

Abstract
Beta-adrenoceptor antagonists are used widely to reduce cardiovascular sympathetic tone, but withdrawal is accompanied by sympathetic hyperactivity. Receptor supersensitivity accounts for some but not all aspects of this withdrawal syndrome. Therefore, we investigated effects of β-blockers on sympathetic innervation. Rats received infusions of adrenergic receptor blockers or saline for one week. The nonselective β-blocker propranolol and the β1-antagonist metoprolol both increased myocardial sympathetic axon density. At 2 days following propranolol discontinuation, β-receptor sensitivity and responsiveness to isoproterenol were similar to controls. However, tyramine-induced mobilization of norepinephrine stores produced elevated ventricular contractility consistent with enhanced sympathetic neuroeffector properties. In addition, rats undergoing discontinuation showed exaggerated increases in mean arterial pressure in response to air puff or noise startle. In sympathetic neuronal cell cultures, both propranolol and metoprolol increased axon outgrowth but the β2-blocker ICI 118,551did not. Norepinephrine synthesis suppression by α-methyl p-tyrosine also increased sprouting and concurrent dobutamine administration reduced it, confirming that locally synthesized norepinephrine inhibits outgrowth via β1 adrenoceptors. Immunohistochemistry revealed β1 adrenoceptor protein on sympathetic axon terminations. In rats with coronary artery ligation, propranolol reversed heart failure-induced ventricular myocardial sympathetic axon depletion, but did not affect infarct-associated sympathetic hyperinnervation. We conclude that sympathetic neurons possess β1 autoreceptors that negatively regulate axon outgrowth. Chronic β-adrenoceptor blockade disrupts this feedback system, leading to ventricular sympathetic axon proliferation and increased neuroeffector gain, which are likely to contribute to β-blocker withdrawal syndrome.
Keywords: Beta-blocker, outgrowth, adrenergic receptor, cardiovascular, autoreceptors, hyperinnervation
Introduction
Beta adrenergic receptor (AR) antagonists are commonly used clinically to treat conditions associated with excessive effects of norepinephrine (NE) and epinephrine including hypertension, congestive heart failure, arrhythmias and risk of sudden cardiac death (Yusuf et al., 1985; Viskin et al., 1995; Rodriguez-Ospina and Montano-Soto, 2008; Germino, 2009). In the heart, β-blockers inhibit catecholamine interactions with β-adrenergic GPCRs, thus preventing Gs protein activation, formation of cAMP, and activation of protein kinase A (Lefkowitz, 2004). As a result, calcium influx and contractile force are decreased (Wang et al., 2004), thus reducing cardiac inotropy, chronotropy and dromotropy, and decreasing arterial blood pressure and myocardial oxygen demand.
Although β-blockers are generally considered safe and effective, cessation of treatment can be accompanied by chest pain, hypertension, arrhythmias, myocardial infarction and increased risk of mortality (Miller et al., 1975; Harrison and Alderman, 1976; Nattel et al., 1979; Hoeks et al., 2007). Adverse effects are attributed to exaggerated sympatho-adrenal responsiveness, generally ascribed to increased β-receptor density and sensitivity resulting from long-term receptor blockade (Aarons et al., 1980; Heilbrunn et al., 1989). While altered βAR responsiveness certainly contributes to increased sympatho-adrenal tone, it does not explain all facets of the withdrawal syndrome (Dollery and Maling, 1979). For example, following β-blocker discontinuation serum NE is paradoxically elevated (Nattel et al., 1979; Vincent et al., 2009), suggesting increased sympathetic transmitter release.
We hypothesized that, similar to some central neurons (Haydon et al., 1984; Whitaker-Azmitia and Azmitia, 1986), peripheral sympathetic neurons possess prejunctional autoreceptors that inhibit axonal outgrowth. If so, then chronic AR blockade could promote axonal growth, thus leading to increased sympathetic innervation density. Since sympathetic hyperinnervation is believed to underlie enhanced end organ responsiveness (Kondo et al., 1996), this could contribute to exaggerated sympathetic activity after β-blocker withdrawal.
Methods
Adrenoceptor blockade
All experiments conformed to the Guide for the Care and Use of Laboratory Animals (NIH publication no. 85-23, 1996) and were approved by the University of Kansas Medical Center Animal Care and Use Committee. Unless otherwise indicated, drugs and reagents were purchased from Sigma Chemical Co. Female Sprague Dawley rats (Harlan Laboratories) aged 60 days were anesthetized by a mixture of ketamine HCl, atropine sulfate and xylazine (Wernli et al., 2009) and minipumps (Durect Corporation)were implanted. Rats received weeklong infusions of saline, propranolol (13.2 mg/kg/day; Conlon et al., 1991), phentolamine (1 mg/kg/day; Steinle and Smith, 2002) or metoprolol (36 mg/kg/day; Ablad et al., 1975; DiBona and Sawin, 1999). Effectiveness of blockade was confirmed 7 days following minipump implantation in rats anesthetized with urethane (1.5 g/kg supplemented as needed). Phentolamine prevented normal αAR-mediated increases in pupil diameter following application of 1% NE to the eye. Propranolol and metoprolol blocked tachycardic responses to iv isoproterenol by 79 and 45%, respectively, assessed electrocardiographically. Metopropolol’s selectivity for β1AR (Ablad et al., 1975; DiBona and Sawin, 1999) was confirmed by showing that isoproterenol-induced β2AR-mediated increases in gracilis muscle blood flow measured by laser Doppler flowmetry (OxyFlow Oxford Optronix Ltd) were unimpaired.
Cardiac sympathetic neuroeffector function after β-blocker discontinuation
Minipumps containing propranolol or saline were removed with ketamine-xylazine-atropine anesthesia and, 48 hours later, a calibrated catheter (SPR 838, Millar Instruments) for measuring pressure-volume relationships was introduced into the left ventricle (Pacher et al., 2008) under urethane anesthesia. After recording resting cardiac hemodynamics, the autonomic ganglionic blocker chlorisondamine (Ciba Pharmaceuticals, 2.5 mg/kg) was administered iv. Tyramine was administered in graded doses until maximum changes were achieved (70μg/kg), and following return to baseline values, this was repeated using isoproterenol (0.35 μg/kg). Neuroeffector function and receptor sensitivity was calculated using LabChart software (AD Instruments) and PVAN Software (Millar Instruments; Pacher et al., 2008).
Cardiovascular reactivity to stress following discontinuation of βAR blockade was assessed in a second group of awake unanesthetized rats. Two days after pump removal, rats were placed in restrainers set on a warming platform and occluding/pressure sensor cuffs positioned at the base of the tail. After an acclimation period of 40 minutes, rats received an air puff directed towards the whiskers. After a recovery period during which blood pressure returned to baseline, rats were subjected to a loud noise (approximately 110db) generated using an air horn. Systolic and diastolic pressures were recorded during control periods and following startle using a CODA rat blood pressure system (Kent Scientific Coroporation), and data are expressed as mean arterial pressures.
Ventricular sympathetic innervation density
After weeklong minipump infusions, hearts were frozen, cryosectioned at 10 μm, and sympathetic innervation was immunostained with an antibody to dopamine-β-hydroxylase (DBH; Immunostar) following methanol fixation (Wernli et al., 2009). In deidentified sections, images of 6 fields evenly distributed within the left lateral posterior area of the ventricle were captured. Nerve density was measured using a stereological grid point counting approach, as described previously (Wernli et al., 2009) and expressed as the percentage of field area occupied by DBH-immunoreactive axons.
