

Abstract
Custom-designed zinc finger nucleases (ZFNs) – proteins designed to cut at specific DNA sequences – combine the non-specific cleavage domain (N) of Fok I restriction endonuclease with zinc finger proteins (ZFPs). Because the recognition specificities of the ZFPs can be easily manipulated experimentally, ZFNs offer a general way to deliver a targeted site-specific double-strand break (DSB) to the genome. They have become powerful tools for enhancing gene targeting – the process of replacing a gene within a genome of cells via homologous recombination (HR) – by several orders of magnitude. ZFN-mediated gene targeting thus confers molecular biologists with the ability to site-specifically and permanently alter not only plant and mammalian genomes but also many other organisms by stimulating HR via a targeted genomic DSB. Site-specific engineering of the plant and mammalian genome in cells so far has been hindered by the low frequency of HR. In ZFN-mediated gene targeting, this is circumvented by using designer ZFNs to cut at the desired chromosomal locus inside the cells. The DNA break is then patched up using the new investigator-provided genetic information and the cells’ own repair machinery. The accuracy and high efficiency of the HR process combined with the ability to design ZFNs that target most DNA sequences (if not all) makes ZFN technology not only a powerful research tool for site-specific manipulation of the plant and mammalian genomes, but also potentially for human therapeutics in the future, in particular for targeted engineering of the human genome of clinically transplantable stem cells.
Keywords: Zinc finger nucleases, gene targeting, genome engineering, site-specific modification, targeted mutagenesis, gene correction, homologous recombination, non-homologous end-joining
Introduction
A robust and reproducible means of specifically correcting faulty bases (or introducing targeted mutations) in genes has been a long-sought goal for genetic medicine. Current methods to manipulate the plant and mammalian genomes have two limitations: (1) the very low rate of homologous recombination (HR) at the targeted site; and (2) the relatively high rate of random, non-targeted integration elsewhere in the genome by non-homologous end joining (NHEJ). In these cells, the former, HR, occurs at a very low rate compared to the latter, NHEJ (Fig. 1). For most mammalian cells, targeted recombinants via HR are overshadowed by nearly 1000-fold higher random, non-targeted integrants (NHEJ) [1–3]. Cells use the universal process of HR to maintain their genomic integrity, particularly in the repair of a double-strand break (DSB), which otherwise would be lethal. Repair of a DSB in a damaged chromosome by HR is a highly accurate form of repair which uses the homologous DNA from the undamaged chromosomal partner as a template. Gene targeting – the process of modifying a gene by HR – uses an extrachromosomal fragment of donor template DNA and invokes the cell’s HR machinery for sequence exchange [1]. Gene targeting is not a very efficient process in plant and mammalian cells; only ~1 in 106 cells provided with excess template sequences undergo the desired gene modification. However, when a targeted genomic DSB is introduced in cells, it induces HR at that local site to repair the DSB in a larger fraction of the cells compared with spontaneous HR [2, 4]. Thus, generation of specific, desired genomic DSB is the rate-limiting step in homology-directed repair (HR) technology for gene modification.
Figure 1.

Schematic representation of two of several possible mechanisms for a DSB repair (HR and NHEJ) in mammalian cells. The DSB (double-strand break) may arise spontaneously such as by damage from reactive oxygen species during normal metabolism, induced randomly by the exposure to ionizing radiation, or induced specifically by zinc finger nucleases. The repair of the gene by homologous recombination (HR) occurs after the induction of a DSB. During HR, the DSB is processed to form free 3′ single-strand tails, a process that requires the Mre11/Rad50/Nbs1 complex [61]. The HR machinery, through the actions of the strand invasion protein Rad51, then uses the free 3′ ends to invade a homologous repair template/donor. How the machinery identifies a homologous repair donor remains unclear, but it is likely that simple physical proximity plays an important role. In the normal repair of a DSB, the repair donor is the sister chromatid, and thus the template is identical to the damaged allele. In gene targeting, the repair donor would be an extrachromosomal piece of DNA that could have sequence differences. After strand invasion, primed DNA synthesis occurs to generate new undamaged DNA using the undamaged donor DNA template. The process is completed by the annealing of the new strand of DNA with its original partner and subsequent use of that new DNA to template DNA synthesis. During non-homologous end joining (NHEJ), the broken ends from the DSB are bound by specific protein factors to limit nucleolytic degradation. The ends are then bridged together by protein-protein interactions between the protein factors. The sequences are then aligned and ligated [62]. NHEJ often occurs without any homology or even microhomologies between the ends that are ligated. NHEJ is mutagenic by nature because it often introduces small deletions or insertions at the fusion site.
Fortunately, techniques have been developed to enable routine use of HR for gene modification in mouse embryonic stem cells (MESCs). However, in many other cell types, successful modification of a gene is still not achievable or is a tour de force. Several approaches, including combined positive-negative selection, are used to enrich the ratio of targeted recombinants to random integrants. Positive-negative selection works well especially in MESCs [1], but does not work well in many other plant and mammalian cell lines, including primary human stem-progenitor cells (HSPCs), and human embryonic stem cells (HESCs), mainly due to difficulties in rigorously cloning these cell types [5]. Therefore, rare targeted recombinants must be selected by long-term clonal culture, which is not feasible using primary human HSPCs because the in vivo engrafting potential of HPSCs is quickly lost during ex vivo culture. Clonal selection is very difficult in HESCs, mainly because HESCs do not grow effectively from single cells [6].
Zinc finger nucleases
Zinc finger nucleases (ZFNs) were originally developed in our lab for delivering a single, targeted genomic DSB within cells [7]. The modular nature of Fok I endonuclease suggested that it might be feasible to engineer chimeric nucleases by fusing other DNA-binding proteins to the Fok I cleavage domain [8]. Several novel fusion nucleases were generated by linking the non-specific Fok I cleavage domain to other DNA-binding proteins [7, 9, 10]. The most extensively studied group of chimeric nucleases is based on the zinc finger proteins (ZFPs), because individual zinc finger (ZF) motifs within ZFPs can be designed to bind a large range of DNA sites. Cys2His2 ZFPs bind DNA by inserting an α-helix into the major groove of the double helix [11]. Each ZF motif primarily binds to a triplet within the DNA substrate. Key amino acids, at positions –1, 2, 3, and 6 relative to the start of the α-helix, contribute most of the specific interactions by the ZF motifs [11–13]. These amino acids can be changed, while maintaining the remaining amino acids as a consensus backbone, to generate ZFPs with different sequence specificities [14, 15]. Furthermore, the ZFP strategy has the additional advantage that greater specificity can be achieved by adding more ZF motifs to the ZFPs [16–18]. Thus, ZF DNA-binding motifs, because of their modular structure and function, offer an attractive framework for designing ZFNs with custom sequence specificities [18, 19].
