

Abstract
Increased CDI incidence and severity in older adults corresponds with the emergence of the BI/NAP1 strain, making elucidation of the host immune response extremely important. We therefore infected germ-free, aged C57BL/6 mice with a BI strain and monitored for response. Infected mice were moribund 48–72 hours post infection, developed gross and histologic cecitis and colitis, elevated concentrations of KC, IL-1β, MCP-1, and G-CSF, and decreased IFN-γ, IL-12(p40), IL-12(p70), and IL-10 vs. controls. Aged, germ-free C57BL/6 mice are susceptible to fulminant CDI from a BI strain and represent a novel model to further elucidate the host immune response to acute CDI.
Clostridium difficile infection (CDI) incidence, severity, and mortality has increased, especially in those ≥65 years of age, possibly associated with the emergence of the epidemic restriction endonuclease analysis (REA) and pulse field gel electrophoresis BI/NAP1 strain (herein referred to as BI)(1). This strain has been present since the 1980's, but was not previously responsible for outbreaks(2). It hyperproduces toxins A and B, secretes a binary toxin (CDT), hypersporulates, and has developed high level fluoroquinolone resistance suggesting that these traits have led to increased virulence and spread(2).
A murine model of infection is extremely important to help elucidate the host immune response to CDI and to better understand the pathogenesis induced by BI as well as additional C.difficile strains. The traditional animal model of CDI, the Syrian hamster, is limited by an inability to study the host cytokine response due to lack of host specific reagents. Previous germ-free CDI experiments using murine strains other than C57BL/6's, particularly in the 1980's and early 1990's, resulted in minimal intestinal pathogenesis to severe cecal and colon ulceration with 100% mortality(3,4). The majority of these studies, however, utilized a highly toxigenic lab strain, VPI 10463, which has high levels of toxin A and B production, low sporulation rates, and has never been found to cause human colitis. No gnotobiotic murine studies have been performed using the BI strain and it has not yet been determined whether C57BL/6 germ-free mice are susceptible to CDI. Additionally, the murine mucosal cytokine response to CDI has not been documented.
Therefore, we undertook this study to determine if C57BL/6 germ-free mice, a commonly investigated strain which can be bred for transgenic and knockout mouse studies, inoculated with a clinically relevant BI strain, could be a beneficial model to study pathogenesis and host mucosal cytokine responses to acute CDI.
Methods
UVA13, a human C.diffcile isolate previously cultured from a fecal specimen from a patient at the University of Virginia Hospital with CDI and typed as BI by the Hines Reference Laboratory at the Hines Veterans Affairs (Hines, IL) using REA and a previously described protocol(5), was inoculated into chopped meat broth and incubated anaerobically at 38° C over 24 hours.16 germ-free C57BL/6 mice, ranging in age from 7 to 14 months, from the National Gnotobiotic Rodent Resource Center at the University of North Carolina were orally gavaged with 330ul of incubated broth containing a total of 1×108 organisms. Similarly, VPI11186, a non-toxigenic strain of C.difficile, was incubated in broth over 24 hours and 2 germ free mice were orally gavaged with 1×108 organisms. 6 germ-free mice received a similar dosage of un-inoculated broth and served as un-infected controls. Sterility was confirmed in germ-free mice by aerobic and anaerobic culture and gram staining of stool samples, as previously outlined(6).
Mice were monitored every 8 hours after inoculation for clinical signs and symptoms of disease including diarrhea and impaired physical condition and behavior. Weights were obtained as a measure of disease at the time of euthanasia. Mice that were determined to be moribund according to Animal Care and Use Committee (ACUC) policy and approved protocol were weighed and euthanized (Supplement 1).
Cecal and colon segments were collected from euthanized mice and prepared for histology using hematoxylin and eosin (H and E) stain and antibodies directed against myeloperoxidase (MPO). Histology specimens were scored by 2 blinded participants (SWP and RF) on a scale of 0–3 (0=minimal score and 3=maximal score) based on each of the following criteria: inflammatory cell infiltration, mucosal hypertrophy, vascular congestion, epithelial disruption, and submucosal edema (Supplement 2).
