

Abstract
The accurate trafficking of AMPA receptors (AMPARs) to and from the synapse is a critical component of learning and memory in the brain, while dysfunction of AMPAR trafficking is hypothesized to be an underlying mechanism of Alzheimer’s disease. Previous work has shown that ubiquitination of integral membrane proteins is a common post-translational modification used to mediate endocytosis and endocytic sorting of surface proteins in eukaryotic cells. Here we report that mammalian AMPARs become ubiquitinated in response to their activation. Using a mutant of GluA1 that is unable to be ubiquitinated at lysines on its carboxy-terminus, we demonstrate that ubiquitination is required for internalization of surface AMPARs and their trafficking to the lysosome in response to the AMPAR agonist AMPA, but not for internalization of AMPARs in response to the NMDA receptor (NMDAR) agonist NMDA. Through over-expression or RNAi-mediated knockdown, we identify that a specific E3 ligase, Nedd4-1, is necessary for this process. Finally, we show that ubiquitination of GluA1 by Nedd4-1 becomes more prevalent as neurons mature. Together, these data show that ubiquitination of GluA1-containing AMPARs by Nedd4-1 mediates their endocytosis and trafficking to the lysosome. Furthermore, these results provide insight into how hippocampal neurons regulate AMPAR trafficking and degradation with high specificity in response to differing neuronal signaling cues, and suggest that changes to this pathway may occur as neurons mature.
Keywords: AMPA receptor, endocytosis, ubiquitination, lysosome, Nedd4-1, endocytic sorting
Introduction
Changes in synaptic strength are believed to be the basis of learning and memory. These alterations in neuronal communication are controlled in part by the insertion or removal of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs) from the postsynaptic membrane of excitatory synapses. Regulation of AMPAR trafficking to and from the plasma membrane can occur through the C-terminal tails of AMPARs, either through interactions with other proteins or changes in phosphorylation (Malinow and Malenka, 2002; Derkach et al., 2007). When surface AMPARs are internalized, subunit-specific interactions with other proteins and changes in their phosphorylation status allow for their recycling and functional insertion into the plasma membrane (Hirling, 2009). Alternatively, AMPARs have been shown to traffic to the lysosome by a distinct pathway, though the mechanisms regulating this process are not well understood (Ehlers, 2000; Lin et al., 2000; Kessels et al., 2009).
The ubiquitin-proteasome system (UPS) is a highly regulated proteolytic pathway that mediates a majority of protein degradation in eukaryotic cells (Hershko and Ciechanover, 1998). However, many studies have shown that ubiquitination can also serve as an endocytosis signal for plasma membrane proteins (Hicke and Dunn, 2003). While it is not completely understood why ubiquitinated plasma membrane proteins often avoid degradation by the proteasome, one hypothesis is that differing structures of the attached ubiquitin chains can be a factor (Ikeda and Dikic, 2008). Regardless, protein ubiquitination is a highly selective process, dependent on the high specificity E3 ligases, the molecules responsible for attaching ubiquitin to proteins, have for their target substrates (Hershko and Ciechanover, 1998; Nalepa et al., 2006).
The neural-precursor cell-expressed developmentally down-regulated gene 4 (Nedd4) family of ubiquitin ligases are HECT (homology to E6-AP carboxy terminus) E3 ligases originally identified by their high expression in the embryonic mouse central nervous system (Kumar et al., 1997; Kamynina et al., 2001). The yeast homolog of Nedd4-1, Rsp5p, is thought to be the primary E3 ligase in yeast responsible for mediating the endocytosis and endocytic sorting of many plasma membrane proteins (Rotin et al., 2000). More recently, many neuronal proteins have been found to be ubiquitinated by Nedd4 family ligases to control their endocytosis and endocytic sorting (Kamynina et al., 2001; Fotia et al., 2004; Arevalo et al., 2006; Sorkina et al., 2006; Ekberg et al., 2007; Jespersen et al., 2007; Miranda et al., 2007). It is therefore plausible that Nedd4-1 may play an important role in neuronal function by controlling the endocytosis and endocytic sorting of specific synaptic proteins.
Here we report that mammalian GluA1-containing AMPARs are ubiquitinated on C-terminal lysines to mediate their internalization and trafficking to the lysosome in an agonist-dependent manner. This endocytosis and endocytic sorting is mediated by the E3 ligase Nedd4-1. Over-expression of Nedd4-1 causes a loss of surface GluA1-containing AMPARs and promotes their accumulation in the lysosome, while RNAi-mediated knockdown of Nedd4-1 blocks AMPA-but not NMDA-induced ubiquitination and internalization of GluA1-containing AMPARs. These results demonstrate that ubiquitination of AMPARs is a distinct mechanism by which neurons can regulate surface populations and endosomal sorting of AMPARs in the hippocampus.
Materials and Methods
Antibodies and Reagents
Antibodies were obtained as follows: pAb GluA1, pAb GluA2, and pAb Nedd4-1 (Millipore), pAb surface (N-terminal) GluA1 (Calbiochem), mAb ubiquitin (P4D1) and pAb GFP (Santa Cruz), anti-GFP secondary antibody (Invitrogen), mAb HA (Covance), pAb Actin (Cytoskeleton Inc.), mAb Lamp1 (Stressgen), mAb T7 (Sigma), AMPA, NMDA, CNQX, APV, and H-89 (Tocris), BFA (Invitrogen), N-ethylmaleimide (NEM) and leupeptin (Sigma), MG-132 (Biomol).
Hippocampal Cultures
Rat dissociated hippocampal or hippocampal and cortical cultures from postnatal day 1 were plated onto poly-D-lysine coated coverslips, 35 mm dishes (Mattek), or 6 well plastic dishes and maintained in B27 supplemented Neurobasal media (Invitrogen) until 14-38 days in vitro.
Immunoprecipitations
Rat hippocampal tissue or hippocampal cultures were homogenized in precipitation buffer (PB) (in mM: 150 NaCl, 10 Na2HPO4, 2 EDTA) with 1% TX-100 and 0.1–0.2% SDS, 25 μM MG-132, 25mM NEM, and protease inhibitors. HEK293T cells were lysed in RIPA Buffer plus 1% BSA with 25 μM MG-132, 25mM NEM, and protease inhibitors. Homogenates were cleared by centrifugation at 14,000 rpm at 4°C. For immunoprecipitations, cleared lysates were incubated with primary antibodies at 4°C O/N, after which protein A or protein A/G sepharose beads were added for an additional 2 hours (Pierce). Immunoprecipitations from neurons were repeated 3–8 times. Immunoprecipitations from HEK293T cells were repeated 2–4 times. For all immunoprecipitations from transfected cells, equal protein expression was verified in cell lysates by Western blot. For quantification of Western blots, protein band mean intensities were calculated using ImageJ. For Western blots measuring protein ubiquitination, ubiquitin band mean intensities were divided by the mean intensities of the corresponding immunoprecipitated receptor. Ubiquitination values from treated IPs were normalized to values from control IPs. For Western blots measuring GluA1 levels, the GluA1 band mean intensity in each condition was normalized to the actin band mean intensity from the same sample.
Transfections and Infections
HEK293T cells, maintained in DMEM + 10% serum and Pen/Strep, were transfected with Lipofectamine 2000 (Invitrogen) or PEI (Polysciences) using recommended protocols. Hippocampal cultures were infected with Sindbis virion at DIV15-19 and allowed to express for 18–22 hours. For RNAi experiments, hippocampal cultures were transfected with Lipofectamine 2000 at DIV10 and expressed the RNAi constructs for 4–5 days, or infected with lentivirus expressing the RNAi constructs for 5 days. Viral titer and transduction efficiency were monitored for all viruses made to ensure equal expression of constructs.
DNA Constructs
GFP-GluA1 obtained from R. Malinow (UCSD) was mobilized in pcDNA3.1(−) vector. GluA1-4KR, Nedd4-1 CS, and Nedd4-1resist. were created using PCR site-directed mutagenesis. All point mutations were verified by sequencing. For Sindbis viral expression, genes were cloned into the Sindbis virus vector SinRep5. For lentiviral expression, an H1 promoter and RNAi sequence were cloned into the FG-12 vector expressing GFP. HA-Nedd4-1 and HA-Cbl plasmids were purchased from Add gene DNA Depository. HA-E6-AP was obtained from P. Howley (Harvard), T7-Nedd4-1 was obtained from D. Rotin (Sick Kids), myc-NR1-1a and GFP-GluA2 was obtained from A. Ghosh (UCSD), and YFP-Nedd4-2 was obtained from S. Polo (FIRC Institute).
