

Abstract
Enteroviruses (EV) frequently infect the central nervous system (CNS) and induce neurological diseases. Although the CNS is composed of many different cell types, the spectrum of tropism for each EV is considerable. These viruses have the ability to completely shut down host translational machinery and are considered highly cytolytic, thereby causing cytopathic effects. Hence, CNS dysfunction following EV infection of neuronal or glial cells might be expected. Perhaps unexpectedly given their cytolytic nature, EVs may establish a persistent infection within the CNS, and the lasting effects on the host might be significant with unanticipated consequences. This review will describe the clinical aspects of EV-mediated disease, mechanisms of disease, determinants of tropism, immune activation within the CNS, and potential treatment regimes.
Introduction
Enterovirus (EV) infections are a significant cause of morbidity and mortality throughout the world. The EV genus is part of the picornavirus family and includes such notable members as poliovirus (PV), coxsackievirus (CV), and enterovirus-71 (EV-71). EVs have been associated with many human diseases, including myocarditis (Klingel et al., 1992), pancreatitis (Ramsingh, 2008), and chronic inflammatory myopathy (Tam and Messner, 1999). Diseases caused by EV are not restricted to the well-known scourge of mankind throughout history, recognized as poliomyelitis. Many non-polio human EVs are quite common, causing an estimated 10–15 million or more symptomatic infections in the US alone. Non-polio EVs are known to target the central nervous system (CNS) and are responsible for numerous clinical manifestations, including encephalitis, and meningitis (Michos et al., 2007). The long-term consequences of EV infection upon the CNS are largely unknown. However, these viruses are known to persist, and the presence of viral RNA by itself is known to be potentially pathogenic in some cases. Also, EVs have been linked to autoimmune-like diseases, including diabetes, chronic inflammatory myopathy, and chronic myocarditis, perhaps in part due to the long-term presence of viral material. Therefore, EVs may be able to persist within the CNS potentially causing lasting neuropathology.
The original classification of enteroviruses included the four groups: Polioviruses (PVs), Coxsackie A viruses, Coxsackie B viruses, and ECHO (Enteric Cytopathic Human Orphan) viruses. However, the significant level of phylogenetic overlap among the four groups has led to a new classification system of consecutive numbers for the more recently isolated viruses (such as Enterovirus-71, Enterovirus-72, etc.) (Oberste et al., 2002). Our review will cover the most common and more extensively studied types of enteroviruses. However, the large number of circulating strains in human populations alone suggests a potential role for these viruses for unknown or unappreciated human diseases (Victoria et al., 2009). Also, vaccine design against enteroviruses may be difficult based on significant EV genetic variability.
Polioviruses (PVs)
Polioviruses (PVs) are perhaps the most studied and characterized of the enteroviruses, especially given the clinical consequences of infection which can infect motor neurons of the anterior horn of the spinal cord and lead to paralytic poliomyelitis in humans. Although the advent of polio vaccination has proven effective in greatly reducing the incidence of disease, some cases still occur and these cases appear to be on the rise; outbreaks following wild PV importations into previously polio-free countries continue to be an ongoing risk (MMWR, 2010). Over the years, basic research on PV has revealed the mechanics of protein translation and viral replication, immune activation, vaccine development, and viral population dynamics (Cameron et al., 2010; Pfeiffer, 2010). These discoveries have provided valuable information for the ongoing research of non-polio enteroviruses which continue to cause encephalitis and meningitis in humans.
Coxsackieviruses (CVs)
Coxsackieviruses (CVs), members of the enterovirus genus, may cause severe morbidity and mortality, particularly in the very young (Romero, 2008; Tebruegge and Curtis, 2009). CV infection during pregnancy has been linked to an increase in spontaneous abortions, fetal myocarditis (Ornoy and Tenenbaum, 2006), and neurodevelopmental delays in the newborn (Euscher et al., 2001). Infants infected with CV have been shown to be extremely susceptible to myocarditis, meningitis and encephalitis with a subsequent mortality rate as high as 10%. Also, a number of delayed neuropathologies have been associated with previous CV infection, including schizophrenia (Rantakallio et al., 1997; Suvisaari et al., 2003), encephalitis lethargica (Cree et al., 2003), and amyotrophic lateral sclerosis (Woodall and Graham, 2004; Woodall et al., 1994). Of note, few studies have been done to determine the lasting consequences of CV infection upon surviving individuals despite the relatively common occurrence of CV meningo-encephalitis in infants.
Enterovirus-71 (EV-71)
Enterovirus-71 (EV-71) is a major public health issue across Asia and of increasing concern globally, causing hand foot and mouth disease with potential neurological complications (Solomon et al., 2010). The brain stem is the most likely target for infection, and infection may cause serious clinical disease and psychological disorders in young children (Lee et al., 2009). New outbreaks of EV-71 have occurred across Asia, perhaps due to the ability of the virus to rapidly evolve (Solomon et al., 2010).
Echoviruses (ECHO-Vs) and Parechoviruses (P-ECHO-Vs)
Echoviruses (ECHO-Vs) were first isolated from the feces of asymptomatic patients; however, these viruses are now recognized to be associated various human diseases, including aseptic meningitis. ECHO-Vs are highly infectious and preferentially target infants and young children. These viruses can cause a mild, nonspecific illness similar to that of CVs. Also, parechoviruses (P-ECHO-Vs) have been found to be associated with encephalitis and white matter injury in neonates (Gupta et al., 2010; Verboon-Maciolek et al., 2008a), with similarities to EV infections (Verboon-Maciolek et al., 2008b). Although originally described as ECHO-V-22 and ECHO-V-23, their distinct genetic features have led to their re-classification (Harvala and Simmonds, 2009).
Theiler’s Murine Encephalomyelitis Virus (TMEV)
Although the picornavirus, Theiler’s murine encephalomyelitis virus (TMEV), is designated a cardiovirus by phylogenetic analyses, much can be learned from the existing literature on TMEV and CNS disease. Also, many similarities exist between EVs and TMEV in terms of their molecular biology, tropism, ability to persist, and induction of neuropathology. Therefore, we have included relevant information on TMEV and CNS pathology in this review. Two main subtypes of TMEV are studied in animal models. The strain GDVII produces acute encephalomyelitis quickly in mice. The DA, WW, or BeAn strains persist within the CNS and induce demyelinating plaques similar to those seen in multiple sclerosis. More detailed reviews of TMEV tropism, immune response, persistence, and demyelination has been published by other research groups (Brahic et al., 2005; Lipton et al., 2005; Oleszak et al., 2004; Olson et al., 2005; Rodriguez, 2007).
Molecular Biology of Enteroviruses
To better understand EV infection of the CNS and potential consequences upon glial or neuronal host cells, a full comprehension of EV structure and molecular biology is essential. EVs consist of icosahedral, non-enveloped viruses of approximately 30nm in diameter. These viruses generally have the proclivity to survive acidic environment, enabling their passage through the stomach following fecal/oral transmission and eventual invasion into the small intestine. The viral capsid contains the positive-strand RNA genome, ranging in size from ~7.2 – 8.5 kb. The viral genome includes several cis-acting RNA elements which play a role in replication and/or translation. These cis-acting elements include the 5′ and 3′ nontranslated regions (NTRs), the cis-acting replication element (CRE) and the 3′ poly(A) tail (Steil and Barton, 2009).
A single open reading frame (ORF) within the NTRs encodes a long viral protein which undergoes post-translational cleavage into the mature viral proteins. These viral proteins include the four structural proteins, VP1-4 which comprise the viral capsid, and seven non-structural proteins (2A–C and 3A–D) including the primer- and RNA-dependent RNA polymerase (3DPol) (Kitamura et al., 1981). In addition, one of the viral proteins 3B (also known as VPg) covalently attaches to the 5′ end of the viral genome. VPg plays an essential role in viral replication by acting as a primer for RNA synthesis. Utilizing PV as a model, VPg has been shown to undergo uridylylation to VPgpUpU by 3DPol using adenosine residues in the stem-loop structure of CRE, located in the 2C region of the EV ORF, as a template (Goodfellow et al., 2000; Paul et al., 2003). Recent elucidation by NMR suggests that VPg undergoes a structural change upon uridylylation which allows viral replication to be more efficient and selective (Schein et al., 2010). Since uridylylation is not observed during host cell transcription, this process may be a unique therapeutic target against EV infections, given that point mutations that block uridylylation may prevent viral growth (Paul et al., 1998). While VPg plays a role in both positive- and negative-strand RNA synthesis, it is still unknown how approximately 40 to 70 copies of positive-strand genome are produced for every negative-strand genome during an EV infection (Novak and Kirkegaard, 1991). Intriguingly, this ratio decreases upon the establishment of CVB3 persistence within the CNS suggesting that a double-stranded RNA genomic structure might assist in virus stability during persistence (Feuer et al., 2009).
Despite the unanswered questions with regards to the ratio of EV positive to negative strand synthesis during both acute and persistent infection, many other aspects of their translation and replication are well understood. For example, the well-characterized internal ribosome entry site (IRES) in the 5′ NTR directly interacts with host cell ribosomes to initiate translation of the viral genome upon entering the cell (Pelletier and Sonenberg, 1988). Immediate translation is necessary in order to produce the primer- and RNA-dependent RNA polymerase needed for viral replication, as well as the other viral proteins that facilitate replication. The viral genome is translated into a ~250-kDa polyprotein, which undergoes a series of cleavages at predetermined cleavage sites carried out by the viral proteases 2Apro and 3CDpro. During proteolytic processing, several precursor protein intermediates also play important roles during viral infection.
Only after viral protein translation is completed does negative-strand replication begin since translation and replication cannot occur at the same time (Gamarnik and Andino, 1998). A ternary complex then forms to facilitate both positive- and negative-strand synthesis at the 5′ cloverleaf structure of the 5′ NTR that includes the host poly(rC) binding protein (PCBP) and the viral precursor protein 3CD (Vogt and Andino, 2010). This complex associates with poly(A) binding protein 1 (PABP1), which is bound to the poly(A) tail of the genome, and causes the genome to circularize during negative-strand synthesis (Herold and Andino, 2001). Further analysis of the crystal structure of 3CD has shown that the N-terminus is in close proximity to the VPg binding site, and that 3CD may be involved in the uridylylation reaction as part of the replication complex (Marcotte et al., 2007). Moreover, 3AB (also the VPg precursor protein) is a membrane bound protein (Fujita et al., 2007), thus providing a spatial link between viral replication on host membranes, viral priming and the replication complex.
Enteroviruses and Autophagy
Interestingly, EVs have been shown to benefit from, and induce the cellular degradation process known as autophagy (Huang et al., 2009; Suhy et al., 2000; Wong et al., 2008). It is thought that EVs use the autophagosome membrane as a scaffold for viral replication. A recent publication has shown an increase in viral replication linked to the induction of autophagy during CV infection in rat primary neurons (Yoon et al., 2008). Autophagy is known to play an important role in preventing cellular damage in neurons, and this normal cellular process has recently been shown to be highly upregulated in neurons during short-term fasting (Alirezaei et al., 2010; Simonsen et al., 2008).
However, it still remains to be determined if EVs use autophagy for viral replication in all cells type within the CNS. For example, given their unique and critical role in development and CNS cellular homeostasis, neural stem cells might respond differently and uniquely to microbial infection. Furthermore, it will be important to elucidate whether autophagy plays a role in the establishment of viral persistence in the CNS. The induction or inhibition of autophagy in cells harboring viral material may alter EV persistence. Autophagy inhibitors have been shown to preferentially decrease extracellular as compared to intracellular PV production, thus providing an attractive model for non-cytolytic virus release from the cell (Jackson et al., 2005). Also, one might expect that an increase in the level of autophagy which might recycle organelles or other cellular components associated with EV persistence may hasten viral RNA degradation.
Host Cell Translational Shutdown Following Enterovirus Infection
In addition to their roles in viral replication and post-translational processing, several viral proteins have profound effects on the host cell during the course of infection effectively modulating cellular transcription, translation and protein secretion. Host cell transcription is suppressed by the viral protease 3C, which is carried into the nucleus by the nuclear localization signal located on 3D when in its precursor form (Sharma et al., 2004). Additionally, the 3C protein of EV-71 has recently been shown to block polyadenylation of host mRNA by cleaving a critical factor needed for this process (Weng et al., 2009). In turn, viral protease 2A halts host cell cap-dependent translation by cleaving eIF4G, which in a clever twist also enhances the translation of viral mRNA by stabilizing polysomes (Etchison et al., 1982; Kempf and Barton, 2008). Furthermore, viral proteins 2B and 3A inhibit host cell protein secretion, as plasma membrane and secretory protein transport are halted (Doedens and Kirkegaard, 1995).
In general, the host cell most likely faces a grim ultimate fate due the cytolytic nature of EVs. Viral protein 2B is a highly efficient viroporin which can permeablize the host cell membrane, as well as those of nearby cells (Madan et al., 2010). However, the pathways activated in the cell during infection can be somewhat contradictory. In what is thought to be an attempt by the virus to keep the cell alive long enough to complete viral replication and eventually escape its cellular confinements. EVs induce both anti-apoptotic (3A and 2B proteins) and pro-apoptotic effects (VP2, 2A and 3C proteins) on the host cell (Whitton et al., 2005). Furthermore, viral protein synthesis, which is boosted by 2A, has been shown to be necessary for apoptosis (Shih et al., 2008). With several reports demonstrating apoptosis in neurons after EV infection, and the role of 2A in regulating translation, it is interesting to speculate that shutoff of host cell translation by EVs may directly cause apoptotic cell death in neurons. We have shown that CVB3 induced apoptosis within pyramidal neurons in the hippocampus (Feuer et al., 2003). In contrast, TMEV may indirectly induce apoptosis of nearby pyramidal neurons in the hippocampus in non-cell autonomous manner (Buenz et al., 2009).
Virus Entry – Cell Receptors
EVs use a wide array of receptors and entry mechanisms to invade the host cell. In particular, EV-71 has been shown to use several receptors, including sialylated glycans, P-selectin glycoprotein ligand-1 and Scavenger receptor B2 (Nishimura et al., 2009; Yamayoshi et al., 2009; Yang et al., 2009). CVB can utilize both decay accelerating factor (DAF) and the coxsackievirus and adenovirus receptor (CAR) for viral entry, depending on the target cell. For example in polarized cells, CVB elicits a unique strategy by binding to DAF at the apical surface of the cell, followed by CAR binding within the tight junctions in a caveolin-dependent, dynamin-independent manner (Coyne and Bergelson, 2006). Whereas in non-polarized cells, CVB only appears to utilize CAR for entry and undergoes dynamin-dependent and caveolin-independent entry (Patel et al., 2009).
While some EVs are capable of utilizing multiple receptors, PV uses only one, CD155, an adhesion molecule also known as the human PV receptor (hPVR). In an in vitro blood-brain barrier (BBB) model, PV has been show to enter human brain microvascular endothelial cells (hBMECs) using hPVR in a dynamin and caveolin-dependent manner (Coyne et al., 2007). However, when using non-CNS specific immortalized cells lines, PV entry was found to be caveolin-independent. Both reports found tyrosine kinases to be essential for entry (Brandenburg et al., 2007). These studies illustrate the importance of characterizing EV entry in multiple CNS cell types, since key differences may exist in different model systems of infection. Similarly, care must be taken in making generalized conclusions of virus entry based on in vitro cell culture models.
Undoubtedly, receptor expression on potential target cells defines the first barrier to virus entry. A recent study has shown a link between decreased CAR expression in differentiated primary neurons and a reduction in CVB3 infection, suggesting that susceptibility to infection may diminish as the level of virus receptor decreases during the differentiation process (Ahn et al., 2008). Similarly, PV infection and tissue tropism was largely found to be restricted by hPVR expression in the CNS of hPVR transgenic mice (Gromeier et al., 1995). Analysis of receptor distribution in hPVR transgenic mice during development showed high levels of protein expression in the spinal cord anterior horn motor neurons which are known to harbor infection in the mature CNS (Gromeier et al., 2000). Although some EV receptors may be widely expressed in tissues including the CNS, additional cellular determinants may ultimately control tropism. For example, CAR expression is relatively widespread in the murine neonatal CNS (Honda et al., 2000; Hotta et al., 2003; Venkatraman et al., 2005) and utilized by adenovirus - a DNA virus with substantial tropism differences as compared to CVB3. Yet, early CVB3 infection is largely restricted to neural progenitor and stem cells (NPSCs) (Figure 1) or infiltrating myeloid cells during early infection (Feuer et al., 2005) (Figure 2). The proliferative status of these cells may provide an additional level of susceptibility to infection (Feuer et al., 2002; Feuer et al., 2003; Feuer et al., 2005; Feuer and Whitton, 2008).
Figure 1. Neural progenitor and stem cells grown in culture are highly susceptible to coxsackievirus infection.
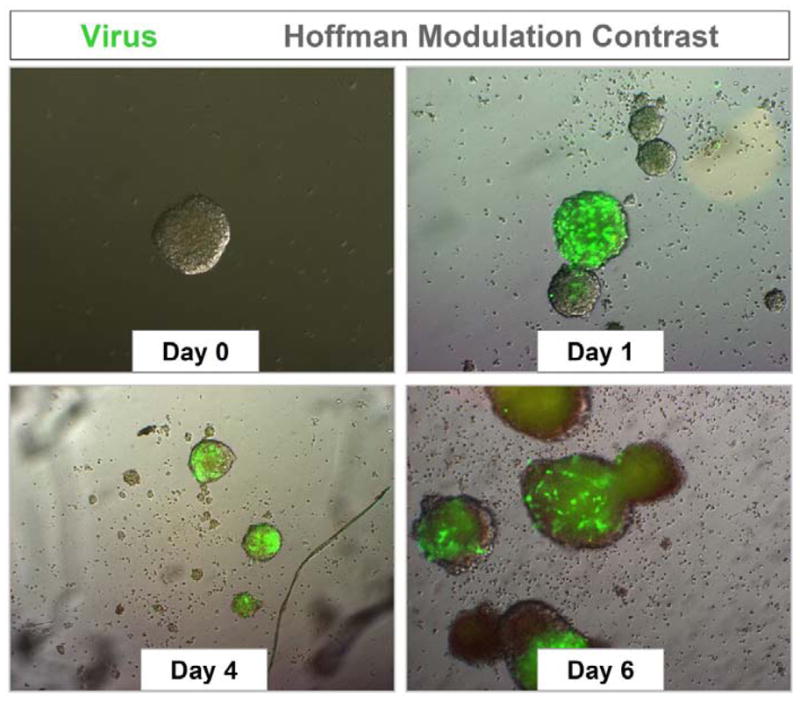
NPSCs were isolated from the cortices of one day-old C57 BL/6 mice, cultured to form neurosphere aggregates, and infected with eGFP-CVB3 (moi = 0.1). Infected neurospheres were observed over time by fluorescence microscopy. Virus protein expression (green) was readily observed by day 1 PI. An increase in viral protein expression was seen until day 4 PI. By day 6 PI, virus protein levels were reduced, and signs of cytopathic effect (cpe) were readily observed at a higher magnification (not shown).
