

Abstract
The intracellular second messenger cAMP is frequently used in induction media to induce mesenchymal stem cells (MSCs) into neural lineage cells. To date, an understanding of the role cAMP exerts on MSCs and whether cAMP can induce MSCs into functional neurons is still lacking. We found cAMP initiated neuron-like morphology changes early and neural differentiation much later. The early phase changes in morphology were due to cell shrinkage, which subsequently rendered some cells apoptotic. While the morphology changes occurred prior to the expression of neural markers, it is not required for neural marker expression and the two processes are differentially regulated downstream of cAMP-activated protein kinase A. cAMP enabled MSCs to gain neural marker expressions with neuronal function, such as, calcium rise in response to neuronal activators, dopamine, glutamate, and potassium chloride. However, only some of the cells induced by cAMP responded to the three neuronal activators and further lack the neuronal morphology, suggesting that although cAMP is able to direct MSCs towards neural differentiation, they do not achieve terminal differentiation.
Keywords: cAMP, Morphology, Apoptosis, Neural differentiation
Introduction
Mesenchymal stem cells (MSCs) are adult stem cells with multipotency to differentiate into mesodermal lineage cells such as osteoblasts [1], adipocytes [2], and chondrocytes [3]. Studies also suggested that these cells have the potential to transdifferentiate into other lineages, such as hepatocytes [4], cardiomyocytes [5], neurons [6], and astrocytes [7]. Several recent studies indicated that the shape of the cell [8] guided by surface cues [9], and matrix elasticity [10] can influence the lineage commitment of stem cells. Thus, these studies suggest that the surface is as important, if not more so, in directing cell lineage and guiding function to follow form. In contrast, we found that cyclic adenosine monophosphate (cAMP) induced the function but not the form.
cAMP is a soluble, biochemical cue that is frequently used either alone [11] or in combination with other factors [12–14] to induce neural differentiation of MSCs. cAMP initiated transient neuron-like morphology changes that lasted only a few hours. This morphology change was the result of cell shrinkage and did not contribute to the later phase neural differentiation. Similarly, studies using β-mercaptoethanol (BME), dimethyl sulfoxide (DMSO), and butylated hydroxyanisole (BHA) to induce neural differentiation of MSCs have attributed the neuron-like morphology to an artifact of cell shrinkage rather than neurite outgrowth [15]. However, unlike BME, which induced changes in morphology for up to 24 h [15], cAMP initiated a transient change in morphology for up to 3 h that decreased over time.
The classical cAMP signaling pathway involves activation of PKA, which is composed of two catalytic subunits, PKAc, and two regulatory subunits, PKAr [16]. Binding of cAMP to the regulatory subunits dissociates PKAr from PKAc, thereby enabling the activation of PKAc [17]. Signaling events initiated by PKAc play important roles in regulating cell death and survival [18, 19], cell movement and structure [20], as well as differentiation [21, 22]. We investigated whether PKA is involved in regulating both the morphology changes and neural differentiation and function and whether neural differentiation is contingent upon the changes in morphology.
Although cAMP has been shown to induce neural marker expression [11, 13], we show that cAMP also induced a calcium rise, an indicator of neural function, that persisted for at least 1 week. We found that MSCs showed differential responses to neuronal activators (i.e., dopamine) despite the lack of neuron-like morphology, thus cAMP is able to facilitate neural differentiation but by itself is not sufficient to induce MSCs to terminally differentiated neurons.
Materials and methods
Materials
Forskolin (Sigma, St. Louis, MO, USA) and isobutylmethylxanthine (IBMX) (Sigma) were used to increase intracellular cAMP levels at concentrations of 10 and 100 μM, respectively. H89 (Sigma) and Rp-cAMPS (Sigma) were used as PKA inhibitors at concentration of 2.5 and 10 μM. Actinomycin D (ActD) (Sigma) was used to inhibit transcription at concentration of 1 μg/ml. Cycloheximide (CHX) (Sigma) was used to inhibit translation at concentration of 10 μg/ml. Paclitaxel (Ptx) (Sigma) was used to stabilize microtubules at concentration of 0.4 uM. The three neuronal activators used during calcium imaging were: 100 μM dopamine (Sigma), 100 μM glutamate (Sigma) and 50 mM KCl (J.T. Baker, Phillipsburg, NJ, USA).
Cell isolation and culture
All procedures in the cell isolation were approved by the Institutional Animal Care and Use Committee at Michigan State University. Bone marrow mesenchymal stem cells were isolated from 6 to 8-week-old Sprague-Dawley female rat as previously described [23]. In brief, femurs and tibias from 6 to 8-week-old rat were dissected and the two ends were cut open. The marrow was flushed out with DMEM using a needle and syringe. The cell suspension was filtered through a 65-μm nylon mesh to remove bone debris and blood aggregates. Cells were cultured in DMEM (Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (Invitrogen), 100 μg/ml streptomycin (Invitrogen) and 100 U/ml penicillin (Invitrogen) and placed in an incubator with a humidified atmosphere containing 5% CO2 at 37°C. Non-adherent cells were removed on the second day after plating. Media was replaced every 3–4 days until the cells reached 80–90% confluence. Confluent cells were detached by 0.25% trypsin–EDTA (Invitrogen) and plated for further experiments.
Primary cortical neurons were isolated as described in [24]. In brief, animal heads were decapitated from 1-day-old Sprague-Dawley rat pups. Cortical neurons were obtained from the brain and cultured on poly-l-lysine-coated plates in cortical media [DMEM (Invitrogen) supplemented with 10% horse serum (Sigma), 2 mM glutamine (Invitrogen), 100 μg/ml streptomycin (Invitrogen) and 100 U/ml penicillin (Invitrogen)] in an incubator with a humidified atmosphere containing 5% CO2 at 37°C. Cells were used within 3 days after isolation.
Live cell imaging
Cells were cultured in a four-well chambered glass bottom plate (Thermo Fisher Scientific, Rochester, NY, USA). Before taking images, the media was changed to 0.5 ml Leibovitz (Liz) media (Sigma). The plate was mounted in a temperature-controlled chamber set at 37°C on the microscope station and then 0.5 ml Liz media containing 20 μM forskolin and 200 μM IBMX was added to the chambered well to achieve a final concentration of 10 μM forskolin and 100 μM IBMX. Phase-contrast images were captured by confocal microscope Olympus FluoView 1000 at 5-min intervals.
cAMP assay
Intracellular cAMP levels were measured by a competitive immunoassay from Assay Designs (Assay Designs, Ann Arbor, MI, USA) according to the manufacturer's instructions. In brief, cells were lysed with 0.1 M HCl and the supernatant collected. The cAMP in the samples or standards was allowed to bind to a polyclonal cAMP antibody in a competitive manner with alkaline phosphatase-conjugated cAMP. Cleavage of a substrate by the alkaline phosphatase is inversely proportional to the cAMP level in the samples or standards. Colorimetric readings were taken by SPECTRAmax plus384 from Molecular Device at 405 nm. All the readings were normalized to protein levels (μg/ml) by Bradford assay.
Caspase 3 activity assay
Caspase 3 activity was measured by a kit from BIOMOL (BIOMOL, Plymouth Meeting, PA, USA) according to the manufacturer's instructions. Briefly, cell extracts were incubated with substrate Ac-DEVD-AMC. The cleavage of the substrate generates fluorescence which is proportional to the concentration of active caspase 3 in the cell extracts. Fluorescence was measured by Spectra MAX GEMINI EM plate reader at excitation of 360 nm and emission of 460 nm. All the readings were normalized to protein levels (μg/ml) by Bradford assay.
Western blot
Whole-cell extracts lysed with CelLytic (Sigma) were assayed for protein concentrations by Bradford assay (Bio-Rad, Hercules, CA, USA). Protein samples of 15–30 μg were separated by 10% Tris-HCl gel and transferred to nitrocellulose membrane. Membranes were then blocked in 5% milk and 0.05% Tween 20–TBS (Tris buffered saline) (USB corporation, Cleveland, OH, USA) for 1 h and incubated with primary antibodies, NSE (neuron-specific enolase) (BIOMOL, Plymouth Meeting, PA, USA), Tuj1 (βIII-tubulin) (Millipore, Billerica, MA, USA), GFAP (Glial fibrillary acidic protein) (DAKO, Carpinteria, CA, USA), GAPDH (Cell Signaling, Danvers, MA, USA), PKAc (R&D Systems, Minneapolis, MN, USA), pPKAc (Cell Signaling), ser133 phosphorylated CREB (pCREB) (EMD Chemicals, Gibbstown, NJ, USA) and ICER (a kind gift from Dr. Carlose Molina) overnight at 4°C. Anti-mouse or anti-rabbit HRP-conjugated secondary antibody (Thermo Scientific, Rockford, IL, USA) were added the second day after primary antibody incubation. The blots were incubated for 1 h and then washed three times with 0.05% Tween 20–TBS. The blots were then visualized by SuperSignal west femto maximum sensitivity substrate (Thermo Scientific).
