

Abstract
Phospholipase A2 (PLA2) catalyzes the release of arachidonic acid for generation of lipid mediators of inflammation and is crucial in diverse inflammatory processes. The functions of the secretory PLA2 enzymes (sPLA2), numbering 9 members in humans, are poorly understood, though they have been shown to participate in lipid mediator generation and the associated inflammation. To further understand the roles of sPLA2 in disease, we quantified the expression of these enzymes in the synovial fluid in rheumatoid arthritis and used gene-deleted mice to examine their contribution in a mouse model of autoimmune erosive inflammatory arthritis. Contrary to expectation, we find that the group V sPLA2 isoform plays a novel anti-inflammatory role that opposes the proinflammatory activity of group IIA sPLA2. Mechanistically, group V sPLA2 counter-regulation includes promotion of immune complex clearance by regulating cysteinyl leukotriene synthesis. These observations identify a novel anti-inflammatory function for a PLA2 and identify group V sPLA2 as a potential biotherapeutic for treatment of immune-complex-mediated inflammation.
Keywords: secreted phospholipase A2, arthritis, autoimmunity, inflammation
Phospholipases A2 (PLA2) comprise a diverse family whose members share the capacity to hydrolyze the sn-2 position of membrane glycerophospholipids, releasing fatty acids and lysophospholipids. There are over 25 mammalian PLA2 isoforms that have been grouped into three major classes, namely the calcium-dependent and -independent intracellular enzymes, and calcium-dependent secreted PLA2 (sPLA2) (Schaloske & Dennis, 2006; Valentin & Lambeau, 2000). The best characterized member of the PLA2 family is cytosolic group IV PLA2α, which is constitutively expressed in most tissues. Deletion of group IV cPLA2α confirms its essential role in parturition and fertility (Bonventre et al, 1997; Kudo & Murakami, 2002; Uozumi et al, 1997) and its contributions to diverse inflammatory processes (Hegen M, 2006; Hegen et al, 2003).
The sPLA2 family members are strikingly diverse. They are typically ~14–19 kDa heavily-disulfide bridged proteins found not only in mammals, but also in insects, snake venoms, plants, bacteria, fungi and viruses (Kini, 2003; Lambeau & Gelb, 2008; Nagiec et al, 2004; Soragni et al, 2001; Zadori et al, 2001). Sequence homology analysis led to the identification of 10 mammalian enzymatically active sPLA2s (9 of which are expressed in humans) and two sPLA2-like proteins devoid of catalytic activity.
sPLA2 enzymes have been implicated in physiological functions, host-defense and inflammation. Group IB sPLA2 is present in pancreatic secretions and exhibits a role in dietary phospholipid digestion (Arnesjo et al, 1967; Huggins et al, 2002). Group IIA sPLA2 demonstrates a predilection for negatively charged membranes such as those present in bacterial cell walls and has been implicated in host defense against Gram-positive bacteria (Foreman-Wykert et al, 1999; Menschikowski et al, 2006; Piris-Gimenez et al, 2005). In a mechanistically distinct manner, group V sPLA2 also plays a role in host defense by promoting phagocytosis and killing of fungal species (Balestrieri et al, 2006; Balestrieri et al, 2009).
sPLA2 activity has been demonstrated in multiple inflammatory disease states including rheumatoid arthritis (RA), sepsis, psoriasis, pancreatitis and cancer (Funakoshi et al, 1993; Green et al, 1991; Mounier et al, 2008; Pruzanski et al, 1985). The majority of these studies have focused on the role of group IIA sPLA2 in these processes. However, many of these studies were performed prior to the characterization of the multiple sPLA2 isoforms and before the development of isoform-specific reagents. Recent descriptions of the pro-inflammatory role of group V sPLA2 in allergic airway inflammation (Munoz et al, 2007), acute lung injury (Munoz et al, 2009) and atherosclerosis (Bostrom et al, 2007) prompted us to reexamine the presence of sPLA2 isoforms in RA synovial fluid and to evaluate the role of group V sPLA2 in arthritis. Surprisingly, our data indicate a counter-regulatory role for group V sPLA2 in a mouse model of immune complex-mediated arthritis. Further, we delineate a novel mechanism, shared in mice and humans, in which group V sPLA2 promotes the phagocytosis of immune complexes by macrophages to ameliorate inflammation. This group V sPLA2 function depends on its enzymatic activity and the generation of cysteinyl leukotrienes. Interestingly, we also confirm the pro-inflammatory role of group IIA sPLA2 in arthritis.
These findings highlight the complexity of sPLA2 species’ participation in inflammation, reveal a previously unappreciated and unanticipated anti-inflammatory function for group V sPLA2, underscore the importance of developing selective inhibitors of pro-inflammatory sPLA2 members for use in therapy, and identify group V sPLA2 as a novel potential biotherapeutic in arthritis.
RESULTS
Diverse sPLA2 are detected in RA synovial fluids
Given recent insights into distinct functional activities for sPLA2 species, and since most analyses of PLA2 enzymatic activity in synovial fluid were performed prior to cloning the complete family of sPLA2 isoforms (Pruzanski et al, 1992; Pruzanski et al, 1985; Seilhamer et al, 1989a; Vadas et al, 1985), we quantified the concentration of all catalytically active human sPLA2 in synovial fluid from 45 subjects with RA using a specific immunoassay that distinguishes the different sPLA2 isoforms (Figure 1 A) (Nevalainen et al, 2005). Interestingly, all sPLA2 isoforms could be detected, although typically each isoform was expressed in only a subgroup of subjects.
Figure 1. Expression of sPLA2 in the synovial fluid of patients with RA and healthy controls.
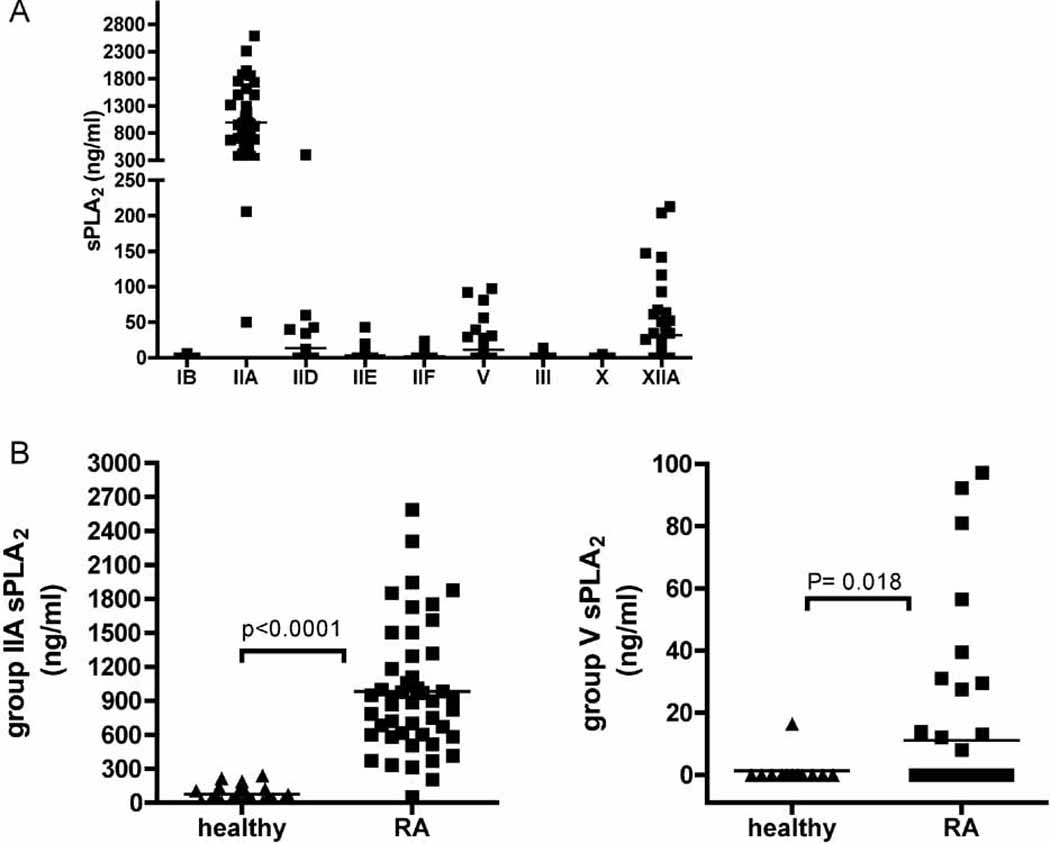
(A) The full set of human sPLA2 isoforms were quantified by time-resolved immunofluorescence analysis in synovial fluid from patients with RA (n=45). (B) Group IIA and V sPLA2 concentrations in synovial fluid obtained from healthy volunteers (n=12).
To examine disease-associated differences in sPLA2 isoform expression, we also quantified sPLA2 levels in synovial fluid from healthy volunteers. Here, due to limitations in synovial fluid volumes obtained from healthy individuals, we confined our analyses to group IIA and V sPLA2. Group IIA sPLA2 was more prominently expressed than group V sPLA2 in both healthy and RA subjects, and both isoforms were present in synovial fluid of RA subjects at levels significantly higher than observed in synovial fluid of healthy volunteers (Figure 1 B). Examination of co-expression of group IIA and V sPLA2 demonstrates no statistically significant correlation between isoform levels in RA synovial fluid (r= 0.3497, P= 0.1326), suggesting that the expression of the two enzymes is independently regulated.