Surface Live-labeling and Endocytosis
Dissociated hippocampal neurons (DIV15-20) or HEK293T cells were live-labeled with anti-GFP secondary or anti-GluA1 antibodies for 15 min at 37°C, and then washed with PBS-MC (1X PBS, 1mM MgCl2, 0.1mM CaCl2). Cells were then fixed for 5–10 min. with 4% paraformaldehyde/4% sucrose at room temperature. Surface GluA1 receptors were then labeled with secondary Alexa antibodies (Molecular Probes) in PBS-MC containing 2% BSA. For endocytosis experiments, cells were pre-treated with TTX (2 μM) for 1 hour before surface labeling. After surface labeling, cells were washed with PBS and replaced with conditioned media containing either vehicle or AMPA (100 μM) and APV (25μM), or NMDA (25μM) and glycine (20μM) for 10–15 min. at 37°C. Cells were then washed with PBS-MC and fixed. Surface receptors were labeled with untagged secondary antibodies in PBS-MC before the cells were permeabilized with PBS-MC, 2% BSA, and 0.2% Triton X-100 for 20 min. The internalized receptors were labeled with fluorophore-conjugated secondary antibodies. For endocytosis experiments involving co-staining with Lamp1 antibody, cells were permeabilized in PBS containing 2% NGS, 1% BSA, and 0.1% saponin for 1 hour at RT. For endocytosis experiments, the fluorescence from the GluA1-N terminal GFP molecule was negligible post fixation as its fluorescence was determined to be detectable only at exposure times ten times greater than those used to detect antibody-labeled GFP.
Colocalization
To quantify the amount of internalized GluA1 immunofluorescence co-localized with Lamp1 immunostaining, images of Lamp1-labeled dendrites were converted to thresholded masks using ImageJ. Thresholded images of internalized GluA1-labeled dendrites were pasted over these masks and the remaining intensity of internalized GluA1 signal was quantified. To verify that colocalization was specific, colocalization between internalized GluA1 and Lamp1 in Nedd4-1+leu cells was calculated as described above where Lamp1 staining was first randomized by rotating the image 180° before creating a mask.
RNAi
To knockdown expression of Nedd4-1 in hippocampal neurons, the oligo GCCACAAATCAAGAGTTAA was synthesized and inserted into the pSuper-eGFP vector or the FG-12 vector. A scramble oligo (GCAGACAAACCTATGAATA) was also created. Dissociated neuronal cultures were transfected with pSuper-Nedd4-1 or infected with FG-12-Nedd4-1 RNAi at DIV10 and experiments were conducted 4–5 days later. To create Nedd4-1resist., six silent point mutations were introduced into T7-Nedd4-1 using PCR mutagenesis into the region targeted by RNAi.
Electrophysiology of dissociated hippocampal neurons
For whole-cell recordings of miniature excitatory postsynaptic currents (mEPSCs) from GFP, GFP + Nedd4-1 WT, GFP + Nedd4-1 CS, and shRNA-expressing hippocampal neurons, cells were perfused at room temperature in a bicarbonate buffered recording solution containin: (in mM) 124 NaCl, 5 KCl, 26 NaHCO3, NaH2PO4, 2 MgCl2, 3 CaCl2, 0.5 TTX, 20 picrotoxin, 10 glucose, bubbled constantly with 95% O2/5% CO2. The electrode recording solution contained (in mM): 10 CsCl, 105 CsMeSO3, 0.5 ATP, 0.3 GTP, 10 HEPES, 5 glucose, 2 MgCl2, 1 EGTA, 0.2 QX-314, pH 7.3. Electrode resistances ranged from 4 to 7 MΩ, access resistances ranged from 10 to 25 M and were monitored for consistency throughout the recordings. Cells with a leak current >100 pA were excluded from analysis. Signals were amplified, filtered to 2 or 5 kHz and digitized at 10 kHz sampling frequency. Holding potential for all traces was −65mV. All recordings were taken from time points after mEPSC frequency and amplitude had reached steady state, greater than 10 min following perfusion of TTX and picrotoxin. mEPSCs were analyzed using custom software in Igor Proor MiniAnalysis.
Confocal Microscopy and Image Analysis
All images were taken with a Leica DMI6000 inverted microscope outfitted with a Yokogawa Nipkon spinning disk confocal head, an Orca ER high resolution B&W cooled CCD camera (6.45 μm/pixel at 1X), Plan Apochromat 40X/1.25na and 63X/1.4na objective, and an argon/krypton 100mW air-cooled laser for 488/568/647 nm excitations. All images were acquired in the dynamic range of 8 bit or 12 bit acquisition. Maximum projected confocal Z-stacks were analyzed with NIH ImageJ. For experiments analyzing surface or internalized GFP or GluA1 immunofluorescence, images were background subtracted and thresholded equally, and the integrated density of each puncta was measured using a modified ImageJ particle analysis macro. The average particle integrated density for each cell was normalized to cell size. This raw data value for each cell was divided by the average raw data value of untreated control cells to obtain a normalized value. Values from the same treatments over multiple experiments were then combined and averaged.
Statistical Analysis
Two-tailed unpaired Student’s t test with an alpha=.05 or ANOVA with Tukey’s or Fisher LSD post-hoc analysis was used for determining statistical significance. Results >0.05 were not considered significant.
Results
AMPARs Undergo Activity-Mediated Ubiquitination
Activation of AMPARs has been shown to induce their endocytosis and subsequent trafficking into endosomal/lysosomal sorting pathways in hippocampal neurons (Ehlers, 2000; Lin et al., 2000; Lee et al., 2004). Since ubiquitination is a well-studied post-translational modification that mediates endocytosis and endocytic sorting of surface receptors, we asked if activation of AMPARs induced their ubiquitination. To examine this, we treated dissociated neuronal cultures (DIV>14) with AMPA (100μM, 10min.), an AMPAR agonist, and immunoprecipitated the resulting lysates with anti-GluA1 antibodies. The IPs were resolved by SDS-PAGE, transferred to nitrocellulose, and probed with anti-ubiquitin antibodies. We found that AMPA caused GluA1 to be ubiquitinated (5.0±0.60 for AMPA to 1±0.23 for control)(Figure 1A, 1B). Significant increases in ubiquitination were not observed for the AMPAR GluA2 subunit (2.1±0.49 for AMPA to 1±0.40 for control) or the N-methyl D-aspartate receptor (NMDAR) NR1 subunit (1.2±0.15 for AMPA to 1±0.21 for control), suggesting AMPA-induced ubiquitination occurs primarily on the GluA1 subunit. To ensure preferential enrichment of GluA1 in the IPs, a buffer containing high amounts of detergents (1% TX-100, .2% SDS) was used to aid the dissociation of AMPAR tetramers and other associated post-synaptic density (PSD) proteins. Quantification of the amount of GluA1 or GluA2 in the GluA1 and GluA2 specific IPs showed enrichment for those specific subunits (1±0.07 of GluA1 to 0.6±0.04 GluA2 in GluA1 IPs; 1±0.07 GluA2 to 0.4±0.05 GluA1 in GluA2 IPs)(Figure 1C). Also, a silver stain of the GluA1 IP showed isolation of a single band which migrated at the predicted size of GluA1 (~117kD), with only moderate amounts of another unidentified protein visible (~80kD) plus antibody heavy chain (~55kD)(Supplemental Figure 1A). On lower exposures of the Western blot probed with anti-GluA1 antibody, the majority of immunoprecipitated GluA1 appears to be unmodified based on size. However, upon higher exposures, high molecular GluA1 reactivity is visible at the size predicted for ubiquitinated GluA1 in AMPA-treated IPs, but absent from untreated IPs (Supplemental Figure 1A). The ubiquitin immunoreactivity was specific, as exposing the ubiquitin antibody to ubiquitin-bound agarose prior to probing the Western blot resulted in a loss of ubiquitin immunoreactivity (Supplemental Figure 1B). Together, these results suggest that the ubiquitination visible by Western blot after application of AMPA occurs on the GluA1 subunit. After AMPA stimulation, ubiquitinated GluA1 consistently appeared as two distinct bands slightly larger in size than unmodified GluA1 on Western blots. This suggests GluA1 is ubiquitinated by single ubiquitin molecules or short ubiquitin chains which cause a modest shift in GluA1 molecular weight. To estimate the molecular weight size of this population of GluA1, we plotted the electrophoretic mobilities of the molecular weight standards from each Western blot against their known molecular weights as previously described (Supplemental Figure 1C–1E)(Weber and Osborn, 1969). After deriving a logarithmic trend line through these points, we used the resulting trend line equation to calculate the predicted size of the two main ubiquitinated GluA1 bands based on their electrophoretic mobility (Supplemental Figure 1D). The estimated size of these bands was found to be highly similar over multiple experiments. The ubiquitinated GluA1 band immediately above unmodified GluA1, called GluA1-Ub3, was estimated to be 24 kD larger than GluA1 (Supplemental Figure 1E). As ubiquitin is approximately 8 kD, we hypothesize that this band represents GluA1 with three ubiquitin molecules attached. A slightly larger ubiquitinated GluA1b and, called GluA1-Ub4, was estimated to be 33 kD larger than unmodified GluA1, suggestive of GluA1 with four ubiquitin molecules attached. These ubiquitin molecules could be attached to distinct lysine residues of GluA1 or combined to form short ubiquitin chains. Together, these findings indicate that application of AMPA induces the ubiquitination of the GluA1 subunit of AMPARs.