Several 3-finger ZFPs, each recognizing a 9 base pair (bp) sequence, have been fused to the non-specific endonuclease domain of Fok I to form ZFNs [9, 20]. The binding specificity of a ZFP correlates directly with the cleavage specificity of the corresponding ZFN [21]. It has been shown that Fok I restriction endonuclease must dimerize via the nuclease domain in order to cleave and induce a DSB [22, 23]. Similarly, ZFNs also require dimerization of the nuclease domain in order to cut DNA [24, 25]. Dimerization of the ZFN, and hence double-strand cleavage, is facilitated by two adjacent, oppositely oriented binding sites [25]. Since a pair of 3-finger ZFNs requires two copies of the 9-bp recognition sites in a tail-to-tail orientation with an optimal spacer sequence in order to dimerize and produce a DSB, they effectively have an 18-bp recognition site, which is long enough to specify a unique genomic address in mammals. In principle, the binding sites need not be identical, provided the ZFNs that bind both sites are present (Fig. 2). We have shown that two 3-finger ZFNs with different sequence specificities collaborate to produce a DSB when their binding sites are appropriately placed and oriented with respect to each other [25]. Urnov et al. have shown that the specificity could be further increased by using a pair of 4-finger ZFNs that recognize a 24-bp sequence, with a decrease in cytotoxicity at the same time [4].
Figure 2.
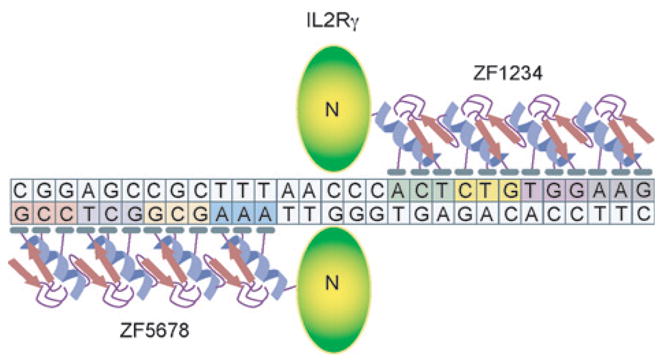
Designer molecular scissors for genome surgery. A schematic representation of a pair of 4-finger ZFNs (zinc finger nucleases) bound to their cognate sites in the IL2Rγ gene [4]. Since a pair of ZFNs requires two copies of the 12-bp recognition sites in a tail-to-tail orientation, they effectively have a 24-bp recognition site, which is long enough to specify a unique genomic address in human cells. Since the ZFN-binding sites in the IL2Rγ gene are not identical, both ZFNs that bind these sites need to be introduced into the cell to induce a targeted genomic DSB. The ZFN sites in the IL2Rγ gene are separated by 5 bp [4].
ZFP design and selection strategies
The creation of highly specific ZFNs that recognize and cleave any target sequence depends on the reliable design and/or selection of ZFPs that can recognize and bind that chosen target with high affinity and specificity. The design and various selection approaches, including the phage display method, are discussed in detail elsewhere [20, 26–30] and are not the subject of this review. The ZF motif designs that specifically recognize each and every one of the possible 64 DNA triplets are not yet available. A wealth of literature data based on design and phage display methodologies is available for the ZF modules that specifically recognize 5′-GNN-3′ and 5′-ANN-3′ triplets, but to a lesser extent the ZF motif preferences for 5′-CNN-3′ and 5′-TNN-3′ triplets are known [26, 31–34]. Currently, two Web-based ZF design softwares are available and can be accessed at URLs http://www.zincfingertools.org [35] and http://www.zincfingers.org/software-tools.htm. The above limits the range of DNA triplets that could be targeted using a design strategy. Since a DSB delivered several hundred base pairs away from the chosen site induces HR, almost all the genes encoded in the genome of a cell are amenable to ZFN-mediated gene targeting. Recent developments, including several cell-based selection strategies using bacterial one-hybrid and two-hybrid systems, to produce highly specific ZFPs have been reported in the literature [36–38]. Hurt et al. have shown that highly specific ZFPs can be obtained by directed domain shuffling and cell-based selection [37]. Both DNA binding affinity and specificity are important determinants for cellular function [37]. Ideally, all ZF motifs in the engineered ZFP need to be optimized concurrently since co-operative and neighboring context-dependent contacts can affect DNA recognition. The simultaneous selection of key contacts in even a 3-finger ZFP requires screening a large number of mutant library members. The strategy of using domain shuffling and cell-based selection appears to provide a general approach for optimizing multi-finger ZFPs.
More recently, we have reported on two bacterial one-hybrid systems for interrogating and/or selecting ZFP-DNA interactions [36]. Our systems utilize two plasmids: a ZF plasmid containing the gene for the ZFP fused to a fragment of the α subunit of RNA polymerase and a reporter plasmid where the ZFP-binding site is located upstream of a reporter gene –either the gene encoding the green fluorescent protein (GFP) or chloramphenicol acetyltransferase (CAT). Upon binding of the ZFP to the target binding site, a 10-fold increase in chloramphenicol resistance was observed with the CAT reporter system, and an 8- to 22-fold increase in total cell fluorescence with the GFP reporter system. The CAT reporter allows for sequence-specific ZFPs to be isolated in a single selection step, whereas the GFP reporter enables quantitative evaluation of libraries using flow cytometry and allows for both negative and positive selection. Both systems have been used to select for ZFPs that have affinity for the site 5′-GGGGCAGAA-3′ from a library of approximately 2 × 105 variants. The systems have been engineered to report on ZFP-DNA binding with dissociation constants less than about 1 μM in order to be most applicable for evaluating binding specificity in an in vivo setting [36].
ZFN-mediated gene targeting: the approach
ZFN-mediated gene targeting methodology entails the following steps:
Identify ZFN target sites near the targeted locus within the Gene X of interest [20, 39, 40].
Design and/or select ZFPs that recognize the chosen ZFN target sites [20, 26, 39, 40].
Convert the designed and/or selected ZFPs into ZFNs [20, 39, 40].
Deliver ZFNs alone to induce a targeted DSB in Gene X of normal cells and stimulate NHEJ to generate a pool of mutants (mGene X), some of which will be frame-shift mutations resulting in functional deletion of Gene X, i.e. knock-outs of Gene X (Fig. 3). Alternatively, deliver ZFNs and the mutant Gene X donor fragment into normal cells to induce a targeted DSB and stimulate HR to generate a specific Gene X mutant, i.e. knockouts or knock-ins of Gene X as the case may be (Fig. 3). To achieve gene editing (or gene correction), deliver ZFNs and the correcting donor Gene X fragment into mutant cells (Fig. 3).
Monitor for gene correction (or mutagenesis) at the targeted gene loci as per the investigator-provided donor Gene X template [4].
It is also critical to show that the donor DNA fragment has not integrated elsewhere within the genome of the cell by using Southern [4].
Figure 3.

