

Abstract
Endocannabinoid signalling is distributed throughout the brain, regulating synaptic release of both excitatory and inhibitory neurotransmitters. The presence of endocannabinoid signalling within stress-sensitive nuclei of the hypothalamus, as well as upstream limbic structures such as the amygdala, suggests it may play an important role in regulating the neuroendocrine and behavioral effects of stress. The evidence reviewed here demonstrates that endocannabinoid signalling is involved in both activating and terminating the hypothalamic-pituitary-adrenal (HPA) axis response to both acute and repeated stress. In addition to neuroendocrine function, however, endocannabinoid signalling is also recruited by stress and glucocorticoid hormones to modulate cognitive and emotional processes such as memory consolidation and extinction. Collectively, these data demonstrate the importance of endocannabinoid signalling at multiple levels as both a regulator and effector of the stress response.
The Endocannabinoid System
Cannabis has been used as a recreational drug in a multitude of cultures for centuries (Russo, 2007). Typically, cannabis produces anxiolytic and mood elevating effects that promote relaxation and stress-reduction, which likely contributes to its high level of recreational use (Green et al., 2003). In the late 1980’s, it was discovered that delta-9-tetrahydrocannabinol (THC), the psychoactive constituent of cannabis, bound to specific receptors within brain tissue (Devane et al., 1988), setting the stage for the subsequent discovery of the endogenous cannabinoid system. In the early 1990’s, the genetic characterization and neuroanatomical localization of the cannabinoid receptor (termed the CB1 receptor) was mapped in the rodent brain (Matsuda et al., 1990; Herkenham et al., 1991). Soon after, the endogenous cannabinoid ligands (termed endocannabinoids, eCBs) N-arachidonylethanolamine (AEA; named anandamide, after anand, the Sanskrit word for bliss; Devane et al., 1992) and 2-arachidonoylglycerol (2-AG; Sugiura et al., 1995) were discovered. While quite distinct in structure from THC, both AEA and 2-AG share the lipophilicity of THC, being arachidonate-derived neuroactive signalling lipids. Unlike canonical neurotransmitters, both AEA and 2-AG are post-synaptically synthesized signalling molecules that are not stored in vesicles, but are generated on demand and then liberated to act in a retrograde fashion on presynaptically localized CB1 receptors (Wilson and Nicoll, 2002). The signalling lifespan of AEA and 2-AG, in turn, is primarily regulated by yet ill-defined uptake processes and intracellular hydrolysis by fatty acid amide hydrolase (FAAH) and monoacylglycerol lipase (MAG-L), respectively (Hillard, 2000). Recent observations also implicate serine hydrolase alpha-beta-hydrolase domain 6 (ABHD6) as an essential regulator of 2-AG and thus a key player in the control of eCB efficacy (Marrs et al., 2010). The CB1 receptor is coupled to Gi/o proteins and functions to suppress adenylate cyclase activity and calcium influx, resulting in the suppression of neurotransmitter release. Accordingly, the primary function of the eCB system is to gate and regulate neurotransmitter release, particularly GABA and glutamate, thus fine tuning the strength of a particular synapse (Freund et al., 2003).
The Stress Response: A Role for Endocannabinoids?
Stress is most readily defined as any stimulus that presents a challenge to homeostasis - typically a real or perceived threat to an organism’s well being. Activation of the hypothalamic-pituitary-adrenal (HPA) axis is a well-characterized response to stress. Activation of the HPA axis results in the release of CRH from neurosecretory cells of the paraventricular nucleus of the hypothalamus (PVN) into the portal vessels, where it is then transported to the anterior pituitary gland and stimulates the release of adrenocorticotropic hormone (ACTH) into the general circulation (Herman et al, 2002). In turn, ACTH causes the release of glucocorticoid hormones (primarily corticosterone in rodents and cortisol in humans) from the adrenal cortex into the circulation. A major function of glucocorticoids during stressful experiences is to mobilize energy stores, which allows the organism to mount the appropriate response to a threat. In addition, glucocorticoids produce a range of effects on cardiovascular, immune, metabolic and neural systems that facilitate optimal responses to aversive stimuli (Pecoraro et al., 2006). Excessive and unchecked secretion of glucocorticoids, however, can produce detrimental effects on health and increase the susceptibility of the organism to an array of disease states, ranging from mood disorders to type II diabetes. To circumvent this, glucocorticoids exert potent negative feedback inhibition of the HPA axis, such that elevated circulating levels of glucocorticoids rapidly suppress HPA axis activity. Negative feedback of the HPA axis occurs at multiple levels in the periphery (e.g. adrenal and pituitary glands) and the brain. Within the PVN, glucocorticoids exhibit both rapid and delayed suppression of HPA axis activity (Dallman, 2005). In addition, extrahypothalamic limbic structures that communicate with the PVN exert both positive and negative regulation of the HPA axis (Ulrich-Lai and Herman, 2009). The amygdala has been identified as one of the primary limbic structures involved in activating the HPA axis in response to stressful stimuli. The hippocampus and prefrontal cortex, on the other hand, have been identified as inhibiting the HPA axis, and are also involved in the extrahypothalamic glucocorticoid-mediated negative feedback of the HPA axis.
Given the pronounced effects of cannabis on emotionality and the processing of stressful information, it is not surprising that eCB signalling is widely distributed throughout corticolimbic circuits that are linked to the stress response. With respect to the central nervous system, CB1 receptors are expressed and both AEA and 2-AG are synthesized in the hypothalamus, including locally within the PVN (Malcher-Lopes et al., 2006), as well as in upstream structures such as the amygdala, hippocampus and prefrontal cortex (Herkenham et al., 1991; Hill et al., 2008). As cannabis consumption is known to produce anxiolytic and stress-reducing effects in humans, the question arose as to whether eCB signalling itself was involved in regulating the stress response and subsequent changes in neuroendocrine function and emotional behavior. Following is a review of a mini-symposium that summarizes the current state of knowledge regarding how eCB signalling integrates into the regulation of the HPA axis in response to stress, and in turn, how stress-induced regulation of eCB signalling is involved in both glucocorticoid feedback and changes in emotional behavior.
