

Abstract
The host's immune response to the Hepatitis C virus (HCV) can result in the selection of characteristic mutations (adaptations) that enable the virus to escape this response. The ability of the virus to mutate at these sites is dependent on the incoming virus, fitness cost incurred by the mutation and the benefit to the virus in escaping the response. Studies examining viral adaptation in chronic HCV infection have shown that these characteristic immune escape mutations can be observed at the population level as human leucocyte antigen (HLA)-specific viral polymorphisms. We examined 63 individuals with chronic HCV infection who were infected from a single HCV genotype 1b source. Our aim was to determine the extent to which the host's immune pressure affects HCV diversity and how the sequence of the incoming virus, including pre-existing escape mutations, can influence subsequent mutations in recipients and infection outcome.
Conclusion
HCV sequences from these individuals revealed 29 significant associations between specific HLA types within the new hosts and variations within their viruses which likely represent new viral adaptations. These associations do not overlap with previously reported adaptations for genotype 1a and 3a, possibly reflecting a combination of constraint due to the incoming virus and genetic distance between the strains. However, these sites only accounted for a portion of sites where viral diversity was observed in the new hosts. Furthermore, pre-existing viral adaptations in the incoming (source) virus were likely to have influenced outcome in the new hosts.
Keywords: HCV, viral adaptation, CD8+ T-cells, viral diversity, single source outbreak
Introduction
Following infection with the Hepatitis C virus (HCV), outcome is variable with spontaneous resolution of infection observed in approximately 30% of individuals, but for others chronic infection develops. Factors such as age, gender and host genetic variants have been associated with different infection outcomes (1,2, reviewed in 3). Study cohorts that capture all individuals exposed to the virus, such as HCV single source outbreak cohorts (4,5) and cohorts that consist of individuals that have a high-risk of HCV exposure (6), have been particularly important in delineating relevant viral and host factors associated with the outcome of HCV infection. Such studies corroborate with others that indicate a host's T-cell response to HCV, including genes involved in regulating these responses, are an important correlate of infection outcome (7-11).
T-cell immune responses are stimulated by the presentation of processed viral peptides (epitopes) by human leucocyte antigen (HLA) molecules to CD4+ and CD8+ T-cells. This host-viral interaction is dependent on the sequence of the viral epitope and surrounding regions which play a role in peptide processing and presentation to T-cells. Viral adaptations can reduce the binding affinity of the peptide to the HLA molecule, result in poor peptide cleavage or poor T-cell recognition; factors that can subvert host immune control (reviewed in 12). The importance of immune control in HCV infection is illustrated in studies that show mutations in CD8+ T-cell epitopes contribute to viral persistence in both chimpanzees and humans (13,14). Accordingly, the extent to which the virus can adapt to the host's immune response is likely to be an important factor in determining infection outcome. These adaptations are dependent on the sequence of the incoming virus and the balance between fitness cost incurred by these mutations (15) and their benefit to the virus due to immune escape.
It is unclear how much genetic diversity observed in HCV is the result of host immune pressures. Recent studies suggest that viral adaptation can be observed at both the individual (16,17) and population level (18,19). For example, genetic studies that examine HCV sequences in the context of the HLA repertoire of a host population have shown associations between specific polymorphisms across the viral genome and HLA types within individuals in a host population (18, 19). These HLA-associated viral polymorphisms are thought to represent viral adaptations and tag regions of the viral genome that are under in-vivo T-cell pressure. However, HCV evolution is shaped by evolutionary forces that include genetic drift and both positive and purifying selection pressures (20,21). It is likely all these factors exert their influence simultaneously on the virus, affecting the ability of the virus to adapt to new selection pressures and/or revert in a new host.
A previous study on the Irish single HCV source cohort showed evidence of immune selection in known T-cell targets (22). In this study, we compared HCV sequences from 63 individuals with genotype 1b infection from this single source outbreak (5) to identify sites that are likely to represent new T-cell targets in the HCV genome and to determine the extent to which the hosts' immune pressures on the virus affect sequence diversity in the cohort. Knowledge of the incoming viral sequence also allows us to determine if pre-existing viral adaptations can predict beneficial or detrimental host HLA alleles within the cohort in relation to infection outcome.
Materials and Methods
Study Population
The study population is part of a cohort of women who had been infected with HCV between May 1977 and November 1978 in Ireland through the administration of anti-D immunoglobulin that had been contaminated with a HCV genotype 1b virus originating from a single individual (5). From this original cohort, we studied 63 individuals with chronic HCV infection of which a subset (n=15) had been selected based on the carriage of HLA-A*03; an allele which has previously been shown to be protective in this cohort (8). A comparison of the HLA alleles found in this cohort to another Irish population is in the supplementary material.
Serum samples from the subjects were collected between 1996 and 2002 and stored at -80°C. Written informed consent was obtained from participants and local institutional review board approval was obtained by all centers contributing to the study.
Viral RNA extraction
Viral RNA was extracted from sera samples using the QIAamp Viral RNA Mini Kit (QIAGEN) or the COBAS AMPLICOR HCV Specimen Preparation Kit v2.0 (Roche) according to manufacturers' instructions.
HLA genotyping
Two-digit resolution HLA Class I (HLA-A, -B and -C) typing was performed at St James Hospital, Dublin Ireland (8).
IL28B genotyping
Genotyping of the single nucleotide polymorphism (SNP) rs12979860 upstream of the IL28B gene was performed for 34 subjects as previously described (23).