Figure 2. Induction of CCL12 and the progressive extravasation of infected myeloid cells through the basement membrane as determined by confocal microscopy and Imaris 3D analysis.

Three day-old C57 BL/6 mice were intra-cranially infected with a recombinant coxsackievirus B3 expressing eGFP (eGFP-CVB3; 107 pfu). The brains were harvested 12 hours later and inspected by confocal fluorescence microscopy. Myeloid cells responding to CVB3 infection were observed entering through the tight junctions of the choroid plexus epithelium. As these myeloid cells entered the lateral ventricle, they expressed high levels of viral protein (green). Also, the chemokine CCL12 (red), a potential chemoattractant molecule for infiltrating myeloid cells, was expressed within the choroid plexus and surrounding subventricular zone. The kinetics of myeloid cell migration through the basement membrane (outlined by laminin staining in red) was observed by confocal microscopy and IMARIS 3D analysis. Immunofluorescence images showed infected myeloid cell migration (green) through the tight junctions of the choroid plexus epithelium. Grey scale images of all three colors (virus-green; laminin-red, DAPI-blue) at 12 hours PI revealed the intensity and organization of the myeloid cell infiltration in greater detail. In order to better visualize myeloid cell entry, IMARIS 3D with diminishing laminin label (diminishing red) was carried out, and the methodology revealed the extravasation of infected myeloid cells through the basement membrane (white arrow). Original images were obtained with a 63X Plan-Aprochromat objective at 0.3μm interval step slices.
Coxsackievirus Infection of Neural Progenitor and Stem Cells
What might be the benefit of CVB3 targeting NPSCs for infection? We expect that the benefits may be many fold in terms of viral replication, persistence, dissemination, and transmission. First, NPSCs actively proliferate not only during neonatal development, but also occasionally within the adult host. Infection of NPSCs may assist in expanding the tropism of CVB3 upon their differentiation into the three neural lineages - neurons, astrocytes, and oligodendrocytes. Previously, we determined that CVB3 preferentially replicates within actively proliferating cells (Feuer et al., 2002). Hence, NPSCs may be considered ideal host cells for infection.
Second, NPSCs migrate upon their differentiation into particular regions within the CNS, including the hippocampus and the olfactory bulb. The olfactory bulb might be considered an escape route for the virus upon anterograde transport across the olfactory neuroepithelium. Third, NPSCs might be altered upon their differentiation into neurons. Alterations in neuronal function may lead to behavioral modifications within the host, which may act to maximize virus transmission. Fourth, targeting of NPSCs might be a strategy of CVB3 to establish persistent infection with sporadic reactivation whenever quiescent stem cells become activated to generate downstream progenitor cells (Figure 3). The majority of primary neural stem cells at any moment in time might be expected to remain within a quiescent state. Therefore, CVB3 replication may be temporarily arrested in the host cell until a later point in time when the production of virions may be of benefit to viral transmission, perhaps through the olfactory neuroepithelium. Finally, normal immune responses may be suppressed within neurogenic regions, either due to the immunopriviledged status of the CNS, or potential reduced antiviral responses in these crucial neural stem cells.
Figure 3. Potential reactivation of a persistent coxsackievirus infection within quiescent neural stem cell.
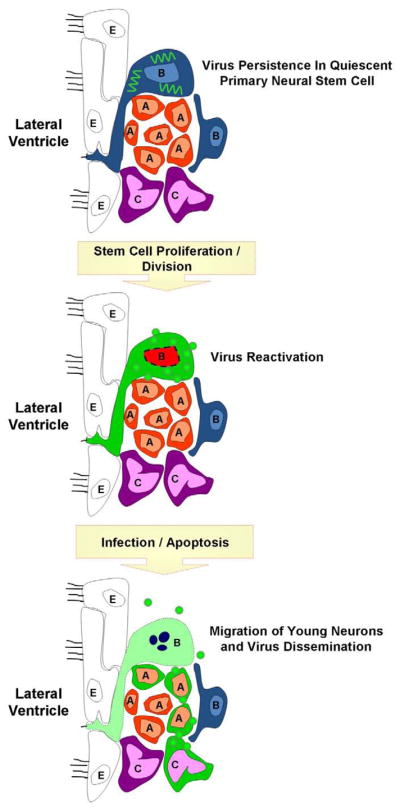
Coxsackieviral RNA (squiggly green lines) may persist in a latent state within quiescent type B neural stem cells in the CNS. Following cellular division reactivation of viral RNA may lead to viral protein expression and sporadic infectious virus production. The production of infectious virions may infect nearby progenitor cells. Alternatively, asymmetric division of infected type B stem cells may generate downstream progenitor cells or immature neurons that remained infected and migrate to other regions of the CNS. The outcome over the long-term of continuous virus reactivation may be virus-mediated neuropathology and virus dissemination.
Enterovirus Dissemination – Hitch-Hiking on Migratory Immune Cells
Perhaps surprisingly, polarized epithelial cells are not considered primary target cells for CVB3 infection in vivo, despite their expression of intrajunctional proteins. Rather, the major targets of infection have been found to include the acinar cells of the pancreas, cardiomyocytes, activated lymphocytes within the marginal regions of the spleen, infiltrating nestin+ myeloid cells, proliferating NPSCs, and immature neurons of the CNS. Instead, targeting of intrajunctional proteins such as CAR by CVB3 might be a sophisticated viral strategy to specifically infect migratory immune cells as they undergo diapedesis through the tight junctions. In this regard, we have recently shown the ability of CVB3 to infect nestin+ myeloid cells upon their infiltration across the tight junctions of the choroid plexus epithelial cells comprising the blood-cerebral spinal fluid-barrier in response to early CNS infection (Tabor-Godwin et al., 2010). These infected myeloid cells eventually migrate into the parenchyma of the brain, thereby assisting in virus dissemination.
The benefits to virus dissemination by targeting intrajunctional proteins may be many fold; first, virions temporarily attached to sequestered intrajunctional proteins might find themselves in a perfect location to “hitch-hike” onto a migratory immune cell as these cells enter sites of inflammation or injury in response to early infection. Second, immune cells responding to inflammation or injury tend to be highly activated or undergoing proliferation, and each virus may induce unique chemokine profiles early after infection to attract their immune cells of choice. CVB3 has been shown previously to preferentially replicate within activated or proliferating cells. Third, virions carried internally by immune cells might temporarily shield the virus from neutralizing antibodies upon dissemination. Neutralizing antibodies are thought to play a major role in limiting EV infections, and hypogamma-globulinemic patients deficient in humoral immunity are highly susceptible to EV infection. Perhaps not surprisingly, other viruses have been shown to bind to intrajunctional proteins (Bergelson, 2009), suggesting that the proposed mechanism above might be a general feature of virus dissemination within the host. Therefore, rather than being a hindrance to virus entry - which might be expected for receptors sequestered within highly inaccessible areas - virus evolution may have optimized virion attachment to intrajunctional proteins to maximize viral spread using immune target cell “Trojan horses” which become infected during their migration across tight junctions. Supporting this hypothesis, evidence for viral targeting of lymphocytes or monocytes for spread into primary target organs and maximal viral replication has been described for CVB3 (Mena et al., 1999) and other viruses (Noda et al., 2006).
Routes of Enterovirus Entry into the CNS
Since EVs, such as PV, are transmitted via the fecal-oral route of infection, a detailed understanding of how the virus travels from the gut to the brain is essential in designing strategies to limit EV disease. Despite the extensive research that has been performed to understand CNS invasion, many questions remain. Multiple pathways may actually work in combination to complete this phenomenon. The two main models of PV entry into the CNS both involve viremia, or the presence of virus in the blood, after the virus has replicated in the lymphatic tissues of the gastrointestinal tract. One hypothesis simply proposes the direct movement of the virus through the blood-brain-barrier (BBB) in an hPVR-independent manner (Yang et al., 1997). Perhaps direct entry across the BBB via early PV infection of hBMECs may occur first, thus weakening or exposing the BBB and allowing for secondary virus entry into the CNS regardless of receptor expression.
A second model of EV entry proposes PV spread from muscle to the CNS along neural pathways (Ren and Racaniello, 1992). These neural pathways were more clearly defined by Gromeier and Wimmer in 1998 as retrograde axonal transport of the virus from damaged muscle to the CNS (Gromeier and Wimmer, 1998). Since muscle injury has been shown to induce neurological PV infection, the model is typically referred to as “provocation poliomyelitis”. Initially, it was proposed that hPVR was necessary for retrograde axonal transport of the virus along microtubules (Ohka et al., 2004). However more recently, retrograde transport has been shown to occur independently of the hPVR (Ohka et al., 2009).
Another emerging model of CNS entry involves the use of EV-infected immune cells invading the CNS in a “Trojan horse” fashion, as described above. Of note, PV has been shown to infect monocytes, macrophages and dendritic cells in an hPVR-dependent manner (Freistadt et al., 1993; Wahid et al., 2005). In addition, lymphocytes may play a role in monocyte infection by inducing their activation and facilitating viral replication (Eberle et al., 1995). Further investigation has revealed a possible link between PV pathogenesis in the CNS and monocyte infection, as neurovirulent strains were found to replicate more efficiently in monocytes (Freistadt and Eberle, 1996). As described above, CVB3 and also EV-71 have been shown to replicate in immune cells, B-lymphocytes, T-lymphocytes and cells of the myeloid lineage (Haddad et al., 2004; Vuorinen et al., 1996). Our laboratory has recently revealed the rapid infiltration of a novel population of nestin+ myeloid cells into the neonatal CNS following CVB3 infection (Tabor-Godwin et al., 2010). These cells are highly susceptible to CVB3 infection and move into the brain parenchyma over time, suggesting a role in viral dissemination. The growing number of studies demonstrating EV infection of immune cells suggests that furthers investigation of the “Trojan horse” model of CNS invasion may be warranted.
Enterovirus Quasispecies
Another essential factor in CNS invasion may be the involvement of EV quasispecies, or genotypic variants of virus populations, due to mutational errors accumulated during viral replication. These errors result from the low fidelity of the RNA-dependent RNA polymerase whereby at least one incorrect nucleotide is incorporated per genome duplication (Ward and Flanegan, 1992; Ward et al., 1988). Other contributing factors during viral replication may also increase viral diversity. For example, a recent in vitro study performed with PV 3DPol found that the replication fidelity may be relatively high; yet other events during replication, such as template switching, may actually contribute to the errors found in the genome (Freistadt et al., 2007).
Regardless of the nature of mutation rate, the existence of a swarm of viral variants, or a quasispecies cloud, has been shown to contribute to EV spread to the CNS. Recent elegant work utilizing a PV variant encoding a mutation in 3DPol isolated during serial passage in the presence of ribavirin has revealed the critical nature of viral quasispecies in contributing to invasion of the CNS. The PV isolate (G64S) was shown to increase the fidelity of the viral polymerase and decrease quasispecies diversity (Vignuzzi et al., 2006). The G64S PV isolate was less neurovirulent than wild type virus, and CNS invasion failed to occur unless the isolate was co-inoculated in combination with a diverse quasispecies population. These intriguing results demonstrated that cooperation between variants within a viral population was necessary for CNS invasion and the ensuing neuropathology. Cooperation among viral genotypes might include evasion of the immune response simultaneously with CNS invasion.
Genetic bottleneck effects have also been observed as viral variants enter the CNS. As the first few viruses enter cellular routes into the CNS, antiviral responses become initiated which block entry for the remaining variants (Pfeiffer and Kirkegaard, 2006). This antiviral genetic bottleneck effect, also referred to as the “burned-bridge” model, may contribute to the rare transmission of PV into the CNS which occurs in approximately 1 to 2% of cases (Gromeier and Wimmer, 1998). Later studies have added to the “burned-bridge” model whereby multiple barriers may contribute to genetic bottleneck effects of quasispecies. These barriers which may limit neurovirulence include the integrity of the gut, the induction of a protective innate immune response, and inefficiency in neuronal transport of virus (Kuss et al., 2008; Lancaster and Pfeiffer, 2010). Thus, EV entrance into the brain appears to be a complicated interplay not only between the virus and the host, but also among viral variants within the quasispecies population.
Enterovirus Tropism
EVs are inherently neurotropic, yet each member of the virus genus targets different regions of the CNS (Table 1). A recent in vivo study by Nagata et al. examined the localization of PV and EV-71 following intravenous infection of monkeys. The authors identified PV-induced lesions primarily within the pyramidal tract of the CNS causing a total loss of motor neurons in the anterior horn of the spine. In contrast, EV-71 induced only limited damage in both the extrapyramidal and pyramidal tracts, leaving many motor neurons in the anterior horn of the spine intact (Nagata et al., 2004). Furthermore, the authors were able to isolate PV from the entire CNS, including the dorsal root ganglia and the trigeminal. In contrast, EV-71 could not be isolated from either of the latter two regions of the CNS. These results illustrate the broad range of neuronal target cells for PV, as compared to the more restricted neuronal targets for EV-71.
Table 1.
Enterovirus tropism in the CNS
| Virus | CNS Localization | Cell Types Infected - Acute | Cell Types Infected - Persistence |
|---|---|---|---|
| Poliovirus | entire CNS | neurons, astrocytes, oligodendrocytes | neurons |
| Enterovirus-71 | CNS - but not dorsal root ganglia or trigemina | neurons, astrocytes | persistence documented; site unknown |
| Coxsackievirus | choroid plexus, neurogenic regions (SVZ, SGZ), hippocampus, cortex | nestin+ myeloid cells, NPSCs, neurons | NPSCs (in culture) |
| TMEV | grey matter (acute) white matter (persistence) | neurons, astrocytes, oligodendrocytes | macrophages, microglia, oligodendrocytes |
In yet another example of neuronal targeting of a picornavirus, TMEV was shown to initially infect gray matter regions of the CNS, yet virus persisted in the white matter following acute infection (Roussarie et al., 2007). Echovirus type 1 (ECHO-1-V) has been shown to cause cellular necrotic lesions in the cerebral cortices of transgenic mice expressing human integrin very late antigen 2, leading to paralysis and wasting (Hughes et al., 2003). In contrast, early CVB3 infection was found to be localized to specific neurogenic regions of the brain, including the subventricular zone (SVZ) (Feuer et al., 2005).
Each EV genus member clearly prefers distinct regions of the CNS. Yet, what factors determine preferential tropism at the cellular level? Tropism at the cellular level can viewed as an interplay of at least three general factors: the ability of the virus to infect and replicate in the specific host tissues; the capability of the host cell to clear the virus; and the capacity of the virus to avoid clearance by the host. Potential differences in the magnitude of the interferon α/β response may greatly influence PV tissue tropism (Ida-Hosonuma et al., 2005; Yoshikawa et al., 2006). The tropism of PV for neurons has previously been shown to be also determined by the PV Internal Ribosomal Entry Site (IRES) (Kauder and Racaniello, 2004). Kauder et al. initially found that the tropism of PV towards nervous tissues could not be fully explained by organ-specific differences in IRES-mediated translation within age-matched samples. They investigated alterations in PV tropism by generating recombinant viruses engineered to contain the IRES from human rhinovirus (Kauder et al., 2006). In this manner, the investigators observed IRES-mediated, organ-specific translational differences between EVs in adult versus neonatal infection. They concluded that these differences might be attributed to IRES trans-acting Factors (ITAFs) differentially expressed in various cell types, including neurons.
Ires Trans-Acting Factors (ITAFs)
ITAFs have been found to play a key role in EV IRES-mediated translation initiation. Some IRESs require specific ITAFs, while others require few additional proteins (Fitzgerald and Semler, 2009). Hence, tissue specific ITAFs may help to explain the tropism of EVs towards particular tissue types. Neurons may be more susceptible to infection due to the availability of specific ITAFs needed to initiate IRES-mediated translation. Investigation of TMEV and FMDV IRESs suggested that cell-specific differences in ITAFs may explain why some viruses are able to replicate in neurons, while others cannot (Pilipenko et al., 2000). Furthermore, translation initiation of different EVs IRES may depend on a combination of different ITAFs and eukaryotic initiation factors which may have different cell-specific levels of abundance (Boussadia et al., 2003). Recently, the Far Upstream Element (FUSE) binding protein (FBP2), originally identified to associate with a protein complex involved in intronic c-src neuronal specific silencing enhancer, was discovered as a critical ITAF for EV-71 infection (Lin et al., 2009a).
While the IRES and ITAFs have largely been associated with viral replication in neurons, EVs are capable of infecting other cell types in the CNS. For example, PV can infect astrocytes and oligodendrocytes (Couderc et al., 2002). Also, it has been known for some time that TMEV can infect astrocytes, yet persist in oligodendrocytes (Rodriguez et al., 1983). EV-71 has been found to readily infect and replicate in astrocytoma cultures (Kung et al., 2007) and cultures of rat brain astrocytes (Tung et al., 2010). We have shown that CVB3 preferentially infects NPSCs (Feuer and Whitton, 2008), and more recently, a novel population of nestin+ myeloid cells infiltrating into the CNS (Tabor-Godwin et al., 2010). Parechoviruses (P-ECHO-Vs) are suspected to be taken up by microglia which leads to the activation of these resident immune cells (Volpe, 2008). Also, microglia, and glial cells may be potential sites of EV persistence. For example, macrophages and microglia have been found to be the main reservoir for harboring TMEV during persistent infection (Roussarie et al., 2007). Respiratory EV, including rhinoviruses (RVs), have not been historically found infect the CNS. However, a recent study suggested that recombination of respiratory EV to genetically similar viruses, such as CV and PV, may eventually lead to respiratory EVs with tropism for the central nervous system (Tapparel et al., 2009). This possibility is not implausible, especially with the recent findings suggesting the contribution of viral variants or quasispecies to PV spread into the CNS (Vignuzzi et al., 2006).
