

Abstract
Refractive index dispersion is an intrinsic optical property and a useful source of contrast in biological imaging studies. In this report, we present the first dispersion phase imaging of living eukaryotic cells. We have developed quantitative dispersion microscopy based on the principle of quantitative phase microscopy. The dual-wavelength quantitative phase microscope makes phase measurements at 310 nm and 400 nm wavelengths to quantify dispersion (refractive index increment ratio) of live cells. The measured dispersion of living HeLa cells is found to be around 1.088, which agrees well with that measured directly for protein solutions using total internal reflection. This technique, together with the dry mass and morphology measurements provided by quantitative phase microscopy, could prove to be a useful tool for distinguishing different types of biomaterials and studying spatial inhomogeneities of biological samples.
1. Introduction
Refractive indices of biomolecules are fundamental properties that play key roles in a number of optical imaging and microscopy techniques, which include optical coherence tomography, confocal reflectance microscopy, light scattering spectroscopy, and quantitative phase microscopy [1]. The refractive index variation within cells and tissue provides the source of contrast for these different optical imaging modalities, which can be further related to the structural or morphological features of the sample. The refractive index of a material is wavelength dependent, which is known as the dispersion property of the material. The importance of dispersion is widely recognized in multiphoton microscopy and optical coherence tomography because of the large bandwidth of the light source used [2, 3]. However, most often dispersion is viewed as a deleterious effect that requires compensation, either by optics or numerical post-processing. Nonetheless, there have been a few interesting studies utilizing the refractive index dispersion of biomolecules to quantify molecular concentration. For example, the dispersion of hemoglobin was used to extract the concentration of hemoglobin in intact red blood cell [4]. In another study, the dispersion of an exogenous dye was used to decouple refractive index measurement from height measurement of cells in digital holographic microscopy [5]. However, most biomolecules do not have significant dispersion in the visible wavelength region due to their inherently low absorption [6]. It would be of great interest to directly measure the dispersion of biomaterials, including cells, to study their biochemical compositions.
The majority components of a eukaryotic cell are proteins, nucleic acids, lipids, and polysaccharides. Both proteins and nucleic acids have strong absorption in the middle UV region. This characteristic has previously been successfully used to map the quantities of proteins and nucleic acids in living cells [7]. However, imaging directly at the absorption peak poses significant challenges because physical and chemical damage of the cell is unavoidable [8], thus long term observation of the cell is extremely difficult. This constraint also places stringent requirements on the UV transmission of the system and the detection sensitivity of the charge coupled device (CCD). Moreover, the scattering contribution of cellular components could interfere with the absorption of biomolecules and lead to erroneous analysis. According to the Kramers-Kronig relationship, the spectrum of refractive index is much broader than that of absorption. Therefore, it is possible to image refractive index dispersion of biological cells in the near UV (NUV, 300–400nm) while minimizing the damage caused by strong protein and nucleic acid absorptions. With that objective in mind, we designed a dual-wavelength quantitative phase microscope to study the refractive index dispersion of live cells.
Previously we have shown that proteins exhibit substantial dispersion near their absorption peak at 280 nm [9]. Here we report the dispersion imaging of living eukaryotic cells, for the first time to our knowledge, in the NUV range using dual wavelength quantitative phase microscopy. The dispersion parameter is typically defined as dn/dλ. However, because we only measure at two wavelengths, for simplicity we used an alternative parameter to characterize dispersion: α2/α1, where α2 and α1 are the refractive index increment of biomolecules (will be discussed in details in the experimental results section) at the two measured wavelengths. This definition will be used throughout the whole manuscript. The dispersion of protein and DNA molecules is also calibrated independently using a total internal reflection (TIR) method [10]. We show that the dispersion of live HeLa cells agrees well with that measured for pure proteins solutions using the TIR method.