Cecal and colon segements were homogenized and assayed for the cytokines G-CSF, IL-1β, KC, TNF-α, IFN-γ, MCP-1, IL-12(p40), IL-12(p70), IL-6, IL-17, IL-13, and IL-10 using bead-based Luminex immunoanalysis (Bio-Rad, Hercules, CA).
Mean differences in cecal and colon histopathologic scores and cytokine levels were analyzed with SPSS software and evaluated using an independent t-test. P≤0.05 was determined to be significant.
Results
BI infected mice display severe CDI symptoms
All mice administered UVA13 died or were moribund necessitating euthanasia within 72 hours, while mice given VPI 11186 or broth were asymptomatic. Germ-free mice administered UVA13 developed diarrhea and/or wet tail, accompanied by weight loss. 11 of 16 (69%) UVA13 infected mice were euthanized and processed for histology and cytokine analysis (5 UVA13 infected mice died and could not be processed). Broth controls (N=6) and non-toxigenic infected mice (N=2) were euthanized at 72 hours, processed, and assessed for comparison of histology scores and cytokine levels.
BI infection leads to gross and histopathologic changes
Grossly, UVA13 infected mice had shorter ceca as compared to controls with evidence of purulent, hemorrhagic, or loose cecal contents, with less pronounced alterations in colon segments. Histopathology scores were significantly higher in UVA13 infected mice, with evidence of both cecal and colon ulceration, loss of mucosal architecture, epithelial exfoliation, inflammatory cell infiltration, edema, and hemorrhage in the lamina propria vs. control mice (Figure 1). While neutrophilic infiltration was demonstrated utilizing MPO staining, it was relatively minor with evidence of MPO within neutrophils, and also extravasated within the lumen (Figure 1).
Figure 1.
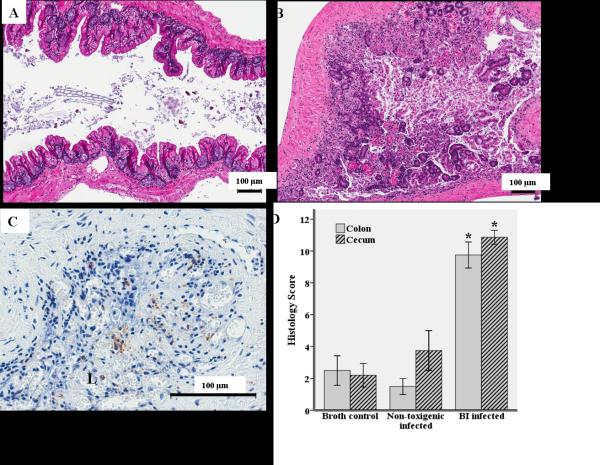
Histopathology of cecal and colon segments 72 hours after oral inoculation with 1×108 CFU's UVA13 (BI strain of C.difficile),VPI 11186 (non-toxigenic C. difficile strain), or broth. A, Hematoxylin and eosin (H&E) stain of a cecal segment from a germ-free mouse infected with the non-toxigenic strain. B, C, H&E and myeloperoxidase (MPO) stain of a cecal segment from a germ-free mouse infected with the BI strain (brown indicates MPO activity). D, Ceca and colon histopathologic differences between BI infected (N=11), non-toxigenic infected (N=2), and broth controls (N=6) as determined by inflammatory cell infiltration, mucosal hypertrophy, vascular congestion, epithelial disruption, and submucosal edema, with a maximum score of 15. * P≤0.001, BI infected compared with broth controls. L=lumen
BI infection leads to varied cytokine pattern
We next examined cecal and colon segments to determine mucosal cytokine concentrations produced after infection. UVA13 infection led to elevated cecal and colonic KC, MCP-1, IL-1β, and G-CSF concentrations (pg cytokine/mg protein) as compared to controls, with greater concentrations in the cecum than in the colon (Figure 2). Additionally, BI infection led to lower levels of cecal and colonic IL-12(p40), IL-12(p70), IFN-γ, and IL-10 (Figure 2). There were no differences in the concentrations of the pro-inflammatory cytokines TNF-α, IL-6, IL-13, and IL-17.
