

Abstract
It is well-established that LTP, a paradigm for learning and memory, results in a stable enlargement of potentiated spines associated with recruitment of additional GluA1-containing AMPARs. Although regulation of the actin cytoskeleton is involved, the detailed signaling mechanisms responsible for this spine expansion are unclear. Here we used cultured mature hippocampal neurons stimulated with a Glycine-Induced, synapse-specific form of chemical LTP (GI-LTP). We report that the stable structural plasticity (i.e., spine head enlargement and spine length shortening) that accompanies GI-LTP was blocked by inhibitors of NMDARs (APV) or CaM-kinase kinase (STO-609), the upstream activator of CaM-kinase I (CaMKI), as well as by transfection with dominant-negative (dn) CaMKI but not dnCaMKIV. Recruitment of GluA1 to the spine surface occurred following GI-LTP and was mimicked by transfection with constitutively-active CaMKI. Spine enlargement induced by transfection of GluA1 was associated with synaptic recruitment of Ca2+-permeable AMPARs (CP-AMPARs) as assessed by an increase in the rectification index of mEPSCs and their sensitivity to IEM-1460, a selective antagonist of CP-AMPARs. Furthermore, the increase in spine size and mEPSC amplitude resulting from GI-LTP itself was blocked by IEM-1460, demonstrating involvement of CP-AMPARs. Downstream signaling effectors of CP-AMPARs, identified by suppression of their activation by IEM-1460, included the Rac/PAK/LIM-kinase pathway that regulates spine actin dynamics.
Taken together, our results suggest that synaptic recruitment of CP-AMPARs via CaMKI may provide a mechanistic link between NMDAR activation in LTP and regulation of a signaling pathway that drives spine enlargement via actin polymerization.
Keywords: LTP, spine, AMPA receptors, protein kinase, actin
Introduction
Dendritic spines are highly dynamic structures that receive the vast majority of excitatory synaptic input within the brain. A long-held hypothesis states that functional changes in synaptic efficacy accompany changes in synapse/spine morphology (Cajal, 1911; Hebb, 1949). Particularly, changes in spine structure and number have been associated with long-term potentiation (LTP) (reviewed in Carlisle and Kennedy, 2005; Tada and Sheng, 2006; Bourne and Harris, 2008), a candidate cellular model for learning and memory. Indeed, several types of mental retardation and cognitive dysfunction are associated with abnormalities in spine density and morphology (Purpura, 1974; Kaufmann and Moser, 2000; Fiala et al., 2002).
Alterations in spine morphology are tightly coupled to changes in filamentous actin present throughout the spine (Fifkova and Delay, 1982; Matus et al., 1982; Matsuzaki et al., 2004) where it also anchors many postsynaptic channels and signaling proteins (Oertner and Matus, 2005). Spine actin is modulated largely by Rac1-activated PAK that activates LIM-kinase (LIMK) (reviewed in Boda et al., 2006) to promote actin polymerization by phosphorylating ADF/cofilin, thereby inhibiting actin depolymerizing activity (Bamburg et al., 1999). Activated PAK1 and PAK3 are concentrated in dendritic spines (Penzes et al., 2003; Hayashi et al., 2004), and genetic disruption (Allen et al., 1998) or small peptide inhibitors of PAK result in altered spine phenotype and impaired memory consolidation (Hayashi et al., 2004), whereas constitutively-active (ca) PAK promotes spine formation (Hayashi et al., 2007). LIMK-/- mice show reduced levels of phosphorylated cofilin, abnormal spine morphology, and deficits in activity-dependent synaptic plasticity and spatial learning (Meng et al., 2002), consistent with LIMK's role in stabilizing F-actin in spines. Notably, genetic mutations of PAK-3 and LIMK-1 have been implicated in non-syndromic X-linked mental retardation (Allen et al., 1998) and Williams syndrome (Tassabehji et al., 1996) respectively.
Prior work establishes that actin-mediated spine enlargement associated with LTP requires increased intracellular calcium (Fifkova, 1985; Oertner and Matus, 2005; Vlachos et al., 2009). It has been assumed that the major source of this calcium is due to activity-dependent activation of NMDARs. However, we and others have recently shown that LTP induced by certain protocols (e.g., theta-burst) is associated with the transient incorporation of calcium-permeable AMPARs (CP-AMPARs) into the synapse (Plant et al., 2006; but see Adesnik and Nicoll, 2007; Gray et al., 2007; Lu et al., 2007; Guire et al., 2008)). We found that synaptic incorporation of CP-AMPARs, which can significantly enhance synaptic conductance, requires the activation of CaM-kinase I (CaMKI) (Guire et al., 2008), but a role for CP-AMPRs in structural plasticity of spines has not previously been reported. We previously demonstrated that spinogenesis during early development requires CaMK1-mediated phosphorylation of βPIX to activate Rac1 (Saneyoshi et al., 2008).
Here we have demonstrated that synaptic recruitment of CP-AMPARs by CaMKI contributes to actin-dependent structural plasticity induced by GI-LTP. We also show that synaptic CP-AMPARs are essential for downstream activation of the Rac/PAK/LIMK pathway necessary for actin-mediated spine enlargement and associated synaptic potentiation.
Methods
Reagents and constructs
D-(-)-2-Amino-5-phosphonopentanoic acid (D-APV), 1,8-Naphthoylene benzimidazole-3-carboxylic acid (STO-609), N,N,N,-Trimethyl-5-[(tricyclo[3.3.1.13,7]dec-1-ylmethyl )amino]-1-pentanaminiumbromide hydrobromide (IEM-1460), [R-(R*,S*)]-5-(6,8-Dihydro-8-oxofuro[3,4-e]-1,3-benzodi oxol-6-yl)-5,6,7,8-tetrahydro-6,6-dimethyl-1,3-dioxolo[ 4,5-g] isoquinolinium iodide (Bicuculline methiodide), strychnidin-10-one hydrochloride (Strychnine), 6-Cyano-7-nitroquinoxaline-2,3-dione disodium (CNQX), and octahydro-12-(hydroxymethyl)-2-imino-5,9:7,10a-dimethan-o-10aH-[1,3]dioxocino[6,5-d]pyrimidine-4,7,10,11,12-pen tol (TTx) were purchased from Tocris Bioscience. Latrunculin A was purchased from Calbiochem.
Constitutively active CaMKI (caCaMKI), dominant-negative CaMKI (dnCaMKI), dnCaMKK, CaMKKins, mRFP-βactin, dominant-negative PAK1 (dnPAK1), and myc-tagged Rac1 (Rac1) have been described previously (Wayman et al., 2004; Wayman et al., 2006; Saneyoshi et al., 2008; Impey S, 2010).
Cell Culture and Neuronal Transfection
Hippocampal neurons were isolated from P0–2 Sprague-Dawley rats as described previously (Wayman et al., 2006). After harvesting, neurons were plated on poly-l-lysine (Sigma; molecular weight 300,000) coated 12 mm glass coverslips at a density of 4.0 × 104 cells per square centimeter or coated plastic 35 mm wells at 4.5 × 105 cells per square centimeter. Neurons were maintained in Neurobasal A media (Invitrogen) supplemented with B27 (Invitrogen) and 0.5 mM L-glutamine with 5 μM cytosine-D-arabinofuranoside (AraC) added at 2 DIV. Neurons were cultured for 13 – 16 days before being transfected using LipofectAMINE 2000 (Invitrogen) and/or treated with indicated pharmacological reagents. DNA, transfection reagent, and transfection duration were optimized to minimize toxicity and maximize transfection efficiency (0.5 – 3% of neurons between 13 and 16 DIV).
Quantification of spine density and morphology
For experiments used in the quantification of spine morphology hippocampal neurons were first transfected with mRFP-βactin between 12 - 14 DIV to aid in the visualization of dendritic spines (Saneyoshi et al., 2008) +/- test plasmids. Expression of low levels of βactin has been shown to have no significant effect on either spine density or size (Saneyoshi et al., 2008). Prior to imaging neurons were fixed for 15 min at room temperature in pre warmed (37 °C) fixative (4% paraformaldehyde, 4% sucrose in phosphate-buffered saline, (PBS)). Fluorescent images were acquired using a cooled CCD camera (Hamamatsu Photonics, Shizuoka, Japan) attached to a Zeiss Axiplan2 (Carl Zeiss, Inc.) inverted microscope with a 63× oil immersion lens. Morphometric measurements consisting of spine head area, length, head width, and density were carried out using NIH Image J software (Abramoff, 2004). Spines were defined as having head widths that were at least twice the diameter of the spine neck. Three 50 μm sections of secondary dendrite from each neuron were analyzed. The number of spines and coverslips quantified for each experiment is reported within the corresponding figure legend. Each experiment was repeated at least three times with independent culture preparations.
Immunocytochemistry
Hippocampal neurons were fixed in 4% paraformaldehyde, 4% sucrose in PBS, and 50 mM HEPES pH 7.5 at 37 °C for 15 min. Neurons were rinsed 3 times for 10 min in PBS and blocked for 1 h in blocking buffer (PBS containing 3% BSA) at room temperature (22 °C). Neurons were then stained for surface-expressed GluA1 using a rabbit anti-GluA1 N-terminal antibody (Calbiochem, 1:100), which recognizes amino acids 271-285 of rat GluA1, in blocking buffer overnight at room temperature and washed 3 times for 10 min in blocking buffer. Surface GluA1 was subsequently detected by incubating coverslips in blocking buffer containing anti-rabbit Alexa Fluor-488 (Molecular Probes, 1:2000) for 40 min at room temperature. Hoechst dye (Molecular probes) was also added to the secondary buffer to allow assessment of cell viability. Coverslips were then washed quickly in PBS and mounted onto slides using Elvanol mounting medium.