Bulk viral sequencing
HCV sequencing was performed as previously described (18,19). Briefly, three RT-PCRs were performed to cover the Core to Nonstructural (NS) 5B region. The first-round products were used as templates in nested second round PCRs containing generic or genotype specific primers. Amplicons were bulk sequenced using the BigDye® Terminator v3.1 cycle sequencing Kit (Applied Biosystems) according to manufacturer's recommendations and electropherograms edited using Assign™ (Conexio Genomics). Mixtures were identified where the secondary peak was >20% of the main peak.
HCV sequences in this study have been submitted to GenBank (accession numbers HM106522 to HM106981). Supplementary Table 1 lists the mean sequence coverage by protein.
An analysis of the viral sequences to test the single source nature of this outbreak can be found in the supplementary material.
Ultra-deep sequencing
To identify minor quasispecies below the detection threshold of bulk sequencing methods, ultra-deep sequencing was carried-out using the 454 Life Sciences platform (Roche Applied Science) for two individuals (HLA-A*03+/-B*08- and HLA-A*03-/-B*08-). PCR templates using the amplification method described above were obtained that covered NS3 (positions 3494-4530) and NS5A to NS5B (positions 7335–8356). Amplicons were quantified and pooled for each individual. Library preparation and sequencing was performed according to manufacturer's protocol. Data was collected and analysed using Roche and public license software programs. All sequence reads were aligned to the source sequence (AF313916) using GS Reference Mapper software (Roche). Threshold for mixtures was set at 1% with ≥100-fold coverage.
HLA-associated viral polymorphisms
Associations between HLA alleles and amino acid distribution at each residue of the HCV proteins were assessed via Fisher's exact tests for classification as consensus vs. non-consensus amino acid. A false discovery rate analysis was carried-out and q-values obtained as in (19). Only sequences with ≥50% sequence coverage for each respective protein were used. Analyses were carried-out using TIBCO Spotfire S+ 8.1 (Somerville, MA). Associations with p-value≤0.01 for the Fisher's exact test of consensus vs non-consensus are reported. Assessment of possible confounding by founder effects via viral cluster stratification and the Mantel-Haenszel procedure as described in (19) indicated no correction for significant associations was necessary, consistent with the sequences originating from a single source. In addition, since p-values associated with relatively small frequencies can be affected by small numbers of misclassified cases, we restricted to associations for which there were ≥5 non-consensus amino acids and ≥5 carriers of the HLA allele.
Sliding window analysis
In order to identify viral escape that may not be captured using a single amino acid approach, an analysis was conducted as described above with the exception that “adaptation” was defined as non-consensus at any residue within sliding windows of nine amino acids, representing typical peptide sizes for HLA Class I molecules. Significant sites of associations were identified as strings of “significant” values while the window slides over any residues containing strong associations or combinations of associations. We restricted the analysis to cases that had all amino acids in the window. Associations with p≤0.01 were reported.
Co-variation
The assessment of residue co-variation utilised Fisher's exact tests for classification as consensus vs non-consensus amino acid. Co-variation based on sequence with ≥90% coverage was reported where co-varying sites have a p≤0.001 for amino acid versus amino acid comparison and p≤0.0001 for amino acid versus nucleotide comparison. Given the exploratory nature of this part of the analysis, no adjustment was made for multiple comparisons.
Peptide prediction for HLA-associated viral polymorphism sites
Flanking sequences of the identified HLA-associated viral polymorphisms and sites of common divergence from the source sequence were entered into the epitope prediction software SYFPEITHI (24) to identify putative epitopes based on a cut-off score of 20 with the highest scoring peptide reported. HLA-associated viral polymorphism sites were compared against published genotype 1 epitopes found in the Immune Epitope Database (http://www.immuneepitope.org/).
Viral sequence diversity
Sequence diversity from the source sequence (AF313916) was determined using the program Highlighter (available from http://www.hiv.lanl.gov) for NS3 and NS5B to identify sites of synonymous and nonsynonymous substitutions for sequences with >50% sequence coverage. Genetic diversity was determined using the Kimura-2-parameter model and differences in rate of nonsynonymous and synonymous change (ds-dn) were obtained using the Modified Nei and Gojobori method with MEGA v3.1 (25).
IL28B-associated viral polymorphisms
We assessed associations between the presence or absence of the minor allele rs12979860 and consensus vs. non-consensus amino acid at each residue of the HCV proteins via Fisher's exact test. Given the smaller number of subjects with typing available for this part of the analysis, no assessment of false discovery rates was made and p≤0.01 was used to indicate significance.
Results
HLA-associated viral polymorphisms: putative viral adaptations in the new hosts reflecting sites of immune pressure
We determined if there were associations between the expression of particular HLA alleles in subjects in this cohort and specific polymorphisms in their viral sequences (putative viral adaptations) reflecting areas under in-vivo T-cell immune pressure. We identified 29 HLA-associated viral polymorphisms with p ≤0.01 for 23 sites along the HCV genome (Table 1 and Supplementary Figure 3). In some instances HLA alleles from different loci were associated with the same site and we have previously shown (18) these associations can, in part, be explained by the linkage disequilibrium that is observed within the Major Histocompatiblity Complex (MHC). Of those associations shown in Table 1, three HLA-B/C combinations are associated with common MHC haplotypes. The q values for associations within some of the proteins are high relative to others (particularly E2), possibly reflecting smaller sample sizes in these proteins (Supplementary Table 1).