Figure 1. GluA1-Containing AMPARs Undergo Activity-Mediated Ubiquitination.

(A) Dissociated neuronal cultures were treated with AMPA (100μM, 10min.) or left untreated prior to immunoprecipitation of resulting lysates with anti-GluA1, anti-GluA2, or anti-NR1 antibodies. IPs were resolved by Western blot and probed with anti-ubiquitin antibodies and antibodies against each receptor subunit to confirm equal levels of protein in each IP. (B) Quantification of normalized mean ubiquitin intensity. (C) Quantification of IP receptor intensity divided by input receptor intensity for GluA1 and GluA2 IPs. n=3–4 IPs for each condition. *p<0.05, unpaired Student’s t-test. Error bars=s.e.m.
It was previously reported that in response to AMPA, AMPARs were endocytosed and targeted to the lysosome for degradation (Ehlers, 2000). However, since ubiquitination of certain proteins can mediate their degradation by the proteasome, we confirmed that application of AMPA caused AMPAR degradation by the lysosome and not the proteasome. Total GluA1 in neuronal cultures decreased with increased exposure to AMPA (100μM, 5–60 min.)(Supplemental Figure 2A–2B). This loss was not blocked by co-application of the proteasomal inhibitor MG-132 (25μM) but was blocked by the lysosome inhibitor leupeptin (200μg/ml). Therefore, application of AMPA induces the ubiquitination and endocytosis of GluA1, as well as its trafficking to the lysosome for degradation.
Ubiquitination of AMPARs is Dependent on AMPAR Activation and Calcium
Previous work has suggested that the relative activation of AMPARs by direct or indirect stimuli differentially manipulates their endocytosis and endocytic sorting (Beattie et al., 2000; Ehlers, 2000; Lin et al., 2000). To determine if GluA1 ubiquitination was specific to AMPAR activation, we treated neuronal cultures with AMPA in the presence of the AMPAR antagonist 6-cyano-7-nitroquinoxaline-2,3-dione(CNQX, 40μM, 30 min.). We found that CNQX significantly attenuated GluA1 ubiquitination (4.5±0.61 for AMPA and 2.5±0.53 for CNQX+AMPA)(Figure 2A, 2B). In contrast, blocking NMDARs with the NMDAR antagonist DL-2-Amino-5-phosphonopentanoic acid (APV, 50μM, 30 min.) had no effect on AMPA-induced ubiquitination of GluA1 (4.5±0.61 for AMPA and 5.0±0.89 for APV+AMPA). Furthermore, stimulating NMDARs directly with the agonist NMDA (25μM, 10 min.) did not induce GluA1 ubiquitination (1.7±0.51 for NMDA). This indicates NMDAR activation is not sufficient for AMPAR ubiquitination. Finally, removing calcium, a key regulator of many synaptic signaling pathways, from the cell media prior to application of AMPA completely abolished GluA1 ubiquitination (4.5±0.61 for AMPA to 1.5±0.40 for Ca2+-free AMPA). These results indicate that the rapid ubiquitination of GluA1 in response to AMPA is dependent on the direct activation of AMPARs and requires external calcium entry into the neurons, most likely through voltage-gated calcium channels or sources other than NMDARs.
Figure 2. Ubiquitination of GluA1-containing AMPARs is Dependent on AMPAR Activation and Calcium.

(A) Dissociated neuronal cultures were treated with AMPA (100μM), CNQX (40μM)+AMPA, Ca2+free media+AMPA, APV (50μM)+AMPA, NMDA (25μM), or left untreated prior to IP with anti-GluA1 antibodies. IPs were resolved by Western blot and probed with anti-ubiquitin and anti-GluA1 antibodies. (B) Quantification of normalized mean ubiquitin. n=4–8 IPs for each condition. *p<0.05, ANOVA with Tukey’s post hoc test. Error bars=s.e.m.
Ubiquitination of GluA1 C-terminal Lysines Regulates Their Surface Accumulation and Internalization
Surface GluA1 contains three transmembrane loops and a C-terminal tail that are exposed to ubiquitination machinery inside the cell. Of these domains, only the C-tail contains lysine residues (at amino acids 813, 819, 822, and 868) that could serve as sites for ubiquitin attachment. Since AMPA-induced GluA1 ubiquitination consistently appears as multiple distinct bands (Figures 1 and 2), and the size of these bands is suggestive of GluA1 with three to four ubiquitins attached (Supplemental Figure 1E), we hypothesized that GluA1 may be ubiquitinated at multiple sites. To explore this, we mutated the C-terminal tail lysine residues of GFP-tagged GluA1 (GluA1-WT) to arginine so they could no longer be ubiquitinated, creating GluA1-4KR (Figure 3A).
Figure 3. Ubiquitination at C-terminal Sites is Necessary for AMPA-mediated but not NMDA-mediated GluA1 Endocytosis.

(A) A schematic of GFP-tagged GluA1 C-terminal highlights lysine residues that are potential sites of ubiquitination. GFP-GluA1-4KR has all four C-terminal lysines mutated to arginines. (B) Representative images of dissociated hippocampal neurons infected with GluA1-WT or GluA1-4KR virus. Surface (red) and internal (green) GFP-GluA1 populations were discretely labeled with anti-GFP antibodies. n=28 cells for WT and n=30 cells for 4KR over 4 experiments. (C) Representative images of internalized GluA1-WT or GluA1-4KR in neurons treated with AMPA (100μM) or NMDA (25μM) for 5–10min. (D) Quantification of internalized GluA1-WT or GluA1-4KR intensity in soma or dendrites of infected neurons. n=30–45 cells per treatment over 4 experiments. *p<0.05, ANOVA with Fisher’s LSD post hoc test. Error bars=s.e.m. Scale bar=10μm.
We next asked if the loss of C-terminal ubiquitination sites would alter surface expression levels of GluA1. To do this, we expressed GFP-GluA1-WT or GluA1-4KR in mature hippocampal neurons using Sindbis virus as previously reported (Shi et al., 1999). We limited Sindbis virus expression to ~18–22 hours to maintain cell viability. After infection, neurons were live-labeled with anti-GFP Alexa594 fluorophore to visualize surface GluA1, permeabilized, and labeled with anti-GFP Alexa488 fluorophore to visualize internal GluA1 before imaging by confocal microscopy. We observed a significant increase in the intensity of surface GluA1-4KR compared to GluA1-WT in both the soma and dendrites of infected neurons (1±0.15 for GluA1-WT to 2.1±0.24 for GluA1-4KR in soma, 1±0.08 for GluA1-WT to 1.8±0.02 for GluA1-4KR in dendrites, *p<0.001, unpaired Student’s t-test)(Figure 3B). We determined this result was not due to increased insertion of newly synthesized proteins in the membrane by treating infected neurons with Brefeldin A (BFA). BFA is an antibiotic that inhibits intracellular protein transport by disrupting trafficking from the trans-Golgi complex. After BFA treatment (5μg/ml, 45min.), we observed that surface GluA1-4KR immunofluorescence was much more stable than GluA1-WT (1±0.08 for GluA1-WT to 0.5±0.07 for GluA1-WT+BFA; 1±0.07 for GluA1-4KR to 0.8±0.10 for GluA1-4KR+BFA)(Supplemental Figure 3A). These results are consistent with GluA1-4KR having increased surface levels due to decreased internalization and lysosomal degradation.
