

Abstract
Autoimmune autonomic neuropathy (AAN) is an acquired, often severe, form of dysautonomia. Many patients with AAN have serum antibodies specific for the neuronal ganglionic nicotinic acetylcholine receptor (AChR). Rabbits immunized with a fusion protein corresponding to the N-terminal extracellular domain of the ganglionic AChR α3 subunit produce ganglionic AChR antibodies and develop signs of experimental AAN (EAAN) that recapitulate the cardinal autonomic features of AAN in man. We now demonstrate that EAAN is an antibody-mediated disorder by documenting sympathetic, parasympathetic, and enteric autonomic dysfunction in mice injected with rabbit IgG containing ganglionic AChR antibodies. Recipient mice develop transient gastrointestinal dysmotility, urinary retention, dilated pupils, reduced heart rate variability, and impaired catecholamine response to stress. The autonomic signs are associated with a reversible failure of nicotinic cholinergic synaptic transmission in superior mesenteric ganglia. Mice injected with IgG from two patients with AAN (of three tested) demonstrated a milder phenotype with evidence of urinary retention and gastrointestinal dysmotility. The demonstration that ganglionic AChR-specific IgG causes impaired autonomic synaptic transmission and autonomic failure in mice implicates an antibody-mediated pathogenesis for AAN. The antibody effect is potentially reversible, justifying early use of immunomodulatory therapy directed at lowering IgG levels and abrogating IgG production in patients with AAN.
Keywords: neuronal nicotinic acetylcholine receptor, sympathetic, parasympathetic, gastrointestinal dysmotility, electrophysiology, pupil
Introduction
Numerous neurological disorders are associated with serum autoantibody markers, but few have been shown to be antibody mediated by directly demonstrating pathogenicity of the marker antibody. Proof that antibodies specific for the muscle nicotinic acetylcholine receptor (AChR) were the cause of myasthenia gravis required developing animal models of experimental autoimmune myasthenia gravis (Patrick and Lindstrom, 1973; Lennon et al., 1975). Passive transfer of muscle AChR antibodies to small rodents reproduces the clinical phenotype and the characteristic electrophysiological defect of neuromuscular junction transmission (Lindstrom et al., 1976; Toyka et al., 1977; Lennon and Lambert, 1980).
Fast synaptic transmission through autonomic ganglia is mediated by neuronal nicotinic AChRs that are structurally similar to the muscle AChR. The ganglionic AChR is a pentameric ion channel receptor typically composed of two α3 subunits in combination with β4 subunits (Patrick et al., 1993). Transgenic mice that are homozygous for null mutations in the α3 gene lack ganglionic AChRs and have profound autonomic dysfunction (Xu et al., 1999).
The autonomic nervous system regulates cardiovascular, thermoregulatory, gastrointestinal (GI), and neuroendocrine function. Patients with autoimmune autonomic neuropathy (AAN) typically present with orthostatic hypotension, gastrointestinal dysmotility (commonly gastroparesis and severe constipation), anhidrosis, impaired pupillary light responses, dry eyes and dry mouth (sicca complex), and bladder dysfunction (Suarez et al., 1994; Klein et al., 2003). In clinical studies, serum autoantibodies specific for the neuronal ganglionic AChR are found in ~50% of patients with AAN. The serum level of ganglionic AChR antibody correlates significantly with the severity of dysautonomia (Vernino et al., 2000). These clinical observations suggest that ganglionic AChR antibodies are directly involved in the pathogenesis of AAN.
We developed an animal model of ganglionic AChR autoimmunity by immunizing rabbits with a recombinant neuronal AChR α3 subunit fusion protein (Lennon et al., 2003). In response to a single immunization, rabbits develop experimental autoimmune autonomic neuropathy (EAAN), a chronic dysautonomic syndrome with prominent gastrointestinal dysmotility, urinary retention, impaired pupillary light reflex, reduced lacrimation, hypotension, impaired heart rate variability, and low plasma catecholamine levels (Lennon et al., 2003; Vernino et al., 2003). To directly investigate whether or not autoimmune autonomic neuropathy is an antibody-mediated disorder, we examined the clinical, autonomic, and electrophysiological phenotype of mice injected with ganglionic AChR IgG prepared from serum of rabbits with EAAN and from serum of patients with AAN.
Materials and Methods
Experimental methods
Immunization.