GluA1 surface biotinylation
Biotinylation experiments were performed as previously described (Oh et al., 2006). Briefly, following treatments neuronal cell cultures were washed with cold ACSF (pH adjusted to 8.2) to fully protonate amine groups prior to biotinylation. After removing the wash buffer cells were incubated with ice cold ACSF (pH 8.2) containing 0.8 mg/ml NHS-SS-biotin (Soltec Ventures) for 30 min on a shaker at 4°C. Cells were then quenched with ACSF containing 50 mM Tris-HCl for 10 min on a shaker at 4°C. Cells were then flash frozen using liquid nitrogen and stored at -80 °C prior to being subjected to an avidin pulldown.
For avidin pulldowns cells were thawed on ice and lysed in ice cold RIPA lysis buffer. Lysates were then centrifuged for 10 min at 14K at 4°C to pellet insoluble material. Neutravidin beads (20 μl, Pierce) were added to the supernatant. Lysates containing avidin beads were rotated at 4°C for 2 hrs and then washed 2 times with ice cold lysis buffer. Biotinylated proteins were extracted with 2× SDS sample buffer supplemented with 50 mM DTT for 30 min at 50°C prior to being subjected to a western blot. Western blotting was performed as previously described (Davare et al., 2004). Biotinylated GluA1 and total GluA1 (cell lysate) were probed with αGluA1 (courtesy of Dr. Wenthold) and detected with IR700- and/or IR800 conjugated secondary antibodies. Blots were quantified using software supplied with the Odyssey Infrared System (LI-COR Biosciences).
GST-PAK-PBD pulldowns
Hippocampal neurons were rapidly harvested with ice-cold PAK-PBD lysis buffer containing 1% Triton-X-100, and (in mM) 50 HEPES pH 7.4, 150 NaCl, and 25 MgCl2 that was supplemented just prior to use with 1 mM DTT and protease and phosphatase inhibitors (Leupeptin, Aprotinin, Benzamidine, Pepstatin-A and Antipain, Cantharidic Acid and Microcystin A). Cell lysates were centrifuged at 20,000g for 10 minutes to clear insoluble material. Cleared lysates were applied to pre-loaded glutathione sepharose beads containing 30 μg GST-PAK-PBD. Samples were incubated for 1 to 1.5 hours on a rocker at 4°C. Resins were washed two times with cold lysis buffer and extracted with 2× SDS sample buffer. Activated endogenous Rac1 or transfected Myc-tagged Rac1 bound to PAK-PBD was detected by western blotting with anti-Rac1 monoclonal antibody (BD Transduction, 1:1000) or 9E10 monoclonal anti-myc tag antibody (Developmental Studies Hybridoma Bank, 1:1000).
Electrophysiology
Whole-cell voltage clamp recordings were performed on cultured hippocampal neurons as described above using an Axopatch-200b amplifier (Molecular Devices, Sunnyvale, CA). Cells were continuously perfused (1 ml·min−1) with normal ACSF (nACSF) that contained (in mM): 125 NaCl, 2.5 KCl, 2 CaCl2, 1 MgCl2, 5 Hepes and 33 glucose; pH was adjusted to 7.3 using NaOH. Osmolarity was adjusted to 290 mosmol·l−1. For isolating miniature excitatory postsynaptic currents (mEPSCs), gabazine (10 μM), strychnine (3 μM), and tetrodotoxin (0.5 μM) were added to the external buffer to block GABAA receptor, glycine receptor, and Na channel activity, respectively. Patch electrodes were pulled from thin-walled borosilicate glass capillaries (tip resistance ranged from 4–6 MΩ) and filled with internal buffer solution that contained (in mM): 100 cesium methanesulfonate, 25 CsCl, 2 MgCl2, 4 Mg2+-ATP, 0.4 Na-GTP, 10 phosphocreatine, 0.4 EGTA, and 10 Hepes (pH 7.4; 284 mosmol·l−1). All experiments were carried out at room temperature (22 °C). Whole-cell recordings were only established after a high-resistance seal (> 2 GΩ) was achieved. Only cells that had an input resistance of > 150 MΩ and resting membrane potentials < −50 mV were considered for experiments. Resting membrane potentials were measured immediately upon breaking into whole-cell mode by setting the current to 0 pA. Cells were then voltage clamped at a holding potential of -70 mV unless otherwise noted. LTP was induced by switching perfusate to ACSF containing (in mM): 125 NaCl, 2.5 KCl, 2 CaCl2, 5 Hepes, 33 glucose, 0.2 glycine, 0.02 bicuculline, and 0.003 strychnine for 10 min at room temp before returning back to nACSF. Access resistance (Ra) was monitored at the beginning and end of each experiment with small voltage pulses and typically ranged between 10 and 15 MΩ and was not compensated. Cells were rejected from analysis if Ra increased by more than 15% during the course of the experiment or if the input resistance fell below 150 MΩ.
Results
CaMKK is required for NMDAR-dependent structural plasticity of hippocampal dendritic spines
Recent evidence from cultured hippocampal neurons suggests formation of spines and synapses in early development is enhanced by CaMKK and CaMKI signaling downstream of NMDARs (Saneyoshi et al., 2008). In this study, we examined whether structural plasticity in mature cultured neurons (14-21 DIV), specifically, the persistent (> 30 min) spine expansion accompanying NMDAR-dependent LTP, also requires CaMKK/CaMKI signaling. This stable component of spine expansion, in contrast to the transient and larger initial spine enlargement, has been shown to be inhibited by the general CaMK inhibitors, KN-62 (Matsuzaki et al., 2004) and KN-93 (Steiner et al., 2008). To better define the role of CaMKs, we utilized a highly selective CaMKK inhibitor, STO-609 (Tokumitsu et al., 2002). STO-609 potently inhibits CaMKK at 5-10 μM with little or no effect on CaMKI, CaMKIV, CaMKII, PKA, PKC or Erk1 (Tokumitsu et al., 2002; Tokumitsu et al., 2003). For example, in theta-burst LTP STO-609 blocks activation of CaMKI by CaMKK with no effect on CaMKII activation or its phosphorylation of GluA1 (Ser831) (Schmitt et al., 2005). In this case, synaptic potentiation itself is suppressed approximately 50%, indicating a potential role of CaMKK/CaMKI in theta-burst LTP (Schmitt et al., 2005; Guire et al., 2008).
We induced NMDAR-dependent synaptic plasticity in our hippocampal cultures using a chemical LTP (c-LTP) protocol. There are two major c-LTP protocols commonly used; treatment with forskolin/rolipram (Otmakhov et al., 2004) and Glycine-Induced LTP (GI-LTP) (Shahi and Baudry, 1993; Lu et al., 2001) - both exhibit many features characteristic of CA1 stimulus-induced LTP. However, whereas the forskolin/rolipram treatment strongly activates PKA throughout the neuron, both pre- and post-synaptically, we chose to utilize GI-LTP because it specifically stimulates NMDARs only at synapses receiving spontaneous release of glutamate. Thus, GI-LTP should more closely mimic stimulus-induced synaptic potentiation (see supplemental Table I and Fig. S1). Furthermore, GI-LTP is associated with an increase in mEPSC amplitude (Lu et al., 2001; Oh and Derkach, 2005) and NMDAR-dependent spine expansion (Park et al., 2006; Korkotian and Segal, 2007) and synapse formation (Ovtscharoff et al., 2008). Stimulus-inducted LTP in region CA1 requires autonomous CaMKII activity due to autophosphorylation of Thr286 (Silva et al., 1992) and GI-LTP also results in CaMKII autophosphorylation (Oh and Derkach, 2005). Moreover, infusion of CaMKIINtide, the active peptide derived from the specific CaMKII inhibitor protein CaMKIIN (Chang et al., 1998, 2001), via the recording electrode blocks synaptic potentiation by GI-LTP (Fig. S2). Furthermore, transfection of cultured neurons with CaMKIIN blocked the formation of constitutively-active (i.e., Thr286 autophosphorylated) CaMKII that occurs in GI-LTP and suppressed spine head enlargement (Fig. S2). Thus, GI-LTP closely mimics stimulus-inducted LTP including a requirement for CaMKII activation.
To quantify changes in spine morphology, hippocampal neurons were transfected with mRFP-βactin at least 24 hrs prior to inducing GI-LTP. As shown in Figure 1A, βactin is highly enriched in spines, and expression of βactin itself does not affect spine morphology (Saneyoshi et al., 2008). In order to isolate the persistent spine expansion of distal dendritic spines, neurons were fixed 40 min following the induction of GI-LTP, individual transfected neurons were imaged, and dendritic spine head widths and lengths were measured. GI-LTP induced a significant increase in spine head width (Control = 1.03 ± 0.02 μm; GI-LTP = 1.42 ± 0.03 μm) and decrease in spine length (Control = 5.25 ± 0.23 μm; GI-LTP = 3.15 ± 0.11 μm), consistent with the formation of mushroom-shaped spines that form synapses (Fig. 1B). Moreover, cumulative probability plots of spine head width and length indicate that the structural plasticity was not limited to a subset of spines (Fig. 1C). This alteration in spine morphology induced by GI-LTP was dependent upon NMDARs as it was prevented by application of the NMDAR antagonist, APV (50 μM) (Fig. 1B,C). More importantly, spine head enlargement was completely suppressed whereas spine length shortening was partially blocked (~50%) by inhibition of CaMKK with STO-609 (10 μM). To further confirm the specificity of STO-609, we induced GI-LTP in neurons co-transfected with a mutant form of CaMKK (L233F) that is 100-fold less sensitive to inhibition by STO-609 (Tokumitsu et al.). Expression of this STO-insensitive CaMKK mutant prevented the STO-dependent suppression of spine morphology following GI-LTP (Fig 1). These results strongly implicate a role for CaMKK in NMDAR-dependent structural plasticity. In addition to inducing changes in spine morphology, GI-LTP also resulted in an increase in spine density (Control = 8.3 ± 0.4 spines/25 μm; GI-LTP = 11.7 ± 0.5μ spines/25 μm, p<0.05 by Student's t-test) that was prevented by pretreatment with APV or STO-609 (data not shown).