2. Experimental setup
The design of the NUV dispersion phase microscope is described in Fig. 1. The principle of measurement is based on diffraction phase microscopy [11]. In order to measure the dispersion, two wavelengths are employed: one at 310 nm and the other at 400nm. The laser source is a frequency doubled tunable optical parametric amplifier (Coherent OPA 9400) pumped by a femtosecond amplified Ti:sapphire laser system (Coherent RegA). We use both the signal output at 310 nm and the frequency doubled 400 nm output. The two beams are combined using a dichroic mirror and sent into a custom-built dual-arm diffraction phase microscope. The UV laser beam is loosely focused onto the sample by a 50 cm lens with spot size around 50 μm. Optical power used for imaging is about 500 μW at 400 nm and 100 μW at 310 nm. The sample is imaged by a 40× NUV objective (Thorlabs, NA = 0.6) and a 200 cm focal length tube lens. After the microscope objective, the beams are separated into two nearly identical imaging arms to separately image the sample at the two wavelengths on the same CCD (JAI Pulnix TM1402-UV). In both imaging arms, a diffraction grating and a custom spatial filter are employed to generate the sample and reference waves that form an interferogram on the CCD, which is then digitally processed to obtain the amplitude and phase image of the sample. The details of implementing the diffraction phase microscope can be found elsewhere [9, 11]. The total magnification of the system is 55.61 at 400 nm and 53.75 at 310 nm. The theoretical lateral resolution of system is 0.81 μm and 0.63 μm for 400 nm and 310 nm illuminations, respectively. To control the exposure time, two separate shutters are placed in the beam path before combing the two wavelengths on the dichroic mirror.
Figure 1.

Schematic diagram of dual wavelength NUV quantitative phase microscope. RegA: Ti: sapphire regenerative amplifier; OPA: optical parametric amplifier; OBJ: objective; G: Ronchi grating; SPF: spatial filter (see the insert).
3. Experimental results
Most proteins have about the same specific refractive index increment, α (the physical parameter that directly relates refractive index, n, to protein concentration, C: n = n0+αC, where n0 is the refractive index of water). For proteins, α is around 0.18–0.19 mL/g in the visible wavelength range. Nucleic acids have similar α at around 0.17–0.18 mL/g [12]. This small difference makes the interpretation of molecular compositions in the cell a very difficult task. We have previously shown that proteins exhibit large dispersion when the wavelength is close to its absorption peak at 280 nm. Although dispersion for nucleic acids was not calibrated, we expect that it will also be large at close to 260 nm. In this study, we chose 310 nm as one of our imaging wavelengths for three reasons: 310 nm is reasonably close to the peak of dispersion but causes much less damage to the cell, due to more than one order of magnitude lower absorption by both proteins and nucleic acids; the CCD camera has relatively good sensitivity at 310 nm; 310 nm has modest transmission in BK7 glass so that samples can be simply prepared on regular microscope coverslips. The other wavelength is chosen at 400 nm, which is readily available from the OPA output and well within the range of objective lens chromatic aberration correction.
We first calibrated the specific refractive index increment of proteins and nucleic acids using a TIR method [10], which has higher sensitivity compared to our previous light-scattering based method. Without loss of generality, we used bovine serum albumin (Sigma-Aldrich) and deoxyribonucleic acid sodium salt from salmon testes (Sigma-Aldrich) as our protein and nucleic acid samples, respectively. Fig. 2 shows refractive index measurements of protein and DNA solutions versus concentrations at the two wavelengths. Through linear regression of refractive index against concentration, the specific refractive index increment α for protein at 310 nm is determined to be 0.208±0.001 mL/g, which is slightly lower than that measured using the light scattering method. At 400 nm, α for protein is determined to be 0.191±0.001 mL/g. For DNA, α at 310 nm and 400 nm are 0.218±0.007 mL/g and 0.186±0.014 mL/g, respectively. The fitting error for the DNA measurements are an order of magnitude larger, due to the fact that the maximum concentration we could obtain is <3% and the refractive index change is much smaller. Nonetheless, DNA exhibits larger dispersion relative to proteins at these two wavelengths. We expect RNA will show similar dispersion behavior because its absorption is similar to that of DNA.
Figure 2.

Refractive index measured at two wavelengths for (a) protein solutions and (b) DNA solutions versus concentrations and their corresponding linear fits to obtain specific refractive index increments, α.