Immune Responses to Enteroviruses
The immune response plays a critical role in protecting the host from viral pathogens by both modulating the release of chemokines and inflammatory cytokines for leukocyte recruitment, and by directly fighting infection via the interferon response. An informative review of chemokine induction in response to neurotropic infections has recently been published by Hosking et al (Hosking and Lane, 2010). These chemoattractant molecules may be especially critical to combating microbes within immunopriviledged sites, such as the CNS. We have demonstrated the induction of numerous chemokines in the CNS following CVB3 infection. One particular chemokine, CCL12, may play an essential role in the recruitment of nestin+ myeloid cells that infiltrate across the blood-CSF-barrier and become infected upon their entry through the choroid plexus (Tabor-Godwin et al., 2010). “Bystander” casualties suffered upon activation of cytolytic T cells during an adaptive immune response within the CNS may be potentially more devastating to neurons than a viral infection itself. Thus, the host may restrict or control immune responses in response to a viral infection within immunopriviledged sites; this restriction may partly explain why certain viruses may preferentially target the CNS.
Innate Immune Response Following Enterovirus Infection
The innate immune response begins with the recognition of the virus by cytoplasmic sensors. EVs have been shown to activate essential innate immune response molecules, including Toll-like Receptor 3 (TLR3), Retinoic Acid Inducible Gene I (RIG-I), and Melanoma Differentiation-Associated Gene 5 (MDA5). TLR3 is located on the membrane of endosomes or the plasma membrane and recognizes double-stranded RNA, a structure generally observed during viral replication of all RNA viruses. Recent studies suggest that TLR3 plays a strong role in cardioprotection against EV infections and may depend on autophagic processes (Gorbea et al., 2010). Studies of the recognition of EVs by cytoplasmic sensors of the innate immune response have largely centered on DExD/H-box-containing RNA helicases, such as RIG-I and MDA5. In examining encephalomyocarditis virus (EMCV) induction of the type I interferon (IFN) response in mouse embryonic fibroblasts, Kato et al concluded that MDA5, but not RIG-I, was critical for picornavirus detection (Kato et al., 2006). However, this does not mean that RIG-I is excluded from picornavirus detection. By Utilizing MDA-5 knockout mice, Papon et al observed that RIG-I was also useful in detecting EMCV (Papon et al., 2009). Both of these sensors may play important roles in EV detection by the host. EVs may circumvent the action of these molecules by encoding proteins that affect their signaling pathways.
Once a pathogen is recognized, the innate immune response may play a substantial role in initiating virus-mediated neuropathology following EV infections. For example, the severity of echovirus and EV infections in the CNS have been associated with higher systemic levels of proinflammatory cytokines like IL-6, IL-1β, and TNF, which can lead to greater cytokine-induced tissue destruction (Lin et al., 2003; Liu et al., 2005). Also, CV activation of the inflammatory response has been shown to cause extensive infiltration of leukocytes into the CNS thereby causing inflammatory lesions and contributing to neuropathology associated with the virus (Feuer et al., 2009).
In addition to the potentially harmful inflammatory responses triggered within the CNS, EV infections may also initiate the type I IFN response pathway. TLR-3, RIG-1, and MDA5 are all able to induce the type I IFN response. TLR-3 induces the production of type I IFNs by activating the transcription factors Interferon Regulatory Factor 3 (IRF3), NF-κB, and AP-I through the adaptor protein TIR-domain-containing Adapter Inducing Interferon β (TRIF) (Matsumoto and Seya, 2008; Wilkins and Gale, Jr., 2010). The RIG-I and MDA-5 pathways are distinct and independent of the TLR-3 pathway. RIG-I and MDA-5 both interact with the adaptor molecule IPS-1 through their CARD domains. IPS-1 transfers the activation signal to downstream kinases, which in turn activate IRF-3 and other transcription factors involved in type I IFN induction (Kato et al., 2006). Malathi et al determined that the helicase activity of RIG-I and MDA5 may have important autoregulatory roles and work in concert with Endoribonuclease L (RNase L) to produce small self RNA cleavage products that can interact with RIG-I or MDA5, thereby amplifying the signal (Malathi et al., 2007).
Following their induction, type I IFNs bind to their receptors either on the same cell or on neighboring cells. Binding of type I IFNs to their receptor induces the transcription of antiviral miRNAs and IFN Stimulated Genes (ISGs) through the JAK/STAT pathway. Many of these ISGs encode antiviral products or serve to further upregulate the type I IFN pathway. For example, RNase L is an ISG that contributes to the amplification of the type I IFN response, but also cleaves viral RNA and inhibits their translation. Furthermore, the antiviral activity of RNase L has been found to have a protective role in the CNS against coronavirus-induced demyelination. In comparing wildtype and RNase L−/− mice, Ireland et al found that RNase L deficiencies increased demyelination and axonal damage in the brain following infection, and affected the regulation apoptosis (Ireland et al., 2009). Additional ISGs encode products that can inhibit specific viruses. Additionally, IFN-β has been found to modulate the expression of antiviral microRNAs in response to RNA viruses, including hepatitis C (Pedersen et al., 2007). Hence, it is reasonable to suspect that type I IFN-induced antiviral miRNAs might play an important role in host defense against EVs in the CNS.
EVs have evolved a surprising number of mechanisms for undermining the host innate immune response, attacking at different stages of the IFN induction and response pathways. EV and cardiovirus (such as TMEV) proteins may counter-act host defenses (Agol and Gmyl, 2010). The ability to evade the host antiviral response may be an important contributing factor not only for maximizing acute infection, but also for the successful establishment of viral persistence. PV, rhinovirus (RV), and EV have been found to reduce RIG-I levels and disrupt RIG-I mediated interferon signaling (Barral et al., 2009). A reduction in RIG-I levels has been attributed to the activity of the PV 3C protease. PV and RV Type-1a have also been observed to cleave MDA5 through a caspase and proteasome-dependent mechanism (Barral et al., 2007). Since RIG-I signaling induces further RIG-I expression, and MDA5 provides secondary protection against viral infection in the host cell, EVs may also target the downstream signaling pathways for these molecules. The 3C protease of EV-71 has also been found to disrupt RIG-I signaling through a protease-independent fashion. Instead of cleaving, 3C protease binds to RIG-I and prevents the recruitment of IPS-1 (Lei et al., 2010). In contrast, RV-14 attenuates the type I interferon response by targeting the activation of IRF-3 upon identification by MDA5 (Kotla et al., 2008). Similarly, TMEV has been found to inhibit IRF-3 dimerization (Ricour et al., 2009).
Once the antiviral response has been initiated, viruses have developed ways to combat these protective actions. PV and RV have been found to cleave Nup62 (Park et al., 2010) and degrade Nup153 and Nup98 (Park et al., 2008). By altering the nucleus by degrading nuclear pore complex proteins, viruses can affect host mRNA and protein localization, and shut down host protective factors. The hyperphosphorylation of Nup98 by the L protein during TMEV infection has been shown to block host mRNA export from the nucleus (Ricour et al., 2009). Herpesvirus genomes have been found to encode miRNAs suggesting that they exploit the host miRNA machinery to regulate the expression of host and viral genes (Pfeffer et al., 2005). Although this mechanism has not been demonstrated for EVs, Pelletier et al have found that cells exposed to and cured of PV using RNAi respond more quickly to RNAi treatment, as compared to cells never having been exposed to PV (Pelletier et al., 2010). These intriguing results suggest that cellular miRNA processing machinery plays an antiviral role during EV infections, as well. Furthermore, cells which have successfully cleared the virus may be permanently altered to better respond to future viral infections.
Adaptive Immune Response Following Enterovirus Infection
Numerous clinical case reports involving patients suffering from encephalitis due to EV document the importance of neutralizing antibodies in controlling infection (Xie et al., 2010). The significance contribution of a neutralizing antibody response in controlling infection is also shown by studies describing individuals suffering from agammaglobulimia. The absence of neutralizing antibodies in agammaglobulimic patients results in an increased susceptibility to EV infections of the CNS that can lead to chronic neuropathies (Misbah et al., 1992). Experiments evaluating CVB3 infection in mice lacking B cells (BcKO mice) indicate that antibodies are critical for viral clearance (Mena et al., 1999). The role of B cells in controlling CVB3 infection may be more complex by evidence suggesting B cells may contribute to virus dissemination via the “Trojan horse” hypothesis. Early after infection in normal mice, high levels of viral RNA was observed in proliferating lymphocytes located within the marginal zone of the spleen.
Macrophages and microglia may be involved in EV clearance within the CNS. These phagocytic cells are thought to be early responders to viral infection. The presence of activated microglia and macrophages in response to CVB3 infection has been observed in our neonatal mouse model (Feuer et al., 2009). By confocal microscopy, Iba1+ macrophages/microglia within the CNS were shown actively engulfing virally-infected cells.
T cells also play a critical role in the adaptive immune response to EV infection in the CNS; however, bystander damage following their activation may be significant. Much work in understanding T cell responses to picornavirus infection in the CNS and the ensuing immune-mediated pathology has been described for TMEV during acute and persistent infection. A highly detailed and informative review on TMEV-induced molecular mimicry model of multiple sclerosis has been published (Olson et al., 2005). Infiltration of CD4+ and CD8+ lymphocytes within the CNS has also been shown following EV infection (Lin et al., 2009b). Exacerbation of virally-induced morbidity may occur in mice deficient in CD4+ and CD8+ lymphocytes, indicating that T cells are necessary to combat infection. In contrast, some EVs such as CVB3, have evolved ways to escape detection by CD8+ T cells via the inhibition of antigen presentation by the MHC class I pathway (Kemball et al., 2009). Therefore, activation of the T cell response may vary greatly, depending upon the EV genus (Slifka et al., 2001).
Enterovirus Persistence
Although EV possess multiple mechanisms by which they can evade the host immune response, their success is not absolute, and the struggle between the host and virus can last for very long periods of time. EV persistence appears to be the product of the ongoing attempt of the host to eliminate or suppress virus replication, and the virus’ struggle to remain intact in a hostile cellular environment. Under the selective pressures generated by a successful host immune response, sometimes the best defense for the virus is simply to mutate. As described above, errors in EV replication can lead to the existence of viral quasispecies (Domingo et al., 2008) which may broaden the tropism of EV (Vignuzzi et al., 2006), assist in evasion from the immune response, and lead to persistence. Thus, perhaps it comes as no surprise that EVs may persist in the CNS and avoid clearance by the host.
EV-71, PV, CVB3, and TMEV all have mechanisms for mitigating the host innate immune response. These viruses have been observed to persist long after the initial infection. Evidence of EV-71 persistence in a clinical setting has been observed by the continued detection of virus sequences in the excretions and secretions of patients long after the detection of the initial infection (Han et al., 2010). Whether or not the CNS is a potential site of the EV-71 persistence has not yet to be determined. In the case of TMEV, virus has been found to persist in oligodendrocyte cell cultures, but the main reservoir for persistent TMEV may be macrophages (Roussarie et al., 2007). Upon infection, TMEV is transferred from the neuronal axon to the oligodendrocyte in a myelin-mediated fashion; following the ingestion of infected oligodendrocytes, macrophages become the primary reservoir for TMEV persistence.
PV has previously been demonstrated to persist in primary neural cell cultures (Colbere-Garapin et al., 1998). PV persistence and reactivation has been a suspected cause of Post-Polio Syndrome (PPS) (Baj et al., 2007). Indeed PV RNA could be detected in some patients with PPS, though the persistent virus may be very different from the wild type strain (Baj et al., 2007). The low viral load and the unclear mechanism of reactivation for mutated virus in PPS patients most likely fuel the controversy regarding the potential contribution of persistent PV material to recurring disease. Perhaps PV persists in an attenuated form in peripheral nervous tissues of the host at low replication rates; a reactivation event, such as injury, might trigger higher levels of viral replication with ensuing neurological sequelae. Injury-mediated retrograde axonal transport of PV has been demonstrated to carry PV into the CNS (Gromeier and Wimmer, 1998), and may be a mechanism by which PV can be transported from sites of persistence back to the CNS.
CV persistence in the heart has been the subject of much investigation, and its persistence in the CNS has only recently been described (Feuer et al., 2009). In the heart, CV persistence is associated with chronic myocarditis and dilated cardiomyopathy (Chapman and Kim, 2008). The host innate immune response, in particular the type I IFN response, is extremely critical for controlling CV infection of the heart (Deonarain et al., 2004). The type I IFN response might also create selective pressures resulting in viral genome mutations (Kim et al., 2005b), attenuation of CV, and persistence in the heart. Genome deletions in the 5′ UTR of CVB3 was recently observed upon passage of CVB3 in primary heart cell cultures. These deletions were associated with a reduction in cytopathic effects (cpe) without reductions in viral titers (Kim et al., 2008). Similarly, persistent CVB3 RNA was detected in the CNS of mice following neonatal infection for up to 90 days post-infection (PI), as determined by nested RT-PCR, in the absence of infectious virus by plaque assay (Feuer et al., 2009). Our most recent results suggest that CVB3-infected neurospheres, or NPSCs grown in culture as free-floating structures, may support a persistent or carrier-state infection (manuscript submitted). Based on these results, we hypothesize that neurogenic regions of the CNS may be potential sites of CVB3 in vivo. Perhaps CVB3 reactivation is induced upon occasional proliferation and asymmetric division of resting or quiescent type B neural stem cells harboring persistent virus within the CNS (Figure 3), thereby leading to intermittent reactivation of virus.
The possible persistence and reactivation of EV in a clinical setting is of profound importance, especially with the addition of immunosuppressive drugs, such as Rituximab, given to patients suffering from B cell lymphomas. Immunosuppression, especially with drugs targeting the humoral immune response, may lead to reactivation/increased replication of persistent EV in the CNS with potentially dangerous neurological complications (Schilthuizen et al., 2010; Servais et al., 2010).
Pathophysiology of Enterovirus Infection
EV infection of the CNS have been associated with acute flaccid paralysis (Solomon and Willison, 2003), acute disseminating myelitis and acute transverse myelitis (Agin et al., 2010; Minami et al., 2004). Aseptic meningitis and encephalitis can also result from EV infection (Dalwai et al., 2010; Lewthwaite et al., 2010). Further evidence indicating the extensive distribution and diversity of EVs associated with human disease is demonstrated by a recent informative publication by Victoria et al (Victoria et al., 2009). Their metagenomic analyses suggested the presence of circulating human EV species A (HEV-A) through HEV-C, including other members of the Picornaviridae, such as P-ECHO-Vs, rhinoviruses, and human cardioviruses in South Asian children suffering from acute flaccid paralysis. Metagenomic analysis may be a highly informative technique in determining the distribution and number of circulating EVs in human populations.
Clinical Diagnosis of Enterovirus Infection of the CNS
A variety of differing techniques have been used in the clinical diagnosis of EV infections of the CNS (Table 2); a comprehensive description of which is beyond the scope of this review. The GreeneChip pioneered by Ian Lipkin represents the technological forefront of pathogen identification, but has yet to be employed in the context of EV infections of the CNS (Hunter, 2008). Diagnosis via selective culture using transgenic cell lines is limited and hindered primarily by the absence of cell lines supporting strain-specific replication (Leland and Ginocchio, 2007). In contrast, molecular techniques, such as RT-PCR, represent a sensitive and precise methodology for identification of EV CNS infection. Cerebrospinal fluid (CSF) from afflicted patients is the preferred source of clinical sample upon which these techniques can be performed. Genomic analysis using RT-PCR can be completed within 24 hours of sample collection (Romero, 1999). RT-PCR diagnosis of enteroviral meningitis has been performed using primers recognizing a conserved region within the 5′ UTR (Rotbart, 1990). While genus level identification of EVs is clinically relevant and can be used to guide antiviral regimes, strain-specific genotyping is required in some instances. RT-PCR characterization of the VP1 region has lead to strain-specific phylogenetic classification (Mirand et al., 2008). Sequencing of the VP4 and VP2 regions was performed in instances where molecular typing using the VP1 region was inconclusive. These studies indicate that RT-PCR is an effective and clinically feasible technique for the identification of the specific EV strain present in clinically obtained CSF samples.
Table 2.
Methods used to identify and classify enterovirus CNS infections
| Technique | Method of Identification | Host Isolate | Tools | Specificity |
|---|---|---|---|---|
| Tissue Culture (Leland, 2008) | Presence of Cellular Cytopathic Effect | Cerebral Spinal Fluid | Selective Cell Lines | Genus |
| Reverse-Transcription Polymerase Chain Reaction + Genomic Sequence (Romero, 1999) | Genomic Amplification | Cerebral Spinal Fluid | Enterovirus Specifc Primers – Majority Amplify 5′ UTR (Rotbart, 1990) | Serotype – Nucleotide Mutations (Mirand, 2008) |
| Quantitative Real Time PCR (Dierssen, 2008) | Genomic Amplification | Cerebral Spinal Fluid | Enterovirus Specifc Primers – Majority Amplify 5′ UTR | Genus |
| Immunohistochemistry | Visualization Using Virally Specific Antibodies | Central Nervous System Tissue | Visualization/Microscopy | Cellular Localization |
| In situ Hybridization | Visualization using Virally Specific Probes | Central Nervous System Tissue | Visualization/Microscopy | Cellular Localization |
Quantitative Real-Time PCR (qRT-PCR) may be a more sophisticate molecular method utilized to detect the amount of EV in clinical CSF samples (Dierssen et al., 2008). The majority of qRT-PCR assays identifying EV use the primer pair designed by Robart et al. in 1990, although variations of these primers do exist (Rotbart, 1990). A limitation of the technique involves the absence of amplification curves for samples containing mutations in the probe binding region (Hymas et al., 2008).