Experimental protocols were approved by the Mayo Clinic Animal Care and Use Committee. Rabbit immunization was performed as described previously (Lennon et al., 2003) using a recombinant human neuronal AChR α3 subunit fusion protein, residues 1–205. Serum was pooled from three immunized rabbits with high ganglionic AChR antibody levels and overt clinical signs of EAAN. Serum pooled from three rabbits immunized with an unrelated fusion protein (neuronal calcium channel β4 subunit residues 419–519) served as a control.
IgG preparation.
IgG was isolated from each pool of rabbit serum by adsorption to protein A-Sepharose. Sera from three individual patients with AAN who were seropositive for ganglionic AChR antibody and from one healthy control donor were processed by adsorption to protein-G-Sepharose to isolate IgG. All IgG samples were subsequently eluted in acidic buffer, dialyzed into PBS, concentrated, and sterilized by filtration before administration to mice. IgG protein concentrations were determined by spectrophotometry. Each IgG preparation was tested for reactivity against human or mouse ganglionic AChR using a previously described radioimmunoprecipitation assay (Vernino et al., 1998). Normal levels of ganglionic AChR binding activity in this assay are 0–0.02 nmol/l. Human ganglionic AChR was solubilized from membranes of IMR-32 neuroblastoma cells grown as xenograft tumors in nude mice and was complexed with 125I-epibatidine. Mouse AChR was solubilized from mouse S20 cells (a subclone of C1300 mouse neuroblastoma cell line) prepared the same way. The rabbit IgG pool was highly reactive with both human and mouse ganglionic AChR (54.0 and 1.13 μmol/l, respectively). The human IgG preparations bound less avidly to both human (105, 44, and 24 nmol/l) and mouse ganglionic AChR (1.2, 0.7, and 1.0 nmol/l).
Male C57BL/6 mice were obtained at 6–8 weeks of age (21–29 gm) from Jackson Laboratories (Bar Harbor, ME), had unrestricted access to food and water, and were allowed to acclimate to the research environment for at least 10 d before experimentation. They were weighed and examined daily starting 5 d before IgG injection. Experimental and control groups were matched so that initial body weights did not differ significantly between experimental groups. Mice received a single intraperitoneal injection of IgG (15 mg on experimental day 0). Recipients of rabbit IgG were autopsied 2–27 d later, and recipients of human IgG at 4 d after injection.
Quantitation of heart rate and heart rate variability.
Starting 3 d before injection, electrocardiograms (ECGs) were recorded daily with mice resting in a quiet room in a plastic restrainer. Surface clip electrodes were applied to shaved sites over the dorsal midline and left sternal border with a ground disk electrode at the tail base. The ECG waveform was recorded using a preamplifier and amplifier (Axon Instruments, Union City, CA) and was digitized at 20 kHz for off-line analysis (Axotape software; Axon Instruments). Three minute epochs were analyzed by customized software to detect QRS complexes using a correlation function algorithm (Sahul et al., 2002). Resting heart rate was calculated from mean R-R interval. From differences between successive R-R intervals, we calculated the root mean square of successive differences as a simple time-domain measure of heart rate variability that primarily reflects cardiovagal (parasympathetic) modulation of heart rate (Tuininga et al., 1995; Vernino et al., 2003).
Assessment of GI motility and bladder function.
Gastrointestinal motility was estimated using a simple transit study. Blue dye number 1 (40–50 μl) was administered by transoral gavage using an 18 gauge feeding needle. After 30 min, mice were killed by carbon dioxide inhalation. The small bowel was ligated, removed from the abdomen, and gently pulled straight. Blue dye within the stomach and small bowel was readily observed. The distance from the gastroduodenal junction to the dye front was measured and expressed as a fraction of the total length of the small bowel. A score of 100% was recorded if the dye reached the ileocecal junction, and 0% was recorded if the dye failed to leave the stomach. The stomach and urinary bladder were ligated, removed, and weighed.
Plasma analyses.
At terminal anesthesia (isoflurane or carbon dioxide inhalation), blood was collected by cardiac puncture into tubes containing EDTA and reduced glutathione. The blood was centrifuged at 4°C, and plasma was stored at −80°C. Ganglionic AChR binding antibodies were quantitated by a previously described radioimmunoprecipitation assay (Vernino et al., 1998, 2002). The plasma concentration of norepinephrine was determined by HPLC analysis. Norepinephrine values in mice that were resting for 15–30 min under isoflurane anesthesia were considered to represent baseline resting values. Values in mice subjected to carbon dioxide inhalation were considered to represent levels of norepinephrine induced by metabolic and respiratory stress.
Electrophysiology.