Figure 1. GI-LTP structural plasticity requires NMDARs and CaMKK.

A, Representative fluorescence images of secondary hippocampal dendritic spines visualized using mRFP-βactin for conditions indicated. For some experiments neurons were pretreated with either APV (50 μM, 30 min) or STO-609 (10 μM, 4 hrs) or transfected with plasmid expressing a STO-insensitive mutant of CaMKK (STOins, 24 hrs) prior to GI-LTP treatment (10 min GI-LTP, see Methods). Scale bar is 5 μM. B, Quantitative analysis of spine head width (left panel) and spine length (right panel) for conditions indicated. C, Cumulative distribution plots for spine head width (left panel) and length (right panel) of the population of spines analyzed (n = 75 – 100 spines/neuron; 5 – 6 neurons per coverslip). Error bars indicate SEM (n = 8 – 10 coverslips per condition from 2 – 3 independent cultures). *p<0.05 by Student's t-test.
CaMKI but not CaMKIV is necessary for structural plasticity
CaMKI and CaMKIV are two well-established CaMKK substrates that require phosphorylation of their activation loops for their full catalytic activity. CaMKII does not have an activation loop phosphorylation site and is not activated by CaMKK (Tokumitsu et al., 1995). In fact, there is no known cross-talk between the CaMKK/CaMKI/CaMKIV cascade and CaMKII. Since inhibition of CaMKK with STO-609 blocks GI-LTP spine plasticity (Fig. 1), we determined whether CaMKI or CaMKIV play a role downstream from CaMKK by expressing the dominant negative (dn) forms (i.e., catalytically inactive) of each kinase. We found that expression of dnCaMKI, but not dnCaMKIV, inhibited the structural plasticity associated with GI-LTP (Fig. 2A, B). Expression of dnCaMKI also blocked the increase in spine density following GI-LTP. These data indicate that CaMKI is the relevant substrate downstream of CaMKK during GI-LTP. To further test this hypothesis we assayed for CaMKI activation following GI-LTP using a phospho-specific antibody directed against the CaMKK phosphorylation/activation site in CaMKI. As shown in Figure 2C, GI-LTP induced a rapid phosphorylation of CaMKI by CaMKK that remained elevated for up to 40 min post induction. The activation of CaMKI by GI-LTP was significant but modest (2-fold) compared to KCl stimulation which gave a 14-fold activation (Fig. S3). Thus, GI-LTP may represent a moderate LTP induction protocol. Activation of CaMKI by GI-LTP was also dependent upon NMDAR activation since treatment with APV suppressed its activation (Fig. 2D).
Figure 2. CaMKK signals through CaMKI but not CaMKIV to induce GI-LTP structural plasticity.

Quantitative analysis (left panel) and cumulative distribution plots (right panel) for spine head width A and spine length B for each condition shown. Neurons were transfected with plasmids expressing dominant-negative (dn) CaMKI or CaMKIV 48 hrs before GI-LTP induction. * p<0.05 by Student's t-test. C, Top, representative western blots for pCaMKI and total ERK2 (loading control) for the indicated time points following the 10 min GI-LTP treatment. Bottom, quantification of pCaMKI intensities prior to and following GI-LTP (n = 3 independent experiments). Error bars indicate SEM (n = 3 from 3 independent experiments). *p<0.05 by one-way ANOVA. D, Top, representative western blots of pCaMKI and total ERK2 five minutes after GI-LTP induction with or without APV or STO-609 (STO) treatments as in Fig. 1. Bottom, quantification of pCaMKI intensities for each condition shown (n = 3-5 from 3 independent experiments). Group data shown as mean ± SEM. ** p<0.01 by Student's t-test.
To determine whether expression of CaMKI is sufficient to drive structural changes in dendritic spines, we transfected neurons with a constitutively-active (ca) form of CaMKI (caCaMKI). We also examined the increase in surface GluA1 which is known to traffic into spines following the induction of synaptic plasticity (Hayashi et al., 2000; Lu et al., 2001; Pickard et al., 2001; Shi et al., 2001). We previously demonstrated that active CaMKI infused into neurons enhances synaptic-incorporation of CP-AMPARs that lack the GluA2 subunit (Guire et al., 2008). Surface GluA1 content was assessed by immunofluorescence microscopy utilizing an N-terminal antibody under non-permeabilizing conditions (see Methods). We found that expression of caCaMKI (24 hr) in neurons increased both spine head area as well as surface GluA1 within spines (Fig. 3). These increases were also observed in the presence of APV as would be expected since activated CaMKI is downstream of the NMDAR. Together, these data suggest that CaMKI is a relevant kinase by which NMDARs trigger activity-dependent morphological plasticity and recruitment of GluA1 to spines.
Figure 3. Constitutively active CaMKI mimics structural plasticity induced by GI-LTP and increases surface GluA1.

A, Immunofluorescence images of hippocampal dendritic spines highlighted with mRFP-βactin (left) or superimposed with surface GluA1 pseudo colored in green (right) for controls (top) or neurons transfected with constitutively-active (ca) CaMKI (24 hrs before fixation) without (middle panel) or with (bottom panel) treatment with APV added immediately after transfection and kept present until fixation. Surface GluA1 staining was performed using an N-terminal antibody under non-permeabilizing conditions. Scale bar is 2.5 μm. B, Group data for spine head width and surface GluA1 for conditions illustrated in A (n = 50 – 75 spines per neuron; 5 – 8 neurons per coverslip). C, Surface GluA1 plotted against spine head area for all experiments shown in panel B indicating that the increase in surface GluA1 was not due solely to a volume effect. Error bars indicate SEM (n = 8 coverslips per condition from 3 independent cultures). ***p<0.001 by Student's t-test.
Expression of GluA1 is sufficient to drive spine expansion
It has been proposed that synaptic incorporation of the GluA1 subunit is necessary for stable increases in both spine size and synaptic strength in paradigms of NMDAR-dependent plasticity (Kopec et al., 2007). Using an antibody to the extracellular N-terminus, we found that our GI-LTP paradigm resulted in a robust increase in surface GluA1 (184.4 ± 26.5% compared to control neurons, Fig. 4A, B) that was associated with spine expansion (Fig. 4B, 4C). Biotinylation of receptors in non-permeabilized cells also reflected an increase in surface GluA1 following GI-LTP induction when normalized to total GluA1 in permeabilized cells (Fig. 4 D, E). This increase in surface GluA1 was dependent upon NMDARs and CaMKK as it was inhibited by APV and STO-609, respectively. Inhibition by STO-609 (Fig. 4), combined with the effects of caCaMKI (Fig. 3), suggest that CaMKK/CaMKI regulate both surface trafficking of GluA1 into spines, as well as spine expansion following activation of synaptic NMDARs. AMPAR recruitment by GI-LTP was not specific for GluA1 as there was also an increase in surface GluA2 (Fig. S1).
Figure 4. GI-LTP-induced surface trafficking of GluA1 requires CaMKK.

A, Immunofluorescence images of hippocampal dendritic spines transfected with mRFP-βactin (left) or superimposed with surface GluA1 pseudo colored in green (right) for control and GI-LTP treated neurons. Scale bar is 15 μm and 2.5 μm for panel and inset, respectively. B, Quantification of spine head area and surface GluA1 (n = 75 – 100 spines per neuron; 6 – 8 neurons per coverslip) for control and GI-LTP treated neurons (n = 8 coverslips per condition from 2 independent cultures). C, Scatter plot of surface GluA1 and spine head area for control and GI-LTP treated cover slips. D, Representative western blots of biotinylated surface GluA1 (GluA1bio) and total GluA1 (GluA1tot) for conditions shown. E, Quantification of the ratio of surface biotinylated to total GluA1 for each condition indicated (n = 8 from 5 independent experiments). For GI-LTP treated cultures, neurons were fixed or biotinylated 40 minutes post GI-LTP. Error bars indicate SEM. * p<0.05 by Student's t-test.