A schematic representation of ZFN-mediated gene targeting in human cells. Targeted ZFN-mediated disruption of the Gene X by NHEJ occurs when cells are transfected with designer ZFNs alone. The ZFNs induce a targeted genomic DSB in cells, which stimulates NHEJ that is mutagenic by nature. It gives rise to a pool of different Gene X mutants (depicted as mGene X). Targeted disruption of the Gene X by ZFN-evoked HR occurs when the normal cells are transfected with both designer ZFNs and the homologous donor DNA containing a predetermined Gene X modification to generate a specific desired mutant (depicted as smGene X) of the targeted gene. The ZFNs induce a targeted genomic DSB in cells, which stimulates local HR in the presence of homologous donor DNA, the recombinogenic repair pathway. Alternatively, to achieve targeted gene correction (or gene editing), the mutant Gene X cells are transfected with both designer ZFNs and the homologous correcting donor DNA fragment of the target gene to generate normal cells.
ZFN-mediated gene targeting of plant and mammalian genomes
Reports from several labs including ours have shown that designed 3-finger ZFNs find and cleave their chromosomal targets in frog oocytes [41], Drosophila [42, 43], plant cells [44, 45], Caenorhabditis elegans [46] and human cells [4, 47, 48]; and as expected, they induce local HR at the site of cleavage to repair the DSB. In the absence of HR (for example, if both alleles of a gene are damaged), cells repair the DSB by simple ligation with the addition or deletion of some sequence (via NHEJ); as a result, repair by NHEJ is mutagenic. NHEJ-mediated repair occurs for DSBs induced by ZFNs in Drosophila [42, 43] and Arabidopsis [44]. Thus, to induce targeted gene modifications in a variety of organisms and cells, ZFNs can be used with or without a homologous template DNA sequence (involving HR and NHEJ, respectively). In a recent seminal Nature article, Urnov et al. [4] used designed 4-finger ZFNs in human cells to target an endogenous target site within the IL2Rγ gene [which causes the human X-linked disease severe combined immune deficiency (SCID)]. They achieved highly efficient permanent modification of the IL2Rγ gene in the K562 cell line – a remarkable gene-modification efficiency of 18% of K562 cells had the desired gene replacement without additional cell selection, and 1/3 of these were altered on both X chromosomes. They obtained similar results using primary human T lymphocytes. Thus, designer ZFNs have become powerful molecular tools to deliver a targeted genomic DSB to cells and stimulate local HR, with an exogenously provided DNA template [4, 42, 45, 47–50]. The high targeting efficiency attests to the power of the ZFN-evoked HR for site-specific engineering of the human genome and raises the possibility of developing ZFN-based strategies (1) for laboratory research of gene-modified human cells, to complement what can be done to study gene function in knock-out and knock-in models of mice and other species, and (2) potentially for human gene therapy in the future [4, 28, 47, 48, 50–53]. There is potential, then, to develop efficient ZFN-based strategies as a means of enhancing gene targeting in HSPCs and HESCs for human therapeutics.
Xenopus laevis
The first successful ZFN-mediated gene-targeting experiment was done in Xenopus using an extrachromosomal plasmid substrate, which was designed to undergo an intramolecular HR event upon ZFN-induced DSB [41]. The ZFN (QQR) was composed of three ZF motifs connected by a flexible linker to the Fok I cleavage domain. The QQR-binding sites for the ZFN with various spacer lengths and in different orientations were cloned into a circular plasmid substrate containing a unique Pvu II restriction site. The target DNA was initially injected directly into the nuclei of the oocytes to assemble into chromatin, and 6 h later the ZFN (QQR) was microinjected into the nuclei. When the plasmid DNA was recovered and analyzed by Southern, the results indicated that, as expected, ZFN cleavage had occurred, followed by an intramolecular recombination of the plasmid substrate. Thus, ZFN binds and cleaves its target site in the extrachromosomal plasmid, inducing local HR in cells [41]. Many factors for a successful ZFN cleavage were explored in this study, including the spacer length between the two DNA-binding sites, the orientation of the ZFN-binding sites with respect to each other and the linker length connecting the ZFPs to the Fok I cleavage domain [41]. A pair of tail-to-tail inverted binding site repeats provided the most effective substrate for cleavage by ZFNs and efficient HR. Single copy of the binding site and head-to-head inverted repeats were completely ineffective. The optimal spacer length was determined to be 6-bp between the inverted binding sites for ZFNs without a linker when the cleavage was very specific and highly efficient [41]. Under optimal conditions, greater than 95% of the plasmid substrate that were cleaved, and underwent intramolecular recombination [41]. Thus, this proof-of-principle study firmly established ZFN-mediated gene targeting as a viable option for site specific and permanent modification of genomes of plant and mammalian cells at an endogenous chromosomal locus for subsequent studies.
Drosophila melanogaster
Bibikova et al. extended the study to a Drosophila model system and demonstrated that ZFN-mediated gene targeting is very effective in creating a targeted chromosomal DSB, which leads to mutagenesis via NHEJ in the absence of a donor DNA. In a subsequent study, they showed an increased frequency of ZFN-mediated gene targeting at a chosen chromosomal locus in the presence of a donor DNA [42, 43, 49]. Altogether, three genes in Drosophila, namely, yellow gene, rosy gene and brown gene, have been targeted; all were chosen for the ease of scoring the mutant phenotypes. In the targeted mutagenesis experiment with the yellow gene, a pair of 3-finger ZFNs was used to target the y gene on the X chromosome. The binding site contained a 6-bp spacer, and the two newly engineered ZFNs were cloned separately under the control of a heat-shock promoter. About half of the male progeny showed obvious y patches, indicating somatic mutations in the y gene. Germline mutations were recovered from 5.7% of males, but none from the females tested. This was as anticipated, because in females, the DSB could be repaired by HR using the uncut homolog, thereby significantly lowering the efficiency of ZFN-mediated gene targeting; and thus the second y+ gene would mask the recessive mutation. In males, only NHEJ is available to repair damage. Mutants represented 0.4% of offspring. They also found that expression of one of the ZFNs was lethal when induced at 37 °C. This toxicity was reduced by lowering the heat-shock temperature [43]. In addition, when a linear, extrachromosomal mutant donor DNA was provided, ZFN-stimulated homologous recombination was observed, and recovery of germline mutations was done in both males and females. 18% of males and 13.5% of females gave at least one mutant offspring. More than 2% of offspring from males were mutant, and 63% of them were products of HR, and about 0.5% of offspring from females. No mutant progeny from females were observed without the donor [43]. Subsequently, two more genes were targeted in Drosophila: ry and bw; both function in the pathway leading to red eye pigment. Mutants would result in brown eyes. For targeted mutagenesis at bw, no mutants were recovered from female parents, and 2.4% of males gave mutants, roughly 0.1% of offspring. The mutant yield at ry was much higher: at optimal heat-shock temperature, 89% of females and 67% of males produced mutants, representing 14% and 6.8% of total offspring, respectively. Gene targeting with an extrachromosomal linear mutant donor was also done at ry. More than 90% of induced parents gave at least one mutant offspring [49]. Thus, these studies showed that ZFN-induced HR frequency was dramatically higher than that without the targeted cleavage using ZFNs, and the presence of a linear donor elevated the yield of targeted HR products even higher. However, lethality due to excessive cleavage was also observed when some of the designed ZFNs were overexpressed, demonstrating that the specificity of 3-finger ZFNs needs to be increased and their expression needs to be regulated. In the case of a heat-shock promoter, the toxicity of 3-finger ZFNs could be reduced by lowering the heat shock temperature [42, 43, 49].