Endocannabinoids and Activation of the HPA Axis
The presence of CB1 receptors within corticolimbic circuits regulating the HPA axis and the anti-stress properties of cannabis consumption suggest that eCB signalling should inhibit the stress response. Both genetic disruption and pharmacological blockade of CB1 receptor signalling result in an increase in HPA axis activity under basal conditions and following exposure to acute stress (Patel et al., 2004; Wade et al., 2006; Cota et al., 2007; Steiner and Wotjak, 2008). These data indicate that an eCB tone provides a steady-state inhibition of HPA axis activity, which, when disrupted, results in activation of the HPA axis. This gatekeeper model was supported by subsequent studies revealing a decrease in AEA content in the amygdala, through an increase in FAAH-mediated hydrolysis, in response to acute stress (Patel et al., 2005b; Rademacher et al., 2008; Hill et al., 2009). Preventing this decline in amygdalar AEA by local administration of a FAAH inhibitor into the basolateral nucleus (BLA) attenuates stress-induced activation of the HPA axis (Hill et al., 2009). Similarly, local administration of a CB1 receptor agonist directly into the BLA dramatically reduces stress-induced activation of the HPA axis (Ganon-Elazar and Akirav, 2009; Hill et al., 2009). Given that administration of a CB1 receptor antagonist to unstressed animals increases neuronal activation of the BLA (Singh et al., 2004; Patel et al., 2005a), and that CB1 receptors regulate the release of glutamate within the amygdala (Karst et al., 2010; Kodirov et al., 2009; Azad et al., 2003; Domenici et al., 2006), we propose (see Fig. 1) that under steady-state conditions, a tonic level of AEA/CB1 receptor signalling within the BLA gates glutamatergic inputs to principal neurons in the BLA. In response to stress, FAAH activity increases and AEA levels decline, resulting in a disinhibition of BLA principal neurons and thereby an increase in the outflow of amygdalar neurons and downstream targets of the amygdala, such as the HPA axis (Hill et al., 2009). In support of this model, genetic work has determined that CB1 receptors localized on glutamatergic, and not on GABAergic, neurons are responsible for the ability of eCBs to restrain activation of the HPA axis (Steiner et al., 2008).
Figure 1.
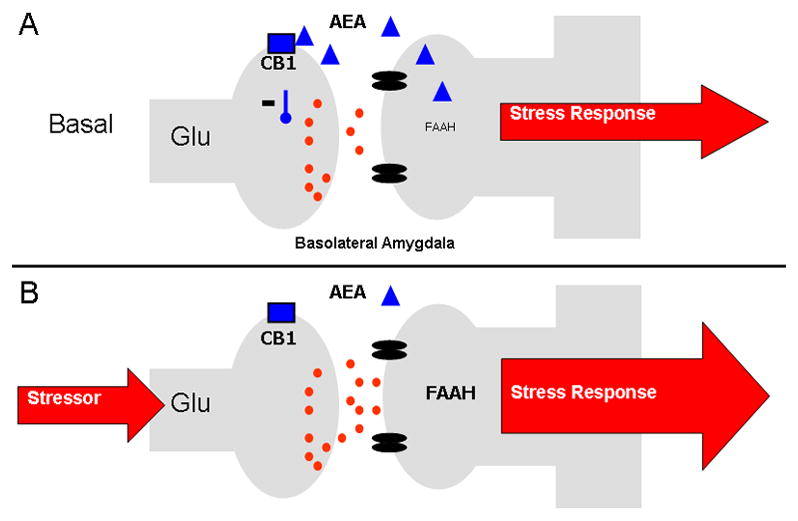
(A) Under steady state conditions, anandamide (AEA) signalling within the basolateral amygdala (BLA) restrains glutamatergic signalling and helps maintain non-stress conditions. In response to stress, fatty acid amide hydrolase (FAAH) activity increases, reducing AEA content and disinhibiting glutamate release onto BLA neurons, facilitating an adequate stress response.
Stress and Glucocorticoid-mediated Regulation of Endocannabinoid Signalling in the Hypothalamus: A Role in Glucocorticoid Feedback
In addition to contributing to the regulation of the HPA axis, eCB signalling is also regulated by the HPA axis. In vitro studies have demonstrated that direct application of glucocorticoids to hypothalamic slices results in a rapid (~10 min) elevation in the tissue content of both AEA and 2-AG (Malcher-Lopes et al., 2006). In vivo studies have supported these findings by demonstrating that peripheral administration to rats of corticosterone results in a rapid (~10 min) increase in both AEA and 2-AG levels in the hypothalamus (Hill et al., 2010a). Similarly, exposure to 30 min of restraint stress results in a significant elevation in hypothalamic 2-AG content, while having no effect on tissue levels of AEA (Evanson et al., 2010). Thus, stress increases hypothalamic eCB signalling through a glucocorticoid-mediated pathway. Furthermore, given the rapid effects of glucocorticoids on hypothalamic eCB signalling, these effects are likely not mediated by the canonical actions of glucocorticoids on gene transcription.
