal of Physiology. 2002;540:743–759. doi: 10.1113/jphysiol.2001.013391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabwine JN, Van Damme P, Eggermont J, De Smedt H, Missiaen L, Van Den Bosch L, Parys JB, Robberecht W, Callewaert G. Long-lasting changes in GABA responsiveness in cultured neurons. Neuroscience Letters. 2004;365:69–72. doi: 10.1016/j.neulet.2004.04.049. [DOI] [PubMed] [Google Scholar]
- Chadderton P, Margrie TW, Hausser M. Integration of quanta in cerebellar granule cells during sensory processing. Nature. 2004;428:856–860. doi: 10.1038/nature02442. [DOI] [PubMed] [Google Scholar]
- Chen L, Wang H, Vicini S, Olsen RW. The gamma-aminobutyric acid type A (GABAA) receptor-associated protein (GABARAP) promotes GABAA receptor clustering and modulates the channel kinetics. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:11557–11562. doi: 10.1073/pnas.190133497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng VY, Martin LJ, Elliott EM, Kim JH, Mount HT, Taverna FA, Roder JC, Macdonald JF, Bhambri A, Collinson N, Wafford KA, Orser BA. α5GABAA receptors mediate the amnestic but not sedative-hypnotic effects of the general anesthetic etomidate. Journal of Neuroscience. 2006;26:3713–3720. doi: 10.1523/JNEUROSCI.5024-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cope DW, Hughes SW, Crunelli V. GABAA receptor-mediated tonic inhibition in thalamic neurons. Journal of Neuroscience. 2005;25:11553–11563. doi: 10.1523/JNEUROSCI.3362-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curmi JP, Premkumar LS, Birnir B, Gage PW. The influence of membrane potential on chloride channels activated by GABA in rat cultured hippocampal neurons. Journal of Membrane Biology. 1993;136:273–280. doi: 10.1007/BF00233666. [DOI] [PubMed] [Google Scholar]
- Dawson GR, Maubach KA, Collinson N, Cobain M, Everitt BJ, MacLeod AM, Choudhury HI, McDonald LM, Pillai G, Rycroft W, Smith AJ, Sternfeld F, Tattersall FD, Wafford KA, Reynolds DS, Seabrook GR, Atack JR. An inverse agonist selective for a5 subunit-containing GABAA receptors enhances cognition. Journal of Pharmacology & Experimental Therapeutics. 2006;316:1335–1345. doi: 10.1124/jpet.105.092320. [DOI] [PubMed] [Google Scholar]
- Doiron B, Longtin A, Berman N, Maler L. Subtractive and divisive inhibition: effect of voltage-dependent inhibitory conductances and noise. Neural Computation. 2001;13:227–248. doi: 10.1162/089976601300014691. [DOI] [PubMed] [Google Scholar]
- During MJ, Spencer DD. Extracellular hippocampal glutamate and spontaneous seizure in the conscious human brain. Lancet. 1993;341:1607–1610. doi: 10.1016/0140-6736(93)90754-5. [DOI] [PubMed] [Google Scholar]
- Everitt AB, Luu T, Cromer B, Tierney ML, Birnir B, Olsen RW, Gage PW. Conductance of recombinant GABA (A) channels is increased in cells co-expressing GABA(A) receptor-associated protein. Journal of Biological Chemistry. 2004;279:21701–21706. doi: 10.1074/jbc.M312806200. [DOI] [PubMed] [Google Scholar]
- Farrant M, Nusser Z. Variations on an inhibitory theme: phasic and tonic activation of GABAA receptors. Nature Reviews Neuroscience. 2005;6:215–229. doi: 10.1038/nrn1625. [DOI] [PubMed] [Google Scholar]
- Fatima-Shad K, Barry PH. Anion permeation in GABA- and glycine-gated channels of mammalian cultured hippocampal neurons. Proceedings of the Royal Society of London - Series B: Biological Sciences. 1993;253:69–75. doi: 10.1098/rspb.1993.0083. [DOI] [PubMed] [Google Scholar]
- Fisher JL. A lysine residue in the β3 subunit contributes to the regulation of GABAA receptor activity by voltage. Molecular & Cellular Neurosciences. 2002;20:683–694. doi: 10.1006/mcne.2002.1143. [DOI] [PubMed] [Google Scholar]