Ventricular DBH content was measured in western blots obtained using sections adjacent to those used for morphometric analysis. The left lateral posterior ventricular tissue from 8 sections per heart was lysed in cell extraction buffer (Invitrogen) with protease and phosphatase inhibitors and phenylmethanesulfonylfluoride. Proteins transferred from PAGE gels were probed with DBH antibody (Immunostar) followed by incubation with donkey anti-rabbit antibody conjugated to HRP (Jackson). Band intensity was normalized to that of total protein visualized with india ink and mean intensity was calculated for each rat.
NGF in cardiomyocyte cultures
Hearts harvested from rat pups were disassociated (Worthington Biochemical) and cardiomyocytes plated in DMEM/F12 (Gibco) with fetal bovine serum and cytosine arabinoside. After 3 days, cultures were incubated with medium alone containing phenylephrine (0.1–10 μM) or isoproterenol (0.1–10 μM). Media was collected after 24 h and proteins precipitated and resolved using SDS-PAGE. Transferred proteins were probed in triplicate with an antibody to NGF (M-20, Santa Cruz Biotechnology) followed by incubation in goat anti-rabbit AP-conjugated (Jackson) and detected (Wernli et al., 2009). NGF antibody specificity was confirmed by stripping a positive membrane (Thermo Scientific) and reprobing it using the antibody after it had been incubated overnight with a 10 fold excess by weight of NGF blocking peptide (Santa Cruz Biotechnology). Some membranes were probed using an antibody directed against an epitope in the pro region of proNGF (Chemicon). Sensitivity of the NGF antibody was demonstrated by loading 0.01–1μg human recombinant β-NGF (Sigma) onto membranes subsequently probed with the NGF antibody. Cultured cardiomyocytes were fixed in 4% paraformaldehyde and stained for actin (Alexa Fluor594-Phalloidin), with the NGF antibody or with a furin antibody (Santa Cruz Biotechnology).
Sympathetic neuronal cultures
Superior cervical ganglion (SCG) neurons were obtained from newborn rats, dissociated with collagenase IA and trypsin, suspended in neurobasal medium (Gibco) with glutamine, B27 (Gibco), primocin (Invivogen), FrdU and uridine, and 10 ng/ml NGF (Alomone Labs). Neurons were plated on glass coverslips (Bellco) coated with poly-D-lysine (MP Biomedicals) and laminin (Gibco) in control medium or medium containing propranolol (10−10 M to 10−8 M) or metoprolol (10−8 M to 10−6 M). Other cultures were conducted with 10−10 to 10−6 M dobutamine, with or without 5 μM α-methyl-p-tyrosine (AMPT, Research Biochemicals International) or with AMPT alone.
After 48h, cultures were fixed in 4% parafomaldehyde and immunostained using a peripherin antibody (Chemicon International). Neurite outgrowth was measured blindly in six captured images distributed equally within each well using a stereological grid with intersections at 25 μm intervals superimposed over each image. Intersections overlying stained neurites were divided by total intersections within the field (fraction of field occupied by neurites) and multiplied by total field area (neurite area per field area). This was divided by the total number of neuronal somata within the field (neurite area per neuron), and values normalized to 1 mm2 (Chakrabarty et al., 2008). Cultures were also double stained for peripherin and β1AR (Santa Cruz Biotechnology).
RT-PCR for β1 receptor mRNA
β1AR gene expression was assessed in SCG cultures by RT-PCR. Fresh heart tissue or SCG neurons were homogenized and total RNA was reverse transcribed (Wernli et al., 2009). Amplification of cDNA was conducted using primers for β1AR (5′-CAACGGCGGGACGACCACTG-3′ sense and 5′-ACACCTTGGACTCGGAGGAGAGC-3′ antisense) and GAPDH (5′-CTCTACCCACGGCAAGTTC-3′ sense and 5′-CTCAGCACCAGCATCACC-3′ antisense) and analyzed by electrophoresis.
Myocardial infarction
To assess the effects of βAR blockers in rats with myocardial infarction, the left descending coronary artery was ligated (CAL) (Wernli et al., 2009) or exposed but left unoccluded. Concurrently, rats received pump infusions of AR antagonists or saline for 7 days. In CAL hearts, images were obtained of 6 fields distributed evenly along the left lateral posterior infarct border region where sympathetic hyperinnervation occurs. In addition, images of 6 fields evenly distributed within the non-ischemic posterior area of the left ventricle were also obtained. Procedures and analyses were as described above.
Statistics
Data were analyzed using one way ANOVA for multiple comparisons followed by Newman-Keuls test. Data are expressed as mean ± SEM. Statistical significance was accepted at P<0.05.
Results
AR blockade increases ventricular sympathetic innervation in vivo
Rats received 7d infusions of saline, the non-selective β-blocker propranolol, the selective β1-blocker metoprolol, or the non-selective α-blocker phentolamine. In rats receiving saline, quantitative analysis of DBH-immunoreactive (-ir) sympathetic axons showed that ventricular innervation occurred at a density similar to that reported previously (Wernli et al., 2009) (Fig. 1A). DBH-ir fibers appeared more abundant in rats receiving propranolol (Fig. 1B) and quantitative analysis revealed a 48% increase in ventricular sympathetic axon density (P<0.05; Fig. 1C). Analysis of adjacent tissue by western blot confirmed that DBH protein content was also significantly increased (Fig. 1D). Because propranolol blocks both β1 and β2ARs, we examined whether selective blockade of β1AR alone increases innervation density. Metoprolol was administered using a comparable regimen, and quantitative analysis showed a 44% increase in sympathetic innervation relative to controls (P<0.01; Fig. 1C). To assess whether other AR antagonists with depressor effects also affect ventricular innervation, we administered the α blocker phentolamine. DBH-ir fiber density after phentolamine treatment was similar to that of controls (Fig. 1C), showing that α-blockade and the associated decrease in blood pressure does not modify cardiac sympathetic innervation.
Figure 1.
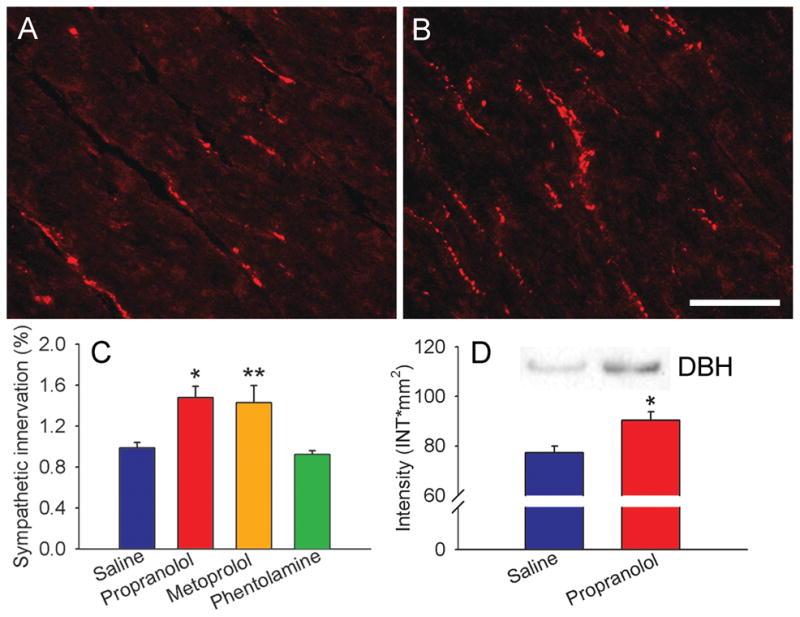
Adrenergic receptor blockade increases ventricular sympathetic innervation. A, Section of ventricular myocardium from a saline-infused rat showing dopamine β-hydroxylase-immunoreactive sympathetic axons. B, Innervation density appears to be increased following a 7 d infusion of propranolol. C, Quantitative analyses of dopamine β-hydroxylase-immunoreactive innervation expressed as percentage of myocardial field area occupied by stained axons. Relative to saline-infused rats (n=12), myocardial innervation density was increased by infusions of propranolol (n=8) and metoprolol (n= 6) but not by phentolamine (n=4). D, ventricular tissue from adjacent sections assessed by western blot shows that propranolol also increases DBH protein levels. Product was analyzed densitometrically and normalized to total protein. Scale bar = 100μm. Results are presented as the mean ± s.e.m. *P<0.05 or **P<0.01 vs saline.