Anti-inflammatory activity of group V sPLA2 in autoimmune arthritis
Given the failed clinical trial of a putative group IIA sPLA2 inhibitor in arthritis (Bradley et al, 2005) and having demonstrated expression of group V sPLA2 in arthritic human synovial fluid, we examined the contribution of group V sPLA2 to inflammatory arthritis. We employed the K/BxN serum transfer model of autoimmune inflammatory arthritis to explore the contributions of group V sPLA2 to the pathophysiology of inflammatory arthritis in vivo. The progressive distal symmetric erosive polyarthritis observed in K/BxN T-cell receptor (TcR) transgenic mice results from recognition of an ubiquitous autoantigen, glucose-6-phosphate isomerase (GPI), presented by the MHC class II Ag7 molecule (Korganow et al, 1999; Kouskoff et al, 1996; Matsumoto et al, 1999). These autoreactive T-cells drive high titer pathogenic autoantibody production. Importantly, arthritis can be induced in recipient mice by passive transfer of arthritogenic IgG autoantibodies (Korganow et al, 1999; Matsumoto et al, 2002). Numerous IgG-driven effector phase mechanisms have been identified in the pathophysiology of this arthritis including multiple innate cellular lineages and soluble mediators (IL-1β, TNF, complement C5a/C5aR, eicosanoids (LTB4 and PGI2) and tryptase) (Boilard et al, 2010; Bruhns et al, 2003; Chen et al, 2008; Chen et al, 2006; Chiba et al, 2005; Corr & Crain, 2002; Ji et al, 2002a; Ji et al, 2002b; Kim et al, 2006; Lee et al, 2002; Shin et al, 2009; Wipke & Allen, 2001). Of particular relevance, since the pathophysiology of the arthritis in the K/BxN serum transfer model includes a contribution from inflammatory eicosanoids (Chen et al, 2008; Chen et al, 2006), it provides an ideal model in which to investigate the role of enzymes involved in eicosanoid biosynthesis.
To assess the in vivo role of group V sPLA2 in inflammatory arthritis, we examined the response of group V sPLA2 null and congenic control mice to adminstration of arthritogenic K/BxN serum. Unexpectedly, rather than showing attenuation of the arthritic response, mice that lack group V sPLA2 demonstrated a significantly more severe autoantibody-driven arthritic response than congenic controls (Figure 2A). Histomorphometric analyses confirm clinical measures of arthritis, with increased leukocytic tissue infiltration, pannus formation and bone and cartilage destruction in group V sPLA2 null mice (Figure 2 B, C).
Figure 2. Group V sPLA2 protects from K/BxN serum-transfer arthritis.

(A) Arthritis response in group V sPLA2-null and congenic control mice mice in a BALB/c group IIA null background. Mice were injected with 20 µl of K/BxN serum at day 0 and 2, and disease development was monitored for 13 days. (B) Histomorphometric quantification of arthritis severity in group V sPLA2-null and congenic control mice at experimental day 13. N=15 mice/group. Data are mean ± SEM pooled from three independent experiments. P < 0.001 for (A). (C) Representative mid-saggital ankle sections from group V sPLA2 null and group V sPLA2-control mice. Upper and lower panels are 25X and 200X magnification respectively (lower panel). White arrows demarcate the hyperplastic synovial lining surrounding a large effusion (upper) while black and white arrowheads highight bone and cartilage erosions respectively. Note the increased leukocytic infiltration, synovial lining hyperplasia and pannus formation in group V sPLA2-null mice (T, tibia; S, synovial space; C, calcaneus). Figures representative of findings in 15 mice/group.
Systemic administration of recombinant group V sPLA2 ameliorates arthritis
To validate the modulating role of endogenous group V sPLA2 in antibody-driven inflammatory arthritis and to provide in vivo proof of concept for therapeutic use of group V sPLA2, we produced highly purified recombinant mouse group V sPLA2 and administered this material parenterally to group V sPLA2 null mice. Mice deficient in group V sPLA2 treated with recombinant group V sPLA2 were substantially protected from K/BxN arthritis (Figure 3 A, C, D). Interestingly, parenteral administration of recombinant group V sPLA2 to WT mice also resulted in reduced clinical and histomorphometric indices of arthritis (Figure 3 B, C). Taken together, these results confirm the counter-regulatory role for group V sPLA2 in inflammatory arthritis that was observed in the genetic studies.
Figure 3. Systemic administration of recombinant group V sPLA2 ameliorates K/BxN serum-transfer arthritis.

Recombinant mouse group V sPLA2 (50 µg in PBS) or control PBS were injected intravenously into group V sPLA2-null mice in a BALB/c group IIA null background (A) or WT BALB/c (B) mice 2 hours prior to administration of K/BxN serum on experimental Day 0 and daily thereafter for 5 days. Arthritis was induced by injection of 35 µl and 20 µl of K/BxN serum on day 0 and 2 respectively; the development of arthritis was monitored for 7 days. Arrows indicate sPLA2-V intravenous injections. (C) Histomorphometric quantification of inflammation in ankle sections from mice injected with recombinant group V sPLA2 or PBS control at experimental day 7. Note that the BALB/c mice express both group V and group IIA sPLA2 and thefore are not the congenic controls to group V sPLA2−/−. N=12 mice/group. Data are mean ± SEM pooled from three independent experiments P < 0.001 for (A–B). (D) Representative mid-saggital ankle sections from group V sPLA2-null mice treated with recombinant group V sPLA2 or its diluent (PBS). Note the decreased leukocytic infiltration, synovial lining hyperplasia and pannus formation in recombinant group V sPLA2 treated mice. Magnification=200X. Figures are representative of findings in 12 mice/group.
Toxicity considerations comprise an important aspect in evaluating both mechanistic activity and therapeutic potential for a novel disease target. Although little is known regarding potential toxic or deleterious activities for group V sPLA2, previous studies in group V sPLA2 transgenic mice have shown that transgenic neonatal pups die from pulmonary distress within 8 hours after birth due to surfactant hydrolysis (Ohtsuki et al, 2006). To assess this potential confounder for our mechanistic and biotherapeutic studies, we examined lung tissues from mice administered recombinant group V sPLA2 and found no evidence for tissue abnormality (Supplementary Figure 1).
Group IIA sPLA2 contributes to synovial inflammation
Since the anti-inflammatory activity of group V sPLA2 was unexpected, we expanded our analyses by investigating whether group IIA sPLA2 displayed an expected pro-inflammatory role in disease. As anticipated, mice with an isolated deficiency in group IIA sPLA2 displayed substantial reduction of clinical signs of arthritis relative to congenic wild-type mice (Figure 4 A, B). Histomorphometric quantification of tissue pathology confirmed clinical measures of arthritis, with decreases in leukocytic infiltration, bone erosion and cartilage destruction by synovial pannus in group IIA sPLA2 null mice (Figure 4 D).
Figure 4. Group IIA sPLA2 contributes to severity of K/BxN serum-transfer arthritis.

Mice were injected with 65 µl and 35 µl K/BxN serum on day 0 and 2, respectively, and the development of arthritis was followed for 13 days. (A) Arthritic response in group IIA sPLA2-null and wild-type congenic BALB/c control mice. (B) Histomorphometric quantification of arthritis severity in group IIA sPLA2-null and congenic BALB/c control mice at experimental day 13. N=15 mice/group. Data are mean ± SEM pooled from three independent experiments. p <0.001 (A). (C) Human group IIA sPLA2 transgenic and wild-type C57BL/6 mice were administered a single 75 µl dose of K/BxN serum on experimental day 0 and development of arthritis was monitored for 13 days. (D) Histomorphometric quantification of arthritis severity in human group IIA sPLA2 transgenic and wild-type control mice at experimental day 13. N=15 mice/group. Data are mean ± SEM pooled from three independent experiments. P < 0.001 for (C).
Although most closely related by sequence homology (Seilhamer et al, 1989b), whether human group IIA sPLA2 is the functional ortholog of murine group IIA sPLA2 remains speculative. To confirm the pro-inflammatory contribution of human group IIA sPLA2 to synovitis, we assessed the severity of K/BxN arthritis in mice expressing a human group IIA sPLA2 transgene (Grass et al, 1996). Because the C57BL/6 (B6) strain contains a spontaneous mutation in group IIA sPLA2 that abrogates expression (Kennedy et al, 1995), we selected human group IIA sPLA2 transgenic mice on this background. Thus, the only group IIA sPLA2 activity in these mice derives from the human transgene. Consistent with a proinflammatory contribution from group IIA sPLA2 to human autoimmune arthritis, group IIA sPLA2 transgenic mice display increased clinical (Figure 4 C) and histological (Figure 4 D) arthritic responses to K/BxN serum transfer.
Group X sPLA2 deficiency does not impact arthritis
Group V sPLA2 and group X sPLA2 are unique among mammalian sPLA2s in that they bind with high affinity to phosphatidylcholine-rich membranes and readily hydrolyze the external leaflet of mammalian cell membranes (Singer et al, 2002). We therefore examined arthritic responses in mice lacking group X sPLA2. We found no contribution of group X sPLA2 in K/BxN serum-induced erosive arthritis (supplementary Figure 2), further confirming separate and distinct functions of individual sPLA2 enzymes in arthritis pathophysiology.
Group V sPLA2 stimulates phagocytic uptake of immune complexes
Several of the known activities of group V sPLA2 activities could plausibly contribute to its impact on inflammatory arthritis. We have previously demonstrated reduced phagocytosis of fungal particles and IgG-coated sheep red blood cells in group V sPLA2 deficient macrophages (Balestrieri et al, 2006; Balestrieri et al, 2009). We therefore hypothesized that group V sPLA2-directed phagocytosis of immune complexes by macrophages or other phagocytes could comprise a novel mechanism by which this isoform modulates arthritis activity. We thus monitored immune complex phagocytosis by primary murine macrophages and found impaired immune complex uptake into cells lacking group V sPLA2 (Figure 5 A).