Transfected GluA1 in cultured neurons has been shown to be incorporated into synapses as demonstrated by its punctate co-localization with surface GluA2 and an increase in current rectification (Shi et al., 1999). Therefore, we next determined whether expressing GluA1 in these neurons could mimic GI-LTP in terms of increasing spine head area. Neurons were co-transfected with wild type GluA1 and mRFP-βactin for 24hrs before fixation, imaging and analysis for spine head area. As shown in Figure 5A, expression of GluA1 led to an increase in spine head area. Cumulative probability plots illustrate that the increase in spine head area was not restricted to a subset of spines but led to a general increase in all spines measured (Fig. 5B). Overall, neurons expressing GluA1 demonstrated a two-fold increase in spine head area (Fig. 5C). To confirm that the expressed GluA1 was incorporated into synapses, we performed whole-cell voltage clamp recordings of AMPA-mediated mEPSCs at two different holding potentials (i.e., -60 and +60 mV) and measured the degree of rectification, a biophysical property associated with GluA2-lacking AMPARs (Kamboj et al., 1995). Since rectification of currents is mediated by endogenous polyamines (i.e., spermine) that can be dialyzed away during whole-cell recordings, we included spermine (100 μM) in the intracellular patch solution (Kamboj et al., 1995). We have previously demonstrated that pyramidal neuron excitatory synapses in our hippocampal cultures under basal conditions do not contain CP-AMPARs (Guire et al., 2008). However, transient expression of GluA1 likely increases the probability that GluA1 homomers will be trafficked into synapses and therefore display increased rectification. It has previously been demonstrated that the induction of NMDAR-dependent forms of synaptic plasticity results in a transient period of rectification (Plant et al., 2006; Lu et al., 2007; Guire et al., 2008). While vector only control neurons demonstrated comparable mEPSC amplitudes at both holding potentials (-60 mV = 9.7 ± 0.8 pA versus 60mV = 8.7 ± 0.3 pA), we found that mEPSCs recorded from GluA1-expressing neurons were less conductive at positive holding potentials (-60 mV = 20.6 ± 2.2 pA versus 60 mV = 10.2 ± 1.9 pA), indicating that they were rectifying (Fig. 5D). To further verify the presence of CP-AMPARs, we compared the amplitude and kinetics of mEPSCs (Vh=60 mV) in these same GluA1 expressing neurons prior to and following bath application of IEM-1460 (30 μM), an inhibitor of CP-AMPARs. At 30 μM IEM-1460 inhibits only GluA2-lacking AMPARs with no effect on NMDARs (Magazanik et al., 1997; Buldakova et al., 1999; Samoilova et al., 1999; Gray et al., 2007; Guire et al., 2008). We have shown previously that IEM-1460 treatment of cultured control cells has no effect on mEPSCs, consistent with their lack of CP-AMPARs (Guire et al., 2008). Treatment of GluA1-transfected neurons with IEM-1460 resulted in a 41.9 ± 8.1% reduction in mEPSC amplitude as compared to controls (Fig. 5E) and an increase in decay time (Baseline = 5.8 ± 0.5 ms; IEM = 6.7 ± 0.5 ms; p<0.05 by Student's paired t-test), both properties indicative of GluA2-lacking AMPARs. Together these data indicate that expression of GluA1 leads to synaptic incorporation of CP-AMPARs which may drive spine expansion.
Figure 5. Overexpression of GluA1 increases spine head area, mEPSC amplitude and synaptic expression of Ca2+-permeable AMPARs (CP-AMPARs).

A, Fluorescence images of dendritic spines expressing mRFP-βactin plus vector (left) or GluA1 (right). B, Cumulative distribution plots for control neurons (black) and neurons expressing GluA1 (gray). C, Mean data for neurons plotted in B. Error bars indicate SEM (n = 8 per condition from 2 independent experiments; **p<0.01 by Student's t-test). D, Top, example of mEPSCs recorded from a vector only (control) or GluA1-expressing neuron at two different holding potentials (-60 and 60 mV). Bottom, mean rectification index, an indicator of CP-AMPARs, for vector only and GluA1 expressing neurons. Rectification index was calculated by dividing the absolute mean peak amplitude recorded at 60 mV by the peak amplitude recorded at -60 mV for individual neurons. Error bars indicate SEM (n = 6 recordings per condition; **p<0.01 by Student's t-test). E, Top, example traces of mEPSCs recorded at -60 mV from a GluA1-expressing neuron illustrating sensitivity toward IEM-1460 (30 μM), an antagonist of CP-AMPARs. Bottom, pooled data for mean mEPSC amplitudes for vector (n = 6), GluA1 (n = 6; **p<0.01 by Student t-test) and GluA1 in the presence of IEM-1460 (IEM; n = 4; **p<0.01 by paired Student's t-test).
Spine expansion induced by GI-LTP requires CP-AMPARs
In order to test whether GI-LTP-induced spine expansion also required CP-AMPARs, we first determined if treatment with IEM-1460, would affect synaptic potentiation. It is well established in hippocampus that increased synaptic strength is highly associated with increases in spine volume (Cingolani and Goda, 2008). As shown in Figure 6, GI-LTP induced a 68.5 ± 9.0% increase in mEPSC amplitude (Fig. 6A) that was largely inhibited by co-application of IEM-1460 (Fig. 6B). Importantly, we found that inhibition of CP-AMPARs during the 10 min glycine treatment also prevented the persistent spine expansion induced by GI-LTP (Fig. 6C, D). IEM-1460 had no effect on CaMKI or CaMKII activation (Fig. S3), indicating that IEM-1460 was not inhibiting LTP induction by non-specifically inhibiting the activation of CaMKI or CaMKII. The fact that CaMKI activation is blocked by APV (Fig. 2) but not IEM-1460 (Fig. S3) indicates the source of Ca2+ for its activation is dependent on NMDARs but not CP-AMPARs. This is consistent with its role in NMDAR-dependent synaptic recruitment of CP-AMPARs (Guire et al., 2008). The conclusion that IEM-1460 is acting via inhibition of AMPARs was further substantiated by the fact that application of the specific AMPAR antagonist GYKI-52466 (20 μM) also blocks spine expansion (data not shown). Interestingly, if IEM-1460 is applied after the 10 min of glycine treatment, there is still a partial suppression of spine enlargement (Fig. S4). These data suggest that CP-AMPARs are necessary to initiate some cellular function(s) during the glycine-treatment, and this function is subsequently required for the persistent spine expansion accompanying GI-LTP. This would not be surprising if prolonged actin remodeling underlies these structural changes.
Figure 6. GI-LTP increases mEPSCs and recruitment of synaptic CP-AMPARs.
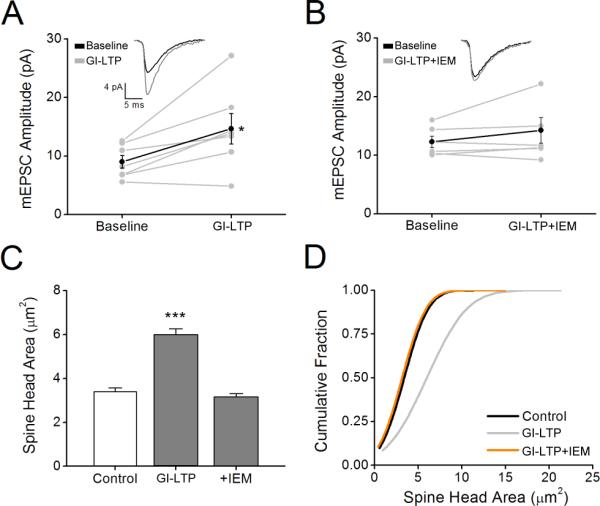
A, Individual mEPSC amplitudes plotted prior to (Baseline) and 40 min post GI-LTP (GI-LTP). Mean amplitudes are denoted by black filled circle. Error bars indicated SEM (n = 7 from 3 independent cultures). *p<0.05 by paired Student's t-test. Inset, example mEPSC traces before and after GI-LTP. Traces are an average of 50 consecutive events. B, Plot of mEPSCs as described in A prior to (Baseline) and following GI-LTP in the presence of IEM-1460 (IEM). Error bars indicate SEM (n = 6 from 3 independent cultures). C, Pooled data of spine head area (n = 50 – 75 spines per neuron; 5 – 8 neurons per coverslip) for controls and neurons following GI-LTP in absence or presence of IEM-1460. Error bars indicate SEM (n = 6 – 8 coverslips/condition). ***p<0.001 by Student's t-test. D, Cumulative distribution of spine head areas for each condition indicated.
It is well established that spine structural plasticity is associated with actin polymerization (Fifkova and Delay, 1982; Matus et al., 1982; Matsuzaki et al., 2004), so we tested the effect of Latrunculin A that binds actin monomers and blocks actin polymerization. Indeed, we found that the addition of 10 μM Latrunculin A prevented the spine enlargement associated with GI-LTP (Fig. 7A). Next, to identify the signaling pathway downstream of CP-AMPARs, we assessed the ability of GI-LTP to activate signaling molecules known to regulate the actin cytoskeleton and whether their activation was suppressed by IEM-1460. We focused on the Rac/PAK/LIMK pathway (Edwards DC, 1999) because of its known regulation of spine actin (Schubert and Dotti, 2007). To measure activation of Rac during GI-LTP, we determined the relative levels of GTP-Rac1 (i.e., active form of Rac1) by affinity pull-down using GST-tagged CRIB domain of PAK1 from control versus GI-LTP-stimulated neuronal lysates. GI-LTP resulted in a small but significant 23.5 ± 1.7% increase in Rac GTP-loading normalized to total Rac protein (Fig. 7B). This is a significant, albeit modest, effect that may represent GTP-loading of Rac only in potentiated spines (Fig. S5). Importantly, the GI-LTP-induced Rac1 activation was inhibited by co-application of IEM-1460 (Fig. 7B). Next, we examined the activities of PAK and its downstream target LIMK using phospho-specific antibodies directed toward the activation/phosphorylation sites of PAK and LIMK, (S141 and T508, respectively). Both of these protein kinases are known regulators of the actin cytoskeleton downstream of Rac (Schubert and Dotti, 2007) and have been implicated in LTP (Allen et al., 1998; Meng et al., 2002; Hayashi et al., 2004). Consistently, we found that GI-LTP increased PAK and LIMK activation by 90.9 ± 24.4% and 153.0 ± 52.7%, respectively (Fig. 8A, B). Notably, the increase in PAK and LIMK activation following GI-LTP was dependent upon CP-AMPARs since it was inhibited by IEM-1460. In cultured hippocampal neurons, phospho-PAK has been shown to be enriched in spines as puncta that co-localizes with the excitatory postsynaptic marker PSD-95 (Zhang et al., 2005). Similarly we found an increase in phosphorylated PAK in spines following GI-LTP (Fig. S5), suggesting that PAK is active locally within spines. Furthermore, transfection of neurons with dnPAK (PAKK299R) blocked the GI-LTP-induced spine expansion (Fig. 8C, D). Taken together these data implicate a role for Rac, PAK and its effector LIMK in the structural plasticity following the induction of GI-LTP. Of particular interest, Rac, PAK and LIMK appear to be downstream of CP-AMPARs, providing the first functional link to a signaling pathway for CP-AMPARs in NMDA-dependent LTP.