Ventricular sympathetic neuroeffector function and cardiovascular reflexes are enhanced after β-blocker discontinuation
To determine whether sympathetic hyperinnervation is associated with altered ventricular sympathetic neuroeffector properties, we assessed cardiac function 48 h after discontinuation of saline or propranolol infusions. Resting heart rate, ventricular pressures, and rates of ventricular contraction and relaxation were similar between groups (Table 1). Elimination of autonomic tone by the ganglionic blocker chlorisondamine reduced all values, and function was comparable between groups for all parameters except for modest increases in dP/dt max and dP/dTmin in rats receiving propranolol.
Table 1.
Cardiac function in response to isoproterenol and tyramine
| Saline (n=8) | Propranolol (n=8) | ||
|---|---|---|---|
| Resting | HR, bpm | 400 ± 14 | 411 ± 13 |
| ESP, mmHg | 124 ± 12 | 131 ± 10 | |
| EDP, mmHg | 7.7 ± 2.8 | 4.0 ± 0.5 | |
| Peak dP/dtmax, mm Hg/s | 11808 ± 1275 | 14497 ± 825 | |
| Peak dP/dtmin, mm Hg/s | 8229 ± 788 | 9081 ± 709 | |
| Chlorisondamine | HR, bpm | 342 ± 10 | 350 ± 8 |
| ESP, mmHg | 60 ± 3 | 68 ± 4 | |
| EDP, mmHg | 6.9 ± 0.9 | 4.8 ± 0.4 | |
| Peak dP/dtmax, mm Hg/s | 2169 ± 145 | 2705 ± 196* | |
| Peak dP/dtmin, mm Hg/s | 2131 ± 263 | 3335 ± 380* | |
| Isoproterenol | HR, bpm | 461 ± 12 | 479 ± 11 |
| ESP, mmHg | 88 ± 5 | 93 ± 5 | |
| EDP, mmHg | 3.9 ± 0.7 | 2.5 ± 0.4 | |
| Peak dP/dtmax, mm Hg/s | 8476 ± 739 | 9552 ± 297 | |
| Peak dP/dtmin,, mm Hg/s | 4894 ± 476 | 5731 ± 779 | |
| Tyramine | HR, bpm | 434 ± 17 | 458 ± 16 |
| ESP, mmHg | 83 ± 3 | 103 ± 8* | |
| EDP, mmHg | 4.9 ± 0.7 | 2.9 ± 0.5* | |
| Peak dP/dtmax, mm Hg/s | 8081 ± 688 | 10922 ± 581** | |
| Peak dP/dtmin, mm Hg/s | 5427 ± 622 | 6881 ± 696 | |
Responses obtained 48 h after discontinuation of a 7 day infusion of saline or propranolol. Values were obtained in the resting state under urethane anesthesia, following ganglionic blockade with chlorisondamine, and after a maximally effective dose of the direct β agonist isoproterenol, or displacement of sympathetic norepinephrine stores by a maximal dose of tyramine. Results are presented as the mean ± s.e.m.
P<0.05 compared to saline infusion,
P<0.01.
We administered the βAR agonist isoproterenol to assess whether receptor sensitivity was altered at 48 h after propranolol discontinuation, and whether cardiac function in response to direct βAR activation was affected. To assess receptor sensitivity, incremental doses of isoproterenol were administered to achieve maximum values for heart rate and dP/dt max. ED50s were comparable in saline- and propranolol-infused rats (0.07±0.02 vs. 0.07±0.02 mg/kg for heart rate, 0.09±0.02 vs. 0.10±0.02 μg/kg for dP/dt max in saline- or propranolol-infused rats, respectively), confirming prior reports that βAR sensitivity in humans and rats is normal at this time following discontinuation (Nattel et al., 1979; Hedberg et al., 1980). All values for heart rate and left ventricular function in response to maximally effective doses of isoproterenol were comparable in saline- and propranolol-infused rats (Table 1).
To determine whether indices of sympathetic neuroeffector transmission are enhanced in ventricles with increased sympathetic axon density, we administered maximally effective doses of tyramine, which acts by displacing sympathetic NE stores. In saline and propranolol infused rats, maximum heart rate was comparable, but rats after β-blockade showed significantly greater ventricular end systolic pressures (despite decreased end diastolic pressure) and significantly increased rate of contraction (Table 1).
To determine if cardiovascular reflexes that are normally mediated by the sympathetic nervous system are also enhanced after βAR blocker discontinuation in unanesthetized rats, we assessed the effects of two common stressors on blood pressure. A puff of air delivered to the facial region of restrained rats resulted in greater increase in blood pressure after propranolol than after saline discontinuation. Similarly, loud noise, also increased blood pressure to a greater extent after propranolol than saline infusion (Fig. 2).
Figure 2.

Cardiovascular responses to stressors are increased after β-blockade discontinuation. Arterial pressure was measured in awake restrained rats 2 days after discontinuation of propranolol (n=5) or saline (n=4) infusion. Mildly restrained animals were subjected to stress induced by a puff of air to the facial region (Air) or sound of approximately 110 db generated by an air horn (Sound). Peak values are presented as mean arterial pressure (MAP). *P<0.05 vs saline.
Adrenoceptors and cardiomyocyte NGF synthesis
βARs modulate NGF synthesis in some cell types (Colangelo et al., 1998), and altered neurotrophin synthesis is a possible mechanism for the observed increase in ventricular sympathetic axon density. To determine if βARs influence cardiac NGF synthesis, we cultured ventricular cardiomyocytes in the presence or absence of AR agonists and measured NGF protein produced and secreted in vitro. Cardiomyocytes in culture showed spontaneous contractions and mature cytoskeletal features (Fig. 3A). These cells were immunoreactive for NGF protein (Fig. 3B), and the endopeptidase furin (Fig. 3C), which converts pro-NGF to mature NGF, the pro-neuritogenic form of this neurotrophin. To assess NGF secretion by these cells we probed western blots using an antibody to NGF. In characterization studies, this antibody detected 10 ng or less of the 14 kDa mature rhNGF, and higher molecular weight forms of NGF at higher concentrations (Fig. 3D). Medium from cultured myocytes showed a dominant band at approximately 40 kDa, which was eliminated preabsorbing the NGF antibody with an excess of blocking peptide (Fig. 3E). Probing membranes with an antibody directed to the non-NGF portion of proNGF confirmed that this band is proNGF (Fig. 3F). To assess whether NGF-related protein secretion is regulated by ARs, cardiomyocytes were incubated with 10−10 to 10−8 M of the α-agonist phenylephrine or the β-agonist isoproterenol. Isoproterenol treatment did not appear to alter the amount or pattern of secreted protein. Similarly, phenylephrine did not have marked effects on NGF-related proteins, although lower molecular weight forms may have been decreased at higher concentrations (Fig. 3G).