Figure 5. Group V sPLA2 promotes clearance of immune complexes in vitro.

Phagocytosis of immune complexes in vitro by (A) peritoneal macrophages from group V sPLA2-null, group V sPLA2-control, and FcγR-null mice or (B) CD14+ cells in RA synovial fluid with or without addition of recombinant group V sPLA2 or its inactive mutant H48Q was quantified cytofluorometrically using FcOxyburst. Data are mean ± SEM pooled from three (A) and six (B) independent experiments. (C) Phagocytosis of immune complexes by human CD14+ cells from peripheral blood incubated with recombinant group V sPLA2 in the presence of either the cyclooxygenase inhibitor indomethacin or the FLAP inhibitor MK886. Data are mean ± SEM pooled from three experiments perfomed in duplicate. (D) CysLTs released by leukocytes from RA SF treated with sPLA2. Group V sPLA2, its mutant H48Q or group IIA sPLA2 were added to leukocytes isolated from RA SF and cysLTs released into the supernatant were quantified by ELISA. Data are mean ± SEM pooled from three experiments performed in duplicate. (E–F) CysLTs promote phagocytosis of immune complexes by CD14+ cells. Indicated concentrations of LTC4 (E) or LTD4 (F) were added to peripheral blood mononuclear cells prior to addition of the FcOxyburst probe and phagocytosis by CD14+ cells was monitored cytofluorometrically. Data are mean ± SEM pooled from three experiments. (G) Phagocytosis of immune complexes by sPLA2-stimulated CD14+ cells from peripheral blood in presence of the cysLT1 antagonist monteleukast. Data are mean ± SEM pooled from three experiments perfomed in duplicate.
To assess a homologous activity for human group V sPLA2 and thereby extend the relevance of our observations to human autoimmune inflammatory disease, we added exogenous human group V sPLA2 to leukocytes present in synovial fluid from inflamed joints of RA patients and monitored phagocytosis of IgG immune complexes by CD14+ macrophages. Consistent with our murine observations, we found that human group V sPLA2 can trigger phagocytosis of IgG immune complexes in this population of cells that are abundant in the diseased joint fluid (Figure 5B).
Group V sPLA2 promotes immune complex phagocytosis via cysteinyl leukotriene generation
We further examined the mechanisms by which group V sPLA2 promotes phagocytic uptake of IgG containing immune complexes. Previous studies have demonstrated that group V sPLA2 activities include both phospholipase enzymatic activity and capacity to interact with the M-type receptor (Rouault et al, 2007). To define a contribution via its enzymatic activity, we mutated the catalytic site of group V sPLA2 to generate an enzymatically inactive protein (group V sPLA2-H48Q)(Lambeau & Gelb, 2008). We found this catalytically inactive mutant incapable of stimulating phagocytosis of IgG immune complexes by human synovial fluid CD14+ monocyte/macrophage cells (Figure 5B).
Having demonstrated a requirement for phospholipid hydrolysis by group V sPLA2, we endeavored to identify which lipid(s) promotes immune complex phagocytosis. Once released from the phospholipidic bilayer by a PLA2, arachidonic acid can be metabolized into several classes of biologically active lipids including prostanoids, leukotrienes, lipoxins, resolvins and others via the cyclooxygenase (COX) or lipoxygenase (LO) pathways. Using pharmacologic inhibitors, we found that inhibition of 5-LO, but not COX, abrogated the promotion of immune complex phagocytosis by group V sPLA2 in human synovial fluid CD14+ monocyte/macrophage cells (Figure 5 C).
To define which 5-LO-dependent eicosanoid mediates group V sPLA2 activity, we next utilized a candidate-based approach. Resolvins and lipoxins have documented anti-inflammatory activities (Haworth et al, 2008; Schwab et al, 2007; Schwab & Serhan, 2006), however, neither lipoxin A4 nor resolvin E1 promoted phagocytosis of immune complexes by macrophages when added exogenously (Supplementary Figure 3 A, B). Since we previously observed an unexplained increase in the severity of arthritis in mice lacking cysteinyl leukotrienes (cysLTs) (Chen et al, 2006) we investigated a role for cysLTs the ability of group V sPLA2 to promote immune complex uptake by phagocytes. Examination of supernatants from RA SF leukocytes or peripheral blood mononuclear cells treated with group V sPLA2 demonstrate significant stimulation of cysLT generation by these populations (Figure 5 D and 6 D). In stark contrast, group IIA sPLA2 and enzymatically inactive group V sPLA2-H48Q lacked the ability to drive cysLT production in these cells (Figure 5 D and 6 D). Confirming studies by others (Mancuso & Peters-Golden, 2000), we found that both LTC4 and LTD4 potently promote immune complex clearance when added exogenously to these cell populations (Figure 5 E, F and supplementary Figure 4). In further confirmation, we found that administration of a cysteinyl leukotriene receptor 1 (CysLTR1) antagonist inhibits promotion by group V sPLA2 of immune complex phagocytosis by CD14+ cells (Figure 5G). In sum, our findings point to a novel pathway in which group V sPLA2 promotes immune complex clearance in monocyte/macrophage cells via stimulating synthesis of CysLTs, which act through CysLTR1 to promote phagocytosis.
Figure 6. Group V sPLA2 promotes clearance of immune complexes in vivo.

Immunofluorescent staining of IgG (Red) and complement C3 (Green) in mid-saggital cryosections of ankle tissues from group V sPLA2-control (A) or group V sPLA2-null (B) ankle joints. Nuclei (blue) are visualized by staining with DAPI. Magnification= 400X. Cartilage tissue and synovial fluid (SF) space as labeled. Mice were injected with 35 µl K/BxN serum at day 0, and ankle tissues were harvested on day 4. Data are representative of 3 independent experiments. (C) ELISA quantification of circulating immune complexes in sera from group V sPLA2 -null and group V sPLA2 control mice 4 days after administration of 35 µl of K/BxN serum. Pooled K/BxN serum and serum from non arthritic wild-type mice were included as controls. N=10 mice/group. Data are mean ± SEM pooled from two independent experiments. P=NS. (D) Quantification of cysLTs released by human peripheral blood mononuclear cells treated with recombinant sPLA2 in the presence (white filled) or absence (black filled) of RBC. Data are mean ± SEM pooled from three experiments. (E) Phagocytosis of immune complexes in the presence of RBC. Peripheral blood mononuclear cells incubated in the presence or absence of RBC were treated with group V sPLA2 and phagocytosis of immune complexes by CD14+ cells was monitored cytofluorometrically. Data are mean ± SEM pooled from three experiments.
Group V sPLA2 promotes clearance of articular immune complexes in vivo
Numerous previous studies demonstrate that IgG immune complexes are found at high levels in joint and synovial fluids of patients with RA (Bonomo et al, 1970; Brandt et al, 1968; Britton & Schur, 1971; Fish et al, 1966; Nydegger et al, 1977; Ruddy & Austen, 1970; Ruddy et al, 1975; Schur et al, 1975). Since the K/BxN model also displays articular deposition and pathogenic contributions from immune complexes (Ji et al, 2002a; Matsumoto et al, 2002), we measured IgG and C3 deposition in joint tissues of mice administered K/BxN serum to demonstrate that group V sPLA2 impacts immune complex clearance in vivo. Consistent with this mechanistic contribution by group V sPLA2 to temper the severity of arthritis, we found significantly more articular immune complexes and C3 deposition in mice lacking group V sPLA2 than their congenic WT littermates (Figure 6 A, B).
To assess whether group V sPLA2 impacts the systemic metabolism of immune complexes, we quantified circulating immune complexes in WT and group V sPLA2 null mice after administration of K/BxN serum. Interestingly, no differences in circulating immune complexes were detectable in these mice, pointing to a selective activity for group V sPLA2 in the joint (Figure 6 C). The basis for this selectivity did not reside in the phagocytic capacity of tissue resident vs circulating phagocytes since we found that group V sPLA2 stimulated comparable phagocytosis of immune complexes by human CD14+ cells, whether from the circulation or from synovial fluid (Figure 5 B, C, G). Since erythrocytes are absent in synovial fluid and abundant in blood, and since the erythrocyte membrane phospholipid composition is well suited for group V sPLA2 binding, we hypothesized that these cells may block group V sPLA2 activity in the circulation. Indeed, addition of small amounts of erythrocytes to the assays wherein group V sPLA2 was administered to phagocytes potently inhibited group V sPLA2 stimulation of cysLT synthesis (Figure 6 D) and immune complex phagocytosis (Figure 6 E).
DISCUSSION
The anticipated finding in our experiments employing isoform-specific antibodies (Nevalainen et al, 2005), human biospecimens, and mice deficient in specific sPLA2 isoforms was an overlapping pro-inflammatory contribution from individual sPLA2 isoforms to inflammatory arthritis. Indeed, group V sPLA2 is potent at releasing arachidonic acid from cell membranes as a substrate for leukotriene synthesis (Kim et al, 2002), and group V sPLA2-deficient cells have impaired eicosanoid synthesis (Kikawada et al, 2007; Satake et al, 2004). Thus, arachidonate release for pro-inflammatory lipid generation was the predominant predicted activity for group V sPLA2 in K/BxN inflammatory arthritis, a model in which eicosanoids contribute (Chen et al, 2008; Chen et al, 2006).