The function of this increase in hypothalamic eCB signalling in response to stress has been highlighted by a series of in vitro and in vivo studies demonstrating a critical role of this process in glucocorticoid-mediated negative feedback. Following activation of the HPA axis in response to stress, elevated circulating glucocorticoids feed back onto the brain to suppress stress-induced HPA activation via both a rapid, transcription-independent inhibitory mechanism and a delayed, transcription-dependent regulation of HPA output. Intracellular recordings from in vitro brain slices have demonstrated a rapid glucocorticoid-induced suppression of glutamatergic excitatory synaptic inputs to CRH neurons of the PVN that is mediated by a putative membrane glucocorticoid receptor (Di et al., 2003). This suppression of excitatory inputs to CRH neurosecretory cells may represent a mechanism of fast-feedback inhibition of the HPA axis, whereby glucocorticoids can rapidly decrease incoming excitatory input and decrease activation of the HPA axis at the level of the PVN. This rapid glucocorticoid suppression of PVN neurons is blocked by inhibiting postsynaptic G protein activity and by blocking CB1 receptors (Di et al., 2003). The glucocorticoid effect is also blocked by inhibiting the activity of diacylglycerol lipase, a synthetic enzyme of 2-AG (Harris and Tasker, unpublished observation), indicating that the glucocorticoid suppression of excitation is mediated by the eCB 2-AG. These findings are consistent with the ability of glucocorticoids to induce 2-AG synthesis rapidly in the hypothalamus in vitro and in vivo (Hill et al., 2010a; Malcher-Lopes et al., 2006). These observations implicate a rapid glucocorticoid-induced suppression of the excitatory synaptic drive to CRH neurons via the dendritic release and retrograde actions of eCBs in the PVN, providing a potential mechanism for the rapid negative feedback effects of glucocorticoids on the HPA axis via a membrane-associated glucocorticoid receptor and non-canonical glucocorticoid signalling mechanism (see Fig. 2).
Figure 2.
Fast feedback inhibition of the HPA axis via glucocorticoid-induced eCB release in the hypothalamus. Left – Stress activation of the HPA axis consists of corticotropin-releasing hormone (CRH) secretion from PVN neurons and CRH-evoked adrenocorticotropic hormone (ACTH) secretion from the pituitary, which, in turn, stimulates corticosteroid (CORT) release from the adrenal cortex and CORT feedback onto the PVN. Right – In PVN CRH neurons, (1) CORT binds to a membrane-associated glucocorticoid receptor (mGR), which causes endocannabinoid (eCB) synthesis in CRH neurons (2) and retrograde eCB release (3); eCB binds to presynaptic CB1 receptors on glutamate terminals (4) and inhibits glutamate release (5) onto the CRH neurons, suppressing the excitatory synaptic drive and decreasing CRH neuron activity and CRH release (6), which suppresses HPA axis activation.
The postulated glucocorticoid-eCB mechanism of rapid HPA axis feedback inhibition has recently been tested in vivo in studies where glucocorticoids and cannabinoid analogs were administered directly into the PVN. Local bilateral PVN administration of glucocorticoids, including a dexamethasone-BSA conjugate that is membrane-impermeant and restricted to the extracellular environment, dampens the restraint stress-induced secretion of ACTH and corticosterone, suggesting a rapid glucocorticoid inhibition of the HPA axis directly at the level of the PVN (Evanson et al., 2010). The rapid glucocorticoid-mediated suppression of HPA axis activation is prevented by the co-application into the PVN of the CB1 receptor antagonist AM251, indicating that the rapid glucocorticoid effect is mediated by eCB actions in the PVN. Again, this is consistent with the observation that both restraint stress and glucocorticoids cause a rapid increase in the level of 2-AG in the hypothalamus (Evanson et al., 2010; Hill et al., 2010a). These findings support a role for the glucocorticoid-induced eCB release in the PVN in the rapid glucocorticoid feedback regulation of the HPA axis.
It has recently been demonstrated that exposure of young rats to repetitive, but not acute, immobilization stress (30 min/day for 5 days) results in a loss of eCB-mediated retrograde signalling in parvocellular neurosecretory cells (PNCs) of the PVN (Wamsteeker et al., 2010). Specifically, following repeated stress, application of dexamethasone to hypothalamic slices no longer causes an eCB-mediated suppression of excitatory inputs to PNCs (Wamsteeker et al., 2010), as was described above. Confirming that this loss of glucocorticoid-mediated suppression of excitation in the PVN is due to an impairment of eCB signalling, bath application of a CB1 receptor agonist to PVN slices from repeatedly stressed animals does not suppress excitatory transmission to PNCs (Wamsteeker et al., 2010). Moreover, the ability of a CB1 receptor agonist to inhibit GABA transmission to PNCs is also compromised following repeated stress (Wamsteeker et al., 2010). This conundrum may underscore the notion that following stress, GABA does not necessarily function as an inhibitory transmitter. In fact, following stress-induced changes in chloride homeostasis in PNCs, GABA can act in an excitatory capacity when synapses are activated by bursts of activity (Hewitt et al., 2009).
This loss of CB1 receptor function in the PVN has important implications for glucocorticoid-mediated feedback inhibition of the HPA. As glucocortioid-mediated induction of eCB signalling within the PVN is integral for fast-feedback inhibition of the HPA axis (see above and Fig. 2), this process may be compromised following chronic stress. In line with this hypothesis, in vivo studies have shown that chronic stress exposure results in impaired glucocorticoid-mediated fast feedback inhibition of the HPA axis (Young et al., 1990).
Taken together, these data indicate that glucocorticoids induce eCB signalling through a rapid, non-genomic process in CRH neurons of the PVN. This induction of eCB signalling inhibits glutamatergic inputs to CRH neurons, and thus decreases the excitatory drive to the HPA axis. This process is believed to subserve glucocorticoid-mediated fast feedback inhibition of the HPA axis. Following chronic stress, eCB signalling in the PVN is compromised, which results in impaired fast feedback inhibition of the HPA axis, and likely contributes to the hypersecretion of glucocorticoids following chronic stress (Hill et al., 2010b).