Contributions of Apoptosis to Enterovirus Disease in the CNS
We have previously described the induction of CNS lesions (Figure 4), inflammation, and apoptosis within CVB3-infected neurons in the cortex and hippocampus (Feuer et al., 2003). TUNEL staining showed colocalization of infection and apoptosis within the CNS following IC inoculation of our neonatal mouse model with a recombinant CVB3 expressing eGFP (eGFP-CVB3). Also, a direct overlap of infection and activated caspase-3 protein revealed infected cells undergoing early stage apoptosis. Also, infection of virally-infected neuronal precursor cells contributed to the generation of lesions within the CNS. We also recently revealed the induction of apoptosis in the choroid plexus (Tabor-Godwin et al., 2010) following CVB3 infection. These results suggest that EV may compromise the function of the choroid plexus, an essential organ involved in CSF production and immune regulation. The observation of hydrocephalus in mice following CVB3 infection may be related to choroid plexus dysfunction following infection. Reports also indicate that PV infection may be associated with induction of apoptosis in the CNS (Girard et al., 1999). DNA oligonucleosomal laddering and enzyme-linked immunosorbent (ELISA) assays performed upon CNS samples obtained from mice exhibiting paralytic poliomyelitis indicated the presence cells undergoing apoptosis. These studies indicate that EVs are capable of inducing apoptosis within the CNS.
Figure 4. Increased susceptibility of young mice to coxsackievirus infection and subsequent CNS pathology.
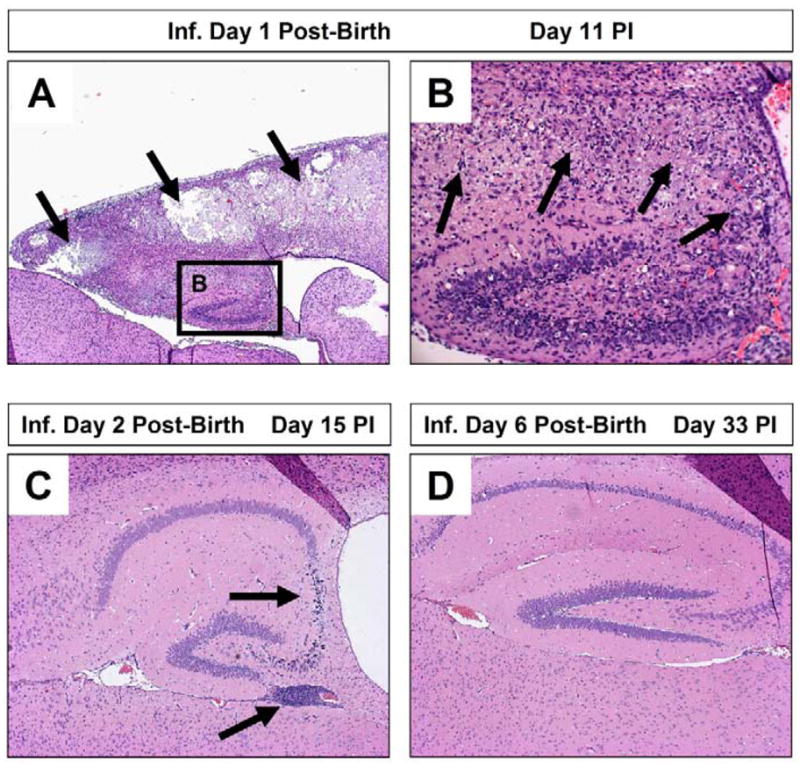
Neonatal SJL mice (day 1, 2, or 6 post-birth) were intra-cranially infected with 100 pfu CVB3. The brains of infected mice were harvested on day 11, 15 or 33 post-infection, respectively. De-paraffinized sections of the brains were stained with hematoxylin and eosin and inspected by microscopy for lesions and inflammatory cells. (A) One day-old mice suffered the greatest level of neuropathology following infection. Lesions were observed within the cortex and hippocampus (black arrows). (B) A higher magnification of (A) revealed the loss of pyramidal neurons in the hippocampus and the presence of inflammatory cells (black arrows). (C) In contrast, two day-old mice infected with an identical amount of CVB3 showed reduce signs of neuronal loss restricted to the CA3 and CA4 field of the hippocampus (black arrow). The presence of immune cell enriched perivascular cuffs were observed near the hippocampus (black arrow). (D) Six day-old mice infected with CVB3 showed little or no signs of CNS disease.
Potential Behavior and Memory Dysfunction Following Enterovirus Infection
The observation of infected neurons and ongoing apoptosis in the hippocampus and other regions of the CNS is highly suggestive that memory dysfunction or behavior changes might be expected following EV infection. Previous studies have suggested a link between EV infection early in childhood and the development of schizophrenia (Rantakallio et al., 1997; Suvisaari et al., 2003). EV infection of the CNS occurring during childhood is also associated with long term neuropathies including delayed neurodevelopment and reduced cognition (Chang et al., 2007). Clinical examination of placental samples from newborns exhibiting neurodevelopmental defects has revealed the presence of EV within the tissue (Euscher et al., 2001). EV-71 has specifically been implicated as the causative agent of developmental defects in the CNS (Huang et al., 2006). Infection of pregnant mice with polyinosinic-polycytidylic acid, a compound that mimics replicating virus, may lead to inhibition of embryonic neuronal stem cell development (De et al., 2010). Therefore, developmental defects observed following EV infection of the CNS might be attributed, at least in part, to the abrogation of normal stem cell replication within the brain.
Experiments using the Morris water maze as an indicator of memory formation found that mice infected with TMEV which also can target the hippocampus exhibited a spatial learning deficit (Buenz et al., 2006). The deficit was correlated with the extent of damage to the hippocampus resulting from TMEV infection. We are currently examining the consequences of CVB3 infection on CNS development and spatial memory dysfunction. We expect that lasting consequences on brain function may result following persistent CVB3 infection. These studies may be highly significant in a clinical setting, especially given that EV infections comprise the great majority of aseptic encephalitis infections in humans.
Antiviral Drugs to Treat Enterovirus Infections - RNAi Based Approaches
The idea that RNAi may act as a potent natural antiviral against RNA viruses in mammalian cells was first shown in 2003 (Gitlin and Andino, 2003). Since then, RNAi has proven to effectively inhibit EV reproduction both in vitro and in vivo in a variety of cell and mouse lines Table 3. Mechanistically, siRNAs inhibit the production of infectious virions via Dicer recognition of the viral dsRNA formed during genomic replication (Aliyari and Ding, 2009). Targeted RNAs are subsequently degraded, effectively hindering the formation of nascent virions.
Table 3.
Compounds used to treat enterovirus infections
| Compound | Target | Stage of Development | BBB Penetration | Resistance | Mechanism |
|---|---|---|---|---|---|
| Ribavirin (Li, 2008) | Viral Genome | Patient Prescription | Conflicting Reports (Hosoya 2001 + Honda 1994) | Yes (Poliovirus Isolates) (Vignuzzi, 2005 + Pfeiffer 2003) | Nucleoside Analog - Genomic Incorporation- Error Catastrophe |
| Pleconaril (Pevear, 1999) | VP1 Protein of Nucleocapsid | Clinical-Phase III-IV (Webster 2005, Desmond 2006) | Yes (Schmidtke 2009) | Yes (CVB3 Nancy Strain) (Schmidtke 2005) | Conformational Change of VP1 Protein - Receptor Attachment - Genomic Uncoating (Chen 2008) |
| RNAi (Multiple Formulations) | Viral Genome – Majority Target Protease 2A + 3C or Polymerase 3D (Yuan, 2005) (Tan 2010) (Tan 2007) | In Vitro/In Vivo (Li, 2008) | Undocumented | Yes (Coxsackie + Poliovirus Isolates) (Merl 2007) (Gitlin 2002) | Viral RNA Degradation via Dicer Recognition (Aliyari 2009) |
The vast majority of siRNAs developed against EV focus upon the degradation of mRNAs coding for the viral RNA-dependent RNA polymerase (RdRp) and proteases. For example, siRNAs directed against protease 2A were the most effective in the inhibition of CVB3 infection in HeLa cells and murine cardiomyocytes (Yuan et al., 2005). Interferon receptor knock-out mice transfected with siRNAs targeting CVB3 protease 2A exhibited increased survival time and attenuated viral replication when challenged with CVB3 (Merl et al., 2005). The 19-mer siRNAs targeting the viral 3Cpro region of coxsackievirus B4 (CVB4) were effective in decreasing viral replication in rhabdomyocarcoma cells (Tan et al., 2010). siRNAs targeted against the 3Dpol of EV-71 has also proven protective against this virus in vivo (Tan et al., 2007). Prophylactic treatment of suckling mice with siRNAs prior to viral inoculation decreased the level of hind limb paralysis and weight loss associated with EV-71 infection. Also, RT-PCR and western blots demonstrated a decrease in viral replication and viral protein expression levels within the intestines of siRNA-treated mice.
EVs are especially prone to the formation of escape mutants due to the absence of an RdRp proofreading mechanism. The low fidelity of the RdRp increases the likelihood of mutants escaping the therapeutic effects of RNAi. The formation of escape mutants resistant to anti-CV siRNAs has been documented (Merl and Wessely, 2007). PV escape variants against siRNAs evolve to incorporate a single nucleotide mutation located in the center of the targeted RNA (Gitlin et al., 2002). One strategy to combat this phenomenon may be the simultaneous administration of multiple siRNAs. The administration of a cocktail consisting of three distinct siRNAs may reduce the number of escape mutant progeny to extremely low levels (Merl and Wessely, 2007). Targeting receptors of viral entry with RNAi may be another strategy of effectively inhibiting EV replication. siRNAs directed against murine CAR (mCAR) decreased CVB3 titers in mice (Werk et al., 2005). RNAi degradation of host genes constitutes a unique approach which may mitigate the formation of potential viral escape mutants by affecting cellular targets, as opposed to viral targets.
The development of therapeutic siRNAs symbolizes a novel and as of yet, undeveloped candidate for the treatment of EV infections of the CNS (Vaishnaw et al., 2010). However, RNAi as antiviral therapy has yet to be employed within the CNS. Once the hurdle of delivering RNAi therapeutic constructs to the CNS has been overcome, siRNAs could potentially be used to treat life-threatening EV infections. EV infections of the CNS rarely occur in the absence of infection of peripheral tissues outside the brain. Therefore, even if a therapeutic agent has not been demonstrated to hinder virus production within the CNS per se; by inhibiting replication in other tissues, an siRNA-based drug might impede virus growth and the eventual progression of disease into the CNS. In summation, RNAi represents an immature yet powerful tool that should not be overlooked in the development of therapeutics designed to treat EV infections of the CNS.
Antiviral Drugs to Treat Enterovirus Infections - Ribavirin
Ribavirin (1-(_-d-ribofuranosyl)-1H-1,2,4-triazole-3-carboxamide) was first synthesized in 1972 by ICN pharmaceuticals. Currently under clinical development as a broad-spectrum antiviral, ribavirin has been shown to inhibit the replication of a variety of EVs. Ribavirin, a nucleoside analog, may act as a mutagen via incorporation into the viral RNA genome (Crotty and Andino, 2002; Crotty et al., 2001; Crotty et al., 2000). The presence of ribavirin may force the afflicted virus into “Error catastrophe” by generating a highly variable noninfectious quasispecies swarm and thereby causing lethal mutagenesis (Vignuzzi et al., 2006). Ribavirin has been found to inhibit both in vitro and in vivo EV-71 replication (Li et al., 2008). Human neuronal and mouse neuronal cell lines treated with ribavirin showed decreased signs of cpe following EV-71 infection. Also, ribavirin-treated mice exhibited decreased mortality, morbidity, and paralysis rates when challenged with EV-71 (Li et al., 2008).
There are conflicting reports on the ability of ribavirin to cross the BBB; a necessary criterion when dealing with the treatment of EV infections of the CNS. Upon treatment of subacute sclerosing panencephalitis (SSPE), a neurological disease caused by persistent measles infection, only direct intracranial (IC) and not IP administration of RBV diminished the effects of this virus (Honda et al., 1994). Combining the drug with the lipophylic carrier molecule, cyclodextrin, may increase the concentration within the CNS following IP injection (Jeulin et al., 2009). Ribavirin has also been combined with IFN-α therapy to treat SSPE (Hosoya et al., 2001). Patients received intravenous (IV) ribavirin, as well as intraventricular IFN-α treatment. Upon treatment with ribavirin (20 mg/kg), high performance liquid chromatography quantification indicated that the compound was present within the cerebral spinal fluid of one patient at a concentration of 7.5 μg/ml, a concentration shown to inhibit the replication of SSPE in both tissue culture and mouse studies (Hosoya et al., 1989; Ishii et al., 1996). Therefore, ribavirin may be capable of crossing the blood brain barrier (BBB) in sufficient quantities and to inhibit viral replication. In our neonatal model of CVB3 infection, IP administration of ribavirin led to brain wet weight recovery (manuscript in preparation) indicating that the compound may cross the BBB in effective quantities to reduce CVB3 replication during persistent infection.
EV resistance may necessitate the development of new antiviral drugs to combat these nefarious pathogens. Resistance against ribavirin by PV has been recently observed (Vignuzzi et al., 2006). PV may combat the effects of ribavirin via a single point mutation in the RdRp, effectively increasing the fidelity and lowering the viral genomic mutation rate (Pfeiffer and Kirkegaard, 2003). Intriguingly, the emergent ribavirin-resistant quasispecies swarm may be less adaptable to a changing environment.
Antiviral Drugs to Treat Enterovirus Infections - Pleconaril
Pleconaril,3-(3,5-dimethyl-4((3-(3-methyl-5-isoxazolyl)propyl]oly)phenyl)-5-(trifluoromethyl)-1,2,4-oxadiazole is currently licensed by Schering Plough and has been developed as an anti-picornaviral drug with demonstrated efficacy against many EVs (Pevear et al., 1999). Pleconaril is able to cross the BBB and remain within the CNS at concentrations that inhibit EV replication (Schmidtke et al., 2009). The mechanism of action for pleconaril against viral pathogens is two-fold; the compound inhibits both viral attachment to the cognate receptor and uncoating of the nucleocapsid during replication (Chen et al., 2008b). Pleconaril inhibits viral attachment by binding to the VP1 protein present in the canyon floor or ‘pocket’ of the nucleocapsid and inducing a conformational change in the protein. This conformational change synergistically inhibits the release of the viral RNA from the nucleocapsid, effectively hindering replication. Tissue culture and animal challenge studies testing the susceptibility of clinical EV isolates to the drug have demonstrated antiviral activity (Pevear et al., 1999). Also, numerous case studies evaluating the efficacy of pleconaril to treat clinically ill patients including immunocompromised individuals have been described (Desmond et al., 2006; Webster, 2005). A previous study has suggested that the Nancy strain of CVB3 may be resistant to pleconaril (Schmidtke et al., 2005). This resistance was found to be due to a point mutation at amino acid 1092 of the CVB3 open reading frame. Resistance was associated with a leucine at this position, whereas isoleucine and valine were associated with susceptibility to pleconaril. In summation, these studies demonstrate that pleconaril may be a valuable compound in the treatment of some EV infections of the CNS.
Intra-venous Immunoglobulins to Treat Enterovirus Infections
The standard therapy for aseptic meningitis caused by EV infection continues to be intra-venous immunoglobulin (IVIg) treatment, although the efficacy has not been proven (Abzug, 2004). IVIg is typically prepared from pools of plasma samples from healthy donors (Cheng et al., 2008). This passive immunization-based therapy might neutralize infectious virus circulating within the host, in addition to other non-specific inflammatory mechanisms (Ooi et al., 2010). Figure 5 illustrates the mechanism of action for each of the antiviral compounds and IVIg, described above. In addition, novel antiviral therapies against EVs continue to be developed (Wu et al., 2010).
Figure 5. Antivirals Against Enteroviruses: Mechanisms of Action.

Pleconaril, ribavirin, intravenous immunoglobulins (IVIg) and RNAi each inhibit the production of infectious virions at different steps. Pleconaril induces a conformational change in the viral VP1 capsid protein that inhibits both genome uncoating and the binding of the virus to its cognate receptor, CAR. Ribavirin, a nucleoside analog, is incorporated into the viral genome. This incorporation results in viral genomic mutations that lead to “error catastrophe”. Virus-specific IVIg may bind to virions extracellularly and inhibit the virus from entering the cell. RNAi molecules target the positive sense viral RNA strand leading to Dicer-mediated degradation of the viral genome. However, escape mutants may form that avoid the effects of RNAi. Positive-sense strand viral RNA is shown in black; negative sense strand viral RNA is shown in green. Antiviral compounds are shown in red. The replication complex is shown in blue.
Vaccines Against Enteroviruses
The historical Salk and Sabin vaccines against PV demonstrate the effectiveness of immunization in protecting the host against EV infections. Many effective vaccines stimulating both the innate and adaptive immune response have been designed to combat the remaining clinically relevant EVs. To combat EV infection, the adaptive immune response employs both T cells and antibodies in the clearance of the virus. Therefore, the most successful vaccines activate both humoral and cell-mediated immunity and induce lasting viral immunity. The administration of novel vaccines to patients comes with potential risks, extensive and costly clinical studies, and a potentially apprehensive public. Unfortunately, acceptance of EV-based vaccines to the public may not materialize unless the potential diseases caused by these pathogens become much more widely appreciated. That said, ongoing EV vaccine studies may help us improve the efficacy and safety of potential vaccine candidates, especially if EV transmission and/or disease manifestations increase in the general population in the future. Also “therapeutic vaccines”, those vaccines given after initial infection, might be more readily accepted in patients suffering from EV-mediated disease. “Therapeutic vaccines” could be envisioned which may enhance or redirect an ongoing immune response in order to reduce viral load during early infection, or after the establishment of viral persistence.
Poliovirus Vaccines
Attenuation of EVs through mutations in the genome has historically led to efficient vaccine production. Perhaps the most well-known attenuated form of PV is that used for the Sabin vaccine, which has decreased neurovirulence in part controlled by two stem loops in the viral IRES (Gromeier et al., 1999). Another mutation that can cause CNS attenuation is located between the 5′ NTR cloverleaf and IRES and reduces the binding of polypyrimidine tract-binding protein (Guest et al., 2004). Other studies have made recombinant viruses that use the IRES from human rhinovirus type 2 (HRV2) to attenuate neurovirulence in the Sabin vaccine strain of PV as well as in herpes simplex virus type 1 (Campbell et al., 2007; Gromeier et al., 1996). Somewhat alarmingly, a vaccine-derived PV and coxsackievirus A17 recombinant has been generated in the laboratory, thus illustrating the possibility of such events occurring naturally (Jegouic et al., 2009).