To explore if GluA1-4KR undergoes normal activity-mediated endocytosis, mature hippocampal neurons were infected with Sindbis GFP-tagged GluA1-WT or GluA1-4KR, live-labeled with anti-GFP antibodies, and exposed to AMPA (100μM) or NMDA (25μM) for 10 min at 37°C. Cells were then fixed and exposed to unlabeled secondary antibodies to block any GFP antibody remaining on the cell surface before permeabilizing. Then, internalized pools of GluA1-WT or GluA1-4KR were labeled with Alexa568-conjugated secondary antibody. As expected, both AMPA and NMDA significantly induced endocytosis of surface GluA1-WT in the soma and dendrites of infected neurons compared to control-untreated neurons (1±0.14 for control to 2.5±0.57 for AMPA and 1.8±0.31 for NMDA in soma; 1±0.12 for control to 1.4±0.16 for AMPA and 1.4±0.15 for NMDA in dendrites)(Figure 3C, 3D). In contrast, AMPA-induced endocytosis was completely abolished for GluA1-4KR (1±0.14 for control to 0.8±0.21 for AMPA in soma; 1±0.11 for control to 0.8±0.14 in dendrites). However, GluA1-4KR was still significantly internalized in response to NMDA (1±0.14 for control to 2.0±0.27 for NMDA in soma; 1±0.11 for control to 1.3±0.10 in dendrites). Since NMDA-induced GluA1-4KR endocytosis was similar to GluA1-WT, this suggests that changing the C-terminal tail lysines to arginines did not affect GluA1’s functionality or ability to interact with proteins that assist in its internalization. These results indicate that ubiquitination at C-terminal lysines is needed for AMPA-but not NMDA-induced internalization of GluA1.
Identification of Nedd4-1 as a Candidate E3 Ligase to Mediate Surface Expression of AMPARs
Nedd4-1 is an E3 ligase whose role in regulating protein trafficking in eukaryotic cells has been well studied (Rotin and Kumar, 2009). In yeast, the homolog of Nedd4-1, Rsp5p, ubiquitinates Ste2p, a G-protein coupled receptor, to mediate its endocytosis and endocytic sorting (Hicke and Dunn, 2003; Dupre et al., 2004). Interestingly, the region of Ste2p targeted by Rsp5p for ubiquitination shares sequence similarity to the C-terminal tail of AMPARs, which suggested to us that Nedd4-1 may interact with AMPARs in mammalian neurons (Burbea et al., 2002). Therefore, we tested whether Nedd4-1 was involved in regulating the trafficking of GluA1. GFP-tagged GluA1-WT and HA-tagged Nedd4-1 were co-transfected in HEK293T cells. Cells were live-labeled with an anti-GFP antibody, permeabilized, labeled with an anti-HA antibody, and imaged by confocal microscopy. Co-expression of Nedd4-1 dramatically decreased surface GluA1 levels (1±0.08 for GluA1-WT to 0.3±0.03 for GluA1-WT+Nedd4-1)(Supplemental Figure 4A, 4B). Co-expression of GluA1 with other HECT domain ligases (Nedd4-2 and E6AP) as well as a RING finger ligase (Cbl) did not decrease surface GluA1 levels (Supplemental Figure 4B). The ability of Nedd4-1 to reduce surface GluA1 levels required its ligase activity, since co-expression of a catalytically-inactive version of Nedd4-1 (Nedd4-1 CS) did not decrease surface GluA1 levels (Supplemental Figure 4A, 4B). Importantly, we found that Nedd4-1 had no effect on surface GluA1 populations when co-transfected with GluA1-4KR (Supplemental Figure 4A, 4B). It also did not affect surface GluA2 levels when co-expressed (1±0.06 for GluA2 to 0.9±0.05 for GluA2 and Nedd4-1). Ubiquitination of surface GluA1-WT by Nedd4-1 was also confirmed biochemically. HEK293T cells were co-transfected with GFP-tagged GluA1-WT or GluA1-4KR, HA-tagged ubiquitin, and Nedd4-1. After 24 hours, these cells were surface labeled with anti-GFP antibody, lysed, precipitated to isolate the antibody-labeled surface GluA1, and resolved by SDS-PAGE. HA-ubiquitin immunoreactivity occurred most abundantly when GluA1-WT was co-expressed with Nedd4-1, while GluA1-4KR showed minimal ubiquitination, even in the presence of Nedd4-1 (Supplemental Figure 4C). Together these data suggest that Nedd4-1 targets GluA1 for internalization through ubiquitination of GluA1 C-terminal lysines.
We also observed a specific interaction between GluA1 and Nedd4-1 when co-expressed in HEK293T cells, while no interaction between Nedd4-1 and GluA2 or NR1 was observed (Supplemental Figure 4D–4F, 4H). Furthermore, GluA1 did not interact with a highly similar Nedd4 ligase family member, Nedd4-2 (Supplemental Figure 4G, 4H). These observations suggest that an interaction between Nedd4-1 and GluA1 is specific and likely direct, as HECT ligases have been shown to interact directly with their targeted substrates. Additional evidence that endogenous GluA1 and Nedd4-1 interact in mature hippocampal neurons came from immunoprecipitation of Nedd4-1 from lysates of mature hippocampal tissue (Figure 4A). When we resolved these IPs on Western blots and probed them with anti-GluA1 antibodies, we found GluA1 to be present in the Nedd4-1 precipitates. Similarly, when we immunoprecipitated GluA1 from mature hippocampal lysates, we found Nedd4-1 to be present in the GluA1 precipitates (Figure 4A). Together, these results indicate that Nedd4-1 associates with GluA1, providing an opportunity for it to ubiquitinate and regulate the trafficking of GluA1-containing AMPARs in hippocampal neurons.
Figure 4. Nedd4-1 Interacts With and Regulates Surface Expression of GluA1.

(A) Nedd4-1 or GluA1 was isolated via IP with anti-Nedd4-1 or anti-GluA1 antibodies from mature hippocampal tissue. IPs were resolved by Western blot and probed with anti-GluA1 or anti-Nedd4-1 antibodies. IPs were repeated 3 times.(B) Representative images of neuronal cultures infected with GFP virus or GFP and HA-Nedd4-1 virus and surface-labeled with anti-GluA1 antibodies. Quantification of surface GluA1 intensity in dendrites of infected neurons was performed. n=50 cells for control, n=65 cells for Nedd4-1, over 6 experiments. Scale bar=10μm for whole cell images, 5μm for straightened dendrites. (C) Representative image of a neuron expressing GFP and Nedd4-1 during mEPSC recording. (D) Example mEPSC traces recorded from GFP (control) or Nedd4-1 infected neurons. (E) Quantification of event amplitudes averaged over all neurons expressing either GFP (control) or Nedd4-1.*p<0.005, unpaired Student’s t-test.(F) Cumulative histogram of event amplitudes for GFP (control) or Nedd4-1 neurons. (G) Quantification of event frequencies averaged over all neurons expressing GFP (control) or Nedd4-1. n=21 cells for control, n=19 cells for Nedd4-1 over 6 experiments. Error bars=s.e.m.
Over-expression of Nedd4-1 Leads to Loss of Surface and Synaptic GluA1-containing AMPARs
Since over-expression of Nedd4-1 led to a significant decrease in surface GluA1 in HEK293T cells (Supplemental Figure 4A, 4B) and Nedd4-1 associates with AMPARs in hippocampal neurons (Figure 4A), we reasoned that over-expression of Nedd4-1 in hippocampal neurons would diminish surface AMPAR populations in those neurons. Neurons, DIV 15–18, were infected with Sindbis virus expressing either GFP alone (control) or co-expressing GFP with HA-tagged Nedd4-1. Infection time was limited to 18–22 hours to limit cell toxicity. Cells were then labeled with antibodies directed against surface GluA1, permeabilized, and labeled with anti-HA antibodies to detect Nedd4-1 positive cells. Compared to uninfected neurons or GFP-control cells, neurons expressing HA-Nedd4-1 showed a significant loss in surface GluA1 immunofluorescence from the dendritic plasma membrane (1±0.03 for GFP, 0.7±0.03 for Nedd4-1, *p<0.001, unpaired Student’s t-test) indicating that increased expression of Nedd4-1 decreased surface populations of GluA1-containing AMPARs (Figure 4B, Supplemental Figure 5B). We demonstrated this result was not caused by increased insertion of newly synthesized receptors by treating infected hippocampal neurons with BFA (5μg/ml, 45min) (Supplemental Figure 3B). After BFA treatment, there was a significant loss in surface GluA1 immunofluorescence in control neurons (1±0.05 for GFP to 0.7±0.04 for GFP+BFA). Untreated Nedd4-1-infected neurons had less surface GluA1 than control neurons, as expected (1±0.05 for GFP to 0.7±0.04 for Nedd4-1). After BFA treatment, however, there was an even larger decrease in surface GluA1 immunofluorescence (0.7±0.04 for Nedd4-1 to 0.5±0.05 for Nedd4-1+BFA). This suggests that over-expression of Nedd4-1 leads to increased endocytosis or decreased recycling of GluA1-containing AMPARs. This loss was specific to Nedd4-1 because over-expression of another HECT ligase, E6-AP, did not change surface GluA1 levels (Supplemental Figure 5A, 5B).