- Fujiwara-Tsukamoto Y, Isomura Y, Imanishi M, Fukai T, Takada M. Distinct types of ionic modulation of GABA actions in pyramidal cells and interneurons during electrical induction of hippocampal seizure-like network activity. European Journal of Neuroscience. 2007;25:2713–2725. doi: 10.1111/j.1460-9568.2007.05543.x. [DOI] [PubMed] [Google Scholar]
- Gaspary HL, Wang W, Richerson GB. Carrier-mediated GABA release activates GABA receptors on hippocampal neurons. Journal of Neurophysiology. 1998;80:270–281. doi: 10.1152/jn.1998.80.1.270. [DOI] [PubMed] [Google Scholar]
- Glykys J, Mody I. Hippocampal network hyperactivity after selective reduction of tonic inhibition in GABAA receptor a5 subunit-deficient mice. Journal of Neurophysiology. 2006;95:2796–2807. doi: 10.1152/jn.01122.2005. [DOI] [PubMed] [Google Scholar]
- Glykys J, Mody I. The main source of ambient GABA responsible for tonic inhibition in the mouse hippocampus. Journal of Physiology. 2007;582:1163–1178. doi: 10.1113/jphysiol.2007.134460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glykys J, Mann EO, Mody I. Which GABAA receptor subunits are necessary for tonic inhibition in the hippocampus? Journal of Neuroscience. 2008;28:1421–1426. doi: 10.1523/JNEUROSCI.4751-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodkin HP, Joshi S, Mtchedlishvili Z, Brar J, Kapur J. Subunit-specific trafficking of GABAA receptors during status epilepticus. Journal of Neuroscience. 2008;28:2527–2538. doi: 10.1523/JNEUROSCI.3426-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray R, Johnston D. Rectification of single GABA-gated chloride channels in adult hippocampal neurons. Journal of Neurophysiology. 1985;54:134–142. doi: 10.1152/jn.1985.54.1.134. [DOI] [PubMed] [Google Scholar]
- Hablitz JJ. Voltage-dependence of GABAA-receptor desensitization in cultured chick cerebral neurons. Synapse. 1992;12:169–171. doi: 10.1002/syn.890120210. [DOI] [PubMed] [Google Scholar]
- Hamann M, Rossi DJ, Attwell D. Tonic and spillover inhibition of granule cells control information flow through cerebellar cortex. Neuron. 2002;33:625–633. doi: 10.1016/s0896-6273(02)00593-7. [DOI] [PubMed] [Google Scholar]
- Houser CR, Esclapez M. Downregulation of the a5 subunit of the GABAA receptor in the pilocarpine model of temporal lobe epilepsy. Hippocampus. 2003;13:633–645. doi: 10.1002/hipo.10108. [DOI] [PubMed] [Google Scholar]
- Isomura Y, Sugimoto M, Fujiwara-Tsukamoto Y, Yamamoto-Muraki S, Yamada J, Fukuda A. Synaptically activated Cl- accumulation responsible for depolarizing GABAergic responses in mature hippocampal neurons. Journal of Neurophysiology. 2003;90:2752–2756. doi: 10.1152/jn.00142.2003. [DOI] [PubMed] [Google Scholar]
- Jones MV, Westbrook GL. Shaping of IPSCs by endogenous calcineurin activity. Journal of Neuroscience. 1997;17:7626–7633. doi: 10.1523/JNEUROSCI.17-20-07626.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi S-y, Hirano T. Sustained structural change of GABAA receptor-associated protein underlies long-term potentiation at inhibitory synapses on a cerebellar purkinje neuron. Journal of Neuroscience. 2007;27:6788–6799. doi: 10.1523/JNEUROSCI.1981-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keros S, Hablitz JJ. Subtype-specific GABA transporter antagonists synergistically modulate phasic and tonic GABAA conductances in rat neocortex. Journal of Neurophysiology. 2005;94:2073–2085. doi: 10.1152/jn.00520.2005. [DOI] [PubMed] [Google Scholar]