However, our results document a novel counter regulatory function for group V sPLA2 in inflammatory arthritis. Our mechanistic investigation was guided by our recent observation that group V sPLA2 promotes the phagocytosis of IgG-coated sheep red blood cells by macrophages (Balestrieri et al, 2009). The demonstration in the current experiments of reduced immune complex phagocytosis in primary macrophages lacking group V sPLA2 and the increase in immune complex deposition in joint tissues of group V sPLA2-null mice are consistent with a an impact of group V sPLA2 on the severity of arthritis by modulating immune complex clearance. This hypothesis was further substantiated by the ability of recombinant group V sPLA2 to stimulate immune complex phagocytosis by CD14+ monocyte/macrophage cells in the circulation and in synovial fluid from subjects with RA. Interestingly, the molecular mechanism through which group V sPLA2 regulates the uptake of immune complexes proceeds via its catalytic activity, which promotes cysLT biosynthesis.
While our studies demonstrate a novel anti-inflammatory function for group V sPLA2 in autoimmune inflammatory arthritis, they also point to anatomically or context dependent actions of this enzyme. On the one hand group V sPLA2 appears to augment early inflammation in acute models of peritonitis (Satake et al, 2004) and in allergic pulmonary inflammation (Munoz et al, 2007). On the other hand, it promotes clearance of pathogens (Balestrieri et al, 2009) and immune complexes. Our observations provide insights that resolve this apparent discrepancy. We find that cysLT generation plays a central role in the promotion of immune complex phagocytosis by group V sPLA2. This is congruent with an earlier study that showed cysLTs stimulated macrophage uptake of IgG-opsonized targets but not of unopsonized particles (Mancuso & Peters-Golden, 2000). Further, we previously documented that synthesis of cysLTs was dispensable to the promotion of macrophage uptake of non-opsonized zymosan yeast particles by group V sPLA2 (Balestrieri et al, 2006). Taken together, an integrated view of these distinct properties is that group V sPLA2 promotes phagocytosis of IgG opsonized particles or pathogens via generation of cysLTs; it participates in the innate immune response to non-opsonized pathogens via yet to be discovered mechanisms. Moreover, in the pathologic context of chronic immune complex-driven arthritis,where the inflammatory response is innappropriate and sustained, group V sPLA2 can counter-regulate disease activity via its capacity for promoting phagocytic removal of an inciting factor in disease.
Our findings also provide further insight into the activities of tissue macrophages in arthritis. Previous studies examining of the role of macrophages in K/BxN serum transfer arthritis have offered evidence that this lineage contributes to development of K/BxN arthritis (Solomon et al, 2005) and that they also have the capacity to diminish arthritis via activation of the Fc receptor, FcγRIIB (Bruhns et al, 2003). Our findings expand our understanding of murine and human macrophage behavior in synovitis by documenting a distinct mechanism by which they can impact disease. In addition to their capacity to elaborate soluble mediators of inflammation, it is now apparent that they can alter disease physiology via their prominent phagocyte function.
The contrasting pro-inflammatory and anti-inflammatory properties of group V sPLA2 also raise concern regarding toxicity from exogenously adminstered enzyme as a potential biotherapeutic. In this context it is notable that adult mice that received exogenous group V sPLA2 did not display overt toxicity and that the pulmonary pathology observed in transgenic mice overexpressing group V sPLA2 (Ohtsuki et al, 2006) was absent in treated mice (supplementary Figure 1). We hypothesize that this lack of overt toxicity may be due in part to the capacity for erythrocytes to abrogate the ability of exogenous group V sPLA2 to stimulate cysLT generation. Although not evident in all strains, recent mouse studies also demonstrate an impact of group V sPLA2 on vascular inflammation (Bostrom et al, 2007; Boyanovsky et al, 2009). Thus, extensive assessment of toxicity with chronic parenteral administration of recombinant group V sPLA2 remains warranted prior to investigations in humans.
Group V sPLA2 shares proteoglycan-binding properties with group IIA sPLA2, which is present in increased quantities in the synovial fluid of individuals with RA (Figure 1). In contrast to the antiinflammatory properties of group V sPLA2, our studies in mice deficient in group IIA sPLA2 confirmed the pro-inflammatory actions of group IIA sPLA2 in arthritis. Furthermore, mice transgenic for the human group IIA sPLA2 had an exaggerated inflammatory response and worse clinical disease. Although group IIA sPLA2 has long been implicated as a pro-inflammatory participant in inflammatory arthritis, to our knowledge, these studies are the first to document its functional contribution using a genetic approach. While the pro-inflammatory activity of group IIA sPLA2 awaits clarification, the absence of cysteinyl leukotriene release and the lack of phagocytosis stimulation (data not shown) observed with exogenous administration of group IIA sPLA2 to human macrophages points to a distinct mechanism of activity from group V sPLA2. We speculate that differences in the interfacial binding domains of group V and group IIA sPLA2 may factor prominently in this functional dichotomy. Group IIA sPLA2 prefers membranes rich in anionic phospholipids whereas group V sPLA2 avidly binds phosphatidylcholine (Singer et al, 2002). The external leaflet of mammalian cells is enriched in phosphatidylcholine, thus providing an environment suitable for group V sPLA2. As noted above, further insight into the binding and function of group IIA sPLA2 awaits future investigation.
We also studied mice deficient in group X sPLA2 because this enzyme shares several biochemical properties with group V sPLA2 and has been shown to contribute to arachidonate release and eicosanoid generation (Lambeau & Gelb, 2008). Furthermore, sequence alignment shows ~40% sequence identity between groups IIA, V and X sPLA2. Structurally, based on the crystal structure for human group IIA and group X sPLA2, all three proteins are thought to share a common interfacial binding surface and three dimensional organization (Lambeau & Gelb, 2008; Winget et al, 2006). Despite these similarities in structure and function between the three enzymes, disruption of the gene encoding group X sPLA2 was without effect (either anti- or pro-inflammatory) in the K/BxN serum transfer model of erosive arthritis (supplementary Figure 2). These results underscore the non-redundant function of sPLA2 isoforms and the unique antiinflammatory participation of group V sPLA2 in autoantibody driven arthritis.
Finally, these studies provide important new insight into therapeutic targeting of sPLA2 isoforms. The involvement of immune complexes and complement in RA has been extensively documented (reviewed in (Nigrovic & Lee, 2006)). Therefore, the ability of group V sPLA2 to ameliorate disease severity via stimulation of immune complex phagocytosis by murine and human macrophages suggests its relevance to the pathophysiology of RA and other diseases impacted by immune complexes. Immune complexes are abundantly present in the circulation and tissues of RA patients, while complement activation is evident both by its deposition in RA joint tissues and by depressed levels of intact complement in RA synovial fluid (Britton & Schur, 1971; Brodeur et al, 1991; El-Ghobarey & Whaley, 1980; Fostiropoulos et al, 1964; Pekin & Zvaifler, 1964; Ruddy & Austen, 1970; Ruddy et al, 1975; Schur et al, 1975; Zvaifler, 1969). Thus, the newly identified mechanistic and functional activity of group V sPLA2 suggests that its therapeutic administration is a potential novel treatment opportunity for those patients in which immune complexes have a prominent contribution to disease. In addition, our studies demonstrate that the most optimal sPLA2 inhibitor for treatment of RA should be highly selective for group IIA sPLA2. It has been suggested that lack of efficacy of a group IIA sPLA2 inhibitor tested in a RA clinical trial was due to insufficient dosing (Bradley et al, 2005). Our studies raise the additional concern that the inhibitor used in human arthritis trials lacked sufficient specificity (Oslund et al, 2008) and blocked both group IIA sPLA2 and group V sPLA2. Together, our observations provide rationale for pursuing two distinct therapeutic approaches targeted at sPLA2: the use of highly selective group IIA sPLA2 inhibitors and administration of recombinant group V sPLA2.
METHODS
Human synovial fluid analysis
Human knee synovial fluids were obtained as discarded material from patients with RA undergoing diagnostic or therapeutic arthrocentesis. RA was diagnosed by an American Board of Internal Medicine certified rheumatologist and/or by review of laboratory, radiologic and clinic notes and by applying ACR classification criteria (Arnett et al, 1988). Synovial fluid from healthy volunteers was obtained from individuals without prior history of knee trauma, chronic knee pain, prior knee surgery, blood dyscrasias, cancer, chondrocalcinosis, corticosteroid injection or non-steroidal anti-inflammatory drug use in the prior eight weeks as described (Gobezie et al, 2007). All studies received Institutional Review Board approval. For time-resolved fluorescence immunoassays of sPLA2s (Nevalainen et al, 2005), 50 µl of synovial fluid were used for all assays except 5 µl was used for group IIA sPLA2. Assay buffer (50 mM Tris, pH 7.8, 0.9% NaCl, 0.02% Tween-20, 0.05% NaN3, filtered through a 0.45 micron membrane) was added to each well to bring the total volume to 100 µl. For assay calibration, various amounts of recombinant human sPLA2 (prepared as described (Singer et al, 2002)) were added to assay buffer to generate a standard curve. Blanks were run that contained 100 µl assay buffer alone.