Neuronal activity in abdominal prevertebral ganglia was recorded and analyzed as described previously (Miller and Szurszewski, 1997). At autopsy, a preparation of superior mesenteric ganglion (SMG) with attached lumbar colonic nerve and segment of colon was dissected rapidly and maintained in oxygenated Krebs solution at 37°C in a two-compartment organ bath. Intracellular recordings from ganglion neurons were made using glass microelectrodes filled with 3 m KCl (input resistance, 60–100 MΩ). Membrane potential was recorded using a Duo 773 amplifier (World Precision Instruments, Sarasota, FL) and stored digitally (Digidata 1322A; Axon Instruments). The attached segment of colon was cannulated at both ends to allow colonic distension and measurement of resulting intraluminal pressure. The colon was distended by injecting warm Krebs solution to activate intestinofugal afferent neurons. The colonic distension stimulus used in these experiments has been shown to predominantly activate fast excitatory nicotinic synaptic input to SMG neurons (Miller and Szurszewski, 1997). Electrophysiological data were analyzed by recording the presence and frequency of spontaneous and distension-evoked fast EPSPs (f-EPSPs) and the presence and amplitude of slow EPSPs (s-EPSPs) evoked by colonic distention.
Statistical analysis.
Descriptive statistics are presented. Because some of the autonomic data were not normally distributed, the nonparametric rank sum test (Wilcoxon rank sum test) was used to compare values for autonomic measures between groups. Electrophysiological data were compared using two-tailed t test. Statistical analyses were performed using JMP software (version 5; SAS Institute, Cary, NC). p values <0.05 were considered significant.
Results
Mice injected with control rabbit IgG exhibited no evidence of autonomic failure. Mice injected with rabbit IgG containing ganglionic AChR antibodies developed reversible EAAN. Clinical deficits were maximal 3–5 d after injection. Spontaneous recovery was apparent by 10 –14 d after injection. Rabbit ganglionic AChR IgG in mouse blood declined with a half-life of 8 d and was still detectable 21 d after injection (Fig. 1a ). Table 1 summarizes the anatomical and physiological abnormalities found in EAAN mice compared with controls.
Figure 1.
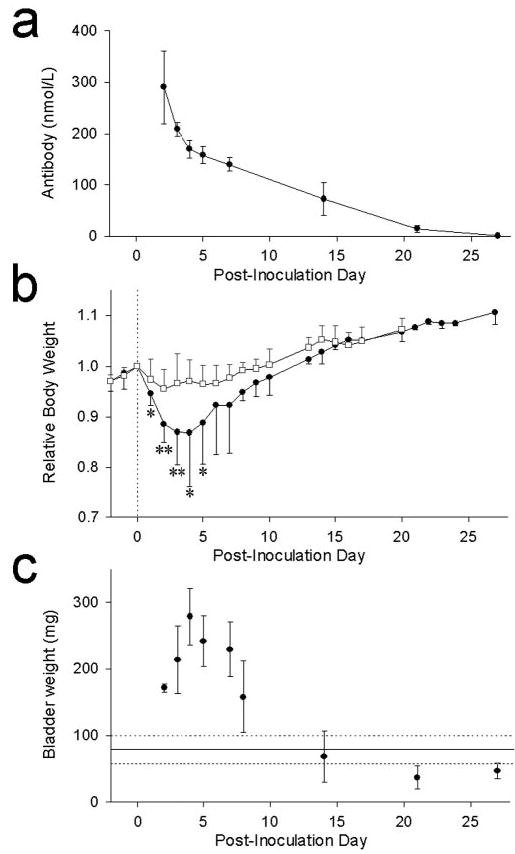
Time course of passively transferred EAAN. IgG was injected intraperitoneally on day 0. Data are expressed as mean with SEM (vertical bar). a, Rabbit IgG specific for ganglionic AChR was detectable in mouse plasma 3 weeks after injection. b, Body weight for each mouse is expressed relative to experimental day 0. Mice injected with ganglionic AChR IgG (filled circles) lose weight rapidly for several days and then regain weight. Asterisks indicate days at which their weights differed from control IgG recipient mice (open squares). *p < 0.01; **p < 0.0001. c, EAAN mice have marked urinary retention. Bladder size is expressed as weight of the bladder and its contents at autopsy. Mean bladder weight of control mice (solid horizontal line) and 95% confidence intervals (broken lines) are shown for comparison.
Table 1.