Figure 7. GI-LTP Structural Plasticity Requires Actin Polymerization and CP-AMPARs Mediated Activation of Rac.

A, Group data of spine head area (n = 50 – 75 spines per neuron; 5 neurons per coverslip) for controls and neurons subjected to GI-LTP in absence or presence of 10 μM Latrunculin A. Error bars indicate SEM (n = 5 coverslips/ condition). **p<0.01 by Student's t-test. B, Top, representative western blot of activated GTP-bound Rac, determined by affinity pull-down using GST-tagged CRIB domain of PAK1, and total Rac for conditions indicated. Bottom, group data plotted as the ratio of GTP-Rac/total Rac for each condition normalized to the mean control. Error bars indicate SEM (n = 4 – 6 independent experiments per condition). *p<0.05 by Student's t-test.
Figure 8. GI-LTP Structural Plasticity Requires the PAK/LIM-kinase Pathway.
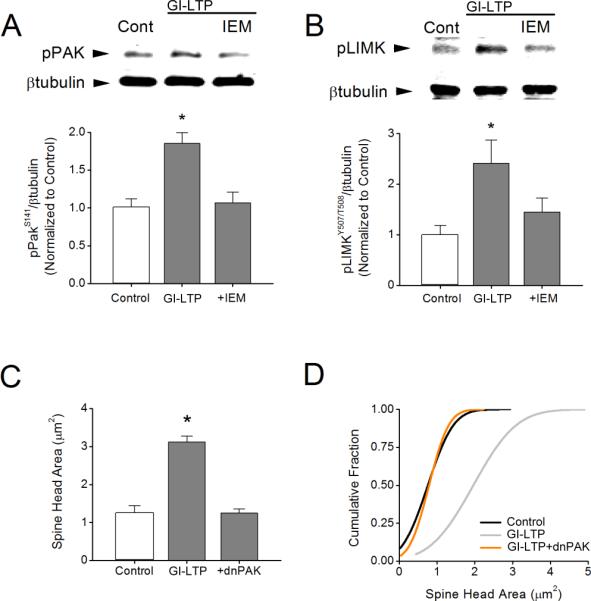
A, Top, western blots of pPAK (S141) and βtubulin (loading control) for conditions indicated. Bottom, group data shown as the ratio of pPAK/βtubulin for each condition normalized to the mean control. B, Top, in these same experiments cell lysates were also probed by western blots for pLIMK (Y507/T508). Bottom, pooled data for ratios of pLIMK/βtubulin for each condition normalized to the mean control. Error bars in A and B indicate SEM (n = 6 – 7 independent experiments). *p<0.05 by Student's t-test. C, Mean spine head area for control and neurons following GI-LTP treatment in the absence or presence of dnPAK (n = 50 – 75 spines per neuron; 5 – 6 neurons per coverslip). Error bars indicate SEM (n = 8 – 10 coverslips per condition from 2 – 3 independent cultures). *p<0.05 by Student's t-test. D, Cumulative distribution of spine head areas for each condition indicated.
Discussion
Induction and maintenance of LTP in the CA1 region of hippocampus involves numerous biological processes including recruitment of AMPARs to synapses, phosphorylation of AMPARs, and formation and/or enlargement of spines/synapses - these require gene transcription, protein synthesis, posttranslational protein modifications and protein trafficking (reviewed in Bramham, 2008; Kerchner and Nicoll, 2008; Kessels and Malinow, 2009; Saneyoshi et al., 2010). The relative contributions of signal transduction pathways regulating these functions to the resulting synaptic potentiation may depend on the system and paradigm used to induce LTP in addition to the prior history of the synapse. This incredible complexity may contribute to conflicting results obtained by different investigators in this intensely investigated field.
Recent studies on activity-dependent stable enlargement of potentiated spines (i.e., structural plasticity) have focused primarily on recruitment of AMPAR subunits, particularly GluA1 and GluA2, and their associated proteins (Kopec et al., 2007; Saglietti et al., 2007). Here we identify a novel signaling pathway responsible for enabling stable spine enlargement following the selective activation of synaptic-NMDARs in cultured hippocampal neurons. Specifically, we find a requirement for NMDAR-dependent activation of CaMKK/CaMKI-mediated synaptic expression of CP-AMPARs. These CP-AMPARs, in turn, are required for the activation of the Rac/PAK/LIMK pathway known to promote F-actin polymerization in spines. Our conclusions are based on multiple, independent experimental approaches. These results are consistent with our previous report that although expressed on the cell surface of dendrites, CP-AMPARs are not present within the synapse under basal conditions (Guire et al., 2008). Importantly, infusion of activated CaMKI alone in cultured neurons drives CP-AMPARs into the synapse and increases synaptic strength. Here we report that transfection of caCaMKI in cultured hippocampal neurons increases the surface expression of GluA1 in spines and promotes stable spine enlargement. Furthermore, induction of TBS LTP in the CA1 region of hippocampal slices results in a partial (~50%) contribution by CP-AMPARs to the synaptic potentiation. However, the contribution of CP-AMPARs to LTP induced by other protocols (e.g., HFS or paired pre/post-synaptic stimulation) remains controversial (Adesnik and Nicoll, 2007; Gray et al., 2007).
Roles for CP-AMPARs in synaptic plasticity are well-established at interneuron synapses (Liu and Zukin, 2007). For example, in the spinal cord synaptic potentiation mediated by CP-AMPARs expressed by GABA-containing interneurons in the dorsal horn contributes to chronic pain (Gu et al., 1996; Hartmann et al., 2004). In contrast, HFS of mossy fiber synapses onto CA3 interneurons induces LTD via CP-AMPARs (Laezza et al., 1999; Lei and McBain, 2004). Surprisingly, very little was previously known about detailed signaling pathways downstream of these unique receptors. In cultured mouse striatal neurons, activation of CP-AMPARs results in stimulation of phosphatidylinositol 3-kinase, MEK/Erk and CREB phosphorylation (Perkinton et al., 1999). In mature cortical neurons and slices, CP-AMPARs form a complex with the Ca2+/CaM-activated Ras/Rac GEF, Ras-GRF1, that is predominantly responsible for Erk activation and CREB phosphoryation (Tian and Feig, 2006). However, in postpubescent mice Ras-GRF1 appears to mediate NMDAR-dependent LTD rather than LTP (Li et al., 2006). It should be noted that the CaMKK/CaMKI pathway can also active the MEK/Erk pathway (Schmitt et al., 2004) resulting in CREB-dependent transcription (Wayman et al., 2006). Furthermore, phosphorylation of Ras-GRF1 (Ser916) upon LTP induction is blocked by inhibitors of CaMKK (Schmitt et al., 2005), implicating that Ras-GRF1 may be downstream of CaMKK.
It is well documented that increased spine size during LTP requires regulation of the actin cytoskeleton (reviewed in Matus, 2000; Hering and Sheng, 2001; Carlisle and Kennedy, 2005; Tada and Sheng, 2006), so we focused on signaling pathways known to regulate actin dynamics. Our results are the first to provide a mechanism by which CP-AMPARs can contribute to synaptic potentiation at excitatory CA1 synapses via spine enlargement through activation of the Rac/PAK/LIMK pathway that regulates the actin cytoskeleton. These results are consistent with previous reports indicating a role for PAK in activity-dependent alterations of spine enlargement (Chen et al., 2007; Rex et al., 2009). Importantly, mutations in PAK have been found in patients with nonsyndromic mental retardation, indicating their importance in cognitive function (Allen et al., 1998; Bienvenu et al., 2000). LIMK is a well-established downstream effector of PAK that enhances formation of F-actin (Edwards DC, 1999) and is also involved in LTP (Meng et al., 2002).
Our results identify an important role for recruitment of the GluA1 subunit of AMPARs to form synaptic CP-AMPARs contributing to stable structural plasticity. Previous studies have identified roles for both the GluA1 and GluA2 subunits. In cultured neurons (22 DIV), expression of GluA2 promotes spine enlargement via interaction of its N-terminus with N-cadherin (Saglietti et al., 2007). Although we focused on the role of GluA1, our GI-LTP protocol also increases surface expression of GluA2 (Fig. S1) that may also contribute to the spine enlargement in our experiments. In contrast to our study, Saglietti et al. did not see any effect of expressing GluA1 on spine size. Although expressed GluA1 in cultured neurons is generally trafficked to the synapse (Shi et al., 1999), this was not actually determined in the Saglietti study. Whether expressed GluA1 is incorporated into synapses may depend on the extent of endogenous neuronal activity present and/or the activation state of CaMKs. In organotypic slices, transiently transfected GluA1 is not localized to synapses unless co-expressed with active CaMKII or the slice is stimulated (Hayashi et al., 2000). Our experience, based on recordings of mEPSCs and measuring the activation states of CaMKII and CaMKI, is that mature cultures of hippocampal neurons exhibit higher levels of endogenous activity than do acute or cultured hippocampal slices (unpublished observations). Thus, the level of endogenous activity in cultured neurons may dictate whether expressed GluA1 is trafficked to synapses.