Mice
We used 6–9 week old mice for all of our studies. All procedures were approved by the Institutional Animal Care and Use Committee of the Dana-Farber Cancer Institute (Boston, MA). Mice were housed in the specific pathogen free animal facility of the Dana-Farber Cancer Institute. The group IIA sPLA2 gene in 129 and C57BL/6 mice has a thymidine insertion that disrupts the open reading frame (Kennedy et al, 1995). Group IIA sPLA2 null mice were produced by backcrossing 129 strain mice (spontaneously group IIA sPLA2 null) to BALB/cJ mice for 10 generations, selecting offspring heterozygous for disruption of the group IIA sPLA2 gene. After 10 backcrosses, heterozygous mice were bred to obtain homozygous congenic group IIA sPLA2 null and wild-type control mice from which breeding colonies were derived. The 129 allele was detected by PCR amplification of genomic DNA followed by DNA sequencing. Mice lacking group V sPLA2 were derived from 129 ES cells that lack expression of group IIA sPLA2, crossed to a BALB/c background for 11 generations as previously described (Satake et al, 2004). Because the genes encoding these enzymes are separated by only ~20 kB, our mice lacking group V sPLA2 are also deficient in group IIA sPLA2 (supplementary Figure 5 and Table 1). To assess the role of group V sPLA2 in inflammatory arthritis, we thus backcrossed our sPLA2-IIA-/V- mice onto the BALB/c background and utilized our group IIA sPLA2 null congenic BALB/c mice as control. The BALB/c and C57BL/6 were obtained from Jackson Laboratory (Bar Harbor, ME). The transgenic human group IIA sPLA2 mice (Grass et al, 1996) (C57BL/6J background) and the FcRγ null mice (Takai et al, 1994) were obtained from Taconic (Hudson, NY). Group X sPLA2 null mice on the C57BL6 background were a generous gift from Dr Nancy Webb (University of Kentucky, Lexington, KY) and will be described elsewhere(Webb).
Table 1.
Mouse strain expression of sPLA2 isoforms
| Mouse strain | Group IIA sPLA2 | Group V sPLA2 |
|---|---|---|
| BALB/c | +/+ | +/+ |
| C57BL/6 | −/− | +/+ |
| 129 | −/− | +/+ |
| group V sPLA2 null (Balb/C background) | −/− | −/− |
| group V sPLA2 congenic control (Balb/c background) | −/− | +/+ |
Recombinant sPLA2
Recombinant sPLA2 enzymes were produced as previously described (Rouault et al, 2007; Singer et al, 2002). Proteins were purified to single peaks by HPLC and both purity as well as appropriate disulfide bond formation was confirmed by SDS-PAGE analysis and by electrospray ionization mass spectrometry.
Serum transfer protocol and arthritis scoring
Arthritogenic K/BxN serum was transferred to recipient mice via intraperitoneal injection on experimental day 0 and 2 to induce arthritis as described (Chen et al, 2008; Korganow et al, 1999). Serum dosing was adjusted based on mouse strain and based on whether the transgenic animals gave an exaggerated or diminished response compared to the wild-type controls. Ankle thickness was measured at the malleoli with the ankle in a fully flexed position, using spring-loaded dial calipers (Long Island Indicator Service, NY). The clinical index of arthritis was graded on a scale 0–12 as described previously (Chen et al, 2008; Chen et al, 2006; Korganow et al, 1999).
Histological examination
For histomorphometric analysis, ankle tissues were fixed for 24 hours in 4% paraformaldehyde in PBS and decalcified for 72 hours with modified Kristensen’s solution. Tissues were then dehydrated, embedded in paraffin, sectioned at 5 µm thickness and stained with hematoxylin and eosin. Histological scoring was performed in a blinded manner as previously described (Chen et al, 2006; Pettit et al, 2001).
Immune complex phagocytosis in vitro
Phagocytosis of immune complexes by murine cells
Mouse macrophages were isolated as previously described (Balestrieri et al, 2006), except in supplementary figure 4 in which F4/80+ macrophages were analysed immediately after isolation from the peritoneal cavity. In brief, peritoneal cavities were flushed thrice with 10 ml RPMI containing calcium and magnesium and 10% FBS. Cells were then centrifuged and placed in culture in the same medium containing 2 mM L-glutamine, 100 U/ml penicillin and 100 µg/ml streptomycin in 6-well plates at 37 °C, 5% CO2 for 3 days. Adherent cells were liberated using GIBCOtm cell dissociation buffer enzyme free (Invitrogen) and resuspended in KRP buffer (PBS containing 1 mM CaCl2, 1.5 mM MgCl2, 5.5 mM glucose pH 7.4). Phagocytosis was quantified using BSA-anti-BSA immune complexes in which BSA is covalently bound to dichlorodyhydrofluorescein (FcOxyburst, Invitrogen). In this method, FcγR-mediated internalization of immune complexes leads to an oxidative burst in the phagosomal vacuole that is monitored cytofluorometrically as described in the manufacturers protocol and is examplified in Supplementary Figure 6. Cells (2 × 106/ml) were incubated at 37 °C with the FcOxyburst probe (60 µg/ml), and development of fluorescence was monitored at the indicated times points. As a negative control, macrophages from FcγR −/− mice were analyzed for phagocytosis. Data are presented as a ratio between the mean fluorescence intensity (MFI) at the indicated time and the MFI at the beginning of the reaction (t=0). For each time point, a minimum of 5000 macrophages was analyzed, in duplicate, in 3 independent experiments.
Phagocytosis of immune complexes by human cells
Human peripheral blood mononuclear cells (PBMC) were isolated by centrifugation of EDTA-anticoagulated human blood obtained from healthy donors on Ficoll-Paque Premiun (GE Healthcare) as described by the manufacturer. PBMC were washed in PBS and resuspended in KRP buffer (4 × 106/ml). For phagocytosis by human cells contained in freshly collected RA synovial fluids, cells were washed in PBS and the cell concentration was adjusted to 4 × 106/ml in KRP buffer. Typically, RA SF comprised ~10× 106/ml leukocytes, 22.9 ± 4% being CD14+. FLAP inhibitor MK886 (5 µM) (Cayman), cyclooxygenases inhibitor indomethacin (1 µM) (Sigma), CysLT1 antagonist montelukast (5 µM) (Cayman) were added 5 minutes prior to sPLA2 addition. Recombinant human group V sPLA2, its inactive mutant H48Q group V sPLA2, and group IIA sPLA2 (5 µg/ml) were added 20 minutes before addition of the FcOxyburst reagent (60 µg/ml). At the indicated time points, cells were transfered to tubes containing 300 µl cold PBS and PE-labeled anti-CD14 (BD Pharmingen) and kept on ice until the cytofluometric analysis were performed. The monocyte/macrophage population was defined by CD14 staining. A minimum of 5,000 cells were analyzed in each of 6 independent experiments.
Phagocytosis in the presence of RBC
Efforts were made to mimic the abundance of RBC in blood and to successfully limit the interference an excess of RBC could have on binding of FcOxyburst probe to macrophages Fc receptors. Autologous RBC washed in PBS (500 RBC: 1 CD14+ cells) were added to purified PBMC and incubated with group V sPLA2 for 20 minutes at 37°C in KRP buffer. Cells were next transferred to 4°C for 10 minutes and incubated further for 25 minutes with the FcOxyburst probe (60 µg/ml) to allow saturation of macrophages Fc receptors. Cells were next transferred to 37°C and the reaction started. At the indicated time points, cells were transfered to tubes containing 300 µl cold PBS and PE-labeled anti-CD14 and kept on ice until the cytoflurometric analysis were performed. The monocyte/macrophage population was defined by CD14 staining. A minimum of 5,000 cells was analyzed in each of 3 independent experiments.
Detection of immune complexes in vivo
Joint immune complexes
Snap frozen ankles were prepared from mice 4 days after injection with K/BxN serum. Cryostat sections from non-fixed, non-decalcified ankle joints were generated using a tape capture technique as described (Ji et al, 2002a; Watts et al, 2005). After blocking with 2% bovine serum albumin and 0.04% Tween in PBS, the sections were incubated with texas-red-conjugated anti-mouse IgG (Jackson), and FITC-conjugated goat anti-mouse C3 (ICN Biomedicals, Costa Mesa, CA) or control IgG (500 ng/section). Fluorescence was detected by microscopy (Nikon Eclipse E800). Yellow staining defines colocalization of C3 and IgG. Nuclei (blue) were counterstained with DAPI (50 ng/section, Molecular Probes). Images were acquired (Camera from Diagnostic Instruments) and processed digitally (Photoshop 6.0).
Circulating immune complexes
Immune complexes in serum were detected by ELISA (Matsumoto et al, 2002). In this method, target C1q (20 µg/ml) (Sigma) in PBS was added to 96-well ELISA plates (Nunc) for 18 hours at 4° C. The wells were blocked with 1% BSA in PBS, after which sera obtained from mice 4 days after the administration of K/BxN serum and diluted as indicated were added. Pooled serum from K/BxN mice was used as a positive control while sera from mice not administered K/BxN serum were used as a negative control. Bound complexes were detected using a HRP-coupled anti-mouse IgG (Jackson Immunoresearch).
Measurements of cysteinyl leukotrienes synthesis
CysLTs were measured in supernatants of cells treated with group V sPLA2, its inactive mutant group V sPLA2-H48Q and group IIA sPLA2 (5 µg/ml) in KRP buffer for 30 minutes at 37°C using a commercial ELISA (Cayman) according to the manufacturer’s instructions.
Statistical analysis
Mouse arthritis experiments are presented as mean ± SEM. The statistical significance for comparisons between groups was determined using two-way ANOVA, followed by Bonferroni correction using Prism software package 4.00 (GraphPAd Software, San Diego, CA). Comparison of sPLA2 content between RA and normal synovial fluids was made by Student’s t-test. P values smaller than 0.05 were considered significant. Spearman rho was calculated to assess correlation between group-IIA and –V sPLA2 in synovial fluid where expression of both isoforms was detected using the Prism software package.