Anatomical and physiological measures of autonomic function in passive transfer EAAN using rabbit IgG
| Control | EAANa | Recoveryb | |
|---|---|---|---|
| Initial body weight (gm; day 0) | 25.1 ± 0.4 (29) | 24.8 ± 0.5 (42) | |
| Weight change (gm; day 3)c | −0.88 ±0.25 (28) | −3.26 ± 0.32 (39)* | |
| Resting heart rate (bpm) | 749 ±3 (21) | 732 ± 8 (23) | 759 ± 4 (5) |
| rMSSD (μsec) | 836 ±53 (21) | 542 ± 34 (23)* | 728 ± 70 (5) |
| GI transit (percentage of small bowel) | 53.3 ± 4.1% (21) | 30.8 ± 2.4% (22)* | 53.4 ± 9.3% (5) |
| Bladder weight (mg) | 79.0 ±10.4 (23) | 246.6 ± 23.2 (31)* | 49.6 ± 14.6 (5) |
| Stomach weight (mg) | 305.4 ± 13.9 (21) | 443.9 ± 20.6 (24)* | 436.4 ± 11.6 (5)* |
| Rest NE (ng/ml) | 3.69 ± 0.96 (9) | 4.89 ± 1.30 (10) | |
| Stress NE (ng/ml) | 19.01 ± 1.33 (22) | 5.18 ± 0.76 (24)* | 11.24 ± 2.25 (5) |
All data are mean ± SEM (number of mice). rMSSD, Root mean square of successive differences in heart period, a measure of heart-rate variability; NE, plasma norepinephrine concentration.
p < 0.0001 compared with control mice (Wilcoxon rank sum test).
Except for body weight, data are from experimental days 2–7.
Data are from EAAN mice examined on experimental days 14–27.
Data are from all mice except those killed before experimental day 3.
Autonomic function
No mouse injected with control rabbit IgG had significant weight loss. In contrast, mice injected with rabbit ganglionic AChR IgG lost up to 20% of total body weight in the 3 d after injection (Fig. 1b ). During this period, the EAAN mice were active and showed no evidence of weakness. In response to bright light, the pupils of EAAN mice remained dilated compared with control mice (Fig. 2a ). From day 5 onward, the EAAN mice regained weight. Within the first 7 d after injection, the stomachs of autopsied EAAN mice were enlarged, the small and large bowels were distended, and there was marked urinary retention (Fig. 2b ). On later days, there was gradual normalization of autopsy findings; by day 14, the urinary bladder size was normal (Fig. 1c ). Gastrointestinal transit studies in the first 7 d showed a reduction of intestinal transit distance in EAAN mice compared with controls. This also normalized at later time points.
Figure 2.
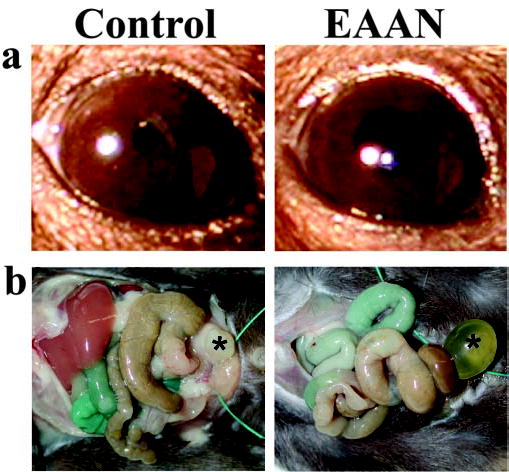
Overt signs of EAAN. a, Pupils of mice receiving control IgG were constricted in response to light. Mice with EAAN (3 d after ganglionic AChR IgG injection) had dilated pupils that responded poorly to light. b, At autopsy on day 4 after injection, mice receiving control IgG have a normal appearing bowel and empty bladder (*). Mice with EAAN show marked urinary retention (*) and dilated loops of large and small bowel. The proximal small bowel is colored resulting from oral administration of blue dye 30 min before autopsy.
The heart rate of EAAN mice, evaluated using surface electrocardiography in awake mice, did not differ significantly from control mice. However, resting heart rate variability was reduced in EAAN mice, consistent with impairment of cardiac autonomic control. Heart rate variability recovered after experimental day 14. Measurements of resting and stress plasma norepinephrine concentrations at the time of autopsy provided a general measure of sympathetic autonomic tone. Resting levels were the same in EAAN and control mice. Norepinephrine levels increased markedly in control mice that were killed in stress conditions, but norepinephrine levels in stressed EAAN mice did not differ significantly from resting levels (Table 1), indicating impairment in sympathetic autonomic reflexes.
