

Abstract
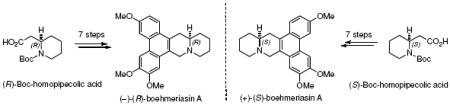
Both enantiomers of boehmeriasin A were synthesized in seven steps each, using a chiral pool approach. Key steps in the synthesis are a one-flask, two-step protocol to generate the quinolizine core and a C-H functionalization reaction between tetrahydroquinolizinones and an aryltrifluoroborate. The natural product (R)-boehmeriasin A demonstrated potent cytotoxicity against several cancer cell lines, whereas the unnatural (+)-(S)-isomer was significantly less potent.
Keywords: natural product synthesis, phenanthroquinolizidine, boehmeriasin A, cytotoxicity, drug resistance
Boehmeriasin A and B were recently isolated from the aqueous ethanolic extract of Boehmeria siamensis via bioassay-guided fractionation (Figure 1).1 The alkaloids were evaluated against a panel of cancer cell lines that included leukemia and cancers of the lung, colon, breast, prostate, and kidney. Boehmeriasin A was found to be more potent than paclitaxel and boehmeriasin B in most cell lines evaluated with GI50 values ranging from 0.80 to 265 nM. In addition, boehmeriasin A potently inhibits the proliferation of the breast cancer cell line MDA-MB-231 through G1 cell cycle arrest and differentiation induction by altering the expression levels of several genes involved with cell proliferation, cell cycle regulation, and apoptosis.2 The synthesis of this natural product was first reported in racemic form leaving the absolute stereochemistry of the natural product initially unknown.3
Figure 1. Structures of boehmeriasin A and B.

While our work on the enantiospecific synthesis of (R)- and (S)-boehmeriasin A was ongoing, an asymmetic synthesis of (R)-boehmeriasin A was reported.4 The synthesis of the natural product was achieved in 13 steps and involved the SAMP-hydrazone method to introduce asymmetry, ring-closing metathesis to form the piperidine ring system, an aldol reaction to prepare the quinolizidine core structure, and a radical reaction to furnish the phenanthrene moiety.
We had become interested in this natural product because of the promising anticancer activity reported and our interest in this class of compounds.5 We now report the total synthesis of both boehmeriasin A enantiomers and data concerning their in vitro anticancer activity. In a retrosynthetic sense (Scheme 1), boehmeriasin A (1) was to be derived from an intramolecular bi-aryl coupling of intermediate 2 which itself was to be accessed from a palladium-mediated cross-coupling reaction. The coupling partner 3 for the cross-coupling was to be obtained by conversion of cyclic enaminone 4 to the triflate 3. This advanced intermediate 4 was envisioned to arise from a novel, palladium(II)-catalyzed C-H functionalization utilizing organotrifluoroborates.6 The coupling partner for this transformation would be obtained from Weinreb amide 6 through methods developed in our laboratories.7
Scheme 1. Retrosynthesis for boehmeriasin A.

The commercially available acids 7 were converted into the corresponding Weinreb amides 6 under standard conditions and subsequently treated with ethynylmagnesium bromide to afford the corresponding ynones 8 in excellent yields (Scheme 2).8,9 These intermediates were subjected to a one-flask, two-step protocol for the cyclization of Boc-ynones to enaminones to afford the desired enaminones 5 in good yields.7
Scheme 2. Synthesis of α-arylated enaminones 4.

With rapid access to the desired enaminones established, our recently reported palladium(II)-catalyzed C-H functionalization using enaminones and potassium organotrifluoroborates was employed.6 This method represents an efficient means of accessing these products, as previous methods required an initial prefunctionalization of the enaminone to the appropriate α-halogenated derivative followed by a Suzuki coupling.10 This protocol eliminates this requirement and thus allows for a more streamlined approach to access these compounds. Utilizing potassium 3,4-dimethoxyphenyltrifluoroborate (9), prepared according to a known procedure,11 in the Pd(II)-catalyzed reactions with enaminones 5 furnished the desired arylated products 4 in good yields (Scheme 2).