Stress and Glucocorticoid-mediated Regulation of Endocannabinoid Signalling in the Amygdala: A Role in Emotional Memory
Glucocorticoid-mediated induction of eCB signalling occurs not only in the hypothalamus, but also in the amygdala. While exposure to stress rapidly reduces tissue levels of AEA in the amygdala (Patel et al., 2005b; Rademacher et al., 2008; Hill et al., 2009), administration of corticosterone in the absence of stress results in a rapid increase in tissue levels of AEA in the amygdala (Hill et al., 2010a). These apparently contradictory findings have lead to the hypothesis that stress, through a glucocorticoid-independent mechanism, rapidly decreases amygdalar AEA content, which is reversible by increasing levels of glucocorticoids following activation of the HPA axis (Hill and McEwen, 2010).
At the functional level, there is growing interest in the putative interactions between glucocorticoids and eCBs within the amygdala. Accumulating evidence demonstrates that glucocorticoid hormones enhance long-term consolidation of emotionally arousing experiences involving rapid actions on intracellular signalling cascades in the BLA (Roozendaal, 2000; McGaugh, 2002). Given the ability of glucocorticoids to rapidly mobilize eCB signalling, the eCB system seems to be a plausible target to mediate these glucocorticoid effects. Bilateral post-training infusions of the CB1 receptor agonist WIN55,212-2 into the BLA induced dose-, time-, and site-dependent enhancement of inhibitory avoidance retention performance that was comparable to performance seen following post-training glucocorticoid administration (Campolongo et al., 2009). In line with these parallels between CB1 receptor activation and glucocorticoids, intra-BLA administration of a CB1 receptor antagonist, at a dose ineffective, per se, on memory consolidation, is capable of blocking the ability of systemically administered corticosterone to facilitate aversive memory consolidation (Campolongo et al., 2009). Taken together, these findings indicate that glucocorticoids recruit eCB signalling in the BLA to modulate aversive memory consolidation.
It is well established that activation of CB1 receptors decreases GABA release (Ohno-Shosaku et al., 2001; Freund et al., 2003) via a rapid inhibition of Ca2+entry into the terminals (Hoffman and Lupica, 2000; Wilson et al., 2001). Furthermore, it has been reported that the amygdalar GABAergic system modulates memory storage (McGaugh, 2002) and that inhibition of GABAergic activity within the BLA enhances memory consolidation by increasing the release of norepinephrine (NE; Hatfield et al., 1999), a neurotransmitter involved in mediating emotional arousal effects on memory consolidation (McGaugh, 2000, 2004). In view of this evidence, the following model is proposed: corticosterone binds to a membrane-bound glucocorticoid receptor (as discussed above), which activates the Gs – cAMP/PKA pathway to induce eCB synthesis, similar to the rapid glucocorticoid signalling in the PVN (Malcher-Lopes et al., 2006). eCBs are released into the synapse, where they bind to CB1 receptors on GABAergic terminals to inhibit GABA release. This inhibition of GABA release leads to the disinhibition of NE release and increases noradrenergic activation of postsynaptic beta adrenoreceptors, increasing the consolidation of emotionally aversive memories (Campolongo et al., 2009; see Hill and McEwen, 2009 for model). As such, these data provide functional evidence of glucocorticoid-mediated recruitment of eCB signalling in the amygdala, demonstrating that this phenomenon is important for complex behavioral processes in addition to glucocorticoid mediated feedback.
A Role for Amygdalar Endocannabinoid Signalling in Adaptation to Stress and Aversive Stimuli
Unlike the hypothalamus, which exhibits a robust increase in 2-AG content following stress or glucocorticoid treatment (Evanson et al., 2010; Hill et al., 2010a; Malcher-Lopes et al., 2006), the amygdala shows no change in 2-AG content in response to an acute stressor (Patel et al., 2005b; Rademacher et al., 2008; Hill et al., 2009). However, following repeated presentation of an aversive stimulus, 2-AG signalling within the amygdala progressively increases (Hill et al., 2010b; Patel et al., 2005b; Rademacher et al., 2008; Patel et al., 2009). This response is temporally restricted, with elevations in 2-AG found within 20–30 min of the onset of stress (Hill et al., 2010b; Patel et al., 2009), but waning within 1 h following the onset of stress (Patel et al., 2009) and reversing completely within 24 h of exposure to stress (Hill et al., 2010b). Based on these data, it has been suggested that repeated stress exposure results in an enhanced capacity to elevate 2-AG levels following subsequent stress exposures (Patel et al., 2009).
Given the prominent role of 2-AG in mediating retrograde eCB signalling at central synapses (Kano et al., 2009), a clear extension of the above findings is that eCB-mediated synaptic suppression would be enhanced after chronic stress exposure. An inherent assumption in this hypothesis is that tissue levels of 2-AG correlate with the magnitude of eCB-mediated synaptic signalling, one that had not been previously tested. In accordance with this hypothesis, a prototypic form of eCB signalling, depolarization-induced suppression of inhibition (DSI), was found to be significantly enhanced in BLA neurons of mice exposed to 10 days of restraint stress (Patel et al., 2009). One day of restraint stress exposure did not affect DSI, consistent with a lack of change in 2-AG levels seen after acute stress exposure (Hill et al., 2009; Patel et al., 2009). These findings support the notion that tissue levels of 2-AG correlate with the magnitude of eCB signalling.
With regard to the functional implications of repeated stress-induced elevations in amygdala 2-AG signalling, Hill and colleagues (2010b) have recently shown that this process is critical for the habituation of the HPA axis that occurs in response to repeated homotypic stressor exposure. Specifically, the elevation of 2-AG within the amygdala following repeated stress correlated with the magnitude of HPA axis suppression, and local administration of a CB1 receptor antagonist into the BLA reversed the expression of stress habituation (Hill et al., 2010b). Accordingly, increased recruitment of 2-AG signalling within the BLA appears to be essential for adaptation of the HPA axis to repeated stress. Given the aforementioned model by which eCB signalling in the BLA gates glutamatergic inputs to principal neurons, the most parsimonious explanation of these data is that a transient augmentation of 2-AG signalling upon repeated stressor exposure dampens excitatory inputs to the BLA, thereby decreasing the outflow of the amygdala, which would include stimulation of the HPA axis [(Hill et al., 2010b); see Fig 3 for model]. This model is in accordance with a recent report demonstrating that corticosterone inhibits glutamatergic inputs to the BLA through an eCB-mediated mechanism, but only in animals that have a history of previous stress exposure (Karst et al., 2010).