Caenorhabditis elegans
No efficient gene targeting procedure was available for C. elegans until recently, when gene targeting using 3-finger ZFNs was shown to be very effective [46]. Both a synthetic extrachromosomal sequence as well as an endogenous genomic site on the X chromosome were successfully targeted using corresponding pairs of engineered 3-finger ZFNs [46]. In the former experiment, a previously characterized ZFN (QQR) was cloned in an expression plasmid, under the control of a heat-shock promoter. The extrachromosomal target contained two inverted QQR-binding sites separated by a unique Mlu I restriction site. The QQR expression plasmid and the target plasmid were co-injected to generate extrachromosomal array, typically consisting of tens to hundreds of copies of injected plasmids, and the larvae were heat-shocked to induce ZFN (QQR) expression. When the PCR-amplified DNA surrounding the QQR target site was analyzed by digesting with Mlu I restriction enzyme, the result indicated that 26% of targets examined contained QQR-induced mutations [46]. In the latter experiment an endogenous chromosomal locus was targeted, to demonstrate that a pair of newly designed ZFNs cleave the chromosomal target site and NHEJ ensues in the absence of donor DNA, introducing mutations [46]. The latter experiment was also repeated using a lig-4 (ok716) strain, a C. elegans mutant lacking DNA ligase IV, to show that DNA ligase IV was required for efficient end joining. Thus, this study confirmed ZFN-mediated gene targeting to be a powerful and versatile tool for targeted genome modification of many different organisms including those, which are not amenable to gene targeting by existing technologies and methods [46]. It must be pointed out that no ZFN expression has yet been achieved in the germline.
Plants
Efficient and commonly usable methods for targeted modification of genomes are not available for plants. In 2005, however, ZFN-mediated gene targeting was successfully applied for targeted genome modification of plants. In this study the target site, QEQ, for a previously reported 3-finger ZFN (QQR) was first introduced into the genome of Arabidopsis thaliana [44]. The QEQ sequence is a synthetic 24-bp oligo-nucleotide that contains two inverted binding sites for QQR separated by a unique EcoR I restriction site. A construct containing ZFN (QQR) driven by the heat-shock promoter was then introduced into A. thaliana [44] cells containing the target QEQ site. After transformation, seven single-locus cell lines were identified. The seedlings from these lines were heat-shocked, and DNA was isolated from the seedlings. The PCR-amplified DNA surrounding the inserted QEQ site was analyzed by EcoR I digestion with the expectation that the EcoR I site would be lost if mutations are generated at the QEQ locus. Undigested PCR-amplified fragments were found in heat-shocked seedlings, but were absent in control seedlings, which were not heat-shocked. The results showed that, as expected, induction of ZFN expression resulted in mutations at the chromosomal target QEQ locus at frequencies as high as 0.4 mutations per cell, which was much higher than the typical rate of less than 10−7 gene-targeting events per cell without ZFNs.
More recently, ZFN-mediated gene targeting was successfully applied to tobacco plants to increase the frequency of localized HR [45]. In this system, HR frequency was measured by restoring function to a defective GUS:NPTII reporter gene integrated at various chromosomal sites in 10 different transgenic tobacco lines. Normal GUS:NPTII gene was a fusion of β-glucuronidase (GUS) and neomycin phosphotransferase (NPTII). Cells expressing this fusion are resistant to kanamycin (Kan) and turn blue when incubated in appropriate substrates. The target GUS:NPTII gene was first made non-functional by a 600-bp deletion with concomitant insertion of a 3-finger ZFN target site at the site of deletion. The donor was about 5 kb containing the GUS:NPTII gene, including the 600 bp missing from the target gene. The target gene was introduced into the tobacco chromosome. Protoplasts were prepared from 10 plants which had incorporated the target gene at various locations. When these cells were exposed to both donor and ZFN DNA, more than 1 HR event for every 10 illegitimate recombination events was observed, which was a 104–105-fold improvement from the typical rate of 105–106 illegitimate events per HR event [45]. And 1 in 5 characterized gene targeting events was free of mutations, indicating the fidelity of gene targeting was about 20% [45].
Human cells
A gene-targeting GFP reporter system designed to study the efficiency and efficacy of gene targeting by ZFNs in human cells was reported by three different labs, first by Porteus and Baltimore [48] and followed later by Urnov et al. [4] and Alwin et al. [47]. A similar system was used previously to study homology-directed repair of DNA damage [54]. In the Porteus and Baltimore study, a non-functional eGFP gene was integrated in the genome of HEK 293 cells. When a wild-type eGFP donor fragment alone was introduced into these cells, 1–2 cells of 500,000 treated cells were corrected for GFP. However, when an expression construct encoding ZFNs targeting the mutant GFP locus was introduced along with the donor, 2.2% of asynchronously growing cells and 10.2% of cells arrested for 30 h at the G2/M cell cycle boundary, respectively, had their eGFP function restored [48]. Once the potential of ZFNs for gene correction was established using the GFP reporter system, engineered ZFNs were designed to target the human IL2Rγ gene, mutations in which cause X-linked SCID (severe combined immunodeficiency). Unlike earlier reports, Urnov et al. [4] used optimized 4-finger ZFNs to modify the sequence at the IL2Rγ gene locus in the K562 cell line and in primary CD4+ T lymphocytes. The exogenous donor template plasmid used in their study contained a fragment of the IL2Rγ gene with a silent point mutation that creates a novel restriction enzyme recognition site in the exon 5 sequence. ZFN-induced HR introduced this restriction site into the cognate chromosomal location. Four days after nucleofection of K562 cells with donor template plasmid plus ZFN plasmids, the HR frequency was 18%. The donor template-specified restriction site was confirmed by sequencing the IL2Rγ gene locus in the gene-modified cells. Southern blotting failed to detect ZFN-induced donor plasmid misintegration or any aberrant rearrangements of the IL2Rγ locus in cell samples harvested after 4 days or 1 month of cell expansion. Notably, HR frequency of cells harvested at 4 days after transfection was the same as that of cells harvested 1 month after transfection, in contrast to published studies using 3-finger ZFNs, which showed a decline [48]. This is promising because it suggested that the 4-finger ZFNs may be less toxic to cells than the 3-finger ZFNs; however, a direct comparison of the efficiency of gene targeting versus cytotoxicity for overlapping pairs of 3- and 4-finger ZFNs is yet to be done. Furthermore, by doing limiting dilution, single clones were isolated and genotyped. The results indicated that 13.2% of the clones had a single allele mutated, and 6.6% achieved biallelic gene modification [4]. Using the same protocol for IL2Rγ targeting by ZFNs in human CD4+ T lymphocytes, they observed an HR frequency of 5%. This was comparable to the HR frequency in K562 cells when adjusted for the lower transfection efficiency in T lymphocytes (30%). Thus, the Urnov et al. [4] study demonstrates that designed ZFNs can evoke HR to generate permanent and precise genome modification at the IL2Rγ gene locus in ~20% of treated cells, without selection.