Immunocytochemistry
For staining against Tuj1, cells were fixed in PBS containing 3.7% formaldehyde for 15 min and permeabilized with 0.5% Triton X-100 (Research Products Internationals, Mt. Prospect, IL, USA) for 20 min at room temperature. After washing with PBS three times, the cells were blocked in 1% BSA (bovine serum albumin) (US Biological, Swampscott, MA, USA) for 20 min and incubated with Tuj1 (Millipore) antibody at room temperature for 1 h. Cells were then washed with PBS three times and incubated with Alexa Fluor 488-conjugated anti-mouse IgG secondary antibody (Invitrogen) for 1 h at room temperature. Stained glass coverslips were washed three times with PBS and mounted in ProLong Gold (Invitrogen). Fluorescence images were taken by confocal microscope Olympus FluoView 1000.
Triple staining for actin filaments, microtubules and nucleus was performed as previously described [25]. In brief, actin filaments were stained with Texas Red-X phalloidin (Invitrogen), microtubules were stained with α-tubulin (Invitrogen) primary antibody followed by Alexa Fluor 488-conjugated anti-mouse IgG secondary antibody (Invitrogen), and the nucleus was stained with DAPI (4′, 6-diamidino-2-phenylindole) (Invitrogen). Stained glass coverslips were mounted in ProLong Gold (Invitrogen). Fluorescence images were taken by confocal microscope Olympus FluoView 1000.
Annexin V and PI (propidium iodide) staining
Apoptosis and necrosis were measured by the annexin V and PI (propidium iodide) staining kit (Invitrogen), respectively, according to the manufacturer's instructions. In brief, cells were stained with Alexa Fluor 488-conjugated annexin V and PI in 1× annexin-binding buffer for 15 min at room temperature and then subjected to flow cytometry analysis by BD FACSVantage. Early apoptotic cells were identified as those stained by Alexa Fluor 488 but not PI, late apoptotic cells were those stained by both Alexa Fluor 488 and PI, and necrotic cells were those stained by PI but not Alexa Fluor 488.
Calcium imaging
Calcium imaging was performed according to the protocol described in [26]. Cells were cultured in four-well chambered cover-glass (Thermo Fisher Scientific). After the desired treatment, the cells were loaded with 4 μM Fluo-4 (Invitrogen) in ACSF–HEPES (artificial cerebral spinal fluid with HEPES: 119 mM NaCl, 2.5 mM KCl, 1.3 mM MgCl2, 2.5 mM CaCl2, 1 mM NaH2PO4, 26.2 mM NaHCO3, 11 mM dextrose, 10 mM HEPES, pH = 7.4) for 30 min at 37°C. Excess dye was removed by washing cells with PBS twice placing into a 37°C chamber on the stage of Olympus FluoView 1000. Then 0.5 ml ACSF–HEPES was added to the well to begin imaging. Images were captured every 1.137 s and fluorescence intensity is represented by a spectral table (warmer colors represent higher intensity whereas cooler colors represent lower intensity). After 15–20 images, 0.5 ml ACSF–HEPES buffer containing the following drugs was added: 200 μM glutamate (final concentration 100 μM), 200 μM dopamine (final concentration 100 μM), 100 mM KCl (final concentration 50 mM), or 200 μM ATP (final concentration 100 μM). A total of 200–300 images were recorded and the data was analyzed by the FluoView 100 software. Changes in the fluorescence intensity of the Ca2+ signal are represented as F/F0. The percent of responsive cells is calculated as the number of cells with a F/F0 signal greater than 20% divided by the total number of cells.
Cell counting
Cells were trypsinized by 0.25% trypsin–EDTA (Invitrogen, Carlsbad, CA, USA) and an equal volume of media was added to inactivate the trypsin. Number of cells was determined by diluting the cell suspension 1:1 with 0.4% trypan blue (Sigma) and then counted on a hemocytometer.
Stable cell lines express dominant negative CREB
Mesenchymal stem cells were transfected with the empty control pCMV vector containing neomycin resistance and the dominant negative CREB mutant (serine 133 mutated to alanine) M1-CREB (a kind gift from Dr. David Ginty) using Lipofectamine 2000 (Invitrogen). Twenty-four hours after transfection, the cells were trypsinized and replated at low density in media containing 500 μg/ml Geneticin (Invitrogen) for selection. The Geneticin-containing media was replaced every 3 days for 2 weeks. Colonies formed from surviving cells were isolated by cloning cylinders (Sigma) and maintained in culture media containing Geneticin.
Statistical analysis
All experiments were performed at least three times and results are shown as mean ± standard deviation. Statistical analysis was carried out by an unpaired, two-tail Student's t test. Asterisks indicate p < 0.05; double asterisks indicate p < 0.01; and triple asterisk indicate p < 0.001.
Results
cAMP induced early phase neuron-like morphology changes
Deng et al. [11] showed that upon exposure of human MSC to cAMP elevating agents, 1 mM dibutyryl cyclic AMP (dbcAMP) and 0.5 mM IBMX, for 2 days, the cells exhibit neuron-like morphology. However, we found that the neuron-like morphology occurred much earlier than previously reported. Uninduced MSCs exhibited flat-like morphology (Fig. 1a), whereas MSCs induced with 10 μM forskolin and 100 μM IBMX (abbreviated as FI) showed neuron-like morphology within an hour of induction (Fig. 1b). These MSCs isolated from rat were characterized as described previously [23]. They have the ability to self-renew as well as undergo multilineage differentiation to other cell lineages such as adipocytes and osteoblasts (Supplementary Fig. S1). A recent study attributed the morphology change to an artifact of cell shrinkage rather than neurite outgrowth [15]. We imaged live cells to determine whether the neuronlike morphology induced by cAMP was also a result of cell shrinkage. As the induction time increases, the cytoskeleton progressively retracts towards the cell center (Fig. 1c–f). Microtubules and actin filaments staining confirmed the reorganization and retraction of the cell body towards the cell center. The retraction appears incomplete, with partial disruption of the cytoplasm in some of the cells (Fig. 1b; Supplementary Fig. S2, arrows). As with the previous study, the cAMP-induced neurite-like structure is due to a disruption in the cytoskeleton and cell shrinkage rather than neurite outgrowth.
Fig. 1.
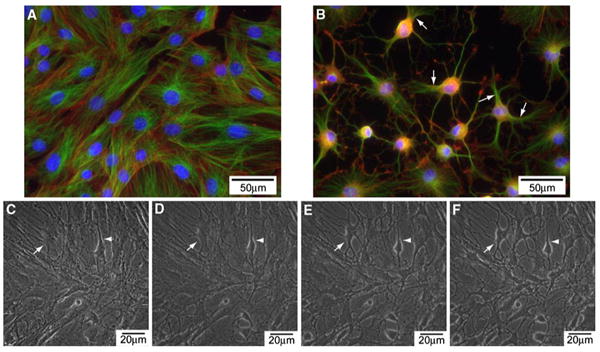
a Morphology of uninduced MSCs. Microtubules (shown in green); actin filaments (in red); nucleus (in blue). b Morphology of MSCs treated with 10 μM forskolin and 100 μM IBMX (FI) for 1 h. c–f Live cell images of cells treated with FI for 15 min (c), 30 min (d), 45 min (e) and 60 min (f)
Neuron-like morphology decreases along time
A large percentage of cells developed neuron-like morphology within an hour after FI treatment (Fig. 2a, denoted by the arrowhead), with an appreciable number still showing neuron-like morphology 3 h later (Fig. 2b, k). However, unlike the previous study with BME, which showed continuous changes in morphology [15], the population of cells with altered morphology decreased with increasing treatment time (Fig. 2k), with fewer cells showing neuron-like morphology 6 (Fig. 2c), 12 (Fig. 2d), and 24 h (Fig. 2e) after FI treatment. By the second day, even with fresh induction media, essentially no change in morphology was observed (Fig. 2f, g, k), which suggested that the neuron-like morphology cannot be re-induced by FI. Similar neuron-like morphology was initiated by the apoptosis inducer staurosporine (Supplementary Fig. S3), therefore we evaluated whether the morphology changes induced by FI treatment were due to apoptosis. However, the rise in apoptotic cells did not occur within the first 3 h after FI treatment (Fig. 2l), during which maximal changes in morphology resulted (Fig. 2k), suggesting the change in morphology was not likely due to apoptosis.