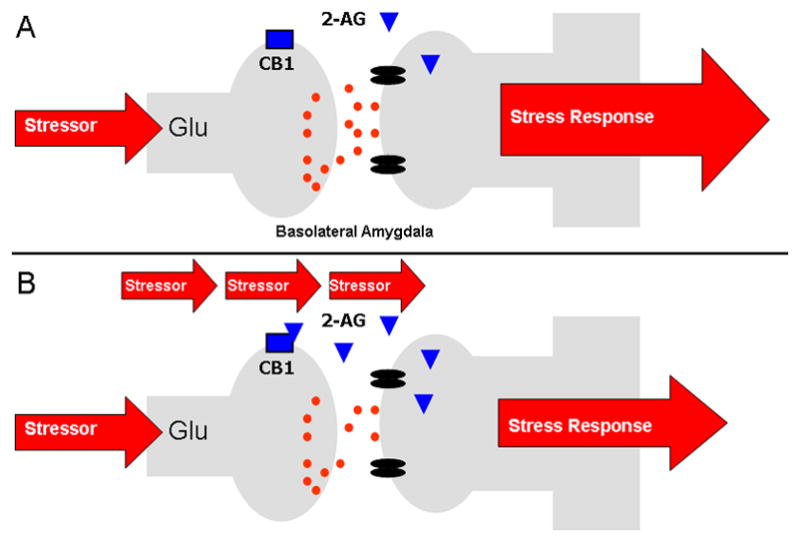
Figure 3.

(A) In response to acute stress, there is little to no change in 2-arachidonoylglycerol (2-AG) within the BLA. (B) Repeated stress exposure, however, primes 2-AG signalling within the BLA such that in response to stressor presentation, there is a phasic and limited increase in 2-AG signalling within the BLA which suppresses glutamatergic inputs to the BLA, decreasing the outflow of the amygdala and driving habituation of the stress response. A similar phenomenon may also subserve behavioral plasticity, such as fear extinction, to aversive stimuli.
In addition to contributing to adaptation of the HPA axis to stressful stimuli, eCB signalling in the amygdala is also important for behavioral adaptation to aversive stimuli. In response to an acutely presented aversive stimulus, an organism must recognize a potential threat and adequately react to it, including fear and anxiety responses. These behavioral responses rapidly vanish when the threat is removed. Such cognitive-emotional flexibility allows an organism to both respond and adapt to threats. Recent observations suggest an important role of the eCB system in this complex adaptive process. Employing classical associative fear conditioning and extinction paradigms, it was revealed that during extinction training, but not initial fear conditioning, eCB levels in the amygdala, but not in the prefrontal cortex, were elevated (Marsicano et al., 2002). The relevance of this increase in eCB signalling was highlighted by subsequent behavioral studies demonstrating that mice lacking the CB1 receptor exhibited prolonged expression of fear behaviors during extinction training, indicating that this augmentation of amygdalar eCB signalling is essential for the adaptation of fear behaviors (Marsicano et al., 2002).
This observation was subsequently extended to non-associative fear, where mice exposed to a brief inescapable electric foot shock showed a considerable fear response to a subsequently presented neutral tone (Kamprath and Wotjak, 2004). However, whereas this fear behavior was rapidly suppressed in wild-type mice, CB1 receptor-deficient mice failed to suppress the conditioned fear response (Kamprath et al., 2006). Specifically, mice lacking CB1 receptors showed persistent fear on repeated tone exposures, indicating that the CB1 receptor is essential not only for within-, but also for between-session habituation (Kamprath et al., 2006). The fear dampening effects of eCBs become evident only in highly aversive situations and are independent of CRH and corticosterone action (Kamprath et al., 2009).
Similar to the hypothesized mechanism of HPA axis regulation, experiments with conditional mouse CB1 receptor mutants reveal that eCBs exert their effects on fear adaptation primarily via CB1 receptors expressed on cortical glutamatergic neurons (Kamprath et al., 2009). As increased eCB signalling within the amygdala is coupled to both fear extinction (Marsicano et al., 2002) and HPA axis habituation (Hill et al., 2010b), these data suggest that CB1 receptor-mediated regulation of cortical glutamatergic inputs to BLA neurons is likely the nexus of the eCB contribution to both neuroendocrine and behavioral adaptation to stress.
Summary
The material reviewed in this mini-symposium demonstrates an increasingly complex role of eCB signalling in multiple facets of the stress response. With respect to the HPA axis, eCB regulation of glutamatergic transmission in both the PVN and the BLA appears to be critical for the suppression of the HPA axis under conditions of fast feedback inhibition and habituation following repeated stress, respectively. Temporal studies demonstrate an interesting dual component role of eCBs within the HPA axis. Specifically, a rapid loss of AEA within the BLA following stress exposure promotes activation of the HPA axis, whereas a glucocorticoid-mediated elevation in 2-AG in the PVN contributes to fast feedback inhibition of the HPA axis. Adding to this complexity is the emerging role of eCB signalling in the BLA in the habituation of the HPA axis response to repeated stress. The eCB system, therefore, is clearly intertwined with HPA axis function, under both acute and chronic conditions.
In addition to regulation of the HPA axis, however, eCB signalling also contributes to higher order responses to stress, such as emotional and cognitive responses. As an extension of the role of eCB signalling in the adaptation of the HPA axis to repeated stress, eCBs are also required for the suppression of behavioral responses to aversive stimuli following their removal or repeated presentation. Furthermore, the recruitment of eCB signalling within the amygdala by glucocorticoids mediates their effects on the consolidation of emotional memory.