Several studies have implicated roles for the GluA1 subunit in increasing synaptic strength associated with LTP (reviewed in Barry and Ziff, 2002; see Kopec et al., 2007; Makino and Malinow, 2009). One study, using organotypic hippocampal slices, identified the C-terminus of GluA1, which contains an essential PDZ interaction site as well as multiple phosphorylation sites, as the essential domain for spine expansion (Kopec et al., 2007). Surprisingly, they found that expression of GluA1 with a mutation in the pore that obviates channel permeation still promotes increased spine size. However, we find that activity-dependent blockade of CP-AMPARS by IEM-1460 suppresses spine enlargement induced by GI-LTP. Kopek et al. note, “Although our study argues against a requirement for calcium entry through GluA2-lacking AMPA receptors, it may be that our LTP induction protocol, which is 16 min long, provides ample Ca2+ influx through NMDA receptors to both drive GluA1 receptors into synapses and subsequently stabilize them there.” Their c-LTP treatment protocol of forskolin/rolipram gives synaptic potentiation that mimics standard strong (e.g., HFS) LTP protocols (Otmakhov et al., 2004), and it results in global activation of PKA throughout the neuron including presynaptic and postsynaptic compartments. In our study we took advantage of a GI-LTP protocol that requires spontaneous release of glutamate from opposing terminals to specifically activate postsynaptic NMDARs and CaMKs.
In the same report, (Kopec et al., 2007) showed that spine enlargement precedes by several minutes the recruitment of AMPARs. This would appear to be at odds with our results where synaptic incorporation of CP-AMPARs was required for spine expansion. Since overexpression of GluA1 was sufficient by itself to promote spine enlargement that was blocked by IEM-1460, our data strongly indicates that recruitment of some CP-AMPARs is required prior to structural plasticity. Due to the unique properties of CP-AMPARS (e.g., high unitary conductance, stabilization in high conductance state by CaMKII phosphorylation), only a very small number (<5% of total synaptic AMPARs) (Guire et al., 2008) need be recruited to the synapse to have a large effect on synaptic current and the Ca2+ influx that may be required for triggering the Rac/PAK/LIMK pathway. This very small number of CP-AMPARs would not be detectable by most methods used to quantify GluA1 trafficking. Thus, there may be initial recruitment of a small number of synaptic CP-AMPARs that precedes and triggers spine expansion followed by a subsequent bulk recruitment of AMPARs (perhaps GluA1/GluA2) that stabilizes the enlarged spine.
Previous studies have provided evidence for roles of the CaMKK/CaMKI pathway in numerous aspects of neuronal development, such as axon formation (Ageta-Ishihara et al., 2009; Davare et al., 2009) and outgrowth (Wayman et al., 2004), dendritic arborization (Wayman et al., 2006) and spine/synapses formation (Saneyoshi et al., 2008), as well contributing to TBS LTP (Schmitt et al., 2005) through recruitment of CP-AMPARs (Guire et al., 2008). The current study adds to this list of neuronal functions, namely spine enlargement in mature neurons during NMDAR-dependent structural plasticity, regulated by CaMKK/CaMKI. Specifically, we demonstrate that GI-LTP, via NMDAR activation of CaMKK/CaMKI, promotes synaptic expression of CP-AMPARs. CP-AMPARs have not previously been implicated in promoting spine enlargement, and this may account for the role of CP-AMPARs in TBS LTP (Guire et al., 2008). Furthermore, our results establish an essential role for CP-AMPARs in the activation of the Rac/PAK/LIMK pathway essential for modulating activity-dependent actin dynamics within hippocampal dendritic spines.
Supplementary Material
Acknowledgements
This work was supported by NIH grant NS027037 (TRS)
References
- Abramoff MD, Magelhaes PJ, Ram SJ. Image Processing with ImageJ. Biophotonics International. 2004;11:36–42. [Google Scholar]
- Adesnik H, Nicoll RA. Conservation of glutamate receptor 2-containing AMPA receptors during long-term potentiation. Journal of Neuroscience. 2007;27:4598–4602. doi: 10.1523/JNEUROSCI.0325-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ageta-Ishihara N, Takemoto-Kimura S, Nonaka M, Adachi-Morishima A, Suzuki K, Kamijo S, Fujii H, Mano T, Blaeser F, Chatila TA, Mizuno H, Hirano T, Tagawa Y, Okuno H, Bito H. Control of cortical axon elongation by a GABA-driven Ca2+/calmodulin-dependent protein kinase cascade. Journal of Neuroscience. 2009;29:13720–13729. doi: 10.1523/JNEUROSCI.3018-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen KM, Gleeson JG, Bagrodia S, Partington MW, MacMillan JC, Cerione RA, Mulley JC, Walsh CA. PAK3 mutation in nonsyndromic X-linked mental retardation. Nature Genetics. 1998;20:25–30. doi: 10.1038/1675. [DOI] [PubMed] [Google Scholar]
- Bamburg JR, McGough A, Ono S. Putting a new twist on actin: ADF/cofilins modulate actin dynamics. Trends in Cell Biology. 1999;9:364–370. doi: 10.1016/s0962-8924(99)01619-0. [DOI] [PubMed] [Google Scholar]
- Barry MF, Ziff EB. Receptor trafficking and the plasticity of excitatory synapses. Current Opinion in Neurobiology. 2002;12:279–286. doi: 10.1016/s0959-4388(02)00329-x. [DOI] [PubMed] [Google Scholar]
- Bienvenu T, des Portes V, McDonell N, Carrie A, Zemni R, Couvert P, Ropers HH, Moraine C, van Bokhoven H, Fryns JP, Allen K, Walsh CA, Boue J, Kahn A, Chelly J, Beldjord C. Missense mutation in PAK3, R67C, causes X-linked nonspecific mental retardation. American Journal of Medical Genetics. 2000;93:294–298. doi: 10.1002/1096-8628(20000814)93:4<294::aid-ajmg8>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- Boda B, Nikonenko I, Alberi S, Muller D. Central nervous system functions of PAK protein family: from spine morphogenesis to mental retardation. Molecular Neurobiology. 2006;34:67–80. doi: 10.1385/mn:34:1:67. [DOI] [PubMed] [Google Scholar]
- Bourne JN, Harris KM. Balancing structure and function at hippocampal dendritic spines. Annual Review of Neuroscience. 2008;31:47–67. doi: 10.1146/annurev.neuro.31.060407.125646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bramham CR. Local protein synthesis, actin dynamics, and LTP consolidation. Current Opinion in Neurobiology. 2008;18:524–531. doi: 10.1016/j.conb.2008.09.013. [DOI] [PubMed] [Google Scholar]
- Buldakova SL, Vorobjev VS, Sharonova IN, Samoilova MV, Magazanik LG. Characterization of AMPA receptor populations in rat brain cells by the use of subunit-specific open channel blocking drug, IEM-1460. Brain Research. 1999;846:52–58. doi: 10.1016/s0006-8993(99)01970-8. [DOI] [PubMed] [Google Scholar]
- Cajal RyS. Histologie du systeme nerveux de l'homme et des vertbres. Maloine; Paris: 1911. [Google Scholar]
- Carlisle HJ, Kennedy MB. Spine architecture and synaptic plasticity. Trends in Neurosciences. 2005;28:182–187. doi: 10.1016/j.tins.2005.01.008. [DOI] [PubMed] [Google Scholar]
- Chang BH, Mukherji S, Soderling TR. Characterization of a calmodulin kinase II inhibitor protein in brain. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:10890–10895. doi: 10.1073/pnas.95.18.10890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang BH, Mukherji S, Soderling TR. Calcium/calmodulin-dependent protein kinase II inhibitor protein: localization of isoforms in rat brain. Neuroscience. 2001;102:767–777. doi: 10.1016/s0306-4522(00)00520-0. [DOI] [PubMed] [Google Scholar]
- Chen LY, Rex CS, Casale MS, Gall CM, Lynch G. Changes in synaptic morphology accompany actin signaling during LTP. Journal of Neuroscience. 2007;27:5363–5372. doi: 10.1523/JNEUROSCI.0164-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani LA, Goda Y. Actin in action: the interplay between the actin cytoskeleton and synaptic efficacy. Nat Rev Neurosci. 2008;9:344–356. doi: 10.1038/nrn2373. [DOI] [PubMed] [Google Scholar]
- Davare MA, Saneyoshi T, Guire ES, Nygaard SC, Soderling TR. Inhibition of calcium/calmodulin-dependent protein kinase kinase by protein 14-3-3. Journal of Biological Chemistry. 2004;279:52191–52199. doi: 10.1074/jbc.M409873200. [DOI] [PubMed] [Google Scholar]
- Davare MA, Fortin DA, Saneyoshi T, Nygaard S, Kaech S, Banker G, Soderling TR, Wayman GA. Transient receptor potential canonical 5 channels activate Ca2+/calmodulin kinase Igamma to promote axon formation in hippocampal neurons. Journal of Neuroscience. 2009;29:9794–9808. doi: 10.1523/JNEUROSCI.1544-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards DCSL, Bokoch GM, Gill GN. Activation of LIM-kinase by Pak1 couples Rac/Cdc42 GTPase signalling to actin cytoskeletal dynamics. Nat Cell Biol. 1999;1:E115–117. doi: 10.1038/12963. [DOI] [PubMed] [Google Scholar]