Fig. 2.
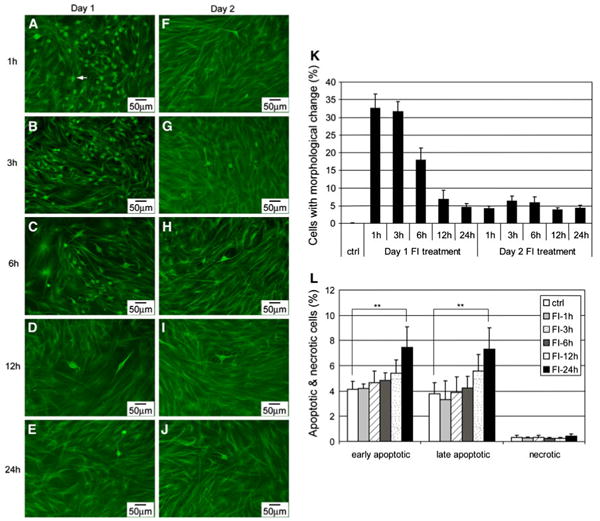
a–j Morphology of MSCs induced with 10 μM forskolin and 100 μM IBMX (FI) for different amount of time. Cells with changed morphology appear as green dots (as indicated by the arrowheads) at the magnification noted. a–e On day 1, cells treated with FI for 1, 3, 6, 12, and 24 h, respectively. f–j One day after FI treatment, cells treated again with FI for 1, 3, 6, 12, and 24 h, respectively. k Quantification of the percentage of cells with changed morphology (n = 3). l Apoptotic and necrotic labeling by PI (propidium iodide) and Alexa Fluor 488-conjugated annexin V, respectively, of cells treated with FI for 1, 3, 6, 12, and 24 h. Early apoptotic cells, PI− annexin V+ cells; late apoptotic cells, PI+ annexin V+ cells; necrotic cells, PI+ annexin V− cells (n = 3). **p<0.01
Morphology changes induced subsequent apoptosis
Changes in cell morphology and cytoskeletal structure can switch cells from surviving to apoptotic [27, 28]. Disruption of the cytoskeletal structure can lead to cell rounding and even detachment, which can result in anchorage-dependent apoptosis called anoikis [29]. Since cAMP elevation induced a disruption of the cytoskeletal structure in the MSCs (Supplementary Fig. S2), we assessed whether the morphology changes led to apoptosis. Initially, FI treatment disrupted the cytoskeletal structure in a large number of cells (Fig. 2a–c). However, most of the cells with changes in morphology remained attached and apoptosis or necrosis was not observed within the first few hours (Fig. 2l). As FI treatment continued, some cells that underwent morphology changes began to round up (Supplementary Fig. S4, arrows) and detach from the surface, likely due to a loss in their ability to anchor (Supplementary Fig. S4, arrowhead). The cells that round up (Fig. 3a, arrows) also showed positive staining against annexin-V (Fig. 3b, c, arrows), indicating that they have become apoptotic. The number of detached cells increased after 12 h, with cells floating after 24 h of treatment (data not shown), corresponding to the time at which the cells stained for apoptosis (Fig. 2l). Apoptosis increased significantly after 24 h (Fig. 2l) and was further enhanced after 48 h of FI treatment (denoted as day 2), albeit not statistically (Fig. 3d). Since additional morphology changes did not occur on the second day of FI treatment (Fig. 2k), i.e., very little cell rounding and detachment, correspondingly, increases in apoptosis was not observed (Fig. 3d). Concomitantly, caspase-3 activity, another indicator of apoptosis, increased significantly upon FI treatment but remained constant during the second day of treatment (Fig. 3e). Accordingly, these results suggest that a disruption of the cytoskeletal structure, induced upon cAMP elevation, resulted in subsequent apoptosis of ∼10% of the MSCs.
Fig. 3.
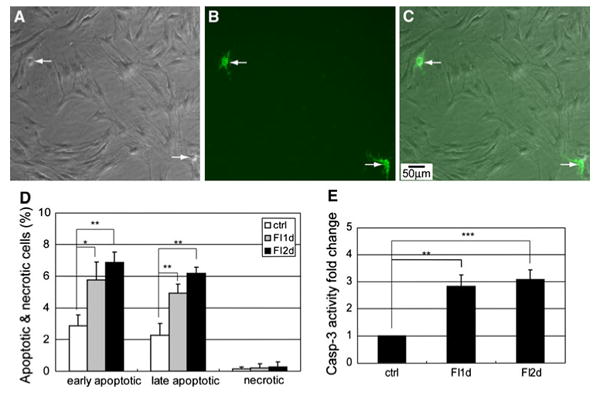
a–c Phase-contrast and fluorescence images of apoptotic cells labeled with Alexa Fluor 488-conjugated annexin V. Cells were treated with 10 μM forskolin and 100 μM IBMX (FI) for 24 h. a Phase-contrast image of MSCs treated with FI for 24 h. b Apoptotic staining with Alexa Fluor 488-conjugated annexin V. c Overlay image of (a) and (b). d Apoptotic and necrotic labeling by Alexa Fluor 488-conjugated annexin V and PI (propidium iodide), respectively, of control cells and cells treated with FI for 1 and 2 days. Early apoptotic cells, PI− annexin V+ cells; necrotic cells, PI+ annexin V− cells (n = 3). e Caspase-3 activity of control cells and cells treated with FI for 1 and 2 days. *p<0.05, **p<0.01, ***p<0.001
cAMP induced late phase neural differentiation
Unlike the early phase changes in morphology, which occurred quickly, neural differentiation took much longer. A previous study using DMSO/BHA to induce neural differentiation of MSCs did not observe an increase in mRNA level of neuron-specific enolase (NSE) [15], while we observed an increase in both mRNA (Supplementary Fig. S5) and protein levels (Fig. 4a) of neural markers. FI treatment increased the expression of neuron markers, NSE, and neuron-specific class β-III tubulin (Tuj1), as well as astrocytic marker, glial fibrillary acidic protein (GFAP) (Fig. 4a, c), but not within the first 6 h (Fig. 4b, d), whereas the changes in morphology peaked within the first 3 h (Fig. 2k). An increase in the expression of NSE and Tuj1 was detected 12 h after FI treatment (Fig. 4d) whereas an increase in GFAP was not detected until well after 12 h (Fig. 4a), suggesting that neural marker expression is a late-phase response as compared to the morphology changes. Immunostaining for the neuronal marker Tuj1 further supports the gradual increase in the neural marker expression. Untreated cells do not express Tuj1 (Fig. 4e), but a few cells gained Tuj1 expression after 2 days of FI treatment (Fig. 4f), with more cells expressing Tuj1 a week after FI treatment (Fig. 4g). Nonetheless, the cells that expressed Tuj1 look distinctly different from primary neurons (Fig. 4h).
Fig. 4.