With a route to the α-arylated intermediates established, the final synthetic sequence was undertaken as shown in Scheme 3. Enaminones 4 were treated with L-Selectride and the resultant enolates were trapped with Comins' reagent (N-(5-chloropyridin-2-yl)-1,1,1-trifluoro-N-(trifluoromethylsulfonyl)methanesulfonamide) to arrive at the desired triflates 3 in good yields.12 A Negishi cross-coupling was then employed to furnish the desired intermediates 2 in near quantitative yields.13 The synthesis was completed utilizing an oxidative bi-aryl ring closure mediated by VOF3 to afford (R)- and (S)-boehmeriasin A in good yields.14 The final products were crystallized from CH2Cl2/MeOH and the crystal structure for the (R)-antipode was determined.15 It is important to note that little racemization occurred throughout the course of the synthesis to afford (R)- and (S)-boehmeriasin A in 97.5:2.5 er and 98:2 er as determined by chiral HPLC.16 In addition, the specific rotation of the (R)-enantiomer gave a matching sign and magnitude with those found in the literature (lit. −80, MeOH, c = 0.10; obs. −86, MeOH, c = 0.10).1,4
Scheme 3. Completion of the synthesis of (R)- and (S)-1.

(R)- and (S)-Boehmeriasin A and synthetic intermediates were subjected to in vitro cytotoxicity assays to confirm the reported biological activity and establish an initial SAR for boehmeriasin A in breast (MCF7), drug-resistant ovarian (NCI-ADR-RES), and colon (COLO-205) cancer cell lines (Table 1). Arylated enaminone 3, and seco-boehmeriasin A (2) were devoid of any cytotoxic activity in the cell lines evaluated, which indicates that a full phenanthrene ring system is required for potent cytotoxic activity. This is in accord with other studies of this class of natural products and their analogues.17 Furthermore, (−)-boehmeriasin A ((−)-1·HCl) was more potent than its antipode, (+)-1·HCl, in all of the cell lines evaluated, indicating that the (R)-configuration is essential for potent cytotoxic activity. Most significantly, the natural product showed activity in the drug resistant cancer cell line, NCI-ADR-RES, where paclitaxel is inactive.
Table 1.
Cytotoxicity evaluation of (−)-(R)- and (+)-(S)-boehmeriasin A in comparison to paclitaxel.
In summary, the total syntheses of (−)-(R)- and (+)-(S)-boehmeriasin A were accomplished in seven steps from commercially available material with an overall yield of 33% and the absolute stereochemistry of the natural product was verified to be of the (R)-configuration. The synthesis showcases the utility of the enaminone chemistry and the palladium(II)-mediated C-H functionalization developed in our laboratories. When evaluated for cytotoxic activity, (−)-(R)-boehmeriasin A demonstrated potent cytotoxicity in several cancer cell lines including a drug-resistant cancer cell line where paclitaxel is inactive. The (S)-enantiomer was significantly less potent. (R)-Boehmeriasin A will serve as a lead compound for further development and studies in this regard are currently underway in our laboratories.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health Grants GM081267 and the University of Minnesota through the Vince and McKnight Endowed Chairs. We also wish to thank Dr. Victor Young for X-ray analysis, Dr. Defeng Tian for conducting the cytotoxicity assays, and Bryant Gay for the synthesis of the methyl ester of racemic 7 and (R)-7 and their analysis by chiral HPLC.