To determine if the Nedd4-1-induced decrease in surface AMPARs affected synaptic AMPARs, we recorded spontaneous miniature excitatory postsynaptic currents (mEPSCs) from GFP (control) or Nedd4-1 infected neurons. We observed a significant decrease in mEPSC amplitude in neurons expressing Nedd4-1 compared to control neurons (23.4±1.4 pA for GFP, 17.6±1.3pA for Nedd4-1)(Figure 4C–4F) while them EPSC frequency was not significantly different between conditions (0.69±0.24 s inter-event interval (IEI) for GFP, 0.59±0.15 s for Nedd4-1)(Figure 4G). Also, mEPSC amplitude recordings from neurons infected with catalytically inactive Nedd4-1 (Nedd4-1 CS) were unchanged from control neurons (Supplemental Figure 5C). We confirmed similar expression of HA-Nedd4-1 WT and Nedd4-1 CS in infected neurons by immunostaining infected neurons with anti-HA antibodies (Supplemental Figure 5D). Additionally, we observed that mEPSC event onset and decay kinetics were slower in Nedd4-1 expressing cells (10–90% slope of mEPSC onset: 22.5±2.4 and 15.0 ±2.1 pA/ms for GFP and Nedd4-1 expressing cells, respectively; mean±SEM, p = 0.04, t-test; single exponential decay constant: 6.7±0.64ms and 8.5±0.84ms for GFP and Nedd4-1 expressing cells, respectively; mean±SEM, p = 0.11, t-test). This is consistent with a predicted increase in NMDAR to AMPAR mediated transmission at synapses due to a decrease in synaptic AMPARs resulting from over-expression of Nedd4-1. Together, this data suggests that over-expression of Nedd4-1 in hippocampal neurons causes a decrease in surface AMPARs populations that includes synaptic AMPARs.
Because our data suggest GluA1 ubiquitination mediates AMPAR trafficking to the lysosome, we hypothesized that over-expression of Nedd4-1 may cause increased trafficking of surface AMPARs to the lysosome. To test this, we briefly surface labeled hippocampal neurons with anti-GluA1 antibodies prior to the addition of GFP or Nedd4-1-expressing Sindbis virus. To one set of infected neurons, we also applied leupeptin (200μg/ml) to block lysosomal degradation. After 18–22 hours, we fixed the neurons, blocked any remaining surface GluA1 antibody with unconjugated secondary antibodies, permeabilized the cells, and labeled the population of internalized GluA1. GFP-expressing neurons, with or without leupeptin, showed minimal levels of internalized GluA1 immunofluorescence (Figure 5A, 5B). However, in neurons expressing Nedd4-1 where lysosomal degradation was inhibited by leupeptin, there was a dramatic accumulation of internalized GluA1 in both the soma and dendrites. This accumulation did not occur in Nedd4-1-infected neurons if leupeptin was not added (1.8±0.18 for N4-1+leu to 1±0.13 for control, 1±0.07 for control+leu, and 0.9±0.10 for N4-1). A significant portion of internalized GluA1 puncta co-localized with late-endosomal/lysosomal compartments, visualized with anti-Lamp1 antibodies, in Nedd4-1 expressing neurons when compared to control neurons (2.8±0.45 for N4-1+leu to 1±0.22 for control+leu and 0.9±0.12 for N4-1+leu with random Lamp1 signal)(Figure 5C). These results indicate that expression of Nedd4-1 in hippocampal neurons targets endogenous AMPARs to late endosomal/lysosomal compartments.
Figure 5. Over-expression of Nedd4-1 Causes Increased Trafficking of Surface GluA1-containing AMPARs to the Lysosome.

(A) Neuronal cultures were surface-labeled with anti-GluA1 antibodies prior to infection with GFP (control) or Nedd4-1 virus, in the absence or presence of leupeptin. Representative images of internalized GluA1 (red) in infected neurons 18–22 hours post-infection and straightened dendrites co-labeled with the late endosome/lysosome antibody Lamp1 (green). Scale bar=10μm for whole cell images, 5μm for straightened dendrites. (B) Quantification of internalized GluA1 intensity in dendrites of infected neurons. (C) Quantification of internalized GluA1 intensity co-localized with Lamp1 staining in dendrites of infected neurons. n=30–45 cells per condition over 3 experiments. *p<0.05, ANOVA with Tukey’s post hoc test. Error bars=s.e.m.
Loss of Nedd4-1 Inhibits AMPA-but not NMDA-Mediated Endocytosis of GluA1-Containing AMPARs
We additionally explored the function of Nedd4-1 in regulating GluA1 endocytosis and endocytic sorting by examining the effects of RNAi-mediated knockdown of Nedd4-1 in mature hippocampal neurons. We designed a small hairpin RNA (shRNA) directed against Nedd4-1 that sufficiently knocked down expression of Nedd4-1 protein compared to scramble or empty vector controls (Figure 6A). We also generated a version of Nedd4-1, called Nedd4-1resist., which was resistant to RNAi-mediated knockdown. Nedd4-1resist. also contained a T7 tag which was used to confirm its expression in cells. Neurons were transfected at DIV10 with a control vector (pSuper-GFP or pSuper-Scramble) or Nedd4-1 shRNA (pSuper-RNAi). After 4–5 days of expression, neurons were labeled with antibodies specific to surface GluA1, imaged by confocal microscopy, and analyzed for any changes in surface GluA1 populations. We observed no change in surface GluA1 immunofluorescence or synaptic GluA1 levels detected by mEPSC recordings (data not shown). Our previous findings suggested, however, that ubiquitination of GluA1 occurs under conditions when GluA1-containing AMPARs are directly activated and endocytosis is induced. Therefore, we hypothesized that loss of Nedd4-1 may prevent the internalization of AMPARs in response to AMPA. To test this, we examined AMPA-induced internalization of AMPARs in control and Nedd4-1 shRNA-expressing hippocampal neurons. As expected, AMPA produced robust internalization of GluA1 in control neurons (1±0.10 for control to 1.6±0.19 for control+AMPA)(Figure 6B, 6C). In contrast, Nedd4-1 shRNA-expressing neurons showed a significant inhibition of AMPA-induced AMPAR internalization (0.7±0.12 for RNAi and 0.5±0.07 for RNAi+AMPA). Strikingly, RNAi-expressing neurons were still able to internalize GluA1 in response to NMDA at levels similar to control neurons exposed to NMDA (1±0.10 for RNAi to 1.3±0.10 for RNAi+NMDA and 1.4±0.10 for control+NMDA)(Figure 6B, 6D). Importantly, co-expression of pSuper-RNAi with Nedd4-1resist. rescued the neurons’ ability to endocytose GluA1-containing AMPARs in response to AMPA (1±0.12 for Nedd4-1resist. to 1.4±0.16 for Nedd4-1resist.+AMPA,*p<0.05, unpaired Student’s t-test)(Figure 6B). This indicates that Nedd4-1 mediates AMPAR internalization following direct AMPAR activation, but is not required for NMDA-dependent endocytosis, and suggests that Nedd4-1 is crucial for mediating AMPAR internalization and trafficking to the lysosome.
Figure 6. Loss of Nedd4-1 Inhibits AMPA-mediated but not NMDA-mediated Endocytosis of GluA1-containing AMPARs.

(A) Representative Western blot of lysates from HEK293T cells co-transfected with T7-Nedd4-1 or Nedd4-1resist. and pSuper-scramble or pSuper-Nedd4-1-RNAi. (B) Transfected neurons were surface-labeled prior to a 10 min. application of AMPA (100μM) or NMDA (25μM). Representative images are of internalized GluA1 (red) after each treatment in neurons expressing control or Nedd4-1 RNAi vectors (green) alone, or with Nedd4-1resist. (blue). Scale bar=10μm. (C–D) Quantification of internalized GluA1 intensity in control or RNAi-transfected neurons treated with AMPA (C) or NMDA(D). n=30–40 cells per condition over 3–4 experiments. *p<0.05, ANOVA with Tukey’s post hoc test. Error bars=s.e.m.