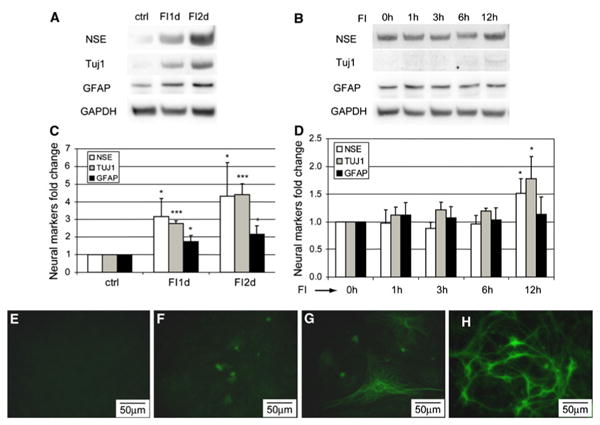
Expression of neural markers, NSE, Tuj1, and GFAP. a Protein levels of NSE, Tuj1, GFAP, and GAPDH of uninduced cells (ctrl), cells treated with 10 μM forskolin and 100 μM IBMX (FI) for 1 day (FI1d) and 2 days (FI2d). b Protein levels of NSE, Tuj1, GFAP, and GAPDH of cells treated with FI for 0, 1, 3, 6, and 12 h. c Quantification of NSE, Tuj1, and GFAP levels of the blots shown in (a) (n = 3). d Quantification of NSE, Tuj1, and GFAP levels of the blots shown in (b) (n = 3). *p<0.05; ***p<0.001. e–h Tuj1 staining of uninduced MSCs (e), MSCs treated with FI for 2 days (f) and 7 days (g) and primary neurons (h)
The induced cells also gained some neuronal function despite their distinct lack of neuronal morphology and features. Neuronal function was assessed with several neuronal activators, (a) dopamine, which stimulates Ca2+ signal through the dopamine receptors [30] or through potentiation of the N-methyl-d-aspartic acid (NMDA) receptors [31], (b) KCl, which stimulates Ca2+ signal through voltage-gated ion channels [32], and (c) glutamate, which induces Ca2+ signal through the glutamate receptors [33]. Figure 5a–d are representative images of FI treated cells upon stimulation with 100 μM dopamine, with time-dependent fluorescence intensity profiles of four individual cells shown in Fig. 5b. Intracellular Ca2+ levels remained fairly constant before stimulation (Fig. 5a, e, position denoted by A). Shortly after dopamine supplementation (Fig. 5e, arrow), some cells showed a Ca2+ signal within 10 s (Fig. 5b, e, Cell-2 and Cell-4, position denoted by B), and others peaked more slowly, taking up to 34 s (Fig. 5c, e, Cell-1 and Cell-3). The pattern of Ca2+ signaling also varied, with some cells responding with a single Ca2+ peak (Fig. 5e, Cell-1 and Cell-3) and other cells being repeatedly excited (Fig. 5e, Cell-2 and Cell-4). Uninduced cells and cells treated with FI for 1, 2, and 7 days were quantified for their neuronal function upon stimulation by dopamine, KCl, and glutamate (Fig. 5f). Around 40% of the uninduced MSCs showed a response to dopamine, which increased to 60% after 1 day and 80% after 7 days of FI treatment (Fig. 5f). Only 12% of the uninduced MSCs responded to KCl stimulation, which increased to 40% after FI treatment (Fig. 5f). Few uninduced cells responded to glutamate, with about 20% of the cells are glutamate responsive after a week of FI induction (Fig. 5f). The different percentage of cells that were responsive to dopamine, KCl, and glutamate suggest that some of the cells are able to respond to all three neuronal activators while others respond to one or two. These results suggest that FI induction enabled MSCs to gain some neuronal function. However, the heterogeneity in their response to neuronal activators (Fig. 5f), and the lack of neuronal morphology (Fig. 4g) indicate that the cells are not terminally differentiated.
Fig. 5.
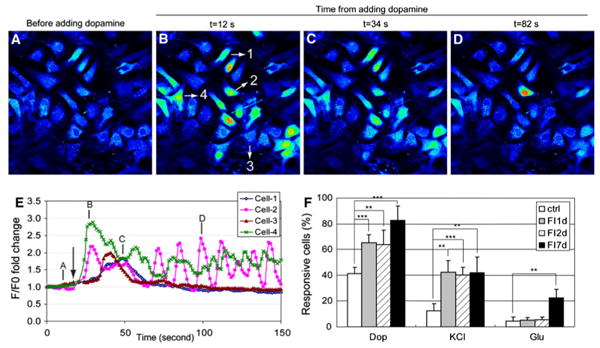
Calcium imaging in response to neuronal modulators, dopamine, glutamate, and KCl. a–d MSCs induced with 10 μM forskolin and 100 μM IBMX (FI) for 1 day and stimulated with 100 μM dopamine. Fluorescence images represented by a spectral table; warmer colors indicate higher fluorescence intensities and cooler colors indicate lower fluorescence intensities. Images were captured before dopamine stimulation or 12, 34, and 82 s after dopamine stimulation. e Fluorescence intensity profiles of four selected cells shown in (b) over time. Arrow indicates the point of dopamine addition. Annotation of A, B, C, D corresponds to the images in (a–d). f Quantification of the percentage of uninduced MSCs, FI induced MSCs for 1, 2, and 7 days that respond to 100 μM dopamine, 100 μM glutamate and 50 mM KCl (n = 4). **p<0.01, ***p<0.001
PKA regulates both the changes in morphology and neural marker expression
As shown above, the percentage of cells with changes in morphology peaked an hour after FI treatment and decreased with treatment time (Fig. 2k). The intracellular cAMP levels paralleled the changes in morphology, peaking also an hour after FI treatment and decreasing over the next few hours to a constant level that was maintained for the next 12 h (Fig. 6a). Although fresh media containing FI was added on the second day, the intracellular cAMP level did not increase (Fig. 6a), perhaps, because of desensitization of the agonist-induced receptor [34]. Since cAMP can trigger a variety of intracellular events through the activation of the classical downstream component protein kinase A (PKA) [16], we therefore assessed whether PKA activation is involved in regulating the changes in morphology and neural marker expressions. The activity of PKA can be determined by the level of phosphorylation of threonine 197 on the PKA catalytic subunit (PKAc) [35, 36]. Upon FI treatment, phosphorylated PKA (pPKAc) increased transiently and then returned to below basal level, whereas the total PKAc level dropped continuously, with both remaining stable after 1 day (Fig. 6b, c). Inhibiting PKA activity with H89 prevented the FI-induced morphology changes (Fig. 6d, e) as well as the neural marker expressions (Fig. 6f, g). Similar results were obtained using another PKA inhibitor Rp-cAMPS (Supplementary Fig. S6), suggesting that PKA mediates the onset of both the morphology changes and neural marker expression.
Fig. 6.