Next, we studied the dispersion of live HeLa cells. To prepare the sample, HeLa cells were plated onto a coverslip that is fixed onto a homemade aluminum chamber and cultured in Dulbecco's modified eagle medium (DMEM) containing 10% Fetal bovine serum (FBS), penicillin and streptomycin in the incubator (37°, 5% CO2) for 8 hours. Immediately preceding the experiment, the cells were washed with Phosphate buffered saline (PBS) three times and the cell chamber was then sealed with another coverslip. Because the 400 nm beam is much less damaging to cells, it was used to locate the cell and record the first interferogram; the second interferogram was then recorded at 310 nm. The time interval between these two interferograms is typically <5 s, which can be further shortened to sub-second if the shutters are synchronized with the CCD. To correct for the background phase of the optical system, a background interferogram of a blank area is also taken for both wavelengths. We used the Hilbert transform to obtain the quantitative phases ϕ of the cell, which can be converted to optical path lengths (OPL) using the following equation:
| (1) |
where λ is the wavelength, αn the specific refractive index of one type of biomolecule with average concentration Cn, and d the thickness of the cell [13]. We note that Eq. (1) is valid only when the medium that the cell immersed in does not contain biomolecules. This is the case in our experiment because all cells are measured in PBS. It is also important to point out that only OPL difference in the x–y plane is spatially resolved and all the contributions along axial direction is integrated and cannot be separated with current technique. Fig. 3 (a)–(b) show the processed OPL images of four cells at 310 nm and 400 nm, respectively. As expected, they show very similar features, with slight difference in OPL due to dispersion. The integration of Cnd over space is the dry mass Mn of that particular type of biomolecule:
| (2) |
Therefore OPL images can be directly related to the dry mass of the cell if the composition of the cell is known. Using the wavelength dependence of the specific refractive index α of different types of biomolecules, it is theoretically possible to solve for their dry mass separately. However, due to the complicated composition of cells and insufficient calibration data for all of the biomolecules, calculating the exact dry mass of proteins, nucleic acids, lipids, and polysaccharides would be challenging. Instead, we simply calculate the ratio of the integrated OPL at the two wavelengths and compare that to the calibrated data for proteins and nucleic acids, using the following equation:
| (3) |
Figure 3.
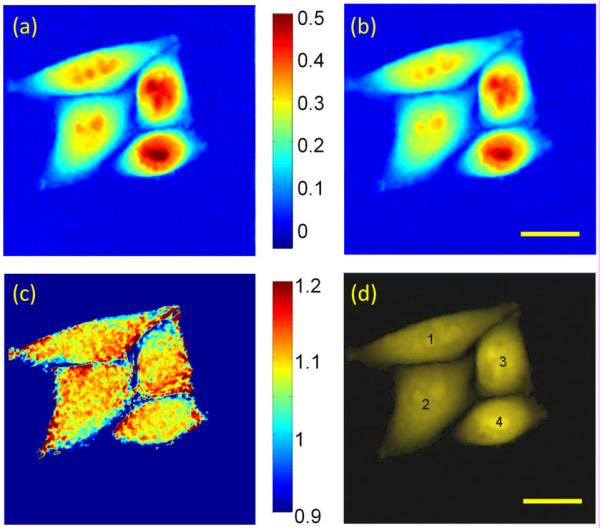
Optical path length image of HeLa cells at (a) 310 nm and (b) 400 nm; (c) ratio image of magnification corrected OPL images at 310 nm and 400 nm; (d) composite image of corrected OPL images using red and green channels for 310 nm and 400 nm images, respectively. The number indicates the auto-segmented cell number and the gray dashed line depicts the borders of each cell. Scale bar: 20 μm.