Taken together, these data clearly point to the integration of eCBs into the stress response. Ongoing research will help us to delineate the extent to which eCB signalling is engaged by stress in different stress-related CNS structures, and how deficits in this system may contribute to stress-related mental disorders, such as depression and post-traumatic stress disorder (Hill and Gorzalka, 2009).
Acknowledgments
The writing of this review, and the research described herein, was supported by an operating grant from the Canadian Institute of Health Research (CIHR) to JSB and a postdoctoral fellowship from CIHR to MNH, as well as NIH grant K08MH090412 to SP and NIMH grant 2R01 MH066958 to JGT. Additionally, funding support was provided by the Catherine and Hunter Pierson Chair in Neuroscience Tulane University Research Enhancement Fund to JGT.
References
- Azad SC, Eder M, Marsicano G, Lutz B, Zieglgansberger W, Rammes G. Activation of the cannabinoid receptor type 1 decreases glutamatergic and GABAergic synaptic transmission in the lateral amygdala of the mouse. Learn Mem. 2003;10:116–128. doi: 10.1101/lm.53303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campolongo P, Roozendaal B, Trezza V, Hauer D, Schelling G, McGaugh JL, Cuomo V. Endocannabinoids in the rat basolateral amygdala enhance memory consolidation and enable glucocorticoid modulation of memory. Proc Natl Acad Sci U S A. 2009;106:4888–4893. doi: 10.1073/pnas.0900835106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cota D, Steiner MA, Marsicano G, Cervino C, Herman JP, Grubler Y, Stalla J, Pasquali R, Lutz B, Stalla GK, Pagotto U. Requirement of cannabinoid receptor type 1 for the basal modulation of hypothalamic-pituitary-adrenal axis function. Endocrinology. 2007;148:1574–1581. doi: 10.1210/en.2005-1649. [DOI] [PubMed] [Google Scholar]
- Dallman MF. Fast glucocorticoid actions on brain: back to the future. Front Neuroendocrinol. 2005;26:103–108. doi: 10.1016/j.yfrne.2005.08.001. [DOI] [PubMed] [Google Scholar]
- Devane WA, Dysarz FA, 3rd, Johnson MR, Melvin LS, Howlett AC. Determination and characterization of a cannabinoid receptor in rat brain. Mol Pharmacol. 1988;34:605–613. [PubMed] [Google Scholar]
- Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, Gibson D, Mandelbaum A, Etinger A, Mechoulam R. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science. 1992;258:1946–1949. doi: 10.1126/science.1470919. [DOI] [PubMed] [Google Scholar]
- Di S, Malcher-Lopes R, Halmos KC, Tasker JG. Nongenomic glucocorticoid inhibition via endocannabinoid release in the hypothalamus: a fast feedback mechanism. J Neurosci. 2003;23:4850–4857. doi: 10.1523/JNEUROSCI.23-12-04850.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domenici MR, Azad SC, Marsicano G, Schierloh A, Wotjak CT, Dodt HU, Zieglgansberger W, Lutz B, Rammes G. Cannabinoid receptor type 1 located on presynaptic terminals of principal neurons in the forebrain controls glutamatergic synaptic transmission. J Neurosci. 2006;26:5794–5799. doi: 10.1523/JNEUROSCI.0372-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evanson NK, Tasker JG, Hill MN, Hillard CJ, Herman JP. Fast Feedback Inhibition of the HPA Axis by Glucocorticoids Is Mediated by Endocannabinoid Signalling. Endocrinology. 2010 doi: 10.1210/en.2010-0285. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freund TF, Katona I, Piomelli D. Role of endogenous cannabinoids in synaptic signalling. Physiol Rev. 2003;83:1017–1066. doi: 10.1152/physrev.00004.2003. [DOI] [PubMed] [Google Scholar]
- Ganon-Elazar E, Akirav I. Cannabinoid receptor activation in the basolateral amygdala blocks the effects of stress on the conditioning and extinction of inhibitory avoidance. J Neurosci. 2009;29:11078–11088. doi: 10.1523/JNEUROSCI.1223-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green B, Kavanagh D, Young R. Being stoned: a review of self-reported cannabis effects. Drug Alcohol Rev. 2003;22:453–460. doi: 10.1080/09595230310001613976. [DOI] [PubMed] [Google Scholar]
- Hatfield T, Spanis C, McGaugh JL. Response of amygdalar norepinephrine to footshock and GABAergic drugs using in vivo microdialysis and HPLC. Brain Res. 1999;835:340–345. doi: 10.1016/s0006-8993(99)01566-8. [DOI] [PubMed] [Google Scholar]
- Herkenham M, Lynn AB, Johnson MR, Melvin LS, de Costa BR, Rice KC. Characterization and localization of cannabinoid receptors in rat brain: a quantitative in vitro autoradiographic study. J Neurosci. 1991;11:563–583. doi: 10.1523/JNEUROSCI.11-02-00563.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman JP, Cullinan WE, Ziegler DR, Tasker JG. Role of the paraventricular nucleus microenvironment in stress integration. Eur J Neurosci. 2002;16:381–385. doi: 10.1046/j.1460-9568.2002.02133.x. [DOI] [PubMed] [Google Scholar]
- Hewitt SA, Wamsteeker JI, Kurz EU, Bains JS. Altered chloride homeostasis removes synaptic inhibitory constraint of the stress axis. Nat Neurosci. 2009;12:438–443. doi: 10.1038/nn.2274. [DOI] [PubMed] [Google Scholar]