Reversible impairment of ganglionic cholinergic synaptic transmission
To directly assess the effects of ganglionic AChR IgG on cholinergic synaptic transmission, we recorded activity from neurons in the SMG. These neurons receive excitatory cholinergic synaptic input from colonic intestinofugal afferent neurons, which produce fast EPSPs mediated by nicotinic AChRs (Miller and Szurszewski, 1997). Some neurons in the mouse SMG also show slow noncholinergic depolarizations in response to colonic distension, which are mediated primarily by neuropeptides (Miller and Szurszewski, 1997).
Electrical activity in SMG neurons of mice injected with rabbit ganglionic AChR IgG was significantly reduced compared with controls (Fig. 3, Table 2). All SMG neurons in control mice showed ongoing electrical activity consisting of fast EPSPs and action potentials (APs), and colonic distension increased the frequency of this activity. Neurons in ganglia of EAAN mice examined 2–3 d after administration of ganglionic AChR IgG had normal resting membrane potential, but only half showed spontaneous or distension-evoked fast EPSPs, whereas the number of neurons showing slow evoked EPSPs was not different from control. Among SMG neurons that did have fast EPSP activity, the frequency of EPSPs and action potentials was significantly reduced compared with controls (Table 2). These observations indicate a selective impairment of fast cholinergic synaptic activity in autonomic ganglia of EAAN mice. This cholinergic impairment was reversible (Fig. 3, Table 2). In ganglia of EAAN mice examined on day 27 after injection, all SMG neurons showed spontaneous and distension-evoked fast EPSPs. The frequency of fast EPSP and action potential discharges in ganglia of recovered mice was significantly higher than in control ganglia.
Figure 3.
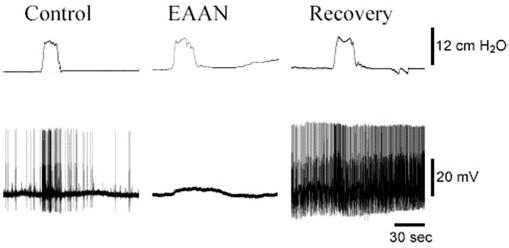
Autonomic neurophysiology in EAAN. Membrane potential of SMG neurons recorded using intracellular sharp microelectrodes. All SMG neurons from mice injected with rabbit control IgG (Control) showed spontaneous f-EPSPs and APs. When the attached segment of colon is distended (colonic intraluminal pressure is shown in the top trace), the frequency of f-EPSPs and APs increased. SMG neurons from mice injected 2–3 d earlier with rabbit ganglionic AChR IgG (EAAN) had normal resting membrane potential, but many showed no spontaneous or distension-evoked f-EPSPs or APs. Distention of the colon did elicit a slow membrane depolarization. In SMG neurons tested 27 d after injecting ganglionic AChR IgG (Recovery), spontaneous and distension-evoked f-EPSPs were present, and the frequencies of f-EPSPs and APs were higher than in control ganglia.
Table 2.
Electrophysiology of SMG neurons in mice injected with rabbit IgG
| Control | EAAN (days 2 and 3) | Recovered (day 27) | |
|---|---|---|---|
| Spontaneous f-EPSPs | 30/30 | 17/34 | 15/15 |
| Evoked f-EPSPsa | 30/30 | 17/34 | 15/15 |
| Evoked s-EPSPa | 19/30 | 21/34b | 11/15 |
| Spontaneous f-EPSP frequency (min−1) | 197.5 ± 50.5 (30) | 33.2 ± 7.7 (17)* | 380.5 ± 55.0 (15)* |
| Evoked f-EPSP frequency (min−1)b | 576.6 ± 93.8 (30) | 173.5 ± 38.5 (17)* | 1020.9 ± 92.1 (15)* |
| Amplitude of evoked s-EPSPs (mV)b | 2.5 ± 0.3 (19) | 2.8 ± 0.4 (21) | 3.5 ± 0.5 (11) |
Data are expressed as neurons responding per number tested, or as mean ± SEM (number of cells analyzed).
p < 0.05 compared with control (t test).
Responses evoked by colonic distention.
Ten neurons exhibited s-EPSPs but no f-EPSPs in response to colonic distention. The difference in number of s-EPSP responses compared with control mice was not statistically significant.
Effects of human IgG
Mice were injected with IgG prepared from three patients with AAN who were seropositive for ganglionic AChR antibodies and from one healthy control subject. No mouse exhibited clinical signs of dysautonomia, and none had significant weight loss. Mice receiving IgG from patient 1 were no different from control mice in any autonomic parameter studied. However, autonomic function assessment and autopsy findings were abnormal in mice injected with IgG from patients 2 and 3. On experimental day 4, these mice had bladder distention and reduced gastrointestinal transit compared with mice injected with control human IgG (Fig. 4). Compared with control mice, those injected with IgG from patient 2 or patient 3 had reduced heart rate variability and lower stress levels of plasma norepinephrine, but these differences were not statistically significant.