Mutations causing changes in virulence and/or attenuation can also exist in other regions of the genome. Recent publications have revealed Sabin vaccine strains that in rare instances caused paralytic poliomyelitis, have mutations in the capsid protein VP1 representing a mutational hot-spot (Blomqvist et al., 2010; Rahimi et al., 2007). Furthermore, defective interfering particles generated through mutations in the capsid region of PV in vitro have been used to investigate viral replication (Hagino-Yamagishi and Nomoto, 1989). Newer studies have used cleavage site mutants to learn more about PV replication and encapsidation, which appear to be separate steps in the viral life cycle (Oh et al., 2009).
Both inactivated and attenuated forms of the historical PV vaccines have been used to induce confer immunity to the virus. The original formalin-inactivated form of the vaccine (IPV) was developed by Jonas Salk and licensed in 1955 (SALK et al., 1954). In 1963, an orally administered live attenuated PV vaccine (OPV) was formulated (SABIN, 1957). The usage of these vaccines and optimized versions has severely reduced the incidence of poliomyelitis, although the pathogen has yet to be eradicated worldwide. Preference for the administration of the IPV over the OPV is due to the discovery of circulating vaccine-derived PVs (cVDPV) associated with the OPV. cVDPVs are virulent PVs derived from the OPV that occur in a small minority of vaccine recipients. The reversion of cVDPVs from attenuation to virulence is a direct consequence of the genetic instability of the vaccine. In 2000, cVDPVs were responsible for a polio outbreak in the Dominican Republic and Haiti (Olen Kew 2002). Sequencing determined that the cVDPVs were recombinant viruses. The derivation of neurovirulent polio strains from the OPV has led to the “OPV paradox,” which is based upon the idea that complete eradication of poliomyelitis is contingent upon the elimination of the attenuated form of the vaccine (Dowdle et al., 2003).
Enterovirus-71 Vaccines
There have been a variety of effective vaccines designed against EV-71. A formaldehyde-inactivated whole virus preparation has been shown to protect mice from encephalomyelitis caused by EV-71 infection (Ong et al., 2010). Histopathological examination using EV-71 specific antibodies revealed an absence of the virus within the brain stem of immunized mice. Plaque assays using brain stem and spinal cord homogenates confirmed the absence of infectious virus within these tissues. Using a unique strategy, transgenic mice were developed to secrete the VP1 capsid protein of EV-71 into milk secretions. When the milk from transgenic animals was fed to neonatal mice, these neonates developed serologically specific antibodies against EV-71 and did not exhibit weight loss when challenged with the virus (Chen et al., 2008a). Also, virus-like particles, which resemble EV-71 in capsid structure and protein composition, have been found to elicit a protective humoral antibody response in animals (Chung et al., 2008). In addition, passive immunization protected neonatal mice from EV-71 associated morbidity when confronted with a lethal dose of virus. A DNA vaccine coding for VP1 has also been developed (Wu et al., 2001). But in contrast to the aforementioned vaccines, the VP1-based vaccine was only partially protective, as shown by a 40% survival rate of mice inoculated with a lethal dose of EV-71 following vaccination.
Coxsackievirus Vaccines
As with EV-71, many researchers have developed effective vaccines against CV, although no clinically available vaccine currently exists. Progress has been made using a variety of vaccines strategies utilizing either DNA plasmids expressing viral proteins, inactivated virus, or live attenuated forms of virus. A safe and effective RNA vaccine against CVB3 that produces noninfectious virus has been recently produced (Hunziker et al., 2004). Genomic mutation via PCR mutagenesis of the 3CDpro cleavage site present between CVB3 proteins 2A and 2B resulted in an RNA viral genome incapable of producing infectious virions, yet conferring partial protective immunity. RNA-immunized mice exhibited lower viral titers within the pancreas and prolonged survival when challenged with CVB3. RNA-based vaccines are advantageous as compared to DNA vaccines due to their inability to integrate into host DNA.
Attenuating mutations have also been found in CVB viral capsid proteins, and administering these attenuated viruses was found to be protective against lethal re-challenge in the pancreas and heart; although the CNS was not examined (Dan and Chantler, 2005). Another successful vaccine in mice designed against CVB3 involved the generation of recombinant plasmids expressing capsid proteins. Two independent plasmids encoded the VP1 and VP3 capsid epitopes induced the production of virally specific antibodies following in vivo electroporation (Park et al., 2009). Although vaccinated mice displayed a reduction in virally induced heart injury, the induced antibodies failed to neutralize infectious CVB3 in culture. Attenuated CVB3 strains have also been used as potential vaccines and may represent the forefront of the field (Kim et al., 2005a). Whether any of the aforementioned vaccine candidates are capable of preventing CV pathology within the CNS has yet to be confirmed.
Studies using EVs have also been used as a model system for the development of innovative vaccine strategies. “High fidelity” RNA viruses isolated upon serial passage in the presence of low concentrations of ribavirin might be a general strategy for viral attenuation by decreasing the level of viral quasispecies (Vignuzzi et al., 2008). Another original approach to vaccine design involved encoding a mutant PV that contained hundreds to thousands of under-represented codon pairs in the PV non-structural proteins (Coleman et al., 2008). This codon-altered PV had the same amino-acid sequence as wild-type PV, yet suffered from a decrease in the virus protein translation rate. Thus, the codon-altered PV could illicit a protective immune response despite its attenuation (Wimmer et al., 2009). Both of these approaches greatly decreased the ability of the attenuated “vaccine strain” to revert to wild-type PV. Importantly both strategies potentially could be developed quickly for emerging RNA viruses, or for other viruses whereby vaccine development has been deemed problematic.
Gene Delivery in the CNS Utilizing Enterovirus-Based Vectors
Due to their remarkable ability to infect the CNS, attenuated EVs have been explored for use in gene delivery to cells of the CNS. Novel PV replicons that express green fluorescent protein (GFP) have been shown to deliver GFP to motor neurons without causing pathogenesis (Jackson et al., 2001). Also, recombinant PVs designed by the Gromeier laboratory may be of clinical use for viral gene delivery into the CNS, or as oncolytic viruses to target glioblastoma cells and other tumorigenic cells (Goetz et al., 2010). Attenuated CV may also represent an attractive option; our laboratory and others have shown the feasibility of generating relatively stable recombinant CVs expressing such gene products as GFP, dsRED, and renilla luciferase (Feuer et al., 2005; Kim et al., 2009). Also with their particular stem cell tropism in mind, attenuated recombinant CVB3 may be of value as a viral gene vector specifically for delivery of stem cell modulators, such as transcription factors inducing differentiation, into neural and embryonic stem cells. However, the high number of circulating antibodies in the population against EVs (Sawyer, 2002) may limit their use as vectors in vivo; In contrast, in vitro use may be of great benefit.
Conclusions and Future Perspectives
In summary, this review has attempted to cover many of the recent advances in EV research with regards to CNS infection and pathogenesis. EVs are among the most common and medically important human pathogens, are a frequent cause of CNS disease (Muir and van Loon, 1997), and may preferentially cause harm in the very young. Much remains to be learned regarding factors influencing EV tropism, activation of the immune response following infection, infiltration of immune cells into the CNS to combat infection, mechanism of pathogenesis, and antiviral/vaccine development (Nathanson, 2008). The large number of EV strains circulating in nature hinders the promise of vaccine design against these viruses. The potential cessation of the globally administered PV vaccine, either due to the successful eradication of poliomyelitis, or due to dwindling World Health Organization resources, may lead to emergence of uncharacterized CVs and other related non-polio EVs previously restrained by cross-reacting antibodies generated through yearly immunizations. These uncharacterized, yet circulating EV strains with potential CNS tropism might constitute a new impediment in our control over a devious viral pathogen. Finally, the ability of EVs to persist in the CNS and target neural stem cells might suggest lasting effects on brain function. Future studies might be directed at inspecting possible behavioral alterations and memory dysfunction following recovery from these challenging, yet successful neurotropic viruses.
Acknowledgments
This work was supported by National Institutes of Health (NIH) award R01 NS054108 (to R.F), National Institutes of Mental Health (NIMH) Minority Research Infrastructure Support Program (M-RISP) Faculty Fellow Award R24 MH065515 at San Diego State University (to R.F.), and two Achievement Rewards for College Scientists (ARCS) Foundation Scholarships (to G.T. and J.M.T-G.).
Footnotes
No conflicts of interest exist between the subject matter and the authors included in the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abzug MJ. Presentation, diagnosis, and management of enterovirus infections in neonates. Paediatr Drugs. 2004;6:1–10. doi: 10.2165/00148581-200406010-00001. [DOI] [PubMed] [Google Scholar]
- Agin H, Apa H, Unalp A, Kayserili E. Acute disseminated encephalomyelitis associated with enteroviral infection. Neurosciences (Riyadh ) 2010;15:46–48. [PubMed] [Google Scholar]
- Agol VI, Gmyl AP. Viral security proteins: counteracting host defences. Nat Rev Microbiol. 2010;8:867–878. doi: 10.1038/nrmicro2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn J, Jee Y, Seo I, Yoon SY, Kim D, Kim YK, Lee H. Primary neurons become less susceptible to coxsackievirus B5 following maturation: the correlation with the decreased level of CAR expression on cell surface. J Med Virol. 2008;80(3):434–440. doi: 10.1002/jmv.21100. [DOI] [PubMed] [Google Scholar]
- Alirezaei M, Kemball CC, Flynn CT, Wood MR, Whitton JL, Kiosses WB. Short-term fasting induces profound neuronal autophagy. Autophagy. 2010;6(6):702–710. doi: 10.4161/auto.6.6.12376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aliyari R, Ding SW. RNA-based viral immunity initiated by the Dicer family of host immune receptors. Immunol Rev. 2009;227:176–188. doi: 10.1111/j.1600-065X.2008.00722.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baj A, Maccari G, Toniolo A. Detection of persistent polioviruses in patients with the post-polio syndrome. In: Méndez-Vilas A, editor. Communicating Current Research and Educational Topics and Trends in Applied Microbiology. Formatex; 2007. pp. 859–867. [Google Scholar]
- Barral PM, Morrison JM, Drahos J, Gupta P, Sarkar D, Fisher PB, Racaniello VR. MDA-5 is cleaved in poliovirus-infected cells. J Virol. 2007;81:3677–3684. doi: 10.1128/JVI.01360-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barral PM, Sarkar D, Fisher PB, Racaniello VR. RIG-I is cleaved during picornavirus infection. Virology. 2009;391:171–176. doi: 10.1016/j.virol.2009.06.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergelson JM. Intercellular junctional proteins as receptors and barriers to virus infection and spread. Cell Host Microbe. 2009;5:517–521. doi: 10.1016/j.chom.2009.05.009. [DOI] [PubMed] [Google Scholar]
- Blomqvist S, Savolainen-Kopra C, Paananen A, El BL, El Maamoon Nasr EM, Firstova L, Zamiatina N, Kutateladze T, Roivainen M. Recurrent isolation of poliovirus 3 strains with chimeric capsid protein Vp1 suggests a recombination hot-spot site in Vp1. Virus Res. 2010;151(2):246–251. doi: 10.1016/j.virusres.2010.05.006. [DOI] [PubMed] [Google Scholar]
- Boussadia O, Niepmann M, Creancier L, Prats AC, Dautry F, Jacquemin-Sablon H. Unr is required in vivo for efficient initiation of translation from the internal ribosome entry sites of both rhinovirus and poliovirus. J Virol. 2003;77:3353–3359. doi: 10.1128/JVI.77.6.3353-3359.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brahic M, Bureau JF, Michiels T. The genetics of the persistent infection and demyelinating disease caused by Theiler’s virus. Annu Rev Microbiol. 2005;59:279–98. 279–298. doi: 10.1146/annurev.micro.59.030804.121242. [DOI] [PubMed] [Google Scholar]
- Brandenburg B, Lee LY, Lakadamyali M, Rust MJ, Zhuang X, Hogle JM. Imaging poliovirus entry in live cells. PLoS Biol. 2007;5(7):e183. doi: 10.1371/journal.pbio.0050183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenz EJ, Rodriguez M, Howe CL. Disrupted spatial memory is a consequence of picornavirus infection. Neurobiol Dis. 2006;24:266–273. doi: 10.1016/j.nbd.2006.07.003. [DOI] [PubMed] [Google Scholar]
- Buenz EJ, Sauer BM, Lafrance-Corey RG, Deb C, Denic A, German CL, Howe CL. Apoptosis of hippocampal pyramidal neurons is virus independent in a mouse model of acute neurovirulent picornavirus infection. Am J Pathol. 2009;175:668–684. doi: 10.2353/ajpath.2009.081126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron CE, Suk OH, Moustafa IM. Expanding knowledge of P3 proteins in the poliovirus lifecycle. Future Microbiol. 2010;5:867–881. doi: 10.2217/fmb.10.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell SA, Mulvey M, Mohr I, Gromeier M. Attenuation of herpes simplex virus neurovirulence with picornavirus cis-acting genetic elements. J Virol. 2007;81(2):791–799. doi: 10.1128/JVI.00714-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang LY, Huang LM, Gau SS, Wu YY, Hsia SH, Fan TY, Lin KL, Huang YC, Lu CY, Lin TY. Neurodevelopment and cognition in children after enterovirus 71 infection. N Engl J Med. 2007;356:1226–1234. doi: 10.1056/NEJMoa065954. [DOI] [PubMed] [Google Scholar]
- Chapman NM, Kim KS. Persistent coxsackievirus infection: enterovirus persistence in chronic myocarditis and dilated cardiomyopathy. Curr Top Microbiol Immunol. 2008;323:275–292. doi: 10.1007/978-3-540-75546-3_13. [DOI] [PubMed] [Google Scholar]
- Chen HL, Huang JY, Chu TW, Tsai TC, Hung CM, Lin CC, Liu FC, Wang LC, Chen YJ, Lin MF, Chen CM. Expression of VP1 protein in the milk of transgenic mice: a potential oral vaccine protects against enterovirus 71 infection. Vaccine. 2008a;26:2882–2889. doi: 10.1016/j.vaccine.2008.03.041. [DOI] [PubMed] [Google Scholar]
- Chen TC, Weng KF, Chang SC, Lin JY, Huang PN, Shih SR. Development of antiviral agents for enteroviruses. J Antimicrob Chemother. 2008b;62:1169–1173. doi: 10.1093/jac/dkn424. [DOI] [PubMed] [Google Scholar]
- Cheng MF, Chen BC, Huang TS, Hsieh KS, Chen SN, Liu YC. Clinical application of reverse-transcription polymerase chain reaction and intravenous immunoglobulin for enterovirus encephalitis. Jpn J Infect Dis. 2008;61:18–24. [PubMed] [Google Scholar]
- Chung YC, Ho MS, Wu JC, Chen WJ, Huang JH, Chou ST, Hu YC. Immunization with virus-like particles of enterovirus 71 elicits potent immune responses and protects mice against lethal challenge. Vaccine. 2008;26:1855–1862. doi: 10.1016/j.vaccine.2008.01.058. [DOI] [PubMed] [Google Scholar]
- Colbere-Garapin F, Duncan G, Pavio N, Pelletier I, Petit I. An approach to understanding the mechanisms of poliovirus persistence in infected cells of neural or non-neural origin. Clin Diagn Virol. 1998;9:107–113. doi: 10.1016/s0928-0197(98)00009-9. [DOI] [PubMed] [Google Scholar]
- Coleman JR, Papamichail D, Skiena S, Futcher B, Wimmer E, Mueller S. Virus attenuation by genome-scale changes in codon pair bias. Science. 2008;320(5884):1784–1787. doi: 10.1126/science.1155761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couderc T, Guivel-Benhassine F, Calaora V, Gosselin AS, Blondel B. An ex vivo murine model to study poliovirus-induced apoptosis in nerve cells. J Gen Virol. 2002;83:1925–1930. doi: 10.1099/0022-1317-83-8-1925. [DOI] [PubMed] [Google Scholar]
- Coyne CB, Bergelson JM. Virus-induced Abl and Fyn kinase signals permit coxsackievirus entry through epithelial tight junctions. Cell. 2006;124(1):119–131. doi: 10.1016/j.cell.2005.10.035. [DOI] [PubMed] [Google Scholar]
- Coyne CB, Kim KS, Bergelson JM. Poliovirus entry into human brain microvascular cells requires receptor-induced activation of SHP-2. EMBO J. 2007;26(17):4016–4028. doi: 10.1038/sj.emboj.7601831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cree BC, Bernardini GL, Hays AP, Lowe G. A fatal case of coxsackievirus B4 meningoencephalitis. Arch Neurol. 2003;60:107–112. doi: 10.1001/archneur.60.1.107. [DOI] [PubMed] [Google Scholar]