Several labs, including ours, are currently working towards efficient ZFN-evoked targeted disruption of the CCR5 gene locus in human cells. HIV-1 entry into cells involves specific interactions between the viral envelope glycoprotein and two target cellular proteins, namely CD4 and either of the two chemokine receptors. Macrophage (M)-tropic viruses require the chemokine receptor CCR5 for entry. Several studies suggest that CCR5-expressing cells are the critical first targets for HIV-1 infection and that the CCR5 expression levels in patients correlate strongly with disease progression. Importantly, individuals with a homozygous 32-bp deletion (Δ32) in their CCR5 gene lack functional CCR5 expression; these individuals, who are otherwise healthy, are highly protected against HIV-1 infection. Individuals who are heterozygous for CCR5Δ32 have reduced levels of CCR5, and their disease progression to AIDS is delayed by 1–2 years [55]. Thus, the CCR5Δ32 mutation appears to function as a harmless polymorphism which is protective against HIV infection and progression. Our translational goal is to induce directed mutagenesis at the endogenous chromosomal sites of the CCR5 gene in human cells that are valuable for laboratory investigations and also potentially transplantable –primary human CD34+ HSPCs and HESCs. A major aim of this study is to provide proof of principle for use of ZFNs to modify genes in human stem cells that can be used for functional studies in the laboratory, e.g. to make possible in primary human CD34+ HSPCs and HESCs the kinds of studies that are already done using MESCs. The ultimate future goal of this study is to apply ZFN targeting of CCR5 clinically to individuals who are already HIV infected or at very high risk for HIV infection. Furthermore, the chromosomal CCR5 locus could possibly be an ideal safe harbor site within the human genome for targeted insertion of other therapeutic transgenes for ectopic expression in human cells, in future studies. Like us, other labs are also pursuing research along similar lines to achieve ZFN-mediated functional deletion of the CCR5 gene in human cells.
More recently, Moehle et al. [51] showed efficient incorporation of novel DNA sequences into a specified location in the genome of living human cells using designed ZFNs. A precisely placed DSB induced by designed 4-finger ZFNs stimulated integration of long DNA stretches at a predetermined genomic locus, resulting in site-specific high-efficiency gene addition. The authors showed targeted integration frequencies of 15, 6 and 5%, respectively, for the extrachromosomal donor DNA carrying a 12-bp tag, a 900-bp ORF (open reading frame) or a 1.5-kb promoter-transcription unit flanked by locus-specific homology arms after 72 h of treatment, without selection for desired recombinants. They also showed that ZFNs can drive the addition of an 8-kb sequence comprising three distinct promoter-transcription units into an endogenous locus at a frequency of 6%, without selection. Thus, the recent published reports from several labs have firmly established ZFN-mediated gene targeting as a powerful research tool for site-specific and precise modification not only of plant and mammalian genomes (including the human genome) but also of many other organisms. Furthermore, the literature reports on the high efficiency of ZFN-mediated gene targeting in human cells are especially exciting and encouraging because of their promise and great potential considering the ultimate goal of applying ZFN-based strategies for human therapeutics, that is, to use ZFN-mediated gene targeting as a form of gene therapy to treat monogenic human diseases.
Designer ZFNs: What is next?
Published research reports suggest that the ZFN-evoked HR process may be universal and that it works well in many cell types and organisms. Despite the potential benefits and promise of the ZFN technology, three technical issues prevent it from becoming a routine tool for widespread use in biological and biomedical applications: (i) Existing strategies for designing and/or selecting ZFPs with high affinity and exquisite sequence specificity for the desired target sites are quite laborious and cumbersome [15, 29, 37, 38, 56, 57]. New methods are being developed to streamline the design and/or selection of ZFPs with high affinity and specificity. (ii) Even though 3-finger ZFN designs based on our original construct have been successfully used to target cleavage of specific sites in cells, sustained ectopic expression of 3-finger ZFNs appears to be toxic to cells, most likely due to binding at secondary sites, which leads to non-targeted cleavage [48]. It is plausible to posit that the higher the specificity of the designer ZFNs, the lower should be their cellular toxicity. Therefore, we hypothesize that highly specific ZFNs will greatly improve and increase the efficiency of ZFN-driven targeted gene correction or targeted mutagenesis. This could be achieved by refining and optimizing the specificity and affinity of individual ZF motifs within the initially designed ZFPs [4]. An alternate approach to address the issue of cytotoxicity caused by non-targeted cleavage by 3-finger ZFNs would be to increase the number of ZF binding motifs within the ZFNs from 3 to 4 fingers, thereby increasing the binding specificity from a 9-bp to a 12-bp sequence, which will lead to highly specific ZFNs. The Nature publication by Urnov et al. on their SCID study appears to indicate this to be the case [4]. However, no direct comparison of the efficiency of gene targeting using 3-finger versus 4-finger ZFNs has yet been reported. Thus, it is critical to design and/or select ZFP proteins that bind their targets with high affinity and sequence specificity. The ZFPs in turn are needed to create highly specific ZFNs. This critical step is also the slow step of the ZFN-mediated gene targeting approach. (iii) Sustained expression of ZFNs in cells likely also contributes to cellular toxicity. Methods to regulate or control the expression of ZFNs in cells using inducible promoters (e.g. heat-shock promoters, drug-inducible promoters etc.) would likely circumvent this problem [42, 43].
Targeted engineering of the human genome in stem cells for therapeutics
The use of ZFN-based strategies as a form of gene therapy for human therapeutics would involve the following steps: (i) Modify patient-specific stem cells by gene transfer ex vivo, (ii) Isolate and characterize individual gene-modified cells. In most cases, this step of isolating individual gene-modified cells may not be necessary, particularly if the efficacy of the ZFN-based strategies has been firmly established and the efficiency of ZFN-evoked gene targeting is very high in the desired cell types. (iii) Expand the desired gene-modified cells in culture to generate enough cells to re-infuse into the patient [50, 58]. Primary human CD34+ HSPCs and HESCs provide attractive models, both for basic science investigations and for the potential translational development of future therapeutics. For a large number of human diseases, in particular for monogenic diseases, the defective genes and the mutations that cause the diseases have been identified. It would be a great achievement in genetic medicine if the effects of such defective genes could be countered by modifying the genome of some stem cells of the body so that they can carry out the needed function. For example, a patient could be treated by this approach for a blood disease caused by a mutant gene producing an abnormal protein. Primary HSPCs are in wide use not only in laboratory studies on molecular mechanisms of hematopoiesis and leukemogenesis, but also in clinical bone marrow transplantation for hematologic malignancies and certain other acquired and genetic diseases (tens of thousands of cases yearly). Primary HSPCs are also used for gene therapy. Such a treatment would normally involve gene addition approaches, where a normal gene is inserted into immature precursor bone marrow cells or human embryonic stem cells like HSPCs or HESCs that could ultimately develop into blood cells [50, 58]. In this way, the normal protein will be made in place of or along with the mutant protein. The hope here is that the genetically altered blood precursor cells or embryonic stem cells would then reconstitute the patient’s haematopoietic system to cure the patient’s disease.