Figure 4.
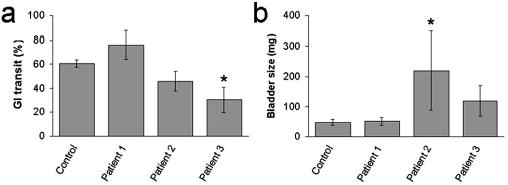
Passive transfer of human IgG. a, b, Gastrointestinal transit ( a) and bladder size ( b) were assessed 4 d after a single injection of human IgG. Mice injected with IgG from patient 2 (n = 5) and mice injected with IgG from patient 3 (n = 4) had reduced transit and increased bladder size. *p < 0.05.
Discussion
Mice injected systemically with IgG containing potent ganglionic AChR antibodies exhibit clinical, functional, and electrophysiological abnormalities consistent with failure of autonomic ganglionic cholinergic transmission. The disturbances of sympathetic, parasympathetic, and enteric autonomic function that we have documented in mice receiving IgG prepared from rabbits immunized with the major extracellular domain of recombinant α3 AChR subunit recapitulate the dysautonomia seen in patients with AAN (Vernino et al., 2000; Klein et al., 2003) and in rabbits with EAAN induced by active immunization with ganglionic AChR protein (Lennon et al., 2003; Vernino et al., 2003). The pathophysiological abnormalities in mice injected with ganglionic AChR IgG are also similar to the autonomic deficits documented in transgenic mice that lack ganglionic AChRs (Xu et al., 1999). Like α3 null mice, mice with EAAN induced by α3 subunit-reactive IgG have severe bowel hypomotility, dilated pupils, and extreme bladder distention associated with a selective deficit in ganglionic cholinergic neurotransmission.
Thus far, IgG prepared from serum of patients with AAN and tested as a single injection in mice has been considerably less pathogenic than immune rabbit IgG. Ganglionic AChR binding capacity in the human IgG samples was <1% of the capacity of the rabbit IgG pool. The human IgGs were also less reactive with mouse than with human ganglionic AChR. Nevertheless, two of the three human ganglionic AChR IgG preparations that we tested induced mild but significant signs of dysautonomia, affecting the gut or urinary bladder. Interestingly, the human IgG with highest binding capacity for mouse and human AChR had no effect. This likely reflects reactivity with epitopes that are accessible in solubilized AChR but not to extracellular IgG in vivo. Polyclonal human autoantibodies reactive with synaptic ion channels may have limited reactivity with mouse antigens and may require multiple injections over days or weeks to cause objectively measurable deficits of synaptic transmission (Toyka et al., 1977; Lang et al., 1981; Lambert and Lennon, 1988; Lennon and Griesmann, 1989). We anticipate that future protocols using multiple injections and the screening of IgG from a larger number of patients will demonstrate more severe dysautonomia in mice (Lambert and Lennon, 1988). The data we present here support the concept that IgG is the cause of dysautonomia in at least some human cases of AAN.
Our electrophysiological findings demonstrate that IgG-induced impairment of synaptic transmission in prevertebral ganglia is selective for nicotinic cholinergic activity (slow non-cholinergic EPSP activity was not affected) and is reversible. The reversibility of the electrophysiological deficit suggests that IgG acts by reducing the number of synaptic AChR through cross-linking, internalization, and accelerated degradation rather than by cytotoxic destruction of preganglionic or postganglionic autonomic neurons. A similar mechanism occurs at the neuromuscular junction with muscle AChR antibodies in experimental autoimmune myasthenia gravis (Lennon, 1977; Drachman et al., 1978) and with calcium channel antibodies in the Lambert–Eaton myasthenic syndrome (Fukunaga et al., 1983). We demonstrated in rabbits with EAAN induced by active immunization that impairment of cholinergic ganglionic synaptic transmission is associated with retention of viability and electrical excitability in the nonresponsive postganglionic autonomic neuron but a marked reduction of AChR in its surface membrane (Lennon et al., 2003; Vernino et al., 2003).
Allosteric interference by IgG binding near the acetylcholine-binding site would be an alternative pathophysiological effect. This seems an unlikely mechanism of action, however, because the autonomic deficits did not peak until 3 d after IgG injection and also because the rabbit ganglionic AChR IgG preparation used in these experiments did not block binding of 125I-labeled agonist (epibatidine) to ganglionic AChR (data not shown) in an in vitro blocking assay (Vernino et al., 2000).