Table 1. HLA Class I -associated viral polymorphisms.
| Protein | Residue | Source AA | Cohort AA# | Variant AA in Cohort | HLA | Odds Ratio | p value | q value | Sliding Window* | Predicted Epitope | Published Epitopeˆ |
|---|---|---|---|---|---|---|---|---|---|---|---|
| E1 | 299 | E | E | V/R/Q/L/K | C*16 | 24.00 | 0.003 | 0.298 | 291, 292, 298, 299 | ||
| 373 | I | I | V | C*16 | 18.00 | 0.006 | 0.357 | 365 | |||
| E2 | 393 | A | A | G | C*06 | 11.00 | 0.008 | 0.739 | |||
| 397 | R | S | H/R/L/F/S/N | A*01 | 0.05 | 0.010 | 0.739 | ||||
| 400 | A/Tˆˆ | T | A/V | C*07 | 0.08 | 0.005 | 0.730 | ||||
| 403 | F | F | L | A*02 | 30.00 | 0.004 | 0.730 | SLLAPGAKQNV | |||
| 405 | S | S | T/A | C*16 | 0.04 | 0.006 | 0.730 | ||||
| 577 | D | D | N/B | A*01 | 0.12 | 0.010 | 0.739 | ||||
| 625 | T | T | S | B*08 | 11.00 | 0.006 | 0.730 | ||||
| P7 | 790 | F | F | l/I | B*07 | 11.00 | 0.001 | 0.037 | 786-794 | ||
| NS2 | 834 | H | H | Q | B*08 | 15.00 | 0.001 | 0.045 | 836-838 | SPHYKVFL | |
| 951 | D | D | N | A*02 | 12.00 | 0.003 | 0.108 | 950-955 | |||
| C*05 | 11.00 | 0.004 | 0.148 | ||||||||
| NS3 | 1040 | L | L | F | B*07 | 12.00 | 0.007 | 0.162 | 1036-1038 | ||
| 1087 | T | T | A | A*03 | 17.00 | 0.001 | 0.030 | 1083-1091 | TVYHGAGTK | ||
| 1088 | K | K | R | A*03 | 57.00 | 2.86×10-6 | 0.000 | ||||
| 1130 | L | L | P/M | B*14 | 17.00 | 0.009 | 0.245 | ||||
| C*08 | 36.00 | 0.001 | 0.035 | 1126-1132 | |||||||
| 1282 | V | V | I | A*02 | 25.00 | 0.005 | 0.128 | 1278 | NIRTGVRTI | ||
| B*14 | 35.00 | 0.001 | 0.035 | 1282, 1283 | |||||||
| C*08 | 26.00 | 0.002 | 0.060 | 1282, 1283 | |||||||
| 1370 | I | I | T/V/D | B*07 | 0.09 | 0.002 | 0.066 | ||||
| NS4B | 1958 | K | K | R | C*08 | 10.00 | 0.009 | 0.059 | 1960 | ||
| NS5A | 2143 | E | E | D | C*16 | 17.00 | 0.008 | 0.553 | |||
| NS5B | 2518 | K | K | R | A*03 | 11.00 | 0.001 | 0.032 | 2520, 2521 | SLTPPHSAK | |
| C*08 | 9.70 | 0.005 | 0.203 | ||||||||
| 2609 | S | S | P | B*35 | 29.00 | 9.30×10-5 | 0.000 | 2605-2613 | |||
| C*04 | 19.00 | 3.82×10-4 | 0.019 | 2608-2613 | |||||||
| 2821 | R | R | K | B*18 | 21.00 | 0.005 | 0.203 | 2821-2825 |
AA = amino acid; numbering from the start of the polyprotein.
AA difference in two source sequences (AF313916 and DQ061375-DQ061378 separated by forward slash).
Median point.
Consensus amino acid. Bold and italicized HLA alleles are likely to form part of an MHC haplotype. Underlined AA is site of interest. Note E2 contains HLA-associated sites that fall within the hypervariable region (393, 397, 400, 403, 405).
Two HLA-associated viral polymorphisms fell within previously published epitopes (HLA-A*02 epitope in E2 404 SLLAPGAKQNV and HLA-A*03 epitope in NS5B 2518 SLTPPHSAK; Table 1). Furthermore, three HLA-associated viral polymorphisms fell within predicted epitopes as determined by the peptide binding prediction program, SYFPEITHI (24) (Table 1). The limited number of matches between known epitopes and putative viral adaptation sites may be the result of the small number of published HCV epitopes in the literature and their focus on common HLA types. Several of the putative viral adaptations are associated with HLA-C alleles for which there are either no or few known HLA-restricted epitopes or characterized binding properties.
None of the associations shown in Table 1 overlap with our previous studies that have examined HLA-associated viral polymorphisms for genotype 1 (18,19). However, the previous study had a much larger number of genotype 1a sequences in the dataset than 1b and given that the sequences in this single source cohort are all genotype 1b it is likely that we will observe differential escape profiles similar to what we have seen between genotype 1a and 3a but to a lesser extent between genotype 1 subtypes (1a and 1b). Furthermore, the subjects in this study have been infected from a single source strain in contrast to the subjects in the previous cross-sectional studies.