- Fiala JC, Spacek J, Harris KM. Dendritic spine pathology: cause or consequence of neurological disorders? Brain Research - Brain Research Reviews. 2002;39:29–54. doi: 10.1016/s0165-0173(02)00158-3. [DOI] [PubMed] [Google Scholar]
- Fifkova E. A possible mechanism of morphometric changes in dendritic spines induced by stimulation. Cellular & Molecular Neurobiology. 1985;5:47–63. doi: 10.1007/BF00711085. [DOI] [PubMed] [Google Scholar]
- Fifkova E, Delay RJ. Cytoplasmic actin in neuronal processes as a possible mediator of synaptic plasticity. Journal of Cell Biology. 1982;95:345–350. doi: 10.1083/jcb.95.1.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray EE, Fink AE, Sarinana J, Vissel B, O'Dell TJ. Long-term potentiation in the hippocampal CA1 region does not require insertion and activation of GluR2-lacking AMPA receptors. Journal of Neurophysiology. 2007;98:2488–2492. doi: 10.1152/jn.00473.2007. [DOI] [PubMed] [Google Scholar]
- Gu JG, Albuquerque C, Lee CJ, MacDermott AB. Synaptic strengthening through activation of Ca2+-permeable AMPA receptors. Nature. 1996;381:793–796. doi: 10.1038/381793a0. [DOI] [PubMed] [Google Scholar]
- Guire ES, Oh MC, Soderling TR, Derkach VA. Recruitment of calcium-permeable AMPA receptors during synaptic potentiation is regulated by CaM-kinase I. Journal of Neuroscience. 2008;28:6000–6009. doi: 10.1523/JNEUROSCI.0384-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann B, Ahmadi S, Heppenstall PA, Lewin GR, Schott C, Borchardt T, Seeburg PH, Zeilhofer HU, Sprengel R, Kuner R. The AMPA receptor subunits GluR-A and GluR-B reciprocally modulate spinal synaptic plasticity and inflammatory pain. Neuron. 2004;44:637–650. doi: 10.1016/j.neuron.2004.10.029. [DOI] [PubMed] [Google Scholar]
- Hayashi K, Ohshima T, Hashimoto M, Mikoshiba K. Pak1 regulates dendritic branching and spine formation. Developmental Neurobiology. 2007;67:655–669. doi: 10.1002/dneu.20363. [DOI] [PubMed] [Google Scholar]
- Hayashi ML, Choi SY, Rao BS, Jung HY, Lee HK, Zhang D, Chattarji S, Kirkwood A, Tonegawa S. Altered cortical synaptic morphology and impaired memory consolidation in forebrain- specific dominant-negative PAK transgenic mice. Neuron. 2004;42:773–787. doi: 10.1016/j.neuron.2004.05.003. [DOI] [PubMed] [Google Scholar]
- Hayashi Y, Shi SH, Esteban JA, Piccini A, Poncer JC, Malinow R. Driving AMPA receptors into synapses by LTP and CaMKII: requirement for GluR1 and PDZ domain interaction. Science. 2000;287:2262–2267. doi: 10.1126/science.287.5461.2262. [DOI] [PubMed] [Google Scholar]
- Hebb DO. Organisation of Behaviour. Wiley; New York: 1949. [Google Scholar]
- Hering H, Sheng M. Dendritic spines: structure, dynamics and regulation. Nature Reviews Neuroscience. 2001;2:880–888. doi: 10.1038/35104061. [DOI] [PubMed] [Google Scholar]
- Impey SDM, Lasiek A, Fortin D, Ando H, Varlamova O, Obrietan K, Soderling TR, Goodman RH, Wayman GA. An activity-induced microRNA controls dendritic spine formation by regulating Rac1-PAK signaling. Mol Cell Neurosci. 2010;43:146–156. doi: 10.1016/j.mcn.2009.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamboj SK, Swanson GT, Cull-Candy SG. Intracellular spermine confers rectification on rat calcium-permeable AMPA and kainate receptors. Journal of Physiology. 1995;486:297–303. doi: 10.1113/jphysiol.1995.sp020812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann WE, Moser HW. Dendritic anomalies in disorders associated with mental retardation. Cerebral Cortex. 2000;10:981–991. doi: 10.1093/cercor/10.10.981. [DOI] [PubMed] [Google Scholar]
- Kerchner GA, Nicoll RA. Silent synapses and the emergence of a postsynaptic mechanism for LTP. Nat Rev Neurosci. 2008;9:813–825. doi: 10.1038/nrn2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessels HW, Malinow R. Synaptic AMPA receptor plasticity and behavior. Neuron. 2009;61:340–350. doi: 10.1016/j.neuron.2009.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopec CD, Real E, Kessels HW, Malinow R. GluR1 links structural and functional plasticity at excitatory synapses. Journal of Neuroscience. 2007;27:13706–13718. doi: 10.1523/JNEUROSCI.3503-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korkotian E, Segal M. Morphological constraints on calcium dependent glutamate receptor trafficking into individual dendritic spine. Cell Calcium. 2007;42:41–57. doi: 10.1016/j.ceca.2006.11.006. [DOI] [PubMed] [Google Scholar]
- Laezza F, Doherty JJ, Dingledine R. Long-term depression in hippocampal interneurons: joint requirement for pre- and postsynaptic events. Science. 1999;285:1411–1414. doi: 10.1126/science.285.5432.1411. [DOI] [PubMed] [Google Scholar]
- Lei S, McBain CJ. Two Loci of expression for long-term depression at hippocampal mossy fiber-interneuron synapses. Journal of Neuroscience. 2004;24:2112–2121. doi: 10.1523/JNEUROSCI.4645-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Tian X, Hartley DM, Feig LA. The environment versus genetics in controlling the contribution of MAP kinases to synaptic plasticity. Current Biology. 2006;16:2303–2313. doi: 10.1016/j.cub.2006.10.028. [DOI] [PubMed] [Google Scholar]
- Liu SJ, Zukin RS. Ca2+-permeable AMPA receptors in synaptic plasticity and neuronal death. Trends in Neurosciences. 2007;30:126–134. doi: 10.1016/j.tins.2007.01.006. [DOI] [PubMed] [Google Scholar]
- Lu W, Man H, Ju W, Trimble WS, MacDonald JF, Wang YT. Activation of synaptic NMDA receptors induces membrane insertion of new AMPA receptors and LTP in cultured hippocampal neurons. Neuron. 2001;29:243–254. doi: 10.1016/s0896-6273(01)00194-5. [DOI] [PubMed] [Google Scholar]
- Lu Y, Allen M, Halt AR, Weisenhaus M, Dallapiazza RF, Hall DD, Usachev YM, McKnight GS, Hell JW. Age-dependent requirement of AKAP150-anchored PKA and GluR2-lacking AMPA receptors in LTP. EMBO Journal. 2007;26:4879–4890. doi: 10.1038/sj.emboj.7601884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magazanik LG, Buldakova SL, Samoilova MV, Gmiro VE, Mellor IR, Usherwood PN. Block of open channels of recombinant AMPA receptors and native AMPA/kainate receptors by adamantane derivatives. Journal of Physiology. 1997;505:655–663. doi: 10.1111/j.1469-7793.1997.655ba.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makino H, Malinow R. AMPA receptor incorporation into synapses during LTP: the role of lateral movement and exocytosis. Neuron. 2009;64:381–390. doi: 10.1016/j.neuron.2009.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki M, Honkura N, Ellis-Davies GC, Kasai H. Structural basis of long-term potentiation in single dendritic spines. Nature. 2004;429:761–766. doi: 10.1038/nature02617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matus A. Actin-based plasticity in dendritic spines. Science. 2000;290:754–758. doi: 10.1126/science.290.5492.754. [DOI] [PubMed] [Google Scholar]
- Matus A, Ackermann M, Pehling G, Byers HR, Fujiwara K. High actin concentrations in brain dendritic spines and postsynaptic densities. Proceedings of the National Academy of Sciences of the United States of America. 1982;79:7590–7594. doi: 10.1073/pnas.79.23.7590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng Y, Zhang Y, Tregoubov V, Janus C, Cruz L, Jackson M, Lu WY, MacDonald JF, Wang JY, Falls DL, Jia Z. Abnormal spine morphology and enhanced LTP in LIMK-1 knockout mice. Neuron. 2002;35:121–133. doi: 10.1016/s0896-6273(02)00758-4. [DOI] [PubMed] [Google Scholar]
- Oertner TG, Matus A. Calcium regulation of actin dynamics in dendritic spines. Cell Calcium. 2005;37:477–482. doi: 10.1016/j.ceca.2005.01.016. [DOI] [PubMed] [Google Scholar]
- Oh MC, Derkach VA. Dominant role of the GluR2 subunit in regulation of AMPA receptors by CaMKII. Nature Neuroscience. 2005;8:853–854. doi: 10.1038/nn1476. [DOI] [PubMed] [Google Scholar]
- Oh MC, Derkach VA, Guire ES, Soderling TR. Extrasynaptic membrane trafficking regulated by GluR1 serine 845 phosphorylation primes AMPA receptors for long-term potentiation. Journal of Biological Chemistry. 2006;281:752–758. doi: 10.1074/jbc.M509677200. [DOI] [PubMed] [Google Scholar]
- Otmakhov N, Tao-Cheng JH, Carpenter S, Asrican B, Dosemeci A, Reese TS, Lisman J. Forskolin-induced LTP in the CA1 hippocampal region is NMDA receptor dependent. J Neurophysiol. 2004;91:1955–1962. doi: 10.1152/jn.00941.2003. [DOI] [PubMed] [Google Scholar]