To further confirm a direct role for Nedd4-1 in AMPA-mediated GluA1 ubiquitination, we created a lentivirus expressing Nedd4-1 RNAi, which allowed us to knockdown endogenous Nedd4-1 expression in large populations of dissociated neuron cultures. The virus also expressed GFP to allow for identification of infected neurons. We infected neurons at DIV9 with Nedd4-1 RNAi lentivirus or lentivirus expressing GFP alone as a control. At DIV14, neurons were treated with AMPA (100μM, 10min.), lysed, and GluA1 was isolated by immunoprecipitation (Figure 7A, 7B). While AMPA induced significant ubiquitination of GluA1 in DIV14 control neurons as previously observed (Figure 1), ubiquitination of GluA1 was completely abolished in cultures lacking Nedd4-1 (3.0±0.4 for DIV14 AMPA to 1±0.2 for DIV14 control and 1.3±0.4 for DIV14 RNAi+AMPA). While performing these experiments, we observed that in older neurons (DIV35), GluA1 was significantly ubiquitinated under control conditions, at levels similar to ubiquitination of GluA1 induced by application of AMPA in younger neurons (3.4±1 for DIV35 control and 2.8±0.7 for DIV35 AMPA to 3.0±0.4 for DIV14 AMPA). Furthermore, this increase in GluA1 ubiquitination in older neurons was still dependent on the presence of Nedd4-1, as loss of Nedd4-1 due to expression of Nedd4-1 RNAi lentivirus for five days resulted in significantly decreased GluA1 ubiquitination levels similar to that of younger untreated neurons (1. ±0.3 for DIV35 RNAi+AMPA to 1±0.2 for DIV14 control). These results suggest that factors, such as increased AMPAR activation or Nedd4-1 ligase activity, may change in neurons as they age, resulting in increased Nedd4-1 mediated AMPAR ubiquitination.
Figure 7. Ubiquitination of GluA1-containing AMPARs is Upregulated in Aged Neurons but Blocked by Loss of Nedd4-1.

(A) Dissociated neuronal cultures (DIV14 or DIV35) infected with lentivirus expressing GFP or GFP and Nedd4-1 RNAi for five days were treated with AMPA (100μM, 10min.) or left untreated prior to immunoprecipitation of resulting lysates with anti-GluA1 antibodies. IPs were resolved by Western blot and probed with anti-ubiquitin antibodies and antibodies against GluA1 to confirm equal levels of protein in each IP. Lysates were also resolved and probed with anti-Nedd4-1 and anti-actin antibodies to confirm Nedd4-1-specific knockdown. (B) Quantification of mean ubiquitin intensity for each IP. n=4–6 IPs for each condition. *p<0.05,**p<0.01, ANOVA with Tukey’s post hoc test. Error bars=s.e.m.
Discussion
In this study, we have reported that direct activation of GluA1-containing AMPARs induces their ubiquitination. This process is not dependent on NMDAR activation, but does require calcium. Ubiquitination occurs on C-terminal lysines of GluA1 and is mediated by the E3 ligase Nedd4-1. Ubiquitination at these sites mediates the endocytosis and lysosomal trafficking of GluA1-containing AMPARs, but may not be required for their endocytosis and recycling back to the plasma membrane (Figure 8).
Figure 8. Model for Ubiquitin-Mediated Endocytic Trafficking of AMPARs.
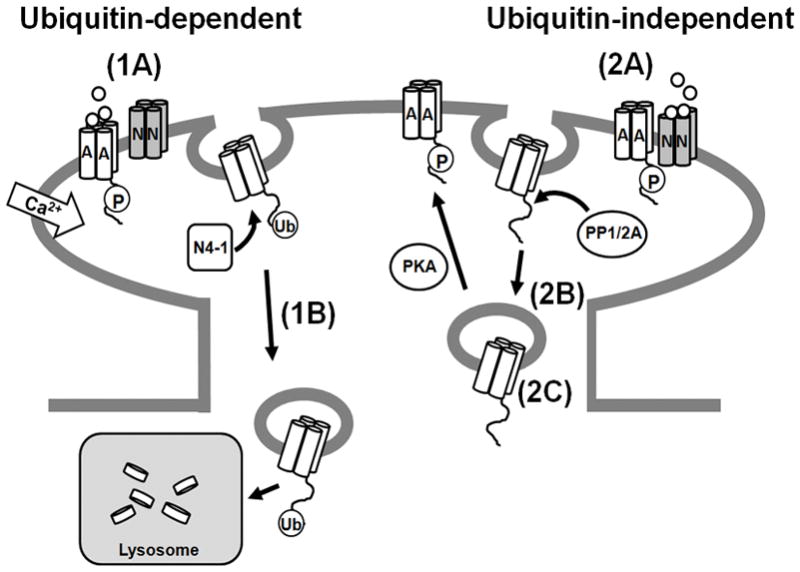
(1A) A distinct endocytosis/lysosomal sorting pathway that is dependent on ubiquitination can be stimulated by direct activation of AMPARs with the agonist AMPA and may be up-regulated during aging. Application of AMPA causes an influx of calcium into hippocampal neurons that may activate the E3 ligase Nedd4-1 to ubiquitinate GluA1-containing AMPARs, most likely in conjunction with phosphatases and other machinery to assist in endocytosis. (1B) Ubiquitination of GluA1-containing AMPARs mediates their trafficking to the lysosome and eventual degradation. (2A) AMPARs can be recruited to a separate endocytosis/recycling pathway upon exposure to NMDA that is not ubiquitin-dependent. (2B) Application of NMDA causes GluA1 to become de-phosphorylated at serine 845 and internalized. (2C) If GluA1 is re-phosphorylated at serine 845 by PKA, it is trafficked to recycling endosomes and returned to the plasma membrane (Ehlers, 2000).
While our data shows that ubiquitination of GluA1 by Nedd4-1 is required for its endocytosis under specific conditions, it is likely that other proteins assist in this process. Indeed, in many eukaryotic cell types, the interaction between ubiquitination and clathrin machinery is a well studied and common occurrence in the regulation of endocytosis and sorting of proteins (Traub, 2009). Ubiquitinated receptors can be recruited to clathrin-coated pits via interactions with the adaptor proteins epsin and eps15 (Traub and Lukacs, 2007). Also, ubiquitination of receptors may aid their interaction with the adaptor protein AP2 (Kumar et al., 2007). Both epsin and AP2 have characterized roles in neurons for regulating protein endocytosis (Carroll et al., 1999; Lee et al., 2002; Sorkina et al., 2006; Jakobsson et al., 2008). Furthermore, GLR-1, the C. elegans homolog of GluA1, has been shown to undergo ubiquitination and removal from the synapse in manner dependent on the clathrin adaptor protein AP180 (Burbea et al., 2002). However, it’s also intriguing to speculate that distinct machinery could regulate ubiquitin-dependent and independent AMPAR internalization. Indeed, this phenomenon has been observed for the EGF receptor (EGFR). Depending on the level of ligand exposure and receptor activation, EGFRs can undergo varying amounts of ubiquitination which differentially regulate their entrance into clathrin-dependent or independent endocytosis pathways (Sigismund et al., 2005). It is interesting to hypothesize that the relative activation of AMPARs and NMDARs could differentially recruit clathrin and ubiquitination machinery to regulate the specificity of endocytosis and endocytic sorting of AMPARs.
Our studies are the first to show that GluA1-containing AMPARs undergo ubiquitinationin mature hippocampal neurons, and through multiple experiments, we have identified Nedd4-1 as the E3 ligase responsible for mediating GluA1 endocytosis and sorting to the lysosome. Interestingly, two other studies have recently shown that Nedd4-1 is crucial for neuronal development (Drinjakovic et al.; Kawabe et al.). Specifically, Kawabe et al. created both a traditional and conditional Nedd4-1 knockout mouse to identify that Nedd4-1, the serine/threonine kinase TNIK, and Rap2A form a complex to mediate Rap2A ubiquitination and ultimately dendrite formation. They also show that loss of Nedd4-1 in these mice did not cause a change in surface AMPAR expression, measured via immunofluorescence and electrophysiology of autaptic neurons. Though this may seem contradictory to our results, we also observed that surface AMPAR levels were unchanged in neurons lacking Nedd4-1 under control conditions (data not shown). Most likely, this is because while AMPARs are unable to internalize and traffic to the lysosome without Nedd4-1, they are still able to internalize and recycle back to the plasma membrane. Furthermore, several key differences in the methodologies of Kawabe et al. and our experiments strongly suggest that we have uncovered a distinct and novel role for Nedd4-1 in mature hippocampal neurons that may be unrelated to their findings. A significant difference is that Kawabe et al. focused on the role of Nedd4-1 very early in neuronal development, while we examined Nedd4-1’s role in mature neurons. Indeed, Nedd4-1 expression is highly upregulated during nervous system development before stabilizing to moderate levels in mature neurons, suggesting that its role in neurons may change over time (Kumar et al., 1997; Kamynina et al., 2001). In addition, several neuronal proteins have already been identified as Nedd4-1 targets, again indicating that Nedd4-1 most likely has several distinct roles in neurons (Kwak et al.; Liu et al., 2009). In fact, Kawabe et al. show that Nedd4-1 associates with TNIK and Rap2A in the perinuclear region of neurons, and that these three proteins co-segregate with the Golgi and ER. However, our studies identify a role for Nedd4-1 in mediating AMPAR endocytosis from the plasma membrane of the soma and dendrites of neurons, in support of our findings that Nedd4-1 is important for balancing surface and synaptic AMPARs populations in mature neurons. These differences point out the interesting possibility that Nedd4-1 has not only temporal and substrate-specificity, but possibly even spatially distinct roles in neurons.