We note that the magnifications for the 310 nm and 400 nm images are slightly different and also that there is a spatial offset due to chromatic aberration and imperfect alignment of the two imaging arms. To compensate, the 400 nm phase image was demagnified and spatially shifted to match the phase image at 310 nm. Fig. 3(c) shows the ratio of the two images at the two wavelengths after compensation, while Fig. 3(d) shows the composite image at both wavelengths using different color labels: the 310 nm image is labeled with red color and the 400 nm image is labeled with green color. From Fig. 3(c), there is some inhomogeneity (with a standard deviation of 0.01–0.02) within the cell but we do not observe the location of the nucleus. The reason for this might be that although DNA is only present in the nucleus, RNA (with a much higher dry mass) is dispersed throughout the cell. Another reason is that laser speckle induced noise in the quantitative phase measurements contributes to fluctuations in the ratio. It is hard to disentangle the contribution of biomolecule spatial distribution and laser speckle noise with the current setup. This problem could potentially be alleviated with phase measurement based on a spatially incoherent source which has much less speckle noise [14].
From the matched phase images, we used a custom written image processing script based on watershed algorithm in Matlab (Mathworks) to automatically identify cell borders and calculate the dry mass M, occupied area A and dispersion ratio R of the cells in each image (the dry mass is calculated using 400 nm OPL images assuming all biomolecules have the same α of 0.191 mL/g). We analyzed altogether 54 cells and the statistics of these cells are shown in Fig. 4 as box plots. Both the dry mass and the projected area of the imaged cells have very large variability, with dry mass being more tightly confined compared to the area. This probably occurs because the dry mass of the cell is a well-regulated cell growth parameter, while the cell area depends also on the 3D shape of the cell which could be highly variable among cells. The refractive index increment ratio, R, is a simplified parameter that reflects the biochemical composition of the cell and therefore has a much narrower distribution. The average dispersion R of the 54 measured HeLa cells is 1.088 with a standard deviation of 0.013. This number is very close to the dispersion of pure protein solutions at a ratio of 1.089. Considering that mammalian cells typically have approximately 60% of their dry mass composed of proteins, this result is not surprising [15]. Even though we expect that the presence of nucleic acids might increase the dispersion ratio based on calibration results in the previous section, due to the fact that the amount of total nucleic acids present in mammalian cells is typically less than 5%, this increase is negligible and could be further masked by the smaller dispersion of other biomolecules such as lipids and polysaccharides. For a full characterization of the biochemical composition of cells, a much more detailed calibration of the dispersion of all biomolecules present and quantitative phase images at several wavelengths would be required.
Figure 4.

Scatter and box plots of the (a) dry mass (b) projected area (c) dispersion of 54 HeLa cells. The box indicates 25%–75% range and whiskers indicates 5%–95% range.
4. Conclusion and discussion
In summary, we report, to the best of our knowledge, the first direct dispersion measurement of live eukaryotic cells. A dual-wavelength quantitative phase microscope is built to measure quantitative phase images of cells at 310 nm and 400 nm. We obtained phase images of 54 HeLa cells and calculated the dry mass, projected area, and dispersion of each cell. The dispersion ratio R is tightly bounded around 1.088 with a standard deviation of 0.013. It shows excellent agreement with the value of protein dispersion measured by a total internal reflection method. This correlates well with the fact that more than 60% percent of the cell dry mass is composed of proteins. Our dispersion phase microscopy provides an additional intrinsic parameter that could prove to be valuable in distinguishing different types of biomaterials or for imaging the spatial inhomogeneity of thin biological samples. However, the current setup utilized a laser source and suffered from large laser speckle noise, which could mask the spatial distribution of the biomolecules. The standard deviation of dispersion ratio R within a single cell is about 0.01–0.02, almost equal to or higher than that of cell to cell variations. A large contribution of this variation might come from laser speckle noise which gives rises to phase noise in the measurements. In order to observe the spatial distribution of biomolecules based on R, the signal to noise ratio (SNR) in the phase images needs to be improved at least by 2–3 times. Quantification of concentration based on R will require even higher SNR. It is possible to significantly reduce the speckle noise with incoherent source based phase measurement or tomographic phase measurement. Further characterization of the dispersion of other biomolecules and multi-wavelength phase imaging is also important for quantitative analysis of biochemical composition of cells or other biological materials.
Acknowledgements
This work was funded by the National Center for Research Resources of the National Institutes of Health (P41-RR02594-24), the National Science Foundation (DBI-0754339), and Hamamatsu Corporation
Footnotes
OCIS codes: (120.3180) Interferometry; (180.3170) Interference Microscopy; (170.3880) Medical and biological imaging.