The most commonly used gene therapy vectors are based on Moloney murine leukemia virus, a highly leukemogenic retrovirus, that in nature can transform cells by the action of its strongly trans-activating long terminal repeat (LTR) enhancer on cellular genes [58]. The gene addition approaches to compensate for defective genes by randomly inserting a new working copy into the human genome of precursor stem cells for in vivo administration of viral vectors suffer from the following problems: (1) Many viral proteins are capable of eliciting an immune response. (2) Multiple cell types in the patient are at risk of being infected. (3) Risk of germline transduction is possible by viral vectors, although it has not been reported so far. (4) The risk of insertional activation of proto-oncogenes is real. The recent occurrence of leukemias due to insertional mutagenesis in a French gene therapy trial [59, 60] has added emphasis to the importance of discovering approaches, like ZFN-mediated gene targeting, that might avoid the need for integrating viral vectors and/or target the therapeutic transgene insertion to defined safe loci in the human genome. HESC lines have become available to researchers relatively recently, but gene targeting has proved far more difficult than in MESCs [5]. Problems in adopting standard HR techniques that are so powerful in MESCs include the approximately 10-fold slower growth rate of HESCs than MESCs, the far lower transfection efficiency of HESCs than MESCs and, most important, the extremely low efficiency of HESC growth from single cells with an accompanying high risk of selecting for spontaneous mutations. Thus, to reap the full potential benefits of HESCs for basic investigation and therapeutic possibilities, these problems with modifying genes in HESCs must be solved or circumvented. Thus, any stem cell-based gene therapy approach that aims to treat human haematopoietic diseases still has to overcome three major technical hurdles associated with stem cell biology. The first is our inability to expand individual haematopoietic stem cells in sufficient numbers in vitro. The second is the general problem of turning HESCs into tissue-specific stem cells that can integrate themselves into the patient. The third deals with the method for most efficient delivery of ZFNs and donor DNA into various cell types. Although lipofection and nucleofection protocols appear to work well for many cell types, they may not transfect the majority of primary HSPCs and/or HESCs. Integration-deficient lentiviral vectors encoding ZFNs and donor DNA offer an attractive alternative approach to gain transduction levels in these cell types. However, any alternative delivery method should avoid prolonged activity of ZFNs in cells, since this may be toxic to HSPCs and HESCs. Ideally, one would like to express ZFNs for only a short while within cells, where hopefully they will find their target and induce a DSB, and then disappear via protein degradation pathways.
Thus, an important question that still remains to be answered is: Can ZFN-mediated gene targeting be used for highly efficient and permanent modification of endogenous genes in human HESCs and primary human HSPCs? The obvious next step is then to explore and develop ZFN-mediated gene targeting using highly specific ZFNs targeted against various genes in primary human CD34+ HSPCs and HESCs for laboratory research and future clinical applications. Work is under way in many labs, including ours, to address this issue.
Future outlook
ZFN-mediated gene targeting is rapidly becoming a powerful and versatile tool for targeted genome engineering of many different organisms and cells, including human cells. ZFN-based strategies as a form of gene therapy, in particular by modifying human stem cells ex vivo, may provide a new paradigm for treating monogenic human diseases by correcting the causative genetic defect. Many of the difficulties associated with gene therapy are likely to be overcome if one could insert the corrected version of the mutation at the precise location of the genetic defect within the human genome. Current gene therapy vectors lack the requisite sequence specificity necessary for targeted correction of the defective site within the human genome. ZFN-based strategies for gene correction of human stem cells may provide a viable option to treat monogenic human disease in the future. ZFN technology is still in its infancy, and many technical difficulties need to be addressed and overcome before it can become a clinically useful approach for treating monogenic human diseases. Recent literature reports suggest that ZFN technology has the potential to fulfill its promise for human therapeutics, in particular of curing monogenic diseases, and make it a reality over the next decade.
Acknowledgments
We thank Dr. Curt Civin of Johns Hopkins University for helpful suggestions and discussions on stem cells. The research on ZFNs in our lab has been supported by various grants from National Institutes of Health, USA, over the past 13 years, including the current grant (GM077291 from NIGMS).
References
- 1.Capecchi MR. Altering the genome by homologous recombination. Science. 1989;244:1288–1292. doi: 10.1126/science.2660260. [DOI] [PubMed] [Google Scholar]
- 2.Jasin M. Genetic manipulation of genomes with rare-cutting endonucleases. Trends Genet. 1996;12:224–228. doi: 10.1016/0168-9525(96)10019-6. [DOI] [PubMed] [Google Scholar]
- 3.Vasquez KM, Marburger K, Intody Z, Wilson JH. Manipulating the mammalian genome by homologous recombination. Proc Natl Acad Sci USA. 2001;98:8403–8410. doi: 10.1073/pnas.111009698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Urnov FD, Miller JC, Lee YL, Beausejour CM, Rock JM, Augustus S, Jamieson AC, Porteus MH, Gregory PD, Holmes MC. Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature. 2005;435:646–651. doi: 10.1038/nature03556. [DOI] [PubMed] [Google Scholar]
- 5.Zwaka TP, Thomson JA. Homologous recombination in human embryonic stem cells. Nat Biotechnol. 2003;21:319–321. doi: 10.1038/nbt788. [DOI] [PubMed] [Google Scholar]