Window analysis identifies additional areas under T-cell pressure
Areas under HLA-specific immune pressure that can accommodate more than one site of variation may not be detected by our initial single amino acid approach. Accordingly, a sliding window analysis (with a size reflective of a typical HLA Class I epitope) was also performed to examine areas under HLA-specific immune pressure in which more than one site may be relevant for escape. As expected, several of the HLA-associated viral polymorphisms identified using a single site analysis were identified using the window analysis (Table 1). However, the single site associations found in highly variable regions (HVR) in E2 were not identified in the window analysis, probably due to the higher level of variation found in this region compared to other proteins that may occur in some cases when variation is not related to adaptation (as tested for here) and hinder the ability to find specific HLA associations with any change(s) within a window. There were three examples (E2 and HLA-C*06 with median position 537, odds radio (OR)=28; NS2 and HLA-B*08 within windows 875-878, OR 0.026-0.039; NS5A and HLA-B*08 with median position 2132, OR=26) in which the window analysis identified HLA-associated substitutions that were not found to be significant in the single site analysis. These cases suggest multiple sites within a target region may be under immune pressure (Supplementary Figure 4). This observation is consistent with our own, and other, studies that show different escape profiles within epitopes including the immunodominant HLA-B*08 epitope (1395-1403) in NS3 (17) and the protective HLA-B*27 epitope (2841-2849) in NS5B (11).
Overall, the number of associations found either with the single site analysis or sliding window analysis represents only a portion of the 184 variable sites across the viral genome that fit into the inclusion criteria described in the methods (18/163 if HVR in E2 is excluded given that this area is likely to also be under other strong selective pressures).
Source and causes of viral adaptation
We then examined the pattern of synonymous and nonsynonymous changes in these sequences to determine if purifying selection was acting across the HCV genome, potentially restricting the ability of the virus to adapt to new selection pressures or revert to non-adapted forms. Figure 1 shows the pattern of these changes in each individual relative to the source within the NS3 and NS5B proteins. It is apparent that there are a greater number of synonymous changes relative to nonsynonymous changes in this region (indicating purifying or negative selection). Similar results are observed for other proteins (data not shown).
Figure 1. Highlighter plot of synonymous and nonsynonymous substitutions in NS3 and NS5B in relation to source sequence (AF313916).

Plot created by Highlighter (available from www.lanl.gov). Red lines denote nonsynonymous substitutions, green lines indicate synonymous substitutions and grey regions show un-sequenced sections.
Co-varying sites in genome likely to reflect networks within HCV genome
As previously suggested, purifying selection may reflect the existence of co-varying sites in the HCV genome (26). Here we identified sites of covariance by assessing amino acid sites in a pair-wise manner per protein and genome-wide for sequences with greater than 90% sequence coverage. Only results with p<0.001 were reported as adjustment for multiple comparisons was not made in this analysis. 13/25 paired sites of significant covariance were within the same protein, while 12/25 fell in different proteins. For the majority of pairs of covariant sites, one or both sites fell at a reported HLA-associated viral polymorphism site, within a known epitope or at a common site of reversion from the source. Four of the 25 paired sites fell at a HLA-associated site in Table 1. In particular, two HLA-A*03-associated sites at positions 1087 and 1088 in NS3 fall within a confirmed HLA-A*03 epitope in which variation at both sites is required to restore replicative efficacy (Fitzmaurice et al, manuscript submitted), reflecting the potential compensatory nature of these co-variations.
Figure 2 shows a linear trend for many co-varying sites suggesting that many fell in close proximity to one another but not necessarily in the same protein. Interestingly, clusters of co-varying sites appeared to connect sites across the genome, particularly other proteins with NS5A. One group contains sites in only one protein (NS3 sites 1644F/Y, 1647A/T and 1656A/T) while another group contained sites in three proteins (NS2 908R/K, NS3 1173S/L and NS5A 2279R/K). These links may further restrict the ability of the virus to adapt or revert quickly and suggests critical interactions between the HCV proteins. We extended this analysis to assess co-variation at amino acid and synonymous sites to identify potential constraint on codon usage (and subsequent amino acid changes) and identified 4 amino acid sites associated with synonymous changes in other proteins.
Figure 2. Co-varying sites (p<0.001) in the HCV genome represented as coordinates.

Open diamond denotes one or both sites fall within an epitope or at an association site, and dark diamonds denote the sites do not fall within either. Many co-variant sites fall in close proximity to one another in the genome (illustrated by the linear trend) however there are groupings that suggest strong co-variation between residues within NS5A and residues in other proteins. Sequence coverage was not found to be a function of co-variant site identification.
Relevance of viral adaptations in the new hosts and pre-existing in source in infection outcome
Although the host immune pressure is one of several forces shaping HCV diversity, it is likely that only a small number of selected viral adaptations in the sequence may affect infection outcome. In this cohort, HLA-A*03 has been shown to be protective (8) and chronic HCV infected individuals with HLA-A*03 were selected for this study to identify viral adaptations in these individuals that may have affected their infection outcome. Three viral polymorphisms were associated with HLA-A*03 in this study (Table 1). Two of the associations were in NS3 at positions 1087 and 1088 within a predicted epitope for HLA-A*03. As mentioned above, this epitope has subsequently been shown to be a true in-vivo target of the immune response (NS3 - 1080 TVYHGAGTK) (Fitzmaurice et al, manuscript submitted) (Figure 3A) and reflects a drop in the SYFPEITHI predicted binding score from 34 for the wildtype to 21 for the putative escape peptide. Another HLA-A*03 associated viral polymorphism at position 2518 in NS5B is within a previously characterized genotype 1a epitope SLTPPHSAK (Figure 3B). Half of the HLA-A*03 individuals had a polymorphism at these sites in both regions. These results suggest that these two viral epitopes are important immune targets and escape within the targets may influence outcome.