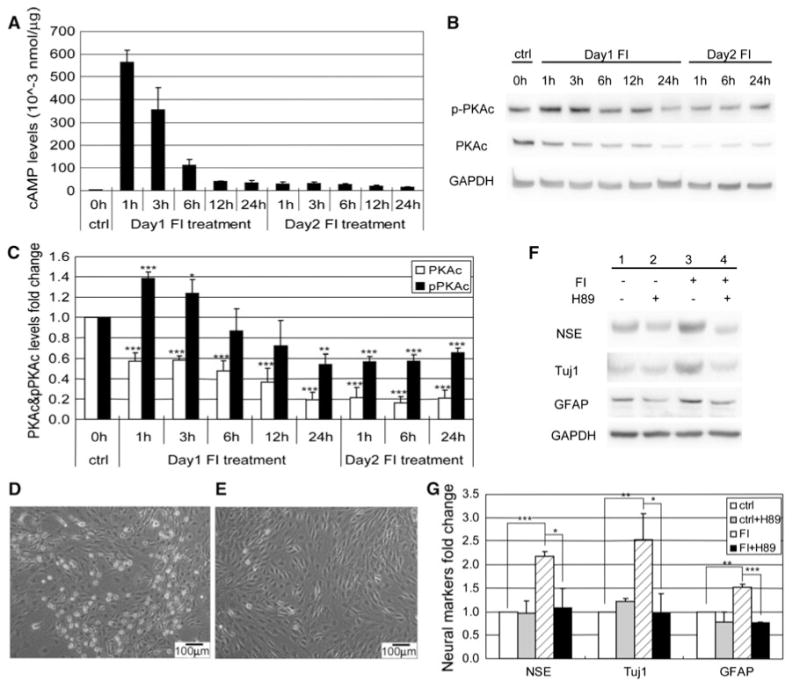
PKA regulates both morphology changes and neural markers expressions. a Intracellular cAMP levels of control MSCs, MSCs induced with 10 μM forskolin, and 100 μM IBMX (FI) for 1, 3, 6, 12, and 24 h (Day 1), and MSCs induced again with FI for 1, 3, 6, 12, and 24 h, following a 24-h FI induction period (day 2) (n = 3). b Western blotting results of PKAc and pPKAc levels of control MSCs, MSCs induced with FI for 1, 3, 6, 12, and 24 h, and MSCs induced with FI for 1, 6, and 24 h following a 24-h FI induction period. c Quantification of PKAc and threonine 197 phosphorylated PKAc (pPKAc) levels as in (b) (n = 4). *p<0.05, **p<0.01, ***p<0.001 as compared with the corresponding control condition. d Phase-contrast image of MSCs induced with FI for 1 h. e Phase-contrast image of MSCs induced with FI in the presence of 2.5 μM PKA inhibitor H89 for 1 h. f Expression of neural markers, NSE, Tuj1, and GFAP, in control MSCs, MSCs induced with 10 μM forskolin and 100 μM IBMX (FI) for 1 day in the absence or presence of 2.5 μM PKA inhibitor H89. g Quantification of Western-blot results as shown in (a) (n = 3). *p<0.05, **p<0.01, ***p<0.001
Morphology changes and neural marker expression are differentially regulated
Although the changes in both morphology and neural marker expression are regulated by PKA, the neuron-like morphology occurs early and very quickly, whereas the neural marker expressions appear much later and more slowly. Since the morphology changes appear prior to changes in neural marker expression (Figs. 1, 4), we assessed whether the morphology changes are required and thus precede the gain of neural marker expression. The microtubule stabilizer paclitaxel (Ptx) was used to inhibit the changes in morphology. Microtubules are dynamic polymers that undergo polymerization and depolymerization powered by energy from GTP hydrolysis [37]. Binding of Ptx to the microtubules prevents the depolymerization and stabilizes the microtubules [38]. Since dynamic polymerization and depolymerization are required for the assembly of the mitotic spindles, applying Ptx may cause mitosis inhibition and subsequent apoptosis [39, 40]. To minimize the toxicity induced by Ptx while still inhibiting the onset of morphology changes, the cells were treated with FI in the presence of 0.4 μM Ptx for only 3 h and the media was subsequently changed to FI media for another 21 h. Stabilized cytoskeletal structure was maintained upon removal of Ptx from the FI media (Supplementary Fig. S7). This could be due to the inability of the media to initiate another intracellular cAMP peak (Supplementary Fig. S8A) required for regulating the morphology changes (Figs. 2k, 6a). The number of viable cells 21 h after replacing the induction media (a total of 24 h including the 3-h pretreatment) remained constant, thus Ptx exposure did not adversely affect cell viability (Supplementary Fig. S8B). Although Ptx inhibited changes in morphology (Fig. 7a, b), it did not prevent the increase in neural marker expression (Fig. 7c, d), suggesting neuron-like morphology is not required for neural markers expression.
Fig. 7.
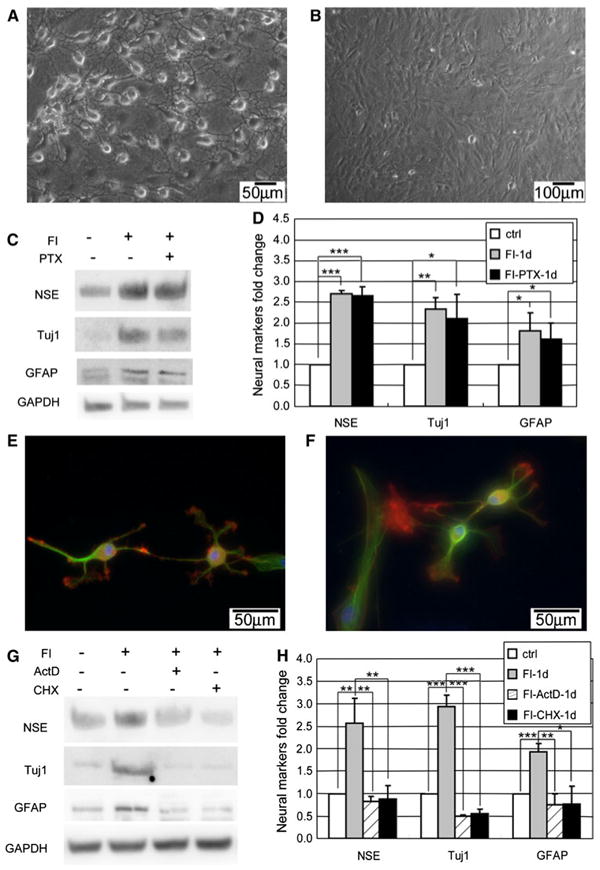
Morphology changes and neural markers are regulated differentially. a Appearance of neuron-like morphology of MSCs induced with 10 μM forskolin and 100 μM IBMX (FI) for 1 h. b Cell morphology of MSCs induced with FI in the presence of 0.4 μM microtubule stabilizer paclitaxel (Ptx) for 1 h. c–d Expression of neural markers, NSE, Tuj1, and GFAP, in control cells, cells induced with FI for 1 day in the absence or presence of Ptx (n = 3). For the FI treatment, cells were treated with FI or FI plus Ptx for 3 h and then changed to fresh FI media to reduce toxicity from Ptx treatment. *p<0.05, **p<0.01, ***p<0.001. e Appearance of neuron-like morphology of MSCs induced with 10 μM forskolin and 100 μM IBMX (FI) for 1 h in the presence of 1 μg/ml Actinomycin D (ActD). f Appearance of neuron-like morphology of MSCs induced with FI for 1 h in the presence of 10 μg/ml Cycloheximide (CHX). g–h Expression of neural markers, NSE, Tuj1, and GFAP, in control cells, cells induced with FI for 1 day, in the presence or absence of ActD or CHX. For the FI treatment, cells were treated with FI or FI plus ActD/CHX for 3 h and then changed to fresh FI media to reduce toxicity from ActD/CHX treatment. *p< 0.05, **p<0.01, ***p<0.001
The morphology changes occur very quickly (beginning minutes after FI treatment), thus it is unlikely that gene or protein synthesis is required for this event. Indeed, inhibiting transcription with actinomycin D (ActD) and translation with cycloheximide (CHX) did not impact the morphology changes (Fig. 7e, f), but did reduce the neural marker expression upon treatment with FI (Fig. 7g, h). Thus, transcription and translation are required for neural marker expression. ActD and CHX affects transcription and translation on a global level and treatment for a long period may cause cell death. Therefore, as with the paclitaxel treatment, the media supplemented with ActD or CHX was replaced after 3 h and the viability of the cells was assessed after 24 h of treatment. Supplementing the FI treatment with ActD or CHX did not significantly change the total number of viable cells (Supplementary Fig. S8B).
Taken together, the early phase neuron-like morphology changes do not rely on gene and protein synthesis, whereas the late-phase neural marker expressions do. Correspondingly, changes in morphology occur prior to but are not required for the expression of neural markers. Therefore, although both the morphology changes and the neural marker expressions are regulated by PKA, they are differentially regulated downstream of PKA.
Discussion
cAMP has been shown to play a positive role in the regeneration of the central nervous system (CNS) [41–43]. A decrease in the neuronal cAMP level has been associated with loss in neuronal regenerative capacity [41], whereas an increase in cAMP levels can promote axonal regeneration [44]. In addition to directly stimulating neuronal regeneration for neuronal repair, cAMP also can indirectly facilitate neuronal repair by inducing adult stem cells, such as MSCs, to differentiate into neural lineage cells [11]. cAMP has been suggested to be one of the best inducers of MSCs neural differentiation [13]. However, the assessment of neural differentiation has been largely based on the appearance of neuron-like morphology and neural marker expression [11, 13]. MSCs treated with cAMP-increasing agents (FI) achieved neuron-like morphology (Fig. 1b), which was due to disruption of the cytoskeleton and cell shrinkage (Fig. 1c–f) rather than neurite outgrowth. Thus, the morphology changes are not always a reliable assessment of neural differentiation. In contrast to the early onset of changes in morphology, expression of neural markers occurred much later. An increase in the neural markers, such as NSE, Tuj1, and GFAP became apparent a day after induction (Fig. 4a, c) with some degree of neuronal function, i.e., rise in calcium signaling in response to neuronal activators (Fig. 5). The gain of neuronal function by MSCs upon induction with cAMP alone has not been previously reported. Although our results showed that cAMP enabled MSCs to obtain neuronal function, the cells are at different stages with respect to their ability to respond to the different neuron activators. Some cells show a calcium rise upon stimulation by all three neuronal activators, while others respond to only one or two, or even none of the activators (Fig. 5f). Since these cells do not show the morphology of primary neurons (Fig. 4g), cAMP alone is unable to terminally differentiate the MSCs into neural lineage cells. The argument of whether MSCs can differentiate into functional neurons in vitro and in vivo is rarely addressed. A couple of transplantation studies indicated that MSCs can differentiate towards neural lineage cells in vivo [7, 45–48] and some of the differentiated cells were able to gain neuronal functionality [48]. However, whether these functional neurons are generated by MSCs through differentiation or fusion of MSCs with existing neurons remains unclear [49].