- Ovtscharoff W, Jr., Segal M, Goldin M, Helmeke C, Kreher U, Greenberger V, Herzog A, Michaelis B, Braun K. Electron microscopic 3D-reconstruction of dendritic spines in cultured hippocampal neurons undergoing synaptic plasticity. Developmental Neurobiology. 2008;68:870–876. doi: 10.1002/dneu.20627. [DOI] [PubMed] [Google Scholar]
- Park M, Salgado JM, Ostroff L, Helton TD, Robinson CG, Harris KM, Ehlers MD. Plasticity-induced growth of dendritic spines by exocytic trafficking from recycling endosomes. Neuron. 2006;52:817–830. doi: 10.1016/j.neuron.2006.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penzes P, Beeser A, Chernoff J, Schiller MR, Eipper BA, Mains RE, Huganir RL. Rapid induction of dendritic spine morphogenesis by trans-synaptic ephrinB-EphB receptor activation of the Rho-GEF kalirin. Neuron. 2003;37:263–274. doi: 10.1016/s0896-6273(02)01168-6. [DOI] [PubMed] [Google Scholar]
- Perkinton MS, Sihra TS, Williams RJ. Ca(2+)-permeable AMPA receptors induce phosphorylation of cAMP response element-binding protein through a phosphatidylinositol 3-kinase-dependent stimulation of the mitogen-activated protein kinase signaling cascade in neurons. Journal of Neuroscience. 1999;19:5861–5874. doi: 10.1523/JNEUROSCI.19-14-05861.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickard L, Noel J, Duckworth JK, Fitzjohn SM, Henley JM, Collingridge GL, Molnar E. Transient synaptic activation of NMDA receptors leads to the insertion of native AMPA receptors at hippocampal neuronal plasma membranes. Neuropharmacology. 2001;41:700–713. doi: 10.1016/s0028-3908(01)00127-7. [DOI] [PubMed] [Google Scholar]
- Plant K, Pelkey KA, Bortolotto ZA, Morita D, Terashima A, McBain CJ, Collingridge GL, Isaac JT. Transient incorporation of native GluR2-lacking AMPA receptors during hippocampal long-term potentiation. Nature Neuroscience. 2006;9:602–604. doi: 10.1038/nn1678. [DOI] [PubMed] [Google Scholar]
- Purpura DP. Dendritic spine “dysgenesis” and mental retardation. Science. 1974;186:1126–1128. doi: 10.1126/science.186.4169.1126. [DOI] [PubMed] [Google Scholar]
- Rex CS, Chen LY, Sharma A, Liu J, Babayan AH, Gall CM, Lynch G. Different Rho GTPase-dependent signaling pathways initiate sequential steps in the consolidation of long-term potentiation. Journal of Cell Biology. 2009;186:85–97. doi: 10.1083/jcb.200901084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saglietti L, Dequidt C, Kamieniarz K, Rousset MC, Valnegri P, Thoumine O, Beretta F, Fagni L, Choquet D, Sala C, Sheng M, Passafaro M. Extracellular interactions between GluR2 and N-cadherin in spine regulation. Neuron. 2007;54:461–477. doi: 10.1016/j.neuron.2007.04.012. [DOI] [PubMed] [Google Scholar]
- Samoilova MV, Buldakova SL, Vorobjev VS, Sharonova IN, Magazanik LG. The open channel blocking drug, IEM-1460, reveals functionally distinct alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionate receptors in rat brain neurons. Neuroscience. 1999;94:261–268. doi: 10.1016/s0306-4522(99)00326-7. [DOI] [PubMed] [Google Scholar]
- Saneyoshi T, Fortin DA, Soderling TR. Regulation of spine and synapse formation by activity-dependent intracellular signaling pathways. Curr Opin Neurobiol. 2010;20:108–115. doi: 10.1016/j.conb.2009.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saneyoshi T, Wayman G, Fortin D, Davare M, Hoshi N, Nozaki N, Natsume T, Soderling TR. Activity-dependent synaptogenesis: regulation by a CaM-kinase kinase/CaM-kinase I/betaPIX signaling complex. Neuron. 2008;57:94–107. doi: 10.1016/j.neuron.2007.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt JM, Wayman GA, Nozaki N, Soderling TR. Calcium activation of ERK mediated by calmodulin kinase I. Journal of Biological Chemistry. 2004;279:24064–24072. doi: 10.1074/jbc.M401501200. [DOI] [PubMed] [Google Scholar]
- Schmitt JM, Guire ES, Saneyoshi T, Soderling TR. Calmodulin-dependent kinase kinase/calmodulin kinase I activity gates extracellular-regulated kinase-dependent long-term potentiation. Journal of Neuroscience. 2005;25:1281–1290. doi: 10.1523/JNEUROSCI.4086-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert V, Dotti CG. Transmitting on actin: synaptic control of dendritic architecture. Journal of Cell Science. 2007;120:205–212. doi: 10.1242/jcs.03337. [DOI] [PubMed] [Google Scholar]
- Shahi K, Baudry M. Glycine-induced changes in synaptic efficacy in hippocampal slices involve changes in AMPA receptors. Brain Research. 1993;627:261–266. doi: 10.1016/0006-8993(93)90329-l. [DOI] [PubMed] [Google Scholar]
- Shi S, Hayashi Y, Esteban JA, Malinow R. Subunit-specific rules governing AMPA receptor trafficking to synapses in hippocampal pyramidal neurons. Cell. 2001;105:331–343. doi: 10.1016/s0092-8674(01)00321-x. [DOI] [PubMed] [Google Scholar]
- Shi SH, Hayashi Y, Petralia RS, Zaman SH, Wenthold RJ, Svoboda K, Malinow R. Rapid spine delivery and redistribution of AMPA receptors after synaptic NMDA receptor activation. Science. 1999;284:1811–1816. doi: 10.1126/science.284.5421.1811. [DOI] [PubMed] [Google Scholar]
- Silva AJ, Stevens CF, Tonegawa S, Wang Y. Deficient hippocampal long-term potentiation in alpha-calcium-calmodulin kinase II mutant mice. Science. 1992;257:201–206. doi: 10.1126/science.1378648. [DOI] [PubMed] [Google Scholar]
- Steiner P, Higley M, Xu W, Czervionke B, Malenka R, Sabatini B. Destabilization of the postsynaptic density by PSD-95 serine 73 phosphorylation inhibits spine growth and synaptic plasticity. Neuron. 2008;60:788–802. doi: 10.1016/j.neuron.2008.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tada T, Sheng M. Molecular mechanisms of dendritic spine morphogenesis. Current Opinion in Neurobiology. 2006;16:95–101. doi: 10.1016/j.conb.2005.12.001. [DOI] [PubMed] [Google Scholar]
- Tassabehji M, Metcalfe K, Fergusson WD, Carette MJ, Dore JK, Donnai D, Read AP, Proschel C, Gutowski NJ, Mao X, Sheer D. LIM-kinase deleted in Williams syndrome. Nature Genetics. 1996;13:272–273. doi: 10.1038/ng0796-272. [DOI] [PubMed] [Google Scholar]
- Tian X, Feig LA. Age-dependent participation of Ras-GRF proteins in coupling calcium-permeable AMPA glutamate receptors to Ras/Erk signaling in cortical neurons. Journal of Biological Chemistry. 2006;281:7578–7582. doi: 10.1074/jbc.M512060200. [DOI] [PubMed] [Google Scholar]
- Tokumitsu H, Enslen H, Soderling TR. Characterization of a Ca2+/calmodulin-dependent protein kinase cascade. Molecular cloning and expression of calcium/calmodulin-dependent protein kinase kinase. Journal of Biological Chemistry. 1995;270:19320–19324. doi: 10.1074/jbc.270.33.19320. [DOI] [PubMed] [Google Scholar]
- Tokumitsu H, Inuzuka H, Ishikawa Y, Kobayashi R. A single amino acid difference between alpha and beta Ca2+/calmodulin-dependent protein kinase kinase dictates sensitivity to the specific inhibitor, STO-609. Journal of Biological Chemistry. 2003;278:10908–10913. doi: 10.1074/jbc.M213183200. [DOI] [PubMed] [Google Scholar]
- Tokumitsu H, Inuzuka H, Ishikawa Y, Ikeda M, Saji I, Kobayashi R. STO-609, a specific inhibitor of the Ca(2+)/calmodulin-dependent protein kinase kinase. J Bio Chem. 2002;277:15813–15818. doi: 10.1074/jbc.M201075200. [DOI] [PubMed] [Google Scholar]
- Vlachos A, Korkotian E, Schonfeld E, Copanaki E, Deller T, Segal M. Synaptopodin regulates plasticity of dendritic spines in hippocampal neurons. Journal of Neuroscience. 2009;29:1017–1033. doi: 10.1523/JNEUROSCI.5528-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayman GA, Impey S, Marks D, Saneyoshi T, Grant WF, Derkach V, Soderling TR. Activity-dependent dendritic arborization mediated by CaM-kinase I activation and enhanced CREB-dependent transcription of Wnt-2. Neuron. 2006;50:897–909. doi: 10.1016/j.neuron.2006.05.008. [DOI] [PubMed] [Google Scholar]
- Wayman GA, Kaech S, Grant WF, Davare M, Impey S, Tokumitsu H, Nozaki N, Banker G, Soderling TR. Regulation of axonal extension and growth cone motility by calmodulin-dependent protein kinase I. Journal of Neuroscience. 2004;24:3786–3794. doi: 10.1523/JNEUROSCI.3294-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Webb DJ, Asmussen H, Niu S, Horwitz AF. A GIT1/PIX/Rac/PAK signaling module regulates spine morphogenesis and synapse formation through MLC. Journal of Neuroscience. 2005;25:3379–3388. doi: 10.1523/JNEUROSCI.3553-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.