Figure 3. HLA-A*03-associated viral polymorphisms at positions (A) NS3 1087, 1088 and (B) NS5B 2518.
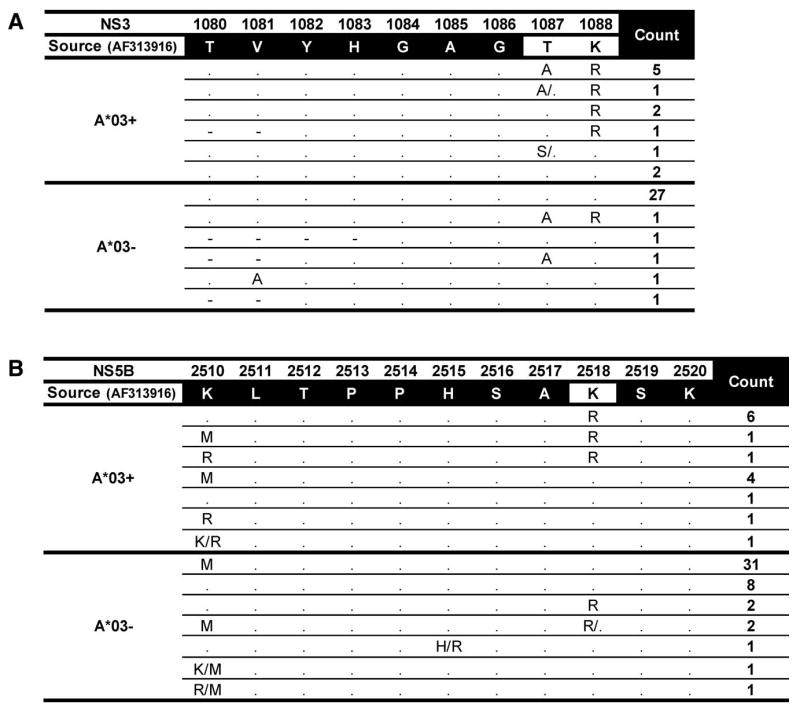
Sequences in regions of interest (from Table 1) are displayed for HLA-A*03 positive and negative subjects. Sequence identity with the source sequence identified by dot. Amino acid mixtures at a site are separated by a forward slash. Number of individuals with a particular sequence is shown in the count column. The lysine (K) to arginine (R) substitution at 2518 (8/15 HLA-A*03+ subjects versus 4/47 HLA-A*03- subjects) results in a change in the SYFPEITHI predicted binding score from 27 to 21. Only one HLA-A*03 individual with chronic infection did not have a polymorphism at either the 1087 or 1088 sites in NS3 or the 2518 site in NS5B.
Further analysis of the quasispecies at the NS3 1087 and 1088 sites in HLA-A*03 positive and negative subjects was performed using ultra-deep sequencing. Table 2 reveals the lack of source sequence at amino acid position NS3 1088 in the HLA-A*03 subject with complete amino acid replacement but 100% retainment of the source sequence in the HLA-A*03 negative subject. The two subjects have the same amino acid at position 1087 (non-adapted) but codon usage is different between the two.
Table 2. Ultra-deep sequencing reveals lack of source sequence at putative viral adaptation sites (NS3 1087 and 1088) in a subject with HLA-A3 but 100% maintenance of source sequence in an HLA-A3 negative subject&.
| Sequence | Position | HLAˆ | ||||||
|---|---|---|---|---|---|---|---|---|
| 1087 | 1088 | A*03+ | A*03- | |||||
| Source | T | K | 0 | 346 | ||||
| A | C | A | A | A | G | |||
| Variant 1 | T | K | 0 | 685 | ||||
| A | C | T | A | A | G | |||
| Variant 2 | T | R | 249 | 0 | ||||
| A | C | A | A | G | G | |||
| Variant 3 | T | R | 1111 | 0 | ||||
| A | C | G | A | G | G | |||
The HLA-A3+ subject only carries species that differ at the amino acid level to the source. Although the two subjects have the same amino acid at position 1087, codon usage is different. Region had more than 1000 fold coverage.
Indicates number of sequence reads with corresponding variant or source combination.
Previous studies have found other HLA alleles to be associated with chronic infection that have been specific to this cohort, for example the alleles HLA-A*01, -B*08 and -C*07 (8) (these alleles most likely correspond to a single MHC haplotype). It has been suggested that the association between infection outcome and specific HLA alleles may be due to pre-existing viral adaptations in the incoming virus that may facilitate the evasion of host immune responses with the corresponding HLA types (27). Here, we test this hypothesis by examining the source sequence for escape mutations within known epitopes as well as putative viral adaptations identified in our previous genetic study of chronic HCV infection (18,19).
Initially, we examined the immunodominant epitope for HLA-B*08 in NS3 (1395 HSKKKCDEL) and the protective HLA-B*27 epitope in NS5B (2841 ARMILMTHF). The region in the source containing the HLA-B*27 epitope in NS5B has the non-mutated form. However, the HLA-B*08 epitope in NS3 in the source sequence has a pre-existing viral adaptation in the epitope (arginine at position 3), which has subsequently reverted in 8/11 subjects without HLA-B*08 and been retained in 5/8 subjects that express HLA-B*08. Although the numbers in the two groups are not significantly different (p=0.18), it supports other studies that show reversion from an arginine to lysine at position 3 in this epitope when there is no immune pressure; suggestive of a fitness cost (15). This HLA-B*08 epitope has been previously studied in this cohort with similar results (15,22). The fitness cost of this substitution is further supported by the results from the ultra-deep sequencing of two HLA-B*08 negative subjects in this region, showing complete reversion from the source escape mutation at position 3 of the epitope (Table 3).