Our results point to differential regulation of the morphology and neural marker expression downstream of PKA. With respect to morphology, PKA can activate the Src homology domain (SH)2-containing phosphotyrosine phosphatase (SHP2) [50], which can then dephosphorylate the focal adhesion protein paxillin and result in disassembly of the focal adhesion and subsequent loss of actin stress fibers and cell rounding [50, 51]. Alternatively, the cAMP-PKA signaling pathway may disrupt the cytoskeleton by inhibiting the Rho family GTPases, which play important roles in modulating the cytoskeletal structure. Rho GTPases can promote actin polymerization and stress fiber formation by activating the polymerization factors actin-related protein 2/3 (Arp2/3) and formin, and inhibiting the depolymerization factor cofilin [52]. They can also regulate microtubule stability by inhibiting the microtubule disassembly factor oncoprotein 18 (Op18) and activating the mammalian Diaphanous-related (mDia) formins [52, 53]. cAMP can promote cytoskeleton disruption in mela-nocytes [54] mediated by RhoA inhibition [55], through phosphorylation of RhoA at serine 188 by PKA [56]. Therefore, the cAMP-PKA pathway could modulate the cytoskeleton by altering the phosphorylation status of focal adhesion proteins or the Rho GTPases.
Signaling events are rapid as compared to gene or protein synthesis. Thus in contrast, expression of neural markers takes longer and requires transcription and translation (Fig. 7g, h), suggesting that transcription factors activated by the cAMP-PKA signaling pathway may be involved. One such downstream factor is the cAMP response element binding protein (CREB), which can be phosphorylated by PKA at serine 133 [16]. FI treatment transiently increased nuclear PKAc and pCREB levels (Fig. 8a, b). Phosphorylation of CREB regulates neuro-genesis by promoting survival and differentiation of newborn neurons [57, 58]. Over-expressing the dominant negative form of CREB, M1-CREB (the serine 133 residue is mutated to alanine, therefore it can no longer be phosphorylated) in MSCs led to loss in their response to neuronal activators, such as dopamine (Fig. 8c, d). This suggests that phosphorylation of CREB plays a critical role in regulating MSC differentiation towards the neural lineage cells. Interestingly, MSCs stably expressing M1-CREB were much smaller than the normal MSCs after a week of FI induction (Fig. 8d), indicating that activated CREB is an important determinant in regulating cell size. Indeed, mice expressing dominant negative CREB are reduced in size during embryonic development [59]. Therefore, although the early onset of morphology changes induced by FI do not appear to require transcription (Fig. 7a), the transcription factor CREB is still required for regulating cell shape during MSC differentiation (Fig. 8d), hinting as possible cross-talk downstream of PKA.
Fig. 8.
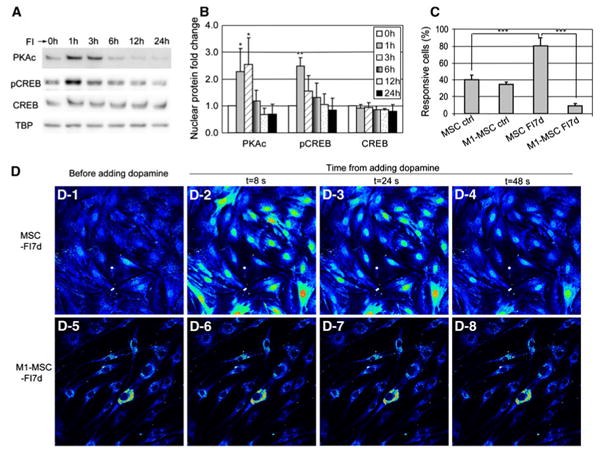
a Nuclear protein levels of PKAc, pCREB (ser133 phosphorylated CREB), and CREB in control cells and cells treated with 10 μM forskolin and 100 μM IBMX (FI) for 1, 3, 6, 12, and 24 h. b Quantification of nuclear PKAc, pCREB, and CREB levels (n = 3). *p<0.05; **p<0.01 as compared with the corresponding ‘0 h’ conditions. c Quantification of responsive cells stimulated with 100 μM dopamine. MSCs (expressing control vector) and M1-MSCs (expressing M1-CREB) were either in control media or induced with FI for 7 days (n = 3, ***p < 0.001). d A representative example of calcium signal in response to neuronal activator dopamine. D-1 to D-4 MSCs (expressing control vector) induced with FI for 1 week and stimulated with 100 μM dopamine. D-5 to D-8 M1-MSCs (expressing M1-CREB) induced with FI for 1 week and stimulated with 100 μM dopamine
Conclusions
The present study showed that cAMP induced changes in morphology early and neural differentiation and function much later and these events are regulated by cAMP-activated PKA, but diverge in their regulation downstream. While the morphology changes are likely due to cell shrinkage, which rendered some cells to be subsequently apoptotic, nonetheless the neural differentiation induced by cAMP enabled the MSCs to gain neuronal function.
Supplementary Material
Acknowledgments
We thank Dr. Carlose Molina at New Jersey Medical School for kindly providing the ICER antibody and Dr. David Ginty at Johns Hopkins University for kindly providing the M1-CREB plasmid. This study was supported in part by the National Science Foundation (CBET 0941055), the National Institute of Health (R01GM079688 and R21RR024439), the MUCI, and the MSU Foundation and the Center for Systems Biology.
Abbreviations
- cAMP
Cyclic adenosine monophosphate
- MSCs
Mesenchymal stem cells
- PKA
Protein kinase A
- PKAc
PKA catalytic subunit
- pPKAc
Threonine 197 phosphorylated PKAc
- SCI
Spinal cord injury
- IBMX
Isobutylmethylxanthine
- FI
Forskolin and IBMX
- ActD
Actinomycin D
- CHX
Cycloheximide
- Ptx
Paclitaxel
- PI
Propidium iodide
- NSE
Neuron-specific enolase
- Tuj1
β-III Tubulin
- GFAP
Glial fibrillary acidic protein
- CREB
cAMP response element binding protein
- pCREB
Serine 133 phosphorylated CREB
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s00018-010-0497-1) contains supplementary material, which is available to authorized users.
Contributor Information
Linxia Zhang, Department of Chemical Engineering and Materials Science, Michigan State University, 2527 EB, East Lansing, MI 48824, USA.
Linsey C. Seitz, Department of Chemical Engineering and Materials Science, Michigan State University, 2527 EB, East Lansing, MI 48824, USA
Amy M. Abramczyk, Department of Chemical Engineering and Materials Science, Michigan State University, 2527 EB, East Lansing, MI 48824, USA
Li Liu, Department of Microbiology and Molecular Genetics, Michigan State University, East Lansing, MI, USA.
Christina Chan, Email: krischan@egr.msu.edu, Department of Chemical Engineering and Materials Science, Michigan State University, 2527 EB, East Lansing, MI 48824, USA; Department of Biochemistry and Molecular Biology, Michigan State University, East Lansing, MI, USA.