Figure 3.

Adrenergic receptors do not modulate cardiomyocyte NGF secretion. Dissociated cardiomyocytes were cultured for 3 days prior to treatment with phenylephrine or isoproterenol. Cardiomyocytes in culture show typical actin cytoskeleton after staining with phalloidin (A). Cardiomyocytes express NGF protein (B) and the pro-neurotrophin convertase, furin (C). Scale bars = 50μm. Staining of western blots loaded with different amounts of recombinant human β-NGF showed that antibody sensitivity is at least 10 ng/lane (D). Following culture for 48 hours, western blot analysis of the culture medium revealed immunoreactive NGF, mainly as pro-NGF with a molecular mass of about 40 kDa (E left) and preincubation of the antibody with an NGF blocking peptide resulted in absence NGF detection (E right). Probing with an antibody recognizing a non-NGF epitope in the pro region of the protein confirmed that the 40 kDa band is proNGF (F). Treatment of the cardiomyocyte cultures with 0.1–10 μM concentrations of the α-agonist phenylephrine (PE) or the β-agonist isoproterenol (IS) did not alter NGF content within the medium (G).
AR blockade increases sympathetic neuritogenesis in vitro
To determine if βAR blockers act directly on sympathetic neurons to increase axon outgrowth, we used an in vitro assay system consisting of neonatal SCG sympathetic neurons cultured in defined media. After 48 h, SCG neurons in control cultures had elaborated many neurites (Fig. 4A). Neurons cultured with 10−10 M propranolol appeared to have greater neurite outgrowth (Fig. 4B), and quantitative analysis confirmed a significant increase in neurite area per neuron (Fig. 4C). However, cultures containing propranolol at the greater concentration of 10−8 M yielded outgrowth comparable to controls (Fig. 4C).
Figure 4.

β1 adrenoceptors promote sympathetic neurite outgrowth. Superior cervical sympathetic ganglion neurons cultured without treatment extend many neurites (A). Addition of propranolol at a concentration of 10−10 M appeared to increase neurite outgrowth (B) and this was confirmed by quantitative analysis of peripherin-immunoreactive neurite area (C; n=12 per condition). Cultures treated in the presence of 10−8M metoprolol (D) showed increased neurite outgrowth similar to propranolol, and this was confirmed by quantitative analysis (E; n=12). Treatment with the β2 antagonist ICI 118,551 had no effect on neurite outgrowth (F; n=3). Counts of neuronal somas per unit area showed no differences between control cultures and those treated with propranolol (G) or metoprolol (H). Scale bar in D = 50μm for all micrographs. Results are presented as the mean ± s.e.m. *P<0.05 compared to control.
To assess whether the propranolol-induced increase in outgrowth is due to blockade of β1AR, we cultured SCG neurons in the presence of metoprolol. Cultures containing 10−8 M metoprolol showed substantial neurite outgrowth (Fig. 4D) similar to that of propranolol. Quantitative analysis confirmed that this concentration of metoprolol increased neurite outgrowth by 29% (Fig. 4E). Cultures containing a higher concentration of 10−6 M metoprolol again showed outgrowth comparable to controls (Fig. 4E).
Sympathetic axons contain β2ARs that are known to enhance NE release (Deegan et al., 1995). We assessed whether these receptors play a role in modulating sympathetic neurite outgrowth by incubating SCG neurons with the β2AR antagonist ICI 118,551. Sympathetic outgrowth was comparable to control cultures at all concentrations tested (Fig. 4F).
To confirm that outgrowth mediated by β1 blockade is not caused by increased neuronal survival, numbers of somas per mm2 were compared. We observed no differences in neuron numbers with different concentrations of propranolol (Fig. 4G) or metoprolol (Fig. 4H). Similarly, ICI 118,551 did not alter neuron numbers (data not shown).
SCG neurons express β1AR mRNA and protein in culture
To confirm that sympathetic neurons express β1AR mRNA, we performed RT-PCR analysis of SCG neurons after 48h in culture. The sensitivity of our primers was confirmed using ventricular myocardium as a control, which showed a strong band at the predicted product size. RT-PCR of SCG neurons also yielded PCR product of the correct size, albeit of less intensity than that of cardiac muscle (Fig. 5A). The presence of the β1AR protein in somata and neurites was evaluated in cultures stained for β1AR protein (Fig. 5B). Cultures were costained with the neuronal marker peripherin (Fig. 5C) and merged images showed that β1AR protein is distributed widely throughout soma and neurites (Fig. 5D). β1AR immunostaining of cells not known to express this protein was negligible (e.g., glia, inset of Fig. 5B).
Figure 5.
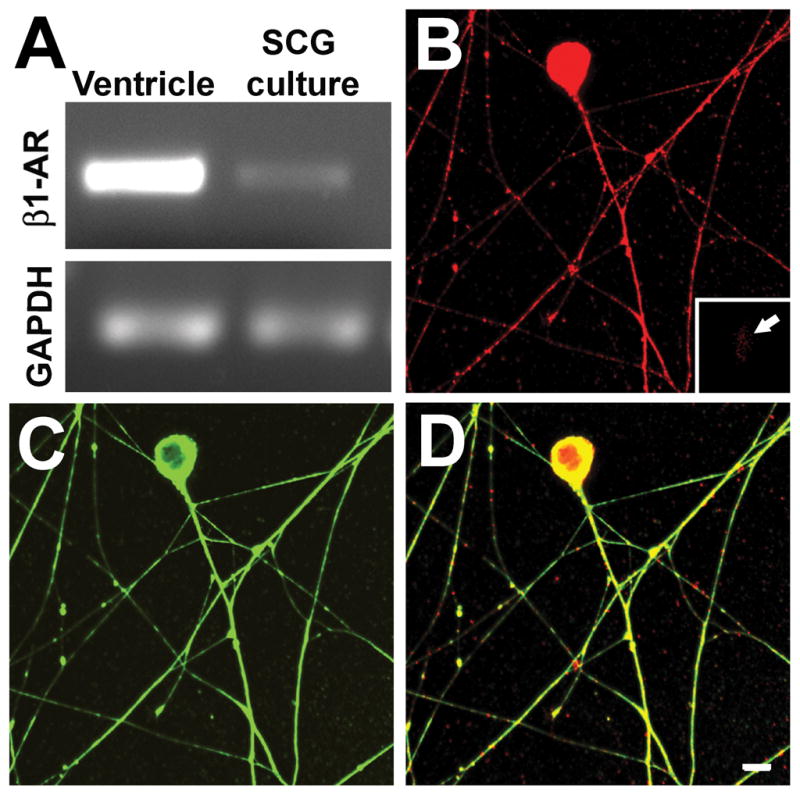
β1 adrenoceptor protein and gene expression in cultured sympathetic neurons. A, mRNA extracted from purified superior cervical ganglion neuronal cultures contains sequences corresponding to the β1 adrenoceptor gene, as revealed by RT-PCR. Ventricular myocardium was used as a positive control and GAPDH was used to show the relative level of receptor expression. B, Immunostaining for β1 adrenoceptor show that this protein is present in sympathetic neurons, as revealed by peripherin staining (C). Inset in B shows a glial cell that is negative for β1AR immunoreactivity. D, A merged image shows that β1 adrenoceptor protein is present throughout the soma and processes including axon terminations. Scale bar = 10μm.