- Crotty S, Andino R. Implications of high RNA virus mutation rates: lethal mutagenesis and the antiviral drug ribavirin. Microbes Infect. 2002;4:1301–1307. doi: 10.1016/s1286-4579(02)00008-4. [DOI] [PubMed] [Google Scholar]
- Crotty S, Cameron CE, Andino R. RNA virus error catastrophe: direct molecular test by using ribavirin. Proc Natl Acad Sci U S A. 2001;98:6895–6900. doi: 10.1073/pnas.111085598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crotty S, Maag D, Arnold JJ, Zhong W, Lau JY, Hong Z, Andino R, Cameron CE. The broad-spectrum antiviral ribonucleoside ribavirin is an RNA virus mutagen. Nat Med. 2000;6:1375–1379. doi: 10.1038/82191. [DOI] [PubMed] [Google Scholar]
- Dalwai A, Ahmad S, Al-Nakib W. Echoviruses are a major cause of aseptic meningitis in infants and young children in Kuwait. Virol J. 2010;7:236–236. doi: 10.1186/1743-422X-7-236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dan M, Chantler JK. A genetically engineered attenuated coxsackievirus B3 strain protects mice against lethal infection. J Virol. 2005;79(14):9285–9295. doi: 10.1128/JVI.79.14.9285-9295.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De MJ, Yaddanapudi K, Hornig M, Villar G, Serge R, Lipkin WI. Induction of Toll-Like Receptor 3-Mediated Immunity during Gestation Inhibits Cortical Neurogenesis and Causes Behavioral Disturbances. MBio. 2010;1:e00176–10. doi: 10.1128/mBio.00176-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deonarain R, Cerullo D, Fuse K, Liu PP, Fish EN. Protective role for interferon-beta in coxsackievirus B3 infection. Circulation. 2004;110:3540–3543. doi: 10.1161/01.CIR.0000136824.73458.20. [DOI] [PubMed] [Google Scholar]
- Desmond RA, Accortt NA, Talley L, Villano SA, Soong SJ, Whitley RJ. Enteroviral meningitis: natural history and outcome of pleconaril therapy. Antimicrob Agents Chemother. 2006;50:2409–2414. doi: 10.1128/AAC.00227-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dierssen U, Rehren F, Henke-Gendo C, Harste G, Heim A. Rapid routine detection of enterovirus RNA in cerebrospinal fluid by a one-step real-time RT-PCR assay. J Clin Virol. 2008;42:58–64. doi: 10.1016/j.jcv.2007.11.016. [DOI] [PubMed] [Google Scholar]
- Doedens JR, Kirkegaard K. Inhibition of cellular protein secretion by poliovirus proteins 2B and 3A. EMBO J. 1995;14(5):894–907. doi: 10.1002/j.1460-2075.1995.tb07071.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domingo E, Martin V, Perales C, Escarmis C. Coxsackieviruses and quasispecies theory: evolution of enteroviruses. Curr Top Microbiol Immunol. 2008;323:3–32. doi: 10.1007/978-3-540-75546-3_1. [DOI] [PubMed] [Google Scholar]
- Dowdle WR, De GE, Kew OM, Pallansch MA, Wood DJ. Polio eradication: the OPV paradox. Rev Med Virol. 2003;13:277–291. doi: 10.1002/rmv.401. [DOI] [PubMed] [Google Scholar]
- Eberle KE, Nguyen VT, Freistadt MS. Low levels of poliovirus replication in primary human monocytes: possible interactions with lymphocytes. Arch Virol. 1995;140(12):2135–2150. doi: 10.1007/BF01323236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etchison D, Milburn SC, Edery I, Sonenberg N, Hershey JW. Inhibition of HeLa cell protein synthesis following poliovirus infection correlates with the proteolysis of a 220,000-dalton polypeptide associated with eucaryotic initiation factor 3 and a cap binding protein complex. J Biol Chem. 1982;257(24):14806–14810. [PubMed] [Google Scholar]
- Euscher E, Davis J, Holzman I, Nuovo GJ. Coxsackie virus infection of the placenta associated with neurodevelopmental delays in the newborn. Obstet Gynecol. 2001;98:1019–1026. doi: 10.1016/s0029-7844(01)01625-8. [DOI] [PubMed] [Google Scholar]
- Feuer R, Mena I, Pagarigan R, Slifka MK, Whitton JL. Cell cycle status affects coxsackievirus replication, persistence, and reactivation in vitro. J Virol. 2002;76:4430–4440. doi: 10.1128/JVI.76.9.4430-4440.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feuer R, Mena I, Pagarigan RR, Harkins S, Hassett DE, Whitton JL. Coxsackievirus B3 and the neonatal CNS: the roles of stem cells, developing neurons, and apoptosis in infection, viral dissemination, and disease. Am J Pathol. 2003;163:1379–1393. doi: 10.1016/S0002-9440(10)63496-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feuer R, Pagarigan RR, Harkins S, Liu F, Hunziker IP, Whitton JL. Coxsackievirus targets proliferating neuronal progenitor cells in the neonatal CNS. J Neurosci. 2005;25:2434–2444. doi: 10.1523/JNEUROSCI.4517-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feuer R, Ruller CM, An N, Tabor-Godwin JM, Rhoades RE, Maciejewski S, Pagarigan RR, Cornell CT, Crocker SJ, Kiosses WB, Pham-Mitchell N, Campbell IL, Whitton JL. Viral persistence and chronic immunopathology in the adult central nervous system following Coxsackievirus infection during the neonatal period. J Virol. 2009;83:9356–9369. doi: 10.1128/JVI.02382-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feuer R, Whitton JL. Preferential coxsackievirus replication in proliferating/activated cells: implications for virus tropism, persistence, and pathogenesis. Curr Top Microbiol Immunol. 2008;323:149–73. 149–173. doi: 10.1007/978-3-540-75546-3_7. [DOI] [PubMed] [Google Scholar]
- Fitzgerald KD, Semler BL. Bridging IRES elements in mRNAs to the eukaryotic translation apparatus. Biochim Biophys Acta. 2009;1789:518–528. doi: 10.1016/j.bbagrm.2009.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freistadt MS, Eberle KE. Correlation between poliovirus type 1 Mahoney replication in blood cells and neurovirulence. J Virol. 1996;70(9):6486–6492. doi: 10.1128/jvi.70.9.6486-6492.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freistadt MS, Fleit HB, Wimmer E. Poliovirus receptor on human blood cells: a possible extraneural site of poliovirus replication. Virology. 1993;195(2):798–803. doi: 10.1006/viro.1993.1433. [DOI] [PubMed] [Google Scholar]
- Freistadt MS, Vaccaro JA, Eberle KE. Biochemical characterization of the fidelity of poliovirus RNA-dependent RNA polymerase. Virol J. 2007;4:44. doi: 10.1186/1743-422X-4-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita K, Krishnakumar SS, Franco D, Paul AV, London E, Wimmer E. Membrane topography of the hydrophobic anchor sequence of poliovirus 3A and 3AB proteins and the functional effect of 3A/3AB membrane association upon RNA replication. Biochemistry. 2007;46(17):5185–5199. doi: 10.1021/bi6024758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamarnik AV, Andino R. Switch from translation to RNA replication in a positive-stranded RNA virus. Genes Dev. 1998;12(15):2293–2304. doi: 10.1101/gad.12.15.2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girard S, Couderc T, Destombes J, Thiesson D, Delpeyroux F, Blondel B. Poliovirus induces apoptosis in the mouse central nervous system. J Virol. 1999;73:6066–6072. doi: 10.1128/jvi.73.7.6066-6072.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitlin L, Andino R. Nucleic acid-based immune system: the antiviral potential of mammalian RNA silencing. J Virol. 2003;77:7159–7165. doi: 10.1128/JVI.77.13.7159-7165.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitlin L, Karelsky S, Andino R. Short interfering RNA confers intracellular antiviral immunity in human cells. Nature. 2002;418:430–434. doi: 10.1038/nature00873. [DOI] [PubMed] [Google Scholar]
- Goetz C, Everson RG, Zhang LC, Gromeier M. MAPK signal-integrating kinase controls cap-independent translation and cell type-specific cytotoxicity of an oncolytic poliovirus. Mol Ther. 2010;18:1937–1946. doi: 10.1038/mt.2010.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodfellow I, Chaudhry Y, Richardson A, Meredith J, Almond JW, Barclay W, Evans DJ. Identification of a cis-acting replication element within the poliovirus coding region. J Virol. 2000;74(10):4590–4600. doi: 10.1128/jvi.74.10.4590-4600.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbea C, Makar KA, Pauschinger M, Pratt G, Bersola JL, Varela J, David RM, Banks L, Huang CH, Li H, Schultheiss HP, Towbin JA, Vallejo JG, Bowles NE. A role for Toll-like receptor 3 variants in host susceptibility to enteroviral myocarditis and dilated cardiomyopathy. J Biol Chem. 2010;285:23208–23223. doi: 10.1074/jbc.M109.047464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gromeier M, Alexander L, Wimmer E. Internal ribosomal entry site substitution eliminates neurovirulence in intergeneric poliovirus recombinants. Proc Natl Acad Sci U S A. 1996;19;93(6):2370–2375. doi: 10.1073/pnas.93.6.2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gromeier M, Bossert B, Arita M, Nomoto A, Wimmer E. Dual stem loops within the poliovirus internal ribosomal entry site control neurovirulence. J Virol. 1999;73(2):958–964. doi: 10.1128/jvi.73.2.958-964.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gromeier M, Lu HH, Wimmer E. Mouse neuropathogenic poliovirus strains cause damage in the central nervous system distinct from poliomyelitis. Microb Pathog. 1995;18(4):253–267. doi: 10.1016/S0882-4010(05)80002-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gromeier M, Solecki D, Patel DD, Wimmer E. Expression of the human poliovirus receptor/CD155 gene during development of the central nervous system: implications for the pathogenesis of poliomyelitis. Virology. 2000;273(2):248–257. doi: 10.1006/viro.2000.0418. [DOI] [PubMed] [Google Scholar]
- Gromeier M, Wimmer E. Mechanism of injury-provoked poliomyelitis. J Virol. 1998;72:5056–5060. doi: 10.1128/jvi.72.6.5056-5060.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guest S, Pilipenko E, Sharma K, Chumakov K, Roos RP. Molecular mechanisms of attenuation of the Sabin strain of poliovirus type 3. J Virol. 2004;78(20):11097–11107. doi: 10.1128/JVI.78.20.11097-11107.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, Fernandez D, Siddiqui A, Tong WC, Pohl K, Jungbluth H. Extensive white matter abnormalities associated with neonatal Parechovirus (HPeV) infection. Eur J Paediatr Neurol. 2010;14:531–534. doi: 10.1016/j.ejpn.2009.12.007. [DOI] [PubMed] [Google Scholar]
- Haddad A, Nokhbeh MR, Alexander DA, Dawe SJ, Grise C, Gulzar N, Dimock K. Binding to decay-accelerating factor is not required for infection of human leukocyte cell lines by enterovirus 70. J Virol. 2004;78(6):2674–2681. doi: 10.1128/JVI.78.6.2674-2681.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagino-Yamagishi K, Nomoto A. In vitro construction of poliovirus defective interfering particles. J Virol. 1989;63(12):5386–5392. doi: 10.1128/jvi.63.12.5386-5392.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, Ma XJ, Wan JF, Liu YH, Han YL, Chen C, Tian C, Gao C, Wang M, Dong XP. Long persistence of EV71 specific nucleotides in respiratory and feces samples of the patients with Hand-Foot-Mouth Disease after recovery. BMC Infect Dis. 2010;10:178. doi: 10.1186/1471-2334-10-178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvala H, Simmonds P. Human parechoviruses: biology, epidemiology and clinical significance. J Clin Virol. 2009;45:1–9. doi: 10.1016/j.jcv.2009.03.009. [DOI] [PubMed] [Google Scholar]
- Herold J, Andino R. Poliovirus RNA replication requires genome circularization through a protein-protein bridge. Mol Cell. 2001;7(3):581–591. doi: 10.1016/S1097-2765(01)00205-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda T, Saitoh H, Masuko M, Katagiri-Abe T, Tominaga K, Kozakai I, Kobayashi K, Kumanishi T, Watanabe YG, Odani S, Kuwano R. The coxsackievirus-adenovirus receptor protein as a cell adhesion molecule in the developing mouse brain. Brain Res Mol Brain Res. 2000;77:19–28. doi: 10.1016/s0169-328x(00)00036-x. [DOI] [PubMed] [Google Scholar]
- Honda Y, Hosoya M, Ishii T, Shigeta S, Suzuki H. Effect of ribavirin on subacute sclerosing panencephalitis virus infections in hamsters. Antimicrob Agents Chemother. 1994;38:653–655. doi: 10.1128/aac.38.4.653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosking MP, Lane TE. The role of chemokines during viral infection of the CNS. PLoS Pathog. 2010;6:e1000937. doi: 10.1371/journal.ppat.1000937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosoya M, Shigeta S, Mori S, Tomoda A, Shiraishi S, Miike T, Suzuki H. High-dose intravenous ribavirin therapy for subacute sclerosing panencephalitis. Antimicrob Agents Chemother. 2001;45:943–945. doi: 10.1128/AAC.45.3.943-945.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosoya M, Shigeta S, Nakamura K, De CE. Inhibitory effect of selected antiviral compounds on measles (SSPE) virus replication in vitro. Antiviral Res. 1989;12:87–97. doi: 10.1016/0166-3542(89)90072-7. [DOI] [PubMed] [Google Scholar]
- Hotta Y, Honda T, Naito M, Kuwano R. Developmental distribution of coxsackie virus and adenovirus receptor localized in the nervous system. Brain Res Dev Brain Res. 2003;143:1–13. doi: 10.1016/s0165-3806(03)00035-x. [DOI] [PubMed] [Google Scholar]
- Huang MC, Wang SM, Hsu YW, Lin HC, Chi CY, Liu CC. Long-term cognitive and motor deficits after enterovirus 71 brainstem encephalitis in children. Pediatrics. 2006;118:e1785–e1788. doi: 10.1542/peds.2006-1547. [DOI] [PubMed] [Google Scholar]
- Huang SC, Chang CL, Wang PS, Tsai Y, Liu HS. Enterovirus 71-induced autophagy detected in vitro and in vivo promotes viral replication. J Med Virol. 2009;81(7):1241–1252. doi: 10.1002/jmv.21502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes SA, Thaker HM, Racaniello VR. Transgenic mouse model for echovirus myocarditis and paralysis. Proc Natl Acad Sci U S A. 2003;100:15906–15911. doi: 10.1073/pnas.2535934100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter P. Viral vigilance. New surveillance strategies and methods help to identify dangerous pathogens earlier: a prerequisite for efficient countermeasures. EMBO Rep. 2008;9:948–950. doi: 10.1038/embor.2008.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunziker IP, Harkins S, Feuer R, Cornell CT, Whitton JL. Generation and analysis of an RNA vaccine that protects against coxsackievirus B3 challenge. Virology. 2004;330:196–208. doi: 10.1016/j.virol.2004.09.035. [DOI] [PubMed] [Google Scholar]
- Hymas WC, Aldous WK, Taggart EW, Stevenson JB, Hillyard DR. Description and validation of a novel real-time RT-PCR enterovirus assay. Clin Chem. 2008;54:406–413. doi: 10.1373/clinchem.2007.095414. [DOI] [PubMed] [Google Scholar]
- Ida-Hosonuma M, Iwasaki T, Yoshikawa T, Nagata N, Sato Y, Sata T, Yoneyama M, Fujita T, Taya C, Yonekawa H, Koike S. The alpha/beta interferon response controls tissue tropism and pathogenicity of poliovirus. J Virol. 2005;79:4460–4469. doi: 10.1128/JVI.79.7.4460-4469.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ireland DD, Stohlman SA, Hinton DR, Kapil P, Silverman RH, Atkinson RA, Bergmann CC. RNase L mediated protection from virus induced demyelination. PLoS Pathog. 2009;5:e1000602. doi: 10.1371/journal.ppat.1000602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii T, Hosoya M, Mori S, Shigeta S, Suzuki H. Effective ribavirin concentration in hamster brains for antiviral chemotherapy for subacute sclerosing panencephalitis. Antimicrob Agents Chemother. 1996;40:241–243. doi: 10.1128/aac.40.1.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson CA, Cobbs C, Peduzzi JD, Novak M, Morrow CD. Repetitive intrathecal injections of poliovirus replicons result in gene expression in neurons of the central nervous system without pathogenesis. Hum Gene Ther. 2001;12(15):1827–1841. doi: 10.1089/104303401753153893. [DOI] [PubMed] [Google Scholar]
- Jackson WT, Giddings TH, Jr, Taylor MP, Mulinyawe S, Rabinovitch M, Kopito RR, Kirkegaard K. Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol. 2005;3(5):e156. doi: 10.1371/journal.pbio.0030156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jegouic S, Joffret ML, Blanchard C, Riquet FB, Perret C, Pelletier I, Colbere-Garapin F, Rakoto-Andrianarivelo M, Delpeyroux F. Recombination between polioviruses and co-circulating Coxsackie A viruses: role in the emergence of pathogenic vaccine-derived polioviruses. PLoS Pathog. 2009;5(5):e1000412. doi: 10.1371/journal.ppat.1000412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeulin H, Venard V, Carapito D, Finance C, Kedzierewicz F. Effective ribavirin concentration in mice brain using cyclodextrin as a drug carrier: evaluation in a measles encephalitis model. Antiviral Res. 2009;81:261–266. doi: 10.1016/j.antiviral.2008.12.006. [DOI] [PubMed] [Google Scholar]
- Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ, Yamaguchi O, Otsu K, Tsujimura T, Koh CS, Reis e Sousa, Matsuura Y, Fujita T, Akira S. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006;441:101–105. doi: 10.1038/nature04734. [DOI] [PubMed] [Google Scholar]