Based on our findings and previous work exploring the activity-dependent sorting of AMPARs, we hypothesize that the relative activation of NMDARs and AMPARs can differentially lead to the recruitment of phosphorylation or ubiquitination machinery which determine the fate of internalized AMPARs (Ehlers, 2000). This is an especially appealing hypothesis as Nedd4-1’s activity and recruitment to the plasma membrane has been found to be regulated by Ca2+ in non-neuronal eukaryotic cells (Plant et al., 1997; Wang et al., 2010). Specifically, Nedd4-1 contains a C2 domain, which was first identified in Ca2+-sensitive isoforms of PKC and has also been shown to be important for the function of the neuronal protein synaptotagmin. C2 domains are thought to be “calcium sensors” which can regulate the activity of a protein and assist in its binding to lipids in a calcium-dependent manner (Lemmon, 2008). Therefore, it is plausible that Nedd4-1 maybe recruited to ubiquitinate GluA1 during specific Ca2+–dependent signaling events in neurons. If and how Nedd4-1 is activated by Ca2+ in neurons is completely unknown, but future experiments exploring this phenomenon would be of great interest.
Our results indicate that AMPA-mediated ubiquitination and lysosomal degradation occur to a population of surface GluA1-containing AMPARs when directly activated by agonist. Meanwhile ubiquitination machinery is not recruited by rapid NMDAR activation, which stimulates a fast, but reversible, removal of synaptic AMPARs. The mechanisms described in this paper provide new insight into how neurons simultaneously and discretely regulate the endocytosis, recycling, and degradation of AMPARs. The amount of ubiquitinated AMPARs we were able to detect in our experiments was distinct but small compared to total AMPAR populations in hippocampal neurons (Figure 1, Supplemental Figure 1). Detection of ubiquitinated proteins is challenging because ubiquitination itself is a rapid and reversible process (Hershko and Ciechanover, 1998). Therefore, it is possible that the amount of ubiquitinated GluA1 visible by Western blot in our experiments is an under-representation of ubiquitinated AMPAR populations in neurons. Furthermore, our data suggests that surface AMPARs are targeted for ubiquitination, and it has been reported that only 15–20% of total AMPARs in hippocampal neurons reside at the plasma membrane (Shi et al., 1999). In addition, many of these surface AMPARs are recycled back to the plasma membrane in a presumably ubiquitin-independent process, suggesting that AMPAR ubiquitination and lysosomal trafficking may only occur to small populations of AMPARs at any given time (Ehlers, 2000). However, we observed that ubiquitination of AMPARs under basal conditions increased as neurons matured. These results suggest that as time passes, factors, such as increased activation of AMPARs or increased Nedd4-1 activity, may change in neurons that cause increased endocytosis and lysosomal degradation of AMPARs. In this regard, the findings presented here may be of interest with relation to other studies focusing on the molecular mechanisms of age-associated neurodegenerative diseases such as Alzheimer’s disease, where both increases in AMPAR internalization and lysosomal dysfunction have separately been implicated (Hsieh et al., 2006; Shepherd and Huganir, 2007; Nixon et al., 2008). Indeed, further research focusing on the circumstances under which AMPAR ubiquitination occurs will likely provide important insight into our understanding of synaptic function under normal and diseased conditions in neurons.
Supplementary Material
Acknowledgments
We are grateful to A. Ghosh, Y. Jin, D. Berg, and members of the Patrick lab for helpful discussion. We thank P. Slesinger and R. Nassirpour for technical assistance. Some of these experiments were initiated in the lab of E. Schuman. This work was funded by a Ray Thomas Edward Career Award, UCSD Startup Funds, and NIH funding (NS054732) to G.N.P., Tulane School of Science and Engineering Startup Funds to B.J.H., and a National Science Foundation Graduate Research Fellowship to L.S.
References
- Arevalo JC, Waite J, Rajagopal R, Beyna M, Chen ZY, Lee FS, Chao MV. Cell survival through Trk neurotrophin receptors is differentially regulated by ubiquitination. Neuron. 2006;50:549–559. doi: 10.1016/j.neuron.2006.03.044. [DOI] [PubMed] [Google Scholar]
- Beattie EC, Carroll RC, Yu X, Morishita W, Yasuda H, von Zastrow M, Malenka RC. Regulation of AMPA receptor endocytosis by a signaling mechanism shared with LTD. Nat Neurosci. 2000;3:1291–1300. doi: 10.1038/81823. [DOI] [PubMed] [Google Scholar]
- Burbea M, Dreier L, Dittman JS, Grunwald ME, Kaplan JM. Ubiquitin and AP180 regulate the abundance of GLR-1 glutamate receptors at postsynaptic elements in C. elegans. Neuron. 2002;35:107–120. doi: 10.1016/s0896-6273(02)00749-3. [DOI] [PubMed] [Google Scholar]
- Carroll RC, Beattie EC, Xia H, Luscher C, Altschuler Y, Nicoll RA, Malenka RC, von Zastrow M. Dynamin-dependent endocytosis of ionotropic glutamate receptors. Proc Natl Acad Sci U S A. 1999;96:14112–14117. doi: 10.1073/pnas.96.24.14112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derkach VA, Oh MC, Guire ES, Soderling TR. Regulatory mechanisms of AMPA receptors in synaptic plasticity. Nat Rev Neurosci. 2007;8:101–113. doi: 10.1038/nrn2055. [DOI] [PubMed] [Google Scholar]
- Drinjakovic J, Jung H, Campbell DS, Strochlic L, Dwivedy A, Holt CE. E3 ligase Nedd4 promotes axon branching by downregulating PTEN. Neuron. 65:341–357. doi: 10.1016/j.neuron.2010.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupre S, Urban-Grimal D, Haguenauer-Tsapis R. Ubiquitin and endocytic internalization in yeast and animal cells. Biochim Biophys Acta. 2004;1695:89–111. doi: 10.1016/j.bbamcr.2004.09.024. [DOI] [PubMed] [Google Scholar]
- Ehlers MD. Reinsertion or degradation of AMPA receptors determined by activity-dependent endocytic sorting. Neuron. 2000;28:511–525. doi: 10.1016/s0896-6273(00)00129-x. [DOI] [PubMed] [Google Scholar]
- Ekberg J, Schuetz F, Boase NA, Conroy SJ, Manning J, Kumar S, Poronnik P, Adams DJ. Regulation of the voltage-gated K(+) channels KCNQ2/3 and KCNQ3/5 by ubiquitination. Novel role for Nedd4-2. J Biol Chem. 2007;282:12135–12142. doi: 10.1074/jbc.M609385200. [DOI] [PubMed] [Google Scholar]
- Fotia AB, Ekberg J, Adams DJ, Cook DI, Poronnik P, Kumar S. Regulation of neuronal voltage-gated sodium channels by the ubiquitin-protein ligases Nedd4 and Nedd4-2. J Biol Chem. 2004;279:28930–28935. doi: 10.1074/jbc.M402820200. [DOI] [PubMed] [Google Scholar]
- Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998;67:425–479. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- Hicke L, Dunn R. Regulation of membrane protein transport by ubiquitin and ubiquitin-binding proteins. Annu Rev Cell Dev Biol. 2003;19:141–172. doi: 10.1146/annurev.cellbio.19.110701.154617. [DOI] [PubMed] [Google Scholar]
- Hirling H. Endosomal trafficking of AMPA-type glutamate receptors. Neuroscience. 2009;158:36–44. doi: 10.1016/j.neuroscience.2008.02.057. [DOI] [PubMed] [Google Scholar]