- Kullmann DM, Semyanov A. Glutamatergic modulation of GABAergic signaling among hippocampal interneurons: novel mechanisms regulating hippocampal excitability. Epilepsia. 2002;5:174–178. doi: 10.1046/j.1528-1157.43.s.5.12.x. [DOI] [PubMed] [Google Scholar]
- Leidenheimer NJ, Robinson LC. Society for Neuroscience. New Orleans: 2003. Depolarization-mediated recovery of desensitized GABAA receptors. [Google Scholar]
- Maguire JL, Stell BM, Rafizadeh M, Mody I. Ovarian cycle-linked changes in GABAA receptors mediating tonic inhibition alter seizure susceptibility and anxiety. Nature Neuroscience. 2005;8:797–804. doi: 10.1038/nn1469. [DOI] [PubMed] [Google Scholar]
- Marsden KC, Beattie JB, Friedenthal J, Carroll RC. NMDA receptor activation potentiates inhibitory transmission through GABA receptor-associated protein-dependent exocytosis of GABAA receptors. Journal of Neuroscience. 2007;27:14326–14337. doi: 10.1523/JNEUROSCI.4433-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellor JR, Randall AD. Voltage-dependent deactivation and desensitization of GABA responses in cultured murine cerebellar granule cells. Journal of Physiology. 1998;506:377–390. doi: 10.1111/j.1469-7793.1998.377bw.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell SJ, Silver RA. Shunting inhibition modulates neuronal gain during synaptic excitation. Neuron. 2003;38:433–445. doi: 10.1016/s0896-6273(03)00200-9. [DOI] [PubMed] [Google Scholar]
- Oh DJ, Dichter MA. Desensitization of GABA-induced currents in cultured rat hippocampal neurons. Neuroscience. 1992;49:571–576. doi: 10.1016/0306-4522(92)90227-s. [DOI] [PubMed] [Google Scholar]
- Pavlov I, Savtchenko LP, Kullmann DM, Semyanov A, Walker MC. Outwardly Rectifying Tonically Active GABAA Receptors in Pyramidal Cells Modulate Neuronal Offset, Not Gain. Journal of Neuroscience. 2009;29:15341–15350. doi: 10.1523/JNEUROSCI.2747-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Z, Huang CS, Stell BM, Mody I, Houser CR. Altered expression of the d subunit of the GABAA receptor in a mouse model of temporal lobe epilepsy. Journal of Neuroscience. 2004;24:8629–8639. doi: 10.1523/JNEUROSCI.2877-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poisbeau P, Cheney MC, Browning MD, Mody I. Modulation of synaptic GABAA receptor function by PKA and PKC in adult hippocampal neurons. Journal of Neuroscience. 1999;19:674–683. doi: 10.1523/JNEUROSCI.19-02-00674.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pytel M, Mozrzymas JW. Membrane voltage differently affects mIPSCs and current responses recorded from somatic excised patches in rat hippocampal cultures. Neuroscience Letters. 2006;393:189–193. doi: 10.1016/j.neulet.2005.09.082. [DOI] [PubMed] [Google Scholar]
- Pytel M, Mercik K, Mozrzymas JW. Membrane voltage modulates the GABAA receptor gating in cultured rat hippocampal neurons. Neuropharmacology. 2006;50:143–153. doi: 10.1016/j.neuropharm.2005.08.001. [DOI] [PubMed] [Google Scholar]
- Radcliffe KA, Fisher JL, Gray R, Dani JA. Nicotinic modulation of glutamate and GABA synaptic transmission of hippocampal neurons. Annals of the New York Academy of Sciences. 1999;868:591–610. doi: 10.1111/j.1749-6632.1999.tb11332.x. [DOI] [PubMed] [Google Scholar]
- Richerson GB, Wu Y. Dynamic equilibrium of neurotransmitter transporters: not just for reuptake anymore. Journal of Neurophysiology. 2003;90:1363–1374. doi: 10.1152/jn.00317.2003. [DOI] [PubMed] [Google Scholar]