Supplementary Material
Acknowledgments
Financial support
DML NIH P01 AI065858, Arthritis Foundation Investigator Award and the Cogan Family Foundation
JPA NIH R01HL070946, and bridge grants from the American Academy of Allergy Asthma and Immunology and the Brigham and Women’s Hospital Biomedical Research.
MHG NIH R37HL036235
HR, NIH A172143
GL, CNRS and Association pour la Recherche sur le Cancer
EB, Arthritis Foundation
BB, NIH K08AI064226
Footnotes
Author Contributions
Eric Boilard, Study conception, Study design, Acquisition of data, Analyses and interpretation of data, Manuscript preparation, Statistical analyses
Ying Lai, Acquisition of data, Analyses and interpretation of data,
Katherine Larabee, Acquisition of data, Analyses and interpretation of data, Manuscript preparation,
Barbara Balestrieri, Generated critical reagent (group IIA sPLA2-null mice on a BALB/c backgound), Manuscript preparation
Farideh Ghomashchi, Acquisition of data, Analyses and interpretation of data,
Daisuke Fujioka, Contributed important reagent, Manuscript preparation,
Reuben Gobezie, Acquisition of data, Manuscript preparation
Jonathan S. Coblyn, Acquisition of data, Manuscript preparation
Michael E. Weinblatt, Acquisition of data, Manuscript preparation
Elena M. Massarotti, Acquisition of data, Manuscript preparation
Thomas S. Thornhill, Acquisition of data, Manuscript preparation
Maziar Divangahi, Acquisition of data, Manuscript preparation
Heinz Remold, Analyses and interpretation of data, Manuscript preparation,
Gérard Lambeau, Analyses and interpretation of data, Manuscript preparation,
Michael H. Gelb, Study design, Acquisition of data, Analyses and interpretation of data, Manuscript preparation,
Jonathan P. Arm, Study design, Analyses and interpretation of data, Manuscript preparation,
David M. Lee, Study conception, Study design, Acquisition of data, Analyses and interpretation of data, Manuscript preparation,
Literature cited
- Arnesjo B, Barrowman J, Borgstrom B. The zymogen of phospholipase A2 in rat pancreatic juice. Acta Chem Scand. 1967;21(10):2897–2900. doi: 10.3891/acta.chem.scand.21-2897. [DOI] [PubMed] [Google Scholar]
- Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, Healey LA, Kaplan SR, Liang MH, Luthra HS, et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988;31(3):315–324. doi: 10.1002/art.1780310302. [DOI] [PubMed] [Google Scholar]
- Balestrieri B, Hsu VW, Gilbert H, Leslie CC, Han WK, Bonventre JV, Arm JP. Group V secretory phospholipase A2 translocates to the phagosome after zymosan stimulation of mouse peritoneal macrophages and regulates phagocytosisc. J Biol Chem. 2006;281(10):6691–6698. doi: 10.1074/jbc.M508314200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balestrieri B, Maekawa A, Xing W, Gelb MH, Katz HR, Arm JP. Group V secretory phospholipase A2 modulates phagosome maturation and regulates the innate immune response against Candida albicans. J Immunol. 2009;182(8):4891–4898. doi: 10.4049/jimmunol.0803776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boilard E, Nigrovic PA, Larabee K, Watts GF, Coblyn JS, Weinblatt ME, Massarotti EM, Remold-O'Donnell E, Farndale RW, Ware J, Lee DM. Platelets amplify inflammation in arthritis via collagen-dependent microparticle production. Science. 2010;327(5965):580–583. doi: 10.1126/science.1181928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonomo L, Tursi A, Trizio D, Gillardi U, Dammacco F. Immune complexes in rheumatoid synovitis: a mixed staining immunofluorescence study. Immunology. 1970;18(4):557–563. [PMC free article] [PubMed] [Google Scholar]
- Bonventre JV, Huang Z, Taheri MR, O'Leary E, Li E, Moskowitz MA, Sapirstein A. Reduced fertility and postischaemic brain injury in mice deficient in cytosolic phospholipase A2. Nature. 1997;390(6660):622–625. doi: 10.1038/37635. [DOI] [PubMed] [Google Scholar]
- Bostrom MA, Boyanovsky BB, Jordan CT, Wadsworth MP, Taatjes DJ, de Beer FC, Webb NR. Group v secretory phospholipase A2 promotes atherosclerosis: evidence from genetically altered mice. Arterioscler Thromb Vasc Biol. 2007;27(3):600–606. doi: 10.1161/01.ATV.0000257133.60884.44. [DOI] [PubMed] [Google Scholar]
- Boyanovsky B, Zack M, Forrest K, Webb NR. The capacity of group V sPLA2 to increase atherogenicity of ApoE−/− and LDLR−/− mouse LDL in vitro predicts its atherogenic role in vivo. Arterioscler Thromb Vasc Biol. 2009;29(4):532–538. doi: 10.1161/ATVBAHA.108.183038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley JD, Dmitrienko AA, Kivitz AJ, Gluck OS, Weaver AL, Wiesenhutter C, Myers SL, Sides GD. A randomized, double-blinded, placebo-controlled clinical trial of LY333013, a selective inhibitor of group II secretory phospholipase A2, in the treatment of rheumatoid arthritis. J Rheumatol. 2005;32(3):417–423. [PubMed] [Google Scholar]
- Brandt KD, Cathcart ES, Cohen AS. Studies of immune deposits in synovial membranes and corresponding synovial fluids. J Lab Clin Med. 1968;72(4):631–647. [PubMed] [Google Scholar]
- Britton MC, Schur PH. The complement system in rheumatoid synovitis. II. Intracytoplasmic inclusions of immunoglobulins and complement. Arthritis Rheum. 1971;14(1):87–95. doi: 10.1002/art.1780140111. [DOI] [PubMed] [Google Scholar]
- Brodeur JP, Ruddy S, Schwartz LB, Moxley G. Synovial fluid levels of complement SC5b-9 and fragment Bb are elevated in patients with rheumatoid arthritis. Arthritis Rheum. 1991;34(12):1531–1537. doi: 10.1002/art.1780341209. [DOI] [PubMed] [Google Scholar]
- Bruhns P, Samuelsson A, Pollard JW, Ravetch JV. Colony-stimulating factor-1-dependent macrophages are responsible for IVIG protection in antibody-induced autoimmune disease. Immunity. 2003;18(4):573–581. doi: 10.1016/s1074-7613(03)00080-3. [DOI] [PubMed] [Google Scholar]
- Chen M, Boilard E, Nigrovic PA, Clark P, Xu D, Fitzgerald GA, Audoly LP, Lee DM. Predominance of cyclooxygenase 1 over cyclooxygenase 2 in the generation of proinflammatory prostaglandins in autoantibody-driven K/BxN serum-transfer arthritis. Arthritis Rheum. 2008;58(5):1354–1365. doi: 10.1002/art.23453. [DOI] [PubMed] [Google Scholar]
- Chen M, Lam BK, Kanaoka Y, Nigrovic PA, Audoly LP, Austen KF, Lee DM. Neutrophil-derived leukotriene B4 is required for inflammatory arthritis. J Exp Med. 2006;203(4):837–842. doi: 10.1084/jem.20052371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiba A, Kaieda S, Oki S, Yamamura T, Miyake S. The involvement of V(alpha)14 natural killer T cells in the pathogenesis of arthritis in murine models. Arthritis Rheum. 2005;52(6):1941–1948. doi: 10.1002/art.21056. [DOI] [PubMed] [Google Scholar]
- Corr M, Crain B. The role of FcgammaR signaling in the K/B x N serum transfer model of arthritis. J Immunol. 2002;169(11):6604–6609. doi: 10.4049/jimmunol.169.11.6604. [DOI] [PubMed] [Google Scholar]
- El-Ghobarey A, Whaley K. Alternative pathway complement activation in rheumatoid arthritis. J Rheumatol. 1980;7(4):453–460. [PubMed] [Google Scholar]
- Fish AJ, Michael AF, Gewurz H, Good RA. Immunopathologic changes in rheumatoid arthritis synovium. Arthritis Rheum. 1966;9(2):267–280. [Google Scholar]
- Foreman-Wykert AK, Weinrauch Y, Elsbach P, Weiss J. Cell-wall determinants of the bactericidal action of group IIA phospholipase A2 against Gram-positive bacteria. J Clin Invest. 1999;103(5):715–721. doi: 10.1172/JCI5468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fostiropoulos G, Austen KF, Bloch KJ. Total hemolytic complement and second component of complement activity in serum and synovial fluid of patients with rheumatic diseases. Arthritis Rheum. 1964;7:308. doi: 10.1002/art.1780080206. [DOI] [PubMed] [Google Scholar]
- Funakoshi A, Yamada Y, Migita Y, Wakasugi H. Simultaneous determinations of pancreatic phospholipase A2 and prophospholipase A2 in various pancreatic diseases. Dig Dis Sci. 1993;38(3):502–506. doi: 10.1007/BF01316506. [DOI] [PubMed] [Google Scholar]