Table 3. Ultra-deep sequencing reveals lack of source sequence at position 1397 in immunodominant HLA-B*08 epitope in NS3 (HSKKKCDEL) in two HLA-B*08 negative subjects.
| Sequence | Position | HLAˆ | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1397 | 1398 | 1399 | A*03+ /B*08- | A*03-/B*08- | |||||||
| Source | R | K | K | 0 | 0 | ||||||
| A | G | G | A | A | A | A | A | A | |||
| Variant 1 | K | K | K | 526 | 358 | ||||||
| A | A | G | A | A | G | A | A | A | |||
Indicates number of sequence reads with corresponding variant or source combination.
Viral adaptation in the source sequence at a site in the HLA-B*08 immunodominant epitope likely to incur a fitness cost suggests that the source may have been an HLA-B*08 positive individual. We would suggest that this could potentially reduce the ability of hosts with HLA-B*08 to control the virus via the reduction of good immune targets; reflecting the association of this allele with poor outcome in this cohort. Additional association sites with HLA-B*08+ individuals found in this study may represent alternative targets for HLA-B*08 along the HCV genome. Furthermore, Table 1 and Figure 1 lists HLA-associated viral polymorphisms that have an odds ratio less than 1, representing the maintenance of the consensus sequence (for most sites in Table 1 same as source) for the specific HLA type, possibly reflecting that the source sequence is pre-adapted at these sites. Interestingly, this occurs for alleles within the MHC haplotype HLA-A*01, -B*08, -C*07 associated with poor outcome.
Other selective pressures likely to affect HCV evolution
In order to determine how other host immune pressures may affect HCV evolution we assessed possible associations between HCV polymorphisms in this cohort and a single nucleotide polymorphism (SNP) which tags the IL28B gene that encodes IFN-lambda 3 and which has recently been associated with infection outcome (2). We found one significant association between homozygosity for the major allele of rs12979860 (associated with good outcome) and variation at position 849 in NS2 (p=0.006). We also tested for additional effects of the IL28B SNP on the HLA-associated polymorphisms. After adjusting for HLA, among the positions identified in Table 1, IL28B was associated with polymorphism (p=0.036) only at position 2609 of NS5B which harbours the strong HLA-B*35/-C*04 association. The significance of the HLA-B*35 association with non-consensus after adjusting for the IL28B SNP is p=0.00004 while for HLA-B*35 alone p=0.0001. There was no significant interaction between effects of HLA-B*35 and IL28B (p>0.9), suggesting they act independently. Further studies examining the association between variations that tag IL28B and HCV evolution are warranted and should be performed on larger cohorts that include subjects with different treatment and infection outcomes.
Discussion
Here we illustrate that incoming viral sequence, host immune pressure and co-variation play an important role in shaping HCV viral diversity. Specifically, we identified 29 significant HLA-associated viral polymorphisms (p≤0.01;23 sites) within the cohort which likely reflect viral adaptations. Some of these sites fall within published and/or predicted T-cell epitopes. The use of a sliding window analysis that accounts for more than a single escape variant within a T-cell target identified a small number of additional potential regions under T-cell pressure, supporting other studies that show that escape can require the accumulation of escape mutations (28) or that viral escape sites are often mutually exclusive due to fitness cost (15,18).
The number of significant HLA-associated viral polymorphism sites identified in this study was only a small proportion (23/184) of the sites across the HCV genome that showed variation in the cohort, possibly due to the relatively small sample size or suggesting that the host immune pressure has a targeted influence on HCV diversity. This would be expected as the immune system sees the viral polyprotein as a set of peptides and of these peptides only a small number are likely to be presented to the immune system. Furthermore, the lack of significant overlap with previously reported adaptations for genotypes 1a and 3a, likely reflects constraint of incoming virus and differential viral adaptation pathways on genotype 1b versus the other circulating genotypes due to the genetic distance between these strains. It should be noted that although we did not show HLA Class II-associated viral polymorphisms it is likely that some of the variation may, in addition to what we observe for HLA Class I alleles, correspond to the expression of specific HLA Class II alleles.
To appreciate the extent to which both positive and purifying selections influence HCV diversity, we examined the number of synonymous and nonsynonymous changes across the genome for this single source cohort. An abundance of synonymous changes indicated purifying selection that will to some extent limit the plasticity of HCV. Co-variations which become fixed across the HCV genome may also restrict the ability of HCV to adapt to the host's immune response as well as revert upon entering a new non-HLA matched host. We examined the genome for co-varying sites and showed that although co-variation did occur locally within proteins, there were also a number of sites that were linked to sites more distant in the genome. Furthermore, several of these sites were putative viral adaptation sites.
Access to the source viral sequence from this single source cohort allowed for the identification of pre-existing escape mutations across the genome. A known escape mutation at position 3 of the immunodominant HLA-B*08 NS3 epitope was found in the source sequence. This mutation was for the most part retained in HLA-B*08 subjects but had reverted in most HLA-B*08 negative subjects. Furthermore, deep sequencing revealed no traces of the escape mutant in two B*08 negative individuals, supporting the fitness cost that may be incurred by the escape mutation. Importantly, existing adaptation in the incoming virus may affect infection outcome in individuals that express the appropriate HLA type. The pre-adaptation of the source sequence to HLA-B*08 may account for the observed lack of protection of HLA-B*08 in this cohort.