References
- 1.Kuznetsov SA, Krebsbach PH, Satomura K, Kerr J, Riminucci M, Benayahu D, Robey PG. Single-colony derived strains of human marrow stromal fibroblasts form bone after transplantation in vivo. J Bone Miner Res. 1997;12:1335–1347. doi: 10.1359/jbmr.1997.12.9.1335. [DOI] [PubMed] [Google Scholar]
- 2.Herbertson A, Aubin JE. Cell sorting enriches osteogenic populations in rat bone marrow stromal cell cultures. Bone. 1997;21:491–500. doi: 10.1016/s8756-3282(97)00197-x. [DOI] [PubMed] [Google Scholar]
- 3.Berry L, Grant ME, McClure J, Rooney P. Bone-marrow-derived chondrogenesis in vitro. J Cell Sci. 1992;101(Pt 2):333–342. doi: 10.1242/jcs.101.2.333. [DOI] [PubMed] [Google Scholar]
- 4.Lee KD, Kuo TK, Whang-Peng J, Chung YF, Lin CT, Chou SH, Chen JR, Chen YP, Lee OK. In vitro hepatic differentiation of human mesenchymal stem cells. Hepatology. 2004;40:1275–1284. doi: 10.1002/hep.20469. [DOI] [PubMed] [Google Scholar]
- 5.Makino S, Fukuda K, Miyoshi S, Konishi F, Kodama H, Pan J, Sano M, Takahashi T, Hori S, Abe H, Hata J, Umezawa A, Ogawa S. Cardiomyocytes can be generated from marrow stromal cells in vitro. J Clin Invest. 1999;103:697–705. doi: 10.1172/JCI5298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deng J, Petersen BE, Steindler DA, Jorgensen ML, Laywell ED. Mesenchymal stem cells spontaneously express neural proteins in culture and are neurogenic after transplantation. Stem Cells. 2006;24:1054–1064. doi: 10.1634/stemcells.2005-0370. [DOI] [PubMed] [Google Scholar]
- 7.Kopen GC, Prockop DJ, Phinney DG. Marrow stromal cells migrate throughout forebrain and cerebellum, and they differentiate into astrocytes after injection into neonatal mouse brains. Proc Natl Acad Sci USA. 1999;96:10711–10716. doi: 10.1073/pnas.96.19.10711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McBeath R, Pirone DM, Nelson CM, Bhadriraju K, Chen CS. Cell shape, cytoskeletal tension, and RhoA regulate stem cell lineage commitment. Dev Cell. 2004;6:483–495. doi: 10.1016/s1534-5807(04)00075-9. [DOI] [PubMed] [Google Scholar]
- 9.Kilian KA, Bugarija B, Lahn BT, Mrksich M. Geometric cues for directing the differentiation of mesenchymal stem cells. Proc Natl Acad Sci USA. 2010;107:4872–4877. doi: 10.1073/pnas.0903269107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Engler AJ, Sen S, Sweeney HL, Discher DE. Matrix elasticity directs stem cell lineage specification. Cell. 2006;126:677–689. doi: 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- 11.Deng W, Obrocka M, Fischer I, Prockop DJ. In vitro differentiation of human marrow stromal cells into early progenitors of neural cells by conditions that increase intracellular cyclic AMP. Biochem Biophys Res Commun. 2001;282:148–152. doi: 10.1006/bbrc.2001.4570. [DOI] [PubMed] [Google Scholar]
- 12.Krampera M, Marconi S, Pasini A, Galie M, Rigotti G, Mosna F, Tinelli M, Lovato L, Anghileri E, Andreini A, Pizzolo G, Sbarbati A, Bonetti B. Induction of neural-like differentiation in human mesenchymal stem cells derived from bone marrow, fat, spleen and thymus. Bone. 2007;40:382–390. doi: 10.1016/j.bone.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 13.Jori FP, Napolitano MA, Melone MA, Cipollaro M, Cascino A, Altucci L, Peluso G, Giordano A, Galderisi U. Molecular pathways involved in neural in vitro differentiation of marrow stromal stem cells. J Cell Biochem. 2005;94:645–655. doi: 10.1002/jcb.20315. [DOI] [PubMed] [Google Scholar]
- 14.Dezawa M, Kanno H, Hoshino M, Cho H, Matsumoto N, Itokazu Y, Tajima N, Yamada H, Sawada H, Ishikawa H, Mimura T, Kitada M, Suzuki Y, Ide C. Specific induction of neuronal cells from bone marrow stromal cells and application for autologous transplantation. J Clin Invest. 2004;113:1701–1710. doi: 10.1172/JCI20935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lu P, Blesch A, Tuszynski MH. Induction of bone marrow stromal cells to neurons: differentiation, transdifferentiation, or artifact? J Neurosci Res. 2004;77:174–191. doi: 10.1002/jnr.20148. [DOI] [PubMed] [Google Scholar]
- 16.Daniel PB, Walker WH, Habener JF. Cyclic AMP signaling and gene regulation. Annu Rev Nutr. 1998;18:353–383. doi: 10.1146/annurev.nutr.18.1.353. [DOI] [PubMed] [Google Scholar]
- 17.Mayr B, Montminy M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat Rev Mol Cell Biol. 2001;2:599–609. doi: 10.1038/35085068. [DOI] [PubMed] [Google Scholar]
- 18.Dohi T, Xia F, Altieri DC. Compartmentalized phosphorylation of IAP by protein kinase A regulates cytoprotection. Mol Cell. 2007;27:17–28. doi: 10.1016/j.molcel.2007.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Park SY, Cho SJ, Kwon HC, Lee KR, Rhee DK, Pyo S. Caspase-independent cell death by allicin in human epithelial carcinoma cells: involvement of PKA. Cancer Lett. 2005;224:123–132. doi: 10.1016/j.canlet.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 20.Howe AK. Regulation of actin-based cell migration by cAMP/PKA. Biochim Biophys Acta. 2004;1692:159–174. doi: 10.1016/j.bbamcr.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 21.Yang DC, Tsay HJ, Lin SY, Chiou SH, Li MJ, Chang TJ, Hung SC. cAMP/PKA regulates osteogenesis, adipogenesis and ratio of RANKL/OPG mRNA expression in mesenchymal stem cells by suppressing leptin. PLoS One. 2008;3:e1540. doi: 10.1371/journal.pone.0001540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang TT, Tio M, Lee W, Beerheide W, Udolph G. Neural differentiation of mesenchymal-like stem cells from cord blood is mediated by PKA. Biochem Biophys Res Commun. 2007;357:1021–1027. doi: 10.1016/j.bbrc.2007.04.046. [DOI] [PubMed] [Google Scholar]
- 23.Zhang L, Chan C. Isolation and enrichment of rat mesenchymal stem cells (MSCs) and separation of single-colony derived MSCs. J Vis Exp. 2010 doi: 10.3791/1852. http://www.jove.com/index/details.stp?id=1852. [DOI] [PMC free article] [PubMed]
- 24.Patil S, Melrose J, Chan C. Involvement of astroglial ceramide in palmitic acid-induced Alzheimer-like changes in primary neurons. Eur J Neurosci. 2007;26:2131–2141. doi: 10.1111/j.1460-9568.2007.05797.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bell PB, Jr, Safiejko-Mroczka B. Improved methods for preserving macromolecular structures and visualizing them by fluorescence and scanning electron microscopy. Scanning Microsc. 1995;9:843–857. discussion 858-860. [PubMed] [Google Scholar]
- 26.Tropel P, Platet N, Platel JC, Noel D, Albrieux M, Benabid AL, Berger F. Functional neuronal differentiation of bone marrow-derived mesenchymal stem cells. Stem Cells. 2006;24:2868–2876. doi: 10.1634/stemcells.2005-0636. [DOI] [PubMed] [Google Scholar]
- 27.Chen CS, Mrksich M, Huang S, Whitesides GM, Ingber DE. Geometric control of cell life and death. Science. 1997;276:1425–1428. doi: 10.1126/science.276.5317.1425. [DOI] [PubMed] [Google Scholar]
- 28.Flusberg DA, Numaguchi Y, Ingber DE. Cooperative control of Akt phosphorylation, bcl-2 expression, and apoptosis by cytoskeletal microfilaments and microtubules in capillary endothelial cells. Mol Biol Cell. 2001;12:3087–3094. doi: 10.1091/mbc.12.10.3087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gilmore AP. Anoikis. Cell Death Differ. 2005;12(Suppl 2):1473–1477. doi: 10.1038/sj.cdd.4401723. [DOI] [PubMed] [Google Scholar]
- 30.Bergson C, Levenson R, Goldman-Rakic PS, Lidow MS. Dopamine receptor-interacting proteins: the Ca(2+) connection in dopamine signaling. Trends Pharmacol Sci. 2003;24:486–492. doi: 10.1016/S0165-6147(03)00232-3. [DOI] [PubMed] [Google Scholar]