Norepinephrine synthesis inhibits neurite outgrowth
We postulated that propranolol and metoprolol promote sympathetic axon sprouting by blocking β1 receptors that negatively regulate outgrowth. Accordingly, we sought to determine if activating sympathetic axonal β1ARs would suppress outgrowth in culture. We cultured SCG neurons alone (Fig. 6A) or in the presence of the β1-agonist dobutamine (Fig. 6B, a representative concentration of 10−8 M is shown). Dobutamine did not seem to modulate neurite outgrowth (Fig. 6B) and quantitative analysis confirmed that dobutamine was ineffective in decreasing axonal outgrowth over a wide range of concentrations (Fig. 6C). However, because SCG neurons in culture express catecholamine biosynthetic proteins, it is likely that NE is synthesized and released in these cultures. If so, β1-AR activation may be ongoing, thus masking effects of exogenously applied agonist.
Figure 6.

Sympathetic neurite outgrowth in culture is regulated by locally synthesized norepinephrine. In comparison to control cultures (A), dobutamine at a concentration of 10−8M failed to affect outgrowth (B) and this was confirmed by quantitative analysis of neurite area over a wide range of concentrations (C, n=3 per condition). When norepinephrine synthesis was blocked by α-methyl para-tyrosine (AMPT), neurite outgrowth appeared to be enhanced (D). Addition of dobutamine to cultures in which norepinephrine synthesis was blocked led to diminished outgrowth (E). Scale bar = 50μm. F, Quantitative analysis confirmed that inhibition of NE synthesis by AMPT increases neurite area, and that β1 activation by dobutamine is effective in reducing outgrowth only when NE synthesis is blocked. Results are presented as the mean ± s.e.m. *** P<0.001 vs control (n=9) or vs AMPT+ dobutamine (n=6).
To determine if NE synthesized in culture tonically inhibits axonal outgrowth, we blocked NE synthesis with AMPT, a competitive inhibitor of tyrosine hydroxylase. Neurite outgrowth appeared to be greater in cultures containing AMPT (Fig. 6D), and quantitation confirmed a 35% increase relative to controls (Fig. 6F). To confirm that AMPT acted to increase neurite outgrowth by eliminating tonic β1AR activation, we now added dobutamine to these cultures. This β1 agonist suppressed the AMPT-mediated increase in axon outgrowth to a level comparable to that of controls (Fig. 6E and F), confirming that locally synthesized NE inhibits sympathetic axon outgrowth in culture. Soma counts again confirmed that neurite changes are not the result of differential neuronal survival (data not shown).
AR blockade increases ventricular sympathetic innervation after myocardial infarction
An important clinical application of β-blockers is to reduce complications following myocardial infarction (Cao et al., 2000). To determine if this is associated with changes in ventricular sympathetic axon density, we induced myocardial ischemia by ligating the left anterior coronary artery (CAL) (Hasan et al., 2006; Wernli et al., 2009) and infused saline or an AR antagonist for 7 d; treatments did not affect infarct size (data not shown). In intact myocardium adjacent to the infarct, sympathetic innervation density was lower than that seen in uninjured subjects (P<0.01), consistent with prior reports (Kaye et al., 2000). However, propranolol (but not phentolamine) increased sympathetic axon density (41%, P<0.05, Fig. 7), restoring innervation toward that of control animals (compare with normative values in Fig. 1). Consistent with previous reports (Cao et al., 2000; Hasan et al., 2006; Wernli et al., 2009), the infarct border region showed marked sympathetic hyperinnervation in saline-infused rats but this was unaffected by either propranolol or phentolamine treatment (Fig. 7).
Figure 7.

Adrenergic receptor blockade increases ventricular sympathetic innervation after coronary artery ligation. Quantitative analyses of dopamine β-hydroxylase-immunoreactive innervation shows that regions of the ventricular myocardium remote to the infarct (Non-ischemic) have relatively low innervation density in saline-infused rats (S; n=4), and this was increased by propranolol (Pr; n=4) but not by phentolamine (Ph; n=4). Neither propranolol nor phentolamine altered the relatively high innervation density characteristic of the infarct border. Results are presented as the mean ± s.e.m. *P<0.05 vs saline.
Discussion
Our findings provide evidence that β-blockers increase sympathetic target organ innervation, which contributes to exaggerated sympathetic responses after β-blocker withdrawal. Chronic propranolol administration increased numbers of ventricular DBH-ir axons consistent with sprouting of existing sympathetic projections (Hasan et al., 2006; Wernli et al., 2009). Metoprolol was equally effective, indicating that newer selective β antagonists share this property. Therefore, ventricular sympathetic hyperinnervation appears to be a feature common to at least some βAR blockers that enjoy broad clinical use.
There is precedence for proposing that greater ventricular sympathetic axon density may contribute to enhanced target organ response. Increased sympathetic innervation is associated with smooth muscle hypertrophy and hyper-reactivity (Scott and Pang, 1983; Kondo et al., 1996), and our findings indicate that ventricular neuroeffector function is also exaggerated in hearts with β-blocker induced hyperinnervation. Indices of contractile function in response to tyramine were significantly greater in rats after chronic βAR blockade. Because the magnitude of the tyramine response is determined by the amount of NE that can be displaced from intrinsic sympathetic nerves, this increased response is consistent with greater NE stores in hearts with elevated innervation density. Moreover, βAR sensitivity was normal at 48 h after withdrawal, as reported previously in rats and humans (Nattel et al., 1979; Hedberg et al., 1980), and responses to direct βAR activation with isoproterenol were comparable, indicating that intrinsic ventricular properties during maximal βAR activation are largely unchanged. It is noteworthy, however, that ventricular function following complete ganglionic blockade did show modest increases in the rate of contraction and relaxation, although no such differences were evident in the resting state or following isoproterenol. Whether this reflects ventricular remodeling, or possibly altered preload and afterload induced by chronic β-blockade, remains to be determined. Nonetheless, these changes are unlikely to contribute substantially to the enhanced cardiac function observed with tyramine in chronically blocked subjects. Accordingly, the increased tyramine response appears to represent a functional correlate to ventricular sympathetic hyperinnervation. Interestingly, no differences in heart rate were detected in the resting or stimulated state. Because the sympathetic innervation density of the sinoatrial node is substantially greater than that of the ventricle, it may be the case that β blockade was less effective in increasing axon density in that region, as we find to be the case for peri-infarct hyperinnervation.
While the tyramine response is a reliable indicator of potential sympathetic neuroeffector efficacy, it does not provide evidence that sympathetically mediated behavioral responses are also enhanced. To determine if behavioral indices of sympathetic activation are altered, we assessed cardiovascular responsiveness to stressors in awake rats. Rats show startle responses when subjected to loud noise or a puff of air directed at the face. In both cases, at a time when tyramine response was enhanced but β-AR sensitivity was normal, mean arterial pressure was increased to greater levels in rats undergoing β-blocker withdrawal. This indicates that sympathetic nervous system responsiveness is exaggerated in unanesthetized rats, and that rats, like humans, show increased cardiovascular lability after β-blocker withdrawal.