- Gobezie R, Kho A, Krastins B, Sarracino DA, Thornhill TS, Chase M, Millett PJ, Lee DM. High abundance synovial fluid proteome: distinct profiles in health and osteoarthritis. Arthritis Res Ther. 2007;9(2):R36. doi: 10.1186/ar2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grass DS, Felkner RH, Chiang MY, Wallace RE, Nevalainen TJ, Bennett CF, Swanson ME. Expression of human group II PLA2 in transgenic mice results in epidermal hyperplasia in the absence of inflammatory infiltrate. J Clin Invest. 1996;97(10):2233–2241. doi: 10.1172/JCI118664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green JA, Smith GM, Buchta R, Lee R, Ho KY, Rajkovic IA, Scott KF. Circulating phospholipase A2 activity associated with sepsis and septic shock is indistinguishable from that associated with rheumatoid arthritis. Inflammation. 1991;15(5):355–367. doi: 10.1007/BF00917352. [DOI] [PubMed] [Google Scholar]
- Haworth O, Cernadas M, Yang R, Serhan CN, Levy BD. Resolvin E1 regulates interleukin 23, interferon-gamma and lipoxin A4 to promote the resolution of allergic airway inflammation. Nat Immunol. 2008;9(8):873–879. doi: 10.1038/ni.1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegen MLJ, Shen M, Stedman N, Leach MW, Collins M, Shimizu T, Clark JD, Nicker-Nutter CL. Cytosolic Phospholipase A2α-deficient Mice are Resistant to Arthritis in the K/BxN Serum Transfer Model. The 70th Annual Scientific Meeting of the American College of Rheumatology; Atlanta, GA. Presentation number: 314. [Google Scholar]
- Hegen M, Sun L, Uozumi N, Kume K, Goad ME, Nickerson-Nutter CL, Shimizu T, Clark JD. Cytosolic phospholipase A2alpha-deficient mice are resistant to collagen-induced arthritis. J Exp Med. 2003;197(10):1297–1302. doi: 10.1084/jem.20030016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huggins KW, Boileau AC, Hui DY. Protection against diet-induced obesity and obesity- related insulin resistance in Group 1B PLA2-deficient mice. Am J Physiol Endocrinol Metab. 2002;283(5):E994–E1001. doi: 10.1152/ajpendo.00110.2002. [DOI] [PubMed] [Google Scholar]
- Ji H, Ohmura K, Mahmood U, Lee DM, Hofhuis FM, Boackle SA, Takahashi K, Holers VM, Walport M, Gerard C, Ezekowitz A, Carroll MC, Brenner M, Weissleder R, Verbeek JS, Duchatelle V, Degott C, Benoist C, Mathis D. Arthritis critically dependent on innate immune system players. Immunity. 2002a;16(2):157–168. doi: 10.1016/s1074-7613(02)00275-3. [DOI] [PubMed] [Google Scholar]
- Ji H, Pettit A, Ohmura K, Ortiz-Lopez A, Duchatelle V, Degott C, Gravallese E, Mathis D, Benoist C. Critical roles for interleukin 1 and tumor necrosis factor alpha in antibody-induced arthritis. J Exp Med. 2002b;196(1):77–85. doi: 10.1084/jem.20020439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy BP, Payette P, Mudgett J, Vadas P, Pruzanski W, Kwan M, Tang C, Rancourt DE, Cromlish WA. A natural disruption of the secretory group II phospholipase A2 gene in inbred mouse strains. J Biol Chem. 1995;270(38):22378–22385. doi: 10.1074/jbc.270.38.22378. [DOI] [PubMed] [Google Scholar]
- Kikawada E, Bonventre JV, Arm JP. Group V secretory PLA2 regulates TLR2-dependent eicosanoid generation in mouse mast cells through amplification of ERK and cPLA2alpha activation. Blood. 2007;110(2):561–567. doi: 10.1182/blood-2006-10-052258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim ND, Chou RC, Seung E, Tager AM, Luster AD. A unique requirement for the leukotriene B4 receptor BLT1 for neutrophil recruitment in inflammatory arthritis. J Exp Med. 2006;203(4):829–835. doi: 10.1084/jem.20052349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YJ, Kim KP, Han SK, Munoz NM, Zhu X, Sano H, Leff AR, Cho W. Group V phospholipase A2 induces leukotriene biosynthesis in human neutrophils through the activation of group IVA phospholipase A2. J Biol Chem. 2002;277(39):36479–36488. doi: 10.1074/jbc.M205399200. [DOI] [PubMed] [Google Scholar]
- Kini RM. Excitement ahead: structure, function and mechanism of snake venom phospholipase A2 enzymes. Toxicon. 2003;42(8):827–840. doi: 10.1016/j.toxicon.2003.11.002. [DOI] [PubMed] [Google Scholar]
- Korganow AS, Ji H, Mangialaio S, Duchatelle V, Pelanda R, Martin T, Degott C, Kikutani H, Rajewsky K, Pasquali JL, Benoist C, Mathis D. From systemic T cell self-reactivity to organ-specific autoimmune disease via immunoglobulins. Immunity. 1999;10(4):451–461. doi: 10.1016/s1074-7613(00)80045-x. [DOI] [PubMed] [Google Scholar]
- Kouskoff V, Korganow AS, Duchatelle V, Degott C, Benoist C, Mathis D. Organ-specific disease provoked by systemic autoimmunity. Cell. 1996;87(5):811–822. doi: 10.1016/s0092-8674(00)81989-3. [DOI] [PubMed] [Google Scholar]
- Kudo I, Murakami M. Phospholipase A2 enzymes. Prostaglandins Other Lipid Mediat. 2002;68–69:3–58. doi: 10.1016/s0090-6980(02)00020-5. [DOI] [PubMed] [Google Scholar]
- Lambeau G, Gelb MH. Biochemistry and Physiology of Mammalian Secreted Phospholipases A(2) Annu Rev Biochem. 2008;77:495–520. doi: 10.1146/annurev.biochem.76.062405.154007. [DOI] [PubMed] [Google Scholar]
- Lee DM, Friend DS, Gurish MF, Benoist C, Mathis D, Brenner MB. Mast cells: a cellular link between autoantibodies and inflammatory arthritis. Science. 2002;297(5587):1689–1692. doi: 10.1126/science.1073176. [DOI] [PubMed] [Google Scholar]
- Mancuso P, Peters-Golden M. Modulation of alveolar macrophage phagocytosis by leukotrienes is Fc receptor-mediated and protein kinase C-dependent. Am J Respir Cell Mol Biol. 2000;23(6):727–733. doi: 10.1165/ajrcmb.23.6.4246. [DOI] [PubMed] [Google Scholar]
- Matsumoto I, Maccioni M, Lee DM, Maurice M, Simmons B, Brenner M, Mathis D, Benoist C. How antibodies to a ubiquitous cytoplasmic enzyme may provoke joint-specific autoimmune disease. Nat Immunol. 2002;3(4):360–365. doi: 10.1038/ni772. [DOI] [PubMed] [Google Scholar]
- Matsumoto I, Staub A, Benoist C, Mathis D. Arthritis provoked by linked T and B cell recognition of a glycolytic enzyme. Science. 1999;286(5445):1732–1735. doi: 10.1126/science.286.5445.1732. [DOI] [PubMed] [Google Scholar]
- Menschikowski M, Hagelgans A, Siegert G. Secretory phospholipase A2 of group IIA: is it an offensive or a defensive player during atherosclerosis and other inflammatory diseases? Prostaglandins Other Lipid Mediat. 2006;79(1–2):1–33. doi: 10.1016/j.prostaglandins.2005.10.005. [DOI] [PubMed] [Google Scholar]
- Mounier CM, Wendum D, Greenspan E, Flejou JF, Rosenberg DW, Lambeau G. Distinct expression pattern of the full set of secreted phospholipases A2 in human colorectal adenocarcinomas: sPLA2-III as a biomarker candidate. Br J Cancer. 2008;98(3):587–595. doi: 10.1038/sj.bjc.6604184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz NM, Meliton AY, Arm JP, Bonventre JV, Cho W, Leff AR. Deletion of secretory group V phospholipase A2 attenuates cell migration and airway hyperresponsiveness in immunosensitized mice. J Immunol. 2007;179(7):4800–4807. doi: 10.4049/jimmunol.179.7.4800. [DOI] [PubMed] [Google Scholar]
- Munoz NM, Meliton AY, Meliton LN, Dudek SM, Leff AR. Secretory Group V Phospholipase A2 Regulates Acute Lung Injury and Neutrophilic Inflammation Caused by LPS in Mice. Am J Physiol Lung Cell Mol Physiol. 2009 doi: 10.1152/ajplung.90580.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagiec MJ, Lei B, Parker SK, Vasil ML, Matsumoto M, Ireland RM, Beres SB, Hoe NP, Musser JM. Analysis of a novel prophage-encoded group A Streptococcus extracellular phospholipase A(2) J Biol Chem. 2004;279(44):45909–45918. doi: 10.1074/jbc.M405434200. [DOI] [PubMed] [Google Scholar]
- Nevalainen TJ, Eerola LI, Rintala E, Laine VJ, Lambeau G, Gelb MH. Time-resolved fluoroimmunoassays of the complete set of secreted phospholipases A2 in human serum. Biochim Biophys Acta. 2005;1733(2–3):210–223. doi: 10.1016/j.bbalip.2004.12.012. [DOI] [PubMed] [Google Scholar]
- Nigrovic PA, Lee DM. Immune complexes and innate immunity in rheumatoid arthritis. In: Firestein GS, Panayi GS, Wollheim FA, editors. Rheumatoid Arthritis: new frontiers in pathogenesis and treatment. 2nd edn. Oxford: Oxford University Press; 2006. pp. 135–156. [Google Scholar]