- Kauder S, Kan S, Racaniello VR. Age-dependent poliovirus replication in the mouse central nervous system is determined by internal ribosome entry site-mediated translation. J Virol. 2006;80:2589–2595. doi: 10.1128/JVI.80.6.2589-2595.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauder SE, Racaniello VR. Poliovirus tropism and attenuation are determined after internal ribosome entry. J Clin Invest. 2004;113:1743–1753. doi: 10.1172/JCI21323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemball CC, Harkins S, Whitmire JK, Flynn CT, Feuer R, Whitton JL. Coxsackievirus B3 inhibits antigen presentation in vivo, exerting a profound and selective effect on the MHC class I pathway. PLoS Pathog. 2009;5:e1000618. doi: 10.1371/journal.ppat.1000618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kempf BJ, Barton DJ. Poliovirus 2A(Pro) increases viral mRNA and polysome stability coordinately in time with cleavage of eIF4G. J Virol. 2008;82(12):5847–5859. doi: 10.1128/JVI.01514-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JY, Jeon ES, Lim BK, Kim SM, Chung SK, Kim JM, Park SI, Jo I, Nam JH. Immunogenicity of a DNA vaccine for coxsackievirus B3 in mice: protective effects of capsid proteins against viral challenge. Vaccine. 2005a;23:1672–1679. doi: 10.1016/j.vaccine.2004.10.008. [DOI] [PubMed] [Google Scholar]
- Kim KS, Chapman NM, Tracy S. Replication of coxsackievirus B3 in primary cell cultures generates novel viral genome deletions. J Virol. 2008;82:2033–2037. doi: 10.1128/JVI.01774-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KS, Tracy S, Tapprich W, Bailey J, Lee CK, Kim K, Barry WH, Chapman NM. 5′–Terminal deletions occur in coxsackievirus B3 during replication in murine hearts and cardiac myocyte cultures and correlate with encapsidation of negative-strand viral RNA. J Virol. 2005b;79:7024–7041. doi: 10.1128/JVI.79.11.7024-7041.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YJ, Yun SH, Lim BK, Park KB, Na HN, Jeong SY, Kim DS, Cho YJ, Jeon ES, Nam JH. Systemic analysis of a novel coxsackievirus gene delivery system in a mouse model. J Microbiol Biotechnol. 2009;19(3):307–313. [PubMed] [Google Scholar]
- Kitamura N, Semler BL, Rothberg PG, Larsen GR, Adler CJ, Dorner AJ, Emini EA, Hanecak R, Lee JJ, van der Werf S, Anderson CW, Wimmer E. Primary structure, gene organization and polypeptide expression of poliovirus RNA. Nature. 1981;291(5816):547–553. doi: 10.1038/291547a0. [DOI] [PubMed] [Google Scholar]
- Klingel K, Hohenadl C, Canu A, Albrecht M, Seemann M, Mall G, Kandolf R. Ongoing enterovirus-induced myocarditis is associated with persistent heart muscle infection: quantitative analysis of virus replication, tissue damage, and inflammation. Proc Natl Acad Sci U S A. 1992;89:314–318. doi: 10.1073/pnas.89.1.314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotla S, Peng T, Bumgarner RE, Gustin KE. Attenuation of the type I interferon response in cells infected with human rhinovirus. Virology. 2008;374:399–410. doi: 10.1016/j.virol.2008.01.022. [DOI] [PubMed] [Google Scholar]
- Kung CM, King CC, Lee CN, Huang LM, Lee PI, Kao CL, Kung CM, King CC, Lee CN, Huang LM, Lee PI, Kao CL. Differences in replication capacity between enterovirus 71 isolates obtained from patients with encephalitis and those obtained from patients with herpangina in Taiwan Differences in replication capacity between enterovirus 71 isolates obtained from patients with encephalitis and those obtained from patients with herpangina in Taiwan. J Med Virol. 2007;79:60–68. doi: 10.1002/jmv.20761. [DOI] [PubMed] [Google Scholar]
- Kuss SK, Etheredge CA, Pfeiffer JK. Multiple host barriers restrict poliovirus trafficking in mice. PLoS Pathog. 2008;4(6):e1000082. doi: 10.1371/journal.ppat.1000082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster KZ, Pfeiffer JK. Limited trafficking of a neurotropic virus through inefficient retrograde axonal transport and the type I interferon response. PLoS Pathog. 2010;6(3):e1000791. doi: 10.1371/journal.ppat.1000791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TC, Guo HR, Su HJ, Yang YC, Chang HL, Chen KT. Diseases caused by enterovirus 71 infection. Pediatr Infect Dis J. 2009;28:904–910. doi: 10.1097/INF.0b013e3181a41d63. [DOI] [PubMed] [Google Scholar]
- Lei X, Liu X, Ma Y, Sun Z, Yang Y, Jin Q, He B, Wang J. The 3C protein of enterovirus 71 inhibits retinoid acid-inducible gene I-mediated interferon regulatory factor 3 activation and type I interferon responses. J Virol. 2010;84:8051–8061. doi: 10.1128/JVI.02491-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leland DS, Ginocchio CC. Role of cell culture for virus detection in the age of technology. Clin Microbiol Rev. 2007;20:49–78. doi: 10.1128/CMR.00002-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewthwaite P, Perera D, How OM, Last A, Kumar R, Desai A, Begum A, Ravi V, Shankar MV, Hooi TP, Cardosa MJ, Solomon T. Enterovirus 75 encephalitis in children, southern India. Emerg Infect Dis. 2010;16:1780–1782. doi: 10.3201/eid1611.100672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li ZH, Li CM, Ling P, Shen FH, Chen SH, Liu CC, Yu CK, Chen SH. Ribavirin reduces mortality in enterovirus 71-infected mice by decreasing viral replication. J Infect Dis. 2008;197:854–857. doi: 10.1086/527326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JY, Li ML, Shih SR. Far upstream element binding protein 2 interacts with enterovirus 71 internal ribosomal entry site and negatively regulates viral translation. Nucleic Acids Res. 2009a;37:47–59. doi: 10.1093/nar/gkn901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin TY, Hsia SH, Huang YC, Wu CT, Chang LY. Proinflammatory cytokine reactions in enterovirus 71 infections of the central nervous system. Clin Infect Dis. 2003;36:269–274. doi: 10.1086/345905. [DOI] [PubMed] [Google Scholar]
- Lin YW, Chang KC, Kao CM, Chang SP, Tung YY, Chen SH. Lymphocyte and antibody responses reduce enterovirus 71 lethality in mice by decreasing tissue viral loads. J Virol. 2009b;83:6477–6483. doi: 10.1128/JVI.00434-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton HL, Kumar AS, Trottier M. Theiler’s virus persistence in the central nervous system of mice is associated with continuous viral replication and a difference in outcome of infection of infiltrating macrophages versus oligodendrocytes. Virus Res. 2005;111:214–223. doi: 10.1016/j.virusres.2005.04.010. [DOI] [PubMed] [Google Scholar]
- Liu SC, Lee PI, Lee CY, Wang JD, Chiang BL, Chou MC. Different Cytokine Levels in Enterovirus Meningitis and Encephalitis. Infectious Diseases in Clinical Practice. 2005;13 [Google Scholar]
- Madan V, Redondo N, Carrasco L. Cell permeabilization by poliovirus 2B viroporin triggers bystander permeabilization in neighbouring cells through a mechanism involving gap junctions. Cell Microbiol. 2010;12(8):1144–1157. doi: 10.1111/j.1462-5822.2010.01460.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malathi K, Dong B, Gale M, Jr, Silverman RH. Small self-RNA generated by RNase L amplifies antiviral innate immunity. Nature. 2007;448:816–819. doi: 10.1038/nature06042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcotte LL, Wass AB, Gohara DW, Pathak HB, Arnold JJ, Filman DJ, Cameron CE, Hogle JM. Crystal structure of poliovirus 3CD protein: virally encoded protease and precursor to the RNA-dependent RNA polymerase. J Virol. 2007;81(7):3583–3596. doi: 10.1128/JVI.02306-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto M, Seya T. TLR3: interferon induction by double-stranded RNA including poly(I:C) Adv Drug Deliv Rev. 2008;60:805–812. doi: 10.1016/j.addr.2007.11.005. [DOI] [PubMed] [Google Scholar]
- Mena I, Perry CM, Harkins S, Rodriguez F, Gebhard J, Whitton JL. The role of B lymphocytes in coxsackievirus B3 infection. Am J Pathol. 1999;155:1205–1215. doi: 10.1016/S0002-9440(10)65223-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merl S, Michaelis C, Jaschke B, Vorpahl M, Seidl S, Wessely R. Targeting 2A protease by RNA interference attenuates coxsackieviral cytopathogenicity and promotes survival in highly susceptible mice. Circulation. 2005;111:1583–1592. doi: 10.1161/01.CIR.0000160360.02040.AB. [DOI] [PubMed] [Google Scholar]
- Merl S, Wessely R. Anti-coxsackieviral efficacy of RNA interference is highly dependent on genomic target selection and emergence of escape mutants. Oligonucleotides. 2007;17:44–53. doi: 10.1089/oli.2007.0057. [DOI] [PubMed] [Google Scholar]
- Michos AG, Syriopoulou VP, Hadjichristodoulou C, Daikos GL, Lagona E, Douridas P, Mostrou G, Theodoridou M. Aseptic meningitis in children: analysis of 506 cases. PLoS One. 2007;2:e674. doi: 10.1371/journal.pone.0000674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minami K, Tsuda Y, Maeda H, Yanagawa T, Izumi G, Yoshikawa N. Acute transverse myelitis caused by Coxsackie virus B5 infection. J Paediatr Child Health. 2004;40:66–68. doi: 10.1111/j.1440-1754.2004.00295.x. [DOI] [PubMed] [Google Scholar]
- Mirand A, Bailly JL, Henquell C, Peigue-Lafeuille H. Rapid enterovirus genotyping in cerebrospinal fluids: a two-year prospective study in a virology laboratory setting. Pathol Biol (Paris) 2008;56:471–481. doi: 10.1016/j.patbio.2008.08.005. [DOI] [PubMed] [Google Scholar]
- Misbah SA, Spickett GP, Ryba PC, Hockaday JM, Kroll JS, Sherwood C, Kurtz JB, Moxon ER, Chapel HM. Chronic enteroviral meningoencephalitis in agammaglobulinemia: case report and literature review. J Clin Immunol. 1992;12:266–270. doi: 10.1007/BF00918150. [DOI] [PubMed] [Google Scholar]
- MMWR. Outbreaks following wild poliovirus importations --- Europe, Africa, and Asia, January 2009-September 2010. MMWR Morb Mortal Wkly Rep. 2010;59:1393–1399. [PubMed] [Google Scholar]
- Muir P, van Loon AM. Enterovirus infections of the central nervous system. Intervirology. 1997;40:153–166. doi: 10.1159/000150542. [DOI] [PubMed] [Google Scholar]
- Nagata N, Iwasaki T, Ami Y, Tano Y, Harashima A, Suzaki Y, Sato Y, Hasegawa H, Sata T, Miyamura T, Shimizu H. Differential localization of neurons susceptible to enterovirus 71 and poliovirus type 1 in the central nervous system of cynomolgus monkeys after intravenous inoculation. J Gen Virol. 2004;85:2981–2989. doi: 10.1099/vir.0.79883-0. [DOI] [PubMed] [Google Scholar]
- Nathanson N. The pathogenesis of poliomyelitis: what we don’t know. Adv Virus Res. 2008;71:1–50. doi: 10.1016/S0065-3527(08)00001-8. [DOI] [PubMed] [Google Scholar]
- Nishimura Y, Shimojima M, Tano Y, Miyamura T, Wakita T, Shimizu H. Human P-selectin glycoprotein ligand-1 is a functional receptor for enterovirus 71. Nat Med. 2009;15(7):794–797. doi: 10.1038/nm.1961. [DOI] [PubMed] [Google Scholar]
- Noda S, Aguirre SA, Bitmansour A, Brown JM, Sparer TE, Huang J, Mocarski ES. Cytomegalovirus MCK-2 controls mobilization and recruitment of myeloid progenitor cells to facilitate dissemination. Blood. 2006;107:30–38. doi: 10.1182/blood-2005-05-1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novak JE, Kirkegaard K. Improved method for detecting poliovirus negative strands used to demonstrate specificity of positive-strand encapsidation and the ratio of positive to negative strands in infected cells. J Virol. 1991;65(6):3384–3387. doi: 10.1128/jvi.65.6.3384-3387.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberste MS, Maher K, Pallansch MA. Molecular phylogeny and proposed classification of the simian picornaviruses. J Virol. 2002;76:1244–1251. doi: 10.1128/JVI.76.3.1244-1251.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh HS, Pathak HB, Goodfellow IG, Arnold JJ, Cameron CE. Insight into poliovirus genome replication and encapsidation obtained from studies of 3B–3C cleavage site mutants. J Virol. 2009;83(18):9370–9387. doi: 10.1128/JVI.02076-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohka S, Matsuda N, Tohyama K, Oda T, Morikawa M, Kuge S, Nomoto A. Receptor (CD155)-dependent endocytosis of poliovirus and retrograde axonal transport of the endosome. J Virol. 2004;78(13):7186–7198. doi: 10.1128/JVI.78.13.7186-7198.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohka S, Sakai M, Bohnert S, Igarashi H, Deinhardt K, Schiavo G, Nomoto A. Receptor-dependent and -independent axonal retrograde transport of poliovirus in motor neurons. J Virol. 2009;83(10):4995–5004. doi: 10.1128/JVI.02225-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oleszak EL, Chang JR, Friedman H, Katsetos CD, Platsoucas CD. Theiler’s virus infection: a model for multiple sclerosis. Clin Microbiol Rev. 2004;17:174–207. doi: 10.1128/CMR.17.1.174-207.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson JK, Ercolini AM, Miller SD. A virus-induced molecular mimicry model of multiple sclerosis. Curr Top Microbiol Immunol. 2005;296:39–53. doi: 10.1007/3-540-30791-5_3. [DOI] [PubMed] [Google Scholar]
- Ong KC, Devi S, Cardosa MJ, Wong KT. Formaldehyde-inactivated whole-virus vaccine protects a murine model of enterovirus 71 encephalomyelitis against disease. J Virol. 2010;84:661–665. doi: 10.1128/JVI.00999-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ooi MH, Wong SC, Lewthwaite P, Cardosa MJ, Solomon T. Clinical features, diagnosis, and management of enterovirus 71. Lancet Neurol. 2010;9:1097–1105. doi: 10.1016/S1474-4422(10)70209-X. [DOI] [PubMed] [Google Scholar]
- Ornoy A, Tenenbaum A. Pregnancy outcome following infections by coxsackie, echo, measles, mumps, hepatitis, polio and encephalitis viruses. Reprod Toxicol. 2006;21:446–457. doi: 10.1016/j.reprotox.2005.12.007. [DOI] [PubMed] [Google Scholar]
- Papon L, Oteiza A, Imaizumi T, Kato H, Brocchi E, Lawson TG, Akira S, Mechti N. The viral RNA recognition sensor RIG-I is degraded during encephalomyocarditis virus (EMCV) infection. Virology. 2009;393:311–318. doi: 10.1016/j.virol.2009.08.009. [DOI] [PubMed] [Google Scholar]
- Park JH, Kim DS, Cho YJ, Kim YJ, Jeong SY, Lee SM, Cho SJ, Yun CW, Jo I, Nam JH. Attenuation of coxsackievirus B3 by VP2 mutation and its application as a vaccine against virus-induced myocarditis and pancreatitis. Vaccine. 2009;27:1974–1983. doi: 10.1016/j.vaccine.2009.01.008. [DOI] [PubMed] [Google Scholar]
- Park N, Katikaneni P, Skern T, Gustin KE. Differential targeting of nuclear pore complex proteins in poliovirus-infected cells. J Virol. 2008;82:1647–1655. doi: 10.1128/JVI.01670-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park N, Skern T, Gustin KE. Specific cleavage of the nuclear pore complex protein Nup62 by a viral protease. J Biol Chem. 2010;285:28796–28805. doi: 10.1074/jbc.M110.143404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel KP, Coyne CB, Bergelson JM. Dynamin- and lipid raft-dependent entry of decay-accelerating factor (DAF)-binding and non-DAF-binding coxsackieviruses into nonpolarized cells. J Virol. 2009;83(21):11064–11077. doi: 10.1128/JVI.01016-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul AV, Peters J, Mugavero J, Yin J, van Boom JH, Wimmer E. Biochemical and genetic studies of the VPg uridylylation reaction catalyzed by the RNA polymerase of poliovirus. J Virol. 2003;77(2):891–904. doi: 10.1128/JVI.77.2.891-904.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul AV, van Boom JH, Filippov D, Wimmer E. Protein-primed RNA synthesis by purified poliovirus RNA polymerase. Nature. 1998;393(6682):280–284. doi: 10.1038/30529. [DOI] [PubMed] [Google Scholar]
- Pedersen IM, Cheng G, Wieland S, Volinia S, Croce CM, Chisari FV, David M. Interferon modulation of cellular microRNAs as an antiviral mechanism. Nature. 2007;449:919–922. doi: 10.1038/nature06205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelletier I, Saulnier A, Brisac C, Jegouic S, Vabret N, Tangy F, Blondel B, Colbere-Garapin F. Enhanced gene silencing in cells cured of persistent virus infection by RNA interference. J Virol. 2010;84:6880–6885. doi: 10.1128/JVI.02060-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelletier J, Sonenberg N. Internal initiation of translation of eukaryotic mRNA directed by a sequence derived from poliovirus RNA. Nature. 1988;334(6180):320–325. doi: 10.1038/334320a0. [DOI] [PubMed] [Google Scholar]
- Pevear DC, Tull TM, Seipel ME, Groarke JM. Activity of pleconaril against enteroviruses. Antimicrob Agents Chemother. 1999;43:2109–2115. doi: 10.1128/aac.43.9.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeffer S, Sewer A, Lagos-Quintana M, Sheridan R, Sander C, Grasser FA, van Dyk LF, Ho CK, Shuman S, Chien M, Russo JJ, Ju J, Randall G, Lindenbach BD, Rice CM, Simon V, Ho DD, Zavolan M, Tuschl T. Identification of microRNAs of the herpesvirus family. Nat Methods. 2005;2:269–276. doi: 10.1038/nmeth746. [DOI] [PubMed] [Google Scholar]
- Pfeiffer JK. Innate host barriers to viral trafficking and population diversity: lessons learned from poliovirus. Adv Virus Res. 2010;77:85–118. doi: 10.1016/B978-0-12-385034-8.00004-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer JK, Kirkegaard K. A single mutation in poliovirus RNA-dependent RNA polymerase confers resistance to mutagenic nucleotide analogs via increased fidelity. Proc Natl Acad Sci U S A. 2003;100:7289–7294. doi: 10.1073/pnas.1232294100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer JK, Kirkegaard K. Bottleneck-mediated quasispecies restriction during spread of an RNA virus from inoculation site to brain. Proc Natl Acad Sci U S A. 2006;103(14):5520–5525. doi: 10.1073/pnas.0600834103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilipenko EV, Pestova TV, Kolupaeva VG, Khitrina EV, Poperechnaya AN, Agol VI, Hellen CU. A cell cycle-dependent protein serves as a template-specific translation initiation factor. Genes Dev. 2000;14:2028–2045. [PMC free article] [PubMed] [Google Scholar]