A central question concerns the mechanism by which β-blockers increase ventricular sympathetic axon density. In some cell types, βARs regulate synthesis of NGF, a powerful mediator of sympathetic sprouting (Dal Toso et al., 1987) that could initiate axon outgrowth. However, consistent with other reports (Kaye et al., 2000), our culture studies show that β-ARs do not appear to play a major role in regulating NGF synthesis and release by cardiac myocytes. While treatment with an α-agonist has been reported to suppress cardiac NGF synthesis (Kaye et al., 2000), it did not significantly modify NGF expression in our hands, and our finding that ventricular sympathetic density was unchanged by phentolamine argues against substantial modulation of innervation by αARs. In any event, an increase in NGF synthesis by βAR blockers is unlikely to contribute to sympathetic hyperinnervation.
An alternative mechanism is a direct effect of β antagonists on the sympathetic neuron itself. We demonstrate that β1AR mRNA and protein are present in sympathetic neuronal cell bodies and axons, which extends earlier pharmacological studies inferring the presence of β1 receptors on cardiac sympathetic neurons (Butler et al., 1990; Watson-Wright et al., 1991). Hence, synthesis and distribution of β1 ARs in sympathetic neurons are consistent with their potential role as an autoreceptor.
Our culture studies provide evidence that β-blockers induce sprouting by interfering with activation of the sympathetic β1 autoreceptor. Thus, the non-selective β antagonist propranolol and the selective β1AR antagonist metoprolol both increased neurite outgrowth significantly in cultured sympathetic neurons, indicating direct growth-promoting effects on sympathetic neurons by β1AR inhibition. Support for the selectivity of this effect comes from the finding that blockade of β2 receptors by ICI118,551 is ineffective in altering outgrowth. Interestingly, neuritogenic effects of both propranolol and metoprolol occurred only at the lower concentrations, which most closely approximate estimates of in vivo therapeutic concentrations of these drugs (Abrahamsson et al., 1990; Takahashi et al., 1993), and were lost when the concentration is increased 100-fold. This may not be surprising, as both agents at high concentrations have membrane stabilizing properties (Brunton et al., 2005), and membrane stabilization is known to inhibit axon outgrowth (Ibarretxe et al., 2007).
Findings thus far led us to hypothesize that sympathetic neurons possess β1AR that negatively regulate axon outgrowth. Accordingly, we attempted to demonstrate the activity of these receptors by adding the β1 agonist, dobutamine, to our cultures. However, despite using a wide range of concentrations, dobutamine had no effect on neurite outgrowth. Since sympathetic neurons display features that suggest that they continue to synthesize NE in vitro (Landis, 1978), we postulated that cultured neurons may be releasing NE in quantities sufficient to maximally activate β1ARs, such that additional ligand is ineffective. To test this hypothesis, we used the tyrosine hydroxylase inhibitor AMPT to prevent catecholamine biosynthesis. In cultures where NE synthesis was inhibited, neurite outgrowth was increased to an extent similar to that seen with βAR blockade, indicating that NE synthesized in culture does indeed act to inhibit outgrowth. Now when dobutamine was added, outgrowth was reduced to that of control cultures with intact NE synthesis. Collectively, these studies indicate that, under normal culture conditions, NE tonically inhibits sympathetic neurite outgrowth via β1ARs. Consistent with this hypothesis, addition of dobutamine to cultures where NE synthesis is blocked suppressed outgrowth to levels typical of cultures in which NE is normally synthesized.
Aside from a role in modulating NE release, presynaptic β1ARs have not been implicated in influencing axonal outgrowth. However, there are several reports in other neural systems where transmitters do regulate axonal extension. For example, both dopamine and serotonin suppress elongation of axons in the Helisoma snail (Haydon et al., 1984; McCobb et al., 1988). At least some transmitter-mediated inhibition of axon growth appears to occur via autoreceptors, as serotonin inhibits outgrowth of serotoninergic axons from rat raphe neurons (Whitaker-Azmitia and Azmitia, 1986), and glutamate at high doses reduces axonal outgrowth by immobilizing growth cones of glutaminergic pyramidal neurons (Mattson et al., 1988). Thus, precedents exist for suggesting that sympathetic neurotransmitter autoreceptors could play a critical role in regulating axon outgrowth.
While enhanced sympathetic outgrowth is likely to contribute to β-blocker withdrawal syndrome, it may be relevant to other pathophysiological situations as well. Beta-blockers are commonly administered to patients with myocardial infarction owing to their ability to reduce cardiac excitability, myocardial oxygen consumption, plasma angiotensin II (Ichihara et al., 1995), and sudden death (Hunt, 2005). However, heart failure is accompanied by diminished sympathetic innervation in non-necrotic regions of the ventricle (Himura et al., 1993; Li et al., 2004), which could contribute to cardiac dysfunction. It is interesting to speculate that in heart failure patients, β-blocker therapy could prove beneficial not only by directly altering cardiac properties, but also by restoring cardiac sympathetic innervation. Our findings of normalization of axon numbers following propranolol infusion in the intact myocardium in this rat model of myocardial infarction suggest that this may be the case. Consistent with the idea that sympathetic axon density may be restored in the failing ventricle, l123I-metaiodobenzylguanidine (MIBG) scintigraphic studies in patients receiving β-blockers show increased ventricular catecholamine reuptake (Merlet et al., 1999; Toyama et al., 2003; Cohen-Solal et al., 2005). It is also of interest that, in our model, hyperinnervation induced by infarction was unaffected by propranolol administration. There is abundant evidence that myocardial ischemic injury is associated with proliferation of sympathetic axons in the vicinity of the infarct (Cao et al., 2000; Hasan et al., 2006; Wernli et al., 2009). Somewhat surprisingly, this hyperinnervation was not increased by β-blocker administration, a finding that may suggest an upper limit beyond which additional increases in innervation density are not apparent. On the other hand, it has been reported in rabbits that sympathetic hyperinnervation in the infarct border region is reduced by metoprolol (Jiang et al., 2007). Border region sprouting is likely to involve complex interactions among target-derived trophic factors, inflammatory cells and damaged and regenerating axons, and response variability may not be surprising. Further studies are necessary to determine whether β-blockers alter post-infarct sympathetic innervation patterns in humans.
In summary, we propose that sympathetic neurons possess presynaptic β1-ARs that, when activated by physiological levels of NE or other agonists, inhibit axon extension. We further hypothesize that this is an important mechanism for establishing levels of target innervation density. According to this concept, when innervation density is low, levels of NE will also be low and sympathetic axons will be encouraged to proliferate. This will continue until local levels of NE are sufficient to activate presynaptic β1-ARs, at which point further axon extension would be prevented. Such a feedback system could provide a sensitive mechanism for establishing final levels of target innervation where the set point for innervation density is determined by the local concentration of transmitter. In the presence of a drug that blocks β1ARs, outgrowth inhibition is lost and target organ innervation density is therefore increased.
Acknowledgments
We thank Timothy Donohue, Dora Krizsan-Agbas, Zhaohui Liao, Elza Kharatyan, Michelle Winter and Kenneth McCarson for their assistance. This work was supported by NIH HL079652 with core support from HD002528.
Footnotes
Conflict of Interest: none declared.