- Hill MN, Gorzalka BB. The endocannabinoid system and the treatment of mood and anxiety disorders. CNS Neurol Disord Drug Targets. 2009;8:451–458. doi: 10.2174/187152709789824624. [DOI] [PubMed] [Google Scholar]
- Hill MN, McEwen BS. Endocannabinoids: The silent partner of glucocorticoids in the synapse. Proc Natl Acad Sci U S A. 2009;106:4579–4580. doi: 10.1073/pnas.0901519106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill MN, McEwen BS. Involvement of the endocannabinoid system in the neurobehavioural effects of stress and glucocorticoids. Prog Neuropsychopharmacol Biol Psychiatry. 2010;34:791–797. doi: 10.1016/j.pnpbp.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill MN, Carrier EJ, McLaughlin RJ, Morrish AC, Meier SE, Hillard CJ, Gorzalka BB. Regional alterations in the endocannabinoid system in an animal model of depression: effects of concurrent antidepressant treatment. J Neurochem. 2008;106:2322–2336. doi: 10.1111/j.1471-4159.2008.05567.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill MN, Karatsoreos IN, Hillard CJ, McEwen BS. Rapid elevations in limbic endocannabinoid content by glucocorticoid hormones in vivo. Psychoneuroendocrinology. 2010a doi: 10.1016/j.psyneuen.2010.03.005. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill MN, McLaughlin RJ, Morrish AC, Viau V, Floresco SB, Hillard CJ, Gorzalka BB. Suppression of amygdalar endocannabinoid signalling by stress contributes to activation of the hypothalamic-pituitary-adrenal axis. Neuropsychopharmacology. 2009;34:2733–2745. doi: 10.1038/npp.2009.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill MN, McLaughlin RJ, Bingham B, Shrestha L, Lee TT, Gray JM, Hillard CJ, Gorzalka BB, Viau V. Endogenous cannabinoid signalling is essential for stress adaptation. Proc Natl Acad Sci U S A. 2010b;107:9406–9411. doi: 10.1073/pnas.0914661107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillard CJ. Biochemistry and pharmacology of the endocannabinoids arachidonylethanolamide and 2-arachidonylglycerol. Prostaglandins Other Lipid Mediat. 2000;61:3–18. doi: 10.1016/s0090-6980(00)00051-4. [DOI] [PubMed] [Google Scholar]
- Hoffman AF, Lupica CR. Mechanisms of cannabinoid inhibition of GABA(A) synaptic transmission in the hippocampus. J Neurosci. 2000;20:2470–2479. doi: 10.1523/JNEUROSCI.20-07-02470.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamprath K, Wotjak CT. Nonassociative learning processes determine expression and extinction of conditioned fear in mice. Learn Mem. 2004;11:770–786. doi: 10.1101/lm.86104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamprath K, Plendl W, Marsicano G, Deussing JM, Wurst W, Lutz B, Wotjak CT. Endocannabinoids mediate acute fear adaptation via glutamatergic neurons independently of corticotropin-releasing hormone signalling. Genes Brain Behav. 2009;8:203–211. doi: 10.1111/j.1601-183X.2008.00463.x. [DOI] [PubMed] [Google Scholar]
- Kamprath K, Marsicano G, Tang J, Monory K, Bisogno T, Di Marzo V, Lutz B, Wotjak CT. Cannabinoid CB1 receptor mediates fear extinction via habituation-like processes. J Neurosci. 2006;26:6677–6686. doi: 10.1523/JNEUROSCI.0153-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kano M, Ohno-Shosaku T, Hashimotodani Y, Uchigashima M, Watanabe M. Endocannabinoid-mediated control of synaptic transmission. Physiol Rev. 2009;89:309–380. doi: 10.1152/physrev.00019.2008. [DOI] [PubMed] [Google Scholar]
- Karst H, Berger S, Erdmann G, Schutz G, Joels M. Metaplasticity of amygdalar responses to the stress hormone corticosterone. Proc Natl Acad Sci U S A. 2010;107:14449–14454. doi: 10.1073/pnas.0914381107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodirov SA, Jasiewicz J, Amirmahani P, Psyrakis D, Bonni K, Wehrmeister M, Lutz B. Endogenous cannabinoids trigger the depolarization-induced suppression of excitation in the lateral amygdala. Learn Mem. 2009;17:43–49. doi: 10.1101/lm.1663410. [DOI] [PubMed] [Google Scholar]
- Malcher-Lopes R, Di S, Marcheselli VS, Weng FJ, Stuart CT, Bazan NG, Tasker JG. Opposing crosstalk between leptin and glucocorticoids rapidly modulates synaptic excitation via endocannabinoid release. J Neurosci. 2006;26:6643–6650. doi: 10.1523/JNEUROSCI.5126-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrs WR, et al. The serine hydrolase ABHD6 controls the accumulation and efficacy of 2-AG at cannabinoid receptors. Nat Neurosci. 2010;13:951–957. doi: 10.1038/nn.2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsicano G, Wotjak CT, Azad SC, Bisogno T, Rammes G, Cascio MG, Hermann H, Tang J, Hofmann C, Zieglgansberger W, Di Marzo V, Lutz B. The endogenous cannabinoid system controls extinction of aversive memories. Nature. 2002;418:530–534. doi: 10.1038/nature00839. [DOI] [PubMed] [Google Scholar]
- Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature. 1990;346:561–564. doi: 10.1038/346561a0. [DOI] [PubMed] [Google Scholar]
- McGaugh JL. Memory--a century of consolidation. Science. 2000;287:248–251. doi: 10.1126/science.287.5451.248. [DOI] [PubMed] [Google Scholar]