Figure 2.

Ceca and colon cytokine concentrations (pg cytokine/mg protein) 72 hours after oral inoculation with 1×108 CFU's UVA13 (BI strain of C.difficile), VPI 11186 (non-toxigenic strain), or broth. A, B, Ceca and colon segments were homogenized in a proteinase inhibitor cocktail and assayed for the cytokines G-CSF, MCP-1, KC, IL-1β, IL-12(p40), IL-12(p70), IFN-γ, IL-10, TNF-α, IL-6, IL-17, and IL-13 using Luminex immunoanalysis (Bio-rad, Hercules, CA). There were no significant differences in IL-6, IL-17, or IL-13 concentrations between the groups (not shown). * P≤0.05; ** P≤0.001, BI infected (N=11) compared with broth controls (N=6).
Discussion
The results of this study indicate that acute infection with a clinically relevant BI C.difficile strain leads to weight loss, symptomatic disease, cecal and colonic ulceration, a relatively minor neutrophil infiltration, a measurable cytokine response, and eventual death in monoassociated C57BL/6 mice. The clinical and histopathology findings are in agreement with those of Vernet and colleagues, who previously observed that NIH germ-free mice infected with C.difficile that secreted elevated levels of toxin A in vitro developed cecal and colon ulceration and 100% mortality(4). Published studies demonstrate that the BI strain hyperproduces toxins A and B in vitro, and additional studies as well as work in our lab, indicate that VPI 10463, used in the majority of previous studies involving CDI in germ-free mice, produces even greater levels of toxin A in vitro as compared to UVA13(2).
Previous studies of the host cytokine response to toxins A and B have been limited to cell culture data and small intestinal loops directly exposed to purified toxins A or B, and have shown a predominant inflammatory response to toxin exposure. Binding of the toxins to their respective receptor leads to induction of MAP kinase, translocation of NF-κB into the nucleus, inhibition of the Rho family of proteins, and activation of submucosal neurons, all of which have an effect of activating pro-inflammatory cytokines, recruitment of neutrophils, epithelial cell apoptosis, and disruption of tight junctions. Not only does toxin exposure lead to the secretion of some pro-inflammatory cytokines, it may also lead to decreased protective cytokines such as TGF- β(7). We demonstrated increased production of many, but not all, proinflammatory cytokines produced by innate immune cells and epithelial cells, as well as decreased mucosal IL-10 production, another protective cytokine. We did not measure TGF- β concentrations.
The elevations of IL-1β, KC (human IL-8 equivalent), and MCP-1 in our study of BI CDI are in agreement with cell culture and murine intestinal loop studies which show elevations in the same cytokines in response to toxin exposure. It is also in agreement with cytokine data in human studies, which show elevated fecal IL-8 levels in patients with clinically severe CDI(8). Similarly, decreased levels of IL-10, normally thought to play a role in maintaining the host barrier defense and suppression of inflammation, in our BI infected mice are in agreement with our preliminary data in rabbit intestinal loops showing decreased IL-10 levels after toxin A exposure (CAW, personal communication). These similarities of cytotoxic profiles further validate our model is clinically relevant.
Interestingly, there was no difference in the pro-inflammatory cytokine TNF-α, which has previously been shown to be increased in toxin A enteritis(9). In addition, the significantly lower levels of IFN-γ in our severely infected mice is in contrast to studies in small intestinal loops exposed to purified toxin A by Ishida and colleagues, who hypothesized that IFN-γ is the crucial mediator of toxin-induced enteritis after noting attenuated disease in IFN-γ KO mice(9). The interrelationship among cytokines likely explains the low levels of TNF-α, as IFN-γ is partially responsible for TNF-α expression in murine intestinal loops exposed to toxin A(9). Additionally, IL-12, which was significantly lower in our BI infected mice, has been shown to induce IFN-γ expression in macrophages, T cells, and in intestinal loops, and therefore may be partially responsible for the low levels of IFN- γ found(9–11).