Unexpectedly, the frequency of spontaneous and distension-evoked synaptic activity in the SMG after recovery was increased relative to control recordings. This observation suggests that the electrophysiological properties of the enteric neural network can adapt to impairment in autonomic function. There are many possible explanations for this observation, including increased activity of the preganglionic intestinofugal afferent neurons, enhanced efficiency of ganglionic synaptic transmission, and changes in dendritic or synaptic architecture. One example of plasticity of ganglionic cholinergic synaptic transmission that has been described previously is the long-term potentiation response to tetanic stimulation (Briggs and McAfee, 1988; Bennett, 1994). Additional studies are needed to characterize the nature of the adaptive changes that occur in the autonomic nervous system in response to IgG-mediated disruption of ganglionic synaptic transmission.
The reversibility of antibody-mediated dysautonomia in EAAN suggests that AAN in humans may also be reversible, at least in its early stages. Strategies to remove the pathogenic antibodies, to improve ganglionic synaptic transmission, and to repopulate the synapse with AChR would be expected to improve autonomic function in patients.
Footnotes
This work was supported by National Institutes of Health Grants NS02247 (S.V.), DK17632 (J.H.S.), NS22352 (P.A.L.), and NS32352 (P.A.L.) and by the Mayo Foundation. We acknowledge T. J. Kryzer, J. Thoreson, T. Vikse, and E. E. Swanson for their excellent technical support and K. K. Nickander for providing measurements of plasma norepinephrine.
References
- Bennett MR. Nitric oxide release and long term potentiation at synapses in autonomic ganglia. Gen Pharmacol. 1994;25:1541–1551. doi: 10.1016/0306-3623(94)90353-0. [DOI] [PubMed] [Google Scholar]
- Briggs CA, McAfee DA. Long-term potentiation at nicotinic synapses in the rat superior cervical ganglion. J Physiol (Lond) 1988;404:129 –144. doi: 10.1113/jphysiol.1988.sp017282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drachman DB, Angus CW, Adams RN, Michelson JD, Hoffman GJ. Myasthenic antibodies cross-link acetylcholine receptors to accelerate degradation. N Engl J Med. 1978;298:1116 –1122. doi: 10.1056/NEJM197805182982004. [DOI] [PubMed] [Google Scholar]
- Fukunaga H, Engel AG, Lang B, Newsom-Davis J, Vincent A. Passive transfer of Lambert-Eaton myasthenic syndrome with IgG from man to mouse depletes the presynaptic membrane active zones. Proc Natl Acad Sci USA. 1983;80:7636 –7640. doi: 10.1073/pnas.80.24.7636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein CM, Vernino S, Lennon VA, Sandroni P, Fealey RD, Benrud-Larson L, Sletten D, Low PA. The spectrum of autoimmune autonomic neuropathies. Ann Neurol. 2003;53:752–758. doi: 10.1002/ana.10556. [DOI] [PubMed] [Google Scholar]
- Lambert EH, Lennon VA. Selected IgG rapidly induces Lambert-Eaton myasthenic syndrome in mice: complement independence and EMG abnormalities. Muscle Nerve. 1988;11:1133–1145. doi: 10.1002/mus.880111105. [DOI] [PubMed] [Google Scholar]
- Lang B, Newsom-Davis J, Wray D, Vincent A, Murray N. Autoimmune aetiology for myasthenic (Eaton-Lambert) syndrome. Lancet. 1981;2:224 –226. doi: 10.1016/s0140-6736(81)90474-8. [DOI] [PubMed] [Google Scholar]
- Lennon VA (1977) Immunofluorescence analysis of surface acetylcholine receptors on muscle: modulation by autoantibodies. In: Cholinergic mechanisms and psychopharmacology (DJ Jenden, ed), pp 77–91. New York: Plenum.