- Rahimi P, Tabatabaie H, Gouya MM, Zahraie M, Mahmudi M, Ziaie A, Rad KS, Shahmahmudi S, Musavi T, Azad TM, Nategh R. Characterization of mutations in the VP(1) region of Sabin strain type 1 polioviruses isolated from vaccine-associated paralytic poliomyelitis cases in Iran. J Clin Virol. 2007;39(4):304–307. doi: 10.1016/j.jcv.2007.04.017. [DOI] [PubMed] [Google Scholar]
- Ramsingh AI. CVB-induced pancreatitis and alterations in gene expression. Curr Top Microbiol Immunol. 2008;323:241–58. doi: 10.1007/978-3-540-75546-3_11. [DOI] [PubMed] [Google Scholar]
- Rantakallio P, Jones P, Moring J, Von WL. Association between central nervous system infections during childhood and adult onset schizophrenia and other psychoses: a 28-year follow-up. Int J Epidemiol. 1997;26:837–843. doi: 10.1093/ije/26.4.837. [DOI] [PubMed] [Google Scholar]
- Ren R, Racaniello VR. Poliovirus spreads from muscle to the central nervous system by neural pathways. J Infect Dis. 1992;166(4):747–752. doi: 10.1093/infdis/166.4.747. [DOI] [PubMed] [Google Scholar]
- Ricour C, Delhaye S, Hato SV, Olenyik TD, Michel B, van Kuppeveld FJ, Gustin KE, Michiels T. Inhibition of mRNA export and dimerization of interferon regulatory factor 3 by Theiler’s virus leader protein. J Gen Virol. 2009;90:177–186. doi: 10.1099/vir.0.005678-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez M. Effectors of demyelination and remyelination in the CNS: implications for multiple sclerosis. Brain Pathol. 2007;17:219–229. doi: 10.1111/j.1750-3639.2007.00065.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez M, Leibowitz JL, Lampert PW. Persistent infection of oligodendrocytes in Theiler’s virus-induced encephalomyelitis. Ann Neurol. 1983;13:426–433. doi: 10.1002/ana.410130409. [DOI] [PubMed] [Google Scholar]
- Romero JR. Reverse-transcription polymerase chain reaction detection of the enteroviruses. Arch Pathol Lab Med. 1999;123:1161–1169. doi: 10.5858/1999-123-1161-RTPCRD. [DOI] [PubMed] [Google Scholar]
- Romero JR. Pediatric group B coxsackievirus infections. Curr Top Microbiol Immunol. 2008;323:223–239. doi: 10.1007/978-3-540-75546-3_10. [DOI] [PubMed] [Google Scholar]
- Rotbart HA. Diagnosis of enteroviral meningitis with the polymerase chain reaction. J Pediatr. 1990;117:85–89. doi: 10.1016/s0022-3476(05)82451-5. [DOI] [PubMed] [Google Scholar]
- Roussarie JP, Ruffie C, Brahic M. The role of myelin in Theiler’s virus persistence in the central nervous system. PLoS Pathog. 2007;3:e23. doi: 10.1371/journal.ppat.0030023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SABIN AB. Properties and behavior of orally administered attenuated poliovirus vaccine. J Am Med Assoc. 1957;164:1216–1223. doi: 10.1001/jama.1957.62980110008008. [DOI] [PubMed] [Google Scholar]
- SALK JE, KRECH U, YOUNGNER JS, BENNETT BL, LEWIS LJ, BAZELEY PL. Formaldehyde treatment and safety testing of experimental poliomyelitis vaccines. Am J Public Health Nations Health. 1954;44:563–570. doi: 10.2105/ajph.44.5.563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawyer MH. Enterovirus infections: diagnosis and treatment. Semin Pediatr Infect Dis. 2002;13:40–47. doi: 10.1053/spid.2002.29756. [DOI] [PubMed] [Google Scholar]
- Schein CH, Oezguen N, van der Heden van Noort GJ, Filippov DV, Paul A, Kumar E, Braun W. NMR solution structure of poliovirus uridylyated peptide linked to the genome (VPgpU) Peptides. 2010;31:1441–1448. doi: 10.1016/j.peptides.2010.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilthuizen C, Berenschot HW, Levin MD. Enteroviral encephalitis in a patient with a marginal zone lymphomatreated with rituximab. Neth J Med. 2010;68:221–223. [PubMed] [Google Scholar]
- Schmidtke M, Hammerschmidt E, Schuler S, Zell R, Birch-Hirschfeld E, Makarov VA, Riabova OB, Wutzler P. Susceptibility of coxsackievirus B3 laboratory strains and clinical isolates to the capsid function inhibitor pleconaril: antiviral studies with virus chimeras demonstrate the crucial role of amino acid 1092 in treatment. J Antimicrob Chemother. 2005;56:648–656. doi: 10.1093/jac/dki263. [DOI] [PubMed] [Google Scholar]
- Schmidtke M, Wutzler P, Zieger R, Riabova OB, Makarov VA. New pleconaril and [(biphenyloxy)propyl]isoxazole derivatives with substitutions in the central ring exhibit antiviral activity against pleconaril-resistant coxsackievirus B3. Antiviral Res. 2009;81:56–63. doi: 10.1016/j.antiviral.2008.09.002. [DOI] [PubMed] [Google Scholar]
- Servais S, Caers J, Warling O, Frusch N, Baron F, De PB, Beguin Y. Enteroviral meningoencephalitis as complication of Rituximab therapy in a patient treated for diffuse large B-cell lymphoma. Br J Haematol. 2010;150:379–381. doi: 10.1111/j.1365-2141.2010.08202.x. [DOI] [PubMed] [Google Scholar]
- Sharma R, Raychaudhuri S, Dasgupta A. Nuclear entry of poliovirus protease-polymerase precursor 3CD: implications for host cell transcription shut-off. Virology. 2004;320(2):195–205. doi: 10.1016/j.virol.2003.10.020. [DOI] [PubMed] [Google Scholar]
- Shih SR, Weng KF, Stollar V, Li ML. Viral protein synthesis is required for Enterovirus 71 to induce apoptosis in human glioblastoma cells. J Neurovirol. 2008;14(1):53–61. doi: 10.1080/13550280701798980. [DOI] [PubMed] [Google Scholar]
- Simonsen A, Cumming RC, Brech A, Isakson P, Schubert DR, Finley KD. Promoting basal levels of autophagy in the nervous system enhances longevity and oxidant resistance in adult Drosophila. Autophagy. 2008;4(2):176–184. doi: 10.4161/auto.5269. [DOI] [PubMed] [Google Scholar]
- Slifka MK, Pagarigan R, Mena I, Feuer R, Whitton JL. Using recombinant coxsackievirus B3 to evaluate the induction and protective efficacy of CD8+ T cells during picornavirus infection. J Virol. 2001;75:2377–2387. doi: 10.1128/JVI.75.5.2377-2387.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon T, Lewthwaite P, Perera D, Cardosa MJ, McMinn P, Ooi MH. Virology, epidemiology, pathogenesis, and control of enterovirus 71. Lancet Infect Dis. 2010;10:778–790. doi: 10.1016/S1473-3099(10)70194-8. [DOI] [PubMed] [Google Scholar]
- Solomon T, Willison H. Infectious causes of acute flaccid paralysis. Curr Opin Infect Dis. 2003;16:375–381. doi: 10.1097/00001432-200310000-00002. [DOI] [PubMed] [Google Scholar]
- Steil BP, Barton DJ. Cis-active RNA elements (CREs) and picornavirus RNA replication. Virus Res. 2009;139(2):240–252. doi: 10.1016/j.virusres.2008.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suhy DA, Giddings TH, Jr, Kirkegaard K. Remodeling the endoplasmic reticulum by poliovirus infection and by individual viral proteins: an autophagy-like origin for virus-induced vesicles. J Virol. 2000;74(19):8953–8965. doi: 10.1128/jvi.74.19.8953-8965.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suvisaari J, Mautemps N, Haukka J, Hovi T, Lonnqvist J. Childhood central nervous system viral infections and adult schizophrenia. Am J Psychiatry. 2003;160:1183–1185. doi: 10.1176/appi.ajp.160.6.1183. [DOI] [PubMed] [Google Scholar]
- Tabor-Godwin JM, Ruller CM, Bagalso N, An N, Pagarigan RR, Harkins S, Gilbert PE, Kiosses WB, Gude NA, Cornell CT, Doran KS, Sussman MA, Whitton JL, Feuer R. A novel population of myeloid cells responding to coxsackievirus infection assists in the dissemination of virus within the neonatal CNS. J Neurosci. 2010;30:8676–8691. doi: 10.1523/JNEUROSCI.1860-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam PE, Messner RP. Molecular mechanisms of coxsackievirus persistence in chronic inflammatory myopathy: viral RNA persists through formation of a double-stranded complex without associated genomic mutations or evolution. J Virol. 1999;73:10113–10121. doi: 10.1128/jvi.73.12.10113-10121.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan EL, Tan TM, Tak KCV, Poh CL. Inhibition of enterovirus 71 in virus-infected mice by RNA interference. Mol Ther. 2007;15:1931–1938. doi: 10.1038/sj.mt.6300287. [DOI] [PubMed] [Google Scholar]
- Tan EL, Wong AP, Poh CL. Development of potential antiviral strategy against coxsackievirus B4. Virus Res. 2010;150:85–92. doi: 10.1016/j.virusres.2010.02.017. [DOI] [PubMed] [Google Scholar]
- Tapparel C, Junier T, Gerlach D, Van-Belle S, Turin L, Cordey S, Muhlemann K, Regamey N, Aubert JD, Soccal PM, Eigenmann P, Zdobnov E, Kaiser L. New respiratory enterovirus and recombinant rhinoviruses among circulating picornaviruses. Emerg Infect Dis. 2009;15:719–726. doi: 10.3201/eid1505.081286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tebruegge M, Curtis N. Enterovirus infections in neonates. Semin Fetal Neonatal Med. 2009;14:222–227. doi: 10.1016/j.siny.2009.02.002. [DOI] [PubMed] [Google Scholar]
- Tung WH, Lee IT, Hsieh HL, Yang CM. EV71 induces COX-2 expression via c-Src/PDGFR/PI3K/Akt/p42/p44 MAPK/AP-1 and NF-kappaB in rat brain astrocytes. J Cell Physiol. 2010;224:376–386. doi: 10.1002/jcp.22133. [DOI] [PubMed] [Google Scholar]
- Vaishnaw AK, Gollob J, Gamba-Vitalo C, Hutabarat R, Sah D, Meyers R, de FT, Maraganore J. A status report on RNAi therapeutics. Silence. 2010;1:14. doi: 10.1186/1758-907X-1-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatraman G, Behrens M, Pyrski M, Margolis FL. Expression of Coxsackie-Adenovirus receptor (CAR) in the developing mouse olfactory system. J Neurocytol. 2005;34:295–305. doi: 10.1007/s11068-005-8359-8. [DOI] [PubMed] [Google Scholar]
- Verboon-Maciolek MA, Groenendaal F, Hahn CD, Hellmann J, van Loon AM, Boivin G, de Vries LS. Human parechovirus causes encephalitis with white matter injury in neonates. Ann Neurol. 2008a;64:266–273. doi: 10.1002/ana.21445. [DOI] [PubMed] [Google Scholar]
- Verboon-Maciolek MA, Krediet TG, Gerards LJ, de Vries LS, Groenendaal F, van Loon AM. Severe neonatal parechovirus infection and similarity with enterovirus infection. Pediatr Infect Dis J. 2008b;27:241–245. doi: 10.1097/INF.0b013e31815c1b07. [DOI] [PubMed] [Google Scholar]
- Victoria JG, Kapoor A, Li L, Blinkova O, Slikas B, Wang C, Naeem A, Zaidi S, Delwart E. Metagenomic analyses of viruses in stool samples from children with acute flaccid paralysis. J Virol. 2009;83:4642–4651. doi: 10.1128/JVI.02301-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vignuzzi M, Stone JK, Arnold JJ, Cameron CE, Andino R. Quasispecies diversity determines pathogenesis through cooperative interactions in a viral population. Nature. 2006;439:344–348. doi: 10.1038/nature04388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vignuzzi M, Wendt E, Andino R. Engineering attenuated virus vaccines by controlling replication fidelity. Nat Med. 2008;14(2):154–161. doi: 10.1038/nm1726. [DOI] [PubMed] [Google Scholar]
- Vogt DA, Andino R. An RNA element at the 5′-end of the poliovirus genome functions as a general promoter for RNA synthesis. PLoS Pathog. 2010;6(6):e1000936. doi: 10.1371/journal.ppat.1000936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpe JJ. Neonatal encephalitis and white matter injury: more than just inflammation? Ann Neurol. 2008;64:232–236. doi: 10.1002/ana.21466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuorinen T, Vainionpaa R, Vanharanta R, Hyypia T. Susceptibility of human bone marrow cells and hematopoietic cell lines to coxsackievirus B3 infection. J Virol. 1996;70(12):9018–9023. doi: 10.1128/jvi.70.12.9018-9023.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahid R, Cannon MJ, Chow M. Dendritic cells and macrophages are productively infected by poliovirus. J Virol. 2005;79(1):401–409. doi: 10.1128/JVI.79.1.401-409.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward CD, Flanegan JB. Determination of the poliovirus RNA polymerase error frequency at eight sites in the viral genome. J Virol. 1992;66(6):3784–3793. doi: 10.1128/jvi.66.6.3784-3793.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward CD, Stokes MA, Flanegan JB. Direct measurement of the poliovirus RNA polymerase error frequency in vitro. J Virol. 1988;62(2):558–562. doi: 10.1128/jvi.62.2.558-562.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster AD. Pleconaril--an advance in the treatment of enteroviral infection in immuno-compromised patients. J Clin Virol. 2005;32:1–6. doi: 10.1016/j.jcv.2004.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng KF, Li ML, Hung CT, Shih SR. Enterovirus 71 3C protease cleaves a novel target CstF-64 and inhibits cellular polyadenylation. PLoS Pathog. 2009;5(9):e1000593. doi: 10.1371/journal.ppat.1000593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werk D, Schubert S, Lindig V, Grunert HP, Zeichhardt H, Erdmann VA, Kurreck J. Developing an effective RNA interference strategy against a plus-strand RNA virus: silencing of coxsackievirus B3 and its cognate coxsackievirus-adenovirus receptor. Biol Chem. 2005;386:857–863. doi: 10.1515/BC.2005.100. [DOI] [PubMed] [Google Scholar]
- Whitton JL, Cornell CT, Feuer R. Host and virus determinants of picornavirus pathogenesis and tropism. Nat Rev Microbiol. 2005;3(10):765–776. doi: 10.1038/nrmicro1284. [DOI] [PubMed] [Google Scholar]
- Wilkins C, Gale M., Jr Recognition of viruses by cytoplasmic sensors. Curr Opin Immunol. 2010;22:41–47. doi: 10.1016/j.coi.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wimmer E, Mueller S, Tumpey TM, Taubenberger JK. Synthetic viruses: a new opportunity to understand and prevent viral disease. Nat Biotechnol. 2009;27:1163–1172. doi: 10.1038/nbt.1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong J, Zhang J, Si X, Gao G, Mao I, McManus BM, Luo H. Autophagosome supports coxsackievirus B3 replication in host cells. J Virol. 2008;82(18):9143–9153. doi: 10.1128/JVI.00641-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodall CJ, Graham DI. Evidence for neuronal localisation of enteroviral sequences in motor neurone disease/amyotrophic lateral sclerosis by in situ hybridization. Eur J Histochem. 2004;48:129–134. doi: 10.4081/877. [DOI] [PubMed] [Google Scholar]
- Woodall CJ, Riding MH, Graham DI, Clements GB. Sequences specific for enterovirus detected in spinal cord from patients with motor neurone disease. BMJ. 1994;308:1541–1543. doi: 10.1136/bmj.308.6943.1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CN, Lin YC, Fann C, Liao NS, Shih SR, Ho MS. Protection against lethal enterovirus 71 infection in newborn mice by passive immunization with subunit VP1 vaccines and inactivated virus. Vaccine. 2001;20:895–904. doi: 10.1016/s0264-410x(01)00385-1. [DOI] [PubMed] [Google Scholar]
- Wu KX, Ng MM, Chu JJ. Developments towards antiviral therapies against enterovirus 71. Drug Discov Today. 2010;15:1041–1051. doi: 10.1016/j.drudis.2010.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie J, Jiao Y, Qiu Z, Li Q, Li T. Significant elevation of B cells at the acute stage in enterovirus 71-infected children with central nervous system involvement. Scand J Infect Dis. 2010 doi: 10.3109/00365548.2010.498018. [DOI] [PubMed] [Google Scholar]
- Yamayoshi S, Yamashita Y, Li J, Hanagata N, Minowa T, Takemura T, Koike S. Scavenger receptor B2 is a cellular receptor for enterovirus 71. Nat Med. 2009;15(7):798–801. doi: 10.1038/nm.1992. [DOI] [PubMed] [Google Scholar]
- Yang B, Chuang H, Yang KD. Sialylated glycans as receptor and inhibitor of enterovirus 71 infection to DLD-1 intestinal cells. Virol J. 2009;6:141. doi: 10.1186/1743-422X-6-141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WX, Terasaki T, Shiroki K, Ohka S, Aoki J, Tanabe S, Nomura T, Terada E, Sugiyama Y, Nomoto A. Efficient delivery of circulating poliovirus to the central nervous system independently of poliovirus receptor. Virology. 1997;229(2):421–428. doi: 10.1006/viro.1997.8450. [DOI] [PubMed] [Google Scholar]
- Yoon SY, Ha YE, Choi JE, Ahn J, Lee H, Kweon HS, Lee JY, Kim DH. Coxsackievirus B4 uses autophagy for replication after calpain activation in rat primary neurons. J Virol. 2008;82(23):11976–11978. doi: 10.1128/JVI.01028-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshikawa T, Iwasaki T, Ida-Hosonuma M, Yoneyama M, Fujita T, Horie H, Miyazawa M, Abe S, Simizu B, Koike S. Role of the alpha/beta interferon response in the acquisition of susceptibility to poliovirus by kidney cells in culture. J Virol. 2006;80:4313–4325. doi: 10.1128/JVI.80.9.4313-4325.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J, Cheung PK, Zhang HM, Chau D, Yang D. Inhibition of coxsackievirus B3 replication by small interfering RNAs requires perfect sequence match in the central region of the viral positive strand. J Virol. 2005;79:2151–2159. doi: 10.1128/JVI.79.4.2151-2159.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]