The differences in cytokine expression between our model of CDI and studies involving pure toxin exposure of cell culture and intestinal loops may in part be due to a lack of direct host-pathogen interaction in the latter models. Toxin A−, toxin B+ C.difficile strains lead to severe human CDI despite limited pathogenesis observed when purified toxin B is inoculated into murine, hamster, and rabbit small intestinal loops(5). On the other hand, purified toxin A within murine, hamster, and rabbit small intestinal loops leads to severe disease. Further lending credence to the importance of the pathogen-host interaction is a recent study indicating that toxin B is responsible for pathogenesis as opposed to toxin A, when the authors found that hamsters exposed to a toxin A+, toxin B− C.difficile showed no evidence of disease whereas disease was observed with a toxin A−, toxin B + variant(12). Additionally, C. difficile surface layer proteins lead to pro-inflammatory IL-1β and IL-6 secretion, highlighting the importance of host-pathogen recognition and signaling and the interaction of toxin and the C.difficile-derived components(13).
While undertaking our germ-free studies, Chen and colleagues discovered a murine model of CDI using conventional C57BL/6 mice, which leads to ulcerative cecitis, colitis, and a moribund state after exposure to six antibiotics and VPI 10463, while a BI strain given in a similar manner leads to symptomatic disease, cecal and colon pathogenesis, but no mortality(14). Cecal and colon histology of BI infected mice in their study demonstrate subcutaneous edema, neutrophilic infiltration, but little epithelial disruption, whereas mice given VPI10463 show destruction of the host architecture in addition to edema and neutrophil margination, indicating that epithelial disruption is necessary to induce mortality, similar to that shown in gnotobiotic mice(4,14). Why the BI strain was able to induce epithelial destruction and death in our monoassociated mice, as opposed to Chen and colleagues' conventional mice is unknown, but possibilities include the greater number of organisms used to colonize our mice (1×108 CFU's vs. 1×105 CFU's), age related differences in the host immune response, less developed mucosal responses in germ-free mice, or an influence by the retained host micro-biota after antibiotics(15). While microbiota analysis was not performed on fecal pellets before or after exposure to antibiotics, one can hypothesize that exposure to six different antibiotics led to decreased normal bacterial concentrations and altered composition, although antibiotics do not induce a sterile state.
We have thus shown that BI CDI in an aged germ-free C57BL/6 mouse leads to symptomatic disease, a moribund state, and profound histopathology in the cecum and colon. Additionally, as a result of CDI, certain cecal and colonic cytokines are produced, which had not previously been documented. Differences in cytokine production with in vivo infection compared with small intestinal loop studies and in vitro toxin studies highlight the importance of the host-pathogen relationship in CDI. Use of this model in concurrence with other newly established conventional C57BL/6 models will lead to a greater understanding of the host-pathogen interaction and the role of the host micro-biota in CDI.
Supplementary Material
Acknowledgements
We thank Maureen Bower for her assistance with gnotobiotic animal work.
Funding: This work is supported in part by the NIH/NIAD 5T32 AI 007046-33, UO1 AI075526, P40 RR018603 and P30 DK34987.
Footnotes
Conflicts of interest The authors disclose no conflicts of interest.
This work was presented as an oral presentation at the 2009 IDSA annual meeting, Philadelphia, PA.