- 6.Zambidis ET, Peault B, Park TS, Bunz F, Civin CI. Hematopoietic differentiation of human embryonic stem cells progresses through sequential hematoendothelial, primitive, and definitive stages resembling human yolk sac development. Blood. 2005;106:860–870. doi: 10.1182/blood-2004-11-4522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim YG, Cha J, Chandrasegaran S. Hybrid restriction enzymes: zinc finger fusions to Fok I cleavage domain. Proc Natl Acad Sci USA. 1996;93:1156–1160. doi: 10.1073/pnas.93.3.1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li L, Wu LP, Chandrasegaran S. Functional domains in Fok I restriction endonuclease. Proc Natl Acad Sci USA. 1992;89:4275–4279. doi: 10.1073/pnas.89.10.4275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim YG, Chandrasegaran S. Chimeric restriction endonuclease. Proc Natl Acad Sci USA. 1994;91:883–887. doi: 10.1073/pnas.91.3.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim YG, Smith J, Durgesha M, Chandrasegaran S. Chimeric restriction enzyme: Gal4 fusion to FokI cleavage domain. Biol Chem. 1998;379:489–495. doi: 10.1515/bchm.1998.379.4-5.489. [DOI] [PubMed] [Google Scholar]
- 11.Pavletich NP, Pabo CO. Zinc finger-DNA recognition: crystal structure of a Zif268-DNA complex at 2.1 A. Science. 1991;252:809–817. doi: 10.1126/science.2028256. [DOI] [PubMed] [Google Scholar]
- 12.Elrod-Erickson M, Pabo CO. Binding studies with mutants of Zif268. Contribution of individual side chains to binding affinity and specificity in the Zif268 zinc finger-DNA complex. J Biol Chem. 1999;274:19281–19285. doi: 10.1074/jbc.274.27.19281. [DOI] [PubMed] [Google Scholar]
- 13.Shi Y, Berg JM. A direct comparison of the properties of natural and designed zinc-finger proteins. Chem Biol. 1995;2:83–89. doi: 10.1016/1074-5521(95)90280-5. [DOI] [PubMed] [Google Scholar]
- 14.Desjarlais JR, Berg JM. Use of a zinc-finger consensus sequence framework and specificity rules to design specific DNA binding proteins. Proc Natl Acad Sci USA. 1993;90:2256–2260. doi: 10.1073/pnas.90.6.2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wolfe SA, Greisman HA, Ramm EI, Pabo CO. Analysis of zinc fingers optimized via phage display: evaluating the utility of a recognition code. J Mol Biol. 1999;285:1917–1934. doi: 10.1006/jmbi.1998.2421. [DOI] [PubMed] [Google Scholar]
- 16.Beerli RR, Segal DJ, Dreier B, Barbas CF., 3rd Toward controlling gene expression at will: specific regulation of the erbB-2/HER-2 promoter by using polydactyl zinc finger proteins constructed from modular building blocks. Proc Natl Acad Sci USA. 1998;95:14628–14633. doi: 10.1073/pnas.95.25.14628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim JS, Pabo CO. Getting a handhold on DNA: design of poly-zinc finger proteins with femtomolar dissociation constants. Proc Natl Acad Sci USA. 1998;95:2812–2817. doi: 10.1073/pnas.95.6.2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu Q, Segal DJ, Ghiara JB, Barbas CF., 3rd Design of polydactyl zinc-finger proteins for unique addressing within complex genomes. Proc Natl Acad Sci USA. 1997;94:5525–5530. doi: 10.1073/pnas.94.11.5525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chandrasegaran S, Smith J. Chimeric restriction enzymes: what is next? Biol. Chem. 1999;380:841–848. doi: 10.1515/BC.1999.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mani M, Kandavelou K, Dy FJ, Durai S, Chandrasegaran S. Design, engineering, and characterization of zinc finger nucleases. Biochem Biophys Res Commun. 2005;335:447–457. doi: 10.1016/j.bbrc.2005.07.089. [DOI] [PubMed] [Google Scholar]
- 21.Smith J, Berg JM, Chandrasegaran S. A detailed study of the substrate specificity of a chimeric restriction enzyme. Nucleic Acids Res. 1999;27:674–681. doi: 10.1093/nar/27.2.674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bitinaite J, Wah DA, Aggarwal AK, Schildkraut I. FokI dimerization is required for DNA cleavage. Proc Natl Acad Sci USA. 1998;95:10570–10575. doi: 10.1073/pnas.95.18.10570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wah DA, Bitinaite J, Schildkraut I, Aggarwal AK. Structure of FokI has implications for DNA cleavage. Proc Natl Acad Sci USA. 1998;95:10564–10569. doi: 10.1073/pnas.95.18.10564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mani M, Smith J, Kandavelou K, Berg JM, Chandrasegaran S. Binding of two zinc finger nuclease monomers to two specific sites is required for effective double-strand DNA cleavage. Biochem Biophys Res Commun. 2005;334:1191–1197. doi: 10.1016/j.bbrc.2005.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smith J, Bibikova M, Whitby FG, Reddy AR, Chandrasegaran S, Carroll D. Requirements for double-strand cleavage by chimeric restriction enzymes with zinc finger DNA-recognition domains. Nucleic Acids Res. 2000;28:3361–3369. doi: 10.1093/nar/28.17.3361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Durai S, Mani M, Kandavelou K, Wu J, Porteus MH, Chandrasegaran S. Zinc finger nucleases: custom-designed molecular scissors for genome engineering of plant and mammalian cells. Nucleic Acids Res. 2005;33:5978–5990. doi: 10.1093/nar/gki912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Isalan M, Klug A, Choo Y. A rapid, generally applicable method to engineer zinc fingers illustrated by targeting the HIV-1 promoter. Nat Biotechnol. 2001;19:656–660. doi: 10.1038/90264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kandavelou K, Mani M, Durai S, Chandrasegaran S. Magic scissors for genome surgery. Nat Biotechnol. 2005;23:686–687. doi: 10.1038/nbt0605-686. [DOI] [PubMed] [Google Scholar]
- 29.Pabo CO, Peisach E, Grant RA. Design and selection of novel Cys2His2 zinc finger proteins. Annu Rev Biochem. 2001;70:313–340. doi: 10.1146/annurev.biochem.70.1.313. [DOI] [PubMed] [Google Scholar]
- 30.Segal DJ, Beerli RR, Blancafort P, Dreier B, Effertz K, Huber A, Koksch B, Lund CV, Magnenat L, Valente D, et al. Evaluation of a modular strategy for the construction of novel polydactyl zinc finger DNA-binding proteins. Biochemistry. 2003;42:2137–2148. doi: 10.1021/bi026806o. [DOI] [PubMed] [Google Scholar]
- 31.Dreier B, Beerli RR, Segal DJ, Flippin JD, Barbas CF., 3rd Development of zinc finger domains for recognition of the 5′-ANN-3′ family of DNA sequences and their use in the construction of artificial transcription factors. J Biol Chem. 2001;276:29466–29478. doi: 10.1074/jbc.M102604200. [DOI] [PubMed] [Google Scholar]
- 32.Dreier B, Fuller RP, Segal DJ, Lund CV, Blancafort P, Huber A, Koksch B, Barbas CF., 3rd Development of zinc finger domains for recognition of the 5′-CNN-3′ family DNA sequences and their use in the construction of artificial transcription factors. J Biol Chem. 2005;280:35588–35597. doi: 10.1074/jbc.M506654200. [DOI] [PubMed] [Google Scholar]
- 33.Dreier B, Segal DJ, Barbas CF., 3rd Insights into the molecular recognition of the 5′-GNN-3′ family of DNA sequences by zinc finger domains. J Mol Biol. 2000;303:489–502. doi: 10.1006/jmbi.2000.4133. [DOI] [PubMed] [Google Scholar]
- 34.Liu Q, Xia Z, Zhong X, Case CC. Validated zinc finger protein designs for all 16 GNN DNA triplet targets. J Biol Chem. 2002;277:3850–3856. doi: 10.1074/jbc.M110669200. [DOI] [PubMed] [Google Scholar]