- Nydegger UE, Zubler RH, Gabay R, Joliat G, Karagevrekis CH, Lambert PH, Miescher PA. Circulating complement breakdown products in patients with rheumatoid arthritis. Correlation between plasma C3d, circulating immune complexes, and clinical activity. J Clin Invest. 1977;59(5):862–868. doi: 10.1172/JCI108708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtsuki M, Taketomi Y, Arata S, Masuda S, Ishikawa Y, Ishii T, Takanezawa Y, Aoki J, Arai H, Yamamoto K, Kudo I, Murakami M. Transgenic expression of group V, but not group X, secreted phospholipase A2 in mice leads to neonatal lethality because of lung dysfunction. J Biol Chem. 2006;281(47):36420–36433. doi: 10.1074/jbc.M607975200. [DOI] [PubMed] [Google Scholar]
- Oslund RC, Cermak N, Gelb MH. Highly specific and broadly potent inhibitors of mammalian secreted phospholipases A2. J Med Chem. 2008;51(15):4708–4714. doi: 10.1021/jm800422v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pekin TJ, Jr, Zvaifler NJ. Hemolytic Complement in Synovial Fluid. J Clin Invest. 1964;43:1372–1382. doi: 10.1172/JCI105013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettit AR, Ji H, von Stechow D, Muller R, Goldring SR, Choi Y, Benoist C, Gravallese EM. TRANCE/RANKL knockout mice are protected from bone erosion in a serum transfer model of arthritis. Am J Pathol. 2001;159(5):1689–1699. doi: 10.1016/S0002-9440(10)63016-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piris-Gimenez A, Paya M, Lambeau G, Chignard M, Mock M, Touqui L, Goossens PL. In vivo protective role of human group IIa phospholipase A2 against experimental anthrax. J Immunol. 2005;175(10):6786–6791. doi: 10.4049/jimmunol.175.10.6786. [DOI] [PubMed] [Google Scholar]
- Pruzanski W, Scott K, Smith G, Rajkovic I, Stefanski E, Vadas P. Enzymatic activity and immunoreactivity of extracellular phospholipase A2 in inflammatory synovial fluids. Inflammation. 1992;16(5):451–457. doi: 10.1007/BF00918971. [DOI] [PubMed] [Google Scholar]
- Pruzanski W, Vadas P, Stefanski E, Urowitz MB. Phospholipase A2 activity in sera and synovial fluids in rheumatoid arthritis and osteoarthritis. Its possible role as a proinflammatory enzyme. J Rheumatol. 1985;12(2):211–216. [PubMed] [Google Scholar]
- Rouault M, Le Calvez C, Boilard E, Surrel F, Singer A, Ghomashchi F, Bezzine S, Scarzello S, Bollinger J, Gelb MH, Lambeau G. Recombinant production and properties of binding of the full set of mouse secreted phospholipases A2 to the mouse M-type receptor. Biochemistry. 2007;46(6):1647–1662. doi: 10.1021/bi062119b. [DOI] [PubMed] [Google Scholar]
- Ruddy S, Austen KF. The complement system in rheumatoid synovitis. I. An analysis of complement component activities in rheumatoid synovial fluids. Arthritis Rheum. 1970;13(6):713–723. doi: 10.1002/art.1780130601. [DOI] [PubMed] [Google Scholar]
- Ruddy S, Fearon DT, Austen KF. Depressed synovial fluid levels of properdin and properdin factor B in patients with rheumatoid arthritis. Arthritis Rheum. 1975;18(4):289–295. doi: 10.1002/art.1780180401. [DOI] [PubMed] [Google Scholar]
- Satake Y, Diaz BL, Balestrieri B, Lam BK, Kanaoka Y, Grusby MJ, Arm JP. Role of group V phospholipase A2 in zymosan-induced eicosanoid generation and vascular permeability revealed by targeted gene disruption. J Biol Chem. 2004;279(16):16488–16494. doi: 10.1074/jbc.M313748200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaloske RH, Dennis EA. The phospholipase A2 superfamily and its group numbering system. Biochim Biophys Acta. 2006;1761(11):1246–1259. doi: 10.1016/j.bbalip.2006.07.011. [DOI] [PubMed] [Google Scholar]
- Schur PH, Britton MC, Franco AE, Corson JM, Sosman JL, Ruddy S. Rheumatoid synovitis: complement and immune complexes. Rheumatology. 1975;6:34–42. [PubMed] [Google Scholar]
- Schwab JM, Chiang N, Arita M, Serhan CN. Resolvin E1 and protectin D1 activate inflammation-resolution programmes. Nature. 2007;447(7146):869–874. doi: 10.1038/nature05877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab JM, Serhan CN. Lipoxins and new lipid mediators in the resolution of inflammation. Curr Opin Pharmacol. 2006;6(4):414–420. doi: 10.1016/j.coph.2006.02.006. [DOI] [PubMed] [Google Scholar]
- Seilhamer JJ, Plant S, Pruzanski W, Schilling J, Stefanski E, Vadas P, Johnson LK. Multiple forms of phospholipase A2 in arthritic synovial fluid. J Biochem. 1989a;106(1):38–42. doi: 10.1093/oxfordjournals.jbchem.a122815. [DOI] [PubMed] [Google Scholar]
- Seilhamer JJ, Pruzanski W, Vadas P, Plant S, Miller JA, Kloss J, Johnson LK. Cloning and recombinant expression of phospholipase A2 present in rheumatoid arthritic synovial fluid. J Biol Chem. 1989b;264(10):5335–5338. [PubMed] [Google Scholar]
- Shin K, Nigrovic PA, Crish J, Boilard E, McNeil HP, Larabee KS, Adachi R, Gurish MF, Gobezie R, Stevens RL, Lee DM. Mast cells contribute to autoimmune inflammatory arthritis via their tryptase/heparin complexes. J Immunol. 2009;182(1):647–656. doi: 10.4049/jimmunol.182.1.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer AG, Ghomashchi F, Le Calvez C, Bollinger J, Bezzine S, Rouault M, Sadilek M, Nguyen E, Lazdunski M, Lambeau G, Gelb MH. Interfacial kinetic and binding properties of the complete set of human and mouse groups I, II, V, X, and XII secreted phospholipases A2. J Biol Chem. 2002;277(50):48535–48549. doi: 10.1074/jbc.M205855200. [DOI] [PubMed] [Google Scholar]
- Solomon S, Rajasekaran N, Jeisy-Walder E, Snapper SB, Illges H. A crucial role for macrophages in the pathology of K/B x N serum-induced arthritis. Eur J Immunol. 2005;35(10):3064–3073. doi: 10.1002/eji.200526167. [DOI] [PubMed] [Google Scholar]
- Soragni E, Bolchi A, Balestrini R, Gambaretto C, Percudani R, Bonfante P, Ottonello S. A nutrient-regulated, dual localization phospholipase A(2) in the symbiotic fungus Tuber borchii. EMBO J. 2001;20(18):5079–5090. doi: 10.1093/emboj/20.18.5079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takai T, Li M, Sylvestre D, Clynes R, Ravetch JV. FcR gamma chain deletion results in pleiotrophic effector cell defects. Cell. 1994;76(3):519–529. doi: 10.1016/0092-8674(94)90115-5. [DOI] [PubMed] [Google Scholar]
- Uozumi N, Kume K, Nagase T, Nakatani N, Ishii S, Tashiro F, Komagata Y, Maki K, Ikuta K, Ouchi Y, Miyazaki J, Shimizu T. Role of cytosolic phospholipase A2 in allergic response and parturition. Nature. 1997;390(6660):618–622. doi: 10.1038/37622. [DOI] [PubMed] [Google Scholar]
- Vadas P, Stefanski E, Pruzanski W. Characterization of extracellular phospholipase A2 in rheumatoid synovial fluid. Life Sci. 1985;36(6):579–587. doi: 10.1016/0024-3205(85)90640-x. [DOI] [PubMed] [Google Scholar]
- Valentin E, Lambeau G. Increasing molecular diversity of secreted phospholipases A(2) and their receptors and binding proteins. Biochim Biophys Acta. 2000;1488(1–2):59–70. doi: 10.1016/s1388-1981(00)00110-4. [DOI] [PubMed] [Google Scholar]
- Watts GM, Beurskens FJ, Martin-Padura I, Ballantyne CM, Klickstein LB, Brenner MB, Lee DM. Manifestations of inflammatory arthritis are critically dependent on LFA-1. J Immunol. 2005;174(6):3668–3675. doi: 10.4049/jimmunol.174.6.3668. [DOI] [PubMed] [Google Scholar]
- Webb N. manuscript in preparation. [Google Scholar]
- Winget JM, Pan YH, Bahnson BJ. The interfacial binding surface of phospholipase A2s. Biochim Biophys Acta. 2006;1761(11):1260–1269. doi: 10.1016/j.bbalip.2006.08.002. [DOI] [PubMed] [Google Scholar]
- Wipke BT, Allen PM. Essential role of neutrophils in the initiation and progression of a murine model of rheumatoid arthritis. J Immunol. 2001;167:1601–1608. doi: 10.4049/jimmunol.167.3.1601. [DOI] [PubMed] [Google Scholar]
- Zadori Z, Szelei J, Lacoste MC, Li Y, Gariepy S, Raymond P, Allaire M, Nabi IR, Tijssen P. A viral phospholipase A2 is required for parvovirus infectivity. Dev Cell. 2001;1(2):291–302. doi: 10.1016/s1534-5807(01)00031-4. [DOI] [PubMed] [Google Scholar]
- Zvaifler NJ. Breakdown products of C 3 in human synovial fluids. J Clin Invest. 1969;48(8):1532–1542. doi: 10.1172/JCI106119. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.