- Rossi DJ, Hamann M, Attwell D. Multiple modes of GABAergic inhibition of rat cerebellar granule cells. Journal of Physiology. 2003;548:97–110. doi: 10.1113/jphysiol.2002.036459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scimemi A, Semyanov A, Sperk G, Kullmann DM, Walker MC. Multiple and plastic receptors mediate tonic GABAA receptor currents in the hippocampus. Journal of Neuroscience. 2005;25:10016–10024. doi: 10.1523/JNEUROSCI.2520-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal M, Barker JL. Rat hippocampal neurons in culture: properties of GABA-activated Cl- ion conductance. Journal of Neurophysiology. 1984;51:500–515. doi: 10.1152/jn.1984.51.3.500. [DOI] [PubMed] [Google Scholar]
- Semyanov A, Walker MC, Kullmann DM. GABA uptake regulates cortical excitability via cell type-specific tonic inhibition. Nature Neuroscience. 2003;6:484–490. doi: 10.1038/nn1043. [DOI] [PubMed] [Google Scholar]
- Semyanov A, Walker MC, Kullmann DM, Silver RA. Tonically active GABAA receptors: modulating gain and maintaining the tone. Trends in Neurosciences. 2004;27:262–269. doi: 10.1016/j.tins.2004.03.005. [DOI] [PubMed] [Google Scholar]
- Shen H, Gong QH, Aoki C, Yuan M, Ruderman Y, Dattilo M, Williams K, Smith SS. Reversal of neurosteroid effects at a4b2d GABAA receptors triggers anxiety at puberty. Nature Neuroscience. 2007;10:469–477. doi: 10.1038/nn1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjostrom PJ, Rancz EA, Roth A, Hausser M. Dendritic excitability and synaptic plasticity. Physiological Reviews. 2008;88:769–840. doi: 10.1152/physrev.00016.2007. [DOI] [PubMed] [Google Scholar]
- Staley KJ, Soldo BL, Proctor WR. Ionic mechanisms of neuronal excitation by inhibitory GABAA receptors. Science. 1995;269:977–981. doi: 10.1126/science.7638623. [DOI] [PubMed] [Google Scholar]
- Wanaverbecq N, Semyanov A, Pavlov I, Walker MC, Kullmann DM. Cholinergic axons modulate GABAergic signaling among hippocampal interneurons via postsynaptic a7 nicotinic receptors. Journal of Neuroscience. 2007;27:5683–5693. doi: 10.1523/JNEUROSCI.1732-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss DS. Membrane potential modulates the activation of GABA-gated channels. Journal of Neurophysiology. 1988;59:514–527. doi: 10.1152/jn.1988.59.2.514. [DOI] [PubMed] [Google Scholar]
- Wu Y, Wang W, Richerson GB. GABA transaminase inhibition induces spontaneous and enhances depolarization-evoked GABA efflux via reversal of the GABA transporter. Journal of Neuroscience. 2001;21:2630–2639. doi: 10.1523/JNEUROSCI.21-08-02630.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Wang W, Richerson GB. Vigabatrin induces tonic inhibition via GABA transporter reversal without increasing vesicular GABA release. Journal of Neurophysiology. 2003;89:2021–2034. doi: 10.1152/jn.00856.2002. [DOI] [PubMed] [Google Scholar]
- Wu Y, Wang W, Richerson GB. The transmembrane sodium gradient influences ambient GABA concentration by altering the equilibrium of GABA transporters. Journal of Neurophysiology. 2006;96:2425–2436. doi: 10.1152/jn.00545.2006. [DOI] [PubMed] [Google Scholar]
- Wu Y, Wang W, Diez-Sampedro A, Richerson GB. Nonvesicular inhibitory neurotransmission via reversal of the GABA transporter GAT-1. Neuron. 2007;56:851–865. doi: 10.1016/j.neuron.2007.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon KW. Voltage-dependent modulation of GABAA receptor channel desensitization in rat hippocampal neurons. Journal of Neurophysiology. 1994;71:2151–2160. doi: 10.1152/jn.1994.71.6.2151. [DOI] [PubMed] [Google Scholar]
- Zhang N, Wei W, Mody I, Houser CR. Altered localization of GABAA receptor subunits on dentate granule cell dendrites influences tonic and phasic inhibition in a mouse model of epilepsy. Journal of Neuroscience. 2007;27:7520–7531. doi: 10.1523/JNEUROSCI.1555-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.