- 35.Mandell JG, Barbas CF., 3rd Zinc Finger Tools: custom DNA-binding domains for transcription factors and nucleases. Nucleic Acids Res. 2006;34:W516–W523. doi: 10.1093/nar/gkl209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Durai S, Bosley A, Abulencia AB, Chandrasegaran S, Ostermeier M. A bacterial one-hybrid selection system for interrogating zinc-finger-DNA interactions. Comb Chem High Throughput Screen. 2006;9:301–311. doi: 10.2174/138620706776843147. [DOI] [PubMed] [Google Scholar]
- 37.Hurt JA, Thibodeau SA, Hirsh AS, Pabo CO, Joung JK. Highly specific zinc finger proteins obtained by directed domain shuffling and cell-based selection. Proc Natl Acad Sci USA. 2003;100:12271–12276. doi: 10.1073/pnas.2135381100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Joung JK, Ramm EI, Pabo CO. A bacterial two-hybrid selection system for studying protein-DNA and protein-protein interactions. Proc Natl Acad Sci USA. 2000;97:7382–7387. doi: 10.1073/pnas.110149297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Carroll D, Morton JJ, Beumer KJ, Segal DJ. Design, construction and in vitro testing of zinc finger nucleases. Nat Protoc. 2006;1:1329–1341. doi: 10.1038/nprot.2006.231. [DOI] [PubMed] [Google Scholar]
- 40.Wright DA, Thibodeau-Beganny S, Sander JD, Winfrey RJ, Hirsh AS, Eichtinger M, Fu F, Porteus MH, Dobbs D, Voytas DF, et al. Standardized reagents and protocols for engineering zinc finger nucleases by modular assembly. Nat Protoc. 2006;1:1637–1652. doi: 10.1038/nprot.2006.259. [DOI] [PubMed] [Google Scholar]
- 41.Bibikova M, Carroll D, Segal DJ, Trautman JK, Smith J, Kim YG, Chandrasegaran S. Stimulation of homologous recombination through targeted cleavage by chimeric nucleases. Mol Cell Biol. 2001;21:289–297. doi: 10.1128/MCB.21.1.289-297.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bibikova M, Beumer K, Trautman JK, Carroll D. Enhancing gene targeting with designed zinc finger nucleases. Science. 2003;300:764. doi: 10.1126/science.1079512. [DOI] [PubMed] [Google Scholar]
- 43.Bibikova M, Golic M, Golic KG, Carroll D. Targeted chromosomal cleavage and mutagenesis in Drosophila using zinc-finger nucleases. Genetics. 2002;161:1169–1175. doi: 10.1093/genetics/161.3.1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lloyd A, Plaisier CL, Carroll D, Drews GN. Targeted mutagenesis using zinc-finger nucleases in Arabidopsis. Proc Natl Acad Sci USA. 2005;102:2232–2237. doi: 10.1073/pnas.0409339102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wright DA, Townsend JA, Winfrey RJ, Jr, Irwin PA, Rajagopal J, Lonosky PM, Hall BD, Jondle MD, Voytas DF. High-frequency homologous recombination in plants mediated by zinc-finger nucleases. Plant J. 2005;44:693–705. doi: 10.1111/j.1365-313X.2005.02551.x. [DOI] [PubMed] [Google Scholar]
- 46.Morton J, Davis MW, Jorgensen EM, Carroll D. Induction and repair of zinc-finger nuclease-targeted double-strand breaks in Caenorhabditis elegans somatic cells. Proc Natl Acad Sci USA. 2006;103:16370–16375. doi: 10.1073/pnas.0605633103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Alwin S, Gere MB, Guhl E, Effertz K, Barbas CF, 3rd, Segal DJ, Weitzman MD, Cathomen T. Custom zinc-finger nucleases for use in human cells. Mol Ther. 2005;12:610–617. doi: 10.1016/j.ymthe.2005.06.094. [DOI] [PubMed] [Google Scholar]
- 48.Porteus MH, Baltimore D. Chimeric nucleases stimulate gene targeting in human cells. Science. 2003;300:763. doi: 10.1126/science.1078395. [DOI] [PubMed] [Google Scholar]
- 49.Beumer K, Bhattacharyya G, Bibikova M, Trautman JK, Carroll D. Efficient gene targeting in Drosophila with zinc-finger nucleases. Genetics. 2006;172:2391–2403. doi: 10.1534/genetics.105.052829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Porteus MH. Mammalian gene targeting with designed zinc finger nucleases. Mol Ther. 2006;13:438–446. doi: 10.1016/j.ymthe.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 51.Moehle EA, Rock JM, Lee YL, Jouvenot Y, Dekelver RC, Gregory PD, Urnov FD, Holmes MC. Targeted gene addition into a specified location in the human genome using designed zinc finger nucleases. Proc Natl Acad Sci USA. 2007;104:3055–3060. doi: 10.1073/pnas.0611478104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Porteus MH, Carroll D. Gene targeting using zinc finger nucleases. Nat Biotechnol. 2005;23:967–973. doi: 10.1038/nbt1125. [DOI] [PubMed] [Google Scholar]
- 53.Wilson JH. Pointing fingers at the limiting step in gene targeting. Nat Biotechnol. 2003;21:759–760. doi: 10.1038/nbt0703-759. [DOI] [PubMed] [Google Scholar]
- 54.Pierce AJ, Johnson RD, Thompson LH, Jasin M. XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev. 1999;13:2633–2638. doi: 10.1101/gad.13.20.2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Huang Y, Paxton WA, Wolinsky SM, Neumann AU, Zhang L, He T, Kang S, Ceradini D, Jin Z, Yazdanbakhsh K, et al. The role of a mutant CCR5 allele in HIV-1 transmission and disease progression. Nat Med. 1996;2:1240–1243. doi: 10.1038/nm1196-1240. [DOI] [PubMed] [Google Scholar]
- 56.Greisman HA, Pabo CO. A general strategy for selecting high-affinity zinc finger proteins for diverse DNA target sites. Science. 1997;275:657–661. doi: 10.1126/science.275.5300.657. [DOI] [PubMed] [Google Scholar]
- 57.Rebar EJ, Greisman HA, Pabo CO. Phage display methods for selecting zinc finger proteins with novel DNA-binding specificities. Methods Enzymol. 1996;267:129–149. doi: 10.1016/s0076-6879(96)67010-4. [DOI] [PubMed] [Google Scholar]
- 58.Porteus MH, Connelly JP, Pruett SM. A look to future directions in gene therapy research for monogenic diseases. PLoS Genet. 2006;2:e133. doi: 10.1371/journal.pgen.0020133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hacein-Bey-Abina S, von Kalle C, Schmidt M, Le Deist F, Wulffraat N, McIntyre E, Radford I, Villeval JL, Fraser CC, Cavazzana-Calvo M, et al. A serious adverse event after successful gene therapy for X-linked severe combined immunodeficiency. N Engl J Med. 2003;348:255–256. doi: 10.1056/NEJM200301163480314. [DOI] [PubMed] [Google Scholar]
- 60.Hacein-Bey-Abina S, Von Kalle C, Schmidt M, McCormack MP, Wulffraat N, Leboulch P, Lim A, Osborne CS, Pawliuk R, Morillon E, et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science. 2003;302:415–419. doi: 10.1126/science.1088547. [DOI] [PubMed] [Google Scholar]
- 61.West SC. Molecular views of recombination proteins and their control. Nat Rev Mol Cell Biol. 2003;4:435–445. doi: 10.1038/nrm1127. [DOI] [PubMed] [Google Scholar]
- 62.Hefferin ML, Tomkinson AE. Mechanism of DNA double-strand break repair by non-homologous end joining. DNA Repair (Amst) 2005;4:639–648. doi: 10.1016/j.dnarep.2004.12.005. [DOI] [PubMed] [Google Scholar]