Footnotes
Supporting Information Available: Full experimental procedures, compound characterization, and complete crystallographic data for boehmeriasin A. This information is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Luo Y, Liu Y, Luo D, Gao X, Li B, Zhang G. Cytotoxic Alkaloids from Boehmeria siamensis. Planta Medica. 2003;69:842–845. doi: 10.1055/s-2003-43215. [DOI] [PubMed] [Google Scholar]
- 2.(a) Yan J, Luo D, Luo Y, Gao X, Zhang G. Induction of G1 Arrest and Differentiation in MDA-MB-231 Breast Cancer Cell by Boehmeriasin A, a Novel Compound from Plant. Int J Gynecol Cancer. 2006;16:165–170. doi: 10.1111/j.1525-1438.2006.00291.x. [DOI] [PubMed] [Google Scholar]; (b) Wei H, Yan J, Liu J, Luo D, Zhang J, Gao X. Genes Involved in the Anti-Cancer Effect of a Potent New Compound Boehmeriasin A on Breast Cancer Cell. J Med Plants Res. 2009;3:35–44. [Google Scholar]
- 3.(a) Cui M, Wang Q. Total Synthesis of Phenanthro-Quinolizidine Alkaloids: (±)-Cryptopleurine, (±)-Boehmeriasin A, (±)-Boehmeriasin B and (±)-Hydroxycryptopleurine. Eur J Org Chem. 2009:5445–5451. [Google Scholar]; (b) Wang Z, Wang Q. Highly Efficient Synthesis of Phenanthroquinolizidine Alkaloids via Parham-Type Cycliacylation. Tetrahedron Lett. 2010;51:1377–1379. [Google Scholar]
- 4.Dumoulin D, Lebrun S, Couture A, Deniau E, Grandclaudon P. First Asymmetric Synthesis of Boehmeriasin A. Eur J Org Chem. 2010:1943–1950. [Google Scholar]
- 5.(a) Kimball FS, Tunoori AR, Victory SF, Dutta D, White JM, Himes RH, Georg GI. Synthesis, in vitro and in vivo Cytotoxicity of 6,7-Diaryl-2,3,8,8a-tetrahydroindolizin-5(1H)-ones. Bioorg Med Chem Lett. 2007;17:4703–4707. doi: 10.1016/j.bmcl.2007.05.103. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Niphakis MJ, Georg GI. Synthesis of Tylocrebrine and Related Phenanthroindolizidines by VOF3-Mediated Oxidative Aryl-Alkene Coupling. Org Lett. 2011;13:196–199. doi: 10.1021/ol1023954. [DOI] [PubMed] [Google Scholar]
- 6.Ge H, Niphakis MJ, Georg GI. Palladium(II)-Catalyzed Direct Arylation of Enaminones Using Organotrifluoroborates. J Am Chem Soc. 2008;130:3708–3709. doi: 10.1021/ja710221c. [DOI] [PubMed] [Google Scholar]
- 7.(a) Turunen BJ, Georg GI. Amino Acid-Derived Enaminones: A Study in Ring Formation Providing Valuable Asymmetric Synthons. J Am Chem Soc. 2006;128:8702–8703. doi: 10.1021/ja0609046. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Niphakis MJ, Turunen BJ, Georg GI. Synthesis of 6- and 7-Membered Cyclic Enaminones: Scope and Mechanism. J Org Chem. 2010;75:6793–6805. doi: 10.1021/jo100907u. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Niphakis MJ, Georg GI. Total Syntheses of Arylindolizidine Alkaloids (+)-Ipalbidine and (+)-Antofine. J Org Chem. 2010;75:6019–6022. doi: 10.1021/jo101051w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Both (R)- and (S)-7 are commercially available from PolyPeptide Group.