- 31.Chen G, Greengard P, Yan Z. Potentiation of NMDA receptor currents by dopamine D1 receptors in prefrontal cortex. Proc Natl Acad Sci USA. 2004;101:2596–2600. doi: 10.1073/pnas.0308618100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wulff H, Castle NA, Pardo LA. Voltage-gated potassium channels as therapeutic targets. Nat Rev Drug Discov. 2009;8:982–1001. doi: 10.1038/nrd2983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tikhonov DB, Magazanik LG. Origin and molecular evolution of ionotropic glutamate receptors. Neurosci Behav Physiol. 2009;39:763–773. doi: 10.1007/s11055-009-9195-6. [DOI] [PubMed] [Google Scholar]
- 34.Bunemann M, Lee KB, Pals-Rylaarsdam R, Roseberry AG, Hosey MM. Desensitization of G-protein-coupled receptors in the cardiovascular system. Annu Rev Physiol. 1999;61:169–192. doi: 10.1146/annurev.physiol.61.1.169. [DOI] [PubMed] [Google Scholar]
- 35.Steinberg RA, Cauthron RD, Symcox MM, Shuntoh H. Autoactivation of catalytic (C alpha) subunit of cyclic AMP-dependent protein kinase by phosphorylation of threonine 197. Mol Cell Biol. 1993;13:2332–2341. doi: 10.1128/mcb.13.4.2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cauthron RD, Carter KB, Liauw S, Steinberg RA. Physiological phosphorylation of protein kinase A at Thr-197 is by a protein kinase A kinase. Mol Cell Biol. 1998;18:1416–1423. doi: 10.1128/mcb.18.3.1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Desai A, Mitchison TJ. Microtubule polymerization dynamics. Annu Rev Cell Dev Biol. 1997;13:83–117. doi: 10.1146/annurev.cellbio.13.1.83. [DOI] [PubMed] [Google Scholar]
- 38.Xiao H, Verdier-Pinard P, Fernandez-Fuentes N, Burd B, Angeletti R, Fiser A, Horwitz SB, Orr GA. Insights into the mechanism of microtubule stabilization by Taxol. Proc Natl Acad Sci USA. 2006;103:10166–10173. doi: 10.1073/pnas.0603704103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yvon AM, Wadsworth P, Jordan MA. Taxol suppresses dynamics of individual microtubules in living human tumor cells. Mol Biol Cell. 1999;10:947–959. doi: 10.1091/mbc.10.4.947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ofir R, Seidman R, Rabinski T, Krup M, Yavelsky V, Weinstein Y, Wolfson M. Taxol-induced apoptosis in human SKOV3 ovarian and MCF7 breast carcinoma cells is caspase-3 and caspase-9 independent. Cell Death Differ. 2002;9:636–642. doi: 10.1038/sj.cdd.4401012. [DOI] [PubMed] [Google Scholar]
- 41.Cai D, Qiu J, Cao Z, McAtee M, Bregman BS, Filbin MT. Neuronal cyclic AMP controls the developmental loss in ability of axons to regenerate. J Neurosci. 2001;21:4731–4739. doi: 10.1523/JNEUROSCI.21-13-04731.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rydel RE, Greene LA. cAMP analogs promote survival and neurite outgrowth in cultures of rat sympathetic and sensory neurons independently of nerve growth factor. Proc Natl Acad Sci USA. 1988;85:1257–1261. doi: 10.1073/pnas.85.4.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Goldberg JL, Barres BA. The relationship between neuronal survival and regeneration. Annu Rev Neurosci. 2000;23:579–612. doi: 10.1146/annurev.neuro.23.1.579. [DOI] [PubMed] [Google Scholar]
- 44.Pearse DD, Pereira FC, Marcillo AE, Bates ML, Berrocal YA, Filbin MT, Bunge MB. cAMP and Schwann cells promote axonal growth and functional recovery after spinal cord injury. Nat Med. 2004;10:610–616. doi: 10.1038/nm1056. [DOI] [PubMed] [Google Scholar]
- 45.Mahmood A, Lu D, Wang L, Li Y, Lu M, Chopp M. Treatment of traumatic brain injury in female rats with intravenous administration of bone marrow stromal cells. Neurosurgery. 2001;49:1196–1203. Discussion 1203⌓1194. [PubMed] [Google Scholar]
- 46.Mahmood A, Lu D, Lu M, Chopp M. Treatment of traumatic brain injury in adult rats with intravenous administration of human bone marrow stromal cells. Neurosurgery. 2003;53:697–702. doi: 10.1227/01.neu.0000079333.61863.aa. discussion 702-693. [DOI] [PubMed] [Google Scholar]
- 47.Mezey E, Key S, Vogelsang G, Szalayova I, Lange GD, Crain B. Transplanted bone marrow generates new neurons in human brains. Proc Natl Acad Sci USA. 2003;100:1364–1369. doi: 10.1073/pnas.0336479100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bae JS, Han HS, Youn DH, Carter JE, Modo M, Schuchman EH, Jin HK. Bone marrow-derived mesenchymal stem cells promote neuronal networks with functional synaptic transmission after transplantation into mice with neurodegeneration. Stem cells. 2007;25:1307–1316. doi: 10.1634/stemcells.2006-0561. [DOI] [PubMed] [Google Scholar]
- 49.Weimann JM, Charlton CA, Brazelton TR, Hackman RC, Blau HM. Contribution of transplanted bone marrow cells to Purkinje neurons in human adult brains. Proc Natl Acad Sci USA. 2003;100:2088–2093. doi: 10.1073/pnas.0337659100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rocchi S, Gaillard I, van Obberghen E, Chambaz EM, Vilgrain I. Adrenocorticotrophic hormone stimulates phosphotyrosine phosphatase SHP2 in bovine adrenocortical cells: phosphorylation and activation by cAMP-dependent protein kinase. Biochem J. 2000;352(Pt 2):483–490. [PMC free article] [PubMed] [Google Scholar]
- 51.Han JD, Rubin CS. Regulation of cytoskeleton organization and paxillin dephosphorylation by cAMP. Studies on murine Y1 adrenal cells. J Biol Chem. 1996;271:29211–29215. doi: 10.1074/jbc.271.46.29211. [DOI] [PubMed] [Google Scholar]
- 52.Jaffe AB, Hall A. Rho GTPases: biochemistry and biology. Annu Rev Cell Dev Biol. 2005;21:247–269. doi: 10.1146/annurev.cellbio.21.020604.150721. [DOI] [PubMed] [Google Scholar]
- 53.Palazzo AF, Cook TA, Alberts AS, Gundersen GG. mDia mediates Rho-regulated formation and orientation of stable microtubules. Nat Cell Biol. 2001;3:723–729. doi: 10.1038/35087035. [DOI] [PubMed] [Google Scholar]
- 54.Busca R, Bertolotto C, Abbe P, Englaro W, Ishizaki T, Narumiya S, Boquet P, Ortonne JP, Ballotti R. Inhibition of Rho is required for cAMP-induced melanoma cell differentiation. Mol Biol Cell. 1998;9:1367–1378. doi: 10.1091/mbc.9.6.1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dong JM, Leung T, Manser E, Lim L. cAMP-induced morphological changes are counteracted by the activated RhoA small GTPase and the Rho kinase ROKalpha. J Biol Chem. 1998;273:22554–22562. doi: 10.1074/jbc.273.35.22554. [DOI] [PubMed] [Google Scholar]
- 56.Lang P, Gesbert F, Delespine-Carmagnat M, Stancou R, Pouchelet M, Bertoglio J. Protein kinase A phosphorylation of RhoA mediates the morphological and functional effects of cyclic AMP in cytotoxic lymphocytes. EMBO J. 1996;15:510–519. [PMC free article] [PubMed] [Google Scholar]
- 57.Nakagawa S, Kim JE, Lee R, Chen J, Fujioka T, Malberg J, Tsuji S, Duman RS. Localization of phosphorylated cAMP response element-binding protein in immature neurons of adult hippocampus. J Neurosci. 2002;22:9868–9876. doi: 10.1523/JNEUROSCI.22-22-09868.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nakagawa S, Kim JE, Lee R, Malberg JE, Chen J, Steffen C, Zhang YJ, Nestler EJ, Duman RS. Regulation of neurogenesis in adult mouse hippocampus by cAMP and the cAMP response element-binding protein. J Neurosci. 2002;22:3673–3682. doi: 10.1523/JNEUROSCI.22-09-03673.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sordella R, Classon M, Hu KQ, Matheson SF, Brouns MR, Fine B, Zhang L, Takami H, Yamada Y, Settleman J. Modulation of CREB activity by the Rho GTPase regulates cell and organism size during mouse embryonic development. Dev Cell. 2002;2:553–565. doi: 10.1016/s1534-5807(02)00162-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.