The single source cohort studied here has provided the opportunity to obtain a better understanding of viral diversity and how different forces can shape viral diversity at the population level.
Supplementary Material
Acknowledgments
We thank the patients and clinical staff who participated in this study and our colleagues within the Centre for Clinical Immunology and Biomedical Statistics, Murdoch University.
Grant Support: This work was supported by the National Health and Medical Research Council of Australia (program grant 384702), Wellcome Trust, NIHR Biomedical Research Centre Programme (Oxford), James Martin School for 21st Century (Oxford) and NIH U19 (NIAID 1U19AI082630-01). Contribution to this project by S. Merani was conducted while receiving a University International Stipend award and a Scholarship for International Research Fees award from the University of Western Australia, as well as funding from the Centre for Clinical Immunology and Biomedical Statistics, Murdoch University.
Abbreviations
- HCV
Hepatitis C virus
- HLA
Human Leucocyte Antigen
- NS
Nonstructural
- MHC
Major Histocompatiblity Complex
- OR
odds ratio
- SNP
single nucleotide polymorphism
Footnotes
Conflict of interest: The authors do not have any competing financial interests or conflicts of interest.
All authors have viewed the submitted manuscript and agree with its contents.
Genetic Sequence Data: HCV sequences described in this manuscript have been submitted to GenBank with the following accession numbers: HM106522 to HM106981.
Contributor Information
Shahzma Merani, Email: merans01@student.uwa.edu.au.
Danijela Petrovic, Email: petrovid@tcd.ie.
Ian James, Email: i.james@murdoch.edu.au.
Abha Chopra, Email: a.chopra@iiid.com.au.
Don Cooper, Email: d.cooper@iiid.com.au.
Elizabeth Freitas, Email: liz.freitas@iinet.net.au.
Andri Rauch, Email: andri.rauch@insel.ch.
Julia di Iulio, Email: julia.di-iulio@chuv.ch.
Mina John, Email: m.john@iiid.com.au.
Michaela Lucas, Email: michaela.lucas@health.wa.gov.au.
Karen Fitzmaurice, Email: kfitzmau@tcd.ie.
Susan McKiernan, Email: smckiernan@stjames.ie.
Suzanne Norris, Email: snorris@stjames.ie.
Dermot Kelleher, Email: kellehdp@tcd.ie.
Paul Klenerman, Email: paul.klenerman@ndm.ox.ac.uk.
Silvana Gaudieri, Email: silvana.gaudieri@uwa.edu.au.
References
- 1.Poynard T, Bedossa P, Opolon P. Natural history of liver fibrosis progression in patients with chronic hepatitis C. Lancet. 1997;349:825–832. doi: 10.1016/s0140-6736(96)07642-8. [DOI] [PubMed] [Google Scholar]
- 2.Thomas DL, Thio CL, Martin MP, Qi Y, Ge D, O'hUigin C, et al. Genetic variation in IL28B and spontaneous clearance of hepatitis C virus. Nature. 2009;461:798–U752. doi: 10.1038/nature08463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rauch A, Kutalik Z, Descombes P, Cai T, Iulio J, Mueller T, et al. Genetic variation in IL28β is associated with chronic hepatitis C and treatment failure: A genome wide association study. Gastroenterology. 2010;138:1338–U175. doi: 10.1053/j.gastro.2009.12.056. [DOI] [PubMed] [Google Scholar]
- 4.Wiese M, Grungreiff K, Guthoff W, Lafrenz M, Oesen U, Porst H. Outcome in a hepatitis C (genotype 1b) single source outbreak in Germany - a 25-year multicenter study. J Hepatol. 2005;43:590–598. doi: 10.1016/j.jhep.2005.04.007. [DOI] [PubMed] [Google Scholar]
- 5.Kenny-Walsh E. Clinical outcomes after hepatitis C infection from contaminated anti-D immune globulin. N Engl J Med. 1999;340:1228–1233. doi: 10.1056/NEJM199904223401602. [DOI] [PubMed] [Google Scholar]
- 6.Dore GJ, MacDonald M, Law MG, Kaldor JM. Epidemiology of hepatitis C virus infection in Australia. Aust Fam Physician. 2003;32:796–798. [PubMed] [Google Scholar]
- 7.Missale G, Bertoni R, Lamonaca V, Valli A, Massari M, Mori C, et al. Different clinical behaviors of acute hepatitis C virus infection are associated with different vigor of the anti-viral cell-mediated immune response. J Clin Invest. 1996;98:706–714. doi: 10.1172/JCI118842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McKiernan SM, Hagan R, Curry M, McDonald GSA, Kelly A, Nolan N, et al. Distinct MHC class I and II alleles are associated with hepatitis C viral clearance, originating from a single source. Hepatology. 2004;40:108–114. doi: 10.1002/hep.20261. [DOI] [PubMed] [Google Scholar]
- 9.Fanning LJ, Levis J, Kenny-Walsh E, Wynne F, Whelton M, Shanahan F. Viral clearance in hepatitis C (1b) infection: Relationship with human leukocyte antigen class II in a homogeneous population. Hepatology. 2000;31:1334–1337. doi: 10.1053/jhep.2000.7437. [DOI] [PubMed] [Google Scholar]
- 10.Barrett S, Ryan E, Crowe J. Association of the HLA-DRB1*01 allele with spontaneous viral clearance in an Irish cohort infected with hepatitis C virus via contaminated anti-D immunoglobulin. J Hepatol. 1999;30:979–983. doi: 10.1016/s0168-8278(99)80249-9. [DOI] [PubMed] [Google Scholar]