- Lennon VA, Griesmann GE. Evidence against acetylcholine receptor having a main immunogenic region as target for autoantibodies in myasthenia gravis. Neurology. 1989;39:1069 –1076. doi: 10.1212/wnl.39.8.1069. [DOI] [PubMed] [Google Scholar]
- Lennon VA, Lambert EH. Myasthenia gravis induced by monoclonal antibodies to acetylcholine receptors. Nature. 1980;285:238 –240. doi: 10.1038/285238a0. [DOI] [PubMed] [Google Scholar]
- Lennon VA, Lindstrom JM, Seybold ME. Experimental autoimmune myasthenia: a model of myasthenia gravis in rats and guinea pigs. J Exp Med. 1975;141:1365–1375. doi: 10.1084/jem.141.6.1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lennon VA, Ermilov LG, Szurszewski JH, Vernino S. Immunization with neuronal nicotinic acetylcholine receptor induces neurological autoimmune disease. J Clin Invest. 2003;111:907–913. doi: 10.1172/JCI17429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindstrom JM, Engel AG, Seybold ME, Lennon VA, Lambert EH. Pathological mechanisms in experimental autoimmune myasthenia gravis. II. Passive transfer of experimental autoimmune myasthenia gravis in rats with anti-acetylcholine receptor antibodies. J Exp Med. 1976;144:739 –753. doi: 10.1084/jem.144.3.739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller S, Szurszewski J. Colonic mechanosensory afferent input to neurons in the mouse superior mesenteric ganglion. Am J Physiol. 1997;272:G357–G366. doi: 10.1152/ajpgi.1997.272.2.G357. [DOI] [PubMed] [Google Scholar]
- Patrick J, Lindstrom J. Autoimmune response to acetylcholine receptors. Science. 1973;180:871–872. doi: 10.1126/science.180.4088.871. [DOI] [PubMed] [Google Scholar]
- Patrick J, Seguela P, Vernino S, Amador M, Luetje C, Dani J (1993) Functional diversity of neuronal nicotinic acetylcholine receptors. In: Progress in brain research (Cuello AC, ed), pp 113–120. Amsterdam: Elsevier. [DOI] [PubMed]
- Sahul Z, Black J, Shen W. Accurate QRS detection at times of high artifact during tilt table testing. Clin Auton Res. 2002;12:329. [Google Scholar]
- Suarez GA, Fealey RD, Camilleri M, Low PA. Idiopathic autonomic neuropathy: clinical, neurophysiologic, and follow-up studies on 27 patients. Neurology. 1994;44:1675–1682. doi: 10.1212/wnl.44.9.1675. [DOI] [PubMed] [Google Scholar]
- Toyka KV, Drachman DB, Griffin DE, Pestronk A, Winkelstein JA, Fishbeck KH, Kao I. Myasthenia gravis. Study of humoral immune mechanisms by passive transfer to mice. N Engl J Med. 1977;296:125–131. doi: 10.1056/NEJM197701202960301. [DOI] [PubMed] [Google Scholar]
- Tuininga YS, Crijns HJ, Brouwer J, van den Berg MP, Man in’t Veld AJ, Mulder G, Lie KI. Evaluation of importance of central effects of atenolol and metoprolol measured by heart rate variability during mental performance tasks, physical exercise, and daily life in stable postinfarct patients. Circulation. 1995;92:3415–3423. doi: 10.1161/01.cir.92.12.3415. [DOI] [PubMed] [Google Scholar]
- Vernino S, Adamski J, Kryzer TJ, Fealey RD, Lennon VA. Neuronal nicotinic ACh receptor antibody in subacute autonomic neuropathy and cancer-related syndromes. Neurology. 1998;50:1806 –1813. doi: 10.1212/wnl.50.6.1806. [DOI] [PubMed] [Google Scholar]
- Vernino S, Low PA, Fealey RD, Stewart JD, Farrugia G, Lennon VA. Autoantibodies to ganglionic acetylcholine receptors in autoimmune autonomic neuropathies. N Engl J Med. 2000;343:847–855. doi: 10.1056/NEJM200009213431204. [DOI] [PubMed] [Google Scholar]
- Vernino S, Kryzer T, Lennon V (2002) Autoantibodies in autoimmune autonomic neuropathies and neuromuscular hyperexcitability disorders. In: Manual of clinical laboratory immunology (Rose N, Hamilton R, Detrick B, eds), pp 1013–1017. Washington, DC: ASM.
- Vernino S, Low PA, Lennon VA. Experimental autoimmune autonomic neuropathy. J Neurophysiol. 2003;90:2053–2059. doi: 10.1152/jn.00408.2003. [DOI] [PubMed] [Google Scholar]
- Xu W, Gelber S, Orr-Urtreger A, Armstrong D, Lewis RA, Ou CN, Patrick J, Role L, De Biasi M, Beaudet AL. Megacystis, mydriasis, and ion channel defect in mice lacking the alpha3 neuronal nicotinic acetylcholine receptor. Proc Natl Acad Sci USA. 1999;96:5746 –5751. doi: 10.1073/pnas.96.10.5746. [DOI] [PMC free article] [PubMed] [Google Scholar]