References and links
- 1.Vo-Dinh T. Biomedical Photonics Handbook. CRC Press; 2003. [Google Scholar]
- 2.Wojtkowski M, Srinivasan V, Ko T, Fujimoto J, Kowalczyk A, Duker J. Ultrahigh-resolution, high-speed, Fourier domain optical coherence tomography and methods for dispersion compensation. Opt. Express. 2004;12:2404–2422. doi: 10.1364/opex.12.002404. [DOI] [PubMed] [Google Scholar]
- 3.Zipfel WR, Williams RM, Webb WW. Nonlinear magic: multiphoton microscopy in the biosciences. Nat Biotech. 2003;21:1369–1377. doi: 10.1038/nbt899. [DOI] [PubMed] [Google Scholar]
- 4.Park Y, Yamauchi T, Choi W, Dasari R, Feld MS. Spectroscopic phase microscopy for quantifying hemoglobin concentrations in intact red blood cells. Opt. Lett. 2009;34:3668–3670. doi: 10.1364/OL.34.003668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rappaz B, Charriere F, Depeursinge C, Magistretti PJ, Marquet P. Simultaneous cell morphometry and refractive index measurement with dual-wavelength digital holographic microscopy and dye-enhanced dispersion of perfusion medium. Opt. Lett. 2008;33:744–746. doi: 10.1364/ol.33.000744. [DOI] [PubMed] [Google Scholar]
- 6.Yang CH, Wax A, Dasari RR, Feld MS. Phase-dispersion optical tomography. Opt. Lett. 2001;26:686–688. doi: 10.1364/ol.26.000686. [DOI] [PubMed] [Google Scholar]
- 7.Zeskind BJ, Jordan CD, Timp W, Trapani L, Waller G, Horodincu V, Ehrlich DJ, Matsudaira P. Nucleic acid and protein mass mapping by live-cell deep-ultraviolet microscopy. Nat. Methods. 2007;4:567–569. doi: 10.1038/nmeth1053. [DOI] [PubMed] [Google Scholar]
- 8.Sinha RP, Hader DP. UV-induced DNA damage and repair: a review. Photochemical & Photobiological Sciences. 2002;1:225–236. doi: 10.1039/b201230h. [DOI] [PubMed] [Google Scholar]
- 9.Fu D, Choi W, Sung Y, Oh S, Yaqoob Z, Park Y, Dasari RR, Feld MS. Ultraviolet refractometry using field-based light scattering spectroscopy. Opt. Express. 2009;17:18878–18886. doi: 10.1364/OE.17.018878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jin YL, et al. Refractive index measurement for biomaterial samples by total internal reflection. Phys. Med. Biol. 2006;51:N371. doi: 10.1088/0031-9155/51/20/N02. [DOI] [PubMed] [Google Scholar]
- 11.Popescu G, Ikeda T, Dasari RR, Feld MS. Diffraction phase microscopy for quantifying cell structure and dynamics. Opt. Lett. 2006;31:775–777. doi: 10.1364/ol.31.000775. [DOI] [PubMed] [Google Scholar]
- 12.Barer R. Refractometry and Interferometry of Living Cells. J. Opt. Soc. Am. 1957;47:545–556. doi: 10.1364/josa.47.000545. [DOI] [PubMed] [Google Scholar]
- 13.Popescu G, Park Y, Lue N, Best-Popescu C, Deflores L, Dasari RR, Feld MS, Badizadegan K. Optical imaging of cell mass and growth dynamics. Am. J. Physiol. Cell Physiol. 2008;295:C538–C544. doi: 10.1152/ajpcell.00121.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fu D, Oh S, Choi W, Yamauchi T, Dorn A, Yaqoob Z, Dasari RR, Feld MS. Quantitative DIC microscopy using an off-axis self-interference approach. Opt. Lett. 2010;35:2370–2372. doi: 10.1364/OL.35.002370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P. Molecular Biology of the Cell. Garland Science; New York: 2002. [Google Scholar]