Reference List
- (1).Zilberberg MD, Shorr AF, Kollef MH. Increase in adult Clostridium difficile-related hospitalizations and case-fatality rate, United States, 2000–2005. Emerg Infect Dis. 2008;14(6):929–931. doi: 10.3201/eid1406.071447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).McDonald LC, Killgore GE, Thompson A, Owens RC, Jr., Kazakova SV, Sambol SP, et al. An epidemic, toxin gene-variant strain of Clostridium difficile. N Engl J Med. 2005;353(23):2433–2441. doi: 10.1056/NEJMoa051590. [DOI] [PubMed] [Google Scholar]
- (3).Corthier G, Muller MC, Wilkins TD, Lyerly D, L'Haridon R. Protection against experimental pseudomembranous colitis in gnotobiotic mice by use of monoclonal antibodies against Clostridium difficile toxin A. Infect Immun. 1991;59(3):1192–1195. doi: 10.1128/iai.59.3.1192-1195.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Vernet A, Corthier G, Dubos-Ramare F, Parodi AL. Relationship between levels of Clostridium difficile toxin A and toxin B and cecal lesions in gnotobiotic mice. Infect Immun. 1989;57(7):2123–2127. doi: 10.1128/iai.57.7.2123-2127.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Sambol SP, Merrigan MM, Lyerly D, Gerding DN, Johnson S. Toxin gene analysis of a variant strain of Clostridium difficile that causes human clinical disease. Infect Immun. 2000;68(10):5480–5487. doi: 10.1128/iai.68.10.5480-5487.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Kim SC, Tonkonogy SL, Albright CA, Tsang J, Balish EJ, Braun J, et al. Variable phenotypes of enterocolitis in interleukin 10-deficient mice monoassociated with two different commensal bacteria. Gastroenterology. 2005;128(4):891–906. doi: 10.1053/j.gastro.2005.02.009. [DOI] [PubMed] [Google Scholar]
- (7).Johal SS, Solomon K, Dodson S, Borriello SP, Mahida YR. Differential effects of varying concentrations of clostridium difficile toxin A on epithelial barrier function and expression of cytokines. J Infect Dis. 2004;189(11):2110–2119. doi: 10.1086/386287. [DOI] [PubMed] [Google Scholar]
- (8).Steiner TS, Flores CA, Pizarro TT, Guerrant RL. Fecal lactoferrin, interleukin-1beta, and interleukin-8 are elevated in patients with severe Clostridium difficile colitis. Clin Diagn Lab Immunol. 1997;4(6):719–722. doi: 10.1128/cdli.4.6.719-722.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Ishida Y, Maegawa T, Kondo T, Kimura A, Iwakura Y, Nakamura S, et al. Essential involvement of IFN-gamma in Clostridium difficile toxin A-induced enteritis. J Immunol. 2004;172(5):3018–3025. doi: 10.4049/jimmunol.172.5.3018. [DOI] [PubMed] [Google Scholar]
- (10).Puddu P, Fantuzzi L, Borghi P, Varano B, Rainaldi G, Guillemard E, et al. IL-12 induces IFN-gamma expression and secretion in mouse peritoneal macrophages. J Immunol. 1997;159(7):3490–3497. [PubMed] [Google Scholar]
- (11).Way SS, Havenar-Daughton C, Kolumam GA, Orgun NN, Murali-Krishna K. IL-12 and type-I IFN synergize for IFN-gamma production by CD4 T cells, whereas neither are required for IFN-gamma production by CD8 T cells after Listeria monocytogenes infection. J Immunol. 2007;178(7):4498–4505. doi: 10.4049/jimmunol.178.7.4498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Lyras D, O'Connor JR, Howarth PM, Sambol SP, Carter GP, Phumoonna T, et al. Toxin B is essential for virulence of Clostridium difficile. Nature. 2009;458(7242):1176–1179. doi: 10.1038/nature07822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Ausiello CM, Cerquetti M, Fedele G, Spensieri F, Palazzo R, Nasso M, et al. Surface layer proteins from Clostridium difficile induce inflammatory and regulatory cytokines in human monocytes and dendritic cells. Microbes Infect. 2006;8(11):2640–2646. doi: 10.1016/j.micinf.2006.07.009. [DOI] [PubMed] [Google Scholar]
- (14).Chen X, Katchar K, Goldsmith JD, Nanthakumar N, Cheknis A, Gerding DN, et al. A mouse model of Clostridium difficile-associated disease. Gastroenterology. 2008;135(6):1984–1992. doi: 10.1053/j.gastro.2008.09.002. [DOI] [PubMed] [Google Scholar]
- (15).Umesaki Y, Setoyama H. Structure of the intestinal flora responsible for development of the gut immune system in a rodent model. Microbes Infect. 2000;2(11):1343–1351. doi: 10.1016/s1286-4579(00)01288-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.