- 9.Pirc S, Bevk D, Golobič A, Stanovnik B, Svete J. Transformation of Amino Acids into Nonracemic 1-(Heteroaryl)ethanamines by the Enamino Ketone Methodology. Helv Chim Acta. 2006;89:30–44. [Google Scholar]
- 10.Wang X, Turunen BJ, Leighty MW, Georg GI. Microwave-assisted Suzuki-Miyaura Couplings on a-Iodoenaminones. Tetrahedron Lett. 2007;48:8811–8814. doi: 10.1016/j.tetlet.2007.10.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Molander GA, Biolatto B. Efficient Ligandless Palladium-Catalyzed Suzuki Reactions of Potassium Aryltrifluoroborates. Org Lett. 2002;4:1867–1870. doi: 10.1021/ol025845p. [DOI] [PubMed] [Google Scholar]
- 12.(a) Comins DL, Dehghani A. Pyridine-Derived Triflating Reagents: An Improved Preparation of Vinyl Triflates from Metallo Enolates. Tetrahedron Lett. 1992;33:6299–6302. [Google Scholar]; (b) Fortunato JM, Ganem B. Lithium and Potassium Trialkylborohydrides. Reagents for Direct Reduction of α,β-Unsaturated Carbonyl Compounds to Synthetically Versatile Enolate Anions. J Org Chem. 1976;41:2194–2200. [Google Scholar]
- 13.Comins DL, Chen X, Morgan LA. Enantiopure N-Acyldihydropyridones as Synthetic Intermediates: Asymmetric Synthesis of (−)-Septicine and (−)-Tylophorine. J Org Chem. 1997;62:7435–7438. doi: 10.1021/jo9711495. [DOI] [PubMed] [Google Scholar]
- 14.Bremmer ML, Khatri NA, Weinreb SM. Quinolizidine Alkaloid Synthesis via the Intramolecular Imino Diels-Alder Reaction. epi-Lupinine and Cryptopleurine. J Org Chem. 1983;48:3661–3666. [Google Scholar]
- 15.See Supporting Information.
- 16.Chiralpak AD-H (250 mm × 4.6 mm), 35-80% isopropanol in heptane, 45 min, 0.5 mL/min, 254 nm, (S)-isomer, Rt = 24.2 min, (R)-isomer Rt = 32. 6 min;; Fürstner A, Kennedy JWJ. Total Syntheses of the Tylophora Alkaloids Cryptopleurine, (−)-Antofine, (−)-Tylophorine, and (−)-Ficuseptine C. Chem–Eur J. 2006;12:7398–7410. doi: 10.1002/chem.200600592. [DOI] [PubMed] [Google Scholar]; (c) Racemic 7 was converted to the corresponding methyl ester and the enantiomers were separated via chiral HPLC using a Chiracel OJ column (0.46 × 25 cm) with an isocratic gradient of 98:2 hexanes/i-PrOH and a flow rate of 1 mL/min, 254 nm; R-isomer tr 3.3 min and S-isomer tr 7.5 min. The methyl ester of (R)-7 was also prepared and subjected to the HPLC conditions noted above; tr = 3.5 min. The (S)-7 enantiomer was not observed under these conditions.
- 17.(a) Stærk D, Lykkeberg AK, Christensen J, Budnik BA, Abe F, Jaroszewski JW. In vitro Cytotoxic Activity of Phenanthroindolizidine Alkaloids from Cynanchum vincetoxicum and Tylophora tanakae Against Drug-sensitive and Multidrug-resistant Cancer Cells. J Nat Prod. 2002;65:1299–1302. doi: 10.1021/np0106384. [DOI] [PubMed] [Google Scholar]; (b) Wei L, Brossi A, Kendall R, Bastow KF, Morris-Natschke SL, Shi Q, Lee KH. Antitumor Agents 251: Synthesis, Cytotoxic Evaluation, and Structure-Activity Relationship Studies of Phenanthrene-Based Tylophorine Derivatives (PBTs) as a New Class of Antitumor Agents. Bioorg Med Chem. 2006;14:6560–6569. doi: 10.1016/j.bmc.2006.06.009. [DOI] [PubMed] [Google Scholar]; (c) Chemler SR. Phenanthroindolizidines and Phenanthroquinolizidines: Promising Alkaloids for Anti-Cancer Therapy. Curr Bioact Compd. 2009;5:2–19. doi: 10.2174/157340709787580928. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.