References
- Aarons RD, Nies AS, Gal J, Hegstrand LR, Molinoff PB. Elevation of beta-adrenergic receptor density in human lymphocytes after propranolol administration. J Clin Invest. 1980;65:949–957. doi: 10.1172/JCI109781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ablad B, Borg KO, Carlsson E, Ek L, Johnson G, Malmfors T, Regardh CG. A survey of the pharmacological properties of metoprolol in animals and man. Acta Pharmacol Toxicol (Copenh) 1975;36:7–23. doi: 10.1111/j.1600-0773.1975.tb03318.x. [DOI] [PubMed] [Google Scholar]
- Abrahamsson B, Lucker P, Olofsson B, Regardh CG, Sandberg A, Wieselgren I, Bergstrand R. The relationship between metoprolol plasma concentration and beta 1-blockade in healthy subjects: a study on conventional metoprolol and metoprolol CR/ZOK formulations. J Clin Pharmacol. 1990;30:S46–54. doi: 10.1002/j.1552-4604.1990.tb03495.x. [DOI] [PubMed] [Google Scholar]
- Brunton L, Lazo J, Parker K. Goodman and Gilman’s The Pharmacological Basis of Therapeutics. McGraw-Hill; 2005. [Google Scholar]
- Butler CK, Smith FM, Nicholson J, Armour JA. Cardiac effects induced by chemically activated neurons in canine intrathoracic ganglia. Am J Physiol. 1990;259:H1108–1117. doi: 10.1152/ajpheart.1990.259.4.H1108. [DOI] [PubMed] [Google Scholar]
- Cao JM, Fishbein MC, Han JB, Lai WW, Lai AC, Wu TJ, Czer L, Wolf PL, Denton TA, Shintaku IP, Chen PS, Chen LS. Relationship between regional cardiac hyperinnervation and ventricular arrhythmia. Circulation. 2000;101:1960–1969. doi: 10.1161/01.cir.101.16.1960. [DOI] [PubMed] [Google Scholar]
- Chakrabarty A, Blacklock A, Svojanovsky S, Smith PG. Estrogen elicits dorsal root ganglion axon sprouting via a renin-angiotensin system. Endocrinology. 2008;149:3452–3460. doi: 10.1210/en.2008-0061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen-Solal A, Rouzet F, Berdeaux A, Le Guludec D, Abergel E, Syrota A, Merlet P. Effects of carvedilol on myocardial sympathetic innervation in patients with chronic heart failure. J Nucl Med. 2005;46:1796–1803. [PubMed] [Google Scholar]
- Colangelo AM, Follesa P, Mocchetti I. Differential induction of nerve growth factor and basic fibroblast growth factor mRNA in neonatal and aged rat brain. Brain Res Mol Brain Res. 1998;53:218–225. doi: 10.1016/s0169-328x(97)00296-9. [DOI] [PubMed] [Google Scholar]
- Conlon D, Johnston A, Turner P, O’Malley K, Kilfeather S. Hepatic beta-adrenoceptor adaptation during propranolol administration is impaired in aging rats. Eur J Pharmacol. 1991;208:323–330. doi: 10.1016/0922-4106(91)90078-v. [DOI] [PubMed] [Google Scholar]
- Dal Toso R, De Bernardi MA, Costa E, Mocchetti I. Beta-adrenergic receptor regulation of NGF-mRNA content in rat C6-2B glioma cells. Neuropharmacology. 1987;26:1783–1786. doi: 10.1016/0028-3908(87)90133-x. [DOI] [PubMed] [Google Scholar]
- Deegan R, He HB, Krivoruk Y, Wood AJ, Wood M. Regulation of norepinephrine release by beta 2-adrenergic receptors during halothane anesthesia. Anesthesiology. 1995;82:1417–1425. doi: 10.1097/00000542-199506000-00013. [DOI] [PubMed] [Google Scholar]
- DiBona GF, Sawin LL. Effect of metoprolol administration on renal sodium handling in experimental congestive heart failure. Circulation. 1999;100:82–86. doi: 10.1161/01.cir.100.1.82. [DOI] [PubMed] [Google Scholar]
- Dollery CT, Maling TJ. Beta-blocker withdrawal syndrome. Br Med J. 1979;2:1074–1075. doi: 10.1136/bmj.2.6197.1074-c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germino FW. The management and treatment of hypertension. Clin Cornerstone. 2009;9(Suppl 3):S27–33. doi: 10.1016/s1098-3597(09)60016-8. [DOI] [PubMed] [Google Scholar]
- Harrison DC, Alderman EL. Editorial: Discontinuation of propranolol therapy. Cause of rebound angina pectoris and acute coronary events. Chest. 1976;69:1–2. doi: 10.1378/chest.69.1.1. [DOI] [PubMed] [Google Scholar]
- Hasan W, Jama A, Donohue T, Wernli G, Onyszchuk G, Al-Hafez B, Bilgen M, Smith PG. Sympathetic hyperinnervation and inflammatory cell NGF synthesis following myocardial infarction in rats. Brain Res. 2006;1124:142–154. doi: 10.1016/j.brainres.2006.09.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haydon PG, McCobb DP, Kater SB. Serotonin selectively inhibits growth cone motility and synaptogenesis of specific identified neurons. Science. 1984;226:561–564. doi: 10.1126/science.6093252. [DOI] [PubMed] [Google Scholar]
- Hedberg A, Isaksson O, Lundgren B. Sustained cardiac beta adrenoceptor blockade in vitro and increased vulnerability to aconitine-induced arrhythmias in vivo after propranolol withdrawal in rats. J Pharmacol Exp Ther. 1980;214:664–669. [PubMed] [Google Scholar]
- Heilbrunn SM, Shah P, Bristow MR, Valantine HA, Ginsburg R, Fowler MB. Increased beta-receptor density and improved hemodynamic response to catecholamine stimulation during long-term metoprolol therapy in heart failure from dilated cardiomyopathy. Circulation. 1989;79:483–490. doi: 10.1161/01.cir.79.3.483. [DOI] [PubMed] [Google Scholar]
- Himura Y, Felten SY, Kashiki M, Lewandowski TJ, Delehanty JM, Liang CS. Cardiac noradrenergic nerve terminal abnormalities in dogs with experimental congestive heart failure. Circulation. 1993;88:1299–1309. doi: 10.1161/01.cir.88.3.1299. [DOI] [PubMed] [Google Scholar]
- Hoeks SE, Scholte Op Reimer WJ, van Urk H, Jorning PJ, Boersma E, Simoons ML, Bax JJ, Poldermans D. Increase of 1-year mortality after perioperative beta-blocker withdrawal in endovascular and vascular surgery patients. Eur J Vasc Endovasc Surg. 2007;33:13–19. doi: 10.1016/j.ejvs.2006.06.019. [DOI] [PubMed] [Google Scholar]
- Hunt SA. ACC/AHA 2005 guideline update for the diagnosis and management of chronic heart failure in the adult: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Update the 2001 Guidelines for the Evaluation and Management of Heart Failure) J Am Coll Cardiol. 2005;46:e1–82. doi: 10.1016/j.jacc.2005.08.022. [DOI] [PubMed] [Google Scholar]
- Ibarretxe G, Perrais D, Jaskolski F, Vimeney A, Mulle C. Fast regulation of axonal growth cone motility by electrical activity. J Neurosci. 2007;27:7684–7695. doi: 10.1523/JNEUROSCI.1070-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichihara A, Suzuki H, Murakami M, Naitoh M, Matsumoto A, Saruta T. Interactions between angiotensin II and norepinephrine on renin release by juxtaglomerular cells. Eur J Endocrinol. 1995;133:569–577. doi: 10.1530/eje.0.1330569. [DOI] [PubMed] [Google Scholar]