- Hsieh H, Boehm J, Sato C, Iwatsubo T, Tomita T, Sisodia S, Malinow R. AMPAR removal underlies Abeta-induced synaptic depression and dendritic spine loss. Neuron. 2006;52:831–843. doi: 10.1016/j.neuron.2006.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda F, Dikic I. Atypical ubiquitin chains: new molecular signals. ‘Protein Modifications: Beyond the Usual Suspects’ review series. EMBO Rep. 2008;9:536–542. doi: 10.1038/embor.2008.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakobsson J, Gad H, Andersson F, Low P, Shupliakov O, Brodin L. Role of epsin 1 in synaptic vesicle endocytosis. Proc Natl Acad Sci U S A. 2008;105:6445–6450. doi: 10.1073/pnas.0710267105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jespersen T, Membrez M, Nicolas CS, Pitard B, Staub O, Olesen SP, Baro I, Abriel H. The KCNQ1 potassium channel is down-regulated by ubiquitylating enzymes of the Nedd4/Nedd4-like family. Cardiovasc Res. 2007;74:64–74. doi: 10.1016/j.cardiores.2007.01.008. [DOI] [PubMed] [Google Scholar]
- Kamynina E, Debonneville C, Bens M, Vandewalle A, Staub O. A novel mouse Nedd4 protein suppresses the activity of the epithelial Na+ channel. Faseb J. 2001;15:204–214. doi: 10.1096/fj.00-0191com. [DOI] [PubMed] [Google Scholar]
- Kawabe H, Neeb A, Dimova K, Young SM, Jr, Takeda M, Katsurabayashi S, Mitkovski M, Malakhova OA, Zhang DE, Umikawa M, Kariya K, Goebbels S, Nave KA, Rosenmund C, Jahn O, Rhee J, Brose N. Regulation of Rap2A by the ubiquitin ligase Nedd4-1 controls neurite development. Neuron. 65:358–372. doi: 10.1016/j.neuron.2010.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessels HW, Kopec CD, Klein ME, Malinow R. Roles of stargazin and phosphorylation in the control of AMPA receptor subcellular distribution. Nat Neurosci. 2009;12:888–896. doi: 10.1038/nn.2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar KG, Barriere H, Carbone CJ, Liu J, Swaminathan G, Xu P, Li Y, Baker DP, Peng J, Lukacs GL, Fuchs SY. Site-specific ubiquitination exposes a linear motif to promote interferon-alpha receptor endocytosis. J Cell Biol. 2007;179:935–950. doi: 10.1083/jcb.200706034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Harvey KF, Kinoshita M, Copeland NG, Noda M, Jenkins NA. cDNA cloning, expression analysis, and mapping of the mouse Nedd4 gene. Genomics. 1997;40:435–443. doi: 10.1006/geno.1996.4582. [DOI] [PubMed] [Google Scholar]
- Kwak YD, Wang B, Pan W, Xu H, Jiang X, Liao FF. Functional interaction of PTEN with the E3 ligase NEDD4-1 during neuronal response to zinc. J Biol Chem. doi: 10.1074/jbc.M109.091637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Simonetta A, Sheng M. Subunit rules governing the sorting of internalized AMPA receptors in hippocampal neurons. Neuron. 2004;43:221–236. doi: 10.1016/j.neuron.2004.06.015. [DOI] [PubMed] [Google Scholar]
- Lee SH, Liu L, Wang YT, Sheng M. Clathrin adaptor AP2 and NSF interact with overlapping sites of GluR2 and play distinct roles in AMPA receptor trafficking and hippocampal LTD. Neuron. 2002;36:661–674. doi: 10.1016/s0896-6273(02)01024-3. [DOI] [PubMed] [Google Scholar]
- Lemmon MA. Membrane recognition by phospholipid-binding domains. Nat Rev Mol Cell Biol. 2008;9:99–111. doi: 10.1038/nrm2328. [DOI] [PubMed] [Google Scholar]
- Lin JW, Ju W, Foster K, Lee SH, Ahmadian G, Wyszynski M, Wang YT, Sheng M. Distinct molecular mechanisms and divergent endocytotic pathways of AMPA receptor internalization. Nat Neurosci. 2000;3:1282–1290. doi: 10.1038/81814. [DOI] [PubMed] [Google Scholar]
- Liu Y, Oppenheim RW, Sugiura Y, Lin W. Abnormal development of the neuromuscular junction in Nedd4-deficient mice. Dev Biol. 2009;330:153–166. doi: 10.1016/j.ydbio.2009.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. 2002;25:103–126. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- Miranda M, Dionne KR, Sorkina T, Sorkin A. Three ubiquitin conjugation sites in the amino terminus of the dopamine transporter mediate protein kinase C-dependent endocytosis of the transporter. Mol Biol Cell. 2007;18:313–323. doi: 10.1091/mbc.E06-08-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalepa G, Rolfe M, Harper JW. Drug discovery in the ubiquitin-proteasome system. Nat Rev Drug Discov. 2006;5:596–613. doi: 10.1038/nrd2056. [DOI] [PubMed] [Google Scholar]
- Nixon RA, Yang DS, Lee JH. Neurodegenerative lysosomal disorders: a continuum from development to late age. Autophagy. 2008;4:590–599. doi: 10.4161/auto.6259. [DOI] [PubMed] [Google Scholar]
- Plant PJ, Yeger H, Staub O, Howard P, Rotin D. The C2 domain of the ubiquitin protein ligase Nedd4 mediates Ca2+-dependent plasma membrane localization. J Biol Chem. 1997;272:32329–32336. doi: 10.1074/jbc.272.51.32329. [DOI] [PubMed] [Google Scholar]
- Rotin D, Kumar S. Physiological functions of the HECT family of ubiquitin ligases. Nat Rev Mol Cell Biol. 2009;10:398–409. doi: 10.1038/nrm2690. [DOI] [PubMed] [Google Scholar]
- Rotin D, Staub O, Haguenauer-Tsapis R. Ubiquitination and endocytosis of plasma membrane proteins: role of Nedd4/Rsp5p family of ubiquitin-protein ligases. J Membr Biol. 2000;176:1–17. doi: 10.1007/s00232001079. [DOI] [PubMed] [Google Scholar]
- Shepherd JD, Huganir RL. The cell biology of synaptic plasticity: AMPA receptor trafficking. Annu Rev Cell Dev Biol. 2007;23:613–643. doi: 10.1146/annurev.cellbio.23.090506.123516. [DOI] [PubMed] [Google Scholar]
- Shi SH, Hayashi Y, Petralia RS, Zaman SH, Wenthold RJ, Svoboda K, Malinow R. Rapid spine delivery and redistribution of AMPA receptors after synaptic NMDA receptor activation. Science. 1999;284:1811–1816. doi: 10.1126/science.284.5421.1811. [DOI] [PubMed] [Google Scholar]
- Sigismund S, Woelk T, Puri C, Maspero E, Tacchetti C, Transidico P, Di Fiore PP, Polo S. Clathrin-independent endocytosis of ubiquitinated cargos. Proc Natl Acad Sci U S A. 2005;102:2760–2765. doi: 10.1073/pnas.0409817102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorkina T, Miranda M, Dionne KR, Hoover BR, Zahniser NR, Sorkin A. RNA interference screen reveals an essential role of Nedd4-2 in dopamine transporter ubiquitination and endocytosis. J Neurosci. 2006;26:8195–8205. doi: 10.1523/JNEUROSCI.1301-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traub LM. Tickets to ride: selecting cargo for clathrin-regulated internalization. Nat Rev Mol Cell Biol. 2009;10:583–596. doi: 10.1038/nrm2751. [DOI] [PubMed] [Google Scholar]
- Traub LM, Lukacs GL. Decoding ubiquitin sorting signals for clathrin-dependent endocytosis by CLASPs. J Cell Sci. 2007;120:543–553. doi: 10.1242/jcs.03385. [DOI] [PubMed] [Google Scholar]
- Wang J, Peng Q, Lin Q, Childress C, Carey D, Yang W. Calcium activates Nedd4 E3 ubiquitin ligases by releasing the C2 domain-mediated auto-inhibition. J Biol Chem. 2010;285:12279–12288. doi: 10.1074/jbc.M109.086405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber K, Osborn M. The reliability of molecular weight determinations by dodecyl sulfate-polyacrylamide gel electrophoresis. J Biol Chem. 1969;244:4406–4412. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.