- 11.Neumann-Haefelin C, McKiernan S, Ward S, Viazov S, Spangenberg HC, Killinger T, et al. Dominant influence of an HLA-B27 restricted CD8+ T cell response in mediating HCV clearance and evolution. Hepatology. 2006;43:563–572. doi: 10.1002/hep.21049. [DOI] [PubMed] [Google Scholar]
- 12.Bowden DG, Walker CM. Mutational escape from CD8 cell immunity: HCV evolution, from Chimpanzees to man. J Exp Med. 2005;201:1709–1714. doi: 10.1084/jem.20050808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Erickson AL, Kimura Y, Igarashi S, Eichelberger J, Houghton M, Sidney J, et al. The outcome of hepatitis C virus infection is predicted by escape mutations in epitopes targeted by cytotoxic T lymphocytes. Immunity. 2001;15:883–895. doi: 10.1016/s1074-7613(01)00245-x. [DOI] [PubMed] [Google Scholar]
- 14.Cox AL, Mosbruger T, Mao Q, Liu Z, Want XH, Yang HC, et al. Cellular immune selection with hepatitis C virus persistence in humans. J Exp Med. 2005;201:1741–1742. doi: 10.1084/jem.20050121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Salloum S, Oniangue-Ndza C, Neumann-Haefelin C, Hudson L, Giugliano S, Siepen MAD, et al. Escape from HLA-B*08-restricted CD8 T cells by hepatitis C virus is associated with fitness costs. J Virol. 2008;82:11803–11812. doi: 10.1128/JVI.00997-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cox AL, Mosbruger T, Lauer GM, Pardoll D, Thomas DL, Ray SC. Comprehensive analyses of CD8+ T cell responses during longitudinal study of acute human hepatitis C. Hepatology. 2005;42:104–112. doi: 10.1002/hep.20749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Timm J, Lauer GM, Kavanagh DG, Sheridan I, Kim AY, Lucas M, et al. CD8 epitope escape and reversion in acute HCV infection. J Exp Med. 2004;200:1593–1604. doi: 10.1084/jem.20041006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gaudieri S, Rauch A, Park LP, Freitas E, Herrmann S, Jeffrey G, et al. Evidence of viral adaptation to HLA class I-restricted immune pressure in chronic hepatitis C virus infection. J Virol. 2006;80:11094–11104. doi: 10.1128/JVI.00912-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rauch A, James I, Pfafferott K, Nolan D, Klenerman P, Cheng W, et al. Divergent adaptation of hepatitis C virus genotypes 1 and 3 to human leukocyte antigen-restricted immune pressure. Hepatology. 2009;50:1017–1029. doi: 10.1002/hep.23101. [DOI] [PubMed] [Google Scholar]
- 20.Manzin A, Solforosi L, Debiaggi M, Zara F, Tanzi E, Romano L, et al. Dominant role of host selective pressure in driving hepatitis C virus evolution in perinatal infection. J Virol. 2000;74:4327–4334. doi: 10.1128/jvi.74.9.4327-4334.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kuntzen T, Timm J, Berical A, Lewis-Ximenez LL, Jones A, Nolan B, et al. Viral sequence evolution in acute hepatitis C virus infection. J Virol. 2007;81:11658–11668. doi: 10.1128/JVI.00995-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ray SC, Fanning L, Wang XH, Netski DM, Kenny-Walsh E, Thomas DL. Divergent and convergent evolution after a common-source outbreak of hepatitis C virus. J Exp Med. 2005;201:1753–1759. doi: 10.1084/jem.20050122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Urban TJ, Thomas AJ, Bradrick B, Fellay J, Schuppan D, Cronin KD, et al. IL28 genotyping is associated with differential expression of intrahepatic interferon-stimulated genes in chronic hepatitis C patients. Hepatology. doi: 10.1002/hep.23912. online. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rammensee H, Bachmann J, Emmerich NN, Oskar Alexander Bachor OA, Stevanovic S. SYFPEITHI: Database for MHC ligands and peptide motifs. Immunogenetics. 1999;50:213–219. doi: 10.1007/s002510050595. [DOI] [PubMed] [Google Scholar]
- 25.Kumar S, Nei M, Dudley J, Tamura K. MEGA: A biologist-centric software for evolutionary analysis of DNA and protein sequences. Brief Bioinform. 2008;9:299–306. doi: 10.1093/bib/bbn017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Campo DS, Dimitrova Z, Mitchell RJ, Lara J, Khudyakov Y. Coordinated evolution of the hepatitis C virus. Proc Natl Acad Sci USA. 2008;105:9685–9690. doi: 10.1073/pnas.0801774105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Giugliano S, Ruhl M, Neumann-Haefelin C, Wiese M, Thimme R, Roggendorf M, et al. Differences in the source sequence of two HCV genotype 1b outbreaks within immunodominant CD8 epitopes are associated with differential outcome. 16th International Symposium on Hepatitis C and Related Viruses; Nice, France. 2009. p. 215. [Google Scholar]
- 28.Dazert E, Neumann-Haefelin C, Bressanelli S, Fitzmaurice K, Kort J, Timm J, et al. loss of viral fitness and cross-recognition by CD8+T cells limit HCV escape from a protective HLA-B27-resticted immune response. J Clin Invest. 2009;119:276–286. doi: 10.1172/JCI36587. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.