- Jiang H, Lu Z, Yu Y, Zhao D, Jian X, Yang B, Huang C. Effects of metoprolol on sympathetic remodeling and electrical remodeling at infarcted border zone after myocardial infarction in rabbits. Cardiology. 2007;108:176–182. doi: 10.1159/000096647. [DOI] [PubMed] [Google Scholar]
- Kaye DM, Vaddadi G, Gruskin SL, Du XJ, Esler MD. Reduced myocardial nerve growth factor expression in human and experimental heart failure. Circ Res. 2000;86:e80–e84. doi: 10.1161/01.res.86.7.e80. [DOI] [PubMed] [Google Scholar]
- Kondo M, Fujiwara T, Tabei R. Noradrenergic hyperinnervation in the heart of stroke-prone spontaneously hypertensive rats (SHRSP) Hypertens Res. 1996;19:69–73. doi: 10.1291/hypres.19.69. [DOI] [PubMed] [Google Scholar]
- Landis SC. Growth cones of cultured sympathetic neurons contain adrenergic vesicles. J Cell Biol. 1978;78:R8–14. doi: 10.1083/jcb.78.1.r8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefkowitz RJ. Historical review: a brief history and personal retrospective of seven-transmembrane receptors. Trends Pharmacol Sci. 2004;25:413–422. doi: 10.1016/j.tips.2004.06.006. [DOI] [PubMed] [Google Scholar]
- Li W, Knowlton D, Van Winkle DM, Habecker BA. Infarction alters both the distribution and noradrenergic properties of cardiac sympathetic neurons. Am J Physiol Heart Circ Physiol. 2004;286:H2229–2236. doi: 10.1152/ajpheart.00768.2003. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Dou P, Kater SB. Outgrowth-regulating actions of glutamate in isolated hippocampal pyramidal neurons. J Neurosci. 1988;8:2087–2100. doi: 10.1523/JNEUROSCI.08-06-02087.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCobb DP, Haydon PG, Kater SB. Dopamine and serotonin inhibition of neurite elongation of different identified neurons. J Neurosci Res. 1988;19:19–26. doi: 10.1002/jnr.490190104. [DOI] [PubMed] [Google Scholar]
- Merlet P, Pouillart F, Dubois-Rande JL, Delahaye N, Fumey R, Castaigne A, Syrota A. Sympathetic nerve alterations assessed with 123I-MIBG in the failing human heart. J Nucl Med. 1999;40:224–231. [PubMed] [Google Scholar]
- Miller RR, Olson HG, Amsterdam EA, Mason DT. Propranolol-withdrawal rebound phenomenon. Exacerbation of coronary events after abrupt cessation of antianginal therapy. N Engl J Med. 1975;293:416–418. doi: 10.1056/NEJM197508282930902. [DOI] [PubMed] [Google Scholar]
- Nattel S, Rangno RE, Van Loon G. Mechanism of propranolol withdrawal phenomena. Circulation. 1979;59:1158–1164. doi: 10.1161/01.cir.59.6.1158. [DOI] [PubMed] [Google Scholar]
- Pacher P, Nagayama T, Mukhopadhyay P, Batkai S, Kass DA. Measurement of cardiac function using pressure-volume conductance catheter technique in mice and rats. Nat Protoc. 2008;3:1422–1434. doi: 10.1038/nprot.2008.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Ospina L, Montano-Soto L. Management of chronic stable angina pectoris. Bol Asoc Med P R. 2008;100:39–47. [PubMed] [Google Scholar]
- Scott TM, Pang SC. The correlation between the development of sympathetic innervation and the development of medial hypertrophy in jejunal arteries in normotensive and spontaneously hypertensive rats. J Auton Nerv Syst. 1983;8:25–32. doi: 10.1016/0165-1838(83)90020-6. [DOI] [PubMed] [Google Scholar]
- Steinle JJ, Smith PG. Role of adrenergic receptors in vascular remodelling of the rat choroid. Br J Pharmacol. 2002;136:730–734. doi: 10.1038/sj.bjp.0704771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi H, Ogata H, Kashiwada K, Ohira M, Someya K. Dosing rate-dependent relationship between propranolol plasma concentration and beta-blockade. J Pharmacol Exp Ther. 1993;265:681–689. [PubMed] [Google Scholar]
- Toyama T, Hoshizaki H, Seki R, Isobe N, Adachi H, Naito S, Oshima S, Taniguchi K. Efficacy of carvedilol treatment on cardiac function and cardiac sympathetic nerve activity in patients with dilated cardiomyopathy: comparison with metoprolol therapy. J Nucl Med. 2003;44:1604–1611. [PubMed] [Google Scholar]
- Vincent GM, Schwartz PJ, Denjoy I, Swan H, Bithell C, Spazzolini C, Crotti L, Piippo K, Lupoglazoff JM, Villain E, Priori SG, Napolitano C, Zhang L. High efficacy of beta-blockers in long-QT syndrome type 1: contribution of noncompliance and QT-prolonging drugs to the occurrence of beta-blocker treatment “failures”. Circulation. 2009;119:215–221. doi: 10.1161/CIRCULATIONAHA.108.772533. [DOI] [PubMed] [Google Scholar]
- Viskin S, Kitzis I, Lev E, Zak Z, Heller K, Villa Y, Zajarias A, Laniado S, Belhassen B. Treatment with beta-adrenergic blocking agents after myocardial infarction: from randomized trials to clinical practice. J Am Coll Cardiol. 1995;25:1327–1332. doi: 10.1016/0735-1097(94)00552-2. [DOI] [PubMed] [Google Scholar]
- Wang W, Zhu W, Wang S, Yang D, Crow MT, Xiao RP, Cheng H. Sustained beta1-adrenergic stimulation modulates cardiac contractility by Ca2+/calmodulin kinase signaling pathway. Circ Res. 2004;95:798–806. doi: 10.1161/01.RES.0000145361.50017.aa. [DOI] [PubMed] [Google Scholar]
- Watson-Wright W, Boudreau G, Cardinal R, Armour JA. Beta 1- and beta 2-adrenoceptor subtypes in canine intrathoracic efferent sympathetic nervous system regulating the heart. Am J Physiol. 1991;261:R1269–1275. doi: 10.1152/ajpregu.1991.261.5.R1269. [DOI] [PubMed] [Google Scholar]
- Wernli G, Hasan W, Bhattacherjee A, van Rooijen N, Smith PG. Macrophage depletion suppresses sympathetic hyperinnervation following myocardial infarction. Basic Res Cardiol. 2009;104:681–693. doi: 10.1007/s00395-009-0033-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitaker-Azmitia PM, Azmitia EC. Autoregulation of fetal serotonergic neuronal development: role of high affinity serotonin receptors. Neurosci Lett. 1986;67:307–312. doi: 10.1016/0304-3940(86)90327-7. [DOI] [PubMed] [Google Scholar]
- Yusuf S, Peto R, Lewis J, Collins R, Sleight P. Beta blockade during and after myocardial infarction: an overview of the randomized trials. Prog Cardiovasc Dis. 1985;27:335–371. doi: 10.1016/s0033-0620(85)80003-7. [DOI] [PubMed] [Google Scholar]