- McGaugh JL. Memory consolidation and the amygdala: a systems perspective. Trends Neurosci. 2002;25:456. doi: 10.1016/s0166-2236(02)02211-7. [DOI] [PubMed] [Google Scholar]
- McGaugh JL. The amygdala modulates the consolidation of memories of emotionally arousing experiences. Annu Rev Neurosci. 2004;27:1–28. doi: 10.1146/annurev.neuro.27.070203.144157. [DOI] [PubMed] [Google Scholar]
- Ohno-Shosaku T, Maejima T, Kano M. Endogenous cannabinoids mediate retrograde signals from depolarized postsynaptic neurons to presynaptic terminals. Neuron. 2001;29:729–738. doi: 10.1016/s0896-6273(01)00247-1. [DOI] [PubMed] [Google Scholar]
- Patel S, Cravatt BF, Hillard CJ. Synergistic interactions between cannabinoids and environmental stress in the activation of the central amygdala. Neuropsychopharmacology. 2005a;30:497–507. doi: 10.1038/sj.npp.1300535. [DOI] [PubMed] [Google Scholar]
- Patel S, Roelke CT, Rademacher DJ, Hillard CJ. Inhibition of restraint stress-induced neural and behavioural activation by endogenous cannabinoid signalling. Eur J Neurosci. 2005b;21:1057–1069. doi: 10.1111/j.1460-9568.2005.03916.x. [DOI] [PubMed] [Google Scholar]
- Patel S, Roelke CT, Rademacher DJ, Cullinan WE, Hillard CJ. Endocannabinoid signalling negatively modulates stress-induced activation of the hypothalamic-pituitary-adrenal axis. Endocrinology. 2004;145:5431–5438. doi: 10.1210/en.2004-0638. [DOI] [PubMed] [Google Scholar]
- Patel S, Kingsley PJ, Mackie K, Marnett LJ, Winder DG. Repeated homotypic stress elevates 2-arachidonoylglycerol levels and enhances short-term endocannabinoid signalling at inhibitory synapses in basolateral amygdala. Neuropsychopharmacology. 2009;34:2699–2709. doi: 10.1038/npp.2009.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pecoraro N, Dallman MF, Warne JP, Ginsberg AB, Laugero KD, la Fleur SE, Houshyar H, Gomez F, Bhargava A, Akana SF. From Malthus to motive: how the HPA axis engineers the phenotype, yoking needs to wants. Prog Neurobiol. 2006;79:247–340. doi: 10.1016/j.pneurobio.2006.07.004. [DOI] [PubMed] [Google Scholar]
- Rademacher DJ, Meier SE, Shi L, Ho WS, Jarrahian A, Hillard CJ. Effects of acute and repeated restraint stress on endocannabinoid content in the amygdala, ventral striatum, and medial prefrontal cortex in mice. Neuropharmacology. 2008;54:108–116. doi: 10.1016/j.neuropharm.2007.06.012. [DOI] [PubMed] [Google Scholar]
- Roozendaal B. 1999 Curt P. Richter award. Glucocorticoids and the regulation of memory consolidation. Psychoneuroendocrinology. 2000;25:213–238. doi: 10.1016/s0306-4530(99)00058-x. [DOI] [PubMed] [Google Scholar]
- Russo EB. History of cannabis and its preparations in saga, science, and sobriquet. Chem Biodivers. 2007;4:1614–1648. doi: 10.1002/cbdv.200790144. [DOI] [PubMed] [Google Scholar]
- Singh ME, Verty AN, Price I, McGregor IS, Mallet PE. Modulation of morphine- induced Fos–immunoreactivity by the cannabinoid receptor antagonist SR 141716. Neuropharmacology. 2004;47:1157–1169. doi: 10.1016/j.neuropharm.2004.08.008. [DOI] [PubMed] [Google Scholar]
- Steiner MA, Wotjak CT. Role of the endocannabinoid system in regulation of the hypothalamic-pituitary-adrenocortical axis. Prog Brain Res. 2008;170:397–432. doi: 10.1016/S0079-6123(08)00433-0. [DOI] [PubMed] [Google Scholar]
- Steiner MA, Marsicano G, Wotjak CT, Lutz B. Conditional cannabinoid receptor type 1 mutants reveal neuron subpopulation-specific effects on behavioral and neuroendocrine stress responses. Psychoneuroendocrinology. 2008;33:1165–1170. doi: 10.1016/j.psyneuen.2008.06.004. [DOI] [PubMed] [Google Scholar]
- Sugiura T, Kondo S, Sukagawa A, Nakane S, Shinoda A, Itoh K, Yamashita A, Waku K. 2-Arachidonoylglycerol: a possible endogenous cannabinoid receptor ligand in brain. Biochem Biophys Res Commun. 1995;215:89–97. doi: 10.1006/bbrc.1995.2437. [DOI] [PubMed] [Google Scholar]
- Ulrich-Lai YM, Herman JP. Neural regulation of endocrine and autonomic stress responses. Nat Rev Neurosci. 2009;10:397–409. doi: 10.1038/nrn2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wade MR, Degroot A, Nomikos GG. Cannabinoid CB1 receptor antagonism modulates plasma corticosterone in rodents. Eur J Pharmacol. 2006;551:162–167. doi: 10.1016/j.ejphar.2006.08.083. [DOI] [PubMed] [Google Scholar]
- Wamsteeker JI, Kuzmiski JB, Bains JS. Repeated stress impairs endocannabinoid signalling in the paraventricular nucleus of the hypothalamus. J Neurosci. 2010;30:11188–11196. doi: 10.1523/JNEUROSCI.1046-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson RI, Nicoll RA. Endocannabinoid signalling in the brain. Science. 2002;296:678–682. doi: 10.1126/science.1063545. [DOI] [PubMed] [Google Scholar]
- Wilson RI, Kunos G, Nicoll RA. Presynaptic specificity of endocannabinoid signalling in the hippocampus. Neuron. 2001;31:453–462. doi: 10.1016/s0896-6273(01)00372-5. [DOI] [PubMed] [Google Scholar]
- Young EA, Akana S, Dallman MF. Decreased sensitivity to glucocorticoid fast feedback in chronically stressed rats. Neuroendocrinology. 1990;51:536–542. doi: 10.1159/000125388. [DOI] [PubMed] [Google Scholar]